 Maria Teresa Sabrina Bertilaccio
Maria Teresa Sabrina Bertilaccio Shih-Shih Chen
Shih-Shih Chen- 1Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Institute of Molecular Medicine, The Feinstein Institutes for Medical Research, Manhasset, NY, United States
Although the chronic lymphocytic leukemia (CLL) treatment landscape has changed dramatically, unmet clinical needs are emerging, as CLL in many patients does not respond, becomes resistant to treatment, relapses during treatment, or transforms into Richter. In the majority of cases, transformation evolves the original leukemia clone into a diffuse large B-cell lymphoma (DLBCL). Richter transformation (RT) represents a dreadful clinical challenge with limited therapeutic opportunities and scarce preclinical tools. CLL cells are well known to highly depend on survival signals provided by the tumor microenvironment (TME). These signals enhance the frequency of immunosuppressive cells with protumor function, including regulatory CD4+ T cells and tumor-associated macrophages. T cells, on the other hand, exhibit features of exhaustion and profound functional defects. Overall immune dysfunction and immunosuppression are common features of patients with CLL. The interaction between malignant cells and TME cells can occur during different phases of CLL development and transformation. A better understanding of in vivo CLL and RT biology and the availability of adequate mouse models that faithfully recapitulate the progression of CLL and RT within their microenvironments are “conditio sine qua non” to develop successful therapeutic strategies. In this review, we describe the xenograft and genetic-engineered mouse models of CLL and RT, how they helped to elucidate the pathophysiology of the disease progression and transformation, and how they have been and might be instrumental in developing innovative therapeutic approaches to finally eradicate these malignancies.
Introduction
Chronic lymphocytic leukemia (CLL) is a disease with remarkable complexity that can evolve into Richter transformation (RT), an aggressive lymphoma with a dismal prognosis (Figure 1). CLL cells are enriched not only with old/quiescent cells, but also with a small fraction of recently born/proliferating cells. Division of CLL cells mainly occurs in lymph nodes (LNs) but not in the bone marrow or blood (12), highlighting the importance of the tumor microenvironment (TME) in the pathophysiology of this malignancy. CLL is also a disease with genetic complexity; CLL patients have common mutations involved in driving disease progression (13–15) and RT (6, 7, 16, 17). These genetic abnormalities are already present in CLL patient bone marrow CD34+ hematopoietic stem cells (HSCs) (1, 18, 19) and in the pre-leukemic stage of monoclonal B-cell lymphocytosis (MBL) (20–22). In contrast, the nonmalignant immune cells within TME revealed transcriptional similarity across patients (21, 23).
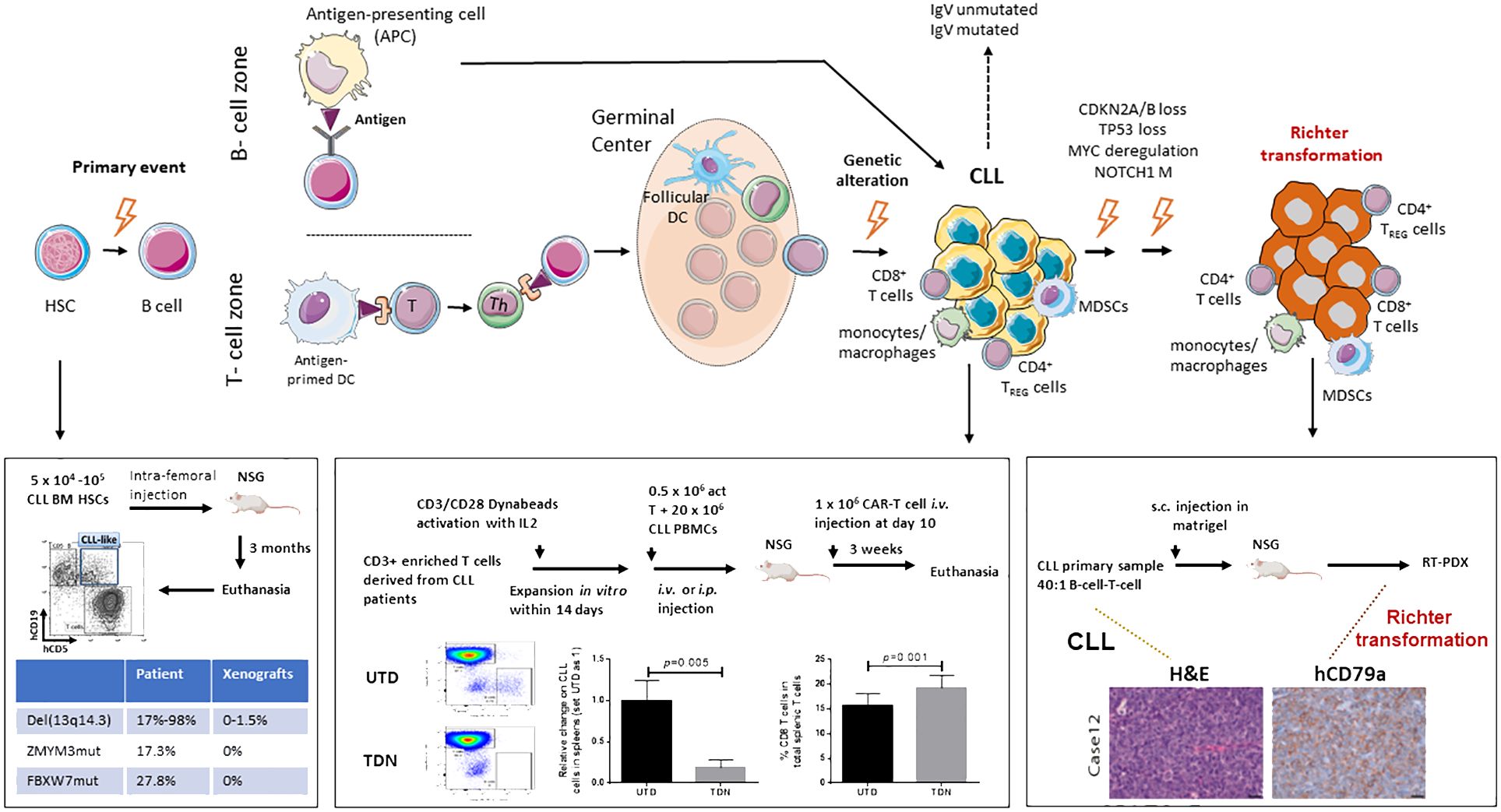
Figure 1 Schematic model of CLL initiation, progression, and Richter transformation. Initial genetic events occur during the B-cell development at the stage of hematopoietic stem cells HSC (1–3). Additional genetic abnormalities, within either a T-cell-dependent or -independent phase, then trigger the progression into CLL (4, 5). Finally, through the acquisition of independent genetic events, Richter transformation (RT) occurs (6–9). A complex network of reactive cells of the immune microenvironment plays a critical role within every phase of the biology of the disease, before and after transformation, either in the lymphoid tissues or peripheral blood (9). HSC, hematopoietic stem cell; DC, dendritic cell; MDSCs, myeloid-derived suppressor cells; TREG, CD4+regulatory T cells; Th, CD4+ helper T cells. Published datasets are from Patten et al. (10), Chiang et al. (11), and Playa-Albinyana et al. (8).
Developing mouse models faithfully mimicking CLL would facilitate the understanding of disease mechanisms, especially those driven by the crosstalk between the tumor and the TME. Preclinical mouse models that closely represent patient disease are also indispensable to improve treatments. Here, we review the recently developed genetic-engineered (GEMMs) and patient-derived xenograft (PDX) mouse models of CLL and RT.
GEMMs have contributed significantly to the field of CLL research. With the recently developed CRISPR-Cas9 technique, multiplexed-GEMMs have been established (11, 24–28). These GEMMs capturing driver mutations of CLL develop de novo tumors. Tumors arising from multiplexed-GEMM mice closely mimic the genetic heterogeneity of their human counterparts (11, 24) and are capable of spontaneously transforming into RT (26–28). Because GEMMs capture both extrinsic factors from TME and the intrinsic properties of CLL, these mice are suitable for in vivo validation of candidate cancer-driven genes and therapeutic agents targeting the crosstalk between tumor and TME. However, the current GEMMs still have drawbacks; for example, none of the GEMMs of CLL recapitulate the development of IgHV-mutated versus unmutated CLL, or are capable of modeling responses to existing treatment history in CLL patients.
The usefulness of PDXs in studying CLL and RT depends on the level of relatedness of the disease characteristics between these models and patients. There are several features of xenografts to consider for the successful translation into clinics. First, they must faithfully recapitulate the spectrum and the heterogeneity of lymphoproliferation observed in patients. Second, xenografts should have the genetic, phenotypic, and clinical features of the human disease. Third, all relevant CLL and RT events occur in permissive tissue microenvironments, and xenograft systems must fully mimic the co-evolution of malignant clones with nonmalignant cell types. This is especially a problem with PDXs that require serial adoptive transfers after the first inoculation. PDX capture clonal selection and evolution in an immunodeficient murine microenvironment that do not reflect human counterparts. Thus, the reliability of such models has been hampered in the past by the availability of proper humanized recipient mice able to fully reconstitute the human tumor immune microenvironment.
Mouse models of CLL
Xenograft models of CLL patient-derived bone marrow CD34+ hematopoietic stem cells
Accumulated evidence suggests that CLL is a stepwise disease, preceded by a pre-leukemic state. Driver mutations such as SF3B1 commonly seen in CLL tumors are also present in CLL patient bone marrow CD34+CD19- HSCs (1, 18, 19, 29) and MBL patient B cells (22, 30, 31). Functionally, BM-HSCs from both early- and late-stage CLL patients display increased protein levels of HIF-1a, GATA-1, PU.1, and GATA-2, and are poorly responsive to colony-forming unit (CFU) assays (2). In 2011, Kikushige et al. (3) injected CLL patient BM-HSCs in NOD-SCID/IL-2Rγnull (NSG) and NOD-Rag1nullIL2rγnull (NRG) mice (Table 1) and found the CLL-like mono- or oligo-clonal B cells in the recipients; however, B cells implanted in mice were not clonally related to the original patient cells. Similarly, in 2022, Chiang et al. intra-femorally injected CLL-BM HSCs in busulfan pre-conditioned NSG xenografts (11) and obtained CLL-like cells with VDJ rearrangements distinct from those of the originally transplanted CLL patient cells. Notably, the renewal and survival of HSCs were dependent on GATA2 and IKZF2 (11). Altogether, current xenografts of CLL BM-HSCs support the differentiation of CLL-like B cells that are clonal unrelated to patients, and none of the mice develop a full-blown CLL disease (Figure 1).
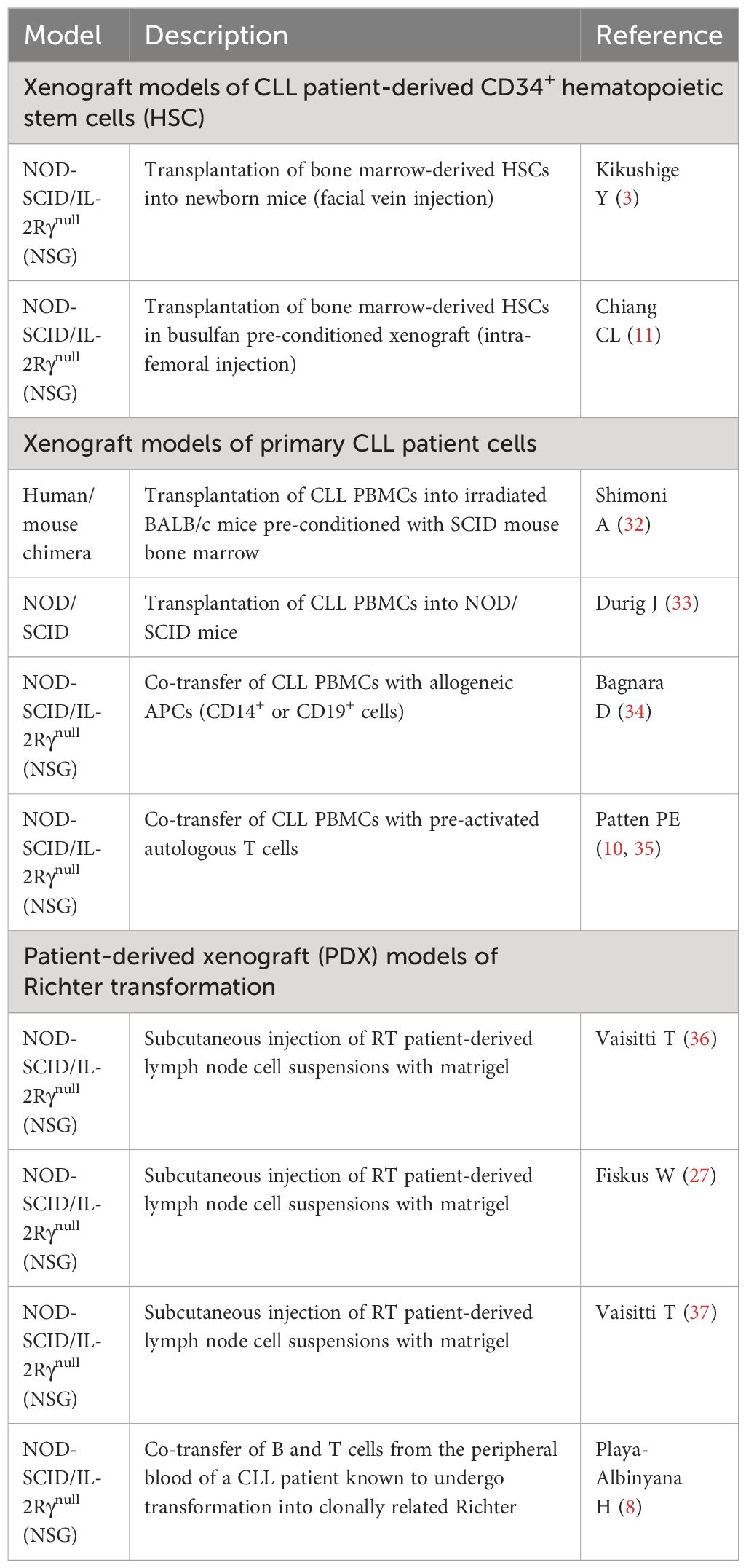
Table 1 Xenograft models of CLL and RT.
Xenograft models of primary CLL patient cells
The first xenografts using primary CLL patient cells were established by Berrebi and Reisner (32, 38) using irradiated BALB/c mice pre-conditioned with SCID mouse bone marrow (Table 1), followed by Dürig et al. using NOD/SCID mice intraperitoneally injected with primary tumors obtained from CLL patient blood mice (33, 39). Both studies evidentiated a disease-stage-dependent CLL cell engraftment; PBMCs from late-stage patients consistently engraft better in mice. Similar results were also observed when Chiorazzi’s group inoculated CLL PBMCs together with autologous T cells in nonobese diabetes/severe combined immunodeficiency/γc (null) mice (34). This model was the first to report the reproducible engraftment of CLL cells in the mice and uncover the requirement of autologous T cells for the growth of CLL B cells (34). Chiorazzi’ s group further modified the model by injecting NSG mice with pre-activated autologous T cells and CLL-PBMCs at the ratio of 1:40, and again discovered T-dependent CLL B-cell proliferation in murine spleens (10, 35). CLL B cells were present in mouse spleens, but the percentage of CLL B cells was decreasing over time. In contrast, T-cell population increased and became the major population of total human lymphocytes (10). The predominant T cells but not CLL B cells at the late stage occurred even in mice injected with CLL PBMCs without pre-activated T cells (10).
Similar results were also shown by Wiestners’ group when they inoculated 60 million CLL PBMCs in NSG mice; again, none of the mice died from CLL (40, 41). However, CLL B cells engrafted in murine spleens highly resembled their original donor LN counterparts for the gene expression profiles, BCR, and NF-kB signal signatures (40, 41).
The feature of T cell-dependent CLL cell growth in NSG xenografts allows one to test novel therapies in the context of CLL B cell–T cell interaction. Clinically, anti-CD19 chimeric antigen receptor (CD19.CAR) T cells reject CLL tumors by overcoming immunological tolerance; however, the efficiency is low compared to other B-cell malignancies (42, 43). CD19. CAR T cells therefore have been tested in NSG-xenografts (42–44). CD19.CAR T cells (TDN) and control untransduced T cells generated from PBMCs obtained from three treatment-naive CLL patients were injected intravenously into NSG xenografts. Compared to the untransduced T-cell cohort, CD19.CAR T cell-treated mice have significantly increased the percentage of CD8 T cells and reduced CLL B cells in the spleens, suggesting that the model is suitable for developing strategies to improve the efficacy of CARs in CLL (Figure 1).
Genetic-engineered mouse models of CLL for studies in TME
Different from xenografts, GEMMs of CLL allow the preclinical TME intervention studies. GEMMs of CLL have shown the critical functions of non-malignant cells such as T cells including Foxp3+ CD4+ T-regulatory cells (TREG), monocytes/macrophages, dendritic cells, and stromal cells, including specialized antigen-presenting cells and follicular dendritic cells (FDCs) (Figure 1). Majority of these studies are based on Eμ-TCL1 transgenic mouse model (Table 2), the mostly utilized GEMM for CLL, characterized by overexpression of human TCL1 specifically in B cells (45). TCL1 oncogene is common in CLL patients (55). These mice develop aggressive disease similar to IGHV-unmutated patients. Importantly, the malignant TCL1 CLL B cells are serial transferrable, allowing one to identify key factors within TME that impact CLL disease progression (56).
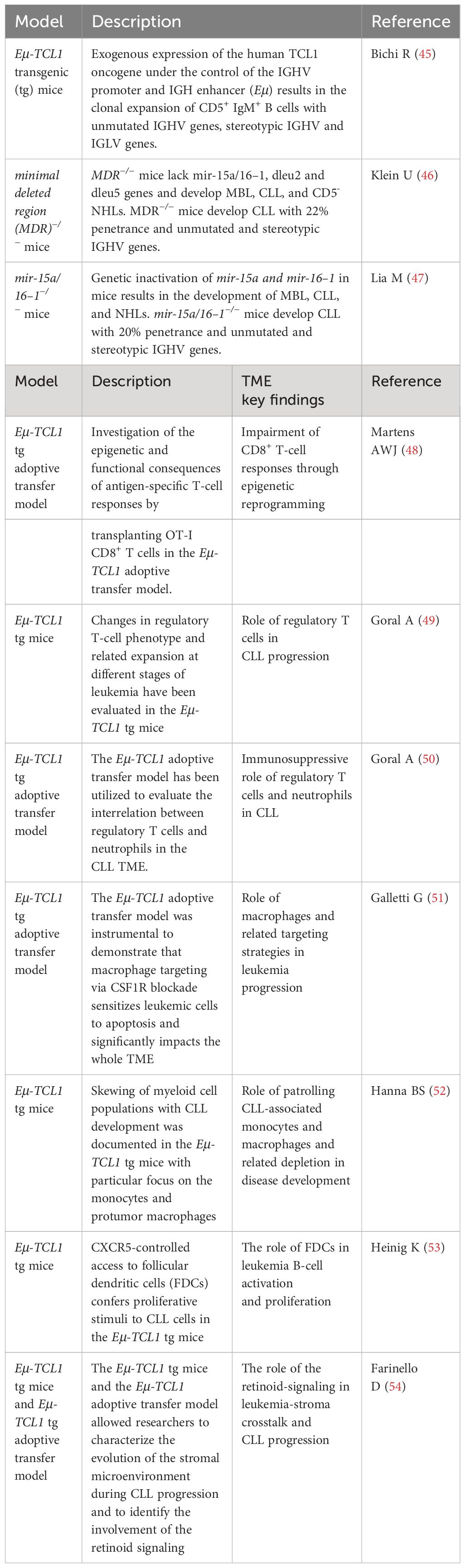
Table 2 Genetic-engineered mouse models (GEMMs) of CLL.
CLL disease is known to have dysfunctional immunity due to impaired activities of myeloid cells, neutrophils, dendritic cells, and T cells. CLL cells impact non-malignant supporting cells to alter their functions and phenotype in favor of leukemic growth. Recently, Martens et al. explored the antigen-specific response of naïve OT-I CD8+ T cells to antigen mCMV-OVA and showed that TCL1 leukemic B cells induced epigenetic modifications and skewing of short-lived effector cells in these antigen-specific T cells (48). CLL infiltration also alters the subsets of T cells. Using the TCL1 mouse system (49), Goral et al. discovered impacted neutrophils and TREG including the subset of CD44lowCD25low TREG after CLL B-cell infiltration; CLL tumors activate TREG to block CD62L and IL-4 receptor expressed on neutrophils, and further suppress neutrophil functions. Depletion of TREG cells restores the impaired neutrophils and induces changes in the CLL TME (50). In TCL1 mice, CLL cells initially accumulate in the peritoneal cavity; this also triggers the infiltration of monocytes and macrophages mainly expressing protumor signature, including CD206, CD124, and ARG-1 molecules in the peritoneum. At later stages, when CLL cells accumulate in the enlarged spleens, patrolling monocytes expressing high levels of PD-L1 were found accumulating in spleens (52). Targeting macrophages sensitizes CLL to apoptosis and delays disease progression (51).
Stromal cells clearly regulate the dynamic behavior of CLL cells, contributing to homing and trafficking in and out of the tissues, even during treatment. In 2014, Heinig et al. (53) demonstrated the key function of FDCs in the Eμ-TCL1 transgenic mouse model. Heinig et al. (53) knocked out CXCR5 in TCL1 CLL B cells and uncovered the CXCR5-regulated access of CLL cells to FDCs; CXCR5-expressing CLL cells further stimulate CXCL13 secretion and stromal cell remodeling. In 2018, Farinello et al. (54) discovered that TCL1 CLL B cells induce CXCL13 expression in the remodeled stromal microenvironment; this process is dependent on the induction of retinoid (RA) signaling in stromal cells; targeting RA signaling delays disease progression and prolongs overall survival. Consistent with these observations, the expression of RA nuclear receptors (54) and plasma levels of CXCL13 (57) correlates with bad prognosis in CLL patients.
Bone marrow niche is the site where CLL malignancy begins with primary genetic mutations followed by antigen-driven expansion (58, 59). The TME of BM is known to contribute not only to the survival of malignant cells (60), but also to the development of drug resistance (61, 62). In CLL, the BM infiltration of CLL cells causes the bone erosion and thinning of the femoral cortex in a xenograft NSG mouse model via the activation of the RANK/RANKL signaling (63). The BM environmental RANKL-RANK signaling provides the survival of CLL cells, shown by Alankus et al. in mice that express hyperactive RANKk240E transgenic gene in B lymphocytes; ex vivo, RANKL-expressed BM stromal cells also support the survival and proliferation of TCL-1 murine CLL cells and MEC-1 cells (64). Although the potential effects of anti-RANKL in counteracting chemoresistance or targeted therapy resistance has not been tested, the contact of CLL cells and stromal cells is known to lead to drug resistance (65, 66). Thus, modulation of the BM microenvironment might provide opportunities to improve treatment outcome. However, a suitable animal experimental model that can recapitulate the significance of BM TME in CLL patients is still lacking.
Besides TCL1 oncogene, deletion of 13q14 (del13q14) is the most frequent genetic lesion in CLL; 60% of CLL patients carry del13q14. The 13q14 region encodes genes highly conserved in human and mice; the minimal deleted region (MDR) includes the DLEU2 long non-coding RNA (ncRNA), and the miR-15a/miR-16–1 cluster. Klein et al. elegantly recapitulated the 13q14 deletion and CLL phenotype in MDR (46) and miR-15a/16–1-deleted mice (47). In 2023, Ten Hacken et al. created del(13q)-Cd19-Cas9 LSK cells, introduced control guide RNAs, and demonstrated CLL development already in mice carrying only del(13q)-B cells (26). In contrast, the generation of single loss-of-function (LOF) lesion using sgRNA targeting Atm, Tp53, Birc3, Chd2, Mga, or Samhd1 was not sufficient to drive CLL disease development (25).
Mouse models of Richter transformation
Xenograft models of Richter transformation
The impact of the TME in CLL progression is more evident when the disease transforms into RT with dramatic LN involvement. RT is characterized by an evolution of CLL into an aggressive lymphoma. Two percent to 10% of patients with CLL develop diffuse large B-cell lymphoma (DLBCL)-RT with a median overall survival of less than 12 months (67). The whole genome, epigenome, and transcriptome of patient-derived RT cells have been extensively investigated by several independent groups. New driver alterations and a B-cell receptor (BCR)LOW-signaling transcriptional axis in RT cells have been identified (6, 7, 68, 69). Targeted therapies have not shown good responses in RT. Though CD19.CAR-T cell therapy is an established treatment for de novo DLBCL (70–72), data on the efficacy of CD19.CAR-T cell therapy in RT are limited and need better investigation (73, 74). High levels of PD1/PD-L1 checkpoint molecules have been observed on selected immune cells and encouraging results recently came from a phase 2 trial based on the combination of nivolumab and ibrutinib with an overall response rate of 42% (75). The evidence that checkpoint inhibitors show clinical activity in patients with RT compared to patients with CLL highlights the critical difference in the TME between RT and CLL that should be better investigated to improve outcomes in patients. Distinct immune signatures have been described in CLL and RT (76, 77). Patients with RT show a more diverse T-cell repertoire, lower T-cell TCR clonality, and increased infiltration of TREG cells compared to patients with CLL (76). CD68+CD163+ protumor macrophages have been found at increased levels in LN sections of patients with RT compared to CLL (76).
The interaction between malignant cells and TME can occur during different phases of CLL progression and RT (Figure 1). How and when selected immune cells become dysfunctional and acquire a protumor phenotype during leukemia progression and whether this phenotype is exacerbated in patients undergoing RT is unexplored.
The availability of mouse models recapitulating the human RT with a fully reconstituted immune microenvironment is crucial to identify and preclinically develop therapeutic strategies for these uncurable malignancy.
To date, two PDX models of RT have been established in NSG mice (Table 1) by two independent groups (27, 36). Vaisitti et al. reported for the first time the development of two PDX models of RT documenting extensive involvement of the spleen (SP), bone marrow (BM), peripheral blood (PB), and extra-nodal organs (36). LN cell suspensions were injected subcutaneously with matrigel, and after the first engraftment, tumor cells were retransplanted in vivo for at least 10 passages to stabilize the PDX models. These models preserved the phenotypes, and the genomic and biomolecular features of the original RT in the patients. Targeted deep sequencing, whole-exome sequencing (WES), and RNA sequencing were exploited to characterize the two models that appeared to share 80% of their transcriptome with the original patient samples. Of note, one of the models maintained in vivo the BTK mutation associated to ibrutinib resistance (p.C481S) found in the original primary sample. Primary and PDX samples shared the same IGHV gene mutational status and were EBV negative, thus ruling out the possibility of EBV-driven non-malignant B-cell proliferation in vivo.
Three additional DLBCL-RT PDX models (HPRT1, HPRT2, and HPRT3) with similar pathophysiology features have been reported more recently in NGS mice (27). Immunoglobulin gene analysis performed on the PDX and the original samples allowed one to identify clonally related or unrelated models; the HPRT3 model was documented as clonally related to the original CLL/RT patient-derived sample, while HPRT2 was defined as clonally unrelated. Additionally, based on a detailed phenotypic characterization, two out of three PDX lines were identified as ABC-DLBCL type due to the expression of MUM/IRF4. The HPRT1 PDX line was described as GCB-DLBCL type expressing high levels of CD10 and BCL6. The RT-DLBCL cells were found growing in the BM, SP, and liver with marked splenomegaly and hepatomegaly (27). These RT-PDXs were found to display active enhancers, and protein expression of IRF4, TCF4, and BCL2, together with high sensitivity to BET inhibitors. Unlike RS9737 and RS13160, HPRT PDXs have been stabilized as cell lines for in vitro cytotoxicity studies. When exploited in survival experiments, these HPRT PDX models allowed one to preclinically test and demonstrate the activity of the combination based on BET-PROTAC and venetoclax, thus uncovering the potentiality of a novel treatment for patients with RT (27).
Two additional PDX models (RS1050 and IP867/17) have been reported by Valsitti et al. (37) using the same experimental strategy (36). IP867/17 was developed from an untreated patient. These PDX models were evaluated in flow cytometry for the expression of the Receptor tyrosine kinase-like orphan receptor 1 (ROR1), a known tumor-specific target (78). The antibody–drug conjugate VLS-101 combining the ROR1 targeting moiety and monomethyl auristatin E (MMAE) has been preclinically tested in these PDX models and showed a favorable impact on the in vivo growth and survival of RS PDX models (37). These results performed in the RT PDX models supported the development of the phase 1 trial NCT03833180 in patients with RT. Of note, further preclinical studies in the RT PDX models RS1316 and IP867/17 helped demonstrate in vivo the synergistic effect of the dual phosphatidylinositol 3-kinase-d/g (PI3K-d/g) inhibitor duvelisib and the Bcl-2 inhibitor venetoclax and allowed the enrollment of patients with RT in the trial NCT03892044 combining the two agents. More recently, Deaglio’s group preclinically evaluated the targeting of the surface antigen CD37 in the all the PDX models developed by her group (79). Three amanitin-based ADC anti-CD37 agents were tested in four established RT PDX models and significantly prolonged the mice survival.
Very recently, Playa-Albinyana H et al. generated an RT-PDX model (Case 12, Figure 1) mimicking the evolution of CLL into RT by injecting B cells and T cells from the peripheral blood of a patient with CLL, known to undergo transformation into clonally related Richter 20 years later after ibrutinib treatment (8). An additional RT-PDX was developed by the same group by transplanting B cells and T cells from a patient with RT (Case 19). Of note, they characterized over time in vivo the dynamics of the subclonal architecture and identified in the xenotransplanted mice the engraftment of a small subclone originally present in the patient RT19 that acquired later in the mice relevant alterations including BCL2 and MYC (8). As in the previous PDX models, they confirmed in the mice the RT transcriptional profile. This study confirms the concept of early seeding of RT subclones in the circulation of patients with CLL and elegantly described in vivo the evolutionary process of transformation (7).
Overall, these models (27, 36) maintain the malignant phenotype, genomic architecture, and biomolecular signature of the original tumors and have been successfully exploited in vivo and in vitro to preclinically test the activity of new agents. However, they do not recapitulate LN dissemination, which is a typical feature of RT in patients in the context of a fully immune reconstituted patient-derived microenvironment.
Genetically engineered mouse models of Richter transformation
In 1992, the first mouse model of RT was documented by E.S. Raveche and her group within the context of the NZB mouse strain (Table 3). Multiple passages through successive F1 recipients of a clonal line originating from an old NZB mouse resulted in a transformed clone localizing to the LNs and liver with the distinct features of the human RT (80, 88, 89). Unlike the original CLL-like clone, the murine secondary transformation recapitulated the pathology of RT with the disruption of the normal tissue architecture and the massive infiltration of the spleen, LNs, and liver by large cells with cleaved nuclei and evident nucleoli (88).
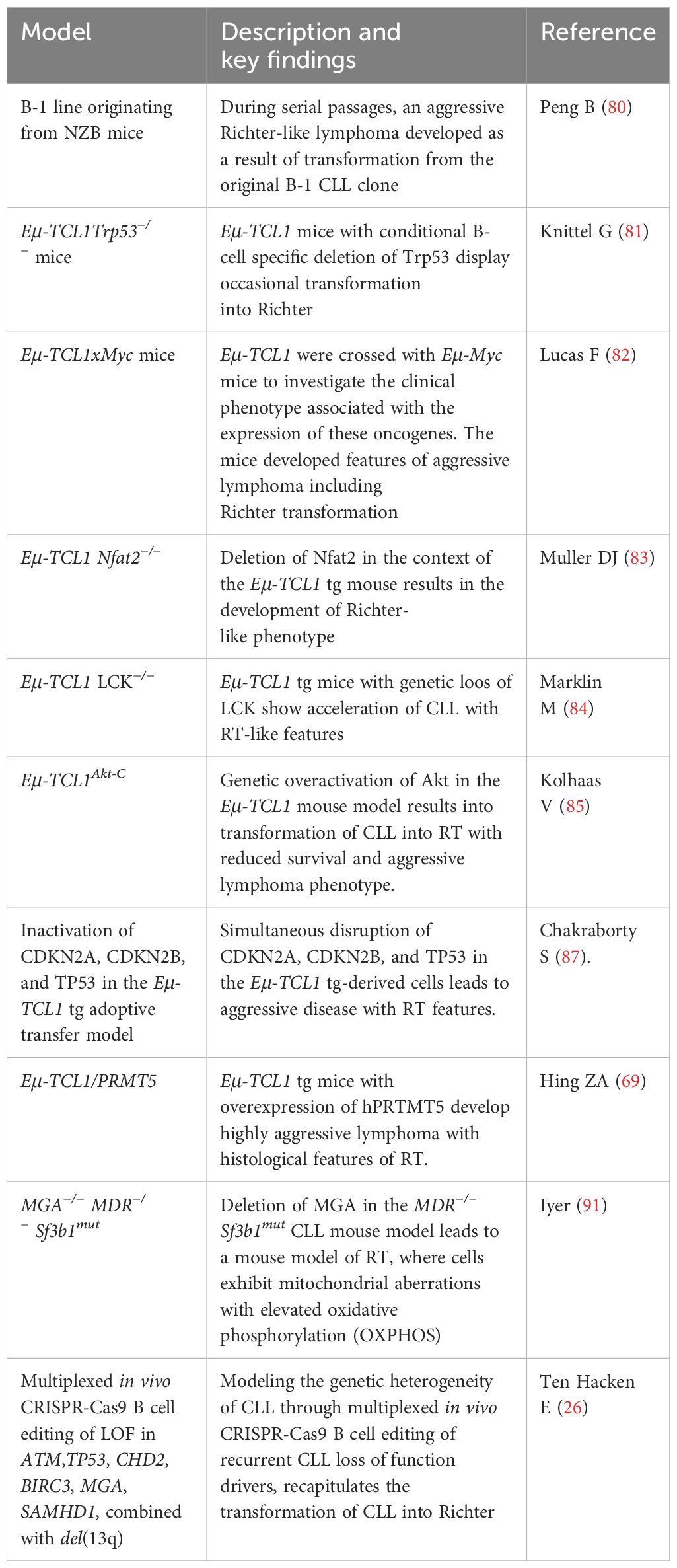
Table 3 Genetic-engineered mouse models (GEMMs) of RT.
Almost two decades later, two TCL1-driven models of high-risk CLL have been generated with the conditional B-cell-specific deletion of Tp53 that displayed occasionally the features of RT with the occurrence of large CD5- blastoid cells in the splenic infiltrates (81).
Then, a mouse model resembling RT, the double transgenic Eµ-TCL1xMyc, has been reported with features of concurrent CLL and highly aggressive lymphoma (82). This model was exploited in preclinical studies to test the BTK inhibitor ARQ531 and helped demonstrate its superior activity over ibrutinib in survival experiments (90).
More recently, the B-cell-specific deletion of either the transcription factor NFAT2 (83) or its target gene tyrosine kinase LCK (84) in the TCL1 transgenic mice was shown to induce the acceleration of CLL and the development of an RT-like phenotype.
RT has been associated with somatic mutations involving TP53, CDKN2, MYC, EGR2, and NOTCH1. Kohlhaas et al. demonstrated that high levels of AKT phosphorylation occur in patients with high-risk CLL and RT with TP53 and NOTCH1 mutations (85). The genetic inactivation of Akt in the TCL1 transgenic mice led to the development of a typical RT phenotype, with mice carrying splenomegaly, emerging large blastoid cells with pleomorphic nuclei, and high levels of lactate dehydrogenase (85). Of note, Kohlhaas et al. showed that Akt-mediated control promotes cell–cell interaction, the induction of CD4+ T cells, and the overexpression of DII1, which induces NOTCH1 activation and facilitates RT transformation. Overall, this model helped to demonstrate that the potential inhibition of PI3K/AKT and NOTCH1 might be a strategy to explore patients with high-risk CLL and RT. This model validates several evidence observed in RT PDX models and an ongoing multicenter trial with the PI3Kδ,γ inhibitor duvelisib (86).
Additionally, an interesting model has been reported by the group of D. Efremov (87). Unlike the above-described models, they mimicked in mice for the first time multiple genetic lesions associated to RT, thus better recapitulating the genetic evolution of the disease. By using the (CRISPR)/Cas9 technology, they demonstrated that the simultaneous inactivation of CDKN2A, CDKN2B, and TP53 in primary TCL1 transgenic-derived murine CLL cells induces proliferation in vitro and accelerates tumor growth in the TCL1 tg transplantation system. The administration of BCR and CDK4/6 inhibitors ibrutinib and palbociclib has a favorable impact on the survival of mice transplanted with the CLL murine cells carrying the CDKN2A, CDKN2B, and TP53 lesions (87). These data gave relevant indications on the treatment of a subset of RT patients with TP53 and CDKN2A/2B abnormalities, suggesting the combination of BCR inhibitor with CDK4/6 inhibitors such as palbociclib. Overall, this evidence highlights the importance of simultaneously mimicking in vivo the genetic lesions observed in distinct subsets of patients with RT to investigate the activity of new combination agents.
PRMT5 is known to regulate oncogenes such as NOTHC1, c-MYC, and P53 that are often dysregulated in patients with RT. Recently, Hing et al. demonstrated that PRMT5 is expressed in patients with RT transformation leading to the hypothesis that it might be involved in the transformation (68). Indeed, they generated PRMT5/TCL1 double transgenic mice developing an aggressive lymphoma with the clinical features of RT, including lymphadenopathy and palpable splenomegaly (68).
Together with TP53, CDKN2A/B deletions, and NOTCH activations, additional genetic lesions have been identified in patients with RT, including the loss-of-function mutations and deletions in Max gene associated (MGA), a MYC transcriptional repressor (7). Iyer et al. established a new model of RT by knocking out Mga in an Sf3b1/Mdr model of CLL (91). In detail, they crossed the murine CLL line CD19cre/+Mdrfl/+Sf3b1 K700Efl/+ with a mouse strain that conditionally expresses Cas9 to obtain a donor mouse line Cd19-Crefl/+Sf3b1fl/+ Mdrfl/+Cas9fl/+. Murine hematopoietic stem cells, Lin-cKit+Sca1+ cells (CD45.2+), were then isolated from these mice and transduced in vitro with lentivirus expressing single guide RNA (sgRNA) targeting Mga. Edited cells were then transplanted into CD45.1+ recipient mice. When total splenic cells were secondarily transplanted into CD45.1+ recipient mice, rapid expansion of B220+ cells with CD5 loss and lymphoid tissue infiltration was observed. Cells became larger and acquired the morphology and phenotype of a more aggressive lymphoma with high proliferation index and expression of CD21 and CD71. Based on the immunoglobulin gene analysis, the CLL-like and RT cells were clonal. Further characterization of this model led to the identification of the MGA-NME1 axis as a driver of RT through the OXPHOS upregulation and uncovered a potential new targeting opportunity for patients with RT based on the simultaneous targeting of MYC and OXPHOS pathways (91).
Very recently, a sophisticated way to model in vivo the genetic complexity of CLL transformation into RT has been reported by Ten Hacken et al. via the multiplexed introduction of well-known loss-of-function CLL driver mutations (including Atm, Trp53, Samhd1, Mga, Birc3, and Chd2) into del(13q) murine B cells (26). Essentially, Lin- cKit+ Sca1+ cells from donor mice expressing homozygous del(13q) were lentivirally transduced with sgRNA targeting six or five loss-of-functions lesions. Trp53 was present or absent in the multiplex to evaluate Trp53 involvement in the transformation. Transduced cells were then transplanted into either immunocompetent or immunodeficient NSG mice. CLL and RT lymphomas were observed either in immunocompetent or NSG mice; however, RT arose mainly in CD45.1+ recipient mice compared to NSG mice. All features of human RT histology were confirmed in murine RT. Further analyses allowed one to identify the co-occurrence of Trp53, Mga, and Chd2 lesions in RT and a tonic PI3K signaling as a characteristic feature of RT. Overall, this approach offers an interesting opportunity to model complex disease phenotypes and opens new venues of preclinical testing in uncurable malignancies (26).
Conclusion
The xenograft models of primary CLL BM-HSCs or CLL PBMCs never gave a full-blown CLL disease, suggesting that additional genetic editing might be required. Recurrent mutations such as NOTCH1, MYC, SF3B1, BRAF, TP53, XPO1, MED12, NFKBIE, and EGR2 are commonly seen in various subsets of CLL patients (14, 29, 92–95). However, because of the technique limitation to transfect primary CLL patient BM-HSCs or primary CLL patient B cells, modeling the driver mutations by the (CRISPR)/Cas9-based platform was only applied in CLL cell lines such as MEC1 cells. These works used (CRISPR)/Cas9-edited cell line-injected NSG mice for in vivo validation of candidate cancer genes of interest and demonstrated the critical roles of high-risk alterations such as del(11q), del(17p), BIRC3, ATM, and TP53 mutations alone or in combination for their biological effects (96, 97), BCR-targeted drug resistance (98), and chimeric antigen receptor (CAR)-T cell therapy responses (99). Thus, future studies on applying (CRISPR)/Cas9-based knock-out and knock-in approaches in the primary CLL patient BM-HSCs or CLL B cells are expected to accelerate the development of novel mouse models of CLL.
To fine-tune human CLL and RT in mice, approaches allowing the engraftment of the entire human immune system are required. Several next generations of humanized mouse strains such as NRG (NOD-Rag2-IL2rgTm1/Rj) and NRGS (NRG-SGM3) mice (100), MISTRG mice (expressing human M-CSF, IL-3/GM-CSF, and THPO) (101), and MISTRG-6 (MISTRG with an additional knock-in of the human IL-6 allele) (102) that express human cytokines supporting the engraftment of human HSCs, myeloid cells, and NK cells might enable the generation of CLL mouse models that give a full-blown disease and allow the dissection of the impact of the TME in vivo. Combining the (CRISPR)/Cas9 approach with the next-generation humanized mouse strains is expected to facilitate the development of mouse models of CLL and RT for mechanistic and preclinical studies.
Author contributions
MB: Writing – original draft, Writing – review & editing. S-SC: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a CLL Global Research Foundation Alliance Grant, MD Anderson Cancer Center Moon Shot Program (to MB), Swim Across America, the Feinstein Institutes AWSM Program, and The Elliott J. Netzer Career Enhancement Award.
Acknowledgments
The authors thank Dr. JM and Dr. Nicholas Chiorazzi for their support on CD19-CAR T-cell preclinical research and their critical reading of the manuscript. The figure was obtained by using Servier Medical Art: www.smart.servier.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Damm F, Mylonas E, Cosson A, Yoshida K, Della Valle V, Mouly E, et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discovery. (2014) 4(9):1088–101. doi: 10.1158/2159-8290.CD-14-0104
2. Manso BA, Zhang H, Mikkelson MG, Gwin KA, Secreto CR, Ding W, et al. Bone marrow hematopoietic dysfunction in untreated chronic lymphocytic leukemia patients. Leukemia. (2019) 33:638–52. doi: 10.1038/s41375-018-0280-0
3. Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. (2011) 20:246–59. doi: 10.1016/j.ccr.2011.06.029
4. Klein U, Dalla-Favera R. New insights into the pathogenesis of chronic lymphocytic leukemia. Semin Cancer Biol. (2010) 20:377–83. doi: 10.1016/j.semcancer.2010.10.012
5. Bosch F, Dalla-Favera R. Chronic lymphocytic leukaemia: from genetics to treatment. Nat Rev Clin Oncol. (2019) 16(11):684–701. doi: 10.1038/s41571-019-0239-8
6. Parry EM, Leshchiner I, Guieze R, Johnson C, Tausch E, Parikh SA, et al. Evolutionary history of transformation from chronic lymphocytic leukemia to Richter syndrome. Nat Med. (2023) 29:158–69. doi: 10.52843/cassyni.mg17p0
7. Nadeu F, Royo R, Massoni-Badosa R, Playa-Albinyana H, Garcia-Torre B, Duran-Ferrer M, et al. Detection of early seeding of Richter transformation in chronic lymphocytic leukemia. Nat Med. (2022) 28:1662–71. doi: 10.1038/s41591-022-01927-8
8. Playa-Albinyana H, Arenas F, Royo R, Giro A, Lopez-Oreja I, Aymerich M, et al. Chronic lymphocytic leukemia patient-derived xenografts recapitulate clonal evolution to Richter transformation. Leukemia. (2023) 38(3):557–67. doi: 10.1038/s41375-023-02095-5
9. Condoluci A, Milan L, Forestieri G, Terzi di Bergamo L, Spina V, Bruscaggin A, et al. Anatomical heterogeneity of residual disease in chronic lymphocytic leukemia treated with ibrutinib. Hematol Oncol. (2022) 40(5):1105–8. doi: 10.1002/hon.3039
10. Patten PEM, Ferrer G, Chen SS, Kolitz JE, Rai KR, Allen SL, et al. A detailed analysis of parameters supporting the engraftment and growth of chronic lymphocytic leukemia cells in immune-deficient mice. Front Immunol. (2021) 12:627020. doi: 10.3389/fimmu.2021.627020
11. Chiang CL, Hu EY, Chang L, Labanowska J, Zapolnik K, Mo X, et al. Leukemia-initiating HSCs in chronic lymphocytic leukemia reveal clonal leukemogenesis and differential drug sensitivity. Cell Rep. (2022) 40:111115. doi: 10.1016/j.celrep.2022.111115
12. Herndon TM, Chen SS, Saba NS, Valdez J, Emson C, Gatmaitan M, et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia. (2017) 31(6):1340–7. doi: 10.1038/leu.2017.11
13. Wang L, Fan J, Francis JM, Georghiou G, Hergert S, Li S, et al. Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia. Genome Res. (2017) 27(8):1300–11. doi: 10.1101/gr.217331.116
14. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. (2013) 152:714–26. doi: 10.1016/j.cell.2013.01.019
15. Sun C, Chen YC, Martinez Zurita A, Baptista MJ, Pittaluga S, Liu D, et al. The immune microenvironment shapes transcriptional and genetic heterogeneity in chronic lymphocytic leukemia. Blood Adv. (2023) 7:145–58. doi: 10.1182/bloodadvances.2021006941
16. Klintman J, Appleby N, Stamatopoulos B, Ridout K, Eyre TA, Robbe P, et al. Genomic and transcriptomic correlates of Richter transformation in chronic lymphocytic leukemia. Blood. (2021) 137:2800–16. doi: 10.1182/blood.2020005650
17. Parry EM, Ten Hacken E, Wu CJ. Richter syndrome: novel insights into the biology of transformation. Blood. (2023) 142:11–22. doi: 10.1182/blood.2022016502
18. Quijada-Álamo M, Hernández-Sánchez M, Robledo C, Hernández-Sánchez J-M, Benito R, Montaño A, et al. Next-generation sequencing and FISH studies reveal the appearance of gene mutations and chromosomal abnormalities in hematopoietic progenitors in chronic lymphocytic leukemia. J Hematol Oncol. (2017) 10:83. doi: 10.1186/s13045-017-0450-y
19. Marsilio S, Khiabanian H, Fabbri G, Vergani S, Scuoppo C, Montserrat E, et al. Somatic CLL mutations occur at multiple distinct hematopoietic maturation stages: documentation and cautionary note regarding cell fraction purity. Leukemia. (2017) 32(4):1040–3. doi: 10.1038/leu.2017.343
20. Quinten E, Sepúlveda-Yáñez JH, Koning MT, Eken JA, Pfeifer D, Nteleah V, et al. Autonomous B-cell receptor signaling and genetic aberrations in chronic lymphocytic leukemia-phenotype monoclonal B lymphocytosis in siblings of patients with chronic lymphocytic leukemia. Haematologica. (2023) 109(3):824–34. doi: 10.3324/haematol.2022.282542
21. Kretzmer H, Biran A, Purroy N, Lemvigh C, Clement K, Gruber M, et al. Preneoplastic alterations define CLL DNA methylome and persist through disease progression and therapy. Blood Cancer Discov. (2021) 2:54–69. doi: 10.1158/2643-3230.BCD-19-0058
22. Klinger M, Zheng J, Elenitoba-Johnson KS, Perkins SL, Faham M, Bahler DW. Next-generation IgVH sequencing CLL-like monoclonal B-cell lymphocytosis reveals frequent oligoclonality and ongoing hypermutation. Leukemia. (2016) 30:1055–61. doi: 10.1038/leu.2015.351
23. Penter L, Gohil SH, Lareau C, Ludwig LS, Parry EM, Huang T, et al. Longitudinal single-cell dynamics of chromatin accessibility and mitochondrial mutations in chronic lymphocytic leukemia mirror disease history. Cancer Discovery. (2021) 11:3048–63. doi: 10.1158/2159-8290.CD-21-0276
24. Monteiro CJ, Heery DM, Whitchurch JB. Modern approaches to mouse genome editing using the CRISPR-cas toolbox and their applications in functional genomics and translational research. Adv Exp Med Biol. (2023) 1429:13–40. doi: 10.1007/978-3-031-33325-5_2
25. Ten Hacken E, Yin S, Redd RA, Hernández Sánchez M, Clement K, Brunsting Hoffmann G, et al. Loss-of-function lesions impact B-cell development and fitness but are insufficient to drive CLL in mouse models. Blood Adv. (2022) 7(16):4514–7. doi: 10.1182/bloodadvances.2022009135
26. Ten Hacken E, Sewastianik T, Yin S, Hoffmann GB, Gruber M, Clement K, et al. In vivo modeling of CLL transformation to richter syndrome reveals convergent evolutionary paths and therapeutic vulnerabilities. Blood Cancer Discov. (2023) 4:150–69. doi: 10.1158/2643-3230.BCD-22-0082
27. Fiskus W, Mill CP, Perera D, Birdwell C, Deng Q, Yang H, et al. BET proteolysis targeted chimera-based therapy of novel models of Richter Transformation-diffuse large B-cell lymphoma. Leukemia. (2021) 35:2621–34. doi: 10.1038/s41375-021-01181-w
28. Martines C, Chakraborty S, Vujovikj M, Gobessi S, Vaisitti T, Deaglio S, et al. Macrophage- and BCR-derived but not TLR-derived signals support the growth of CLL and Richter syndrome murine models in vivo. Blood. (2022) 140:2335–47. doi: 10.1182/blood.2022016272
29. Di Ianni M, Baldoni S, Del Papa B, Aureli P, Dorillo E, De Falco F, et al. NOTCH1 is aberrantly activated in chronic lymphocytic leukemia hematopoietic stem cells. Front Oncol. (2018) 8:105. doi: 10.3389/fonc.2018.00105
30. Agathangelidis A, Ljungström V, Scarfò L, Fazi C, Gounari M, Pandzic T, et al. Highly similar genomic landscapes in monoclonal B-cell lymphocytosis and ultra-stable chronic lymphocytic leukemia with low frequency of driver mutations. Haematologica. (2018) 103:865–73. doi: 10.3324/haematol.2017.177212
31. Barrio S, Shanafelt TD, Ojha J, Chaffee KG, Secreto C, Kortüm KM, et al. Genomic characterization of high-count MBL cases indicates that early detection of driver mutations and subclonal expansion are predictors of adverse clinical outcome. Leukemia. (2017) 31:170–6. doi: 10.1038/leu.2016.172
32. Shimoni A, Marcus H, Dekel B, Shkarchi R, Arditti F, Shvidel L, et al. Autologous T cells control B-chronic lymphocytic leukemia tumor progression in human{->}mouse radiation chimera. Cancer Res. (1999) 59:5968–74
33. Durig J, Ebeling P, Grabellus F, Sorg UR, Mollmann M, Schutt P, et al. A novel nonobese diabetic/severe combined immunodeficient xenograft model for chronic lymphocytic leukemia reflects important clinical characteristics of the disease. Cancer Res. (2007) 67:8653–61. doi: 10.1158/0008-5472.CAN-07-1198
34. Bagnara D, Kaufman MS, Calissano C, Marsilio S, Patten PE, Simone R, et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood. (2011) 117:5463–72. doi: 10.1182/blood-2010-12-324210
35. Patten PE, Ferrer G, Chen SS, Simone R, Marsilio S, Yan XJ, et al. Chronic lymphocytic leukemia cells diversify and differentiate in vivo via a nonclassical Th1-dependent, Bcl-6-deficient process. JCI Insight. (2016) 1(4):e86288. doi: 10.1172/jci.insight.86288
36. Vaisitti T, Braggio E, Allan JN, Arruga F, Serra S, Zamo A, et al. Novel richter syndrome xenograft models to study genetic architecture, biology, and therapy responses. Cancer Res. (2018) 78:3413–20. doi: 10.1158/0008-5472.CAN-17-4004
37. Vaisitti T, Arruga F, Vitale N, Lee TT, Ko M, Chadburn A, et al. ROR1 targeting with the antibody-drug conjugate VLS-101 is effective in Richter syndrome patient-derived xenograft mouse models. Blood. (2021) 137:3365–77. doi: 10.1182/blood.2020008404
38. Marcus H, Shimoni A, Ergas D, Canaan A, Dekel B, Ben-David D, et al. Human/mouse radiation chimera generated from PBMC of B chronic lymphocytic leukemia patients with autoimmune hemolytic anemia produce anti-human red cell antibodies. Leukemia. (1997) 11:687–93. doi: 10.1038/sj.leu.2400645
39. Aydin S, Grabellus F, Eisele L, Möllmann M, Hanoun M, Ebeling P, et al. Investigating the role of CD38 and functionally related molecular risk factors in the CLL NOD/SCID xenograft model. Eur J Haematol. (2011) 87:10–9. doi: 10.1111/ejh.2011.87.issue-1
40. Herman SE, Sun X, McAuley EM, Hsieh MM, Pittaluga S, Raffeld M, et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia. (2013) 27:2311–21. doi: 10.1038/leu.2013.131
41. Zhang D, Harris HM, Chen J, Judy J, James G, Kelly A, et al. NRX-0492 degrades wild-type and C481 mutant BTK and demonstrates in vivo activity in CLL patient-derived xenografts. Blood. (2023) 141:1584–96. doi: 10.1182/blood.2022016934
42. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
43. Todorovic Z, Todorovic D, Markovic V, Ladjevac N, Zdravkovic N, Djurdjevic P, et al. CAR T cell therapy for chronic lymphocytic leukemia: successes and shortcomings. Curr Oncol. (2022) 29:3647–57. doi: 10.3390/curroncol29050293
44. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. (2016) 127:1117–27. doi: 10.1182/blood-2015-11-679134
45. Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci USA. (2002) 99:6955–60. doi: 10.1073/pnas.102181599
46. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16–1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. (2010) 17:28–40. doi: 10.1016/j.ccr.2009.11.019
47. Lia M, Carette A, Tang H, Shen Q, Mo T, Bhagat G, et al. Functional dissection of the chromosome 13q14 tumor-suppressor locus using transgenic mouse lines. Blood. (2012) 119:2981–90. doi: 10.1182/blood-2011-09-381814
48. Martens AWJ, Kavazović I, Krapić M, Pack SM, Arens R, Jongejan A, et al. Chronic lymphocytic leukemia presence impairs antigen-specific CD8(+) T-cell responses through epigenetic reprogramming towards short-lived effectors. Leukemia. (2023) 37:606–16. doi: 10.1038/s41375-023-01817-z
49. Goral A, Firczuk M, Fidyt K, Sledz M, Simoncello F, Siudakowska K, et al. A specific CD44lo CD25lo subpopulation of regulatory T cells inhibits anti-leukemic immune response and promotes the progression in a mouse model of chronic lymphocytic leukemia. Front Immunol. (2022) 13:781364. doi: 10.3389/fimmu.2022.781364
50. Goral A, Sledz M, Manda-Handzlik A, Cieloch A, Wojciechowska A, Lachota M, et al. Regulatory T cells contribute to the immunosuppressive phenotype of neutrophils in a mouse model of chronic lymphocytic leukemia. Exp Hematol Oncol. (2023) 12:89. doi: 10.1186/s40164-023-00452-9
51. Galletti G, Scielzo C, Barbaglio F, Rodriguez TV, Riba M, Lazarevic D, et al. Targeting macrophages sensitizes chronic lymphocytic leukemia to apoptosis and inhibits disease progression. Cell Rep. (2016) 14:1748–60. doi: 10.1016/j.celrep.2016.01.042
52. Hanna BS, McClanahan F, Yazdanparast H, Zaborsky N, Kalter V, Rossner PM, et al. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia. (2016) 30:570–9. doi: 10.1038/leu.2015.305
53. Heinig K, Gatjen M, Grau M, Stache V, Anagnostopoulos I, Gerlach K, et al. Access to follicular dendritic cells is a pivotal step in murine chronic lymphocytic leukemia B-cell activation and proliferation. Cancer Discov. (2014) 4:1448–65. doi: 10.1158/2159-8290.CD-14-0096
54. Farinello D, Wozinska M, Lenti E, Genovese L, Bianchessi S, Migliori E, et al. A retinoic acid-dependent stroma-leukemia crosstalk promotes chronic lymphocytic leukemia progression. Nat Commun. (2018) 9:1787. doi: 10.1038/s41467-018-04150-7
55. Paduano F, Gaudio E, Mensah AA, Pinton S, Bertoni F, Trapasso F. T-cell leukemia/lymphoma 1 (TCL1): an oncogene regulating multiple signaling pathways. Front Oncol. (2018) 8:317. doi: 10.3389/fonc.2018.00317
56. Chen SS, Batliwalla F, Holodick NE, Yan XJ, Yancopoulos S, Croce CM, et al. Autoantigen can promote progression to a more aggressive TCL1 leukemia by selecting variants with enhanced B-cell receptor signaling. Proc Natl Acad Sci USA. (2013) 110:E1500–7. doi: 10.1073/pnas.1300616110
57. Ahmed HA, Nafady A, Ahmed EH, Hassan EEN, Soliman WGM, Elbadry MI, et al. CXC chemokine ligand 13 and galectin-9 plasma levels collaboratively provide prediction of disease activity and progression-free survival in chronic lymphocytic leukemia. Ann Hematol. (2023) 103(3):781–92. doi: 10.1007/s00277-023-05540-8
58. Gale RP, Caligaris-Cappio F, Dighiero G, Keating M, Montserrat E, Rai K. Recent progress in chronic lymphocytic leukemia. Int Workshop chronic Lymphocytic Leukemia Leukemia. (1994) 8:1610–4
59. Giannoni P, Marini C, Cutrona G, Sambuceti GM, Fais F, de Totero D. Unraveling the bone tissue microenvironment in chronic lymphocytic leukemia. Cancers (Basel). (2023) 15(2):5058. doi: 10.3390/cancers15205058
60. Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. (1998) 91:2387–96. doi: 10.1182/blood.V91.7.2387
61. de Jong MME, Chen L, Raaijmakers M, Cupedo T. Bone marrow inflammation in haematological Malignancies. Nat Rev Immunol. (2024). doi: 10.1038/s41577-024-01003-x
62. Burger JA. No cell is an island unto itself: The stromal microenvironment in chronic lymphocytic leukemia. Leuk Res. (2007) 31(7):887–8. doi: 10.1016/j.leukres.2006.12.004
63. Marini C, Bruno S, Fiz F, Campi C, Piva R, Cutrona G, et al. Functional activation of osteoclast commitment in chronic lymphocytic leukaemia: a possible role for RANK/RANKL pathway. Sci Rep. (2017) 7:14159. doi: 10.1038/s41598-017-12761-1
64. Alankus B, Ecker V, Vahl N, Braun M, Weichert W, Macher-Göppinger S, et al. Pathological RANK signaling in B cells drives autoimmunity and chronic lymphocytic leukemia. J Exp Med. (2021) 218(2):e20200517. doi: 10.1084/jem.20200517
65. Boissard F, Fournie JJ, Quillet-Mary A, Ysebaert L, Poupot M. Nurse-like cells mediate ibrutinib resistance in chronic lymphocytic leukemia patients. Blood Cancer J. (2015) 5:e355. doi: 10.1038/bcj.2015.74
66. Fagnano E, Pendharkar S, Colton M, Jones PN, Sallan MC, Klymenko T, et al. Stromal cell inhibition of anti-CD20 antibody mediated killing of B-cell Malignancies. Front Cell Dev Biol. (2023) 11:1270398. doi: 10.3389/fcell.2023.1270398
67. Condoluci A, Rossi D. Biology and treatment of richter transformation. Front Oncol. (2022) 12:829983. doi: 10.3389/fonc.2022.829983
68. Hing ZA, Walker JS, Whipp EC, Brinton L, Cannon M, Zhang P, et al. Dysregulation of PRMT5 in chronic lymphocytic leukemia promotes progression with high risk of Richter's transformation. Nat Commun. (2023) 14:97. doi: 10.1038/s41467-022-35778-1
69. Broseus J, Hergalant S, Vogt J, Tausch E, Kreuz M, Mottok A, et al. Molecular characterization of Richter syndrome identifies de novo diffuse large B-cell lymphomas with poor prognosis. Nat Commun. (2023) 14:309. doi: 10.1038/s41467-022-34642-6
70. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
71. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
72. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
73. Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li DH, et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood. (2020) 135:1650–60. doi: 10.1182/blood.2019002936
74. Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J Clin Oncol. (2017) 35:3010–20. doi: 10.1200/JCO.2017.72.8519
75. Jain N, Senapati J, Thakral B, Ferrajoli A, Thompson PA, Burger JA, et al. A phase 2 study of nivolumab combined with ibrutinib in patients with diffuse large B-cell richter transformation of CLL. Blood Adv. (2022) 7(10):1958–66. doi: 10.1182/bloodadvances.2022008790
76. Wang Y, Sinha S, Wellik LE, Secreto CR, Rech KL, Call TG, et al. Distinct immune signatures in chronic lymphocytic leukemia and Richter syndrome. Blood Cancer J. (2021) 11:86. doi: 10.1038/s41408-021-00477-5
77. Auge H, Notarantonio AB, Morizot R, Quinquenel A, Fornecker LM, Hergalant S, et al. Microenvironment remodeling and subsequent clinical implications in diffuse large B-cell histologic variant of richter syndrome. Front Immunol. (2020) 11:594841. doi: 10.3389/fimmu.2020.594841
78. Kipps TJ. ROR1: an orphan becomes apparent. Blood. (2022) 140:1583–91. doi: 10.1182/blood.2021014760
79. Vaisitti T, Vitale N, Micillo M, Brandimarte L, Iannello A, Papotti MG, et al. Anti-CD37 alpha-amanitin-conjugated antibodies as potential therapeutic weapons for Richter syndrome. Blood. (2022) 140:1565–9. doi: 10.1182/blood.2022016211
80. Peng B, Sherr DH, Mahboudi F, Hardin J, Wu YH, Sharer L, et al. A cultured Malignant B-1 line serves as a model for Richter's syndrome. J Immunol. (1994) 153:1869–80. doi: 10.4049/jimmunol.153.4.1869
81. Knittel G, Rehkamper T, Korovkina D, Liedgens P, Fritz C, Torgovnick A, et al. Two mouse models reveal an actionable PARP1 dependence in aggressive chronic lymphocytic leukemia. Nat Commun. (2017) 8:153. doi: 10.1038/s41467-017-00210-6
82. Lucas F, Rogers KA, Harrington BK, Pan A, Yu L, Breitbach J, et al. Emu-TCL1xMyc: A novel mouse model for concurrent CLL and B-cell lymphoma. Clin Cancer Res. (2019) 25:6260–73. doi: 10.1158/1078-0432.CCR-19-0273
83. Muller DJ, Wirths S, Fuchs AR, Marklin M, Heitmann JS, Sturm M, et al. Loss of NFAT2 expression results in the acceleration of clonal evolution in chronic lymphocytic leukemia. J Leukoc Biol. (2019) 105:531–8. doi: 10.1002/JLB.2AB0218-076RR
84. Marklin M, Fuchs AR, Tandler C, Heitmann JS, Salih HR, Kauer J, et al. Genetic loss of LCK kinase leads to acceleration of chronic lymphocytic leukemia. Front Immunol. (2020) 11:1995. doi: 10.3389/fimmu.2020.01995
85. Kohlhaas V, Blakemore SJ, Al-Maarri M, Nickel N, Pal M, Roth A, et al. Active Akt signaling triggers CLL toward Richter transformation via overactivation of Notch1. Blood. (2021) 137:646–60. doi: 10.1182/blood.2020005734
87. Chakraborty S, Martines C, Porro F, Fortunati I, Bonato A, Dimishkovska M, et al. B-cell receptor signaling and genetic lesions in TP53 and CDKN2A/CDKN2B cooperate in Richter transformation. Blood. (2021) 138:1053–66. doi: 10.1182/blood.2020008276
88. Phillips JA, Mehta K, Fernandez C, Raveche ES. The NZB mouse as a model for chronic lymphocytic leukemia. Cancer Res. (1992) 52:437–43.
89. Mahboudi F, Raveche ES. Analysis of immunoglobulin from hybridomas obtained by fusing spontaneously arising CD5+ B-cell clones. Ann N Y Acad Sci. (1992) 651:369–72. doi: 10.1111/j.1749-6632.1992.tb24637.x
90. Reiff SD, Mantel R, Smith LL, Greene JT, Muhowski EM, Fabian CA, et al. The BTK inhibitor ARQ 531 targets ibrutinib-resistant CLL and richter transformation. Cancer Discov. (2018) 8:1300–15. doi: 10.1158/2159-8290.CD-17-1409
91. Iyer P, Zhang B, Liu T, Jin M, Hart K, Zhang J, et al. MGA deletion leads to Richter's transformation via modulation of mitochondrial OXPHOS. bioRxiv. (2023). doi: 10.1101/2023.02.07.527502
92. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. and their evolution in progression and relapse. Nature. (2015) 526:525–30. doi: 10.1038/nature15395
93. Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. (2015) 526:519–24. doi: 10.1038/nature14666
94. Knisbacher BA, Lin Z, Hahn CK, Nadeu F, Duran-Ferrer M, Stevenson KE, et al. Molecular map of chronic lymphocytic leukemia and its impact on outcome. Nat Genet. (2022) 54:1664–74. doi: 10.1038/s41588-022-01140-w
95. Gruber M, Bozic I, Leshchiner I, Livitz D, Stevenson K, Rassenti L, et al. Growth dynamics in naturally progressing chronic lymphocytic leukaemia. Nature. (2019) 570:474–9. doi: 10.1038/s41586-019-1252-x
96. Quijada-Alamo M, Perez-Carretero C, Hernandez-Sanchez M, Rodriguez-Vicente AE, Herrero AB, Hernandez-Sanchez JM, et al. Dissecting the role of TP53 alterations in del(11q) chronic lymphocytic leukemia. Clin Transl Med. (2021) 11:e304. doi: 10.1002/ctm2.304
97. Quijada-Álamo M, Hernández-Sánchez M, Rodríguez-Vicente AE, Pérez-Carretero C, Rodríguez-Sánchez A, Martín-Izquierdo M, et al. Biological significance of monoallelic and biallelic BIRC3 loss in del(11q) chronic lymphocytic leukemia progression. Blood Cancer J. (2021) 11:127. doi: 10.1038/s41408-021-00520-5
98. Quijada-Alamo M, Hernandez-Sanchez M, Alonso-Perez V, Rodriguez-Vicente AE, Garcia-Tunon I, Martin-Izquierdo M, et al. CRISPR/Cas9-generated models uncover therapeutic vulnerabilities of del(11q) CLL cells to dual BCR and PARP inhibition. Leukemia. (2020) 34:1599–612. doi: 10.1038/s41375-020-0714-3
99. Mancikova V, Peschelova H, Kozlova V, Ledererova A, Ladungova A, Verner J, et al. Performance of anti-CD19 chimeric antigen receptor T cells in genetically defined classes of chronic lymphocytic leukemia. J Immunother Cancer. (2020) 8(1):e000471. doi: 10.1136/jitc-2019-000471
100. Barve A, Casson L, Krem M, Wunderlich M, Mulloy JC, Beverly LJ. Comparative utility of NRG and NRGS mice for the study of normal hematopoiesis, leukemogenesis, and therapeutic response. Exp Hematol. (2018) 67:18–31. doi: 10.1016/j.exphem.2018.08.004
101. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. (2014) 32:364–72. doi: 10.1038/nbt.2858
Keywords: CLL, Richter transformation, mouse model, tumor microenvironment, CRISPR
Citation: Bertilaccio MTS and Chen S-S (2024) Mouse models of chronic lymphocytic leukemia and Richter transformation: what we have learnt and what we are missing. Front. Immunol. 15:1376660. doi: 10.3389/fimmu.2024.1376660
Received: 25 January 2024; Accepted: 16 May 2024;
Published: 06 June 2024.
Edited by:
Christelle Vincent-Fabert, UMR7276 Contrôle des Réponses Immunes B et des Lymphoproliférations (CRIBL), FranceReviewed by:
Sophie Peron, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceKay L. Medina, Mayo Clinic, United States
Copyright © 2024 Bertilaccio and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shih-Shih Chen, Schen9@northwell.edu