 Kazunari Kaneko
Kazunari Kaneko Shohei Akagawa
Shohei Akagawa- Department of Pediatrics, Kansai Medical University, Osaka, Japan
Kawasaki disease (KD) was first described by Dr. Tomisaku Kawasaki in 1967. The etiology of KD has been studied comprehensively but remains largely unknown. The disease seems to result from the interplay of genetic and environmental susceptibility factors with infectious triggers, followed by a subsequent abnormal immune response characterized by increased levels of inflammatory cytokines and chemokines during the acute phase. Evidence has mounted to suggest that an imbalance between T helper 17 cells (Th17s) and regulatory T cells (Tregs) is associated with aberrant immune responses in KD. Recent advances in culture-independent techniques for detection and identification of intestinal commensal bacteria enabled the discovery that Th17 and Treg differentiation are regulated by short chain fatty acids (SCFAs), in particular butyrate, produced by the gut microbiota. This finding provided a mechanistic link between dysbiosis, defined as changes in the composition of the gut microbiota, and various inflammatory diseases. On this basis, we propose that dysbiosis, with reduced production of SCFAs leading to imbalances of Th17s/Tregs, could be involved in the etiology of KD. A pilot study supported this hypothesis, as only fecal concentrations of butyrate were significantly reduced in KD patients among SCFAs. This evolving perspective prompted us to undertake metagenomic analyses of bacterial DNA from the feces of KD patients who were antibiotic-naïve at diagnosis. Simultaneous measurements of Th17s/Tregs in peripheral blood and SCFA concentrations in feces would provide valuable information regarding the association between dysbiosis and dysregulated immune responses in KD.
Introduction
Kawasaki disease (KD), named after Dr. Tomisaku Kawasaki deceased June 5th, 2020, mainly affects young children between the ages of 6 months and 4 years (1). KD is characterized by persistent fever, bilateral conjunctival congestion, changes of the lips and oral cavity, polymorphous exanthema, changes of peripheral extremities, and acute non-purulent cervical lymphadenopathy (2, 3). Although KD was originally reported to be self-limiting and benign (4), it is now recognized as a systemic vasculitis with a specific predilection for forming coronary artery lesions. These develop in up to 25% of children with KD who are not treated with intravenous immunoglobulin (5). Coronary artery lesions associated with KD are the most common causes of pediatric heart disease in developed countries. The incidence of KD in the Japanese population continues to increase and reached 330 cases per 100,000 children aged ≤4 years per year in 2015 (6).
Despite extensive ongoing research into the etiology of KD, the underlying mechanisms of this enigmatic vasculitis are not fully understood (3, 7).
Current Paradigm of the Etiology of KD
The current paradigm of KD pathogenesis is that the disease results from a pathologically amplified immune response against infectious agent(s) in a genetically and environmentally susceptible child (3). This paradigm is based on the following observations.
Proposed Infectious Causes of KD
First, there is clinical overlap between KD and infectious diseases such as adenovirus and streptococcosis. Second, seasonal clustering of KD in the winter and spring mimics that of several viral diseases (8). Third, temporal clusters of epidemics have been reported in Japan, the US, Canada, and Finland (9). Moreover, an outbreak in Japan began in Tokyo and spread throughout the country over a period of 6 months (10). Finally, low incidence in the first 3 months of life suggests at least partial protection from trans-placental antibodies (11). The low incidence of KD in schoolchildren indicates a potential role of common antigen(s) that most children encounter uneventfully in early childhood and against which they mount an appropriate and protective immune response (12). However, efforts to find a single unifying microbiological cause of KD have been, to date, unsuccessful. Standard microbiological techniques, molecular methods and serological investigations have all failed to identify an etiological agent.
It has long been held that infection by one or more widely distributed microorganism(s) might elicit dysregulated immune responses in genetically susceptible children resulting in KD. Candidate pathogens include Epstein-Barr virus (13, 14), human herpes virus (15), human immunodeficiency virus (16), human adenovirus (17), human coronavirus (18), retrovirus (19), human parvovirus B19 (20), human bocavirus (21), Staphylococcus aureus (22), Streptococcus pyogenes (23), Yersinia pseudotuberculosis (24), Bacillus cereus (25), Mycoplasma pneumoniae (26), Mycobacterium spp. (27), Bartonella henselae (28), Coxiella burnetti (29), and Candida spp. (30). Candida albicans (C. albicans) has recently drawn attention, as administration of CAWS (water-soluble extracellular polysaccharide from culture supernatants of C. albicans) induces coronary arteritis similar to KD in mice (31). Several reports suggested that C. albicans plays an important role as an infectious trigger of KD (30, 32, 33). Considering the recent pandemic of coronavirus disease 2019 (COVID-19), potential links between KD and COVID-19 are also deserving of mention (34–37). Clusters of children presenting with KD-like symptoms have been documented in the UK, US, France, and Italy, and some of them were confirmed to have COVID-19. It appears that hyperinflammation associated with COVID-19 could act as primer for KD development in individuals having genetically or environmentally determined predisposition. However, the specific mechanism is not yet defined (34).
Genetic and Environmental Risk Factors for KD
The higher incidence of KD in Asian countries suggests a genetic predisposition for acquiring the disease (9). Increased risk in family members of KD patients in Japanese populations (38) also hint at genetically determined susceptibility. Recent advances have been made in identifying disease-susceptibility genes from genome-wide association studies. Candidate genes contributing to KD susceptibility include B-lymphoid kinase (BLK) (39), caspase-3 (CASP3) (40), low-affinity immunoglobulin gamma Fc region receptor II-a (FCGR2A) (41), human leukocyte antigen (HLA) (39), inositol 1,4,5-triphosphate kinase-C (ITPKC) (42), transforming growth factor (TGF)-β2 (TGFβ2) (43), TGF-β receptor 2 (TGFBR2) (43), SMAD3 (43), CD40 (39), and ORAI1 (44).
Epidemiologic studies have extensively searched for environmental factors that may explain variation and seasonality in KD incidence. Surveys from the US and Japan have demonstrated that higher precipitation and lower temperatures were associated with higher incidence of KD (45, 46). An investigation of the role of the early social environment in KD susceptibility in a Japanese population found that higher household income, smaller family size, and urbanization were associated with increased KD incidence (47). This study, however, did not find a significant association between absence of infectious exposures during early life and KD.
Aberrant Immune Responses in KD
The prominent role played by the immune system in KD has been confirmed by many studies demonstrating activation of neutrophils and other immune cells as well as overproduction of inflammatory cytokines and chemokines such as tumor necrosis factor-α, interleukin (IL)-1, IL-2, IL-6, and IL-8, and monocyte chemotactic protein-1 (3). Levels of inflammatory cytokines and chemokines are reported to be elevated during the acute phase of KD. However, the mechanisms responsible for abnormal immune responses and overexpression of inflammatory cytokines remain unclear. Several lines of evidence have revealed decreased numbers of regulatory T cells (Tregs) and imbalances between T helper 17 cells (Th17s) and Tregs in acute KD (48–50). Jia et al. elegantly demonstrated that Th17 proportions and cytokine (IL-17, IL-6, and IL-23) levels were significantly increased, while Treg proportions and expression of Treg transcription factors (e.g., FoxP3) were significantly decreased in patients with acute KD (49). They concluded that Th17 expansion and Treg depletion were characteristic of acute KD. Furthermore, recent studies suggested that treatment efficacy was associated with decreased Th17 proportions and increased Treg proportions, with both returning closer to a normal range (48, 51). Thus, Th17/Treg imbalances may contribute to exaggerated immune responses in KD patients.
The Importance of Gut Microbiota in Human Health
Growing evidence suggests that disturbances within intestinal communities of commensal bacteria may lead to illness through aberrant immune system development. While the study of gut microbiology related to human health has a centennial history, recent technological advances have enabled us to explore this field in a more sophisticated manner. Current approaches rely primarily on culture-independent methods such as amplification of conserved regions of the 16S rRNA gene present in all bacteria (52). Studies using these techniques have demonstrated that an adult humans harbor 100 trillion gut bacteria comprising more than 1,000 different species and approximately 160 species per person per fecal sample (53). Development of the gut microbiota, defined as its colonization by microorganisms, might begin not at birth but in utero. However, the existence of viable bacteria in the womb was recently questioned (54–56). The maternal microbiota provides the first microbial inoculum, and from birth, microbial diversity increases and converges toward an adult-like microbiome within the first 3–5 years of life (53). The composition of the microbiota in childhood depends on various factors including sanitation, mode of delivery, maturity at birth, infant diet, antibiotic use during infancy, immunizations, and environmental factors such as geography or diet (52, 53, 57, 58). Ethnic and genetic factors may also give rise to dysbiosis (59, 60). The factors that can alter the microbiome are being studied as potential drivers of changing trends in non-communicable diseases. Dysbiosis, defined as changes in the composition of the gut microbiota, may be associated with several clinical conditions including obesity and metabolic diseases (61), cancer (62), autoimmune diseases (63), allergy (64), chronic inflammatory bowel diseases (65, 66), chronic kidney diseases (67), and autistic-spectrum disorders (68). We also found that dysbiosis was present in children with idiopathic nephrotic syndrome (69).
Although research on the mechanism(s) through which dysbiosis impairs human health has just begun, regulation of the gut immune system by microbiota is believed to be involved. Experiments with germ-free animals (deficient in commensal microbiota including gut microbiota) have demonstrated that microbial colonization promotes anatomical development of the intestinal epithelium, increases epithelial cell turnover rates, and initiates the maturation of gut-associated lymphoid tissue (70). Both Tregs and Th17s have received much attention in terms of their role in the gut immune system and its regulation by microbiota (71). Tregs were originally identified as CD4-positive, CD25-positive and Foxp3-positive T cells that exerted inhibitory control over immune responses (72), maintained tolerance to self-antigens, and prevented autoimmune disease (73). There are several immunosuppressive mechanisms mediated by Tregs, including secretion of immunosuppressive cytokines such as IL-10 and TGF-β, functional modification or killing of antigen-presenting cells, and cell contact-dependent suppression via cytotoxic T-lymphocyte- associated protein 4 (72). Tregs mainly arise from naïve T cell precursors following stimulation by short chain fatty acids (SCFAs) such as acetate, propionate or butyrate produced by the gut microbiota (74, 75). Previous studies demonstrated that members of the genus Clostridium were potent inducers of Treg differentiation through butyrate production (76, 77) and that reduced luminal concentrations of SCFAs resulted in impaired development of intestinal Tregs in germ-free mice (74, 76). In these mice, reconstitution with commensal bacteria or administration of SCFAs, especially butyrate, restored Treg frequency (76), supporting the role of bacterial metabolites in Treg development. Therefore, it has been proposed that a decrease in the relative abundance of butyrate-producing microbes may disrupt mucosal immune homeostasis (78). The gut microbiota also plays a crucial role in the induction of effector T cell responses in the intestine. Th17s are a subtype of CD4-positive T cells specialized for mounting immune responses against fungi and some extracellular bacteria. In addition to IL-17A, Th17s produce IL-17F, IL-21, IL-22, and IL-26 (79). Because IL-17 is a potent proinflammatory cytokine that amplifies ongoing inflammation, aberrant regulation of Th17s contributes to development of inflammatory and autoimmune disorders. In germ-free mice, the number of Th17s was markedly decreased. However, the Th17 compartment was restored by reconstitution with conventional microbiota (80), thus indicating a crucial role for gut microbes in Th17 development. Among commensal bacteria, segmented filamentous bacteria (SFB) are one of the most potent inducers of Th17s. Colonization of mice by SFB causes abundant accumulation of Th17s in the small intestine via enhanced production of serum amyloid A (81, 82). The existence of human commensal bacteria equivalent to SFB in rodents is probable because mixtures of bacterial strains isolated from fecal samples ulcerative colitis patients could induce Th17 development (81).
The Gut Microbiota in KD
The gut, the largest interface between microbial factors and the host, contains the largest proportion of bacteria and the largest amount of lymphoid tissue in the body. Thus, it was hypothesized that the intestinal environment might be reshaped in patients with KD. Indeed, KD patients frequently exhibit gastrointestinal symptoms and complications (83). The contribution of the gut microbiota to KD has been evaluated in limited numbers of small cohorts using culture-based methods. Several studies have been performed to identify the causative microbial agent(s) of KD at disease onset. Takeshita et al. showed that the gut microbiomes of KD patients were distinguished by a lack of Lactobacilli during the acute phase (84), while Nagata et al. isolated both HSP60-producing Gram-negative bacteria and Gram-positive cocci capable of inducing Vβ2 T cell expansion from KD patients (85). These results indicated that distinctive gut microbes might be involved in the pathophysiology of KD. However, because these studies of the microbiomes of KD patients were carried out using culture-based methods, microorganisms that cannot be cultured, which constitute more than half of the human gut microbiome, would have been overlooked.
Unlike culture methods, metagenomic analyses can reveal the composition of the intestinal microbiota irrespective of the ability to culture microbes. Kinumaki et al. first reported the results of a metagenomic analysis of feces using culture-independent methods (86). They collected 28 paired fecal samples from children with KD during the acute and non-acute phases and demonstrated that Streptococcus spp., including S. pneumoniae, S. pseudopneumoniae, S. mitis, S. oralis, S. gordonii, and S. sanguinis, were more abundant during the acute phase while the genera Ruminococcus, Roseburia and Faecalibacterium were less abundant. Unfortunately, more than half of subjects were treated empirically with antibiotics during the early phase of KD when the fecal samples were collected because clinical and laboratory findings often do not fulfill the diagnostic criteria of KD (2), but are instead suggestive of bacterial infections (87). As antibiotic administration rapidly perturbs the gut microbiota (88), these results may reflect the effects of antibiotic therapy on the gut microbiota and not dysbiosis associated with KD.
Novel Perspectives on KD Pathogenesis
Here, we would like to focus on a novel viewpoint on the role of the gut microbiota in KD: Dysbiosis, defined as changes in the composition of the gut microbiota and caused by various prenatal and postnatal factors that are not necessarily infectious agent(s), might contribute to genetically and environmentally determined predilection for KD. This perspective is illustrated in Figure 1 and can be explained as follows: [1] various factors during the in utero and postnatal period drive dysbiosis in young children; [2] dysbiosis results in reduced intestinal production of SCFAs including butyrate; [3] reduced levels of SCFAs in the gut cause an imbalance of Th17s/Tregs; and [4] individuals with Th17/Treg imbalances develop hypercytokinemia triggered by ubiquitous infectious agents(s), followed by KD (Figure 1). Recent observations revealed that viral respiratory infections may alter microbial growth in the gut leading to dysbiosis (89). In addition, the gut microbiota has been shown to play an important role in regulating the generation of virus-specific CD4-positive and CD8-positive T cells and antibody responses following influenza virus infection (90). Therefore, we hypothesize that young children with dysbiosis are prone to a vicious cycle of hypercytokinemia following infection by viruses.
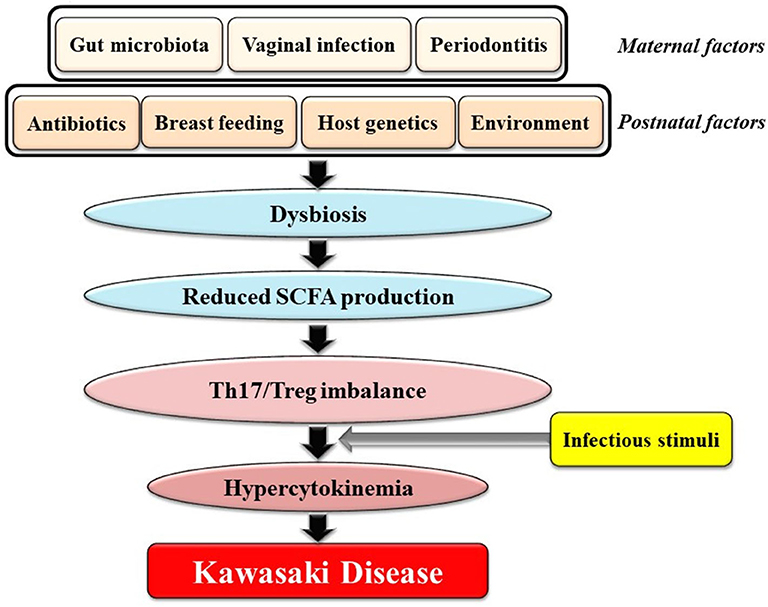
Figure 1. Potential association between gut dysbiosis and Kawasaki Disease (KD). Various factors during both the in utero and postnatal period could drive dysbiosis in infants. Dysbiosis results in reduced intestinal production of SCFAs including butyrate. Reduced SCFA concentrations cause an imbalance of Th17s/Tregs. Individuals with Th17/Treg imbalances develop hypercytokinemia following stimuli from one or more infectious agents(s) followed by KD. SCFAs, short chain fatty acids.
This paradigm for the pathogenesis of KD contrasts with previous hypotheses, which focused on specific microorganisms, toxins, or pathogen-associated molecular patterns (91–93).
As butyrate has been reported to limit Th17 differentiation and promote Treg development (94–96), we hypothesize that KD-associated dysbiosis might be characterized by lower abundance of butyrate-producing bacteria. Interestingly, the genera Roseburia and Faecalibacterium, which were reported to be less abundant in patients with acute KD by Kinumaki et al. (86), are butyrate-producing bacteria (97, 98). Furthermore, the strong association between KD and allergic diseases (99–101) in which dysbiosis plays an important role (102) also supports this perspective. Recent observations of a potential association between previous antibiotic therapy and development of KD also supports our hypothesis (103). In this study, the median interval between the final dose of antibiotics and the onset of KD was 2.5 months, which was insufficient time for restoration of the gut microbiota and complete resolution of dysbiosis caused by antibiotic use (104).
To confirm our hypothesis, further investigations involving metagenomic analysis of bacterial DNA from the feces of a larger number of antibiotic-naïve patients with KD is clearly needed. It would be worthwhile to compare the proportions of specific microbial species such as butyrate-producing bacteria. Simultaneous analysis of Th17/Treg ratios in peripheral blood and measurement of fecal butyrate concentrations, reflecting the intestinal production of SCFAs, would also be helpful.
We have just begun a project to test our hypothesis with approval from our institutional ethics committee (approval no. 2015127) and parental informed consent. Fecal samples will be collected not only from KD patients and healthy children but also from controls with viral infections. This will allow us to specifically characterize dysbiosis in KD because recent observations suggested that viral infection itself may cause dysbiosis (89).
The results of our pilot study of four acute KD patients (median age 1.1 years, range 0.8–2.1 years; 3 boys and 1 girl) and four healthy children (median age 2.1 years, range 1.2–3.7 years, 3 boys and 1 girl) supports our perspective as fecal butyrate concentrations were significantly lower in KD patients (p < 0.05, Mann–Whitney U test). In contrast, fecal concentrations of acetate, lactate, and propionate did not differ between KD patients and healthy children (Figure 2). The KD patients studied had a median body mass index of 14.7 (range: 13.0–18.1), median maximal body temperatures of 39.0°C (range: 38.0–39.7°C), and median maximal C-reactive protein levels of 41 mg/L (range: 20–57 mg/L). All KD patients presented with typical clinical features and fulfilled the diagnostic criteria (2). None reported diarrhea or constipation and none received antibiotics. Fecal samples were provided before intravenous immunoglobulin administration. No cases were complicated by coronary artery lesions.
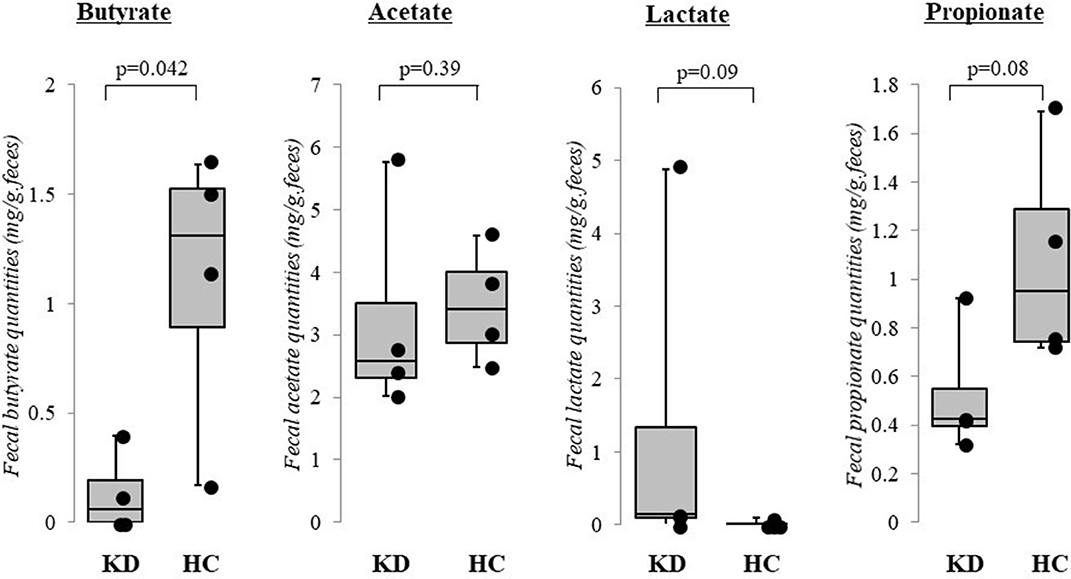
Figure 2. Results of pilot study: fecal organic acid concentrations in Kawasaki disease (KD). Fecal butyrate concentrations were significantly lower in KD patients (p < 0.05, Mann–Whitney U test) while concentrations of acetate, lactate, and propionate did not differ between KD patients and healthy control children. KD: Kawasaki disease; HC: age-matched healthy children. The horizontal lines in the boxes represent the median values, and the bottoms and tops of the boxes represent the 25th and 75th percentiles, respectively. The vertical lines extend from the box to the maximum values and minimum values. Frozen fecal specimens were thawed, and a 0.1 g subsample was placed in a 2.0 mL tube with zirconia beads and suspended in 0.1 mM perchloric acid solution containing 3% phenol. Samples were heated at 80°C for 15 min, vortexed at 5 m/s for 45 s using FastPrep 24 instrument (MP Biomedicals, Irvine, CA, USA), and centrifuged at 15,350 g for 10 min. Supernatants were filtered using 0.45 μm filters. Fecal organic acids including acetate, propionate, butyrate, and lactate were measured using high-performance liquid chromatography (Prominence, Shimadzu, Kyoto, Japan) using a post-column reaction with a detector (CDD-10A, Shimadzu), two columns arranged in tandem (Shim-pack SCR-102(H), 300 × 8 mm ID, Shimadzu) and a guard column (Shim-pack SCR-102(H), 50 × 6 mm ID, Shimadzu). The mobile phase was 5 mM p-toluenesulfonic acid and the reaction solution was 5 mM p-toluenesulfonic acid containing 100 μM ethylenediaminetetraacetic acid and 20 mM Bis-Tris. The flow rate and oven temperature were 0.8 mL/min and 45°C, respectively. The detector cell temperature was kept at 48°C. Measurements were performed at Techno Suruga Lab, Shizuoka, Japan.
What factors perturb the gut microbiota and cause dysbiosis in young children? As shown in Figure 1, both prenatal and postnatal conditions affect the establishment of the intestinal microbiota in infancy. Factors that influence the initial colonization of the gut by microbes include maternal factors such as the maternal gut microbiota, vaginal infection or periodontitis as well as postnatal factors such as cesarean delivery, formula feeding, excessive antibiotic use, host genetics, and the environment (105). Interestingly, formula feeding (106) and social environment factors such as higher household income, smaller family size, and urbanization were associated with both increased risk of dysbiosis and increased KD incidence (47). In addition, the peak age of KD onset ranging from 6 months to 4 years corresponds to the critical period for establishment of the gut microbiota during the first 1,000 days of life (107).
Conclusions
We believe that dysbiosis underlies KD and could contribute to genetically and environmentally determined predilections for KD. Therefore, KD might be included in the growing list of dysbiosis-associated conditions. If our perspective is confirmed, it would be valuable to investigate whether supplying probiotics starting at birth could reduce the risk of KD in infancy.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Kansai Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Edanz Group (https://en-author-services.edanzgroup.com/) for editing a draft of this manuscript.
References
1. Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the committee on rheumatic fever, endocarditis, and kawasaki disease, council on cardiovascular disease in the young, American heart association. Pediatrics. (2004) 114:1708–33. doi: 10.1542/peds.2004-2182
2. Singh S, Jindal AK, Pilania RK. Diagnosis of kawasaki disease. Int J Rheum Dis. (2018) 21:36–44. doi: 10.1111/1756-185X.13224
3. Greco A, De Virgilio A, Rizzo MI, Tombolini M, Gallo A, Fusconi M, et al. Kawasaki disease: an evolving paradigm. Autoimmun Rev. (2015) 14:703–9. doi: 10.1016/j.autrev.2015.04.002
4. Kawasaki T. [Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children]. Arerugi. (1967) 16:178–222.
5. Burns JC, Glodé MP. Kawasaki syndrome. Lancet. (2004) 364:533–44. doi: 10.1016/S0140-6736(04)16814-1
6. Makino N, Nakamura Y, Yashiro M, Kosami K, Matsubara Y, Ae R, et al. Nationwide epidemiologic survey of kawasaki disease in Japan, 2015-2016. Pediatr Int. (2019) 61:397–403. doi: 10.1111/ped.13809
7. Lo MS. A framework for understanding kawasaki disease pathogenesis. Clin Immunol. (2020) 214:108385. doi: 10.1016/j.clim.2020.108385
8. Burns JC, Herzog L, Fabri O, Tremoulet AH, Rodo X, Uehara R, et al. Seasonality of kawasaki disease: a global perspective. PLoS ONE. (2013) 8:e74529. doi: 10.1371/journal.pone.0074529
9. Uehara R, Belay ED. Epidemiology of kawasaki disease in Asia, Europe, and the United States. J Epidemiol. (2012) 22:79–85. doi: 10.2188/jea.JE20110131
10. Yanagawa H, Nakamura Y, Kawasaki T, Shigematsu I. Nationwide epidemic of kawasaki disease in Japan during winter of 1985-86. Lancet. (1986) 2:1138–9. doi: 10.1016/S0140-6736(86)90541-6
11. Nomura Y, Yoshinaga M, Masuda K, Takei S, Miyata K. Maternal antibody against toxic shock syndrome toxin-1 may protect infants younger than 6 months of age from developing kawasaki syndrome. J Infect Dis. (2002) 185:1677–80. doi: 10.1086/340513
12. Burgner D, Harnden A. Kawasaki disease: what is the epidemiology telling us about the etiology? Int J Infect Dis. (2005) 9:185–94. doi: 10.1016/j.ijid.2005.03.002
13. Muso E, Fujiwara H, Yoshida H, Hosokawa R, Yashiro M, Hongo Y, et al. Epstein-barr virus genome-positive tubulointerstitial nephritis associated with kawasaki disease-like coronary aneurysms. Clin Nephrol. (1993) 40:7–15.
14. Kikuta H, Matsumoto S, Osato T. Kawasaki disease and epstein-barr virus. Acta Paediatr Jpn. (1991) 33:765–70. doi: 10.1111/j.1442-200X.1991.tb02606.x
15. Shingadia D, Bose A, Booy R. Could a herpesvirus be the cause of kawasaki disease? Lancet Infect Dis. (2002) 2:310–3. doi: 10.1016/S1473-3099(02)00265-7
16. Johnson RM, Bergmann KR, Manaloor JJ, Yu X, Slaven JE, Kharbanda AB. Pediatric kawasaki disease and adult human immunodeficiency virus kawasaki-like syndrome are likely the same malady. Open Forum Infect Dis. (2016) 3:ofw160. doi: 10.1093/ofid/ofw160
17. Okano M, Thiele GM, Sakiyama Y, Matsumoto S, Purtilo DT. Adenovirus infection in patients with kawasaki disease. J Med Virol. (1990) 32:53–7. doi: 10.1002/jmv.1890320109
18. Esper F, Shapiro ED, Weibel C, Ferguson D, Landry ML, Kahn JS. Association between a novel human coronavirus and kawasaki disease. J Infect Dis. (2005) 191:499–502. doi: 10.1086/428291
19. Shulman ST, Rowley AH, Fresco R, Morrison DC. The etiology of kawasaki disease: retrovirus? Prog Clin Biol Res. (1987) 250:117–24.
20. Nigro G, Zerbini M, Krzysztofiak A, Gentilomi G, Porcaro MA, Mango T, et al. Active or recent parvovirus B19 infection in children with kawasaki disease. Lancet. (1994) 343:1260–1. doi: 10.1016/S0140-6736(94)92154-7
21. Santos RA, Nogueira CS, Granja S, Baptista JB, Ribeiro ML, Rocha MG. Kawasaki disease and human bocavirus–potential association? J Microbiol Immunol Infect. (2011) 44:235–7. doi: 10.1016/j.jmii.2011.01.016
22. Leung DY, Meissner HC, Fulton DR, Murray DL, Kotzin BL, Schlievert PM. Toxic shock syndrome toxin-secreting Staphylococcus aureus in kawasaki syndrome. Lancet. (1993) 342:1385–8. doi: 10.1016/0140-6736(93)92752-F
23. Yoshioka T, Matsutani T, Iwagami S, Toyosaki-Maeda T, Yutsudo T, Tsuruta Y, et al. Polyclonal expansion of TCRBV2- and TCRBV6-bearing T cells in patients with kawasaki disease. Immunology. (1999) 96:465–72. doi: 10.1046/j.1365-2567.1999.00695.x
24. Vollmer-Conna U, Piraino BF, Cameron B, Davenport T, Hickie I, Wakefield D, et al. Cytokine polymorphisms have a synergistic effect on severity of the acute sickness response to infection. Clin Infect Dis. (2008) 47:1418–25. doi: 10.1086/592967
25. Kusuda T, Nakashima Y, Murata K, Kanno S, Nishio H, Saito M, et al. Kawasaki disease-specific molecules in the sera are linked to microbe-associated molecular patterns in the biofilms. PLoS ONE. (2014) 9:e113054. doi: 10.1371/journal.pone.0113054
26. Vitale EA, La Torre F, Calcagno G, Infricciori G, Fede C, Conti G, et al. Mycoplasma pneumoniae: a possible trigger of kawasaki disease or a mere coincidental association? Report of the first four Italian cases. Minerva Pediatr. (2010) 62:605–7.
27. Yokota S, Tsubaki K, Kuriyama T, Shimizu H, Ibe M, Mitsuda T, et al. Presence in kawasaki disease of antibodies to mycobacterial heat-shock protein HSP65 and autoantibodies to epitopes of human HSP65 cognate antigen. Clin Immunol Immunopathol. (1993) 67:163–70. doi: 10.1006/clin.1993.1060
28. Rathore MH, Barton LL, Dawson JE, Regnery RL, Ayoub EM. Ehrlichia chaffeensis and rochalimaea antibodies in kawasaki disease. J Clin Microbiol. (1993) 31:3058–9. doi: 10.1128/JCM.31.11.3058-3059.1993
29. Weir WR, Bouchet VA, Mitford E, Taylor RF, Smith H. Kawasaki disease in European adult associated with serological response to coxiella burneti. Lancet. (1985) 2:504. doi: 10.1016/S0140-6736(85)90440-4
30. Rodó X, Curcoll R, Robinson M, Ballester J, Burns JC, Cayan DR, et al. Tropospheric winds from northeastern China carry the etiologic agent of kawasaki disease from its source to Japan. Proc Natl Acad Sci U S A. (2014) 111:7952–7. doi: 10.1073/pnas.1400380111
31. Ohno N. Murine model of kawasaki disease induced by mannoprotein-beta-glucan complex, CAWS, obtained from Candida albicans. Jpn J Infect Dis. (2004) 57:S9–10.
32. Ishida-Okawara A, Nagi-Miura N, Oharaseki T, Takahashi K, Okumura A, Tachikawa H, et al. Neutrophil activation and arteritis induced by C. albicans water-soluble mannoprotein-beta-glucan complex (CAWS). Exp Mol Pathol. (2007) 82:220–6. doi: 10.1016/j.yexmp.2006.05.006
33. Nagi-Miura N, Okuzaki D, Torigata K, Sakurai MA, Ito A, Ohno N, et al. CAWS administration increases the expression of interferon γ and complement factors that lead to severe vasculitis in DBA/2 mice. BMC Immunol. (2013) 14:44. doi: 10.1186/1471-2172-14-44
34. Xu S, Chen M, Weng J. COVID-19 and kawasaki disease in children. Pharmacol Res. (2020) 159:104951. doi: 10.1016/j.phrs.2020.104951
35. Viner RM, Whittaker E. Kawasaki-like disease: emerging complication during the COVID-19 pandemic. Lancet. (2020) 395:1741–3. doi: 10.1016/S0140-6736(20)31129-6
36. Verdoni L, Mazza A, Gervasoni A, Martelli L, Ruggeri M, Ciuffreda M, et al. An outbreak of severe kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. (2020) 395:1771–8. doi: 10.1016/S0140-6736(20)31103-X
37. Moreira A. Kawasaki disease linked to COVID-19 in children. Nat Rev Immunol. (2020) 20:407. doi: 10.1038/s41577-020-0350-1
38. Uehara R, Yashiro M, Nakamura Y, Yanagawa H. Parents with a history of kawasaki disease whose child also had the same disease. Pediatr Int. (2011) 53:511–4. doi: 10.1111/j.1442-200X.2010.03267.x
39. Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for kawasaki disease. Nat Genet. (2012) 44:517–21. doi: 10.1038/ng.2220
40. Onouchi Y, Ozaki K, Buns JC, Shimizu C, Hamada H, Honda T, et al. Common variants in CASP3 confer susceptibility to kawasaki disease. Hum Mol Genet. (2010) 19:2898–906. doi: 10.1093/hmg/ddq176
41. Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for kawasaki disease. Nat Genet. (2011) 43:1241–6. doi: 10.1038/ng.981
42. Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. (2008) 40:35–42. doi: 10.1038/ng.2007.59
43. Shimizu C, Jain S, Davila S, Hibberd ML, Lin KO, Molkara D, et al. Transforming growth factor-beta signaling pathway in patients with kawasaki disease. Circ Cardiovasc Genet. (2011) 4:16–25. doi: 10.1161/CIRCGENETICS.110.940858
44. Onouchi Y, Fukazawa R, Yamamura K, Suzuki H, Kakimoto N, Suenaga T, et al. Variations in ORAI1 gene associated with kawasaki disease. PLoS ONE. (2016) 11:e0145486. doi: 10.1371/journal.pone.0145486
45. Bronstein DE, Dille AN, Austin JP, Williams CM, Palinkas LA, Burns JC. Relationship of climate, ethnicity and socioeconomic status to kawasaki disease in San Diego county, 1994 through 1998. Pediatr Infect Dis J. (2000) 19:1087–91. doi: 10.1097/00006454-200011000-00012
46. Abrams JY, Blase JL, Belay ED, Uehara R, Maddox RA, Schonberger LB, et al. Increased kawasaki disease incidence associated with higher precipitation and lower temperatures, Japan, 1991-2004. Pediatr Infect Dis J. (2018) 37:526–30. doi: 10.1097/INF.0000000000001838
47. Fujiwara T, Shobugawa Y, Matsumoto K, Kawachi I. Association of early social environment with the onset of pediatric kawasaki disease. Ann Epidemiol. (2019) 29:74–80. doi: 10.1016/j.annepidem.2018.10.010
48. Guo MM, Tseng WN, Ko CH, Pan HM, Hsieh KS, Kuo HC. Th17- and treg-related cytokine and mRNA expression are associated with acute and resolving kawasaki disease. Allergy. (2015) 70:310–8. doi: 10.1111/all.12558
49. Jia S, Li C, Wang G, Yang J, Zu Y. The T helper type 17/regulatory T cell imbalance in patients with acute kawasaki disease. Clin Exp Immunol. (2010) 162:131–7. doi: 10.1111/j.1365-2249.2010.04236.x
50. Furuno K, Yuge T, Kusuhara K, Takada H, Nishio H, Khajoee V, et al. CD25+CD4+ regulatory T cells in patients with kawasaki disease. J Pediatr. (2004) 145:385–90. doi: 10.1016/j.jpeds.2004.05.048
51. Koizumi K, Hoshiai M, Katsumata N, Toda T, Kise H, Hasebe Y, et al. Infliximab regulates monocytes and regulatory T cells in kawasaki disease. Pediatr Int. (2018) 60:796–802. doi: 10.1111/ped.13555
52. Arrieta MC, Stiemsma LT, Amenyogbe N, Brown EM, Finlay B. The intestinal microbiome in early life: health and disease. Front Immunol. (2014) 5:427. doi: 10.3389/fimmu.2014.00427
53. Rodriguez JM, Murphy K, Stanton C, Ross RP, Kober OI, Juge N, et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis. (2015) 26:26050. doi: 10.3402/mehd.v26.26050
54. Rackaityte E, Halkias J, Fukui EM, Mendoza VF, Hayzelden C, Crawford ED, et al. Viable bacterial colonization is highly limited in the human intestine in utero. Nat Med. (2020) 26:599–607. doi: 10.1038/s41591-020-0761-3
55. Leiby JS, McCormick K, Sherrill-Mix S, Clarke EL, Kessler LR, Taylor LJ, et al. Lack of detection of a human placenta microbiome in samples from preterm and term deliveries. Microbiome. (2018) 6:196. doi: 10.1186/s40168-018-0575-4
56. Perez-Muñoz ME, Arrieta MC, Ramer-Tait AE, Walter J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome. (2017) 5:48. doi: 10.1186/s40168-017-0268-4
57. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. (2011) 334:105–8. doi: 10.1126/science.1208344
58. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. (2010) 107:14691–6. doi: 10.1073/pnas.1005963107
59. Stearns JC, Zulyniak MA, de Souza RJ, Campbell NC, Fontes M, Shaikh M, et al. Ethnic and diet-related differences in the healthy infant microbiome. Genome Med. (2017) 9:32. doi: 10.1186/s13073-017-0421-5
60. Kubinak JL, Stephens WZ, Soto R, Petersen C, Chiaro T, Gogokhia L, et al. MHC variation sculpts individualized microbial communities that control susceptibility to enteric infection. Nat Commun. (2015) 6:8642. doi: 10.1038/ncomms9642
61. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. (2006) 444:1027–31. doi: 10.1038/nature05414
62. Biragyn A, Ferrucci L. Gut dysbiosis: a potential link between increased cancer risk in ageing and inflammaging. Lancet Oncol. (2018) 19:e295–e304. doi: 10.1016/S1470-2045(18)30095-0
63. Van Praet JT, Donovan E, Vanassche I, Drennan MB, Windels F, Dendooven A, et al. Commensal microbiota influence systemic autoimmune responses. EMBO J. (2015) 34:466–74. doi: 10.15252/embj.201489966
64. Fyhrquist N. The human microbiota and its relationship with allergies. Gastroenterol Clin North Am. (2019) 48:377–87. doi: 10.1016/j.gtc.2019.04.005
65. Valitutti F, Cucchiara S, Fasano A. Celiac disease and the microbiome. Nutrients. (2019) 11:2403. doi: 10.3390/nu11102403
66. Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset crohn's disease. Cell Host Microbe. (2014) 15:382–92. doi: 10.1016/j.chom.2014.02.005
67. Yang T, Richards EM, Pepine CJ, Raizada MK. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat Rev Nephrol. (2018) 14:442–56. doi: 10.1038/s41581-018-0018-2
68. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. (2013) 155:1451–63. doi: 10.1016/j.cell.2013.11.024
69. Tsuji S, Suruda C, Hashiyada M, Kimata T, Yamanouchi S, Kitao T, et al. Gut microbiota dysbiosis in children with relapsing idiopathic nephrotic syndrome. Am J Nephrol. (2018) 47:164–70. doi: 10.1159/000487557
70. Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. (2008) 456:507–10. doi: 10.1038/nature07450
71. Luo A, Leach ST, Barres R, Hesson LB, Grimm MC, Simar D. The microbiota and epigenetic regulation of T helper 17/regulatory T cells: in search of a balanced immune system. Front Immunol. (2017) 8:417. doi: 10.3389/fimmu.2017.00417
72. Sakaguchi S, Wing K, Yamaguchi T. Dynamics of peripheral tolerance and immune regulation mediated by treg. Eur J Immunol. (2009) 39:2331–6. doi: 10.1002/eji.200939688
73. Kamada N, Nunez G. Regulation of the immune system by the resident intestinal bacteria. Gastroenterology. (2014) 146:1477–88. doi: 10.1053/j.gastro.2014.01.060
74. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/science.1241165
75. Geuking MB, McCoy KD, Macpherson AJ. Metabolites from intestinal microbes shape treg. Cell Res. (2013) 23:1339–40. doi: 10.1038/cr.2013.125
76. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
77. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science. (2011) 331:337–41. doi: 10.1126/science.1198469
78. Fujimoto T, Imaeda H, Takahashi K, Kasumi E, Bamba S, Fujiyama Y, et al. Decreased abundance of faecalibacterium prausnitzii in the gut microbiota of crohn's disease. J Gastroenterol Hepatol. (2013) 28:613–9. doi: 10.1111/jgh.12073
79. Capone A, Volpe E. Transcriptional regulators of T helper 17 cell differentiation in health and autoimmune diseases. Front Immunol. (2020) 11:348. doi: 10.3389/fimmu.2020.00348
80. Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. (2008) 4:337–49. doi: 10.1016/j.chom.2008.09.009
81. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. (2015) 163:367–80. doi: 10.1016/j.cell.2015.08.058
82. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
83. Eladawy M, Dominguez SR, Anderson MS, Glodé MP. Kawasaki disease and the pediatric gastroenterologist: a diagnostic challenge. J Pediatr Gastroenterol Nutr. (2013) 56:297–9. doi: 10.1097/MPG.0b013e3182794432
84. Takeshita S, Kobayashi I, Kawamura Y, Tokutomi T, Sekine I. Characteristic profile of intestinal microflora in kawasaki disease. Acta Paediatr. (2002) 91:783–8. doi: 10.1111/j.1651-2227.2002.tb03327.x
85. Nagata S, Yamashiro Y, Ohtsuka Y, Shimizu T, Sakurai Y, Misawa S, et al. Heat shock proteins and superantigenic properties of bacteria from the gastrointestinal tract of patients with kawasaki disease. Immunology. (2009) 128:511–20. doi: 10.1111/j.1365-2567.2009.03135.x
86. Kinumaki A, Sekizuka T, Hamada H, Kato K, Yamashita A, Kuroda M. Characterization of the gut microbiota of kawasaki disease patients by metagenomic analysis. Front Microbiol. (2015) 6:824. doi: 10.3389/fmicb.2015.00824
87. Han SB, Lee SY. Antibiotic use in children with kawasaki disease. World J Pediatr. (2018) 14:621–2. doi: 10.1007/s12519-018-0157-3
88. Panda S, El khader I, Casellas F, Lopez Vivancos J, Garcia Cors M, Santiago A, et al. Short-term effect of antibiotics on human gut microbiota. PLoS ONE. (2014) 9:e95476. doi: 10.1371/journal.pone.0095476
89. Hanada S, Pirzadeh M, Carver KY, Deng JC. Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front Immunol. (2018) 9:2640. doi: 10.3389/fimmu.2018.02640
90. Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A. (2011) 108:5354–9. doi: 10.1073/pnas.1019378108
91. Lee KY, Hamada H, Arvonen M. Editorial: infection-related immune-mediated diseases and microbiota. Front Pediatrics. (2020) 8:108. doi: 10.3389/fped.2020.00108
92. Rhim JW, Kang HM, Han JW, Lee KY. A presumed etiology of kawasaki disease based on epidemiological comparison with infectious or immune-mediated diseases. Front Pediatrics. (2019) 7:202. doi: 10.3389/fped.2019.00202
93. Esposito S, Polinori I, Rigante D. The gut microbiota-host partnership as a potential driver of kawasaki syndrome. Front Pediatrics. (2019) 7:124. doi: 10.3389/fped.2019.00124
94. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
95. Asarat M, Apostolopoulos V, Vasiljevic T, Donkor O. Short-chain fatty acids regulate cytokines and Th17/Treg cells in human peripheral blood mononuclear cells in vitro. Immunol Invest. (2016) 45:205–22. doi: 10.3109/08820139.2015.1122613
96. Lee JR, Huang J, Magruder M, Zhang LT, Gong C, Sholi AN, et al. Butyrate-producing gut bacteria and viral infections in kidney transplant recipients: a pilot study. Transpl Infect Dis. (2019) 21:e13180. doi: 10.1111/tid.13180
97. Haak BW, Littmann ER, Chaubard JL, Pickard AJ, Fontana E, Adhi F, et al. Impact of gut colonization with butyrate-producing microbiota on respiratory viral infection following allo-HCT. Blood. (2018) 131:2978–86. doi: 10.1182/blood-2018-01-828996
98. Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. (2009) 294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x
99. Hassidim A, Merdler I, Chorin O, Merdler-Rabinowicz R, Dallal I, Perlman M, et al. Atopic predilection among kawasaki disease patients: a cross-sectional study of 1,187,757 teenagers. Int Arch Allergy Immunol. (2016) 170:92–6. doi: 10.1159/000447639
100. Woon PY, Chang WC, Liang CC, Hsu CH, Klahan S, Huang YH, et al. Increased risk of atopic dermatitis in preschool children with kawasaki disease: a population-based study in taiwan. Evid Based Complement Alternat Med. (2013) 2013:605123. doi: 10.1155/2013/605123
101. Hwang CY, Hwang YY, Chen YJ, Chen CC, Lin MW, Chen TJ, et al. Atopic diathesis in patients with kawasaki disease. J Pediatr. (2013) 163:811–5. doi: 10.1016/j.jpeds.2013.03.068
102. Salameh M, Burney Z, Mhaimeed N, Laswi I, Yousri NA, Bendriss G, et al. The role of gut microbiota in atopic asthma and allergy, implications in the understanding of disease pathogenesis. Scand J Immunol. (2020) 91:e12855. doi: 10.1111/sji.12855
103. Fukazawa M Jr, Fukazawa M, Nanishi E, Nishio H, Ichihara K, et al. Previous antibiotic use and the development of kawasaki disease: a matched-pair case-control study. Pediatr Int. (2020) doi: 10.1111/ped.14255. [Epub ahead of print].
104. Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. (2008) 6:e280. doi: 10.1371/journal.pbio.0060280
105. Tamburini S, Shen N, Wu HC, Clemente JC. The microbiome in early life: implications for health outcomes. Nat Med. (2016) 22:713–22. doi: 10.1038/nm.4142
106. Yorifuji T, Tsukahara H, Doi H. Breastfeeding and risk of kawasaki disease: a nationwide longitudinal survey in Japan. Pediatrics. (2016) 137:e20153919. doi: 10.1542/peds.2015-3919
Keywords: Kawasaki disease, regulatory T cell, T helper 17 cell, allergy, gut microbiota, dysbiosis
Citation: Kaneko K, Akagawa S, Akagawa Y, Kimata T and Tsuji S (2020) Our Evolving Understanding of Kawasaki Disease Pathogenesis: Role of the Gut Microbiota. Front. Immunol. 11:1616. doi: 10.3389/fimmu.2020.01616
Received: 28 April 2020; Accepted: 17 June 2020;
Published: 24 July 2020.
Edited by:
Marcello Chieppa, European Biomedical Research Institute of Salerno (EBRIS), ItalyReviewed by:
Francesco Valitutti, Ospedali Riuniti San Giovanni di Dio e Ruggi d'Aragona, ItalyAndy Wullaert, Ghent University, Belgium
Nicolas Lapaque, INRA Centre Jouy-en-Josas, France
Copyright © 2020 Kaneko, Akagawa, Akagawa, Kimata and Tsuji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazunari Kaneko, a2FuZWtva0BoaXJha2F0YS5rbXUuYWMuanA=