 Xin Huang
Xin Huang Bingxuan Wu
Bingxuan Wu Di Wu1
Di Wu1 Min Shen
Min Shen- 1Department of Rheumatology and Clinical Immunology, Chinese Academy of Medical Sciences & Peking Union Medical College, National Clinical Research Center for Dermatologic and Immunologic Diseases (NCRC-DID), Ministry of Science & Technology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital (PUMCH), Key Laboratory of Rheumatology and Clinical Immunology, Ministry of Education, Beijing, China
- 2Department of General Internal Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Science & Peking Union Medical College, Beijing, China
Haploinsufficiency of GATA2, also known as GATA2 deficiency, leads to a wide spectrum of clinical manifestations. Here we described another 28-year-old man with a GATA2 variant who also suffered from hemophagocytic lymphohistiocytosis(HLH), who was finally diagnosed with HLH triggered by Mycobacterium avium bloodstream infection due to primary immunodeficiency. We reviewed GATA2 deficiency patients with HLH and found that GATA2 variants causing loss of zinc finger domains were associated with HLH, and erythema nodosa might be an accompanying symptom.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening syndrome due to immune responses of activated macrophages and lymphocytes, with common clinical features including fever, splenomegaly, cytopenia, elevated aminotransferase and ferritin levels (1). Driven by the aberrant immune response, HLH can occur at all ages. The products of HLH-related genes are involved in cell-mediated cytotoxicity and lymphocytes activation/survival, and immunocompromised patients are susceptible to HLH (1). As an important determinant of multilineage hematopoiesis, GATA-binding protein 2 (GATA2) is a member of the GATA family of transcription factors.
Caused by heterozygous loss-of-function GATA2 gene variants, haploinsufficiency of GATA2, also known as GATA2 deficiency, leads to a wide spectrum of clinical manifestations including mycobacterial infections, viral infections, bone marrow failure, leukemia and lymphedema (2). It has been reported that non-tuberculous mycobacterial infections were the most common infections, while disseminated mycobacteriosis was rare in childhood but becomes more frequent with age, due to the diminishing hematopoietic function of the bone marrow (3–5). GATA2 deficiency has highly variable penetrance, and infections or other factors may affect epigenetic mechanisms to trigger pathogenesis and alter penetrance (6).
In 2021, our group reported a 17-year-old Chinese Han woman with a heterozygous GATA2 variant, who had recurrent HLH and erythema nodosa (7). Here we described another 28-year-old man with a GATA2 variant who also suffered from HLH triggered by non-tuberculous mycobacterial infection, and further functional evaluation was conducted.
Case description
A 28-year-old Chinese Han man was admitted with a history of intermittent fever for 3 years and erythema nodosa for 7 months. He had the first unprovoked onset of fever with a maximum temperature of 40°C lasting for two weeks. He had a second flare after two months and went to the local hospital, where he was diagnosed with hemophagocytosis and right inferior lobar pneumonia, based on bone marrow smear and chest computed tomography (CT). In 2021, he developed bilateral parotid enlargement, recurrent fever, and erythema nodosa. He was treated with various anti-infective agents, including moxifloxacin, linezolid, vancomycin, voriconazole, and cefoperazone-sulbactam, but the fever continued, mostly in the afternoon and at night.
The physical examination revealed the presence of papules on the trunk and upper extremities. His complete blood count indicated pancytopenia with WBC (0.6-1.3)×109/L, neutrophils (0.3-0.8)×109/L, lymphocytes 0.2×109/L, monocytes 0.00×109/L, hemoglobin (72-90)g/L, and platelets (43-64)×109/L. NK cells activity decreased (1.36%, normal range: ≥15.11%), and sCD25 was 6696pg/ml (normal range: <6400pg/ml). His CD107a expression in NK and CTL cells was within the normal range. His NK cell ΔPerforin was 72.56%, which was lower than the normal level. He also had elevated levels of IL-5 (76.2pg/ml, normal range: 0-17pg/ml), IFN-γ (106.3pg/ml, normal range:0-95pg/ml), ferritin (895ng/ml, normal range: 80-130ng/ml), C-reactive protein (CRP) (107mg/L, normal range: 0-8mg/L), and erythrocyte sedimentation rate (ESR) (75mm/h, normal range: 0-15mm/h). Antiphospholipid antibodies, antinuclear antibodies and antineutrophil cytoplasmic antibodies were all negative. Multiple lesions in the bilateral lung fields and multiple hypermetabolic lymphadenopathies in the hilum, mediastinum, and supraclavicular area and enlarged spleen were found by CT and PET/CT (Figure 1A). Mediastinum lymph node biopsy revealed a necrotic background with some plasma cells, eosinophils and lymphocytes infiltration. The pathological biopsy of subcutaneous nodules revealed lymphocyte infiltration scattered around the superficial small blood vessels of the dermis and collagen fiber proliferation. A bone marrow biopsy showed proliferation of erythroid series, predominantly of intermediate or late erythroblasts. In local hospital, the patient underwent blood cultures (including cultures for slowly-growing bacteria/mycobacteria), respiratory infection antigen IgM antibody, T-SPOT.TB, G test, GM test, Mycoplasma pneumoniae serological test, CMV-DNA and EBV-DNA, and the results were all negative. Bronchial brushing including Xpert, cryptococcal antigen, Aspergillus antigen, tuberculous smear, Fungal smear, TB-DNA, GM test were all negative. Next-generation sequencing (NGS) technology was conducted in BALF and detected Klebsiella pneumoniae. A diagnosis of HLH was made based on HLH-2004 criteria (8). He had no relevant family history. He had the heterozygous nonsense variant of the GATA2 gene c.599dupG, p.S201*. His father and sister carried the wild type of the GATA2 gene, and his mother died in an accident (Figure 1B).
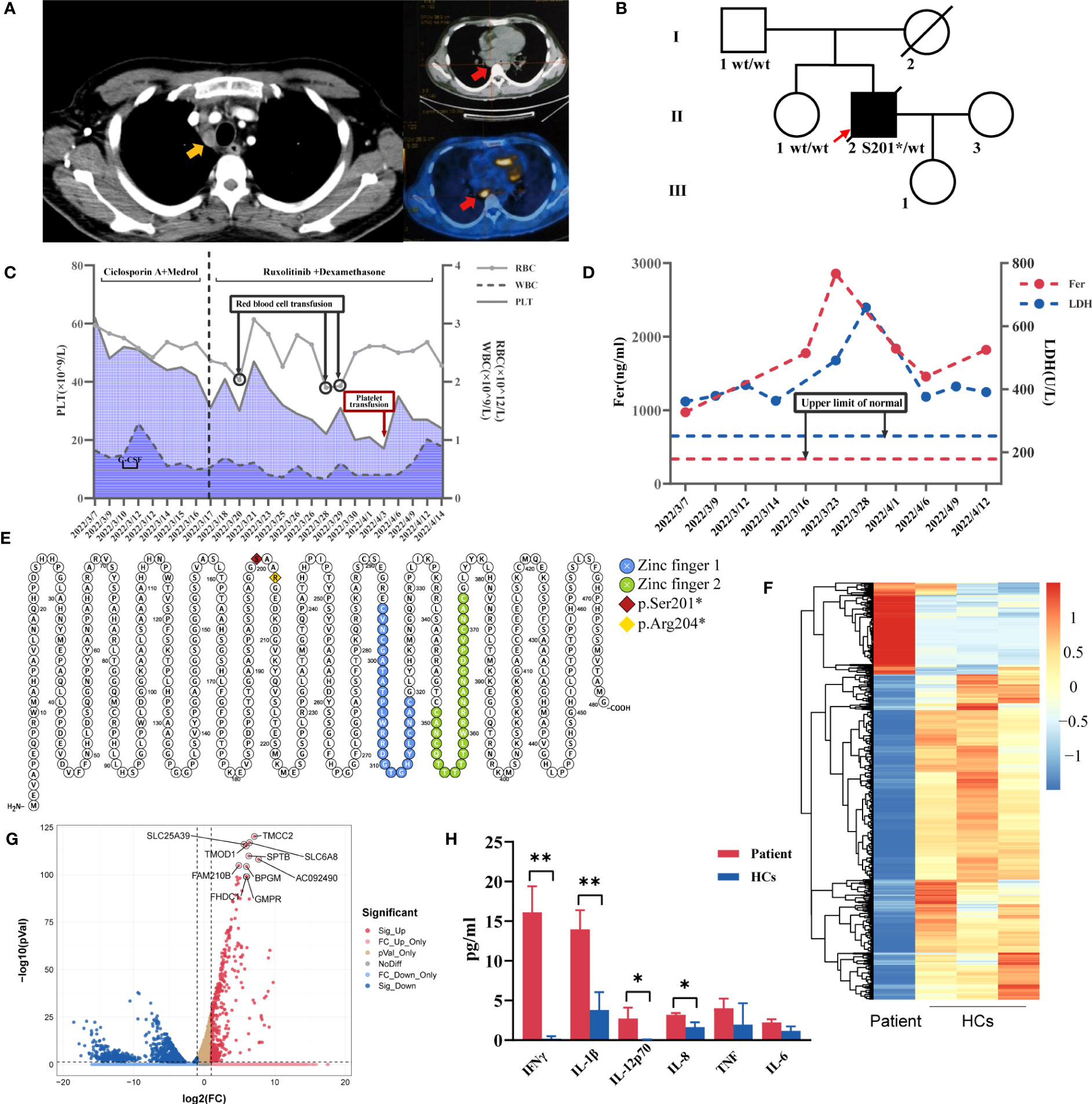
Figure 1 (A) Chest CT showing enlarged mediastinal lymph nodes (yellow arrow) and PET/CT with hypermetabolic mediastinal lymph nodes (red arrow). (B)Pedigree chart of the patient. (C) Trends in red blood cells (RBC), white blood cells (WBC), platelet count (PLT) levels and treatments during the patient’s hospital stay. (D) Changes in ferritin (Fer) and lactate dehydrogenase (LDH) during the hospitalization. (E) GATA2 variants sites of two GATA2 deficiency-related HLH patients in our center (Protter, http://wlab.ethz.ch/protter). Heatmap (F) and volcano plot (G) for differentially expressed genes identified comparing patient and healthy controls (HCs) (volcano plot was performed using the OmicStudio tools at https://www.omicstudio.cn). (H) The comparisons of plasma levels of inflammatory cytokines between the patient and HCs. **P ≤ 0.01; *P ≤ 0.05.
Therapies, changes of his complete blood count, ferritin and lactate dehydrogenase during the hospitalization were shown in Figures 1C, D. His fever and erythema nodosa temporarily improved but relapsed after dexamethasone withdrawal. There are multiple pieces of evidence confirming the effectiveness of ruxolitinib combined with dexamethasone in the treatment of HLH (9, 10), therefore ruxolitinib was added in his treatment with dexamethasone. The blood culture at three weeks after admission revealed the presence of mycobacteria. He was finally diagnosed with HLH triggered by Mycobacterium avium bloodstream infection due to primary immunodeficiency caused by germline GATA2 deficiency. His symptoms did not improve despite 4-drug antituberculosis therapy, combined with dexamethasone and ruxolitinib. Bone marrow biopsy showed the hematopoietic tissue was decreased and myeloid/erythroid ratio was reduced, no typical tuberculosis granuloma or multinucleated giant cells observed. The last bone marrow smear revealed hypoplastic myelopoiesis, myeloid/erythroid ratio=0.63:1, intermediate or late erythroblasts ratio increased. Two weeks after discharge, he was hospitalized in the local hospital due to short of breath, and chest CT scan showed emerging ground glass opacities. He was given antibiotics but died of severe pulmonary infection one month later.
Discussion
GATA2 presents two highly conserved zinc finger domains, playing a critical role during erythroid maturation and hematopoietic development. The GATA2 variant in our patient creates stop codons that result in the absence of both two zinc finger domains (Figure 1E), which is the location of most pathogenic variants (6). Neither the proband’s father nor his sister carried this variant, suggesting the GATA2 variant might be a de novo mutation. This mutation site has not been previously reported to be associated with GATA2 deficiency and HLH.
Current reports of HLH caused by GATA2 deficiency remain scarce, and we would venture to hypothesize that most variants of these HLH patients affected zinc finger domain function or resulted in loss of the zinc finger domain due to premature termination of translation, and the HLH onset was induced by the infections of bacteria or virus (7, 11–17) (Table 1). HLH occurs most commonly in infants and children, and some adults develop the disease may due to mutations with partial residual protein function that compensate for some immunological deficiencies (18). Most of the GATA2 deficiency patients presented with HLH in adulthood (11, 12, 15–17), which can be explained by partial defect in GATA2 protein function. 4 of 9 patients simultaneously experienced two zinc finger domain deletions all developed HLH after the age of 16, and were usually accompanied by rash, pancytopenia, splenomegaly and lymphadenopathy (7, 12, 16), further summary of clinical features requires the increase in the number of cases. Although it was not mentioned in other previous case reports, erythema nodosa appeared in two HLH patients with GATA2 deficiency identified as different infections in our center (7). As we know, erythema nodosa may indicate an underlying infection with non-tuberculous mycobacteria or fungi in GATA2 deficiency (4, 19), but the patient’s nodule biopsy did not reveal evidence of infection. Hence, we assumed erythema nodosa as inflammatory reaction in GATA2 deficiency patients with HLH. Furthermore, it is known that NK cells develop and function under the influence of GATA2. While GATA2 haploinsufficiency leads to a specific loss of CD56bright NK cells (20), lacking activity of NK cells of this patient might be related to the absence of two zinc finger domains, with a reduced function of perforin release. The survival and differentiation potential of NK cells lacking perforin is higher (21), mutations in the perforin-coding gene cause familial HLH (22). The patient’s phenotype may suggest that the defect in the GATA2 zinc finger domain and the function of perforin secretion in NK cells needs further investigation. It was also noted that among the four nonsense mutations in the case reports, three caused loss of both zinc finger domains in GATA2 (7, 16), while one caused loss of C-terminal zinc-finger domain (17). It has been reported that 56% of GATA2 deficiency patients had lung involvement, and nontuberculous mycobacteria was the most common pathogen associated with chronic infection (23). The changes in bone marrow smear in the later stage suggest that HLH may eventually develop into MDS. Taken together, we suggest that primary immunodeficiency such as GATA2 deficiency especially patients who lack zinc finger domains should be considered in HLH patients, erythema nodosa might be an accompanying symptom, and opportunity infections should be highly suspected especially in those affecting cellular immunity (23).
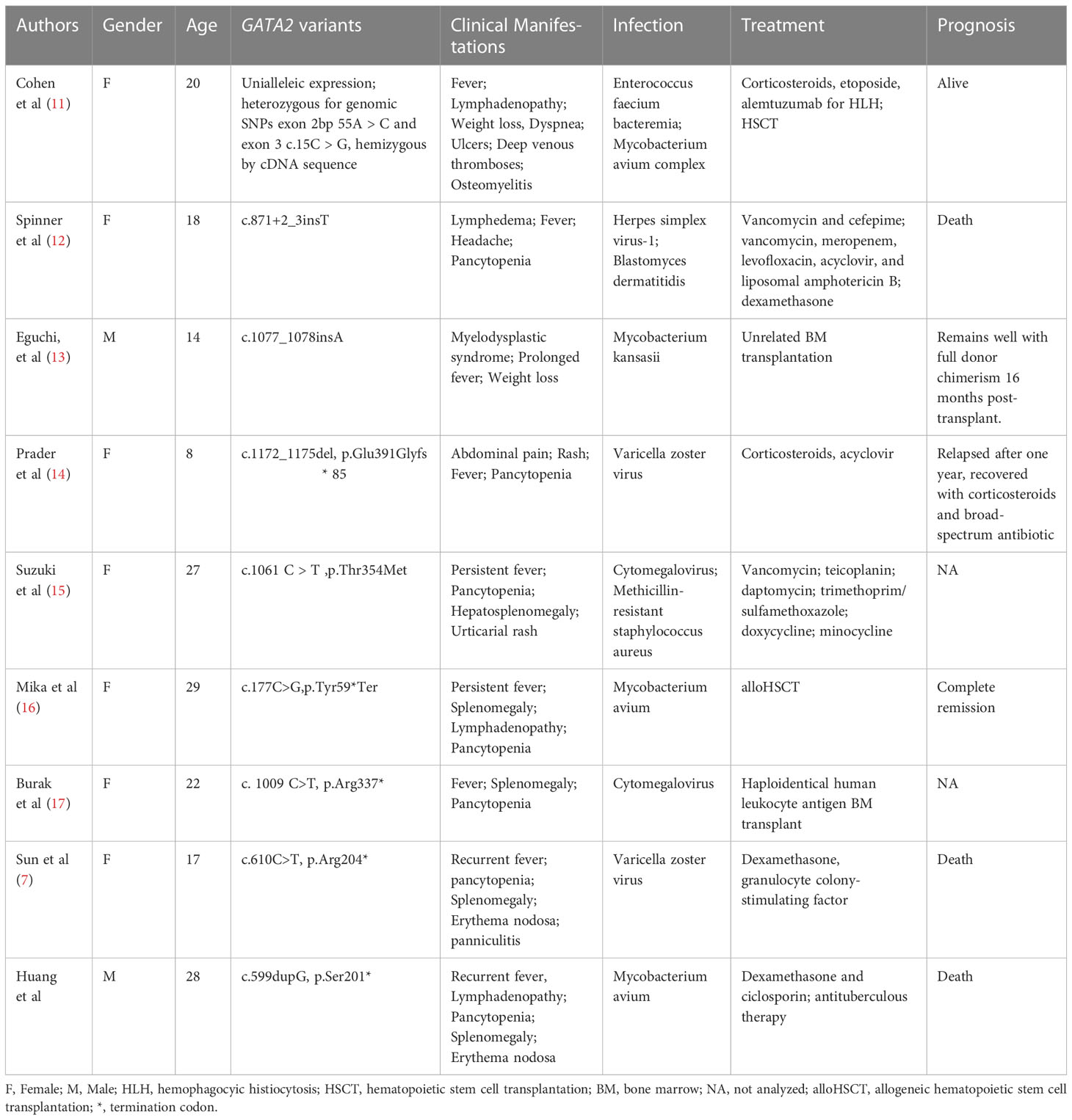
Table 1 Summary of GATA2 deficiency patients with HLH.
RNA-seq was used to analyze gene expression in the proband’s peripheral blood mononuclear cells (Figures 1F, G), revealing that four of the top ten differentially expressed genes SLC6A8, FAM210B, FHDC1 and GMPR were the targets genes of the GATA2 transcription factor in the ChIP-seq datasets from the ENCODE Transcription Factor Targets dataset (24, 25), which shed light on the importance of GATA2 in his phenotype. Given that the patient had an HLH phenotype, we found that it had been reported SLC6A8 could mediate creatine to activate macrophages (26). Plasma levels of his inflammatory cytokines were significantly higher than those of the healthy controls (Figure 1H). Further experiments on GATA2 could not be performed because the patient was deceased.
In two cases from our center, GATA2 deficiency-associated infections were likely to be the occult cause of HLH (7). Neither patient received alloHSCT for various considerations, and respectively died at 1 and 6 months after discharge. It has been documented that alloHSCT may be a modality of a cure for GATA2 deficiency-related HLH, regardless of the presence of active infection (16). The various treatment modalities often resulted in a poor prognosis, suggesting that perhaps early alloHSCT is the treatment of choice. Therefore, having dealt with this severe disease, the only thing can be done is to save time. Early diagnosis and prompt treatment might improve the prognosis of these patients.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://ngdc.cncb.ac.cn/gsa-human, HRA004493.
Ethics statement
The studies involving humans were approved by the ethics committee of Peking Union Medical College Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
XH and BXW, writing - original draft, experiment. DW, XMH, and MS, writing - review and editing, treatment of the patient. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Beijing (Grant No.7192170), the National Key Research and Development Program of China (Grant No.2016YFC0901500; 2016YFC0901501), National High Level Hospital Clinical Research Funding (Grant No.2022-PUMCH-D-002, 2022-PUMCH-B-013).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol (2020) 34(4):101515. doi: 10.1016/j.berh.2020.101515
2. Hsu AP, McReynolds LJ, Holland SM. GATA2 deficiency. Curr Opin Allergy Clin Immunol (2015) 15(1):104–9. doi: 10.1097/ACI.0000000000000126
3. Fabozzi F, Strocchio L, Mastronuzzi A, Merli P. GATA2 and marrow failure. Best Pract Res Clin Haematol (2021) 34(2):101278. doi: 10.1016/j.beha.2021.101278
4. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood (2014) 123(6):809–21. doi: 10.1182/blood-2013-07-515528
5. McReynolds LJ, Calvo KR, Holland SM. Germline GATA2 mutation and bone marrow failure. Hematology/Oncology Clinics North Am (2018) 32(4):713–28. doi: 10.1016/j.hoc.2018.04.004
6. Bresnick EH, Jung MM, Katsumura KR. Human GATA2 mutations and hematologic disease: how many paths to pathogenesis? Blood Adv (2020) 4(18):4584–92. doi: 10.1182/bloodadvances.2020002953
7. Sun L, Xu N, Shen M, Wang R, Sun Y, Zhuang J, et al. GATA2 mutation with recurrent haemophagocytic lymphohistiocytosis and panniculitis: a case report. Rheumatol (Oxf) (2021) 60(7):e229–31. doi: 10.1093/rheumatology/keaa913
8. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48(2):124–31. doi: 10.1002/pbc.21039
9. Zhou D, Huang X, Xie M, Zhu L, Huang X, Li X, et al. Ruxolitinib combined with dexamethasone in adult patients with newly diagnosed Hemophagocytic lymphohistiocytosis: A single-center pilot trial. Am J Hematol (2023) 98(5):E106-E109. doi: 10.1002/ajh.26877
10. Ahmed A, Merrill SA, Alsawah F, Bockenstedt P, Campagnaro E, Devata S, et al. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: an open-label, single-centre, pilot trial. Lancet Haematol (2019) 6(12):e630–7. doi: 10.1016/S2352-3026(19)30156-5
11. Cohen JI, Dropulic L, Hsu AP, Zerbe CS, Krogmann T, Dowdell K, et al. Association of GATA2 deficiency with severe primary Epstein-Barr Virus (EBV) infection and EBV-associated cancers. Clin Infect Dis (2016) 63(1):41–7. doi: 10.1093/cid/ciw160
12. Spinner MA, Ker JP, Stoudenmire CJ, Fadare O, Mace EM, Orange JS, et al. GATA2 deficiency underlying severe blastomycosis and fatal herpes simplex virus-associated hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol (2016) 137(2):638–40. doi: 10.1016/j.jaci.2015.07.043
13. Eguchi K, Ishimura M, Sonoda M, Ono H, Shiraishi A, Kanno S, et al. Nontuberculous mycobacteria-associated hemophagocytic lymphohistiocytosis in MonoMAC syndrome. Pediatr Blood Cancer (2018) 65(7):e27017. doi: 10.1002/pbc.27017
14. Prader S, Felber M, Volkmer B, Trück J, Schwieger-Briel A, Theiler M, et al. Life-threatening primary varicella zoster virus infection with hemophagocytic lymphohistiocytosis-like disease in GATA2 haploinsufficiency accompanied by expansion of double negative T-lymphocytes. Front Immunol (2018) 9:2766. doi: 10.3389/fimmu.2018.02766
15. Suzuki T, Takaya S, Kunimatsu J, Kutsuna S, Hayakawa K, Shibata H, et al. GATA2 mutation underlies hemophagocytic lymphohistiocytosis in an adult with primary cytomegalovirus infection. J Infect Chemother (2020) 26(2):252–6. doi: 10.1016/j.jiac.2019.07.002
16. Mika T, Vangala D, Eckhardt M, La Rosée P, Lange C, Lehmberg K, et al. Case report: hemophagocytic lymphohistiocytosis and non-tuberculous mycobacteriosis caused by a novel GATA2 variant. Front Immunol (2021) 12:682934. doi: 10.3389/fimmu.2021.682934
17. Burak N, Jan N, Kessler J, Oei E, Patel P, Feldman S. Diagnosis of GATA2 deficiency in a young woman with hemophagocytic lymphohistiocytosis triggered by acute systemic cytomegalovirus infection. Am J Case Rep (2021) 22:e927087. doi: 10.12659/AJCR.927087
18. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood (2011) 118(22):5794–8. doi: 10.1182/blood-2011-07-370148
19. Donadieu J, Lamant M, Fieschi C, de Fontbrune FS, Caye A, Ouachee M, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica (2018) 103(8):1278–87. doi: 10.3324/haematol.2017.181909
20. Wang D, Uyemura B, Hashemi E, Bjorgaard S, Riese M, Verbsky J, et al. Role of GATA2 in human NK cell development. Crit Rev Immunol (2021) 41(2):21–33. doi: 10.1615/CritRevImmunol.2021037643
21. Arapović M, Brizić I, Popović B, Jurković S, Jordan S, Krmpotić A, et al. Intrinsic contribution of perforin to NK-cell homeostasis during mouse cytomegalovirus infection. Front Immunol (2016) 7:133. doi: 10.3389/fimmu.2016.00133
22. Steen EA, Hermiston ML, Nichols KE, Meyer LK. Digenic inheritance: evidence and gaps in hemophagocytic lymphohistiocytosis. Front Immunol (2021) 12:777851. doi: 10.3389/fimmu.2021.777851
23. Marciano BE, Olivier KN, Folio LR, Zerbe CS, Hsu AP, Freeman AF, et al. Pulmonary manifestations of GATA2 deficiency. Chest (2021) 160(4):1350–9. doi: 10.1016/j.chest.2021.05.046
24. ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA elements) project. Science (2004) 306(5696):636–40. doi: 10.1126/science.1105136
25. ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PloS Biol (2011) 9(4):e1001046. doi: 10.1371/journal.pbio.1001046
Keywords: hemophagocytic lymphohistiocytosis (HLH), GATA2 deficiency, zinc finger domain, GATA2, Mycobacterium avium
Citation: Huang X, Wu B, Wu D, Huang X and Shen M (2023) Case Report: Missing zinc finger domains: hemophagocytic lymphohistiocytosis in a GATA2 deficiency patient triggered by non-tuberculous mycobacteriosis. Front. Immunol. 14:1191757. doi: 10.3389/fimmu.2023.1191757
Received: 22 March 2023; Accepted: 28 July 2023;
Published: 23 August 2023.
Edited by:
Fabien Touzot, University of Montreal, CanadaReviewed by:
David Boutboul, APHP Hôpital Saint Louis, FranceClaudio Pignata, University of Naples Federico II, Italy
Copyright © 2023 Huang, Wu, Wu, Huang and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Shen, shenmpumch@163.com
†These authors have contributed equally to this work