 Kenneth K. Y. Ting
Kenneth K. Y. Ting- 1Department of Immunology, University of Toronto, Toronto, ON, Canada
- 2Toronto General Hospital Research Institute, University Health Network, Toronto, ON, Canada
The overconsumption of dietary fructose has been proposed as a major culprit for the rise of many metabolic diseases in recent years, yet the relationship between a high fructose diet and neurological dysfunction remains to be explored. Although fructose metabolism mainly takes place in the liver and intestine, recent studies have shown that a hyperglycemic condition could induce fructose metabolism in the brain. Notably, microglia, which are tissue-resident macrophages (Mφs) that confer innate immunity in the brain, also express fructose transporters (GLUT5) and are capable of utilizing fructose as a carbon fuel. Together, these studies suggest the possibility that a high fructose diet can regulate the activation and inflammatory response of microglia by metabolic reprogramming, thereby altering the susceptibility of developing neurological dysfunction. In this review, the recent advances in the understanding of microglia metabolism and how it supports its functions will be summarized. The results from both in vivo and in vitro studies that have investigated the mechanistic link between fructose-induced metabolic reprogramming of microglia and its function will then be reviewed. Finally, areas of controversies and their associated implications, as well as directions that warrant future research will be highlighted.
1 Introduction of microglia
Microglia, which are tissue resident macrophages (Mφs) of the central nervous system, constitute a major component of the innate immune system in the brain. Depending on the area, microglia can comprise 5% to 12% of the total cell populations in the brain (1). Although microglia are classified as tissue resident Mφs, their ontogeny is notably distinct when compared to other types of Mφs. Genetic fate-mapping studies have now demonstrated that microglia are derived from primitive Mφs, which are differentiated from erythro-myeloid precursors in the yolk sac, and they colonize the developing brain as early as embryonic day 9.5 (2–4). Upon colonization, they retain their embryonic origin throughout adult life with no replacement from circulating monocytes.
To provide immune protection and maintain a healthy neural microenvironment, microglia are responsible for monitoring and scavenging the parenchyma continuously through its interaction with environmental cues, such as chemokines, cytokines, and other trophic factors (5–7). Upon detection of changes in the local environment, depending on the type of stimuli, microglia becomes activated and display a diverse profile of phenotypes. Similar to other tissue resident Mφs, microglia can also be divided into M1 and M2 Mφs. As M1 Mφs, they display cytotoxic and pro-inflammatory responses upon recognition of inflammatory stimulus, such as bacterial lipopolysaccharides (LPS). On other hand, they can also be alternatively activated into M2 Mφs, where they can perform repair and regeneration functions (M2a), immune-regulatory functions (M2b) or the acquisition of a deactivating phenotype (M2c) (8, 9). Overall, microglia demonstrate high phenotypic and functional plasticity in response to the wide spectrum of changes that take place in the brain (1, 10). Apart from its scavenging functions, microglia are also now recognized to be involved in other dynamic processes that can modulate brain functions, including the formation and maturation of synapse, brain homeostasis, neurogenesis and regulating neuronal excitability (11).
2 Metabolic reprogramming of microglia
Recent advances in the immunometabolism field have shown that the acquisition of altered metabolic adaptations due to the rewiring of metabolic circuits, known as metabolic reprogramming, is critical for the activation of immune cells and their functions. To support the heterogenous phenotypes and functions of microglia as described above, they are capable of metabolizing a variety of carbon sources, such as glucose (12), fructose (13), free fatty acids (14), lactate (15), and ketone bodies (16), in which the type of fuel it metabolizes can modulate its ability to perform its effector functions. In general, microglia express GLUT3 and GLUT5 to facilitate the uptake of glucose and fructose respectively (17–19). However, under inflammatory conditions, microglia can undergo metabolic reprogramming by transcriptionally activating the expression of glycolytic genes, such as glucose transporters (GLUT1) and lactate transporters (MCT1), to increase the breakdown of glucose (glycolysis) (15, 20, 21). More importantly, it has been shown that LPS-induced activation of microglia cell lines increased the rate of glycolysis while decreased the rate of oxidative phosphorylation (OXPHOS) (22). This shift from OXPHOS to aerobic glycolysis is known as the Warburg effect, which was first observed in cancer cells by Otto Warburg (23). Mechanistically, it is believed that this metabolic shift is due to the activation of mechanistic target of rapamycin (mTOR) complex 1 and hypoxia-induced factor 1α (HIF-1α) (24–26), which directly transactivate the expression of glycolytic and inflammatory genes, along with the activation of HIF-1α co-activators.
The increased flux of glycolysis in activated microglia is directly linked to its ability elicit an inflammatory response as blocking glycolysis with 2-deoxy-glucose impaired its production of pro-inflammatory cytokines, such as Tumor Necrosis Factor alpha (TNF-α) and Interleukin-6 (IL-6), in a NF-kB-dependent manner (27, 28). On the other hand, incubation of microglia with increased concentration of glucose further enhanced its production of TNF-α (29, 30). Although the direct link between the consequence of increased glycolytic flux and production of inflammatory cytokines is an ongoing investigation, recent reports have shown that the induction of glycolysis could lead to increased flux of the pentose phosphate pathway (PPP) and multiple disruptions in the tricarboxylic acid (TCA) cycle of Mφs in general (31–33). While the increased flux of PPP increased the production of NADPH, which is critical for regulating Mφ inflammatory responses (34), the disrupted TCA cycle had led to the accumulation of key metabolites that are linked to inflammation. For instance, the accumulation of succinate due to the inhibition of succinate dehydrogenase is important for the stabilization of HIF-1α and its direct transcription of Il1b transcripts (35, 36). On the other hand, the accumulation of citrate due to the inhibition of isocitrate dehydrogenase (36, 37) can be converted back to acetyl-CoA by ATP citrate lyase and it is critical for histone acetylation of inflammatory genes (38, 39), as well as de novo lipid synthesis, which supports the secretion of pro-inflammatory mediators (40). Apart from metabolites, the increased flux of the PPP also fuels the production of nitric oxide (NO) by nitric oxide synthase, in which the production of NO could inhibit the function of the electron transport chain (ETC) in Mφs (41, 42). The impaired activity of the ETC then induce the reversal of electron flow and drive the production of ROS, which is responsible to inhibit the activity of prolyl hydroxylases and thus stabilizing HIF-1α levels (43). More recently, it has also been revealed that blocking the activity of factor inhibiting HIF (FIH), an enzyme that inhibits the transactivation capacity of HIF-1α, is also critical for fully activating HIF-1α transcriptional function in Mφs (44). Collectively, these studies have shown that glucose metabolism plays a critical role in orchestrating the inflammatory response of activated microglia (Figure 1).
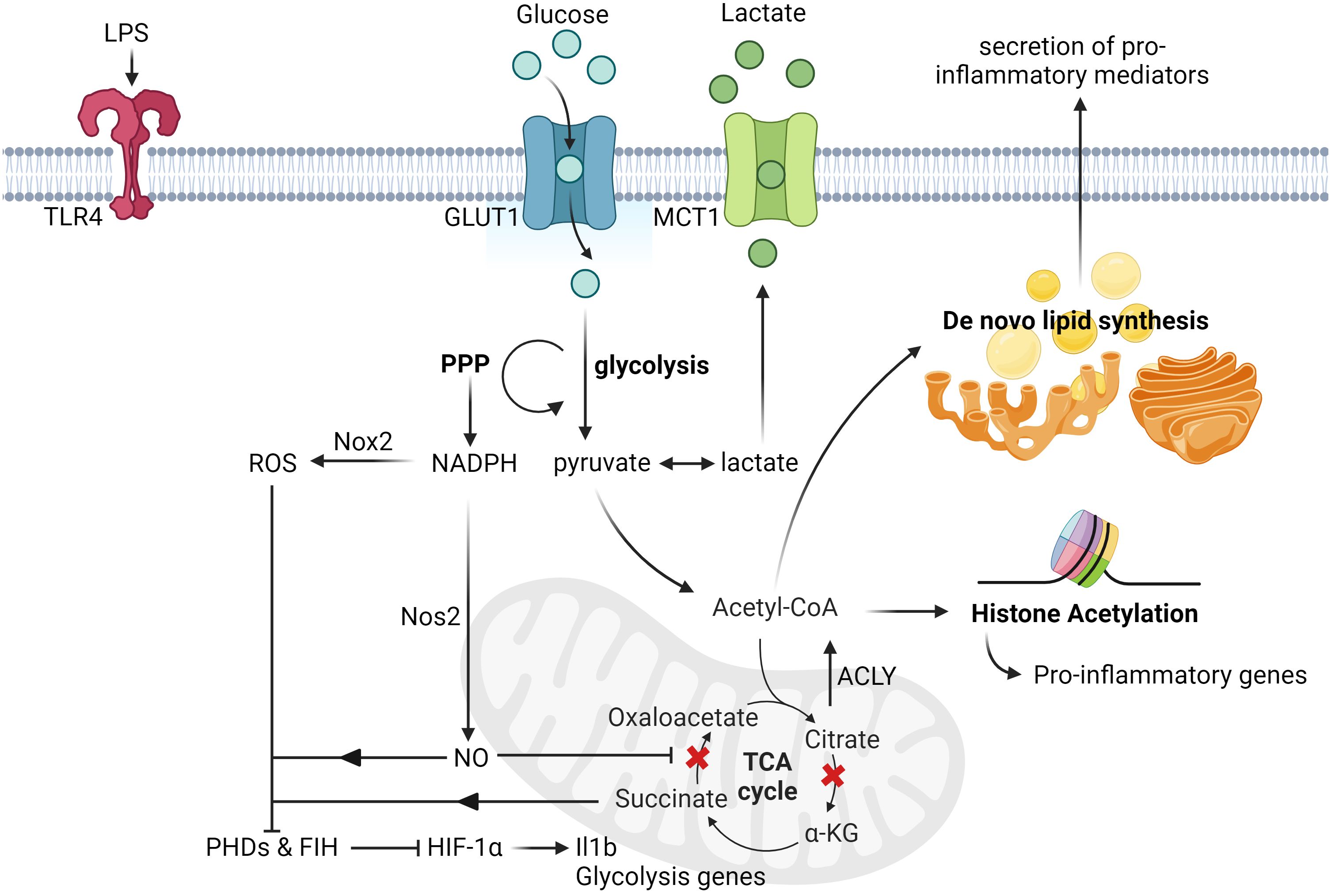
Figure 1 Glucose-induced metabolic reprogramming of activated microglia and macrophages. Upon LPS stimulation, microglia upregulate the expression of glucose transporters (GLUT1) and lactate transporters (MCT1) to increase the rate of glucose metabolism. This induction of glycolysis then leads to the increased rate of PPP, which is responsible for the synthesis of nucleotides and NADPH. The augmented production of NADPH subsequently drives the production of nitric oxide and ROS by Nos2 and Nox2 respectively. Upon nitric oxide-mediated inhibition of succinate dehydrogenase activity, it leads to the accumulation of succinate, which together with ROS, inhibit the activity of both FIH and PHDs. This eventually leads to the stabilization and activation of HIF-1α transactivation capacity, and the transcription of its targeted genes, such as Il1b and other glycolysis genes. Apart from this, another metabolic break happens at the isocitrate dehydrogenase level due to its suppressed expression post LPS stimulation, thereby leading to the accumulation of citrate. The rapid increased levels of citrate can then be converted to acetyl-CoA by ACLY and contribute to histone acetylation and de novo lipid biosynthesis (ex. synthesis of fatty acids), which can promote histone acetylation of pro-inflammatory genes and secretion of pro-inflammatory cytokines respectively. All figures are created with BioRender.com.
On the other hand, under anti-inflammatory conditions, such as stimulation by Interleukin-4 or Interleukin-13, microglia are metabolically adapted to utilize OXPHOS, which led to an attenuated uptake of glucose and production of lactate, and display similar oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) levels as unstimulated microglia (14, 31). Finally, under homeostatic physiological conditions, microglia utilize a combination of oxidative catabolism of glucose and free fatty acids to fuel the TCA cycle and electron transport chain for generating large amounts of ATP, thereby sustaining the energetic needs for their basic surveyance functions (14, 45).
3 Fructose-induced metabolism in the brain and its reprogramming of microglia
It is well-established that glucose metabolism is vital to fuel the inflammatory functions of microglia, yet the role of fructose metabolism in modulating microglia inflammatory response is largely unexplored. In fact, it was previously unclear if dietary fructose consumption could even affect fructose metabolism in the brain as the breakdown of fructose mainly takes place in the intestine and liver (46–49). Until recently, Hwang et al. have used 1H magnetic resonance spectroscopy (MRS) scanning to measure intracerebral fructose levels, and found it was rapidly increased in the human brain under hyperglycemic condition (intravenous injection of 20% dextrose in human subjects) (50). Specifically, the authors found the fructose levels in the brain rose significantly, as early as 20 minutes post dextrose injection, and maintained elevated until the end of the study (240 minutes), with the range of fructose concentration changing from 0 to 0.7mmol/L approximately (50). More importantly, the authors also discovered that the rapid rise of intracerebral fructose levels occurred prior to the increase of plasma fructose levels, suggesting that the hyperglycemic condition could induce endogenous fructose production in the brain (50). Similar findings were also observed in a previous study performed by Hwang et al, where the authors have used gas chromatography-mass spectroscopy to measure fructose concentration in cerebrospinal fluids (CSF) from pregnant women and found that CSF-derived fructose and sorbitol levels were much higher (9-20-fold) than the levels in plasma (51). Taken together, these findings collectively demonstrate that fructose could be produced endogenously in the brain and that the effects of intracerebral fructose extend beyond its dietary consumption.
Understanding that fructose can be endogenously produced in the brain under hyperglycemic condition, and that a high-fructose diet is known to induce hyperglycemia, this then raises the possibility that a high-fructose diet can potentially modulate the metabolic and inflammatory profiles in the brain. Indeed, past reports have already revealed that a high fructose diet increased the expression of GLUT5 (52), as well as pro-inflammatory mediators, such as Tnfa, in the hippocampus (53). Mechanistically, fructose-diet induced Toll-like receptor 4/NF-kB and p38 MAPK/ERK inflammatory phosphorylating signaling cascades in the hippocampus, which eventually led to the increased expression of pro-inflammatory cytokines (54).
To further investigate if dietary fructose-induced inflammation in the hippocampus is due to increased activation of microglia, recent effort has been invested to elucidate the link between fructose-mediated metabolic reprogramming in microglia and its effector functions. Using leptin receptor-deficient type 2 diabetes mellitus (db/db) mice, Li et al reported an upregulation of fructose-related metabolism in the hippocampus, as determined by metabolomic, proteomic, and transcriptomic analysis (13). Single-cell RNA sequencing of hippocampus also revealed that the expression of fructose metabolism-related genes, such as Khk and Slc2a5, as well as ROS generating enzymes, such as NADPH Oxidase 4 (Nox4), were increased in microglia in db/db mice compared to littermate control (13). Mechanistically, selective knockdown of Khk in the hippocampus and microglia resulted in reduced expression of Nox4 in microglia (13). Fructose-mediated Nox4 regulation was also linked to its mitochondrial translocation and that Nox4-induced ROS impaired mitochondrial homeostasis, eventually leading to the damage of synaptic plasticity (13). Similar to this, another study done by Hyer et al also found that high fructose diet induced the activation of microglia in male, but interestingly not in female rats (55). Specifically, male rats fed on a high fructose diet had increased activation of microglia with reduction of their dendritic complexity, which correlated to an impairment of their reverse learning ability (55).
Although the studies above demonstrated that the metabolism and function of microglia in vivo was modulated in response to high fructose diet, in vitro studies that investigated the direct intrinsic effects of fructose on microglia remains to be controversial. For instance, Mizuno et al have treated primary microglia and SIM-A9 cells (murine microglia cell lines) with high concentration of fructose (7.5mM for 24h) and glucose (7.5mM for 24h) separately and found that only glucose has induced inflammation and expression of Slc2a5 (GLUT5) (56). However, on the other hand, Xu et al have treated microglia BV-2 cells with high concentration of fructose (5mM for 24h) and found it induced TLR4/NF-κB activation, as well as pro-inflammatory gene expression, such as Il1b, Il6 and Tnfa (57). Similar findings were also reported by Cigliano et al where they also incubated BV-2 cells with a range of concentration of fructose (0 to 10mM, 24h) and found it increased TNF-α production as measured by ELISA (54). Overall, these studies suggest that the intrinsic inflammatory effect of fructose appears to be dependent on specific experimental condition, and further experiments are needed to resolve the controversies.
Several important points of consideration need to be taken when comparing the results obtained from the presented in vitro and in vivo studies, one of which is the discrepancy between the intracerebral fructose levels quantified in the in vivo experiments, and the experimental fructose levels that were used in the in vitro experiments. For instance, as shown in the MRS scanning study performed by Hwang et al., the intracerebral fructose levels fluctuated only between 0 to 0.7mmol/L post dextrose injection (50). Yet, all the in vitro studies presented in this review have incubated microglia with fructose at a much higher concentration (at least 5mM). This not only raises concerns that the in vitro findings may not demonstrate physiological relevance, but also suggests the need of more in vitro studies that incubate microglia with physiologically relevant levels of fructose. Apart from this, the expression and function of proteins involved in fructose metabolism, such as GLUT5, is not always measured in the in vitro studies discussed in this review, thus making it difficult to draw a mechanistic link between the observed inflammatory phenotypes in microglia and their metabolism of fructose.
4 Conclusion and future perspectives
The overconsumption of dietary fructose has been associated with the rise of chronic inflammatory diseases, such as obesity, diabetes, cardiovascular disease, and cancer (58–60). Furthermore, epidemiological studies have now revealed that high fructose consumption can also induce brain disturbances and negatively affect the development of neural system (61, 62). Early studies that investigated the relationship between the effects of dietary fructose and neural functions have shown that microglia do express fructose transporter (GLUT5) (18) and that fructose metabolism in the brain was stimulated under hyperglycemic condition (50). Understanding that the type of carbon fuel in which microglia metabolize can profoundly shape their effector functions, the focus of recent research has now shifted to the elucidation of a mechanistic link between high fructose consumption and its metabolic effects on microglia activation and inflammatory response. While this topic is still currently under heavy investigation, recent completed studies have generally demonstrated that high fructose-diet induced fructose metabolism in microglia is linked to its increased activation and inflammatory response, which can possibly lead to cognitive dysfunction and impairment (Figure 2). Yet, in vitro studies that directly investigate the intrinsic inflammatory response of fructose in microglia remains to be controversial and warrants future research to further determine the inflammatory signaling cascades that fructose metabolism may enhance, as well as the transcriptional regulation that modulates the expression of fructose metabolism genes.
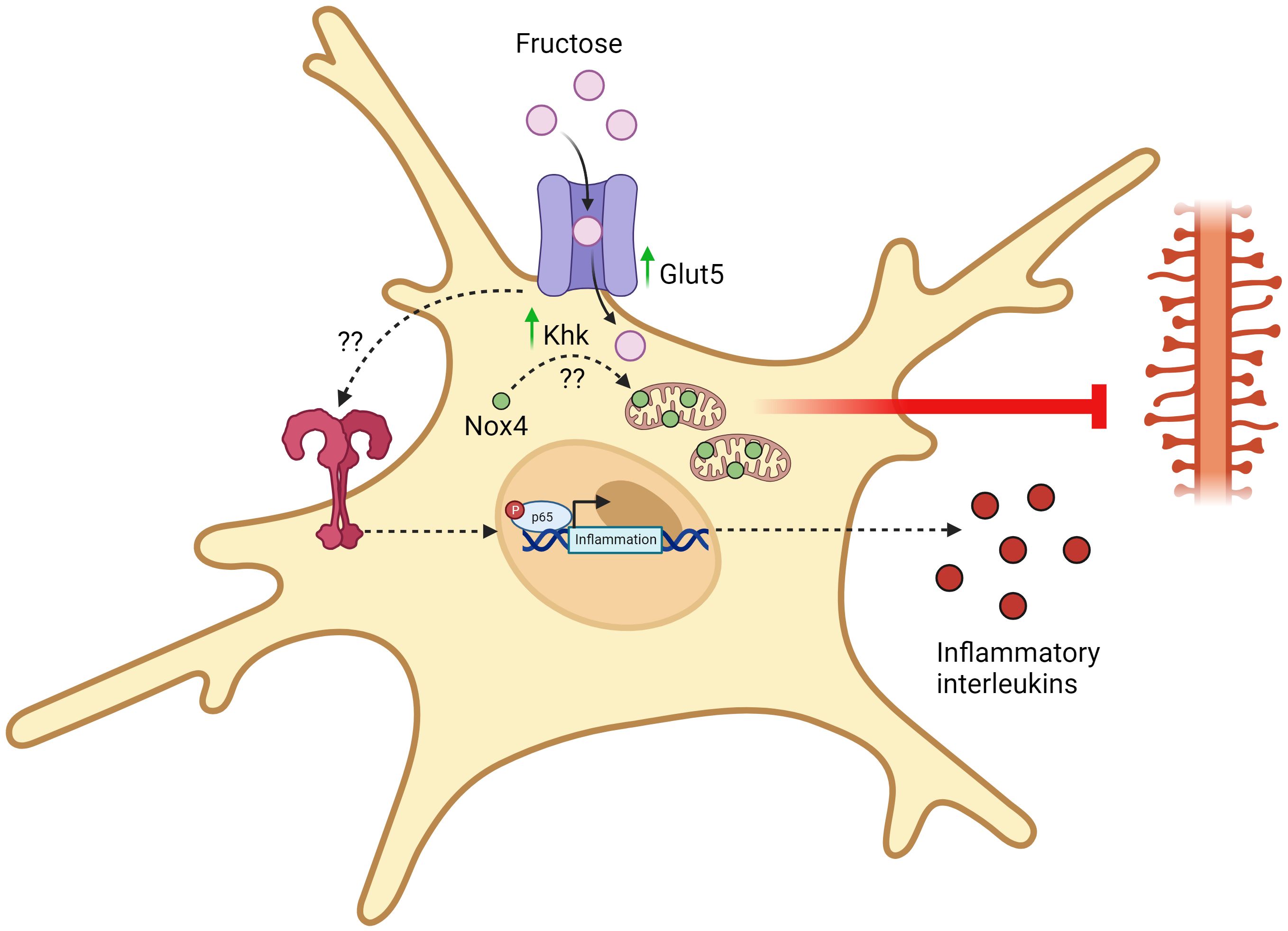
Figure 2 Fructose-induced metabolic reprogramming of activated microglia. Upon chronic exposure of high fructose, microglia upregulates the expression of fructose transporters (GLUT5) and keto-hexokinase (KHK). Through an unidentified mechanism, KHK promotes the translocation of Nox4 into the mitochondria and Nox4-mediated ROS synthesis disrupts mitochondrial homeostasis and eliminates dendrites. This eventually damages synaptic transmission and promotes the development of cognitive disorders. Apart from this, chronic exposure of high fructose also activates TLR4/NF-kB signaling pathways through an unidentified mechanism, thereby leading to the transcription and synthesis of pro-inflammatory mediators. All figures are created with BioRender.com.
The controversial findings observed in vitro could also suggest the existence of other external factors derived from the high fructose diet that could enhance the inflammatory responses of microglia in vivo, such as circulating metabolites that can cross the blood brain barrier. Furthermore, external variables such as sex can also modulate the effects of dietary fructose on neural functions. As described from the study by Hyer et al, cognitive flexibility was only impaired in male, but not female rats fed on high fructose diet, implicating that female sex hormones may play a role in the protection against fructose diets (55). Indeed, past research has shown that females do not develop hyperinsulinemia post fructose feeding with the exception of ovariectomy (63). Understanding the underlying differences between how high fructose consumption differentially affect the neural function of males and females may help to identify novel mechanisms that protect the deleterious effects of fructose in an in vivo setting.
Author contributions
KKYT: Conceptualization, Funding acquisition, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. KKYT was supported by fellowships from Canadian Institutes of Health Research, Ontario Graduate Scholarship, fellowships from the University of Toronto and The Peterborough K. M. Hunter Charitable Foundation.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
2. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
3. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
4. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. (2013) 16:273–80. doi: 10.1038/nn.3318
5. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
6. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
7. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. (2018) 18:225–42. doi: 10.1038/nri.2017.125
8. Chhor V, Le Charpentier T, Lebon S, Oré MV, Celador IL, Josserand J, et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun. (2013) 32:70–85. doi: 10.1016/j.bbi.2013.02.005
9. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. (2013) 39:3–18. doi: 10.1111/nan.12011
10. Schwartz M, Butovsky O, Brück W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. (2006) 29:68–74. doi: 10.1016/j.tins.2005.12.005
11. Lenz KM, McCarthy MM. A starring role for microglia in brain sex differences. Neuroscientist. (2015) 21:306–21. doi: 10.1177/1073858414536468
12. Aldana BI. Microglia-specific metabolic changes in neurodegeneration. J Mol Biol. (2019) 431:1830–42. doi: 10.1016/j.jmb.2019.03.006
13. Li Y, Jiang T, Du M, He S, Huang N, Cheng B, et al. Ketohexokinase-dependent metabolism of cerebral endogenous fructose in microglia drives diabetes-associated cognitive dysfunction. Exp Mol Med. (2023) 55:2417–32. doi: 10.1038/s12276-023-01112-y
14. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. (2016) 173:649–65. doi: 10.1111/bph.13139
15. Moreira TJ, Pierre K, Maekawa F, Repond C, Cebere A, Liljequist S, et al. Enhanced cerebral expression of MCT1 and MCT2 in a rat ischemia model occurs in activated microglial cells. J Cereb Blood Flow Metab. (2009) 29:1273–83. doi: 10.1038/jcbfm.2009.50
16. Huang C, Wang P, Xu X, Zhang Y, Gong Y, Hu W, et al. The ketone body metabolite β-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia. (2018) 66:256–78. doi: 10.1002/glia.23241
17. Kalsbeek MJ, Mulder L, Yi CX. Microglia energy metabolism in metabolic disorder. Mol Cell Endocrinol. (2016) 438:27–35. doi: 10.1016/j.mce.2016.09.028
18. Payne J, Maher F, Simpson I, Mattice L, Davies P. Glucose transporter Glut 5 expression in microglial cells. Glia. (1997) 21:327–31. doi: 10.1002/(ISSN)1098-1136
19. Douard V, Ferraris RP. Regulation of the fructose transporter GLUT5 in health and disease. Am J Physiol Endocrinol Metab. (2008) 295:E227–37. doi: 10.1152/ajpendo.90245.2008
20. Wang L, Pavlou S, Du X, Bhuckory M, Xu H, Chen M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol Neurodegener. (2019) 14:2. doi: 10.1186/s13024-019-0305-9
21. Kong L, Wang Z, Liang X, Wang Y, Gao L, Ma C. Monocarboxylate transporter 1 promotes classical microglial activation and pro-inflammatory effect via 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase 3. J Neuroinflamm. (2019) 16:240. doi: 10.1186/s12974-019-1648-4
22. Voloboueva LA, Emery JF, Sun X, Giffard RG. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. (2013) 587:756–62. doi: 10.1016/j.febslet.2013.01.067
23. Weinhouse S. On respiratory impairment in cancer cells. Science. (1956) 124:267–9. doi: 10.1126/science.124.3215.267
24. Sangineto M, Ciarnelli M, Cassano T, Radesco A, Moola A, Bukke VN, et al. Metabolic reprogramming in inflammatory microglia indicates a potential way of targeting inflammation in Alzheimer's disease. Redox Biol. (2023) 66:102846. doi: 10.1016/j.redox.2023.102846
25. Baik SH, Kang S, Lee W, Choi H, Chung S, Kim J-I, et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer's disease. Cell Metab. (2019) 30:493–507.e6. doi: 10.1016/j.cmet.2019.06.005
26. Bernier L-P, York EM, MacVicar BA. Immunometabolism in the brain: how metabolism shapes microglial function. Trends Neurosci. (2020) 43:854–69. doi: 10.1016/j.tins.2020.08.008
27. Wang Q, Zhao Y, Sun M, Liu S, Li B, Zhang L, et al. 2-Deoxy-d-glucose attenuates sevoflurane-induced neuroinflammation through nuclear factor-kappa B pathway in vitro. Toxicol In Vitro. (2014) 28:1183–9. doi: 10.1016/j.tiv.2014.05.006
28. Cheng J, Zhang R, Xu Z, Ke Y, Sun R, Yang H, et al. Early glycolytic reprogramming controls microglial inflammatory activation. J Neuroinflamm. (2021) 18:129. doi: 10.1186/s12974-021-02187-y
29. Quan Y, Jiang CT, Xue B, Zhu SG, Wang X. High glucose stimulates TNFα and MCP-1 expression in rat microglia via ROS and NF-κB pathways. Acta Pharmacol Sin. (2011) 32:188–93. doi: 10.1038/aps.2010.174
30. Zhang X, Dong H, Zhang S, Lu S, Sun J, Qian Y. Enhancement of LPS-induced microglial inflammation response via TLR4 under high glucose conditions. Cell Physiol Biochem. (2015) 35:1571–81. doi: 10.1159/000373972
31. Gimeno-Bayón J, López-López A, Rodríguez MJ, Mahy N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res. (2014) 92:723–31. doi: 10.1002/jnr.23356
32. Tu D, Gao Y, Yang R, Guan T, Hong JS, Gao HM. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J Neuroinflamm. (2019) 16:255. doi: 10.1186/s12974-019-1659-1
33. Chausse B, Lewen A, Poschet G, Kann O. Selective inhibition of mitochondrial respiratory complexes controls the transition of microglia into a neurotoxic phenotype in situ. Brain Behav Immun. (2020) 88:802–14. doi: 10.1016/j.bbi.2020.05.052
34. Ting KKY, Jongstra-Bilen J, Cybulsky MI. The multi-faceted role of NADPH in regulating inflammation in activated myeloid cells. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1328484
35. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
36. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
37. Williams NC, O'Neill LAJ. A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol. (2018) 9:141. doi: 10.3389/fimmu.2018.00141
38. Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-citrate lyase. Immunity. (2019) 51:997–1011.e7. doi: 10.1016/j.immuni.2019.11.009
39. Ting KKY, Yu P, Iyayi M, Dow R, Hyduk SJ, Floro E, et al. Oxidized low-density lipoprotein accumulation in macrophages impairs lipopolysaccharide-induced activation of AKT2, ATP citrate lyase, acetyl–coenzyme A production, and inflammatory gene H3K27 acetylation. ImmunoHorizons. (2024) 8:57–73. doi: 10.4049/immunohorizons.2300101
40. Yang S, Qin C, Hu Z-W, Zhou L-Q, Yu H-H, Chen M, et al. Microglia reprogram metabolic profiles for phenotype and function changes in central nervous system. Neurobiol Dis. (2021) 152:105290. doi: 10.1016/j.nbd.2021.105290
41. Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. (1998) 95:7631–6. doi: 10.1073/pnas.95.13.7631
42. Van den Bossche J, Baardman J, Otto Natasja A, van der Velden S, Neele Annette E, van den Berg Susan M, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
43. Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (Going)? Trends Immunol. (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
44. Ting KKY, Yu P, Dow R, Floro E, Ibrahim H, Scipione CA, et al. Oxidized low-density lipoprotein accumulation suppresses glycolysis and attenuates the macrophage inflammatory response by diverting transcription from the HIF-1α to the Nrf2 pathway. J Immunol. (2023) 211:1561–77. doi: 10.4049/jimmunol.2300293
45. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab. (2012) 15:432–7. doi: 10.1016/j.cmet.2011.11.013
46. Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, et al. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. (2018) 27:351–61.e3. doi: 10.1016/j.cmet.2017.12.016
47. Lanaspa MA, Ishimoto T, Li N, Cicerchi C, Orlicky DJ, Ruzycki P, et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun. (2013) 4:2434. doi: 10.1038/ncomms3929
48. Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology. (2013) 58:1632–43. doi: 10.1002/hep.26594
49. Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci USA. (2012) 109:4320–5. doi: 10.1073/pnas.1119908109
50. Hwang JJ, Jiang L, Hamza M, Dai F, Belfort-DeAguiar R, Cline G, et al. The human brain produces fructose from glucose. JCI Insight. (2017) 2:e90508. doi: 10.1172/jci.insight.90508
51. Hwang JJ, Johnson A, Cline G, Belfort-DeAguiar R, Snegovskikh D, Khokhar B, et al. Fructose levels are markedly elevated in cerebrospinal fluid compared to plasma in pregnant women. PLoS One. (2015) 10:e0128582. doi: 10.1371/journal.pone.0128582
52. Shu HJ, Isenberg K, Cormier RJ, Benz A, Zorumski CF. Expression of fructose sensitive glucose transporter in the brains of fructose-fed rats. Neuroscience. (2006) 140:889–95. doi: 10.1016/j.neuroscience.2006.02.071
53. Ho CY, Lin YT, Chen HH, Ho WY, Sun GC, Hsiao M, et al. CX3CR1-microglia mediates neuroinflammation and blood pressure regulation in the nucleus tractus solitarii of fructose-induced hypertensive rats. J Neuroinflamm. (2020) 17:185. doi: 10.1186/s12974-020-01857-7
54. Cigliano L, Spagnuolo MS, Crescenzo R, Cancelliere R, Iannotta L, Mazzoli A, et al. Short-term fructose feeding induces inflammation and oxidative stress in the hippocampus of young and adult rats. Mol Neurobiol. (2018) 55:2869–83. doi: 10.1007/s12035-017-0518-2
55. Hyer MM, Dyer SK, Kovalchick LV, Targett I, Burns CM, Kloster A, et al. Fructose-induced cognitive impairment co-occurs with morphological changes in dendrites and microglia of male rats. Psychoneuroimmunol J. (2022) 3:1–12. doi: 10.32371/pnij/246142
56. Mizuno TM, Lew PS, Jhanji G. Regulation of the fructose transporter gene slc2a5 expression by glucose in cultured microglial cells. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms222312668
57. Xu MX, Yu R, Shao LF, Zhang YX, Ge CX, Liu XM, et al. Up-regulated fractalkine (FKN) and its receptor CX3CR1 are involved in fructose-induced neuroinflammation: Suppression by curcumin. Brain Behav Immun. (2016) 58:69–81. doi: 10.1016/j.bbi.2016.01.001
58. Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest. (2018) 128:545–55. doi: 10.1172/JCI96702
59. Herman MA, Birnbaum MJ. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab. (2021) 33:2329–54. doi: 10.1016/j.cmet.2021.09.010
60. Febbraio MA, Karin M. "Sweet death": Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. (2021) 33:2316–28. doi: 10.1016/j.cmet.2021.09.004
61. Beecher K, Alvarez Cooper I, Wang J, Walters SB, Chehrehasa F, Bartlett SE, et al. Long-term overconsumption of sugar starting at adolescence produces persistent hyperactivity and neurocognitive deficits in adulthood. Front Neurosci. (2021) 15:670430. doi: 10.3389/fnins.2021.670430
62. Lien L, Lien N, Heyerdahl S, Thoresen M, Bjertness E. Consumption of soft drinks and hyperactivity, mental distress, and conduct problems among adolescents in Oslo, Norway. Am J Public Health. (2006) 96:1815–20. doi: 10.2105/AJPH.2004.059477
Keywords: microglia, immunometabolism, macrophages, fructose metabolism, GLUT5, inflammation, glycolytic reprogramming, neurological dysfunction
Citation: Ting KKY (2024) Fructose overconsumption-induced reprogramming of microglia metabolism and function. Front. Immunol. 15:1375453. doi: 10.3389/fimmu.2024.1375453
Received: 23 January 2024; Accepted: 18 March 2024;
Published: 26 March 2024.
Edited by:
Fawaz Alzaid, Sorbonne Universités, FranceReviewed by:
Xavier Fioramonti, INRA UMR1286 Laboratoire NutriNeuro, FranceJason M. Miska, Northwestern University, United States
Copyright © 2024 Ting. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth K. Y. Ting, kenneth.ting@mail.utoronto.ca