 Camila Alvarez-Silva1†
Camila Alvarez-Silva1† Robert Schierwagen2,3†
Robert Schierwagen2,3† Alessandra Pohlmann3†
Alessandra Pohlmann3† Fernando Magdaleno3
Fernando Magdaleno3 Frank E. Uschner2,3Patrick Ryan3Maria J. G. T. Vehreschild4Joan Claria5
Frank E. Uschner2,3Patrick Ryan3Maria J. G. T. Vehreschild4Joan Claria5 Eicke Latz6,7,8,9
Eicke Latz6,7,8,9 Benjamin Lelouvier10
Benjamin Lelouvier10 Manimozhiyan Arumugam1,11*
Manimozhiyan Arumugam1,11* Jonel Trebicka2,3,5,11,12*
Jonel Trebicka2,3,5,11,12*- 1Novo Nordisk Foundation Center for Basic Metabolic Research, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 2Department of Internal Medicine I, Goethe University Clinic Frankfurt, Frankfurt, Germany
- 3Department of Internal Medicine I, University of Bonn, Bonn, Germany
- 4Medical Clinic II, University Center for Infectious Diseases, University Hospital Frankfurt, Frankfurt, Germany
- 5European Foundation for the Study of Chronic Liver Failure, Barcelona, Spain
- 6Institute of Innate Immunity, University of Bonn, Bonn, Germany
- 7Division of Infectious Diseases and Immunology, University of Massachusetts Medical School, Worcester, MA, United States
- 8German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany
- 9Centre for Inflammation Research, Norwegian University of Science and Technology, Trondheim, Norway
- 10Vaiomer, Labège, France
- 11Department of Gastroenterology and Hepatology, Odense University Hospital, Odense, Denmark
- 12Institute for Bioengineering of Catalonia, Barcelona, Spain
Background: Acquired dysfunctional immunity in cirrhosis predisposes patients to frequent bacterial infections, especially spontaneous bacterial peritonitis (SBP), leading to systemic inflammation that is associated with poor outcome. But systemic inflammation can also be found in the absence of a confirmed infection. Detection of bacterial DNA has been investigated as a marker of SBP and as a predictor of prognosis. Data is, however, contradictory. Here we investigated whether levels of IL-6 and IL-8 putatively produced by myeloid cells in ascites are associated with systemic inflammation and whether inflammation depends on the presence of specific bacterial DNA.
Methods and Materials: We enrolled 33 patients with decompensated liver cirrhosis from whom we collected paired samples of blood and ascites. IL-6 and IL-8 were measured in serum samples of all patients using ELISA. In a subset of 10 representative patients, bacterial DNA was extracted from ascites and whole blood, followed by 16S rRNA gene amplicon sequencing.
Results: There were significantly higher levels of IL-6 in ascites fluid compared to blood samples in all patients. Interestingly, IL-6 levels in blood correlated tightly with disease severity and surrogates of systemic inflammation, while IL-6 levels in ascites did not. Moreover, patients with higher blood CRP levels showed greater SBP prevalence compared to patients with lower levels, despite similar positive culture results. Bacterial richness was also significantly higher in ascites compared to the corresponding patient blood. We identified differences in microbial composition and diversity between ascites and blood, but no tight relationship with surrogates of systemic inflammation could be observed.
Discussion: In decompensated cirrhosis, markers of systemic inflammation and microbiota composition seem to be dysregulated in ascites and blood. While a relationship between systemic inflammation and microbiota composition seems to exist in blood, this is not the case for ascites in our hands. These data may suggest compartmentalization of the immune response and interaction of the latter with the microbiota especially in the blood compartment.
Introduction
Acquired dysfunctional immunity in cirrhosis predisposes patients to frequent bacterial infections contributing to disease progression and may lead to the development of acute-on-chronic liver failure (ACLF) (1). Especially, spontaneous bacterial peritonitis (SBP) is one of the most frequent infections in cirrhosis and therefore a trigger for ACLF (2). Interestingly, SBP is defined by the number of granulocytes in ascites and not by cultural detection of bacteria (3). This is because many patients with SBP show negative bacterial cultivation from ascites, while in many other patients, viable bacteria can be cultivated in the absence of peritonitis, so-called bacterascites (3). Lack of bacterial cultivation in the presence of SBP can be explained by the concept of dormant microbiota, meaning that bacteria can reside inside circulating and resident immune cells but cannot be cultured and hence detected (4).
The source of infection, especially in the case of SBP, is suspected to be bacterial translocation from the gut (2), which is facilitated by impaired intestinal defense and increased intestinal permeability, processes which are correlated with the stage of cirrhosis (5). Not only viable bacteria (1, 6), but also bacterial components broadly termed pathogen-associated molecular patterns (PAMPs) may induce an immune response and aggravate systemic inflammation (2). As a consequence, the presence of circulating bacterial DNA reflects bacterial translocation in cirrhosis. However, the relationship of bacterial DNA with systemic inflammation is still controversially discussed (7, 8). PAMP-associated immune response requires recognition by innate immune cells and is reflected by synthesis and secretion of cytokines. Especially levels of the cytokines IL-6 and IL-8 are closely related to ACLF and clinical outcome (9). At the same time, the antibacterial capacity of myeloid cells and myelopoiesis is impaired during decompensation and ACLF (10, 11). Interestingly, these effects are not only induced by PAMPs (deriving from pathogens), but in general by bacterial-derived molecules, so-called microbe-associated molecular patterns (MAMPs).
Recent data from our group suggest that specific circulating bacteria or MAMPs in blood are associated with myeloid cell dysregulation and systemic inflammation (12). Therefore, a specific microbial DNA may be more crucial than just detecting any bacterial DNA in circulation. In ascites, the detection of bacterial DNA in ascites does not seem to correlate with SBP or clinical signs of infection (13). Therefore, the question remains, whether specific bacteria in ascites lead to myeloid cell activation with subsequent cytokine release and a systemic inflammatory response. To address this question, we investigated whether the representative cytokine (IL-6, IL-8) levels produced by myeloid cells in ascites reflect systemic inflammation. Moreover, we compared the levels of cytokines in paired ascites and blood samples, together with the general bacterial composition and specific species abundances of the microbiota estimated by 16S ribosomal RNA gene sequencing. Our study focused not only on common pathogens but also on the role of MAMPs in activating myeloid cells of the innate immune response. We hypothesize that DNA from specific bacterial species, but not any bacterial DNA, induces an inflammatory response in cirrhotic patients.
Materials and Methods
Patient Recruitment and Sample Collection
Ascites and blood were collected in 33 cirrhotic patients scheduled for paracentesis. Paracentesis was performed by international standards (14). The study was performed in accordance with the declaration of Helsinki and approved by the local ethics committee at the University of Bonn (Nr. 121/14). Patients gave their written informed consent.
Microbiological Analysis
Microbiological culture of blood and ascites was performed at the clinical microbiology department at the University of Bonn in accordance with Microbiology Procedures Quality Standards of the German Society for Hygiene and Microbiology (15, 16). Ten milliliters of peripheral whole blood or ascitic fluid were transferred into aerobic, anaerobic and fungal blood culture bottles and incubated for up to 5 days using the Bactex FX blood culture system as described previously (17). SBP was diagnosed based on a leucocyte count in ascites >250/μl (3).
Enzyme-Linked Immunosorbent Assay
Ascites and peripheral blood samples (serum, EDTA-plasma and whole-blood) were collected during paracentesis and stored immediately at −80°C until further use. Enzyme-linked immunosorbent assay for cytokines IL-6 and IL-8 (DY206 and DY208, respectively; R&D Systems Inc., Minneapolis, MN, USA) were performed according to the manufacturer's protocols. All samples were measured in duplicates.
16S Ribosomal RNA Gene Sequencing
For extraction of total DNA, 100 μl of whole blood or ascitic fluid were used. Samples were mechanically lysed two times for 30 s at 20 Hz in a bead beater (TissueLyser, Qiagen, Hilden, Germany) with 0.1 mm glass beads (MoBio, CA, USA). DNA extraction was performed using the NucleoSpin Blood Kit (Macherey-Nagel, Düren, Germany). Quality and quantity of extracted nucleic acids were controlled by gel electrophoresis. 16S rRNA gene V3-V4 hypervariable regions were amplified and quantified by qPCR, and sequenced using MiSeq technology (Illumina, San Diego, CA, USA) as described previously producing 2 × 300 bp paired-end reads (18, 19).
Bioinformatics and Statistical Analysis
Paired-ends reads were processed in DADA2 version 1.6.0 (20) using the following parameters: “trimLeft = 10, truncLen = (260;230), maxN = 0, maxEE = (2,5), minOverlap = 20, maxMismatch = 1, pool = TRUE.” The resulting output was an amplicon sequence variant (ASV) table. The ASVs were mapped against SILVA database (21) release 128. Data analysis was performed in R version 3.5, using phyloseq package (22) version 1.24.2. Genus level relative abundances were generated by combining ASVs annotated with the same genus using phyloseq package. The alpha-diversity measures were calculated based on rarefied data at 6,000 reads/sample. Significant differences in alpha-diversity between ascites and blood samples as well as between samples with different inflammation status were determined using t-test.
Beta-diversity was calculated using unweighted and weighted UniFrac measures. Effects of different factors on beta-diversity were determined using Adonis Permutational Multivariate Analysis of Variance (PERMANOVA) test from the vegan package (23) version 2.5.2. For differential abundance analysis, we chose a t-test based on the evaluation using DAtest package (24) version 2.7.11, which showed that, compared with different statistical tests, t-test has the highest area under the curve (AUC = 0.608) while keeping the false discovery rate (FDR) under 0.05.
Statistical analysis of patient characteristics and clinical measures was performed using SPSS version 24 (IBM, Armonk, NY) and Mann–Whitney test for metric variables and Chi-square test for nominal variables. Receiver operator characteristic curve analysis was used to define a cut-off for CRP and divide the cohort into one group with low CRP profile and one group with high CRP profile. P-values were adjusted for multiple testing using Benjamini–Hochberg FDR procedure. An FDR-adjusted P-value below 0.05 was considered statistically significant.
Results
General Description of the Cohort
General characteristics of the patients are summarized in Table 1. The cohort consists of patients with decompensated liver cirrhosis as reflected by the model for end-stage liver disease (MELD) and Child-Pugh (CHILD) scores, which assess severity of liver disease, prognosis and mortality. The median MELD score of 15 represents a value at which patients would benefit from liver transplantation. The median CHILD score of 10 is associated with a 1-year survival below 50%. Seventy-two percent of the patients (24/33) showed ACLF at baseline.
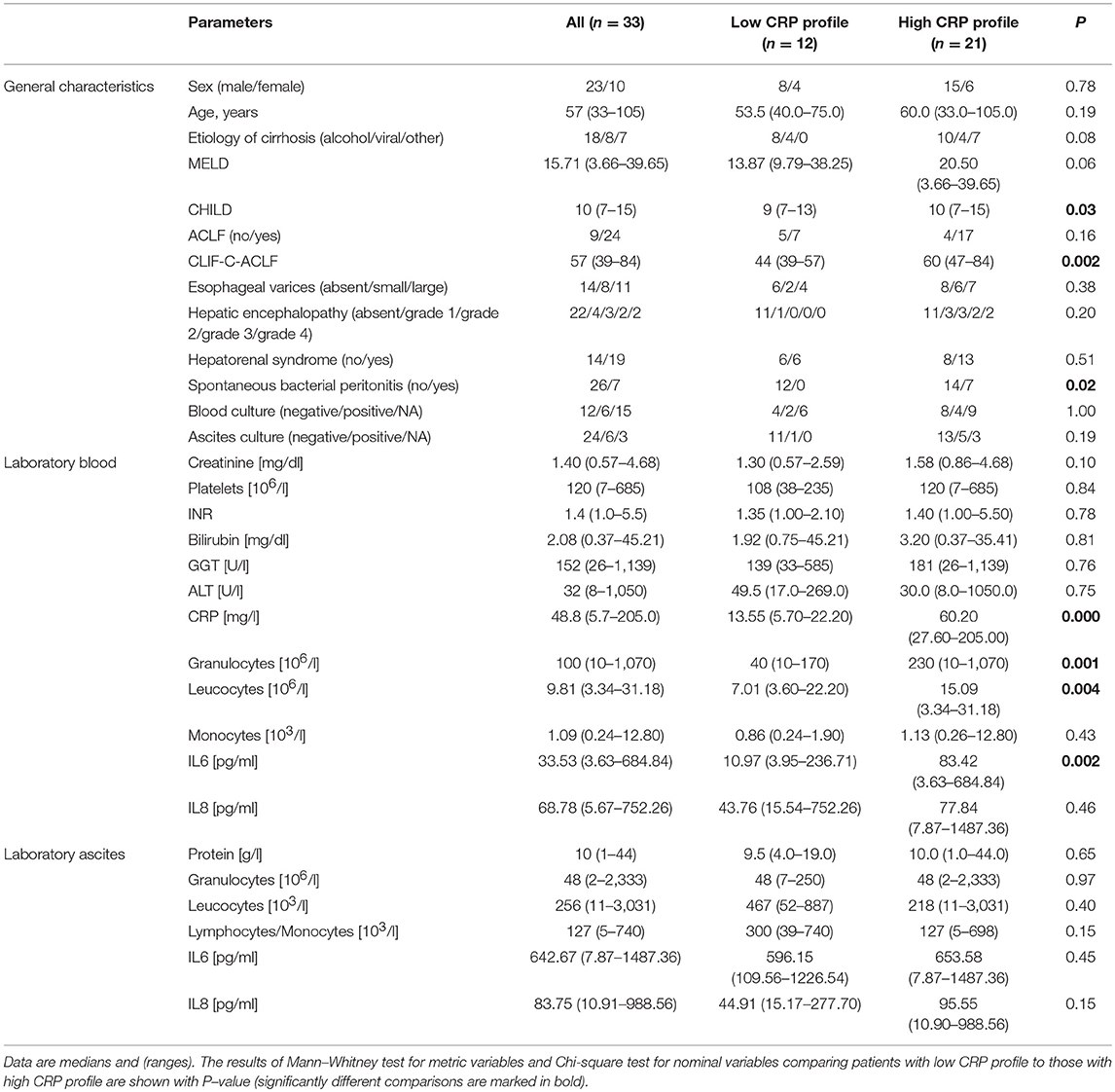
Table 1. Patient characteristics.
The median C-reactive protein (CRP) level of 48.8 mg/l in the blood were compatible with bacterial infections (upper limit of normal 5 mg/l), together with findings of SBP in 21% of the patients (Table 1). CRP performed well to detect SBP using receiver operator characteristic curve analysis (AUC = 0.901, P ≤ 0.001). In blood cultures, we detected members of the bacterial genera Enterococcus, Escherichia, Sporosarcina, and Staphylococcus in five patients. Additionally, we detected Candida spp. in blood culture of one patient. In ascites, we detected the genera Enterococcus, Serratia, and Staphylococcus in five patients and Candida spp. in one patient. None of the patients presented with cultural evidence of bacteria in both, blood and ascites.
There was also evidence of systemic inflammation with elevated levels of cytokines IL-6 (and IL-8, although not statistically significant), which are mainly produced by myeloid cells. The median levels assessed by ELISA assays were 33.53 and 68.78 pg/ml, respectively (Table 1). Reported mean levels in healthy individuals are 1.46 and 12.9 pg/ml, respectively, assessed by the same technique (25, 26). Ascites fluid showed higher median levels of both cytokines, with an almost 20-fold increase of median IL-6 levels in ascites compared to blood from the same patient.
Differences in Patients With Systemic Inflammation
In order to decipher the role of systemic inflammation, we divided the cohort into two groups according to their CRP level. A cut-off of 25 mg/l (sensitivity = 1.0, specificity = 0.54 for detection of SBP) was used to divide the cohort into 12 patients with a low CRP profile and 21 patients with a high CRP profile. Patients with the high CRP profile showed higher MELD, CHILD, and Chronic Liver Failure Consortium (CLIF-C)-ACLF scores (Table 1; P = 0.06, P = 0.03, and P = 0.002, respectively). SBP was detected in 50% of the patients (7 out of 14) with the high CRP profile, but in none of 12 patients with the low CRP profile (P = 0.02). Therefore, the positive predictive value, the probability for patients with high CRP profile to have SBP, is 35%. In contrast, the negative predictive value, the chance for patients with low CRP profile to not have SBP, is 100%. A high negative predictive value in this regards is extremely important since CRP level may be elevated also in absence of infection. Furthermore, patients with the high CRP profile showed a significant increase in blood granulocyte counts (P = 0.001), leucocyte counts (P = 0.004), and IL-6 levels (P = 0.002). However, there was no difference in ascitic cytokine levels between these two groups (Table 1). Similarly, IL-6 levels in blood strongly correlate with MELD, CHILD score, and CLIF-C-ACLF score and surrogate markers of systemic inflammation (leucocytes and CRP). However, blood IL-8 levels only slightly reflected disease stage and did not correlate significantly with markers of inflammation (Figure 1A). Even though median cytokine levels were higher in ascites than in blood (Figure 1B), ascitic cytokine levels reflected neither disease severity nor systemic inflammation (Figures 1A,C). Therefore, ascites cytokine levels did not seem to be affected by systemic inflammation or by the presence of SBP, which was diagnosed in one third of the patients with the high CRP profile (Figure 1C).
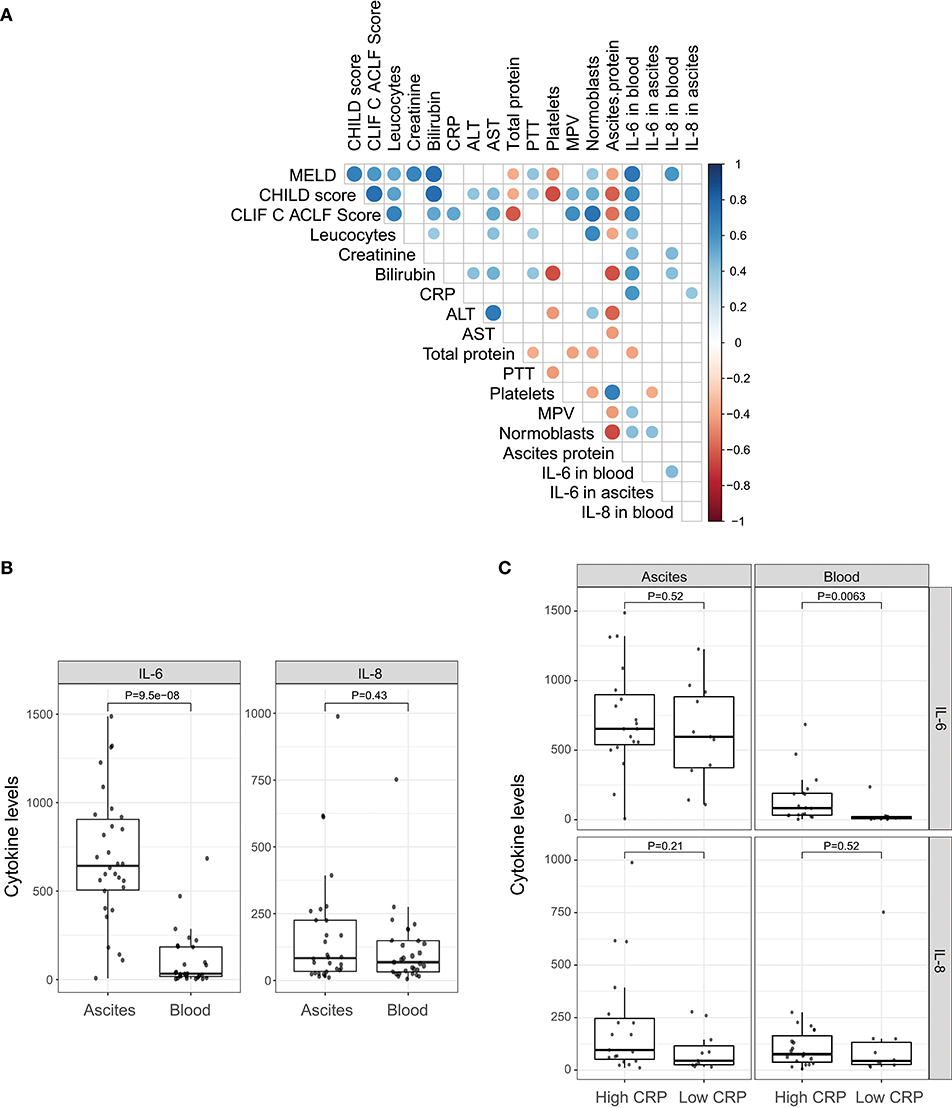
Figure 1. Pro-inflammatory cytokines. (A) Correlation of clinical markers for disease progression and systemic inflammation with cytokines IL-6 and IL-8 in blood and ascites. (B) Comparison between cytokines levels in ascites and peripheral blood. (C) Comparison between cytokines levels in ascites and peripheral blood under different systemic inflammation status (low CRP profile vs. high CRP profile).
The rate of positive blood cultures was identical in patients with the low and high CRP profile. There were three positive blood and three positive ascites cultures in patients with a high CRP against two positive blood and one positive ascites cultures in the patients with lower CRP values.
Characterizing the Microbiome in Ascites and Blood
We selected a subset of 10 representative patients (accounting for etiology and inflammation markers in ascites) from the whole cohort and performed 16S rRNA gene sequencing of ascites and blood to identify differences in microbial composition and diversity between ascites and blood. These 10 patients were also divided into two groups based on CRP levels (n = 4 for high CRP profile and n = 6 for low CRP profile, see Table 2). Their disease profiles with regard to stage of liver cirrhosis and inflammatory markers in blood were comparable to those of the respective subgroups and also of the whole cohort.
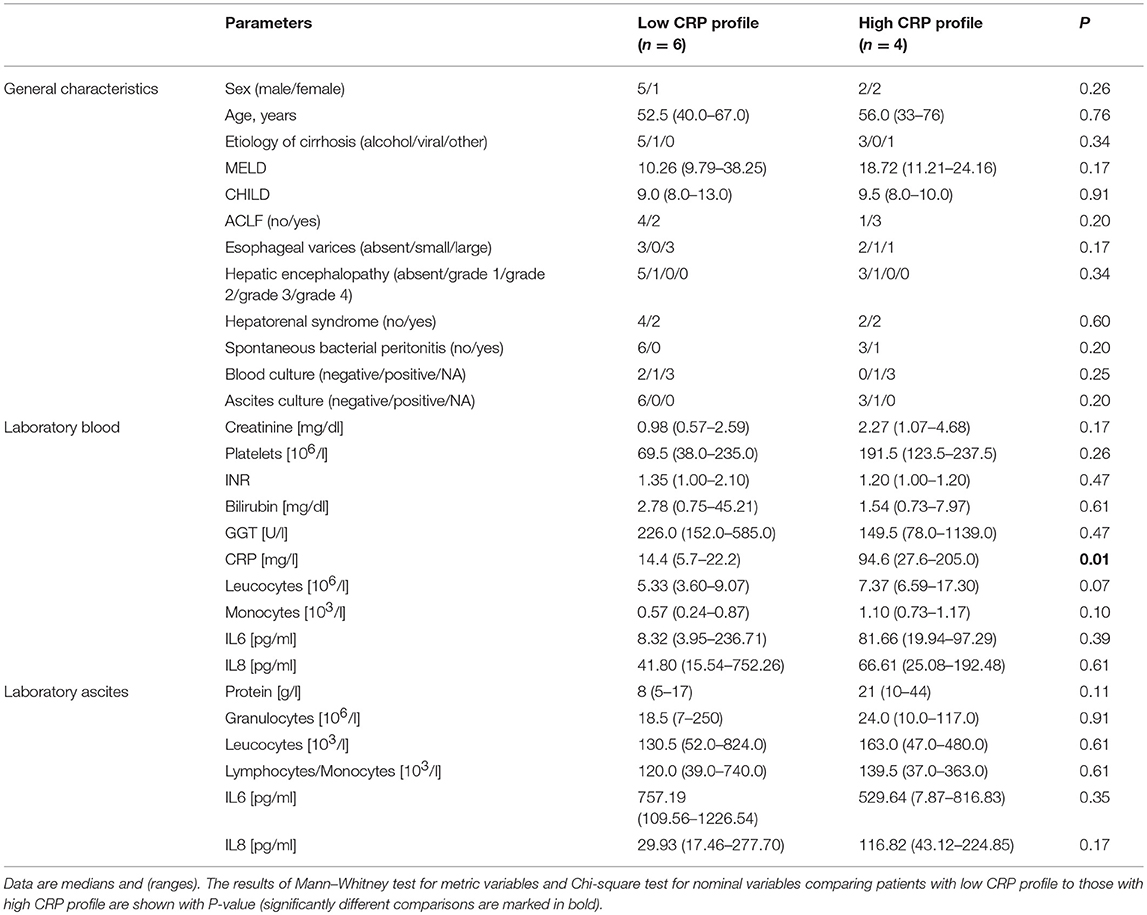
Table 2. Patient characteristics for the subpopulation in which 16S rRNA gene amplicon sequencing was performed in blood and ascites.
Microbiota compositions at the phylum level were in line with previously reported bacterial compositions of ascites (27) and peripheral blood (12). The most abundant phylum detected in ascites samples was Proteobacteria (mean relative abundance 77.7%), followed by Firmicutes (9.4%), Actinobacteria (7.9%), Bacteroidetes (3.9%), and Gemmatimonadetes (0.6%) (Figure 2). The phylum composition was slightly different in the blood samples, with Proteobacteria accounting for 87.4% on average, while Actinobacteria, Firmicutes, and Bacteroidetes accounted for small proportions (5.3, 3.7, and 3.5, respectively; Figure 2). We identified 53 bacterial families in ascites and 48 families in blood samples. Among these, we found a set of six core families detected in all samples in both compartments: Pseudomonadaceae, Oxalobacteriaceae, Neisseriaceae, Enterobacteriaceae, Sphingomonadaceae, and Moraxellaceae. These results were in line with previous studies that reported an increase in potentially pathogenic families such as Enterobacteriaceae on the gut microbiome of liver patients and as well a high prevalence in ascites samples (28).
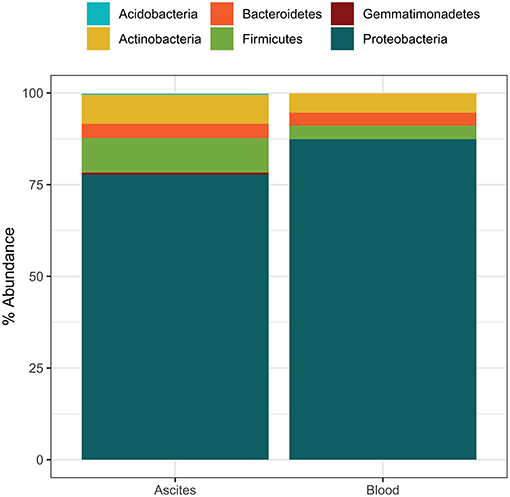
Figure 2. Average microbiome phylum composition in ascites and blood samples (n = 10 each).
We identified 97 bacterial genera from ascites and 89 genera from blood (Figure S2). There were no statistical significant differences between the microbiota compositions of ascites and blood samples at phylum level. At genus level, alpha-diversity indices, such as richness (number of observed genera in a sample) and Shannon diversity (richness as well as evenness of genera in a sample), showed that the ascites microbiome was significantly more diverse than the peripheral blood microbiome (P < 0.01; Figure 3A).
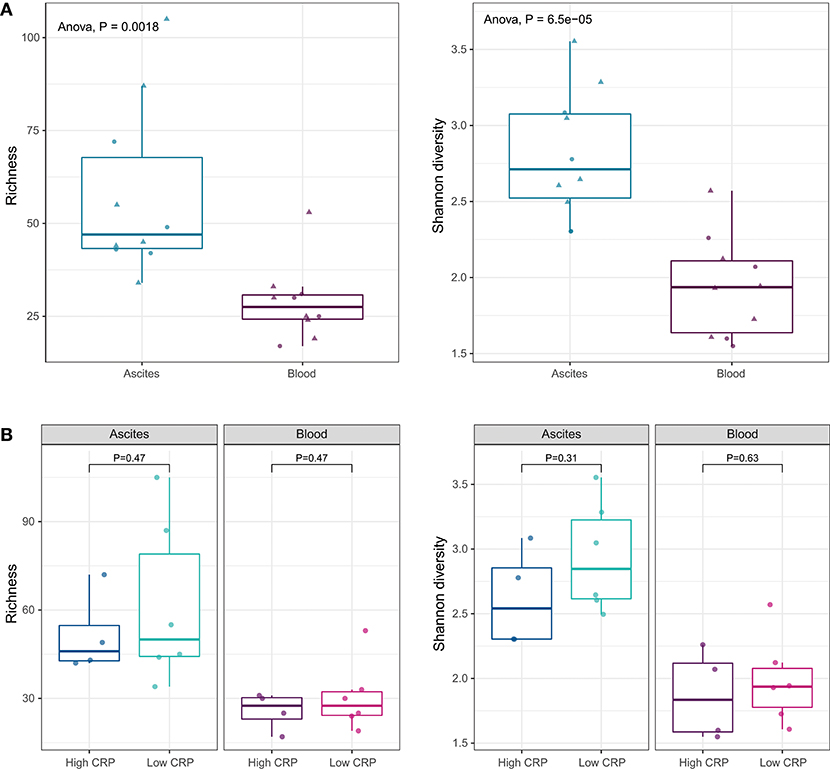
Figure 3. (A) Alpha diversity indices at genus level. Ascites microbiota has higher richness and diversity compared to peripheral blood microbiome. (B) Alpha diversity indices under different systemic inflammation status. The inflammation status does not affect the microbial diversity of the ascites or blood samples.
Translocation of the gut microbiota into the circulatory system and into ascites is increasingly recognized as a driver of complication in advanced liver disease (27, 29). As we showed above, the levels of pro-inflammatory cytokines (IL-6 and IL-8) were higher in ascites compared to blood, suggesting different drivers of inflammation. To determine the relation of the microbiota with cytokine levels and systemic inflammation, we investigated the differences in the microbiome composition of ascites and blood samples, in patients with low and high CRP levels. CRP levels did not appear to affect microbiota diversity of ascites or blood samples (Figure 3B).
Distinct Bacterial Signatures of Ascites and Blood Microbiome
To study the variation in the microbial structure between ascites and peripheral blood samples, we used unweighted and weighted UniFrac metrics (30) as measures of the beta-diversity. Unweighted UniFrac uses a phylogenetic tree taking into account only the phylogenetic affiliation of members to evaluate the community structure and diversity, while weighted UniFrac additionally considers the relative abundance of the members of the community. Both measures clearly separated ascites from blood samples (Figure 4). The sampling compartment explained 11% of the variance in beta-diversity when using unweighted UniFrac metric (PERMANOVA test, P = 0.006), whereas it explained 21.2% of the variance when using weighted UniFrac metric (PERMANOVA test, P = 0.001). Additionally, the first principal coordinate (PC1) of microbiome beta-diversity explained 20.4% of the variance in the samples under unweighted UniFrac metric, whereas it explained 34% of the variance in the samples under weighted UniFrac metric (Figure 4).
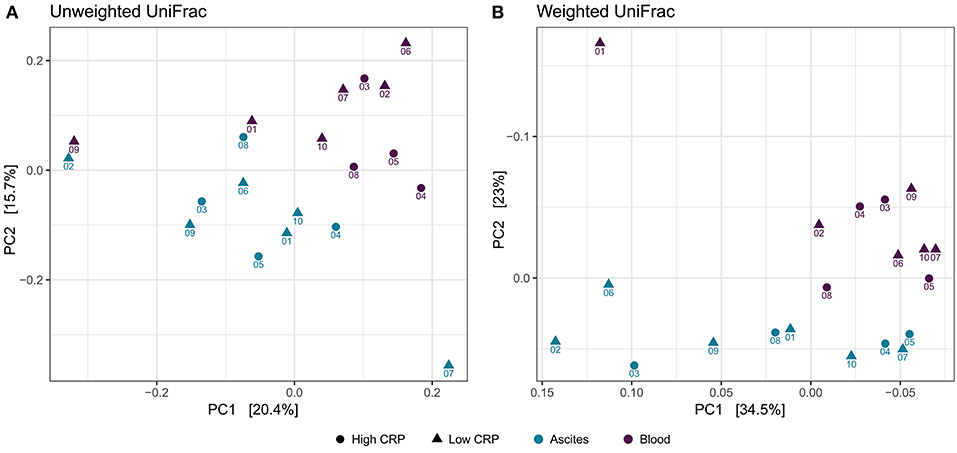
Figure 4. Beta-diversity analysis. Principal coordinate analysis shows clear separation between ascites and peripheral blood samples, but samples do not cluster based on inflammation status. (A) Unweighted UniFrac (B) Weighted UniFrac.
We then investigated specific bacterial signatures that differentiated ascites from peripheral blood samples. We used DAtest (24) to select the most suitable statistical method to identify differentially abundant bacteria. Compared with other statistical methods evaluated by DAtest, t-test had the highest power to distinguish between two simulated sample groups based on our cohort, while keeping the false discovery rate of simulated differential taxa under 0.05. Therefore, we performed t-tests between the ascites and blood microbiomes using the relative abundances of 99 genera. We found that Janthinobacterium, Serratia, and Rugamonas were significantly enriched in ascites, while Escherichia/Shigella and Pseudomonas were enriched in blood (Figure 5, Table 3). These findings were in concordance with our cultivation results, where Serratia was detected in ascites culture and Escherichia in peripheral blood.
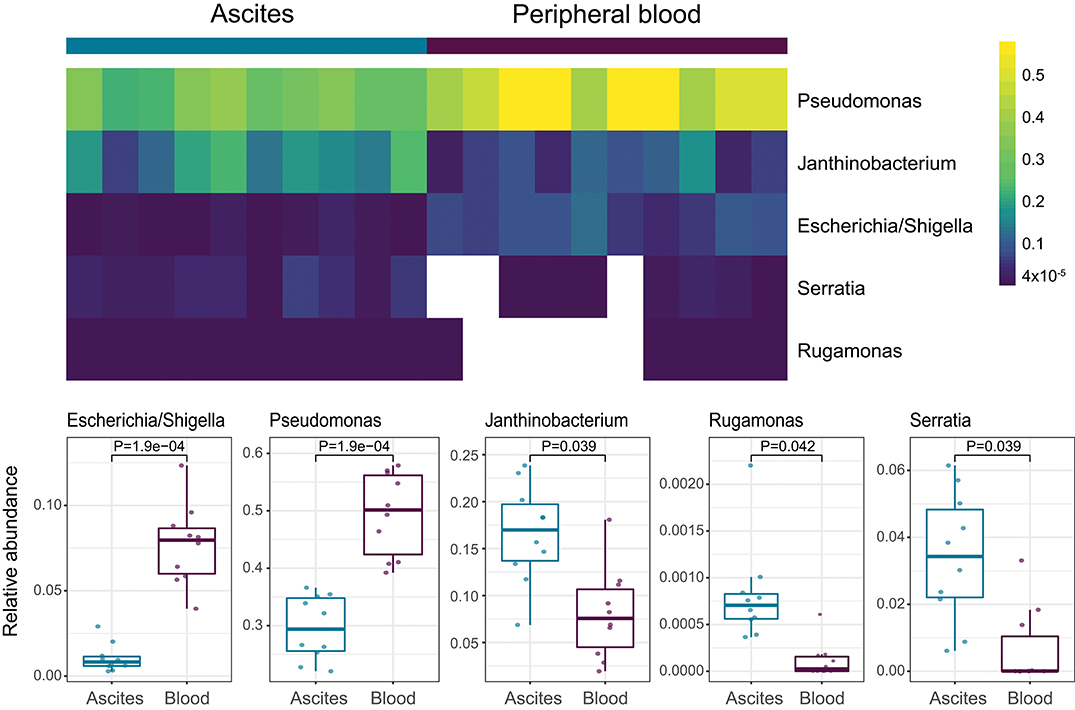
Figure 5. Genera differentially abundant among ascites and blood samples.
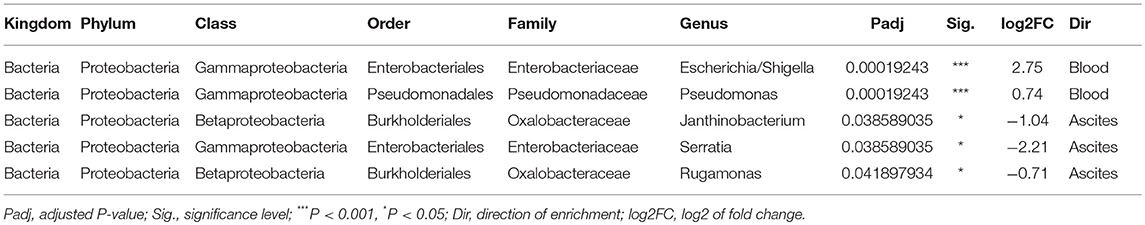
Table 3. Differentially abundant genera between ascites and blood.
Since the presence of bacteria in blood is generally associated with systemic inflammation, we investigated whether there were differences in the bacterial composition of patients with systemic inflammation (high CRP levels, n = 4) and patients without systemic inflammation (low CRP levels, n = 6). There was no significant difference between groups, potentially due to limited sample size. We then examined the 5 bacterial genera that were significantly different between ascites and blood samples to determine their relationship with systemic inflammation. Although there were no statistically significant differences between the samples with and without systemic inflammation, Escherichia/Shigella and Janthinobacterium were more abundant in patients with elevated systemic inflammation, both in ascites and blood samples, while Pseudomonas and Serratia were increased only in the ascites samples with systemic inflammation (Figure S1).
Discussion
The present study demonstrates that the levels of inflammatory cytokines and microbial richness are significantly higher in ascites of patients with decompensated cirrhosis than in their blood compartment. Neither the levels of cytokines nor the overall abundance of bacterial DNA nor microbial richness seemed to be related to the extent of systemic inflammation in these patients.
These findings are particularly striking, since the cell count in circulating blood is several fold higher than in ascites. Still, the levels of inflammatory cytokines were several fold reduced in blood compared to ascites. Moreover, the relationship between the surrogates of systemic inflammation (e.g., leucocyte count, CRP) and severity scores of liver cirrhosis were correlated with the levels of blood cytokines, but no relationship was detected in the same patients in ascites.
A recent study described the role of alcohol consumption for circulating microbiome composition and associated metabolic functions. Even though leucocyte count and endotoxemia increased with the severity of disease in this study, soluble CD14, as a marker for monocyte activation, decreased in severe alcoholic hepatitis underlining the dysregulation of the immune response (31).
Overall, these results may suggest a dysregulation in myeloid cell function in cirrhosis with an excess of cytokine synthesis in ascites. This does, however, not result in at least the two main markers of systemic inflammation investigated here. Although rather descriptive, our study suggests compartmentalization of the immune response in this setting. This compartmental dysregulation may occur as a response to MAMPs, e.g., bacterial DNA.
The reason for this compartmentalization of the immune response remains elusive. Granulocytes, as one of the main producers of cytokines, besides macrophages and monocytes, but crucial for the diagnosis of SBP, were similar or lower in ascites samples than blood samples, although levels of IL-6 and IL-8 were many times higher in ascites. This suggests an external source of these cytokines and inflammation in ascites. Bacterial translocation from the gut is suspected to be the primary source of infection in SBP (2). However, bacteria not associated to the gut can also be found in ascites, suggesting other additional sources of infection (27). Bacterial translocation has been associated with impaired immune response and decompensation of liver cirrhosis, but the efforts to elaborate simple biomarkers such as detection and cut-off levels of bacterial DNA have shown discrepant results. Such discrepancies highlight that the relationship of presence or amount of bacterial DNA is not a strong predictor for decompensation and systemic inflammation in these patients, and probably also does not explain compartmentalization of the immune response. However, recent studies show an association of bacterial DNA levels in ascites and survival (32, 33).
Therefore, we have characterized the microbiome of matched ascites and blood samples from patients with decompensated liver cirrhosis. While ascites and blood harbored the same phyla with marginally different proportions, they showed distinct compositions at genus level. Genus alpha-diversity represented by richness and Shannon's diversity was significantly higher in ascites than in blood. Depending on the choice of the beta-diversity measure, 11% (unweighted UniFrac), or 21.2% (weighted UniFrac) of the variance in beta-diversity was dependent on the sampling compartment. These results suggest that the phylogeny of the members as well as their relative abundance have a significant yet cumulative effect in explaining the differences between ascites and blood microbial communities.
Culture-positive organisms per se do not seem to trigger systemic inflammation assessed by two representative cytokines (9) in patients with cirrhosis as demonstrated by the blood and ascites culture results in the two sub cohorts stratified by their CRP profile. Therefore, we hypothesize that abundance of specific genera must play a prominent role in the induction of systemic inflammation in cirrhosis. This hypothesis is supported by other studies where specific bacterial species correlate with clinical measures (27, 31).
We also identified five bacterial genera that were significantly different between ascites and blood samples, despite the small cohort size. Some of these genera were also cultivated from additional samples of the respective compartments, confirming the viability of at least some of the bacteria reported in our study. Even though high CRP levels were associated with SBP, we did not see a significant difference in microbiome composition between high and low CRP samples, possibly due to reduced statistical power. However, the genera Escherichia/Shigella and Janthinobacterium showed higher abundance in blood samples of patients with systemic inflammation. Escherichia is already known for its pathogenic capabilities via lipopolysaccharides binding to Toll-like receptor 4, especially in individuals with dysfunctional immune response. Janthinobacterium, another Gram-negative bacterium, has not been described in relation to bacteremia previously, but could also induce inflammation via the same mechanism. On the other hand, while relative abundance of Escherichia was decreased in ascites samples, Janthinobacterium was increased. We speculate that the compartmentalization of immune dysfunction might be the reason behind the different behavior of these two bacteria, both of which are Gram-negative, in the different compartments.
In decompensated cirrhosis, representative markers of systemic inflammation and the microbiota seem to be dysregulated in ascites and blood. While a relationship between systemic inflammation and the microbiota seems to exist in blood, this is not found in ascites. These data may suggest the compartmentalization of the immune response and its interaction with the microbiome is especially important and evident in blood, while in ascites not found in our hands.
Author Contributions
CA-S, RS, MA, and JT: conceptualization and methodology. CA-S, RS, AP, MA, and JT: formal analysis. CA-S, RS, AP, FM, FU, and PR: investigation. FM, FU, PR, and EL: resources. BL: data curation. CA-S, RS, EL, BL, JC, MV, MA, and JT: writing—original draft. CA-S, RS, MA, and JT: visualization. MA and JT: supervision.
Conflict of Interest Statement
BL is an employee of Vaiomer. MV has served at the speakers' bureau of Akademie für Infektionsmedizin, Ärztekammer Nordrhein, Astellas Pharma, Basilea, Gilead Sciences, Merck/MSD, Organobalance, and Pfizer, received research funding from 3M, Astellas Pharma, DaVolterra, Evonik, Gilead Sciences, Glycom, MaaT Pharma, Merck/MSD, Morphochem, Organobalance, Seres Therapeutics, Uniklinik Freiburg/Kongress und Kommunikation and is a consultant to Alb-Fils Kliniken GmbH, Ardeypharm, Astellas Pharma, Berlin Chemie, DaVolterra, Ferring, MaaT Pharma, and Merck/MSD. JT has served as speaker or consultant for Gore, Bayer, Alexion, MSD, Gilead, Intercept, Norgine, Grifols, Versantis, and Martin Pharmaceutical.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Gudrun Hack and Silke Bellinghausen for excellent technical assistance. We were supported by grants from the Deutsche Forschungsgemeinschaft (SFB TRR57), Cellex Foundation, and Novo Nordisk Foundation (NNF10CC1016515 and NNF16CC0020896). This study was inspired, but not funded, by European Union's Horizon 2020 research and innovation program's GALAXY study (No 668031). The study was supported by Challenge Grant MicrobLiver grant number NNF15OC0016692 from the Novo Nordisk Foundation. The funders had no influence on study design, data collection and analysis, decision to publish or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00069/full#supplementary-material
References
1. Laleman W, Claria J, Van der Merwe S, Moreau R, Trebicka J. Systemic inflammation and acute-on-chronic liver failure: too much, not enough. Can J Gastroenterol Hepatol. (2018) 2018:1027152. doi: 10.1155/2018/1027152
2. Arroyo V, Moreau R, Kamath PS, Jalan R, Ginès P, Nevens F, et al. Acute-on-chronic liver failure in cirrhosis. Nat Rev Dis Primers (2016) 2:16041. doi: 10.1038/nrdp.2016.41
3. European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu, European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. (2018) 69:406–60. doi: 10.1016/j.jhep.2018.03.024
4. Potgieter M, Bester J, Kell DB, Pretorius E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev. (2015) 39:567–91. doi: 10.1093/femsre/fuv013
5. Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology (2005) 41:422–33. doi: 10.1002/hep.20632
6. Mücke MM, Rumyantseva T, Mücke VT, Schwarzkopf K, Joshi S, Kempf VAJ, et al. Bacterial infection-triggered acute-on-chronic liver failure is associated with increased mortality. Liver Int. (2018) 38:645–53. doi: 10.1111/liv.13568
7. Mortensen C, Jensen JS, Hobolth L, Dam-Larsen S, Madsen BS, Andersen O, et al. Association of markers of bacterial translocation with immune activation in decompensated cirrhosis. Eur J Gastroenterol Hepatol. (2014) 26:1360–6. doi: 10.1097/MEG.0000000000000217
8. González-Navajas JM, Bellot P, Francés R, Zapater P, Muñoz C, García-Pagán JC, et al. Presence of bacterial-DNA in cirrhosis identifies a subgroup of patients with marked inflammatory response not related to endotoxin. J Hepatol. (2008) 48:61–7. doi: 10.1016/j.jhep.2007.08.012
9. Clària J, Stauber RE, Coenraad MJ, Moreau R, Jalan R, Pavesi M, et al. Systemic inflammation in decompensated cirrhosis: characterization and role in acute-on-chronic liver failure. Hepatology (2016) 64:1249–64. doi: 10.1002/hep.28740
10. Hackstein C-P, Assmus LM, Welz M, Klein S, Schwandt T, Schultze J, et al. Gut microbial translocation corrupts myeloid cell function to control bacterial infection during liver cirrhosis. Gut (2017) 66:507–18. doi: 10.1136/gutjnl-2015-311224
11. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature (2016) 535:65–74. doi: 10.1038/nature18847
12. Schierwagen R, Alvarez-Silva C, Madsen MSA, Kolbe CC, Meyer C, Thomas D, et al. Circulating microbiome in blood of different circulatory compartments. Gut (2018). doi: 10.1136/gutjnl-2018-316227. [Epub ahead of print].
13. Appenrodt B, Lehmann LE, Thyssen L, Gentemann M, Rabe C, Molitor E, et al. Is detection of bacterial DNA in ascitic fluid of clinical relevance? Eur J Gastroenterol Hepatol. (2010) 22:1487–94. doi: 10.1097/MEG.0b013e328340c43a
14. European Association for the Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. (2010) 53:397–417. doi: 10.1016/j.jhep.2010.05.004
15. Mauch H. Mikrobiologisch-Infektiologische Qualitätsstandards: MiQ; Qualitätsstandards in der Mikrobiologisch-Infektiologischen Diagnostik. Munich: Elsevier (2007).
16. Vehreschild MJGT, Hamprecht A, Peterson L, Schubert S, Häntschel M, Peter S, et al. A multicentre cohort study on colonization and infection with ESBL-producing Enterobacteriaceae in high-risk patients with haematological malignancies. J Antimicrob Chemother. (2014) 69:3387–92. doi: 10.1093/jac/dku305
17. Lutz P, Goeser F, Kaczmarek DJ, Schlabe S, Nischalke HD, Nattermann J, et al. Relative ascites polymorphonuclear cell count indicates bacterascites and risk of spontaneous bacterial peritonitis. Dig Dis Sci. (2017) 62:2558–68. doi: 10.1007/s10620-017-4637-4
18. Lelouvier B, Servant F, Païssé S, Brunet AC, Benyahya S, Serino M, et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology (2016) 64:2015–27. doi: 10.1002/hep.28829
19. Païssé S, Valle C, Servant F, Courtney M, Burcelin R, Amar J, et al. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion (2016) 56:1138–47. doi: 10.1111/trf.13477
20. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods (2016) 13:581–3. doi: 10.1038/nmeth.3869
21. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. (2013) 41:D590–6. doi: 10.1093/nar/gks1219
22. McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE (2013) 8:e61217. doi: 10.1371/journal.pone.0061217
23. Callahan BJ, Sankaran K, Fukuyama JA, McMurdie PJ, Holmes SP. Bioconductor workflow for microbiome data analysis: from raw reads to community analyses. F1000Res. (2016) 5:1492. doi: 10.12688/f1000research.8986.2
24. Russel J, Thorsen J, Brejnrod AD, Bisgaard H, Sorensen SJ, Burmolle M. DAtest: a framework for choosing differential abundance or expression method. bioRxiv (2018) 2018:241802. doi: 10.1101/241802
25. Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation (2000) 101:1767–72. doi: 10.1161/01.CIR.101.15.1767
26. Arican O, Aral M, Sasmaz S, Ciragil P. Serum levels of TNF-α, IFN-γ, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. (2005) 2005:273–9. doi: 10.1155/MI.2005.273
27. Rogers GB, van der Gast CJ, Bruce KD, Marsh P, Collins JE, Sutton J, et al. Ascitic microbiota composition is correlated with clinical severity in cirrhosis with portal hypertension. PLoS ONE (2013) 8:e74884. doi: 10.1371/journal.pone.0074884
28. Tuomisto S, Pessi T, Collin P, Vuento R, Aittoniemi J, Karhunen PJ. Changes in gut bacterial populations and their translocation into liver and ascites in alcoholic liver cirrhotics. BMC Gastroenterol. (2014) 14:40. doi: 10.1186/1471-230X-14-40
29. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology (2013) 57:601–9. doi: 10.1002/hep.26093
30. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. (2005) 71:8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005
31. Puri P, Liangpunsakul S, Christensen JE, Shah VH, Kamath PS, Gores GJ, et al. The circulating microbiome signature and inferred functional metagenomics in alcoholic hepatitis. Hepatology (2018) 67:1284–302. doi: 10.1002/hep.29623
32. Fagan KJ, Rogers GB, Melino M, Arthur DM, Costello M-E, Morrison M, et al. Ascites bacterial burden and immune cell profile are associated with poor clinical outcomes in the absence of overt infection. PLoS ONE (2015) 10:e0120642. doi: 10.1371/journal.pone.0120642
Keywords: microbiome, systemic inflammation, myeloid cells, cirrhosis, ascites, cytokines, acute-on-chronic liver failure
Citation: Alvarez-Silva C, Schierwagen R, Pohlmann A, Magdaleno F, Uschner FE, Ryan P, Vehreschild MJGT, Claria J, Latz E, Lelouvier B, Arumugam M and Trebicka J (2019) Compartmentalization of Immune Response and Microbial Translocation in Decompensated Cirrhosis. Front. Immunol. 10:69. doi: 10.3389/fimmu.2019.00069
Received: 30 October 2018; Accepted: 11 January 2019;
Published: 08 February 2019.
Edited by:
Hannelie Korf, KU Leuven, BelgiumReviewed by:
Edward Sherwood, Vanderbilt University Medical Center, United StatesGautam Mehta, University College London, United Kingdom
Copyright © 2019 Alvarez-Silva, Schierwagen, Pohlmann, Magdaleno, Uschner, Ryan, Vehreschild, Claria, Latz, Lelouvier, Arumugam and Trebicka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manimozhiyan Arumugam, arumugam@sund.ku.dk
Jonel Trebicka, jonel.trebicka@kgu.de
†These authors share first authorship