 Min Zhao1,2†
Min Zhao1,2† Guixian Chen3†
Guixian Chen3† Shuguang Li4Xiaojun Li3Haoxuan Chen3
Shuguang Li4Xiaojun Li3Haoxuan Chen3 Zhenzhen Lou3
Zhenzhen Lou3 Huiying Ouyang3
Huiying Ouyang3 Yibo Zhan3Chenghao Du3
Yibo Zhan3Chenghao Du3 Yuanqi Zhao1,2*
Yuanqi Zhao1,2*- 1The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangdong Provincial Hospital of Chinese Medicine, Guangzhou, Guangdong, China
- 2State Key Laboratory of Dampness Syndrome of Chinese Medicine, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, China
- 3The Second School of Clinical Medicine, Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, China
- 4School of International Relations, National University of Defense Technology, Nanjing, China
Background: Contactin-1 (CNTN1) antibody-positive nodopathy is rare and exhibits distinct clinical symptoms such as tremors and ataxia. However, the mechanisms of these symptoms and the characteristics of the cerebral spinal fluid (CSF) remain unknown.
Case presentation: Here, we report a case of recurrent CNTN1 antibody-positive nodopathy. Initially, a 45-year-old woman experiencing numbness in the upper limbs and weakness in the lower limbs was diagnosed with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Eleven years later, her symptoms worsened, and she began to experience tremors and ataxia. Tests for serum CNTN1, GT1a, and GQ1b antibodies returned positive. Subsequently, she was diagnosed with CNTN1 antibody-positive nodopathy and underwent plasmapheresis therapy, although the treatment’s efficacy was limited. To gain a deeper understanding of the disease, we conducted a comprehensive literature review, identifying 52 cases of CNTN1 antibody-positive nodopathy to date, with a tremor prevalence of 26.9%. Additionally, we found that the average CSF protein level in CNTN1 antibody-positive nodopathy was 2.57 g/L, with 87% of patients exhibiting a CSF protein level above 1.5 g/L.
Conclusion: We present a rare case of recurrent CNTN1 antibody-positive nodopathy. Our findings indicate a high prevalence of tremor (26.9%) and elevated CSF protein levels among patients with CNTN1 antibody-positive nodopathy.
1 Introduction
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is the most common form of chronic inflammatory neuropathy, characterized by segmental demyelination. Beyond demyelinating or axonal damage, recent studies introduced a concept termed “nodopathy”, referring to microstructural alterations limited to the nodal and paranodal regions, potentially leading to significant nerve dysfunction (1, 2). In patients with CNTN1 antibody positivity, paranodal destruction and axo-glial disjunction have been documented. The cell adhesion molecule neurofascin-186 (NF186) anchors voltage-gated sodium channels at the node, while the glial protein neurofascin-155 (NF155), along with axonal proteins contactin-1 (CNTN1) and contactin-associated protein-1 (CASPR1), constitutes an axoglial complex in the paranodal region (Figure 1) (3). Therefore, CIDP cases with NF155 and CNTN1 antibodies might exhibit similar pathophysiology and clinical manifestations, including distal dominant symptoms and axonal degeneration, differing from the typical CIDP symptoms of proximal and distal muscle weakness with less pronounced axonal degeneration (4).
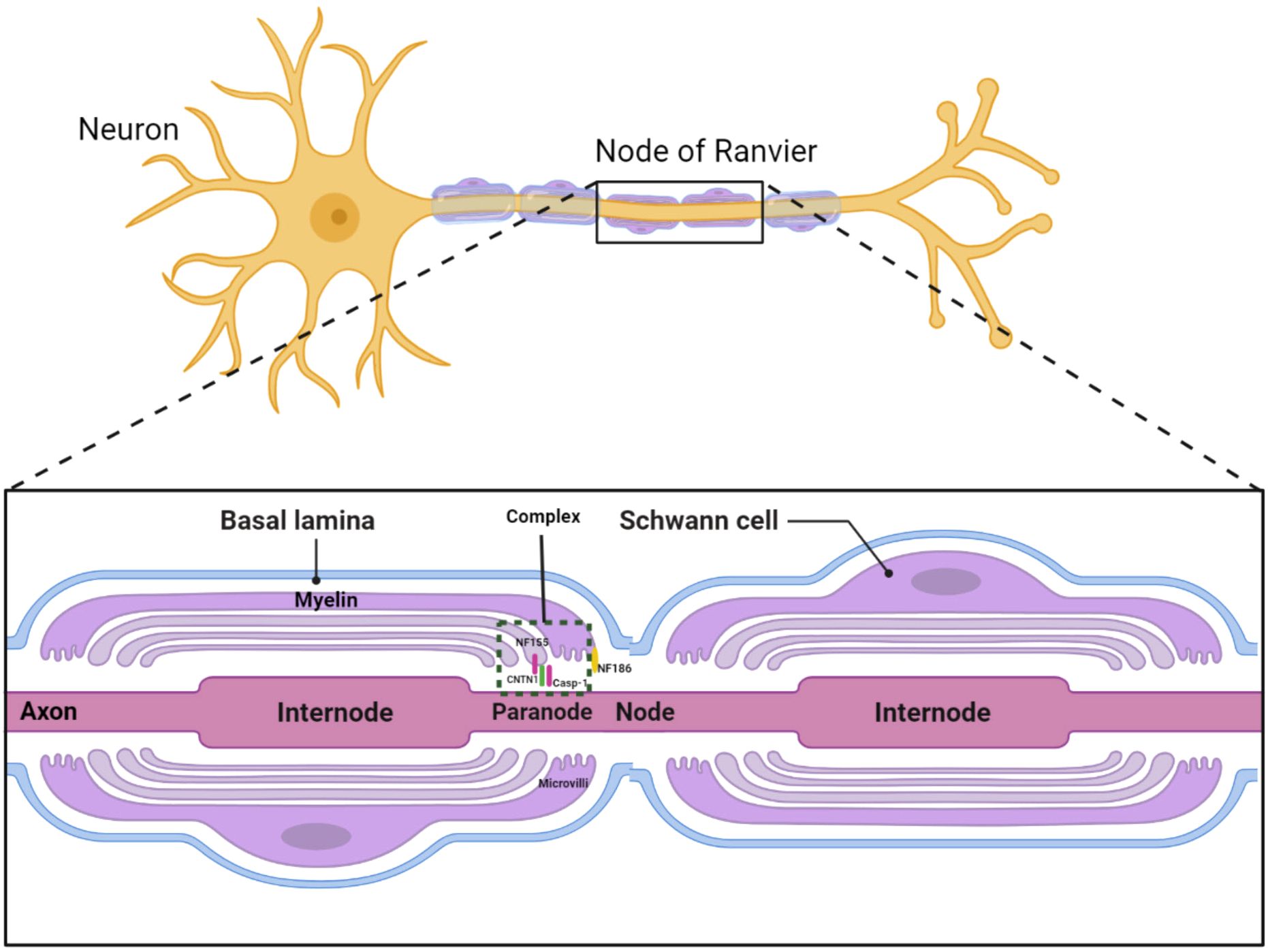
Figure 1 Diagram of the antibody complex in the Ranvier node region.
Approximately 10% of CIDP patients exhibit autoantibodies against nodal and paranodal proteins, with anti-NF155 antibodies present in 4%–18% of patients and antibodies against CNTN1, CASPR1, and the CNTN1-CASPR1 complex reported in 1%–7% of patients (5).
Querol et al. described the characteristics of neuropathy with CNTN1 antibody as including older age at onset, Guillain–Barre syndrome-like acute onset of weakness, sensory ataxia, and early axonal involvement (6), noting a higher prevalence of tremor among those with positive antibodies. The response to corticosteroids and intravenous immune globulin (IVIG) was suboptimal (3).
Given the distinct clinical presentations between typical CIDP and nodopathy, their pathogenic mechanisms might be different. Here, we report a case of recurrent CNTN1 antibody-positive nodopathy and discuss the possible mechanisms of tremor and ataxia, and the CSF characteristics, to deepen our understanding of the disease.
2 Case presentation
2.1 First episode
2.1.1 Medical history and physical examination
On 15 December 2010, a 45-year-old woman was admitted to our hospital with primary complaints of numbness in the upper limbs and weakness in the lower limbs, persisting for 5 months. Neurological examination revealed that the Medical Research Council (MRC) grade (Table 1) for the upper limbs was normal, while the MRC grade for the lower limbs was grade 2. Deep-tendon reflexes across all four limbs were weak. She had a medical history of nephrotic syndrome but no family history of hereditary conditions or infectious diseases.
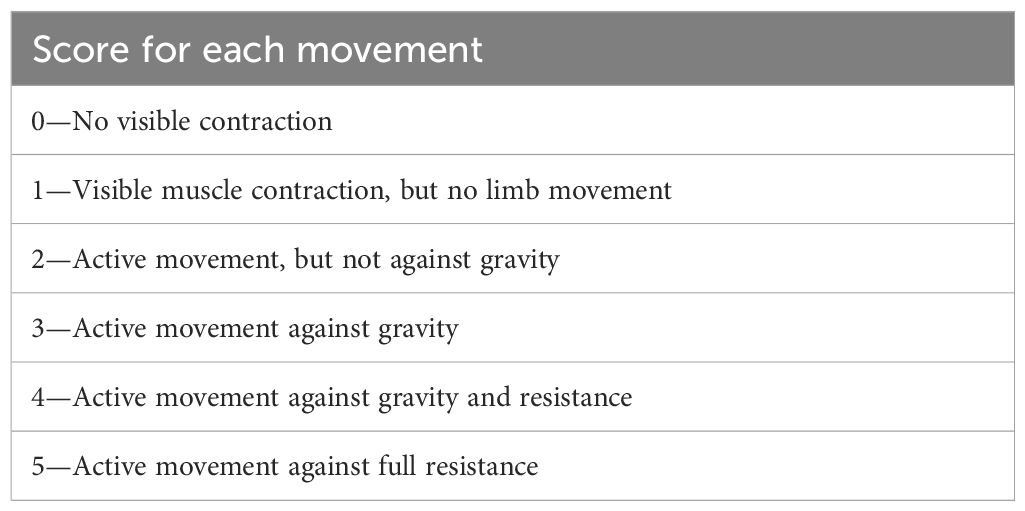
Table 1 Medical research council (MRC) scale for muscle examination.
2.1.2 Auxiliary examination
Electromyography (EMG) results indicated demyelinating alterations and axonal damage in motor and sensory nerves, along with F-wave prolongation (Table 2). CSF analysis revealed a protein concentration of 1,310 mg/L and a white blood cell count of 2 × 106/L.

Table 2 The results of EEG of two episodes.
2.1.3 Diagnosis and treatment
Following the European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the management of chronic inflammatory demyelinating polyradiculoneuropathy (2010) (7), the diagnosis of CIDP was considered. After treatment with intravenous prednisolone, the patient’s symptoms improved, leading to her discharge.
2.2 Second episode
2.2.1 Medical history and physical examination
Eleven years later, the same patient was readmitted to our hospital with numbness and weakness in all four limbs, accompanied by tremor. She required assistance to walk and could not use chopsticks or a pen.
The physical examination revealed bilateral upper limb tremors (Video 1). The MRC grade for the proximal strength in both upper and lower limbs was grade 4, while the distal strength in all four limbs was grade 2. There was noticeable sensory disturbance, including deep sensory disturbances, in the extremities. Tendon reflexes in all four limbs were absent. The patient was unable to perform the finger–nose test and the heel–knee–tibia test successfully. The Babinski sign was absent.
2.2.2 Auxiliary examination
No abnormalities were detected in blood routine tests, coagulation function, interleukin 6 levels, thyroid function, vasculitis testing, or immunoelectrophoresis (blood and urine). CSF analysis showed a protein concentration of 1,344 mg/L and a white blood cell count of 5 × 106/L. The serum immunoglobulin G4 level was 1,283.4 (reference value, 39.2–864).
This time, testing for peripheral ganglioside antibodies and antiparanodal antibodies was conducted. The serum CNTN1 antibody [confirmed by Cytometric Bead Assay (CBA)] tested positive with a titer of 1:1,000+. Additionally, anti-GT1a and anti-GQ1b IgG antibodies in serum were also positive. All tests were performed at Jiangsu Simcere Diagnostic Laboratory (Jiangsu Simcere Diagnostics Co, Ltd, Nanjing 210002, China). Brain MRI, brachial plexus, and lumbosacral MRI showed no evident abnormalities. Electromyography (EMG) indicated worsened demyelinating and axonal damage (Table 2).
2.2.3 Diagnosis and treatment
The patient was diagnosed with CNTN1 antibody-positive nodopathy. Following plasmapheresis therapy, the MRC grade for distal strength in all four limbs improved to 3. However, she remained unable to walk without assistance. Two months post-discharge, no significant improvement was observed.
3 Discussions
Autoantibodies targeting the molecular components of the node of Ranvier proteins, such as neurofascin, contactin-1, and Caspr, have recently been identified in CIDP. The latest guidelines delineate antibody-positive diseases associated with paranodopathy as autoimmune nodopathies (8). Querol first identified the CNTN1 antibody as the target antigen in three patients diagnosed with CIDP (6). These individuals exhibited an aggressive disease phenotype characterized by acute onset, predominantly motor involvement, older age at onset, and, notably, a poor response to IVIg. Subsequently, it was discovered that CNTN1 antibodies were predominantly IgG4, potentially explaining the suboptimal responses to IVIg (9). Diverging from typical CIDP, CNTN1 antibody-positive nodopathy presents distinct clinical features: (1) older age at onset, (2) subacute or chronic onset with progressive development, (3) common occurrences of ataxia and tremor, and (4) frequent deep sensation disturbances (10). Additionally, total cerebrospinal fluid protein levels were elevated. Herein, we report a case of recurrent CNTN1 antibody-positive nodopathy and discuss the possible mechanisms behind tremor and ataxia and the CSF characteristics.
3.1 Tremor and ataxia could be a characteristic symptom of CNTN1 antibody positive nodopathy
Traditionally, tremor and ataxia have been distinct characteristics of NF155 antibody-positive paranodopathy (5, 11, 12). Querol observed that the NF155 antibody binds to the cerebellum, particularly cerebellar neurons, potentially explaining the presence of tremor and ataxia (13).
Initially, Querol identified CNTN1 antibodies in CIDP patients, although none of the three cases exhibited tremor (6). Later, Yumako’s cohort, which included 13 CNTN1 antibody-positive patients out of 533 CIDP patients, reported two instances of tremor (14). Doppler’s cohort described three patients experiencing tremor, both rest and action types. Given the specificity of tremor as a symptom, it may serve as a differential marker between typical CIDP and nodopathy. Further investigations by Doppler and colleagues demonstrated that sera from patients with positive CNTN1 antibodies reacted to the molecular and granular layers of the cerebellum, offering a possible explanation for tremor (10). A review of the literature revealed 52 cases of CNTN1 antibody-positive nodopathy (Table 3), with 14 patients (26.9%) exhibiting tremor, suggesting it as a characteristic symptom of CNTN1-positive nodopathy.

Table 3 Cases of CNTN1 antibody nodopathy.
Tremor is recognized as an accompanying feature of inflammatory-mediated peripheral neuropathies such as immunoglobulin M paraproteinemic neuropathy (25) and CIDP (26, 27). Previous studies have not established a direct relationship between tremor and conduction velocity (28). The speculated mechanism involves central compensation by the cerebellum for delays induced by peripheral neuropathy (29). Moreover, tremor can manifest as a rare symptom of GBS, particularly in specific variants like Miller–Fisher syndrome, where the mechanism is linked to cerebellar function and impaired sensorimotor feedback (30). In the case discussed, the patient did not exhibit tremors during the initial episode nor were ganglioside or paranodal antibodies tested. However, 11 years later, the patient’s symptoms, including tremor and ataxia, worsened. Tests indicated positivity for GT1a, GQ1b, and CNTN1 antibodies. It remains uncertain whether this signifies a change in antibody type or a CNTN1 subtype.
Ataxia has been identified as a common symptom of CNTN1 antibody-positive nodopathy. The mechanisms might involve injury to the Ranvier nodes (31) and the dorsal root ganglia (14, 32). Initially, the CNTN1 and CASPR1 dimer is crucial for maintaining the axon–glial linkage at the paranode and neuronal conduction at the Ranvier nodes (33). In vitro studies have shown that CNTN1 antibodies can disrupt the interaction between the CNTN1/Caspr1 complex and NF155, altering the structure of the Ranvier node region and contributing to symptoms of CNTN1-related CIDP (34). Second, Yumako Miura and colleagues found that CNTN1 is widely expressed in dorsal root ganglion neurons (14). Additionally, the dorsal root ganglia’s blood–nerve barrier is more permeable, allowing CNTN1 antibodies to infiltrate sensory neurons and axons, which may explain sensory ataxia (35). The patient being double-positive for CNTN1 and GT1a and GQ1b antibodies complicates the diagnosis. Given that ataxia is a prevalent sign of Miller–Fisher syndrome, associated with GT1a and GQ1b antibodies (36), it raises the question of whether the ataxia in this case is caused by the CNTN1 antibody or antiganglioside antibodies (GT1a and GQ1b antibodies). At present, the mechanism of tremor and ataxia in CNTN1 antibody-positive nodopathy is still not clear, and further research is needed.
3.2 CSF protein is obviously elevated among CNTN1 antibody-positive nodopathy
In the case of CNTN1 antibody-positive nodopathy, CSF data for eight patients were unavailable. The average CSF protein levels reported were 1.865 g/L in Orazio’s study (21), 1.48 g/L in Cortese’s study (12), and 1.04 g/L in Titulaer’s study (22). Excluding these three studies and the eight patients without CSF data, the average CSF protein level for the remaining 31 patients was 2.57 g/L, with 27 patients (87%) exhibiting a CSF protein level higher than 1.5 g/L (Table 3).
Albuminocytological dissociation, characterized by increased protein levels (>0.45 g/L) in the absence of an elevated white cell count (<50 cells/μL), is a hallmark of GBS (37). High CSF protein levels (>0.45 g/L) have been linked to the demyelinating subtype and proximal or global muscle weakness in patients with GBS. Moreover, a higher CSF protein level has been independently associated with an increased likelihood of poor outcomes in patients with brainstem encephalitis (38). The specific CSF characteristics of paranodopathy, including the relationship between CSF protein levels and prognosis, remain unclear. Through literature review, we identified the CSF characteristic of CNTN1 antibody-positive nodopathy as notably high protein levels, which could serve as a differential diagnostic marker. Future cohort studies should further explore the CSF characteristics of paranodopathy and their correlation with prognosis.
3.3 CNTN1 antibody-positive nodopathy with antiganglioside antibodies
Gangliosides, essential components of peripheral nerves, consist of ceramide bonded to one or more sugars, incorporating sialic acid attached to an oligosaccharide core. Gangliosides are categorized into GM1, GD1a, GT1a, and GQ1b, based on the number and placement of sialic acids. Previous studies identified that antibodies against GM1 and GD1a were linked to acute motor axonal neuropathy (36). The GQ1b antibody, which cross-reacts with the GT1a antibody, has been closely associated with Miller–Fisher syndrome and brainstem encephalitis, characterized by ophthalmoplegia, ataxia, and impaired consciousness (39–41). Theoretically, GQ1b antibodies could explain the sensory ataxia observed in our case. These antibody target molecules have been seldom detected in CIDP patients, and no studies have yet explored the relationship between antiganglioside antibodies and nodopathy. One investigation reported a 12% prevalence of GM1 IgM antibodies among CIDP patients, significantly lower than in multifocal motor neuropathy (60%) (42). A cohort study involving Chinese GBS and CIDP patients identified GM1 as the target antigen in CIDP patients’ sera, noting that the IgM type was more prevalent than IgG in these patients. To our knowledge, this case marks the first instance of CIDP concurrently positive for both CNTN1 antibody and antiganglioside antibodies (anti-GT1a and anti-GQ1b antibodies). The interplay between nodopathy antibodies and antiganglioside antibodies in CIDP and its symptomatic manifestations warrants further investigation.
4 Conclusions
We have reported a rare case of recurrent CNTN1 antibody-positive nodopathy, also positive for GT1a and GQ1b antibodies. Our findings highlight a tremor prevalence of 26.9% among CNTN1 antibody-positive nodopathy cases and suggest that the CSF protein level in such cases may be significantly elevated compared to typical chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Committee of Guangdong Provincial Hospital of Chinese Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
MZ: Writing – review & editing, Writing – original draft, Formal Analysis, Data curation, Conceptualization. GC: Writing – review & editing, Writing – original draft, Methodology, Data curation. SL: Writing – review & editing, Data curation. XL: Writing – review & editing, Investigation. HC: Writing – review & editing, Formal Analysis, Data curation. ZL: Writing – review & editing, Investigation. HO: Writing – review & editing, Investigation. YBZ: Writing – review & editing, Investigation. CD: Writing – review & editing. YQZ: Writing – review & editing, Project administration, Funding acquisition, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This case report was supported by Science and Technology Project of Guangzhou Province (Grant No. 202002020034), Guangdong Bureau of Traditional Chinese Medicine (Grant No. 20225030), NATCM’s Project of High-level Construction of Key TCM Disciplines (zyyzdxk-2023154) and State Key Laboratory of Dampness Syndrome of Chinese Medicine, and The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou 510120, China (Grant Nos. SZ2021ZZ46 and SZ2022KF22). The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Acknowledgments
We thank the patient for her participation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1368487/full#supplementary-material
References
1. Kuwabara S, Misawa S, Mori M. Nodopathy: chronic inflammatory demyelinating polyneuropathy with anti-neurofascin 155 antibodies. J Neurology Neurosurg Psychiatry. (2017) 88:459. doi: 10.1136/jnnp-2016-315170
2. Dziadkowiak E, Waliszewska-Prosół M, Nowakowska-Kotas M, Budrewicz S, Koszewicz Z, Koszewicz M. Pathophysiology of the different clinical phenotypes of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Int J Mol Sci. (2022) 23:179. doi: 10.3390/ijms23010179
3. Vural A, Doppler K, Meinl E. Autoantibodies against the node of ranvier in seropositive chronic inflammatory demyelinating polyneuropathy: diagnostic, pathogenic, and therapeutic relevance. Front Immunol. (2018) 9:1–14. doi: 10.3389/fimmu.2018.01029
4. Kuwabara S, Misawa S, Mori M. Paranodal destruction and axo-glial dysjunction in a subtype of CIDP with anticontaction-1 antibodies. J Neurology Neurosurg Psychiatry. (2015) 86:707. doi: 10.1136/jnnp-2015-310448
5. Bunschoten C, Jacobs BC, Van den Bergh PYK, Cornblath DR, van Doorn PA. Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Lancet Neurol. (2019) 18:784–94. doi: 10.1016/S1474-4422(19)30144-9
6. Luis Q, Gisela N, Ricard R, Eugenia M, Jordi D, Xavier S, et al. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. (2013) 73:370–80. doi: 10.1002/ana.23794
7. Van den Bergh PYK, Hadden RDM, Bouche P, Cornblath DR, Hahn A, Illa I, et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society — First Revision. Eur J Neurol. (2010) 17:356–63. doi: 10.1111/j.1468-1331.2009.02930.x
8. Van den Bergh PYK, van Doorn PA, Hadden RDM, Avau B, Vankrunkelsven P, Allen JA, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force—Second revision. J PERIPHER NERV Syst. (2021) 26:242–68. doi: 10.1111/jns.12455
9. Tackenberg B, Nimmerjahn F, Lünemann JD. Mechanisms of IVIG efficacy in chronic inflammatory demyelinating polyneuropathy. J Clin Immunol. (2010) 30:65–9. doi: 10.1007/s10875-010-9398-1
10. Doppler K, Appeltshauser L, Wilhelmi K, Villmann C, Dib-Hajj SD, Waxman SG, et al. Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J Neurology Neurosurg Psychiatry. (2015) 86:720–8. doi: 10.1136/jnnp-2014-309916
11. Querol L, Nogales-Gadea G, Rojas-Garcia R, Diaz-Manera J, Pardo J, Ortega-Moreno A, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. NEUROLOGY. (2014) 82:879–86. doi: 10.1212/WNL.0000000000000205
12. Cortese A, Lombardi R, Briani C, Callegari I, Benedetti L, Manganelli F, et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP: Clinical relevance of IgG isotype. Neurology: neuroimmunology Neuroinflamm. (2020) 7:e639. doi: 10.1212/NXI.0000000000000639
13. Querol L, Devaux J, Rojas-Garcia R, Illa I. Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications. Nat Rev Neurol. (2017) 13:533–47. doi: 10.1038/nrneurol.2017.84
14. Miura Y, Devaux JJ, Fukami Y, Manso C, Belghazi M, Wong AHY, et al. Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. BRAIN. (2015) 138:1484–91. doi: 10.1093/brain/awv054
15. Delmont E, Manso C, Querol L, Cortese A, Berardinelli A, Lozza A, et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. BRAIN. (2017) 140:1851–8. doi: 10.1093/brain/awx124
16. Lin H, Ho KWD, Chuquilin M. Presence of both anti-contactin 1 and anti-neurofascin 140 antibodies in a case of chronic inflammatory demyelinating polyneuropathy. eNeurologicalSci. (2018) 13:38–9. doi: 10.1016/j.ensci.2018.11.016
17. Zhu J, Liu Y, Tian L, Liu N, Zhang Z. Anti-contactin-1 IgG4 antibody associated chronic inflammatory demyelinating polyradiculoneuropathy:a case report. Chin J Neurol. (2020) 53:1044–8. doi: 10.3760/cma.j.cn113694-20200504-00324
18. Xu Q, Liu S, Zhang P, Wang Z, Chang X, Liu Y, et al. Characteristics of anti-contactin1 antibody-associated autoimmune nodopathies with concomitant membranous nephropathy. Front Immunol. (2021) 12:1–12. doi: 10.3389/fimmu.2021.759187
19. Hou Y, Zhang C, Yu X, Wang W, Zhang D, Bai Y, et al. Effect of low-dose rituximab treatment on autoimmune nodopathy with anti-contactin 1 antibody. Front Immunol. (2022) 13:1–10. doi: 10.3389/fimmu.2022.939062
20. Fehmi J, Davies AJ, Antonelou M, Keddie S, Pikkupeura S, Querol L, et al. Contactin-1 links autoimmune neuropathy and membranous glomerulonephritis. PloS One. (2023) 18:e281156. doi: 10.1371/journal.pone.0281156
21. Liberatore G, De Lorenzo A, Giannotta C, Manganelli F, Filosto M, Cosentino G, et al. Frequency and clinical correlates of anti-nerve antibodies in a large population of CIDP patients included in the Italian database. Neurol Sci. (2022) 43:3939–47. doi: 10.1007/s10072-021-05811-0
22. Broers MC, Wieske L, Erdag E, Gürlek C, Bunschoten C, van Doorn PA, et al. Clinical relevance of distinguishing autoimmune nodopathies from CIDP: longitudinal assessment in a large cohort. J Neurology Neurosurg Psychiatry. (2023) 95:52–60. doi: 10.1136/jnnp-2023-331378
23. Li Q, Chen Q, Zhang T, Xu Y, Kan Y, Zhang J. Case report: Anti-CNTN1 antibody-associated nodopathies disease with asymmetric onset. Front Neurol. (2023) 14:1–7. doi: 10.3389/fneur.2023.1124540
24. Tang Y, Liu J, Gao F, Hao H, Jia Z, Zhang W, et al. CIDP/autoimmune nodopathies with nephropathy: a case series study. Ann Clin Transl NEUR. (2023) 10:706–18. doi: 10.1002/acn3.51754
25. Saifee TA, Schwingenschuh P, Reilly MM, Lunn MPT, Katschnig P, Kassavetis P, et al. Tremor in inflammatory neuropathies. J Neurology Neurosurg Psychiatry. (2013) 84:1282–7. doi: 10.1136/jnnp-2012-303013
26. Smith IS. The natural history of chronic demyelinating neuropathy associated with benign IgM paraproteinaemia. A clinical and neurophysiological study. BRAIN. (1994) 117:949–57. doi: 10.1093/brain/117.5.949
27. Busby M, Donaghy M. Chronic dysimmune neuropathy. J Neurol. (2003) 250:714–24. doi: 10.1007/s00415-003-1068-2
28. Smith I. Tremor in peripheral neuropathy. In: Findler LJ, Koller WC, Warter JM, editors. Handbook of Tremor Disorders. New York: Elsevier Science. (1995).
29. Schwingenschuh P, Saifee TA, Katschnig-Winter P, Reilly MM, Lunn MP, Manji H, et al. Cerebellar learning distinguishes inflammatory neuropathy with and without tremor. NEUROLOGY. (2013) 80:1867–73. doi: 10.1212/WNL.0b013e318292a2b8
30. Rajan R, Anandapadmanabhan R, Vishnoi A, Vishnu VY, Latorre A, Agarwal H, et al. Neuropathic Tremor inGuillain-Barr é Syndrome. Movement Disord Clin Pract. (2023) 10:1333–40. doi: 10.1002/mdc3.13807
31. Stathopoulos P, Alexopoulos H, Dalakas MC. Autoimmune antigenic targets at the node of Ranvier in demyelinating disorders. Nat Rev Neurol. (2015) 11:143–56. doi: 10.1038/nrneurol.2014.260
32. Kanda T. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. J Neurol Neurosurg Psychiatry. (2013) 84:208–12. doi: 10.1136/jnnp-2012-302312
33. Querol L, Illa I. Paranodal and other autoantibodies in chronic inflammatory neuropathies. Curr Opin Neurol. (2015) 28:474–9. doi: 10.1097/WCO.0000000000000233
34. Marilyne L, Bruno H, Gisela N, Luis Q, Isabel I, Catherine F. Specific contactin N-glycans are implicated in neurofascin binding and autoimmune targeting in peripheral neuropathies. J Biol Chem. (2014) 289:7907–18. doi: 10.1074/jbc.M113.528489
35. Plomp JJ, Willison HJ. Pathophysiological actions of neuropathy-related anti-ganglioside antibodies at the neuromuscular junction. J Physiol. (2009) 587:3979–99. doi: 10.1113/jphysiol.2009.171702
36. Shahrizaila N, Lehmann HC, Kuwabara S. Guillain-barré syndrome. Lancet. (2021) 397:1214–28. doi: 10.1016/S0140-6736(21)00517-1
37. Helle A, Alex YD, Amro MS, Sasha AZ, Henning A. CSF findings in relation to clinical characteristics, subtype, and disease course in patients with guillain-barré Syndrome. NEUROLOGY. (2023) 100:e2386–97. doi: 10.1212/WNL.0000000000207282
38. Tan IL, Mowry EM, Steele SU, Pardo CA, McArthur JC, Nath A, et al. Brainstem encephalitis: etiologies, treatment, and predictors of outcome. J Neurol. (2013) 260:2312–9. doi: 10.1007/s00415-013-6986-z
39. Ito M, Kuwabara S, Odaka M, Misawa S, Koga M, Hirata K, et al. Bickerstaff's brainstem encephalitis and Fisher syndrome form a continuous spectrum. J Neurol. (2008) 255:674–82. doi: 10.1007/s00415-008-0775-0
40. Ito M, Matsuno K, Sakumoto Y, Hirata K, Yuki N. Ataxic Guillain-Barre syndrome and acute sensory ataxic neuropathy form a continuous spectrum. J Neurology Neurosurg Psychiatry. (2011) 82:294–9. doi: 10.1136/jnnp.2010.222836
41. Liu JX, Willison HJ, Pedrosa-Domellof F. Immunolocalization of GQ1b and related gangliosides in human extraocular neuromuscular junctions and muscle spindles. Invest Ophthalmol Vis Sci. (2009) 50:3226–32. doi: 10.1167/iovs.08-3333
Keywords: contactin-1, nodopathy, CIDP, tremor, ataxia
Citation: Zhao M, Chen G, Li S, Li X, Chen H, Lou Z, Ouyang H, Zhan Y, Du C and Zhao Y (2024) Recurrent CNTN1 antibody-positive nodopathy: a case report and literature review. Front. Immunol. 15:1368487. doi: 10.3389/fimmu.2024.1368487
Received: 10 January 2024; Accepted: 07 May 2024;
Published: 23 May 2024.
Edited by:
Zhenlong Liu, McGill University, CanadaReviewed by:
Christian Moritz, Centre Hospitalier Universitaire (CHU) de Saint-Étienne, FranceDavid Gillis, Sunshine Coast Hospital and Health Service, Australia
Copyright © 2024 Zhao, Chen, Li, Li, Chen, Lou, Ouyang, Zhan, Du and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanqi Zhao, zyq2022@gzucm.edu.cn
†These authors have contributed equally to this work