 Catalina Argandona Lopez
Catalina Argandona Lopez Amanda M. Brown
Amanda M. Brown- 1Division of Neuroimmunology, Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Division of Neuroimmunology, Department of Neurology and Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD, United States
HIV-infection of microglia and macrophages (MMs) induces neuronal injury and chronic release of inflammatory stimuli through direct and indirect molecular pathways. A large percentage of people with HIV-associated neurologic and psychiatric co-morbidities have high levels of circulating inflammatory molecules. Microglia, given their susceptibility to HIV infection and long-lived nature, are reservoirs for persistent infection. MMs and neurons possess the molecular machinery to detect pathogen nucleic acids and proteins to activate innate immune signals. Full activation of inflammasome assembly and expression of IL-1β requires a priming event and a second signal. Many studies have demonstrated that HIV infection alone can activate inflammasome activity. Interestingly, secreted phosphoprotein-1 (SPP1/OPN) expression is highly upregulated in the CNS of people infected with HIV and neurologic dysfunction. Interestingly, all evidence thus far suggests a protective function of SPP1 signaling through mammalian target of rapamycin (mTORC1/2) pathway function to counter HIV-neuronal injury. Moreover, HIV-infected mice knocked down for SPP1 show by neuroimaging, increased neuroinflammation compared to controls. This suggests that SPP1 uses unique regulatory mechanisms to control the level of inflammatory signaling. In this mini review, we discuss the known and yet-to-be discovered biological links between SPP1-mediated stimulation of mTOR and inflammasome activity. Additional new mechanistic insights from studies in relevant experimental models will provide a greater understanding of crosstalk between microglia and neurons in the regulation of CNS homeostasis.
Introduction
Neurologic and gait disturbances were hallmark features of HIV-1 disease in the 1980s demonstrating the profound negative impact of the virus on central nervous system (CNS) functioning (1). The clinical manifestations of NeuroHIV can include cognitive impairment, depression, anxiety, and deficits in fine motor movements (2, 3). Comprehensive neuropsychological testing is used to identify people with HIV-associated neurocognitive disorder, now more generally known as NeuroHIV, to reflect the changing clinical spectrum of neurologic and psychiatric co-morbidities (4–6). Seminal neuropathology studies on HIV-infected post-mortem human brain tissue identified brain microglia and macrophages (MMs) as the predominant cellular targets of the virus (7–10). Through different mechanisms, HIV-infected monocytes, T-cells, and viral particles cross the blood-brain-barrier, which itself becomes impaired (11–14). Targeted antiretroviral therapies (ART), first introduced in 1996, were highly effective at blocking virus replication and sparing CD4+ T-cell death and immune system dysfunction (15). Many ART regimens reach pharmacological levels in the CSF; however, whether inhibitory concentrations reach regions in the brain parenchyma, where HIV-infected MMs reside, remains unclear (16–18). Additionally, yolk sac-derived microglia are relatively long-lived cells with a turnover of many months, and their capacity for self-renewal provides a sanctuary for HIV in brain tissue (19–21). Even under conditions of low-level HIV gene expression, immune activation in the form of increased circulating pro-inflammatory cytokines and immune markers are present in people with HIV on ART (22, 23).
HIV encodes nine genes that co-opt intrinsic immune cell pathways normally used for growth, metabolism and homeostasis (24, 25). Innate immune signaling is an early detection system meant to thwart pathogen replication by activating the release of inflammatory molecules that, in turn, prime adaptive immunity (26–28). HIV-1 binds to CD4 and chemokine receptors, in a process that initiates fusion of the viral and plasma membranes (Figure 1). Neurons express chemokine receptors that support neuronal development and maturation, but not CD4, and therefore, do not allow HIV entry (33). Viral fusion is followed by the release and trafficking of the preintegration complex (PIC) to the nucleus (Figure 1). The PIC uncoating process within the nucleus was first shown for primary human macrophages years ago (29, 31), but only recently confirmed for T-cells (34). This mechanistic detail has important implications for understanding whether HIV can delay detection by nucleic acid sensors that activate Toll-like receptor (TLR) signaling (24, 30). Importantly, in MMs, virus is packaged in vesicular bodies and buds from the plasma membrane in contrast to the cytopathic release of viral particles from T-cells (35, 36).
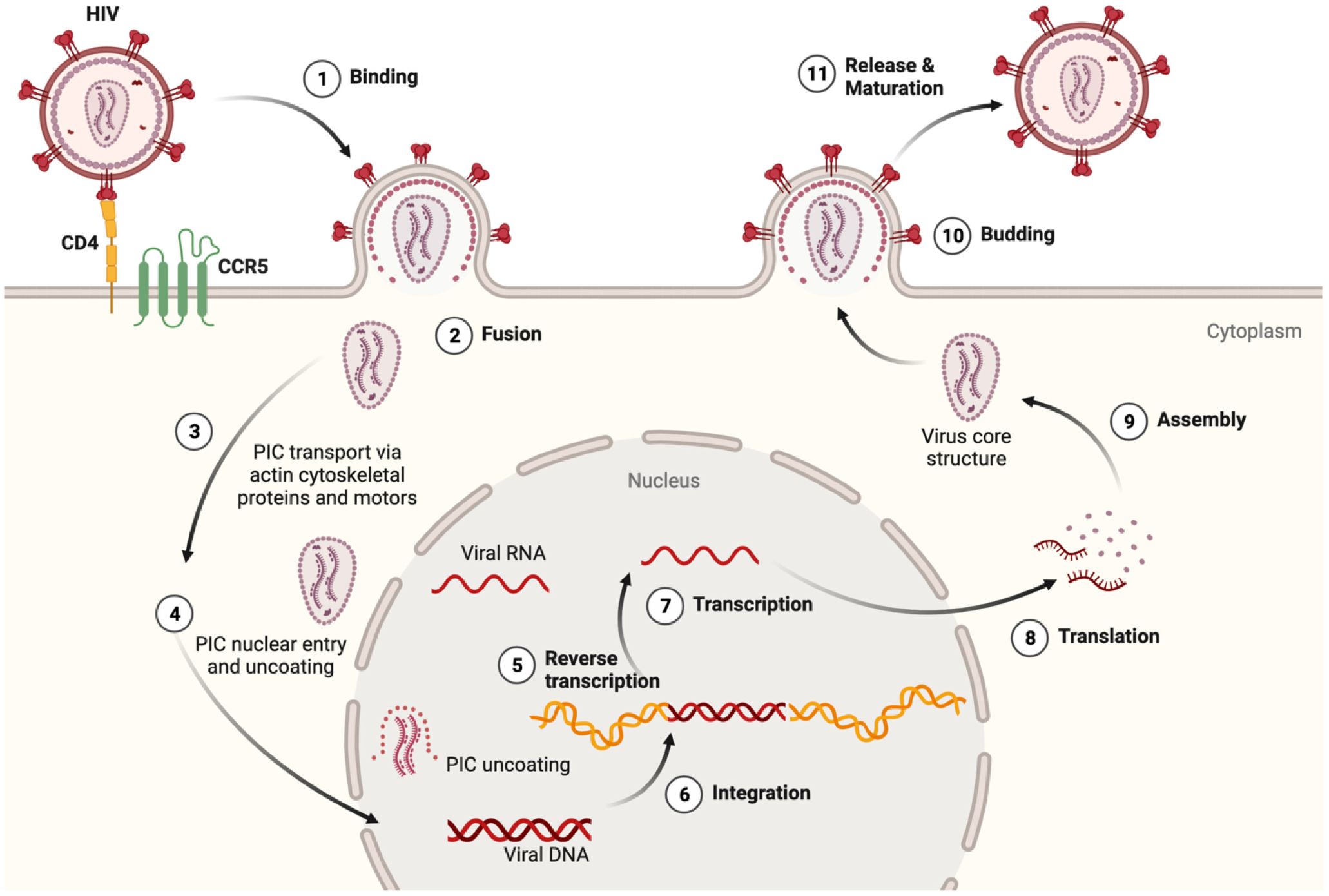
Figure 1 HIV lifecycle and relevance to inflammasome activation. (1) At target cell plasma membrane domains, HIV envelope protein gp120 trimer (red) binds to the CD4 receptor (yellow). Conformational alterations expose binding surfaces for coreceptor CCR5 on the Env trimer (green). (2) Fusion of Env with the cell plasma membrane is followed by uncoating and release of the preintegration complex (PIC) which contains a few molecules of reverse transcriptase, integrase and two copies of HIV RNA (vRNA) (29–31). Should the integrity of the PIC be compromised, viral RNA and proteins could be detected by innate immune sensors and thus initiate an inflammasome priming. (3) The actin cytoskeleton and specific microtubule motors transport the PIC to the nucleus (32). (4) The PIC can enter the nucleus in several ways including direct nuclear import and induced invaginations (24, 30). Degradation of the nucleoprotein coat would expose vRNA outside the nucleus, and provide another opportunity to activate innate antiviral responses. After reverse transcription (5), integration (6), transcription (7), and translation (8), viral proteins, vRNA, and (9) certain host proteins assembled at the inner plasma membrane surface. This mobilizes cytoskeletal proteins and molecular forces that facilitate budding (10), (11) release and maturation of new viral particles (32). Macrophages and microglia unlike T-cells are much more resistant to the cytopathic effects of HIV replication and therefore undergo innate immune activation in a sustained fashion. The figure was created with BioRender.
Microglia not only protect the brain from pathogens and injury, but also serve critical roles in maintaining neuronal viability, proper synaptodendritic function and integrity in development and over the lifespan (20, 21, 37–39). Understanding the mechanisms by which HIV-1 affects microglial innate immune function is key to addressing the brain as a source of pathologic neuroinflammation correlated with neurological and psychiatric comorbidities (18, 23, 40, 41). Below, we discuss what is known about HIV activation of the inflammasome, particularly as it relates to microglia and neurons and the expression of specific pro-inflammatory cytokines that remain elevated in people with NeuroHIV. We then discuss another innate sensor, secreted phosphoprotein-1 (or osteopontin, SPP1/OPN), and its intersection with the mammalian target of rapamycin pathway (mTOR) and potentially the inflammasome to provide a unifying view of putative mechanistic connections and cell-type dependent crosstalk between the pathways.
HIV activation of inflammasome signaling in the CNS
As the exploration of inflammasome function has progressed, NLRP3 is implicated in a variety of neurodegenerative diseases, including NeuroHIV (42–48). The inflammasome is a multiprotein complex involved in the immune and inflammatory response. Different inflammasomes types exist in the nucleotide-binding oligomerization domain, Leucine-rich-containing proteins (NLR) family (49). However, all inflammasomes contain key components including: NALP/NLR protein, PYCARD/ASC (Apoptosis-associated speck-like protein containing a CARD), and an enzyme responsible for pro-inflammatory cytokine activation (50–52). The NLRP3 inflammasome complex interacts with caspase-1 to activate IL-1β and IL-18 (53, 54). Both are pro-inflammatory cytokines that play various roles throughout the body. In microbial infections, the increase in IL-1β secretion is responsible for recruiting innate immune cells. In neurodegenerative diseases, IL-1β levels increase in response to microglial activation and neuronal injury (55, 56). IL-18 induces IFN production in T-cells and natural killer cells, promotes the production of other cytokines, and is suggested to exacerbate demyelination and cellular infiltration (44, 57).
NLRP3 inflammasome assembly needs two signals: a priming and an activating signal (58–60). Of the many ways to prime the inflammasome, the most studied route is through NFkappaB-dependent signals (Figure 2). Many ligands can prime the NLRP3 inflammasome, including lipopolysaccharide (LPS) and TLR inducers like dsRNA (59–61). During reverse transcription, dsRNA can be detected by intracellular, endosome-bound TLR3 (Figure 2) (32, 65). TLR3 ligand binding activates ERK 1/2, MAPK, and NFkappaB-pathways, promoting gene transcription (62). Interestingly, the HIV transactivator of transcription (Tat) protein alone can prime and activate the inflammasome complex (Figure 2) (66). Various ligands such as, ATP, nigericin, aggregated proteins, reactive oxygen species (ROS), and HIV viral proteins activate the NLRP3 inflammasome (46, 49, 58, 61, 66–69). These signals allow for the recruitment of additional proteins like NLRP3, ASC, and caspase-1 that are necessary for oligomerization and subsequent cleavage and maturation of cytokines (62). Caspase-1 also cleaves gasdermin D, leading to cell membrane pore formation, and a type of pro-inflammatory cell death known as pyroptosis (Figure 2) (62).
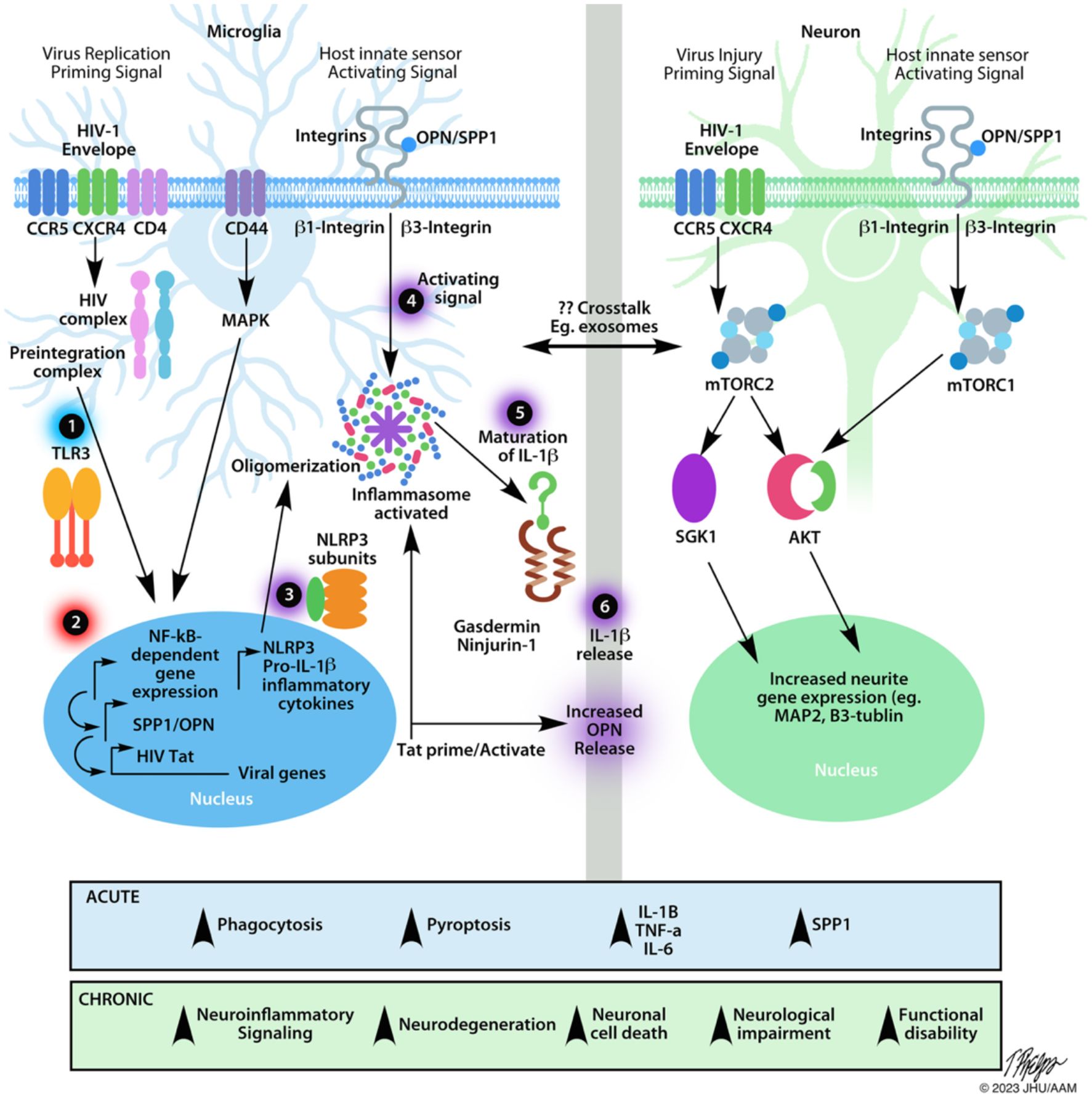
Figure 2 Schematic of the effects of HIV infection on microglia and neurons. The NLRP3 inflammasome is a multi-protein complex implicated in many neurodegenerative diseases including HAND. After HIV crosses the blood-brain barrier, it can bind to CD4+ cells, such as microglia, initiating the fusion of the virus to the plasma membrane, ultimately allowing HIV to enter the cell. After infiltrating the cell, many different aspects can affect the transcriptional activity of microglia via the NF-κβ pathway. Microglia. Step 1 indicates the first step required for inflammasome assembly: the priming step. Many stimuli can prime the NLRP3 inflammasome, including dsRNA for endosome-bound TLR3 (58, 59, 61, 62). Priming of the inflammasome leads to the localization of NF-κβ into the nucleus, indicated by Step 2. Along with host gene transcription, proteins like HIV TAT can be transcribed, which can act to prime/activate the inflammasome. Step 3 indicates the availability of the NLRP3 subunits necessary for the inflammasome to be oligomerized such as the NLRP3 protein, apoptosis-associated speck-like protein (ASC), and pro-caspase-1 (62). Step 4 indicates the activating step in NLRP3 inflammasome activation. Various stimuli, such as extracellular TAT protein, can trigger the activating signal. After receiving an activating signal, the NLRP3 inflammasome can begin its oligomerization and become functional. Pro-interleukin enzymes are recruited to be cleaved into their mature forms. For the NLRP3 inflammasome, IL-1B and IL-18 are cleaved by Caspase-1 and released, as shown by Steps 5, 6. The release of NLRP3-associated pro-inflammatory cytokines occurs via pores formed in the cell membrane. Caspase-1 will also cleave gasdermin D, leading to pyroptosis (62). The release of cytokines and viral proteins can then exacerbate local inflammation, leading to the recruitment of more immune cells and can affect other cell types, such as neurons. Neurons. Considering that HIV is unable to infect neurons directly, there are many examples of HIV-induced neuronal damage. One major contributor is the HIV-1 gp120 (Env). This protein can bind to CXCR4 and CCR5 receptors, expressed on neurons. HIV-1 Env has been shown to damage synaptic connections in cortical neurons when bound to CXCR4 via mTORC2 (63). When neurons were co-treated with HIV-1 Env and OPN/SPP1. Neurons showed signs of activated mTORC1/mTORC2 pathways, suggesting a regulatory feedback loop. Along with the required β1 and β3 integrin receptors, which OPN/SPP1 binds to, OPN/SPP1 acts as a neuroprotective modulator that promotes neurite growth in cortical neurons (β1 integrin) and regulates post-synaptic dendritic spine density in hippocampal neurons (β3 integrin) through mTORC1 (63, 64). Despite the protective effects of OPN/SPP1, over time, HIV-infected individuals present with neuronal degradation. Crosstalk. HIV-infected microglia have increased levels of NLRP3 activity, leading to pyroptosis and the release of highly pro-inflammatory cytokines. Given the role of IL-1β in inflammation, it is important to consider the various impacts it can have on the local microenvironment. Surrounding cells will respond to the inflammatory signal, such as upregulating SPP1/OPN. Regardless of the intent to reduce neuroinflammation, we see that HIV-infected individuals continue having low levels of chronic inflammation while on antiretroviral treatment. When looking at the acute effects, there is an increase in microglial phagocytosis, pyroptosis, pro-inflammatory cytokines, and OPN/SPP1 secretion. Chronically, we begin to see prolonged neuroinflammatory signaling, neurodegeneration, neuronal cell death, neuronal impairment, and functional disability, indicating the urgency to understand better the mechanisms of disease progression, cellular interactions, and regulation of neuroinflammatory pathways in HIV infection.
The NLRP3 inflammasome is robustly expressed in microglia (42, 59). However, whether the same is true for neurons is less well known. Interestingly, neurons undergoing pyroptosis have been documented (70–72). This is important since pyroptosis is strongly associated with NLRP3 inflammasome activation (73–77). The NLRP1 and AIM2 inflammasome complexes of cortical neurons have been the most investigated (70–72). Recently, studies reported that dopaminergic neurons express NLRP3 throughout the progression of Parkinson’s disease (45, 47). However, activation of NLRP3 in microglia contributes to demyelination through IL-1β and IL-18 secretion (44). HIV-positive individuals have increased caspase-1, IL-1β, and IL-18 levels, suggesting NLRP3 inflammasome activation systemically and in the CNS (78–80). Given the association between neurologic disorders, neuroinflammation, and the activation of the NLRP3 inflammasome in microglia and neurons, the potential for crosstalk between these cells is expected.
HIV induced inflammasome activation and mTOR signaling in NeuroHIV
There is renewed interest in mTOR signaling in HIV infection as new roles for this pathway have emerged. Early studies implicated a role for mTORC signaling in promoting virus replication (81–83). Most recently, mTORC-regulated mechanisms in HIV escape from latency in T-cells (84), autophagy (85), apoptosis (86), and the homing of intestinal CCR6+CD4+ T-cells (87) have been reported. Interestingly, in efforts to identify new candidate genes involved in latent HIV infection, a role for pro-inflammatory cytokines and signaling pathways regulated by secreted phosphoprotein-1/osteopontin (SPP1/OPN) were discovered (88). The mTOR pathway is composed of two structurally distinct, multi-subunit protein complexes, mTORC1 and mTORC2 that receive signals about a cell’s metabolic status to fine tune growth and repair processes through activation of relevant transcriptional programs (89, 90). HIV-positive individuals have dysregulated autophagy, indicating upregulated levels of mTOR activity (91). Increases in mTOR activity are associated with reactive microglia, neuronal damage, neurodegeneration, and memory deficits, all characteristics of NeuroHIV (63, 92). Although scarcely investigated, evidence of a regulatory relationships between mTOR and NLRP3 in immune cells and neurons have been reported. Studies have shown that downregulating mTOR activity reduces NLRP3 activation (93–96). With reduced mTOR activity, autophagy removes detrimental pro-inflammatory stimuli, including ROS. Indeed, ROS activates the NLRP3 inflammasome and has been associated with NeuroHIV (97, 98). Another study found that inhibition of mTORC1 leads to decreased secreted IL-1β, indicating post-transcriptional effects on NLRP3 activation (94). A similar regulatory relationship was observed with in vitro and in vivo NLRP3 knock-out studies in which mTOR activity decreased (93, 99). In macrophages an interaction between NLRP3 and mTOR was found, indicating a direct protein-protein interaction and communication between both pathways (93). Lastly, IL-1β can activate mTOR in T-cells and in hippocampal neurons further illustrating NLRP3 cell-specific- and cell-to-cell communication pathways and functional outcomes like neuroinflammation (100–102). The emerging relationships between NLRP3, mTOR, and HIV infection becomes more interesting when considering the function of additional innate immune sensors like SPP1/OPN.
Innate signaling pathways collide: SPP1/OPN and mTOR activation in NeuroHIV
The term neuroinflammation, as it is currently understood, broadly signifies a mix of innate and adaptive responses of resident brain- and circulating immune cells that, if left unregulated, can have damaging short- and long-term consequences (103). In this regard, chronic expression of proinflammatory molecules leads to over activation of the immune system and accumulation of damage and disability with time. Secreted phosphoprotein-1 (SPP1/OPN), by virtue of its modular domain structure, is a multifunctional phosphoprotein implicated in several neurodegenerative diseases (104–110). The expression of SPP1/OPN is markedly elevated in the CNS of humans and non-human primate models of HIV infection (109, 110). However, more recent findings with humanized mice and positron emission tomography neuroimaging demonstrate that SPP1/OPN expression is required to downregulate the microglial inflammatory response (111). How exactly SPP1/OPN modulates the HIV-induced inflammatory response in the brain is not yet understood. However, in cultured primary human macrophages, HIV replication and NF-κβ activity is increased in the presence of SPP1/OPN (110). The degree of neuroinflammation correlated with the extent of HIV replication only in humanized mice expressing SPP1/OPN (111). Neurons cannot be infected with HIV due to their lack of the CD4 receptor, however the presence of certain chemokine coreceptors like CCR5 or CXCR4 makes them vulnerable to excitotoxicity, degeneration and death after binding interactions with HIV Gp120 (33, 112). However, treatment of neurons with recombinant OPN protects hippocampal post-synapses from synaptodendritic injury, and the structural integrity of cortical axons and dendrites via mTORC1/mTORC2 activation (Figure 2) (63, 64). Therefore, in NeuroHIV, increased expression of SPP1/OPN is largely neuroprotective.
The intersection of SPP1/OPN, mTOR and inflammasome signaling in neurodegenerative disorders
We first hypothesized that the overexpression of SPP1/OPN in individuals with NeuroHIV was harmful, but as discussed above the findings thus far point to a neuroprotective function. While there is increasing evidence of linkages between neurodegeneration and cellular repair processes involved in resolving neuronal injury and neuroinflammation, significant gaps in our understanding of the molecular mechanisms remain. SPP1/OPN was identified as a highly-expressed transcript that clustered with a collection of genes termed “disease-associated microglia (DAM) (113–115). Recent studies by Rentsendorj et al., and Qiu et al., beautifully demonstrate using the ADtg and 5XFAD mouse models for AD, respectively roles for specific populations of SPP1+/- expressing monocytes, resident microglia and/or macrophages in the phagocytosis of amyloid and speculate about a role for inflammasome signaling (116, 117). In contrast, in a slow-progressing model of AD (AppNL-F knock-in reporter mice), SPP1+ macrophages and microglia associated with brain blood vessels and those located in the hippocampus were responsible for pathologic microglia-synapse destruction (118, 119). In another example of neuroprotection, regulatory T-cells localized in the brain several weeks after stroke express SPP1/OPN and, through a microglial-β1-integrin-dependent manner, foster repair of white matter axonal damage (120). In a model of glaucoma, a protective role for SPP1/OPN was found (117). Interestingly, in an ischemia model, intranasal delivery of a SPP1/OPN peptide suppressed microglial activation and the release of pro-inflammatory cytokines IL-1β and IL-6, an indication of reduced NLRP3 activity (121). To further support this idea, Zhang et al. demonstrated that SPP1/OPN negatively regulates the NLRP3 inflammasome in ischemic infarction (122). Lastly, in a MS model, NLRP3 knockout, as well as one of its components ASC, reduced mRNA SPP1/OPN expression in splenic CD4+ T cells (123). Whether this same relationship exists in the CNS is unknown, though it is possible that NLRP3 priming lead to NF-kappaB transcription of SPP1/OPN. Given its neuroprotective function, a negative feedback loop may be in place to prevent chronic inflammation via continuous NLRP3 activation. Importantly, as more details on the molecular mechanisms of SPP1/OPN function continue to emerge, the information will help provide a more complete understanding of the correlative findings of clinical studies (124) and toward the design of possible efficacious therapeutic interventions.
Over the last several years, understanding of the direct role of glycolytic metabolism on effector immune cell functions has greatly increased (125–128). As such, there are opportunities for pathogens to alter and/or harness signaling dynamics that feed directly into the mTOR pathway (129–132). Tissue macrophages and microglia assume a variety of activation states in response to local cues, and downstream stimulation of mTOR signaling is implicated in their M2- (anti-inflammatory) or M1-polarization (proinflammatory), respectively (133). Interestingly, inhibition of inflammasome activation is protective against disease progression in a mouse model of multiple sclerosis. In this regard, rapamycin, an immunosuppressive agent, was shown to block antigen presentation by dendritic cells and inflammatory signaling by microglia (133, 134).
The homeostatic balance of the immune system is maintained through direct and indirect interactions and with soluble factors acting locally and over long distances (refs). HIV infection disrupts and hijacks the important cell-to-cell communication network. The virus infects T-cells and MMs robustly and astrocytes in a limited fashion (35), and cells located nearby initiate a signaling cascade that amplifies locally, and recruits additional immune cells from a distance. This idea of cellular crosstalk was investigated by Wang and Gabuzda, who saw that direct contact between neurons and microglia was not necessary for neuronal damage (135). The same study also found that activated astrocytes promoted HIV replication in microglia. In this regard, as discussed above, mTOR signaling in cultured cortical neurons preserves structural integrity, however increased mTOR activity can also be detrimental to cells of the brain (133, 134, 136). Cortical neurons, as well as infected microglia may, in turn, be upregulate and secrete OPN/SPP1 to reduce the inflammatory response by inactivating the NLRP3 inflammasome in microglia, and promoting neuronal survival through mTOR activity. Decreased mTOR activity in astrocytes is primarily beneficial, but negatively affects their ability to differentiate (133, 134, 136). In oligodendrocytes, decreased mTOR activity impairs their differentiation and myelination functions (133, 134, 136). The release of damage signals and proinflammatory molecules from impaired glial cells, activates immune cells and neurons thus amplifying a neuroinflammatory response. An example being the rapid release of IL-1β and IL-18 from microglial pyroptosis (Figure 2). We emphasize the importance of considering that homeostasis in chronic low-level HIV infection is tightly regulated via crosstalk between different cells through secreted pro- and anti-inflammatory cytokines/chemokines. The delicate balance, or lack thereof, of a cellular local environment, can act to exacerbate or ameliorate neuroinflammation. Indeed, HIV utilizes these delicate communication pathways to promote an optimal environment for replication.
Given that microglia have receptors for OPN, it’s possible that signaling by cortical OPN/SPP1 via mTOR acts on microglia to reduce the inflammatory response and increase transcriptional programs involved in preserving neuronal function. Given their opposing, yet collaborative, roles in inflammation, it is important to investigate the relationship between SPP1/OPN, mTOR, and NLRP3 in HIV-induced neuroinflammation and NeuroHIV. In this regard, more research is needed to get a better understanding of the molecular and cellular mechanisms that take place in chronic HIV infection. Doing so would allow us to understand better how HIV manipulates the host’s protective measures, allowing for better treatments aimed to improve the host response to latent HIV infection, guiding us toward a solution to eliminate HIV-associated neuroinflammation and cognitive deficits.
Discussion
There is a greater appreciation that during development and adulthood, dynamic homeostatic regulation of the brain’s neural network is intertwined with and dependent on crosstalk and connectivity with glial. Disruption of the integrity of the brain, as seen in viral infection, leads to activation of what are meant to be protective responses, resulting in a neuroinflammatory response involving resident brain cells and immune sentinels that conduct tissue-level surveillance. As reviewed herein, innate immune signaling, including mTOR, SPP1/OPN, and NLRP3 inflammasome activation, is initiated to monitor and/or alter cell metabolic state, stimulate repair, migration, and other immune effector processes. Given that several myeloid and glial cells and cofactors can contribute and stimulate autocrine and paracrine feedback and feed-forward looping, how are the outputs integrated to restore homeostatic levels of regulation and surveillance? Deeper insight into the physiological, cellular, and molecular mechanisms will help to advance the development of effective interventions to help those suffering from neurological and neuropsychiatric comorbidities related to chronic over-activated innate immune responses in the central nervous system.
Author contributions
AB: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. CA: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institutes of Health, National Institutes of Neurological Disorders and Stroke, R01NS102006 and R21 MH128152 to AB.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Navia BA, Jordan BD, Price RW. The AIDS dementia ocmplex: I. Clinical features. Ann Neurol. (1986) 19:517–24. doi: 10.1002/ana.410190602
2. Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder - pathogenesis and prospects for treatment. Nat Rev Neurol. (2016) 12:234–48. doi: 10.1038/nrneurol.2016.27
3. Rubin LH, Li Y, Fitzgerald KC, Dastgheyb R, Spence AB, Maki PM, et al. Associations between antiretrovirals and cognitive function in women with HIV. J Neuroimmune Pharmacol. (2021) 16:195–206. doi: 10.1007/s11481-020-09910-1
4. Mastrorosa I, Pinnetti C, Brita AC, Mondi A, Lorenzini P, Del Duca G, et al. Declining prevalence of human immunodeficiency virus (HIV)-associated neurocognitive disorders in recent years and associated factors in a large cohort of antiretroviral therapy-treated individuals with HIV. Clin Infect Dis. (2023) 76:e629–37. doi: 10.1093/cid/ciac658
5. Elicer IM, Byrd D, Clark US, Morgello S, Robinson-Papp J. Motor function declines over time in human immunodeficiency virus and is associated with cerebrovascular disease, while HIV-associated neurocognitive disorder remains stable. J Neurovirol. (2018) 24:514–22. doi: 10.1007/s13365-018-0640-6
6. Vance DE, Del Bene VA, Frank JS, Billings R, Triebel K, Buchholz A, et al. Cognitive intra-individual variability in HIV: an integrative review. Neuropsychol Rev. (2021) 32:855–76. doi: 10.1007/s11065-021-09528-x
7. Nottet HS. Interactions between macrophages and brain microvascular endothelial cells: role in pathogenesis of HIV-1 infection and blood - brain barrier function. J Neurovirol. (1999) 5:659–69. doi: 10.3109/13550289909021294
8. Gendelman HE, Orenstein JM, Baca LM, Weiser B, Burger H, Kalter DC, et al. The macrophage in the persistence and pathogenesis of HIV infection. AIDS. (1989) 3:475–96. doi: 10.1097/00002030-198908000-00001
9. Koenig S, Gendelman JM, Orenstein JM, Dal Canto M, Pezeshkpour GH, Yungbluth M, et al. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. (1986) 233:1089–93. doi: 10.1126/science.3016903
10. Meltzer MS, Nakamura M, Hansen BD, Turpin JA, Kalter DC, Gendelman HE. Macrophages as susceptible targets for HIV infection, persistent viral reservoirs in tissue, and key immunoregulatory cells that control levels of virus replication and extent of disease. AIDS Res Hum Retroviruses. (1990) 6:967–71. doi: 10.1089/aid.1990.6.967
11. Langford D, Masliah E. Crosstalk between components of the blood brain barrier and cells of the CNS in microglial activation in AIDS. Brain Pathol. (2001) 11:306–12. doi: 10.1111/j.1750-3639.2001.tb00401.x
12. Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, et al. A model for monocyte migration through the blood-brain barrier during HIV-1 encephalitis. J Immunol. (1997) 158:3499–510. doi: 10.4049/jimmunol.158.7.3499
13. Roberts TK, Buckner CM, Berman JW. Leukocyte transmigration across the blood-brain barrier: perspectives on neuroAIDS. Front Biosci. (2010) 15:478–536. doi: 10.2741/3631
14. Bertrand L, Cho HJ, Toborek M. Blood-brain barrier pericytes as a target for HIV-1 infection. Brain. (2019) 142:502–11. doi: 10.1093/brain/awy339
15. Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. (1996) 271:1582–6. doi: 10.1126/science.271.5255.1582
16. Oliveira MF, Chaillon A, Nakazawa M, Vargas M, Letendre SL, Strain MC, et al. Early antiretroviral therapy is associated with lower HIV DNA molecular diversity and lower inflammation in cerebrospinal fluid but does not prevent the establishment of compartmentalized HIV DNA populations. PloS Pathog. (2017) 13:e1006112. doi: 10.1371/journal.ppat.1006112
17. Livelli A, Vaida F, Ellis RJ, Ma Q, Ferrara M, Clifford DB, et al. Correlates of HIV RNA concentrations in cerebrospinal fluid during antiretroviral therapy: a longitudinal cohort study. Lancet HIV. (2019) 6:e456–62. doi: 10.1016/S2352-3018(19)30143-2
18. Cysique LA, Brew BJ. Comorbid depression and apathy in HIV-associated neurocognitive disorders in the era of chronic HIV infection. Handb Clin Neurol. (2019) 165:71–82. doi: 10.1016/B978-0-444-64012-3.00006-X
19. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
20. Kierdorf K, Masuda T, Jordao MJC, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci. (2019) 20:547–62. doi: 10.1038/s41583-019-0201-x
21. Prinz M, Jung S, Priller J. Microglia biology: one century of evolving concepts. Cell. (2019) 179:292–311. doi: 10.1016/j.cell.2019.08.053
22. Spudich S, Robertson KR, Bosch RJ, Gandhi RT, Cyktor JC, Mar H, et al. Persistent HIV-infected cells in cerebrospinal fluid are associated with poorer neurocognitive performance. J Clin Invest. (2019) 129:3339–46. doi: 10.1172/JCI127413
23. Ellis RJ, Marquine MJ, Kaul M, Fields JA, Schlachetzki JCM. Mechanisms underlying HIV-associated cognitive impairment and emerging therapies for its management. Nat Rev Neurol. (2023) 19:668–87. doi: 10.1038/s41582-023-00879-y
24. Saez-Cirion A, Manel N. Immune responses to retroviruses. Annu Rev Immunol. (2018) 36:193–220. doi: 10.1146/annurev-immunol-051116-052155
25. Moir S, Chun TW, Fauci AS. Pathogenic mechanisms of HIV disease. Annu Rev Pathol. (2011) 6:223–48. doi: 10.1146/annurev-pathol-011110-130254
26. Scully EP, Lockhart A, Garcia-Beltran W, Palmer CD, Musante C, Rosenberg E, et al. Innate immune reconstitution with suppression of HIV-1. JCI Insight. (2016) 1:e85433. doi: 10.1172/jci.insight.85433
27. Spudich SS. Immune activation in the central nervous system throughout the course of HIV infection. Curr Opin HIV AIDS. (2016) 11:226–33. doi: 10.1097/COH.0000000000000243
28. Altfeld M, Gale J. Innate immunity against HIV-1 infection. Nat Immunol. (2015) 16:554–62. doi: 10.1038/ni.3157
29. Chin CR, Perreira JM, Savidis G, Portmann JM, Aker AM, Feeley EM, et al. Direct visualization of HIV-1 replication intermediates shows that capsid and CPSF6 modulate HIV-1 intra-nuclear invasion and integration. Cell Rep. (2015) 13:1717–31. doi: 10.1016/j.celrep.2015.10.036
30. Muller TG, Zila V, Muller B, Krausslich HG. Nuclear capsid uncoating and reverse transcription of HIV-1. Annu Rev Virol. (2022) 9:261–84. doi: 10.1146/annurev-virology-020922-110929
31. Peng K, Muranyi W, Glass B, Laketa V, Yant SR, Tsai L, et al. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. Elife. (2014) 3:e04114. doi: 10.7554/eLife.04114
32. Serrano T, Fremont S, Echard A. Get in and get out: Remodeling of the cellular actin cytoskeleton upon HIV-1 infection. Biol Cell. (2023) 115:e2200085. doi: 10.1111/boc.202200085
33. Nickoloff-Bybel EA, Festa L, Meucci O, Gaskill PJ. Co-receptor signaling in the pathogenesis of neuroHIV. Retrovirology. (2021) 18:24. doi: 10.1186/s12977-021-00569-x
34. Kulkosky J, Culnan DM, Roman J, Dornadula G, Schnell M, Boyd MR, et al. Prostratin: activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood. (2001) 98:3006–15. doi: 10.1182/blood.V98.10.3006
35. Wahl A, Al-Harthi L. HIV infection of non-classical cells in the brain. Retrovirology. (2023) 20:1. doi: 10.1186/s12977-023-00616-9
36. Williams K, Corey S, Westmoreland SV, Pauley DR, Knight HL, deBakker C, et al. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. (2001) 193:905–15. doi: 10.1084/jem.193.8.905
37. Kierdorf K, Prinz M, Geissmann F, Gomez Perdiguero E. Development and function of tissue resident macrophages in mice. Semin Immunol. (2015) 27:369–78. doi: 10.1016/j.smim.2016.03.017
38. Menassa DA, Gomez-Nicola D. Microglial dynamics during human brain development. Front Immunol. (2018) 9:1014. doi: 10.3389/fimmu.2018.01014
39. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. (2013) 155:1596–609. doi: 10.1016/j.cell.2013.11.030
40. Nightingale S, Ances B, Cinque P, Dravid A, Dreyer AJ, Gisslen M, et al. Cognitive impairment in people living with HIV: consensus recommendations for a new approach. Nat Rev Neurol. (2023) 19:424–33. doi: 10.1038/s41582-023-00813-2
41. Valdez AN, Rubin LH, Neigh GN. Untangling the Gordian knot of HIV, stress, and cognitive impairment. Neurobiol Stress. (2016) 4:44–54. doi: 10.1016/j.ynstr.2016.02.005
42. Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ, et al. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. (2014) 10:1761–75. doi: 10.4161/auto.29647
43. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. (2008) 9:857–65. doi: 10.1038/ni.1636
44. Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, et al. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci. (2010) 30:15811–20. doi: 10.1523/JNEUROSCI.4088-10.2010
45. Panicker N, Kam TI, Wang H, Neifert S, Chou SC, Kumar M, et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson's disease. Neuron. (2022) 110:2422–37.e9. doi: 10.1016/j.neuron.2022.05.009
46. Shi F, Yang L, Kouadir M, Yang Y, Wang J, Zhou X, et al. The NALP3 inflammasome is involved in neurotoxic prion peptide-induced microglial activation. J Neuroinflamm. (2012) 9:73. doi: 10.1186/1742-2094-9-73
47. von Herrmann KM, Anderson FL, Martinez EM, Young AL, Havrda MC. Slc6a3-dependent expression of a CAPS-associated Nlrp3 allele results in progressive behavioral abnormalities and neuroinflammation in aging mice. J Neuroinflamm. (2020) 17:213. doi: 10.1186/s12974-020-01866-6
48. Min AK, Fortune T, Rodriguez N, Hedge E, Swartz TH. Inflammasomes as mediators of inflammation in HIV-1 Infection. Transl Res. (2022) 252:1–8. doi: 10.1016/j.trsl.2022.07.008
49. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
50. Aganna E, Martinon F, Hawkins PN, Ross JB, Swan DC, Booth DR, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. (2002) 46:2445–52. doi: 10.1002/art.10509
51. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. (2001) 29:301–5. doi: 10.1038/ng756
52. Manji GA, Wang L, Geddes BJ, Brown M, Merriam S, Al-Garawi A, et al. PYPAF1, a PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NF-kappa B. J Biol Chem. (2002) 277:11570–5. doi: 10.1074/jbc.M112208200
53. Araínga M, Su H, Poluektova LY, Gorantla S, Gendelman HE. HIV-1 cellular and tissue replication patterns in infected humanized mice. Sci Rep. (2016) 6:23513. doi: 10.1038/srep23513
54. Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. (2006) 24:317–27. doi: 10.1016/j.immuni.2006.02.004
55. Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. (1996) 87:2095–147. doi: 10.1182/blood.V87.6.2095.bloodjournal8762095
56. Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. (2019) 11:e10248. doi: 10.15252/emmm.201810248
57. Dinarello CA. IL-18: A TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J Allergy Clin Immunol. (1999) 103:11–24. doi: 10.1016/S0091-6749(99)70518-X
58. Mariathasan S. ASC, Ipaf and Cryopyrin/Nalp3: bona fide intracellular adapters of the caspase-1 inflammasome. Microbes Infect. (2007) 9:664–71. doi: 10.1016/j.micinf.2007.01.017
59. Song L, Pei L, Yao S, Wu Y, Shang Y. NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci. (2017) 11:63. doi: 10.3389/fncel.2017.00063
60. Sim J, Park J, Moon JS, Lim J. Dysregulation of inflammasome activation in glioma. Cell Commun Signal. (2023) 21:239. doi: 10.1186/s12964-023-01255-5
61. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
62. Kawai T, Akira S. TLR signaling. Cell Death Differ. (2006) 13:816–25. doi: 10.1038/sj.cdd.4401850
63. Calvez M, Hseeh G, Benzer S, Brown AM. Osteopontin counters human immunodeficiency virus type 1-induced impairment of neurite growth through mammalian target of rapamycin and beta-integrin signaling pathways. J Neurovirol. (2019) 25:384–96. doi: 10.1007/s13365-019-00729-y
64. Mahmud FJ, Boucher T, Liang S, Brown AM. Osteopontin and integrin mediated modulation of post-synapses in HIV envelope glycoprotein exposed hippocampal neurons. Brain Sci. (2020) 10:346. doi: 10.3390/brainsci10060346
65. Das K, Martinez SE, DeStefano JJ, Arnold E. Structure of HIV-1 RT/dsRNA initiation complex prior to nucleotide incorporation. Proc Natl Acad Sci U S A. (2019) 116:7308–13. doi: 10.1073/pnas.1814170116
66. Chivero ET, Guo ML, Periyasamy P, Liao K, Callen SE, Buch S. HIV-1 tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J Neurosci. (2017) 37:3599–609. doi: 10.1523/JNEUROSCI.3045-16.2017
67. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. (2008) 320:674–7. doi: 10.1126/science.1156995
68. He X, Yang W, Zeng Z, Wei Y, Gao J, Zhang B, et al. NLRP3-dependent pyroptosis is required for HIV-1 gp120-induced neuropathology. Cell Mol Immunol. (2020) 17:283–99. doi: 10.1038/s41423-019-0260-y
69. Mamik MK, Hui E, Branton WG, McKenzie BA, Chisholm J, Cohen EA, et al. HIV-1 viral protein R activates NLRP3 inflammasome in microglia: implications for HIV-1 associated neuroinflammation. J Neuroimmune Pharmacol. (2017) 12:233–48. doi: 10.1007/s11481-016-9708-3
70. Adamczak SE, de Rivero Vaccari JP, Dale G, Brand FJ, Nonner D 3rd, Bullock MR, et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab. (2014) 34:621–9. doi: 10.1038/jcbfm.2013.236
71. Kaushal S, Tamer Z, Opoku F, Forcelli PA. Anticonvulsant drug-induced cell death in the developing white matter of the rodent brain. Epilepsia. (2016) 57:727–34. doi: 10.1111/epi.13365
72. Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, et al. Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer's disease. Cell Death Dis. (2014) 5:e1382. doi: 10.1038/cddis.2014.348
73. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
74. Boise LH, Collins CM. Salmonella-induced cell death: apoptosis, necrosis or programmed cell death? Trends Microbiol. (2001) 9:64–7. doi: 10.1016/S0966-842X(00)01937-5
75. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. (2006) 8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
76. Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. (2007) 9:2562–70. doi: 10.1111/j.1462-5822.2007.01036.x
77. Oladapo A, Jackson T, Menolascino J, Periyasamy P. Role of pyroptosis in the pathogenesis of various neurological diseases. Brain Behav Immun. (2024) 117:428–46. doi: 10.1016/j.bbi.2024.02.001
78. Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG, et al. Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology. (2014) 11:35. doi: 10.1186/1742-4690-11-35
79. Guerville F, Vialemaringe M, Cognet C, Duffau P, Lazaro E, Cazanave C, et al. Mechanisms of systemic low-grade inflammation in HIV patients on long-term suppressive antiretroviral therapy: the inflammasome hypothesis. AIDS. (2023) 37:1035–46. doi: 10.1097/QAD.0000000000003546
80. Feria MG, Taborda NA, Hernandez JC, Rugeles MT. HIV replication is associated to inflammasomes activation, IL-1beta, IL-18 and caspase-1 expression in GALT and peripheral blood. PLoS One. (2018) 13:e0192845. doi: 10.1371/journal.pone.0192845
81. Heredia A, Le N, Gartenhaus RB, Sausville E, Medina-Moreno S, Zapata JC, et al. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc Natl Acad Sci U S A. (2015) 112:9412–7. doi: 10.1073/pnas.1511144112
82. Kuss-Duerkop SK, Wang J, Mena I, White K, Metreveli G, Sakthivel R, et al. Influenza virus differentially activates mTORC1 and mTORC2 signaling to maximize late stage replication. PLoS Pathog. (2017) 13:e1006635. doi: 10.1371/journal.ppat.1006635
83. Thoma C. Infectious disease: Blocking mTOR inhibits HIV-1. Nat Rev Urol. (2015) 12:417. doi: 10.1038/nrurol.2015.185
84. Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, et al. The mTOR complex controls HIV latency. Cell Host Microbe. (2016) 20:785–97. doi: 10.1016/j.chom.2016.11.001
85. Cinti A, Le Sage V, Milev MP, Valiente-Echeverria F, Crossie C, Miron MJ, et al. HIV-1 enhances mTORC1 activity and repositions lysosomes to the periphery by co-opting Rag GTPases. Sci Rep. (2017) 7:5515. doi: 10.1038/s41598-017-05410-0
86. Campbell GR, Bruckman RS, Herns SD, Joshi S, Durden DL, Spector SA. Induction of autophagy by PI3K/MTOR and PI3K/MTOR/BRD4 inhibitors suppresses HIV-1 replication. J Biol Chem. (2018) 293:5808–20. doi: 10.1074/jbc.RA118.002353
87. Planas D, Zhang Y, Monteiro P, Goulet JP, Gosselin A, Grandvaux N, et al. HIV-1 selectively targets gut-homing CCR6+CD4+ T cells via mTOR-dependent mechanisms. JCI Insight. (2017) 2:e93230. doi: 10.1172/jci.insight.93230
88. Dai W, Wu F, McMyn N, Song B, Walker-Sperling VE, Varriale J, et al. Genome-wide CRISPR screens identify combinations of candidate latency reversing agents for targeting the latent HIV-1 reservoir. Sci Transl Med. (2022) 14:eabh3351. doi: 10.1126/scitranslmed.abh3351
89. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
90. Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J. Molecular neurobiology of mTOR. Neuroscience. (2017) 341:112–53. doi: 10.1016/j.neuroscience.2016.11.017
91. Mehla R, Chauhan A. HIV-1 differentially modulates autophagy in neurons and astrocytes. J Neuroimmunol. (2015) 285:106–18. doi: 10.1016/j.jneuroim.2015.06.001
92. Fields J, Dumaop W, Rockenstein E, Mante M, Spencer B, Grant I, et al. Age-dependent molecular alterations in the autophagy pathway in HIVE patients and in a gp120 tg mouse model: reversal with beclin-1 gene transfer. J Neurovirol. (2013) 19:89–101. doi: 10.1007/s13365-012-0145-7
93. Huang XH, Ma Y, Zheng MM, Chen N, Hu MN, Wu LY, et al. NLRP3 and mTOR Reciprocally Regulate Macrophage Phagolysosome Formation and Acidification Against Vibrio vulnificus Infection. Front Cell Dev Biol. (2020) 8:587961. doi: 10.3389/fcell.2020.587961
94. Ko JH, Yoon SO, Lee HJ, Oh JY. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFkappaB pathways in autophagy- and p62-dependent manners. Oncotarget. (2017) 8:40817–31. doi: 10.18632/oncotarget.v8i25
95. Li X, Zhang X, Pan Y, Shi G, Ren J, Fan H, et al. mTOR regulates NLRP3 inflammasome activation via reactive oxygen species in murine lupus. Acta Biochim Biophys Sin (Shanghai). (2018) 50:888–96. doi: 10.1093/abbs/gmy088
96. Yang F, Ye X, Chen M, Li H, Wang Y, Zhong M. Inhibition of NLRP3 inflammsome activation and pyroptosis in macrophages by Taraxasterol is associated with its regulation on mTOR signaling. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.632606
97. Buckley S, Byrnes S, Cochrane C, Roche M, Estes JD, Selemidis S, et al. The role of oxidative stress in HIV-associated neurocognitive disorders. Brain Behav Immun Health. (2021) 13:100235. doi: 10.1016/j.bbih.2021.100235
98. Harijith A, Ebenezer DL, Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol. (2014) 5:352. doi: 10.3389/fphys.2014.00352
99. Marin-Aguilar F, Castejon-Vega B, Alcocer-Gomez E, Lendines-Cordero D, Cooper MA, de la Cruz P, et al. NLRP3 inflammasome inhibition by MCC950 in aged mice improves health via enhanced autophagy and PPARalpha activity. J Gerontol A Biol Sci Med Sci. (2020) 75:1457–64. doi: 10.1093/gerona/glz239
100. Bhaskaran N, Faddoul F, Paes da Silva A, Jayaraman S, Schneider E, Mamileti P, et al. IL-1beta-myD88-mTOR axis promotes immune-protective IL-17A(+)Foxp3(+) cells during mucosal infection and is dysregulated with aging. Front Immunol. (2020) 11:595936. doi: 10.3389/fimmu.2020.595936
101. Cai Y, Xue F, Qin H, Chen X, Liu N, Fleming C, et al. Differential Roles of the mTOR-STAT3 Signaling in Dermal gammadelta T Cell Effector Function in Skin Inflammation. Cell Rep. (2019) 27:3034–3048.e5. doi: 10.1016/j.celrep.2019.05.019
102. Xiao Z, Peng J, Wu L, Arafat A, Yin F. The effect of IL-1beta on synaptophysin expression and electrophysiology of hippocampal neurons through the PI3K/Akt/mTOR signaling pathway in a rat model of mesial temporal lobe epilepsy. Neurol Res. (2017) 39:640–8. doi: 10.1080/01616412.2017.1312070
103. Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, et al. Microglia states and nomenclature: A field at its crossroads. Neuron. (2022) 110(21):3458–83. doi: 10.1016/j.neuron.2022.10.020
104. Shimizu Y, Ota K, Ikeguchi R, Kubo S, Kabasawa C, Uchiyama S. Plasma osteopontin levels are associated with disease activity in the patients with multiple sclerosis and neuromyelitis optica. J Neuroimmunol. (2013) 263:148–51. doi: 10.1016/j.jneuroim.2013.07.005
105. Yim A, Smith C, Brown AM. Osteopontin/secreted phosphoprotein-1 harnesses glial-, immune-, and neuronal cell ligand-receptor interactions to sense and regulate acute and chronic neuroinflammation. Immunol Rev. (2022) 311:224–33. doi: 10.1111/imr.13081
106. Comi C, Carecchio M, Chiocchetti A, Nicola S, Galimberti D, Fenoglio C, et al. Osteopontin is increased in the cerebrospinal fluid of patients with Alzheimer's disease and its levels correlate with cognitive decline. J Alzheimers Dis. (2010) 19:1143–8. doi: 10.3233/JAD-2010-1309
107. Comabella M, Pericot I, Goertsches R, Nos C, Castillo M, Blas Navarro J, et al. Plasma osteopontin levels in multiple sclerosis. J Neuroimmunol. (2005) 158:231–9. doi: 10.1016/j.jneuroim.2004.09.004
108. Vogt MH, Floris S, Killestein J, Knol DL, Smits M, Barkhof F, et al. Osteopontin levels and increased disease activity in relapsing remitting multiple sclerosis patients. J Neuroimmunol. (2004) 155:155–60. doi: 10.1016/j.jneuroim.2004.06.007
109. Brown A, Islam T, Adams R, Nerle S, Kamara M, Eger C, et al. Osteopontin enhances HIV replication and is increased in the brain and cerebrospinal fluid of HIV-infected individuals. J Neurovirol. (2011) 17:382–92. doi: 10.1007/s13365-011-0035-4
110. Burdo TH, Ellis RJ, Fox HS. Osteopontin is increased in HIV-associated dementia. J Infect Dis. (2008) 198:715–22. doi: 10.1086/590504
111. Mahmud FJ, Du Y, Greif E, Boucher T, Dannals RF, Mathews WB, et al. Osteopontin/secreted phosphoprotein-1 behaves as a molecular brake regulating the neuroinflammatory response to chronic viral infection. J Neuroinflamm. (2020) 17:273. doi: 10.1186/s12974-020-01949-4
112. Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. (2001) 410:988–94. doi: 10.1038/35073667
113. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell. (2018) 173:1073–81. doi: 10.1016/j.cell.2018.05.003
114. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. (2017) 169:1276–90.e17. doi: 10.1016/j.cell.2017.05.018
115. Masuda T, Sankowski R, Staszewski O, Prinz M. Microglia heterogeneity in the single-cell era. Cell Rep. (2020) 30:1271–81. doi: 10.1016/j.celrep.2020.01.010
116. Qiu Y, Shen X, Ravid O, Atrakchi D, Rand D, Wight AE, et al. Definition of the contribution of an Osteopontin-producing CD11c(+) microglial subset to Alzheimer's disease. Proc Natl Acad Sci U S A. (2023) 120:e2218915120. doi: 10.1073/pnas.2218915120
117. Rentsendorj A, Sheyn J, Fuchs DT, Daley D, Salumbides BC, Schubloom HE, et al. A novel role for osteopontin in macrophage-mediated amyloid-beta clearance in Alzheimer's models. Brain Behav Immun. (2018) 67:163–80. doi: 10.1016/j.bbi.2017.08.019
118. De Schepper S, Ge JZ, Crowley G, Ferreira LSS, Garceau D, Toomey CE, et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer's disease. Nat Neurosci. (2023) 26:406–15. doi: 10.1038/s41593-023-01257-z
119. Lalwani RC, Volmar C-H, Wahlestedt C, Webster KA, Shehadeh LA. Contextualizing the role of osteopontin in the inflammatory responses of Alzheimer’s disease. Biomedicines. (2023) 11:3232. doi: 10.3390/biomedicines11123232
120. Shi L, Sun Z, Su W, Xu F, Xie D, Zhang Q, et al. Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity. (2021) 54:1527–42.e8. doi: 10.1016/j.immuni.2021.04.022
121. Davaanyam D, Kim ID, Lee JK. Intranasal delivery of RGD-containing osteopontin heptamer peptide confers neuroprotection in the ischemic brain and augments microglia M2 polarization. Int J Mol Sci. (2021) 22:9999. doi: 10.3390/ijms22189999
122. Zhang X, Shu Q, Liu Z, Gao C, Wang Z, Xing Z, et al. Recombinant osteopontin provides protection for cerebral infarction by inhibiting the NLRP3 inflammasome in microglia. Brain Res. (2021) 1751:147170. doi: 10.1016/j.brainres.2020.147170
123. Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, et al. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal. (2012) 5:ra38. doi: 10.1126/scisignal.2002767
124. Lopes KP, Yu L, Shen X, Qiu Y, Tasaki S, Iatrou A, et al. Associations of cortical SPP1 and ITGAX with cognition and common neuropathologies in older adults. Alzheimers Dement. (2024) 20:525–37. doi: 10.1002/alz.13474
125. Cowan M, Petri WA Jr. Microglia: immune regulators of neurodevelopment. Front Immunol. (2018) 9:2576. doi: 10.3389/fimmu.2018.02576
126. Lauro C, Limatola C. Metabolic reprograming of microglia in the regulation of the innate inflammatory response. Front Immunol. (2020) 11:493. doi: 10.3389/fimmu.2020.00493
127. Wenzel TJ, Gates EJ, Ranger AL, Klegeris A. Short-chain fatty acids (SCFAs) alone or in combination regulate select immune functions of microglia-like cells. Mol Cell Neurosci. (2020) 105:103493. doi: 10.1016/j.mcn.2020.103493
128. Ye L, Jiang Y, Zhang M. Crosstalk between glucose metabolism, lactate production and immune response modulation. Cytokine Growth Factor Rev. (2022) 68:81–92. doi: 10.1016/j.cytogfr.2022.11.001
129. Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat Rev Microbiol. (2008) 6:266–75. doi: 10.1038/nrmicro1855
130. Chiramel AI, Best SM. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. (2018) 254:34–40. doi: 10.1016/j.virusres.2017.09.006
131. Karam BS, Morris RS, Bramante CT, Puskarich M, Zolfaghari EJ, Lotfi-Emran S, et al. mTOR inhibition in COVID-19: A commentary and review of efficacy in RNA viruses. J Med Virol. (2021) 93:1843–6. doi: 10.1002/jmv.26728
132. Khalid T, Hasan A, Fatima JE, Faridi SA, Khan AF, Mir SS. Therapeutic role of mTOR inhibitors in control of SARS-CoV-2 viral replication. Mol Biol Rep. (2023) 50:2701–11. doi: 10.1007/s11033-022-08188-1
133. Vakrakou AG, Alexaki A, Brinia ME, Anagnostouli M, Stefanis L, Stathopoulos P. The mTOR signaling pathway in multiple sclerosis; from animal models to human data. Int J Mol Sci. (2022) 23:8077. doi: 10.3390/ijms23158077
134. Xu J, Du YL, Xu JW, Hu XG, Gu LF, Li XM, et al. Neuroligin 3 regulates dendritic outgrowth by modulating Akt/mTOR signaling. Front Cell Neurosci. (2019) 13:518. doi: 10.3389/fncel.2019.00518
135. Wang J, Gabuzda D. Reconstitution of human immunodeficiency virus-induced neurodegeneration using isolated populations of human neurons, astrocytes, and microglia and neuroprotection mediated by insulin-like growth factors. J Neurovirol. (2006) 12:472–91. doi: 10.1080/13550280601039659
Keywords: neuroimmunology, neuroinflammation, microglia, latency, human immunodeficiency virus, neurodegeneration, neurological disorders, integrins
Citation: Argandona Lopez C and Brown AM (2024) Microglial- neuronal crosstalk in chronic viral infection through mTOR, SPP1/OPN and inflammasome pathway signaling. Front. Immunol. 15:1368465. doi: 10.3389/fimmu.2024.1368465
Received: 10 January 2024; Accepted: 25 March 2024;
Published: 05 April 2024.
Edited by:
Wassim Elyaman, Columbia University, United StatesReviewed by:
Kristen E. Funk, University of North Carolina at Charlotte, United StatesMarta Olah, Columbia University, United States
Copyright © 2024 Argandona Lopez and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda M. Brown, abrown76@jhmi.edu