 Paola Adele Lonati1†
Paola Adele Lonati1† Mariangela Scavone2,3†
Mariangela Scavone2,3† Maria Gerosa4
Maria Gerosa4 Maria Orietta Borghi1,4
Maria Orietta Borghi1,4 Francesca Pregnolato1Daniele Curreli1Gianmarco Podda2,3Eti Alessandra Femia2,3
Francesca Pregnolato1Daniele Curreli1Gianmarco Podda2,3Eti Alessandra Femia2,3 Wilma Barcellini5
Wilma Barcellini5 Marco Cattaneo2,3
Marco Cattaneo2,3 Francesco Tedesco1
Francesco Tedesco1 Pier Luigi Meroni1*
Pier Luigi Meroni1*- 1Immunorheumatology Research Laboratory, Istituto Auxologico Italiano, IRCCS, Milan, Italy
- 2Unità di Medicina II, ASST Santi Paolo e Carlo, Milan, Italy
- 3Dipartimento di Scienze Della Salute, University of Milan, Milan, Italy
- 4Dipartimento di Scienze Cliniche e di Comunità, University of Milan, Milan, Italy
- 5UOC Ematologia, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
Antiphospholipid syndrome (APS) is a chronic and disabling condition characterized by recurrent thrombosis and miscarriages mediated by antibodies against phospholipid-binding proteins (aPL), such as beta2glycoprotein I (β2GPI). Complement is involved in APS animal models and complement deposits have been documented in placenta and thrombotic vessels despite normal serum levels. Analysis of circulating blood cells coated with C4d displays higher sensitivity than the conventional assays that measure soluble native complement components and their unstable activation products in systemic lupus erythematosus (SLE). As C4d-coated blood cell count has been reported to be more sensitive than serum levels of complement components and their activation products in systemic lupus erythematosus (SLE) patients, we decided to evaluate the percentage of C4d positive B lymphocytes (BC4d), erythrocytes (EC4d), and platelets (PC4d) in primary APS patients and asymptomatic aPL positive carriers as marker of complement activation in APS. We assessed by flow cytometry the percentages of BC4d, EC4d, and PC4d in primary APS (PAPS; n. 23), 8 asymptomatic aPL positive carriers, 11 APS-associated SLE (SAPS), 17 aPL positive SLE, 16 aPL negative SLE, 8 aPL negative patients with previous thrombosis, 11 immune thrombocytopenia (ITP) patients, and 26 healthy subjects. In addition, we used an in vitro model to evaluate the ability of a monoclonal anti-β2GPI antibody (MBB2) to bind to normal resting or activated platelets and fix complement. EC4d and PC4d percentages were significantly higher in PAPS and aPL carriers as well as aPL positive SLE and SAPS than in aPL negative controls. The highest values were found in PAPS and in SAPS. The EC4d and PC4d percentages were significantly correlated with serum C3/C4 and anti-β2GPI/anti-cardiolipin IgG. In vitro studies showed that MBB2 bound to activated platelets only and induced C4d deposition. The detection of the activation product C4d on circulating erythrocytes and platelets supports the role of complement activation in APS. Complement may represent a new therapeutic target for better treatment and prevention of disability of APS patients.
Introduction
Antiphospholipid syndrome (APS) is a chronic autoimmune disease characterized by recurrent thrombotic events and pregnancy morbidity, in the presence of antibodies targeting anti-phospholipid binding proteins (aPL) (1). APS can be found in patients with no evidence of associated diseases (primary APS, PAPS) or with other autoimmune disorders, mainly systemic lupus erythematosus (SLE) (SAPS) (1).
The major aPL antigenic target is β2-glycoprotein I (β2GPI), a phospholipid binding plasma protein of 50 kDa (2). Upon binding to anionic phospholipids or to membrane receptors of different cell types, β2GPI changes conformation, exposing an immunodominant cryptic epitope located at domain I (D1), which is recognized by aPL. There is sound evidence from in vitro and in vivo models that β2GPI-dependent aPL play a pathogenic role both in thrombosis and pregnancy complications (3, 4). Moreover, epidemiological data support a strong diagnostic/prognostic value of anti-D1 antibodies in APS patients (5).
Complement activation was initially suggested to be involved in APS animal models since the induction of fetal loss or thrombosis by passive infusion of aPL IgG was prevented by treatment with inhibitors of complement activation or the use of animals deficient in complement components (6–10). Moreover, a human monoclonal antibody against β2GPI D1 lacking the complement-fixing portion of the molecule (MBB2ΔCH2), unlike the complement-fixing parent molecule (MBB2) that reacts with the same epitope (11) fails to exhibit pathogenic effect. In contrast, low C3 and C4 serum levels were described in some APS patients only and few studies reported high levels of complement activation products (fragment Bb and anaphylatoxins C4a, C3a, and C5a) with no association with the vascular manifestations of the syndrome (12–15). On the other hand, we recently reported deposition of C1q, C4, C3, and C9 on the endothelium of the vessel wall close to the thrombotic occlusion in a PAPS patient who underwent bypass surgery to treat an arterial thrombotic occlusion. Notably, complement components co-localized with β2GPI and IgG, suggesting that aPL caused complement activation and contributed to the pathogenesis of the thrombotic event (16).
Measurement of serum levels of complement activation products has been reported to be more sensitive than that of native complement components in SLE (17). In particular, the number of C4d-coated B lymphocytes, erythrocytes and platelets in circulating blood of SLE patients with active disease was higher than in controls (17–24).
The number of C4d-bound platelets was associated with lupus disease activity and complement consumption but contrasting results regarding the association with arterial or venous events and aPL were reported (25, 26). This finding is in contrast with the ability of aPL to activate complement and promote binding of complement split products to fixed platelets in vitro (25, 27, 28).
We have investigated the percentage of C4d positive circulating blood cells in PAPS and report a higher number of C4d positive erythrocytes and platelets in aPL positive patients than in controls supporting the hypothesis that complement is activated in vivo. A mechanistic model of complement deposition on platelets in the presence of a human monoclonal antibody specific for D1 of β2GPI was investigated as well.
Materials and Methods
Patients and Controls
We recruited the following patients: 23 PAPS; 11 SAPS, 17 aPL positive SLE, and 16 aPL negative SLE. As controls we included: 11 patients with primary immune thrombocytopenia (ITP), 8 aPL negative patients with previous thrombotic episodes, 8 persistently positive aPL healthy subjects (aPL positive carriers), and 26 normal healthy subjects (NHS). SLE patients were classified according to the 1997 American College of Rheumatology updated criteria for SLE and the new 2012 SLICC SLE criteria (29, 30). Disease activity in SLE patients was assessed by the SELENA version of SLEDAI index in SLE (31). APS patients fulfilled the revised Sapporo criteria (32). The characterization of vascular and obstetric APS was carried out as previously reported (33). Primary ITP was diagnosed according to the International Working Group criteria (34).
All ITP patients were in stable clinical remission. Anti-phospholipid negative patients with thrombosis had previous documented episodes of myocardial infarction, stroke, transient ischemic attack, pulmonary embolism, or deep vein thrombosis.
When entering the protocol, all subjects underwent a detailed clinical evaluation and comprehensive medical history registration. Recorded variables included sex, age, disease duration and therapy (Supplementary Table 1). To avoid any modification due to the acute phase reactants, samples have been collected at least six months after the thrombotic event (35). The protocol was approved by the local Institutional Review Board (IRB) (Comitato Etico Milano area 2–426_2014) and all subjects gave written informed consent.
Blood Samples
Venous blood samples were collected from an antecubital vein using a 21-gauge butterfly needle with minimal stasis. The first 3 mL were collected into K2E EDTA tubes (Becton Dickinson vacutainer, North Ryde, NSW, Australia) and analyzed by coulter hematology analyser for blood cell count (DxH-800, Beckman Coulter, Milano, Italy). Additional blood samples included: 3 mL in K2E EDTA for measurement of C4d deposition on B lymphocytes, red blood cells and platelets by flow cytometry; 3 mL in CTAD vacutainer for lupus anticoagulant (LAC) determination; 5 mL in vacutainer without anticoagulant for serum collection. Five mL of blood were collected into desirudin (Revasc, 25 μg/mL) and used for experiments of C4d deposition on in vitro stimulated and resting platelets.
Serum Complement Determination
Serum concentrations of complement components C3 and C4 were determined by an immunoturbidimetric method (Roche/Hitachi cobas c 701/702): C3 and C4 normal ranges indicated by the manufacturer were 55–180 and 20–50 mg/dL, respectively.
Detection of aPL
Serum anticardiolipin (anti-CL) and anti-β2GPI IgG/IgM autoantibodies were detected as previously described (36). LAC was measured according to international ISTH guidelines (37).
Detection of C4d Bound to Cells by Flow Cytometry
The percentage of C4d bound to B lymphocytes (BC4d), erythrocytes (EC4d), and platelets (PC4d) was measured by flow cytometry after subtraction of background signals. All analyses were performed using a FACS Calibur cytometer and Cell Quest software (BD Biosciences, San Jose, CA). In all experiments, control procedures to establish proper calibration, compensation, and linearity were performed.
C4d Bound to B-Lymphocytes
Erythrocytes lysis was performed by addition of ammonium chloride–based reagent (BD Pharm Lyse; BD Biosciences, San Jose, CA) to EDTA-whole blood (300 μL), left 10 min at 4°C and centrifuged at 800 g at 4°C for 5 min. Cell pellet was suspended in 1 mL of Dulbecco's phosphate buffered saline (DPBS, Sigma-Aldrich, St. Louis, MO) supplemented with 1% heat-inactivated fetal calf serum (FCS) (Sigma-Aldrich, St. Louis, MO) (DPBS/1% FCS) and stained at 2–8°C with 10 μg/mL purified mouse monoclonal antibodies against human C4d (mouse anti-human C4d; Quidel, San Diego, CA) or 10 μg/mL mouse anti-human isotype control IgG1κ (MOPC-21; BD Biosciences, San Jose, CA) for 45 min. Samples were then washed with DPBS/1%FCS and centrifuged at 800 g at 4°C for 5 min. Pellets were suspended in a DPBS/1%FCS solution containing fluorescein isothiocyanate (FITC) conjugated goat anti-mouse antibody (Cappel, MP biomedicals, Santa Ana, CA) (10 μg/mL) and R-phycoerythrin (PE)-conjugated monoclonal antibody against human CD-19 (a 95-Kd type I transmembrane glycoprotein expressed on B cells) (BD Biosciences, San Jose, CA), and stained at 2–8°C in the dark for 45 min. Samples were washed again and suspended in 250 μL cold DPBS/1%FCS. 5,000 events in the lymphocytes gate (based on their size) were acquired at high flow rate. A single fluorochrome dot plot strategy was used to identify B-lymphocytes CD19 positive (SSC vs. CD19-PE) and the percentage of C4d was assessed on B-lymphocytes gate (SSC vs. FITC).
C4d Bound to Erythrocytes
EDTA-whole blood (50 μL) was suspended in 1,5 mL DPBS/1%FCS and centrifuged for 5 min at 800 g at 4°C. The pellet was suspended in 500 μL DPBS/1%FCS; 10 μL of the suspension was subsequently stained with anti-C4d monoclonal antibody or mouse anti-human isotype control IgG1κ at 2–8°C for 45 min. Samples were then washed and cell surface C4d was detected by addition of FITC-conjugated goat anti-mouse antibody at 2–8°C in the dark for 45 min. Samples were washed again and suspended in 250 μL of cold DPBS/1%FCS. 5,000 events in the erythrocytes gate were acquired with a high flow rate. A single fluorochrome dot plot strategy (FSC vs. FITC) was used for quantification of percentage of C4d on erythrocytes. Details regarding the erythrocyte gating strategy are shown in Supplementary Figure 1.
C4d Bound to Resting Platelets
EDTA-whole blood samples (50 μL) were diluted with DPBS/1%FCS (1:2) and stained with anti-C4d monoclonal antibody or mouse anti-human isotype control IgG1κ at 2–8°C for 45 min, followed by staining with FITC-conjugated goat anti-mouse antibody at 2–8°C in the dark for 45 min. A PE–conjugated monoclonal antibody against human CD42b (a 145 kD glycoprotein known as GPIbα) was used to identify platelets. 5,000 events in the platelet gate were acquired at low flow rate. A single fluorochrome dot plot strategy was used to identify CD42b positive platelets (SSC vs. CD42b-PE) and the percentage of C4d was measured (SSC vs. FITC). Details regarding the platelet gating strategy are shown in Supplementary Figure 2.
MBB2 Antibody
The human monoclonal antibody against D1 of β2GPI (MBB2) has been produced and characterized as previously published (11).
In vitro Model of C4d Binding to Normal Platelets
Hirudin-anticoagulated whole blood from healthy blood donors was diluted in home-made PBS (1:15) and incubated with or without 500 μg/mL of MBB2 and/or thrombin receptor activating peptide (TRAP, Sigma-Aldrich, Germany) (20 μM) at 37°C for 20 min. Samples were then diluted with PBS (1:10) and stained with 10 μg/mL mouse anti-human C4d or 10 μg/mL mouse anti-human isotype control IgG1κ at RT for 20 min. Then, FITC-conjugated goat anti-human antibody (Cappel, MP biomedicals, Santa Ana, CA) and/or APC-conjugated goat anti-mouse antibody (ThermoFisher scientific Massachusetts, USA), and/or anti-CD42b-PE, and/or anti-CD62p-Pe/Cy7 (Biolegend, San Diego, CA) were added to the samples and incubated at RT in the dark for 20 min. After incubation, samples were diluted in 300 μL PBS and immediately analyzed by flow cytometry. All analyses were performed using a FACS Verse cytometer and FACS Suite software (BD Biosciences, San Jose, CA). 5,000 events in the platelet gate were acquired with a medium flow rate. Dot plot strategy was used for analysis: first of all, platelets were identified with CD42b-PE vs. SSC, then C4d-APC or MBB2-FITC or CD62p-Pe/Cy7 vs. SSC were used to determine the percentage of C4d and MBB2 platelet bound and of CD62p expression.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software Inc., San Diego, CA, USA). All results were expressed as median with interquartile range. The normality of data was evaluated by the D'Agostino-Pearson test. Comparisons between groups were performed using Kruskal-Wallis or Friedman tests as appropriate followed by Dunn's post-hoc test. Comparison between two groups were performed using Wilcoxon t-test. All tests were two tailed. Correlations between variables were expressed as Spearman's correlation coefficient. p < 0.05 was chosen as the cut-off level for statistical significance.
Results
aPL Profile of Patients and Controls
Normal healthy subjects and pathological controls (ITP, SLE, and patients with previous thrombosis) were negative in all aPL assays. Among the 17 aPL positive SLE patients, three displayed single, 7 double and 7 triple positivity. Four out of 11 SAPS patients were single-positive, 1 double-positive and 6 triple-positive for aPL; 4/8 aPL positive carriers were double- and 4/8 triple-positive; of the 23 PAPS patients, 1 was single-, 5 double-, and 17 triple-positive.
C3 and C4 Serum Levels
aPL positive carriers and PAPS patients displayed lower serum levels of C3 and C4 than normal healthy subjects, albeit their values fell in the normal range (Table 1).
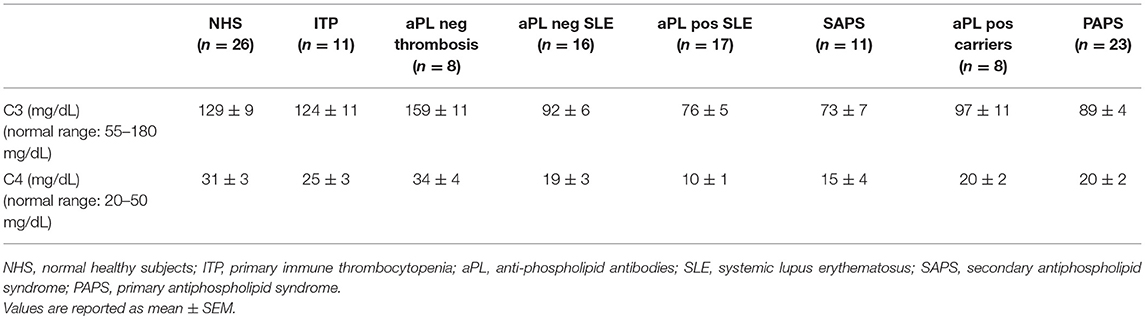
Table 1. Serum levels of C3 and C4.
Measurement of Cell-Bound C4d
Significantly higher percentages of both EC4d and PC4d were found in aPL positive SLE, SAPS and PAPS patients compared to NHS, ITP patients, and aPL negative thrombotic patients; moreover, higher percentages of EC4d and PC4d were detected also in aPL negative SLE and asymptomatic healthy aPL positive carriers (Figures 1A,B). Patients with SLE with or without aPL or full blown APS displayed higher BC4d percentages than PAPS or aPL positive carriers. On the contrary, the percentages of BC4d in aPL positive carriers and PAPS were comparable with the controls (Supplementary Figure 3).
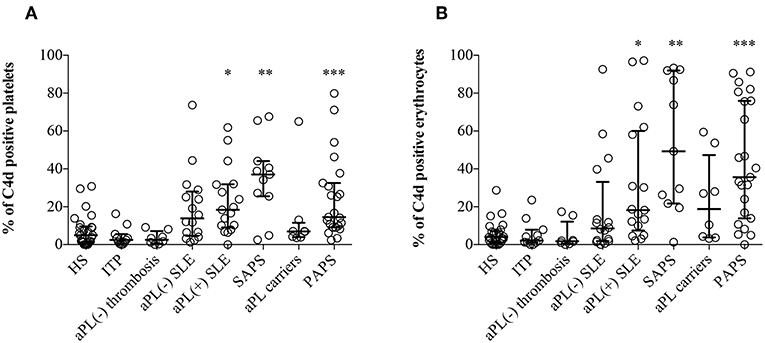
Figure 1. C4d deposition on platelets and erythrocytes. Flow cytometry was performed on EDTA-whole blood samples (n = 120). C4d-positive cells were detected by purified anti-human C4d and FITC conjugated goat anti-mouse antibody on platelets (A) and on erythrocytes (B). Results are expressed as percentage, median with interquartile range, and analyzed by Kruskal-Wallis test and Dunn's multiple comparison post-hoc test. *p < 0.05; **p < 0.01, ***p < 0.001 vs. NHS.
Cell-bound C4d percentages were comparable both in thrombotic and obstetric APS patients, suggesting that they were not associated with the main APS clinical manifestations (Supplementary Figure 4); moreover, we did not find any correlation also with non-classification criteria (e.g., thrombocytopenia; Supplementary Figures 5–7) (32).
Correlations Between the Percentage of PC4d and EC4d With Other Laboratory Parameters
C4 Serum levels were found to be inversely correlated with the percentage of PC4d (r = −0.4682, p < 0.0001; Figure 2A) and EC4d (r = −0.5163, p < 0.0001; Figure 2B).
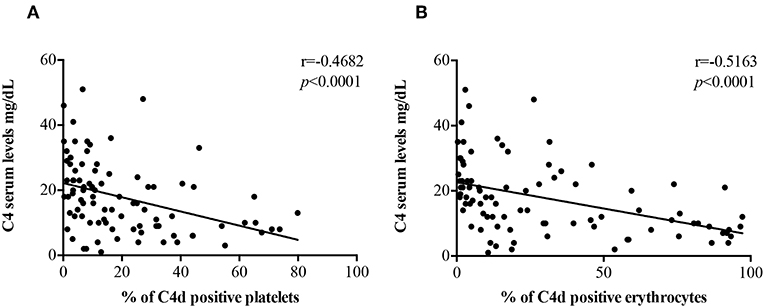
Figure 2. Correlations between C4 serum levels and EC4d and PC4d percentages. C4 serum levels (mg/dL) negatively correlated with the percentages of PC4d (A) and EC4d (B).
There was a positive correlation of both anti-β2GPI IgG (r = 0.2486, p = 0.0157) and anti-CL IgG (r = 0.4537, p < 0.0001) titers with PC4d (Figures 3A,B). The correlation between PC4d and anti-CL IgM was mild, but statistically significant (r = 0.2472, p = 0.0163; Supplementary Figure 8B). In contrast, there was no correlation between PC4d and anti-β2GPI IgM titers (r = 0.09486, p = 0.3631) (Supplementary Figure 8A). EC4d percentages positively correlated with anti-β2GPI IgG (r = 0.3503, p = 0.0005) and anti-CL IgG (r = 0.4805, p < 0.0001) (Supplementary Figures 9A,B). There was also a positive correlation between EC4d and both anti-β2GPI IgM (r = 0.2672, p = 0.0092) and anti-CL IgM (r = 0.4177, p < 0.0001) (Supplementary Figures 9C,D).
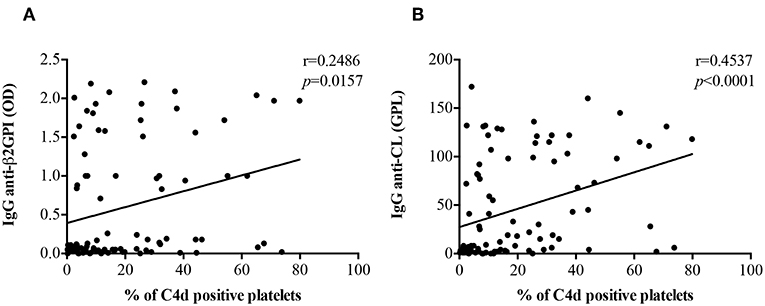
Figure 3. Correlations between anti-β2GPI and anti-CL IgG titers PC4d. PC4d titers positively correlated with both IgG anti-β2GPI (A) and IgG β2GPI-dependent anti-CL titers (B).
In vitro Studies: MBB2 and C4d Deposition on Resting and Activated Platelets
Since β2GPI-dependent aPL may recognize β2GPI adhered on activated platelets (3), we set up an in vitro model to investigate whether the bound antibody was responsible for cell membrane deposition of C4d. To this end, hirudin-anticoagulated whole blood samples from NHS (n = 11) were incubated with TRAP, a platelet agonist. In the absence of exogenous stimuli, the expression of CD62p on platelets was negligible, while it dramatically increased after addition of TRAP. These results indicate that circulating NHS platelets were not activated, offering a suitable in vitro model to investigate the possible role of platelet activation in β2GPI adhesion to cell membranes and antibody binding (Figure 4A). Blood samples, which provided the source of resting platelets, autologous β2GPI and complement, were incubated with MBB2. The binding of MBB2 to resting platelets was negligible (0.4%) but it increased up to 18 times (7.5%; p = 0.002) following platelet stimulation by TRAP (Figure 4B). This finding suggests that pathogenic aPL may recognize autologous β2GPI bound only to activated platelets. We then investigated whether the bound antibody was able to activate complement and to induce C4d deposition on the platelet membranes. The percentages of PC4d increased significantly when the blood samples were simultaneously incubated with TRAP and MBB2, suggesting that MBB2 reacted with adhered β2GPI and activated complement, leading to C4d deposition on activated platelets (Figure 4C).
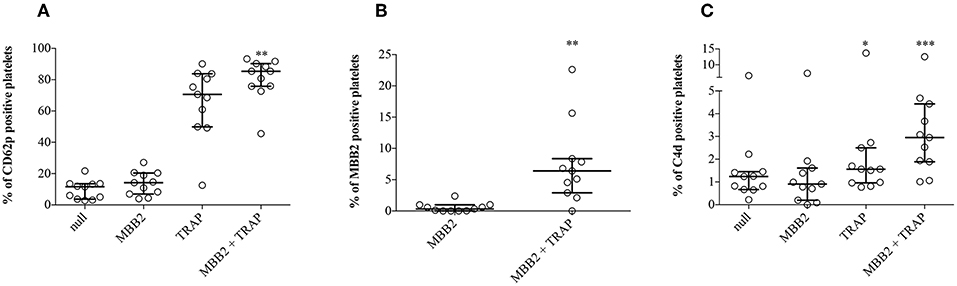
Figure 4. Effects of MBB2 and TRAP, alone and in combination, on the expression of CD62p on platelets. Percentages of CD62p-, MBB2-, and C4d-positive platelets were measured in hirudin-anticoagulated blood from 11 NHS after its incubation with or without MBB2 and/or TRAP at 37°C for 20 min. Data were analyzed by Friedman test and Dunn's multiple comparisons post-hoc test (A,C) and by Wilcoxon matched paired test (B). Results are expressed as percentage, median with interquartile range. *p < 0.05; **p < 0.01, ***p < 0.001 vs. MBB2.
Discussion
Complement has been shown to be activated in several systemic autoimmune diseases and plays a key role in SLE and in SLE-related disorders (38). Although there is ample evidence that thrombosis and fetal loss are complement dependent in experimental models of APS, complement activation in APS patients is still matter of debate (15). Our results show for the first time increased percentages of C4d complement activation product that is stably deposited on cell membranes of platelets and erythrocytes in PAPS.
High percentages of platelet C4d were reported to be specific for SLE (17–24). We confirmed this finding in aPL negative SLE patients included in this study as a pathological control group. The percentages of C4d bound to circulating blood cells, in particular to platelets, display higher sensitivity than the conventional assays that measure soluble complement components and their unstable activation products. Consequently, platelet C4d percentages have been suggested as useful and sensitive tool for mirroring complement activation in SLE patients (17).
The finding of increased percentages of PC4d and EC4d in PAPS and in aPL positive carriers but not in healthy controls and patients with ITP strongly suggests that the presence of aPL is specifically associated with in vivo complement activation. Comparable percentages were found in aPL negative and aPL positive SLE patients with no APS clinical manifestations supporting the hypothesis that SLE and the presence of aPL may be independently linked to complement activation. On the other hand, full blown APS was associated with the highest EC4d and PC4d percentages both in PAPS and in SLE-associated APS. Since the majority of these patients had a history of vascular thrombotic events, we investigated whether a previous thrombosis may affect the percentages of C4d positive cells in the absence of aPL. This possibility was ruled out by the observation that the control group of aPL negative thrombotic patients had normal percentages of C4d positive cells further supporting the specificity of this parameter for in vivo complement activation in APS.
Although the serum levels of C3 and C4 in PAPS and in aPL positive carriers were reduced in comparison with healthy controls and patients with ITP or previous thrombosis without aPL, they were still within the normal range. Moreover, C4d positive cells significantly correlated with both serum C3 and C4 values and titers of anti-β2GPI and β2GPI-dependent anti-CL IgG. These findings are consistent with the hypothesis that the presence of aPL is associated with complement activation in APS.
Few recent studies reported reduced complement levels and increased levels of activation products in some APS patients albeit without clear association with the clinical manifestations of the syndrome (12–14). The high percentages of both EC4d and PC4d in our study further support the involvement of complement in APS. The lack of association with the clinical manifestations of the syndrome was confirmed in our study in spite of the use of a more sensitive and reliable laboratory tool for detection of complement activation.
Complement activation in APS patients should be associated with complement deposition in damaged tissues. There is sound evidence that this is the case for placenta both in animals and in patients (15). A role of complement for the aPL-mediated placenta damage has been suggested in animals while it is still matter of research in humans (15). On the other hand, although less studies looked at vessel complement deposition, the available data are suggestive for a role of complement in endothelial perturbation that may be pivotal for the thrombophilic state (16, 39).
Both circulating and tissue immune complexes are known to play a critical role in activating the complement cascade in SLE (38). Their deposition on the membrane of circulating blood cells explains the increased percentages of C4d positive cells; however additional pathways have been suggested to mediate the binding of complement components to activated platelets without the need of immune complex formation (40). In contrast, the mechanism(s) responsible for complement activation in APS has not been clarified yet. Although circulating anti-β2GPI/β2GPI complexes can be found in a limited proportion of patients (41, 42), they are undetectable in most of them suggesting that tissue bound complexes (e.g., on vessel walls) (16, 39) and/or additional mechanisms play a role in complement activation.
Beta2GPI is the main target for diagnostic and pathogenic aPL and it is general accepted that binds to activated platelets through several candidate membrane receptors or just because of the electrostatic interaction between the cationic PL-binding site of β2GPI and the anionic phospholipid (i.e., phosphatidylserine) that is exposed in the outer cell membrane leaflet upon cell activation (3). Once bound, the adhered β2GPI exposes the immunodominant epitope on D1 that is recognized by pathogenic aPL that in turn may activate complement (3). With this in mind, we set up an in vitro model in which normal platelets were incubated with anti-D1 monoclonal antibody in the presence or absence of a platelet agonist (i.e., TRAP) and autologous serum as a source of β2GPI and complement. Our in vitro results support only in part the role of immune complexes for complement deposition on the platelet cell membrane, because only a small percentage of activated platelets actually displayed membrane C4d. Additional mechanisms that do not require immune complex formation at the platelet surface may play a role as also suggested in SLE patients (40). Moreover, we can speculate that abnormalities in PAPS or SLE platelets themselves may be responsible for the increased complement deposition on their surface.
In conclusion the detection of complement activation products on circulating erythrocytes and platelets using a highly sensitive and specific assay further supports the view that APS is a complement-mediated disorder. Increased EC4d and PC4d percentages are associated with the active inflammatory disease in SLE. It is difficult to translate this finding to APS which is a non-acute inflammatory disorder. We failed to find an association with both the classification and non-classification criteria, including thrombocytopenia. However, we believe that this sensitive tool to evaluate complement activation may offer more information in monitoring the dynamics of the aPL-mediated pathogenic pathways during pregnancy rather than in patients far from the acute thrombotic events.
Ethics Statement
This study was carried out in accordance with the standard of the good clinical practice with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the ethical committee “Milano Area 2 426_2014.”
Author Contributions
PL, MS, and MG wrote the first draft of the manuscript. PL and MS performed the ex-vivo and in-vitro studies and interpreted data. MG, GP, and WB evaluated and recruited the patients. FP performed statistical analysis. DC detected aPL. EAF contributed to developing the in-vitro system. MB, MC, FT, and PM contributed to conception and design of the study. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Rosaria Moia and Elena Bossi for their valuable technical contribution. The study was in part supported by Ricerca Corrente 2018 Istituto Auxologico Italiano, IRCCS, to PM.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00773/full#supplementary-material
References
1. Chighizola CB, Andreoli L, Gerosa M, Tincani A, Ruffatti A, Meroni PL. The treatment of anti-phospholipid syndrome: a comprehensive clinical approach. J Autoimmun. (2018) 90:1–27. doi: 10.1016/j.jaut.2018.02.003
2. Meroni PL. Anti-beta-2 glycoprotein I epitope specificity: from experimental models to diagnostic tools. Lupus. (2016) 25:905–10. doi: 10.1177/0961203316641772
3. Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. (2011) 7:330–9. doi: 10.1038/nrrheum.2011.52
4. Meroni PL, Borghi MO, Grossi C, Chighizola CB, Durigutto P, Tedesco F. Obstetric and vascular antiphospholipid syndrome: same antibodies but different diseases? Nat Rev Rheumatol. (2018) 14:433–40. doi: 10.1038/s41584-018-0032-6
5. Radin M, Cecchi I, Roccatello D, Meroni PL, Sciascia S. Prevalence and thrombotic risk assessment of anti-beta2 glycoprotein I domain I antibodies: a systematic review. Sem Thromb Hemost. (2018) 44:466–74. doi: 10.1055/s-0037-1603936
6. Holers VM, Girardi G, Mo L, Guthridge JM, Molina H, Pierangeli SS, et al. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med. (2002) 195:211–20. doi: 10.1084/jem.200116116
7. Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clini Invest. (2003) 112:1644–54. doi: 10.1172/JCI200318817
8. Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, et al. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood. (2005) 106:2340–6. doi: 10.1182/blood-2005-03-1319
9. Pierangeli SS, Girardi G, Vega-Ostertag M, Liu X, Espinola RG, Salmon J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthr Rheumat. (2005) 52:2120–4. doi: 10.1002/art.21157
10. Thurman JM, Kraus DM, Girardi G, Hourcade D, Kang HJ, Royer PA, et al. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Mol Immunol. (2005) 42:87–97. doi: 10.1016/j.molimm.2004.07.043
11. Agostinis C, Durigutto P, Sblattero D, Borghi MO, Grossi C, Guida F, et al. A non-complement-fixing antibody to beta2 glycoprotein I as a novel therapy for antiphospholipid syndrome. Blood. (2014) 123:3478–87. doi: 10.1182/blood-2013-11-537704
12. Breen KA, Seed P, Parmar K, Moore GW, Stuart-Smith SE, Hunt BJ. Complement activation in patients with isolated antiphospholipid antibodies or primary antiphospholipid syndrome. Thromb Haemost. (2012) 107:423–9. doi: 10.1160/TH11-08-0554
13. Oku K, Atsumi T, Bohgaki M, Amengual O, Kataoka H, Horita T, et al. Complement activation in patients with primary antiphospholipid syndrome. Ann Rheumat Dis. (2009) 68:1030–5. doi: 10.1136/ard.2008.090670
14. Devreese KM, Hoylaerts MF. Is there an association between complement activation and antiphospholipid antibody-related thrombosis? Thromb Haemost. (2010) 104:1279–81. doi: 10.1160/TH10-06-0410
15. Tedesco F, Borghi MO, Gerosa M, Chighizola CB, Macor P, Lonati PA, et al. Pathogenic role of complement in antiphospholipid syndrome and therapeutic implications. Front Immunol. (2018) 9:1388. doi: 10.3389/fimmu.2018.01388
16. Meroni PL, Macor P, Durigutto P, De Maso L, Gerosa M, Ferraresso M, et al. Complement activation in antiphospholipid syndrome and its inhibition to prevent rethrombosis after arterial surgery. Blood. (2016) 127:365–7. doi: 10.1182/blood-2015-09-672139
17. Yang DH, Chang DM, Lai JH, Lin FH, Chen CH. Usefulness of erythrocyte-bound C4d as a biomarker to predict disease activity in patients with systemic lupus erythematosus. Rheumatology (Oxford). (2009) 48:1083–7. doi: 10.1093/rheumatology/kep161
18. Freysdottir J, Sigfusson A. A flow cytometric assay for measuring complement receptor 1 (CR1) and the complement fragments C3d and C4d on erythrocytes. J Immunol Methods. (1991) 142:45–52. doi: 10.1016/0022-1759(91)90291-M
19. Liu CC, Manzi S, Danchenko N, Ahearn JM. New advances in measurement of complement activation: lessons of systemic lupus erythematosus. Curr Rheumatol Rep. (2004) 6:375–81. doi: 10.1007/s11926-004-0012-5
20. Manzi S, Navratil JS, Ruffing MJ, Liu CC, Danchenko N, Nilson SE, et al. Measurement of erythrocyte C4d and complement receptor 1 in systemic lupus erythematosus. Arthr Rheum. (2004) 50:3596–604. doi: 10.1002/art.20561
21. Kao AH, Navratil JS, Ruffing MJ, Liu CC, Hawkins D, McKinnon KM, et al. Erythrocyte C3d and C4d for monitoring disease activity in systemic lupus erythematosus. Arthr Rheum. (2010) 62:837–44. doi: 10.1002/art.27267
22. Kalunian KC, Chatham WW, Massarotti EM, Reyes-Thomas J, Harris C, Furie RA, et al. Measurement of cell-bound complement activation products enhances diagnostic performance in systemic lupus erythematosus. Arthr Rheum. (2012) 64:4040–7. doi: 10.1002/art.34669
23. Dervieux T, Conklin J, Ligayon JA, Wolover L, O'Malley T, Alexander RV, et al. Validation of a multi-analyte panel with cell-bound complement activation products for systemic lupus erythematosus. J Immunol Methods. (2017) 446:54–9. doi: 10.1016/j.jim.2017.04.001
24. Merrill JT, Petri MA, Buyon J, Ramsey-Goldman R, Kalunian K, Putterman C, et al. Erythrocyte-bound C4d in combination with complement and autoantibody status for the monitoring of SLE. Lupus Sci Med. (2018) 5:e000263. doi: 10.1136/lupus-2018-000263
25. Lood C, Tyden H, Gullstrand B, Sturfelt G, Jonsen A, Truedsson L, et al. Platelet activation and anti-phospholipid antibodies collaborate in the activation of the complement system on platelets in systemic lupus erythematosus. PLoS ONE. (2014) 9:e99386. doi: 10.1371/journal.pone.0099386
26. Kao AH, McBurney CA, Sattar A, Lertratanakul A, Wilson NL, Rutman S, et al. Relation of platelet C4d with all-cause mortality and ischemic stroke in patients with systemic lupus erythematosus. Transl Stroke Res. (2014) 5:510–8. doi: 10.1007/s12975-013-0295-9
27. Lood C, Eriksson S, Gullstrand B, Jonsen A, Sturfelt G, Truedsson L, et al. Increased C1q, C4 and C3 deposition on platelets in patients with systemic lupus erythematosus–a possible link to venous thrombosis? Lupus. (2012) 21:1423–32. doi: 10.1177/0961203312457210
28. Peerschke EI, Yin W, Alpert DR, Roubey RA, Salmon JE, Ghebrehiwet B. Serum complement activation on heterologous platelets is associated with arterial thrombosis in patients with systemic lupus erythematosus and antiphospholipid antibodies. Lupus. (2009) 18:530–8. doi: 10.1177/0961203308099974
29. Hochberg MC. Updating the American college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthr Rheum. (1997) 40:1725. doi: 10.1002/art.1780400928
30. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the systemic lupus international collaborating Clinics classification criteria for systemic lupus erythematosus. Arthr Rheum. (2012) 64:2677–86. doi: 10.1002/art.34473
31. Petri M, Kim MY, Kalunian KC, Grossman J, Hahn BH, Sammaritano LR, et al. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med. (2005) 353:2550–8. doi: 10.1056/NEJMoa051135
32. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. (2006) 4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x
33. Ripoll VM, Pregnolato F, Mazza S, Bodio C, Grossi C, McDonnell T, et al. Gene expression profiling identifies distinct molecular signatures in thrombotic and obstetric antiphospholipid syndrome. J Autoimmun. (2018) 93:114–23. doi: 10.1016/j.jaut.2018.07.002
34. Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. (2009) 113:2386–93. doi: 10.1182/blood-2008-07-162503
35. Cugno M, Borghi MO, Lonati LM, Ghiadoni L, Gerosa M, Grossi C, et al. Patients with antiphospholipid syndrome display endothelial perturbation. J Autoimmun. (2010) 34:105–10. doi: 10.1016/j.jaut.2009.07.004
36. Andreoli L, Chighizola CB, Nalli C, Gerosa M, Borghi MO, Pregnolato F, et al. Clinical characterization of antiphospholipid syndrome by detection of IgG antibodies against beta2-glycoprotein i domain 1 and domain 4/5: ratio of anti-domain 1 to anti-domain 4/5 as a useful new biomarker for antiphospholipid syndrome. Arthr Rheum. (2015) 67:2196–204. doi: 10.1002/art.39187
37. Pengo V. ISTH guidelines on lupus anticoagulant testing. Thromb Res. (2012) 130(Suppl. 1):S76–7. doi: 10.1016/j.thromres.2012.08.283
38. Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Ann Rev Immunol. (2004) 22:431–56. doi: 10.1146/annurev.immunol.22.012703.104549
39. Durigutto P, Grossi C, Borghi MO, Macor P, Pregnolato F, Raschi E, et al. New insight into antiphospholipid syndrome: antibodies to beta2glycoprotein I-domain 5 fail to induce thrombi in rats. Haematologica. (2018) 104:819–26. doi: 10.3324/haematol.2018.198119
40. Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol. (2010) 47:2170–5. doi: 10.1016/j.molimm.2010.05.009
41. Perez D, Stojanovich L, Naranjo L, Stanisavljevic N, Bogdanovic G, Serrano M, et al. Presence of immune complexes of IgG/IgM bound to B2-glycoprotein I is associated with non-criteria clinical manifestations in patients with antiphospholipid syndrome. Front Immunol. (2018) 9:2644. doi: 10.3389/fimmu.2018.02644
Keywords: anti-phospholipid syndrome, beta2–glycoprotein I, complement, C4d, platelets, erythrocytes
Citation: Lonati PA, Scavone M, Gerosa M, Borghi MO, Pregnolato F, Curreli D, Podda G, Femia EA, Barcellini W, Cattaneo M, Tedesco F and Meroni PL (2019) Blood Cell-Bound C4d as a Marker of Complement Activation in Patients With the Antiphospholipid Syndrome. Front. Immunol. 10:773. doi: 10.3389/fimmu.2019.00773
Received: 15 January 2019; Accepted: 25 March 2019;
Published: 12 April 2019.
Edited by:
Jean-Christophe Raymond Gris, Centre Hospitalier Universitaire de Nîmes, FranceReviewed by:
Giacomo Emmi, University of Florence, ItalyAlfred Hyoungju Kim, Washington University School of Medicine in St. Louis, United States
Copyright © 2019 Lonati, Scavone, Gerosa, Borghi, Pregnolato, Curreli, Podda, Femia, Barcellini, Cattaneo, Tedesco and Meroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pier Luigi Meroni, pierluigi.meroni@unimi.it
†These authors have contributed equally to this work