 Qiangxing Chen1,2
Qiangxing Chen1,2 Amanda R. Muñoz1†
Amanda R. Muñoz1† Anna A. Korchagina1
Anna A. Korchagina1 Yajun Shou1,2Jensine Vallecer1Austin W. Todd1
Yajun Shou1,2Jensine Vallecer1Austin W. Todd1 Sergey A. Shein1
Sergey A. Shein1 Alexei V. Tumanov1*‡
Alexei V. Tumanov1*‡ Ekaterina Koroleva1*‡
Ekaterina Koroleva1*‡- 1Department of Microbiology, Immunology and Molecular Genetics, University of Texas Health Science Center at San Antonio, San Antonio, TX, United States
- 2Department of Gastroenterology, Second Xiangya Hospital, and Research Center of Digestive Disease, Central South University, Changsha, Hunan, China
The intricate immune mechanisms governing mucosal healing following intestinal damage induced by cytotoxic drugs remain poorly understood. The goal of this study was to investigate the role of lymphotoxin beta receptor (LTβR) signaling in chemotherapy-induced intestinal damage. LTβR deficient mice exhibited heightened body weight loss, exacerbated intestinal pathology, increased proinflammatory cytokine expression, reduced IL-22 expression, and proliferation of intestinal epithelial cells following methotrexate (MTX) treatment. Furthermore, LTβR-/-IL-22-/- mice succumbed to MTX treatment, suggesting that LTβR- and IL-22- dependent pathways jointly promote mucosal repair. Although both LTβR ligands LIGHT and LTβ were upregulated in the intestine early after MTX treatment, LIGHT-/- mice, but not LTβ-/- mice, displayed exacerbated disease. Further, we revealed the critical role of T cells in mucosal repair as T cell-deficient mice failed to upregulate intestinal LIGHT expression and exhibited increased body weight loss and intestinal pathology. Analysis of mice with conditional inactivation of LTβR revealed that LTβR signaling in intestinal epithelial cells, but not in Lgr5+ intestinal stem cells, macrophages or dendritic cells was critical for mucosal repair. Furthermore, inactivation of the non-canonical NF-kB pathway member RelB in intestinal epithelial cells promoted MTX-induced disease. Based on these results, we propose a model wherein LIGHT produced by T cells activates LTβR-RelB signaling in intestinal epithelial cells to facilitate mucosal repair following chemotherapy treatment.
Introduction
Chemotherapy-induced intestinal damage poses a pervasive challenge, affecting up to 90% of patients undergoing chemotherapeutic treatments (1–3). The severity of this issue varies based on factors such as disease type, progression, drug type, and dosing regimen. The resultant gastrointestinal injury manifests in distressing symptoms like nausea, vomiting, diarrhea, and pain (2). Patient-specific risk factors, including age, ethnicity and gender also contribute to the varying susceptibility to intestinal damage during chemotherapy (4, 5). Beyond the immediate physical toll, chemotherapy-induced intestinal damage significantly impacts the quality of life for affected individuals (6). Moreover, it can compromise the effectiveness of treatments, leading to worse clinical outcomes, and potential economic repercussions due to the increased cost of care. Strikingly, reports indicate that 7.5% of deaths in chemotherapy patients result from nonselective toxicity rather than the disease itself (7). Therefore, therapeutic approaches such as combination of drug therapies and fecal microbiota transplantation are being developed to prevent or alleviate intestinal mucositis (8–12). However, despite these efforts, therapeutic targets remain limited, highlighting the need for a deeper understanding of the immune mechanisms governing mucosal repair following chemotherapy.
Due to rapid turnover of intestinal epithelial cells (IEC), the gastrointestinal (GI) tract is particularly sensitive to antineoplastic drugs such as methotrexate (MTX) and 5-Fluorouracil (5-FU) which inhibit cell growth and division (1, 13). MTX is a structural analog of folic acid which prevents folate metabolism via competitive inhibition of dihydrofolate reductase, resulting in the suppression of de novo synthesis of purines and pyrimidines (14). 5-FU mainly suppresses the action of thymidylate synthase but can also induce direct cytotoxicity through incorporation of its products into RNA and DNA (15). Animal models of chemotherapy-induced mucositis utilizing MTX and 5-FU treatments have been developed (2, 16, 17). Although the role of proinflammatory cytokines such as TNF, IL-6, IL-1 and reactive oxygen species (ROS) in pathogenesis of chemotherapy-induced mucositis is well recognized (1, 18), the immune mechanisms controlling the mucosal repair remain poorly understood.
IL-22 is an important cytokine of the interleukin-10 (IL-10) family of cytokines, produced by several hematopoietic cells, including helper T (Th) cells and innate lymphoid cells (ILCs) (19–22). IL-22 signals through the IL-22 receptor (IL-22R) paired with the IL-10Rβ subunit (23, 24). IL-10Rβ is ubiquitously expressed while IL-22R is selectively expressed by IECs and is involved in the regulation of epithelial repair and innate immunity (22, 25, 26). Furthermore, IL-22 can act on epithelial cells to induce secretion of antimicrobial proteins Reg3β and Reg3γ, which have been proposed to suppress inflammation and promote tissue recovery (27, 28). Additionally, IL-22 was shown to act directly on mouse and human intestinal stem cells (ISCs) to induce activation of the signal transducer and activator of transcription 3 (STAT3) to drive ISCs proliferation to increase organoid formation in vitro (26, 29). Moreover, a previous study revealed that group 3 ILCs (ILC3s) safeguard ISCs through production of IL-22 after MTX-induced acute small intestinal damage (30). However, a recent study suggested that ILC3-driven IEC proliferation in response to MTX-induced epithelial injury is independent of IL-22 (31). Furthermore, several studies demonstrated that IL-22 can exacerbate disease in psoriasis (32) and in several models of intestinal inflammation (33–36). Therefore, further understanding of IL-22-dependent and IL-22-independent pathways contributing to mucosal repair following chemotherapy-induced intestinal damage is critical for developing effective therapies.
Lymphotoxin beta receptor (LTβR), a core member of the tumor necrosis factor (TNF) receptor superfamily, exhibits wide expression across non-lymphocyte populations, including epithelial cells, dendritic cells (DCs), macrophages, mast cells, and stromal cells (37–39). LTβR interacts with two ligands: heterotrimeric lymphotoxin (LTα1β2, or LT) and homotrimeric LIGHT (TNFSF14), which are primarily expressed by lymphocytes and ILCs (37, 40). LTβR signaling serves pleiotropic functions, which include the control of lymphoid organ development and maintenance, as well as the regulation of inflammation and protective immunity to infections (38, 41). LTβR signaling activates canonical as well as non-canonical NF-κB signaling pathways to mediate both pro-inflammatory and anti-inflammatory responses (39, 42). Several studies have highlighted the protective role of LTβR signaling, which promotes mucosal healing in chemically-induced and infectious colitis models (43–47). Intriguingly, previous studies revealed the critical role of LTβR signaling in controlling IL-22 production by ILC3s in response to the mucosal bacterial pathogen Citrobacter rodentium (47) as well as in the DSS colitis model (45). Considering the role of ILC3s and IL-22 in MTX-induced mucosal repair (26, 30), we hypothesized that LTβR-dependent regulation of ILC3s and IL-22 mediates protection against chemotherapy-induced intestinal damage.
The goal of this study was to investigate the role of LTβR signaling in chemotherapy-induced intestinal damage using animal models of disease. Our data suggest that LIGHT-expressing T cells interact with LTβR on intestinal epithelial cells to induce non-canonical NF-κB signaling for protection against MTX-induced intestinal damage. Moreover, we show that LTβR and IL-22 pathways jointly protect from MTX-induced injury. Additionally, LTβR signaling also protects against 5-FU induced epithelial damage. These results support a novel role of LTβR signaling in mucosal repair following chemotherapy-induced intestinal injury by controlling cooperation of T cells and intestinal epithelial cells.
Materials and methods
Mice
All animal studies were conducted in accordance with the University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee. 8–14 week old male and female mice were used for experiments. Age and sex matched littermate controls were used for all experiments. C57BL/6 (wild-type, WT) mice, RORγt-/- (48), TCRβδ-/- (49), IL-22-/- (50), RORγt-Cre (48), Villin-Cre, Jax #021504 (51), LysM-Cre (52), CD11c-Cre (53), and Lgr5-EGFP-IRES-CreERT2 mice (54) (all on C57BL/6 background) were purchased from the Jackson Laboratory (Bar Harbor) and bred at the University of Texas Health Science Center at San Antonio. LTβR floxed (45), RelB floxed (55), LTβR-/- (45), LTβ-/- (56) and LIGHT-/- (TNFSF14-/-) (57) mice were described previously. RORγt-LTβ-/- mice were generated by crossing LTβ floxed mice (58) with RORγt-Cre transgenic mice (48). Vil-LTβR-/-, CD11c-LTβR-/-, LysM-LTβR-/- and Lgr5-LTβR-/- mice were generated by crossing LTβR floxed mice with CD11c-Cre (53), LysM-Cre (52), and Lgr5-EGFP-IRES-CreERT2 mice (54), respectively. Lgr5-EGFP-IRES-CreERT2 (54) mice were intercrossed with LTβR-/- mice to generate Lgr5-reporter mice on LTβR-deficient background. To induce Cre-recombination, these mice were treated with 5 mg of tamoxifen for 4 consecutive days by oral gavage. Efficiency of ltb, ltbr, relb targeted gene deletion was validated in previous publications (43, 45, 47, 55, 59). All mice used in this research were housed under specific-pathogen-free conditions in line with National Institutes of Health guidelines.
Intestinal damage models
For MTX-induced intestinal damage, 8–14 week old mice were treated i.p. with 120 mg/kg of Methotrexate (MTX, RPI) on day 0 and 60 mg/kg on day 1. Mice were euthanized and tissues collected on day 2 or 5. For survival studies, mice were weighed daily and euthanized on day 14 or if body weight loss reached 20%. For 5-FU induced colitis, 8–12 week old mice were treated i.p. with 50 mg/kg of 5-fluorouracil (5-FU, Sigma-Aldrich) on days 0, 1, 2, 3. Mice were euthanized on day 5 and small intestine, cecum and colon were removed for analysis.
Assessment of 5-FU-induced colitis
The disease score was determined as an average of body weight loss (0 points, no weight loss; 1 point, weight loss of 1 to 5%; 2 points, weight loss of 5 to 10%; 3 points, weight loss of 10 to 20%; 4 points, weight loss >20%), signs of rectal bleeding (0 points, no blood in feces; 1point, positive hemoccult test; 2 points, dark feces; 3 points, visible blood in feces or traces of blood near anus; 4 points, gross bleeding from anus) and stool consistency (0 points, well-formed pellet; 1 point, soft pellet; 2 points, loose stool; 3 points, diarrhea; 4 points, no stool with dehydration). The scores were added to obtain a disease score ranging from 0 (healthy) to 16 (maximal activity of the disease). If the cecum was included, the cecum appearance score was determined as 0 points (normal), 1 point (slightly abnormal size), 2 points (significantly abnormal size) and 3 points (abnormal size with blood).
Histology
Small intestines, cecums and colons were dissected from mice and fixed in 10% neutral buffered formalin. Paraffin-embedded tissue sections were stained with hematoxylin and eosin (H&E) for tissue pathology evaluation. Images were taken with the Keyence BZ-X800 microscope. Small intestine pathology was scored as previously described (60). Villus, epithelium, inflammation, infiltration, crypt length and abscess, and bleeding, were evaluated on the scale from 0 to 3 and scores were summarized: villus length (0 = normal, 1 = short, 2 = extremely short), villus tops (0 = normal, 1 = damaged, 2 = severely damaged), epithelium (0 = normal, 1 = flattened, 2 = damaged, 3 = severely damaged), inflammation (0 = no infiltration, 1 = mild infiltration, 2 = severe infiltration), crypts (0 = normal, 1 = mild crypt loss, 2 = severe crypt loss), crypt abscesses (0 = none, 1 = present) and bleeding (0 = none, 1 = present). For cecum and colon histopathology score, we used a previously described scoring system (61).
Immunohistochemistry
5-Bromo-2′-deoxyuridine (BrdU, BD Biosciences, 100 mg/kg) was injected i.p. to mice two hours prior to analysis. Small intestines were fixed in 10% neutral buffered formalin and paraffin embedded. Sections were deparaffinized, rehydrated, and treated with 2 M HCl for 30 min at 37°C, and washed 3 times with PBS for 5 minutes, followed by 0.5% Triton X-100 for 30 minutes at room temperature. Tissue sections were blocked with goat serum at 37°C for 30 minutes and incubated with anti-BrdU antibody (Biolegend, clone 3D4) at 1:50 dilution at 4°C overnight. Sections were then incubated with HRP-conjugated goat anti-mouse IgG antibody (Biolegend) at 1:200 dilution at 37°C for 1h. Tissue sections were developed using DAB (Biolegend) and counterstained with hematoxylin. BrdU-positive cells were counted in 4 to 8 crypts per section. For Alcian Blue and Nuclear Fast Red staining slides were deparaffinized using Xylene and hydrated to distilled water. Slides were then incubated in 3% acetic acid for 3 min, stained in Alcian Blue solution pH 2.5 (American MasterTech) for 45 min, washed in running tap water, counter stained in nuclear fast red solution (American MasterTech) for 5 min, washed in running tap water, dehydrated to 100% ethanol, cleared in xylene, and mounted with Cytoseal 60 (Thermo Scientific) mounting medium. Images were taken with the Keyence BZ-X800 microscope.
RNA isolation and real-time reverse transcription PCR analysis
RNA from tissue or cultured cells was extracted using E.Z.N.A. Total RNA Kit I (Omega Bio-tek). RNA from lamina propria and intraepithelial fraction was isolated using RNeasy Micro Kit (QIAGEN). cDNA synthesis and real-time PCR were performed as described previously (43) using Power SYBR Green master mix (Applied Biosystems). Relative mRNA expression of target genes was determined using the comparative 2-ΔΔCt method and normalized to HPRT. Primers used are listed in Supplementary Table 1.
Epithelial cell line CMT-93
CMT-93 cells (mouse rectal carcinoma cell line, ATCC) were cultured in DMEM (Corning) containing 10% FBS. Cells were treated with medium containing 5 μM MTX, or 0.5 μg/ml of agonistic αLTβR antibody (ACH6 clone, provided by Biogen Idec). Cells were incubated for 24 h before being harvested for RNA isolation.
Preparation of epithelial cells, intraepithelial lymphocytes, and lamina propria cells
To isolate epithelial cells, intestines were opened longitudinally, washed, cut, and incubated in DMEM supplemented with 5% FBS, antibiotics and 1mM DTT at 37°C with rotation (170 rpm) for 20 minutes and vortexed for 30 sec. Pieces were then incubated for additional 20 minutes with rotation (37°C) in PBS/15mM EDTA. Crypts were further digested with serum free DMEM with 2 mg/ml of Collagenase D (Roche) for 30 minutes with rotation (37°C). EC suspensions were passed through 70 µm cell strainer, resuspended in complete media and overlaid on the top of a 20%:40% Percoll (GE Healthcare) gradient. Epithelial cells were collected at the interphase of the 20%:40% Percoll gradient, washed and resuspended in DMEM. Intraepithelial lymphocytes (IELs) and lamina propria (LP) lymphocytes were isolated as described previously (62). Briefly, the small intestines were removed, opened longitudinally, and washed in cold PBS to remove fecal material. The whole small intestine or the ileum were cut in 1 cm pieces and incubated in RPMI 1640 media supplemented with 3% FBS, 15mM HEPES, 1 mM penicillin-streptomycin, and 2 mM EDTA with shaking at 150 rpm for 20 min at 37°C to remove epithelium and IEL. IELs were collected in the supernatants and passed through a mesh screen and separated by 40%:80% Percoll gradient. For LP isolation, the remaining tissues were digested in serum-free RPMI media containing 200 μg/ml Liberase TM (Roche) and 0.05% DNAse I (Sigma) on a shaker for 40 min at 37°C. The digested tissue was passed through a mesh strainer, washed with RPMI media containing 3% FBS and separated by a 40%:80% Percoll gradient.
Flow cytometry
For flow cytometry analysis, IELs and LP were preincubated for 20 min with anti-CD16/32 Fc-blocking mAb (2.4G2) and Zombie NIR™ Fixable viability dye (Biolegend) prior to surface staining. For cell surface staining single cell suspensions were incubated on ice with conjugated antibodies in PBS containing 2% of FBS. The following antibodies were used for surface staining: anti-MHCII (M5/114.15.2), anti-CCR2 (475301), anti-CD45 (30-F11), anti-CD8a (53–6.7), anti-NK1.1 (PK136), anti-CD11b (M1/70), anti-CD11c (N418), anti-TCRβ (H57–597), anti-Ly6G (1A8), anti-CD64 (X54–5/7.1), anti-Siglec-F (S17007L), anti-B220 (RA3–6B2), anti-CD4 (GK1.5), anti-CD3 (17A2), anti-CD8b (YTS156.7.7), anti-CD25 (PC61). For the transcriptional factors staining the following antibodies were used: anti-Foxp3 (MF-14) and anti-RORγt (Q31–378). For intracellular staining, cells were fixed and permeabilized with True-Nuclear™ transcriptional factor buffer set (Biolegend) according to the manufacturer’s protocol. For Lgr5-GFP reporter staining, the following antibodies were used: anti-EpCAM (G8.8), anti-TER-119 (TER-119), anti-CD117 (c-Kit) (2B8), anti-CD31 (MEC13.3). All antibodies were purchased from BD Biosciences or Biolegend. Samples were acquired using an FACSCelesta or Cytek Aurora (Cytek Biosciences), and data were analyzed using FlowJo 10 software.
Statistical analysis
All statistics were determined using GraphPad Prism software (v9). Statistical significance was determined using one-way ANOVA or two-way ANOVA with Tukey’s multiple comparison test, Mann-Whitney test, Kruskal Wallis test with Dunn’s correction, or unpaired Student’s t-test, as appropriate. Survival was assessed using the Log-rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests. Not significant, p > 0.05 (ns); p< 0.05 (*); p< 0.01 (**); p< 0.001 (***); p< 0.0001 (****).
Results
LTβR signaling protects from chemotherapy-induced intestinal damage
LTβR signaling is a known regulator of intestinal inflammation (43–45, 63, 64). To investigate the role of LTβR signaling in chemotherapy-induced intestinal damage, we employed an acute epithelial injury model induced by MTX (1, 2) (Figure 1A). Compared to WT mice, LTβR-/- mice exhibited increased weight loss (Figures 1B, M) and increased mortality (Figure 1L) after MTX treatment. Macroscopic examination of small intestines on day 5 revealed severe pathology in LTβR-/- mice compared to control mice (Figure 1C) while the length and weight of the small intestines remained unchanged (Figure 1D). Histological analysis revealed severe destruction of the epithelial layer in LTβR-/- mice characterized by shortened villi, inflammatory cell infiltration, and increased loss of crypts (Figure 1E). Consistently, histopathology scores were significantly increased in the ileum and jejunum of LTβR-/- mice, with the duodenum exhibiting less pronounced damage (Figure 1E). Crypt regenerative capacity was reduced in both WT and LTβR-/- mice at day 2 after MTX administration (Figure 1F). While epithelial cell proliferation, measured by Ki-67 expression and BrdU incorporation, remained reduced in LTβR-/- mice, it was restored in WT mice by day 5 after MTX administration (Figures 1F, G).
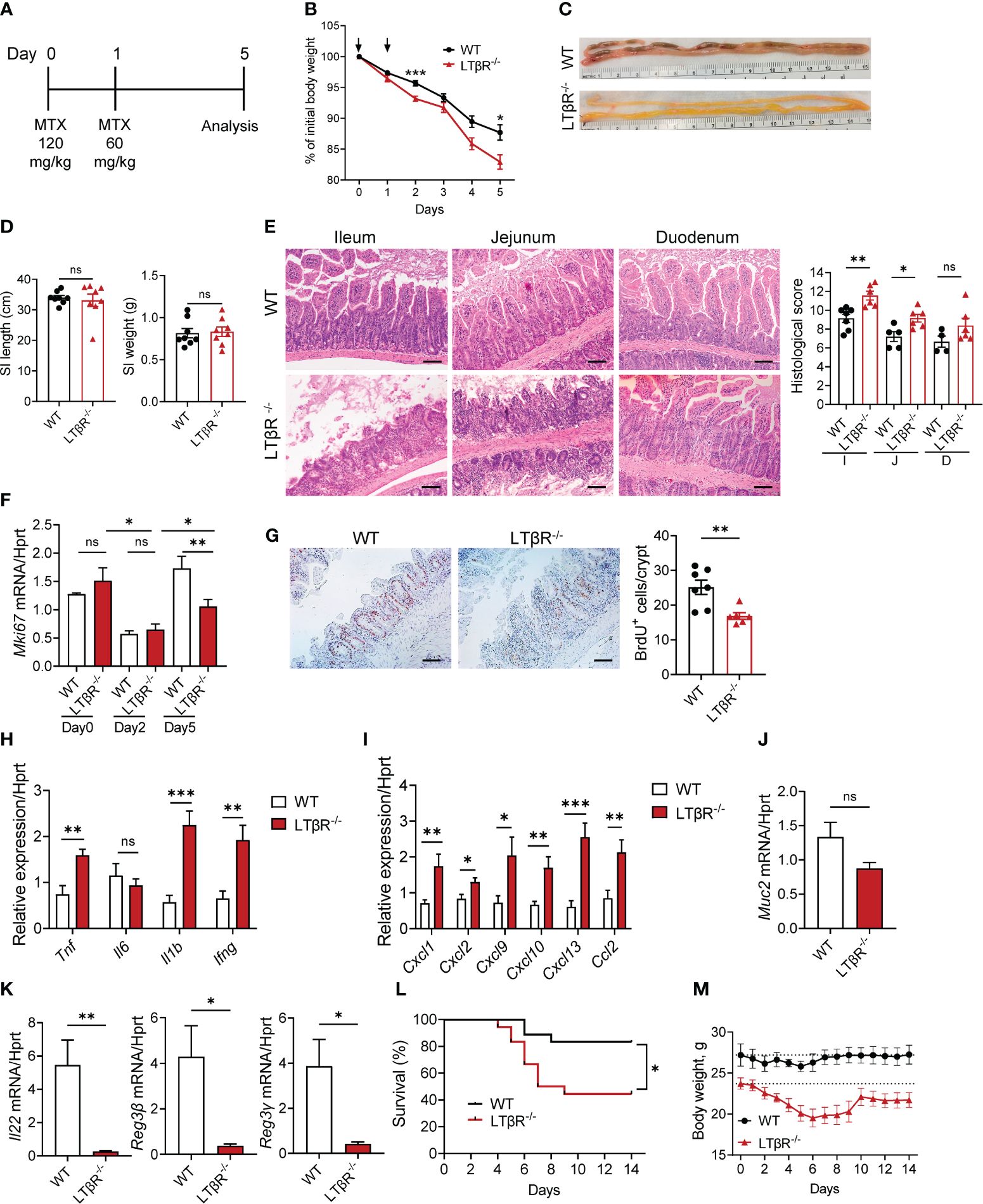
Figure 1 LTβR signaling protects against MTX-induced intestinal damage. (A) Schematic of the experiment. WT and LTβR-/- mice were injected i.p. with MTX on day 0 (120 mg/kg) and day 1 (60 mg/kg), and small intestine (SI) collected at day 5. (B) Body weight change. Black arrows: days of MTX treatment. n=25–28 mice per group. (C) Representative photographs of SI. (D) Measurements of SI. (E) Representative H&E images and histopathology scores. Scale bars, 100μm. I, Ileum; J, Jejunum; D, Duodenum. (F) Ki-67 mRNA expression in ileum at indicated time points. n=4–7 mice per group. (G) Representative images of BrdU+ cells/crypt in the ileum. Scale bars, 100μm. (H–K) Expression of cytokines (H), chemokines (I), Muc2 (J), and IL-22 and antimicrobial proteins (K) in the ileum and quantification of BrdU+ cells measured by real-time PCR. n=7–8 mice per group. (L, M) Survival analysis (n=18 mice per group, L) and long-term body weight analysis (n=5 mice per group, dotted lines represent median starting body weight in each group, M). (C–E) Data represents 1 out of 6 independent experiments with similar results. (B, F–L) Data is combined from 2–6 independent experiments with similar results. Data shown as mean ± SEM. Statistics were determined using two-way ANOVA with Geisser-Greenhouse correction (B, M), unpaired t test (D–K), and Log-rank (Mantel-Cox) test (L). ns, not significant; *p<0.05, **p<0.01, ***p<0.001.
Expression of key proinflammatory cytokines TNF, IL-1β and IFNγ, but not IL-6 was upregulated in the ileum of LTβR-/- mice at day 5 after MTX treatment (Figure 1H), as well as expression of chemokines CXCL1, CXCL2, CXCL9, CXCL10, CXCL13, and CCL2 (Figure 1I). Expression of IL-22 was significantly downregulated in the ileum of LTβR-/- mice compared to WT controls (Figure 1K). Expression of IL-22 dependent antimicrobial proteins Reg3β and Reg3γ in the ileum of LTβR-/- mice was also reduced compared to WT mice (Figure 1K). LTβR signaling is known to promote goblet cell differentiation and production of mucins in the gut during Listeria monocytogenes infection or DSS-induced colitis (45, 59). Interestingly, we did not detect significant reduction of Muc2 expression in the gut of LTβR-/- mice (Figure 1J), suggesting that other LTβR-dependent factors contribute to mucosal repair after MTX-induced injury. Collectively, these data indicate that LTβR signaling is essential for the intestinal repair and control of inflammation after MTX-induced injury.
5-FU is another commonly used chemotherapeutic agent employed in the therapy of various cancers, which can cause damage to intestinal epithelial cells and result in intestinal mucositis (15, 65, 66). To test the role of LTβR signaling in a 5-FU model of chemotherapy-induced intestinal injury, we treated WT and LTβR-/- mice with 50 mg/kg 5-FU daily for 4 days (Supplementary Figure S1). 5-FU treated LTβR-/- mice exhibited aggravated body weight loss (Supplementary Figure S1A), increased clinical disease score (Supplementary Figure S1B) and shortening of the colon (Supplementary Figure S1C). Histological analysis of colon and cecum sections of 5-FU treated LTβR-/- mice revealed severe mucosal damage characterized by loss of goblet cells and decreased crypt density which was accompanied by mass immune cell infiltration (Supplementary Figure S1D). Expression of proinflammatory cytokines TNF, IL-6, IL-1β and IFNγ was upregulated in the colon of 5-FU treated LTβR-/- mice compared to control mice, whereas IL-22 levels were similar (Supplementary Figure S1E). These data indicate that LTβR signaling also contributes to intestinal protection in 5-FU chemotherapy-induced intestinal inflammation.
LTβR signaling controls accumulation of B cells, neutrophils, CD8αα+ and CD4+ T cells in the small intestine early after MTX treatment
To define immune cell types in the small intestine early after MTX administration, we compared SI intraepithelial lymphocytes (IELs) and lamina propria (LP) immune cells in WT mice at steady state and at day 2 after MTX administration by flow cytometry. Gating strategy is shown on Supplementary Figure S2. We found an increased frequency of T cells (CD3+) and non-conventional CD8αα+ T cells in the IEL fraction after MTX administration (Supplementary Figure S3A). Interestingly, in the LP, frequency of Tregs was increased, although we did not find increased frequency of CD3+ T cells (Supplementary Figure S3B). Analysis of myeloid cell populations revealed increased frequency of macrophages and neutrophils (Supplementary Figure S3C) after MTX administration. Gene expression analysis revealed rapid induction of proinflammatory cytokines TNF, IL-6, IL-1β and IFNγ, as well as IL-22 at day 2 (Supplementary Figure S3D). In contrast, by day 5 after MTX administration, expression of these cytokines returned to steady state levels (Supplementary Figure S3D). Expression of IFNγ-induced chemokines CXCL9, CXCL10 (67), neutrophil-recruiting chemokines CXCL1, CXCL2 (68), and CXCL13 and CCL2 chemokines was upregulated on day 2 after MTX administration and reduced to baseline by day 5 (Supplementary Figure S3E). These data indicate that MTX rapidly induces inflammation and promotes immune cell infiltration into the small intestine.
To define the impact of LTβR on the recruitment of immune cells after MTX treatment, we next analyzed immune cells in the SI of LTβR-/- mice at day 2 and compared them to control WT mice. We found an increased frequency of T cells and CD8αα+ T cells in the SI IEL of LTβR-/- mice (Figure 2A). We did not observe a difference in total T cell frequency in the SI LP isolated from LTβR-/- mice; however, the frequency of CD4+ T cells, B cells, DCs, and neutrophils was reduced (Figures 2B, C). Correspondingly, mRNA expression of the neutrophil-recruiting chemokine CXCL2 was reduced in the ileum of LTβR-/- mice on day 2 after MTX treatment (Figures 2C, E), in contrast to the increased levels of CXCL1, CXCL2 at day 5 post MTX treatment (Figure 1I). These results suggest that LTβR signaling controls early neutrophil recruitment after MTX-induced injury but is dispensable at later stages of the disease when inflammation is more pronounced. Similarly, we did not detect increased expression of proinflammatory cytokines TNF and IL-1β in the ileum of LTβR-/- mice at day 2 (Figure 2D) in contrast to day 5 after MTX administration (Figure 1H); however, expression of IFNγ and IFNγ-dependent chemokines CXCL9 and CXCL10 was elevated (Figures 2D, E). Interestingly, LTβR-/- mice failed to upregulate IL-22 expression early after MTX administration (Figure 2D), suggesting that LTβR signaling controls IL-22 production in this model of intestinal inflammation. Collectively, these results suggest that LTβR signaling inhibits inflammation during MTX-induced injury.
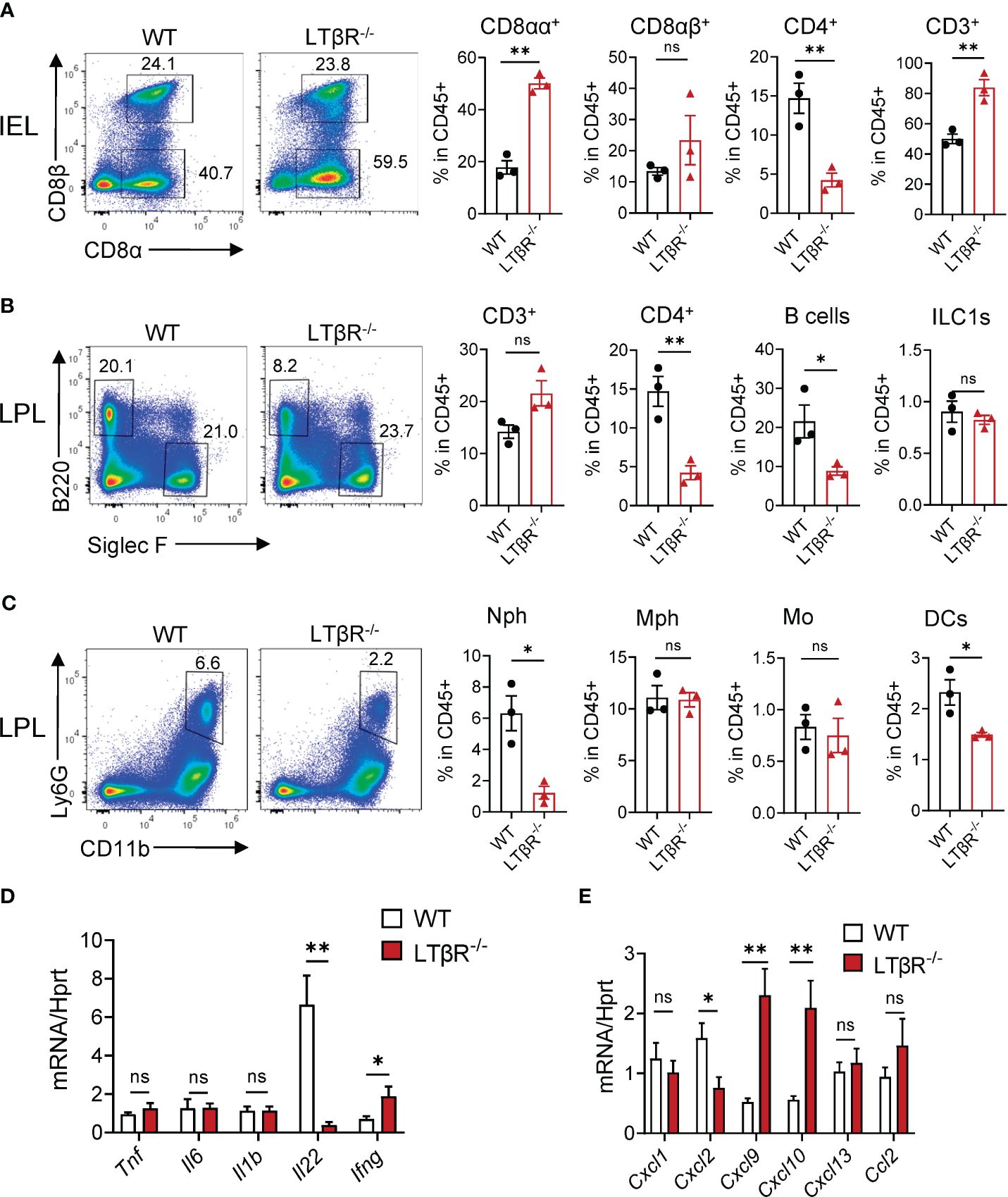
Figure 2 LTβR signaling controls accumulation of B cells, neutrophils and CD4+ T cells in the small intestine early after MTX treatment. WT and LTβR-/- mice were treated as in Figure 1A. Small intestines were collected on day 2 for analysis. (A) Representative flow cytometry plots and frequency of T cell populations in SI IEL. Frequency is calculated in live CD45+ gate. (B, C) Representative flow cytometry plots and frequency of cell populations in LP. B cells (CD45+B220+); CD4+ T cells; CD3+ T cells; ILC1s (CD45+Ly6G-B220-SiglecF-TCRβ-CD64-NK1.1+); Neutrophils (Nph, Ly6G+ CD11b+); Macrophages (Mph, CD11c-Ly6G-SiglecF-CD11b+MHCII+CD64+); Monocytes (Mo, CD45+Ly6G-B220-SiglecF-TCRβ-CD64+MHCII-CD11b+CCR2+); Dendritic cells (DCs, CD45+Ly6G-B220-SiglecF-TCRβ-CD64-MHCII+CD11c+). (D) Cytokine and (E) chemokine expression in the ileum on day 2 measured by real-time PCR. Data is representative from one of two independent experiments with similar results (n=3–6 per group). Data shown as mean ± SEM. ns, not significant, *p<0.05, **p<0.01. Statistics were determined using t test (A, B) or ANOVA with Sidak’s multiple comparison test (D, E). Gating strategy is shown in Supplementary Figure S2.
LTβR ligand LIGHT is necessary for protection from MTX-induced intestinal damage
LTβR signaling can be activated by two ligands, membrane-bound lymphotoxin (LTα1β2) and LIGHT (TNFSF14), both known regulators of intestinal inflammation (38, 40, 47, 64, 69). To test whether MTX treatment regulates expression of LTβR ligands, we analyzed expression of LIGHT and LTβ in the ileum, jejunum, and duodenum of WT mice during MTX treatment. Expression of both LIGHT and LTβ was significantly increased in the ileum on day 2 after MTX treatment and decreased at day 5 during resolution of MTX-induced injury (Figure 3A). Interestingly, expression of LIGHT was also increased in the LP and IEL fractions isolated from total small intestine on day 2 after MTX treatment, while we did not detect induction of LTβ expression (Figure 3B). Expression of LTα followed the same pattern as LTβ (Supplementary Figure S3F, G). To assess which LTβR ligand is essential for protection from MTX-induced injury, we treated WT, LTβ-/- and LIGHT-/- mice with MTX. While body weight loss in LTβ-/- mice followed the same pattern as in WT mice, LIGHT-/- mice lost significantly more body weight (Figure 3C), and all succumbed to the injury induced by MTX (Figure 3G). Consistently, histological analysis showed increased histopathology scores in the ileum of LIGHT-/- mice, but not in LTβ-/- mice, compared to WT controls (Figure 3D). Crypt regenerative capacity measured by expression of Ki-67 was markedly reduced in LIGHT-/- mice (Figure 3E). To further examine the role of LTβR ligands in MTX- induced inflammation, we next measured the expression of proinflammatory cytokines in the ileum of MTX-treated mice on day 5. Expression of TNF and IL-1β was increased in the ileum of LIGHT-/- but not LTβ-/- mice, while IFNγ levels were not changed (Figure 3F). IL-6 expression was not changed in LIGHT-/- mice but reduced in LTβ-/- mice (Figure 3F). We also found that production of IL-22 was reduced in the ileum of both LTβ-/- mice and LIGHT-/- mice (Figure 3F). Collectively, these data suggest that whereas both LIGHT and lymphotoxin are upregulated in the small intestine during MTX-induced injury and both LTβR ligands contribute to IL-22 production, LIGHT, but not LT reduces inflammation and promotes intestinal healing during MTX-induced injury.
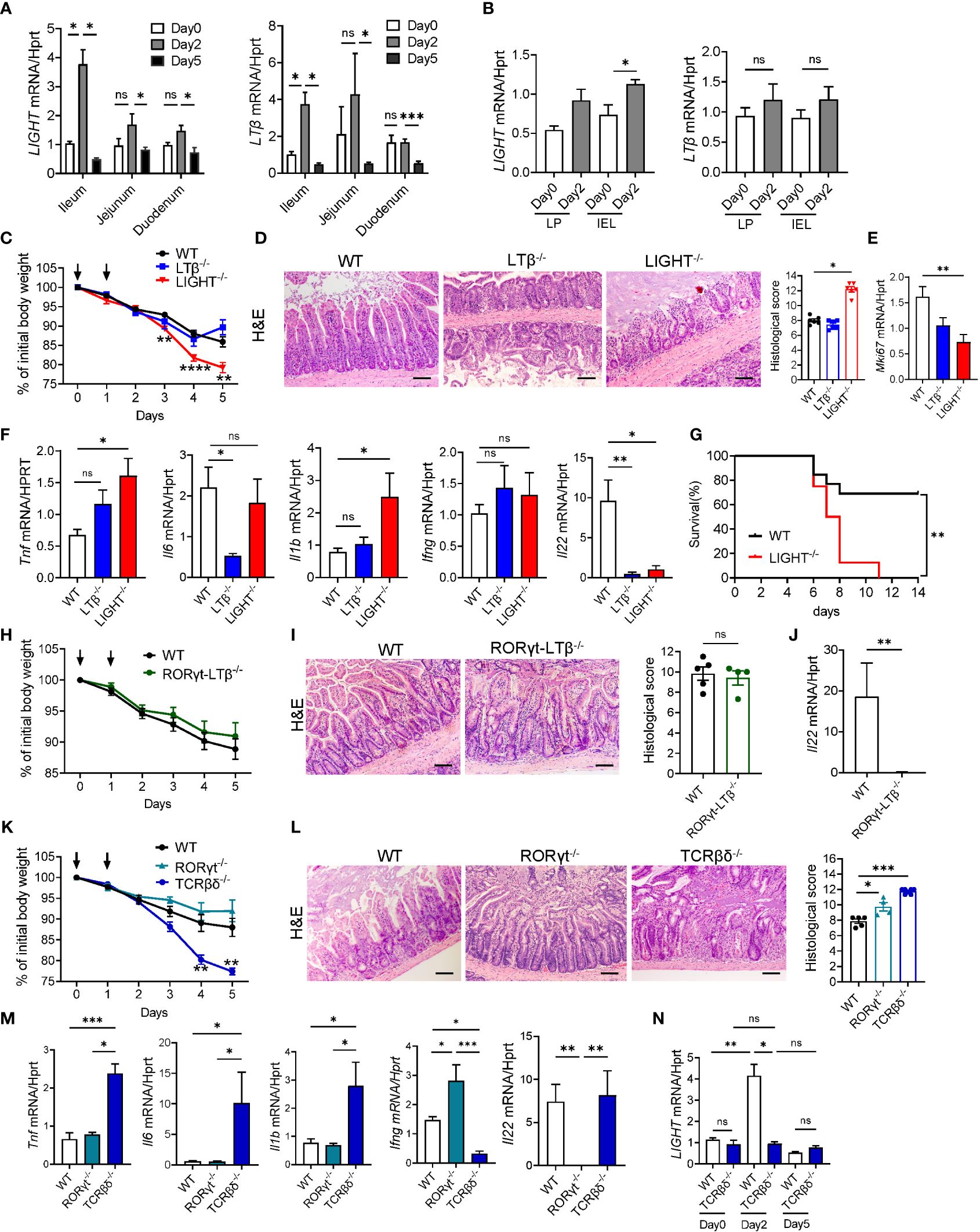
Figure 3 LIGHT and T cells protect against MTX-induced intestinal injury. (A, B) Kinetics of LIGHT and LTβ expression after MTX treatment in (A) ileum, jejunum and duodenum, and (B) LP and IEL from small intestine of WT mice. n=3–4 per group. (C–F) WT, LTβ-/- and LIGHT-/- mice were treated with MTX as in Figure 1A. (C) Body weight loss. Black arrows: days of MTX treatment. n=15–25 mice per group. (D) Representative H&E images (scale bars, 100μm) and histopathology scores; (E) Ki-67 and (F) cytokine expression in the ileum of WT, LTβ-/- and LIGHT-/- mice on day 5 after MTX treatment. n=6–8 mice per group. (G) Survival of LIGHT-/- mice after MTX treatment. n=8–13 mice per group. (H–J) WT and RORγt-LTβ-/- mice were treated with MTX as in Figure 1A. (H) Body weight loss; n=13–15 mice per group. (I) Representative H&E images (Scale bars, 100μm) and histopathology scores; and (J) IL-22 expression in the ileum on day 5 after MTX treatment. n=5 mice per group. (K–M) WT, RORγt-/- and TCRβδ-/- mice were treated with MTX as in Figure 1A. (K) Body weight loss; n=8–14 per group. (L) Representative H&E images (Scale bars, 100μm) and histopathology scores and (M) cytokine expression in the ileum on day 5 after MTX treatment. n= 7 mice per group. (N) LIGHT expression in the ileum of WT and TCRβδ-/- mice at indicated time points after MTX treatment analyzed by real-time PCR. n=4–7 mice per group. H&E images and histopathology scores are representative from 3–4 independent experiments with similar results. Data shown as mean ± SEM. Statistics were determined using two-way ANOVA with Geisser-Greenhouse correction (A, C, K), Mann-Whitney test (B, J), Kruskal-Wallis test (D–F, L, M) or Brown-Forsythe and Welch ANOVA tests (N). ns, not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Previous studies revealed that LT produced by RORγt+ ILC in the intestine is critical for control of IL-22 production and protection of mice against Citrobacter rodentium infection (43, 47). Moreover, depletion of ILCs in Rag1-/- mice resulted in reduced LTβ and IL-22 production in the ileum and diminished crypt proliferation during MTX treatment (30). To test whether LT produced by RORγt+ cells is essential for protection against MTX-induced damage, we utilized mice with specific inactivation of LTβ in RORγt-expressing cells (RORγt-LTβ-/- mice) (47). Surprisingly, we did not find difference in body weight loss or histopathology score between RORγt-LTβ-/- mice and littermate control LTβ floxed mice (Figures 3H, I) despite reduced expression of IL-22 in the ileum of RORγt-LTβ-/- mice (Figure 3J). Thus, these results suggest that although LTβ produced by RORγt+ cells is required for IL-22 production in the gut, it is dispensable for control of intestinal damage during MTX-induced disease.
T cell deficiency aggravates intestinal damage after MTX treatment
Recent studies implicated the role of RORγt+ ILCs in the maintenance of ISCs and intestinal repair following MTX-induced intestinal damage (30, 31). Our data demonstrated that CD3+ T cells are increased in the IEL after MTX treatment (Supplementary Figure S3A). To define the relative contribution of T cells and ILC3s in MTX-induced pathology we treated mice which lack ILC3s (RORγt-/- mice) or T cells (TCRβδ-/- mice) with MTX. RORγt-/- mice displayed 5–10% of body weight loss similar to WT control mice, however histological analysis of the ileum demonstrated increased crypt loss and crypt flattening (Figures 3K, L). Unexpectedly, T cell-deficient mice lost more than 20% of body weight and had to be euthanized by day 5 of MTX treatment (Figure 3K). Histological analysis showed severe loss of crypts, increased inflammation, and bleeding (Figure 3L). Expression of proinflammatory cytokines TNF, IL-6 and IL-1β was increased in the ileum of TCRβδ-/- mice but not RORγt-/- mice (Figure 3M). These results suggest that T cells, but not ILC3s are critical for protection against MTX induced injury. Interestingly, IFNγ expression was very low in the ileum of TCRβδ-/- mice, indicating that T cells are the main producers of IFNγ in the ileum after MTX treatment. We did not find a defect in IL-22 expression in the ileum of TCRβδ-/- mice, however IL-22 transcript was almost undetectable in the ileum of RORγt-/- mice (Figure 3M), implying that RORγt+ ILCs but not T cells are the main source of IL-22 production after MTX-induced injury.
Since LIGHT is mainly produced by activated T cells (37, 40) and LIGHT-/- mice displayed increased intestinal pathology post MTX treatment (Figures 3C–G), we next analyzed kinetics of LIGHT expression in the ileum of TCRβδ-/- mice during MTX treatment. While we did not find difference in LIGHT levels between WT and TCRβδ-/- mice at steady-state, T cell-deficient mice failed to upregulate LIGHT in the ileum at day 2 post MTX treatment (Figure 3N). Thus, these data suggest that T cells are critical for protection from chemotherapy-induced intestinal injury and can serve as the primary source of LIGHT early after MTX-induced damage.
LTβR and IL-22 jointly protect from MTX-induced intestinal damage
IL-22 blockade during MTX-induced intestinal damage led to a significant loss of Lgr5+ stem cells, specifically in the duodenum (30), although crypt proliferation and crypt pathology in the small intestine of IL-22-/- mice after MTX treatment was indistinguishable from WT controls (31). We found that IL-22 expression is induced in the ileum on day 2 after MTX treatment (Supplementary Figure S3D) and that IL-22 is downregulated in LTβR-/- mice (Figures 1K, 2D), suggesting that LTβR signaling regulates production of IL-22 during MTX-induced injury. To determine whether LTβR plays a protective role independently of IL-22, we intercrossed LTβR-/- mice with IL-22-/- mice and compared intestinal pathology in IL-22-/- and LTβR-/- mice with double deficient LTβR-/-IL-22-/- mice after MTX administration. We did not find difference in body weight loss, survival, or intestinal pathology between IL-22-/- and littermate heterozygous control WT mice (Figures 4A–D), consistent with previous studies (31). However, LTβR-/-IL-22-/- mice displayed increased body weight loss, intestinal pathology and exacerbated mortality compared to IL-22-/- mice (Figures 4A–D) suggesting that loss of LTβR exacerbates MTX-induced intestinal pathology in IL-22-/- mice. Interestingly, body weight loss and mortality were exacerbated in LTβR-/-IL-22-/- double deficient mice compared to LTβR-/- mice (Figures 4B, C), suggesting that complete loss of IL-22 exacerbates MTX-induced pathology in LTβR-/- mice. Consistently, LTβR-/-IL-22-/- mice displayed increased levels of proinflammatory cytokines TNF, IL-1β, and IFNγ in the ileum compared to IL-22-/- and WT control mice (Figure 4E). These results imply that LTβR and IL-22 jointly protect from MTX-induced intestinal damage and that LTβR may control both IL-22 dependent and IL-22 independent pathways for mucosal protection.
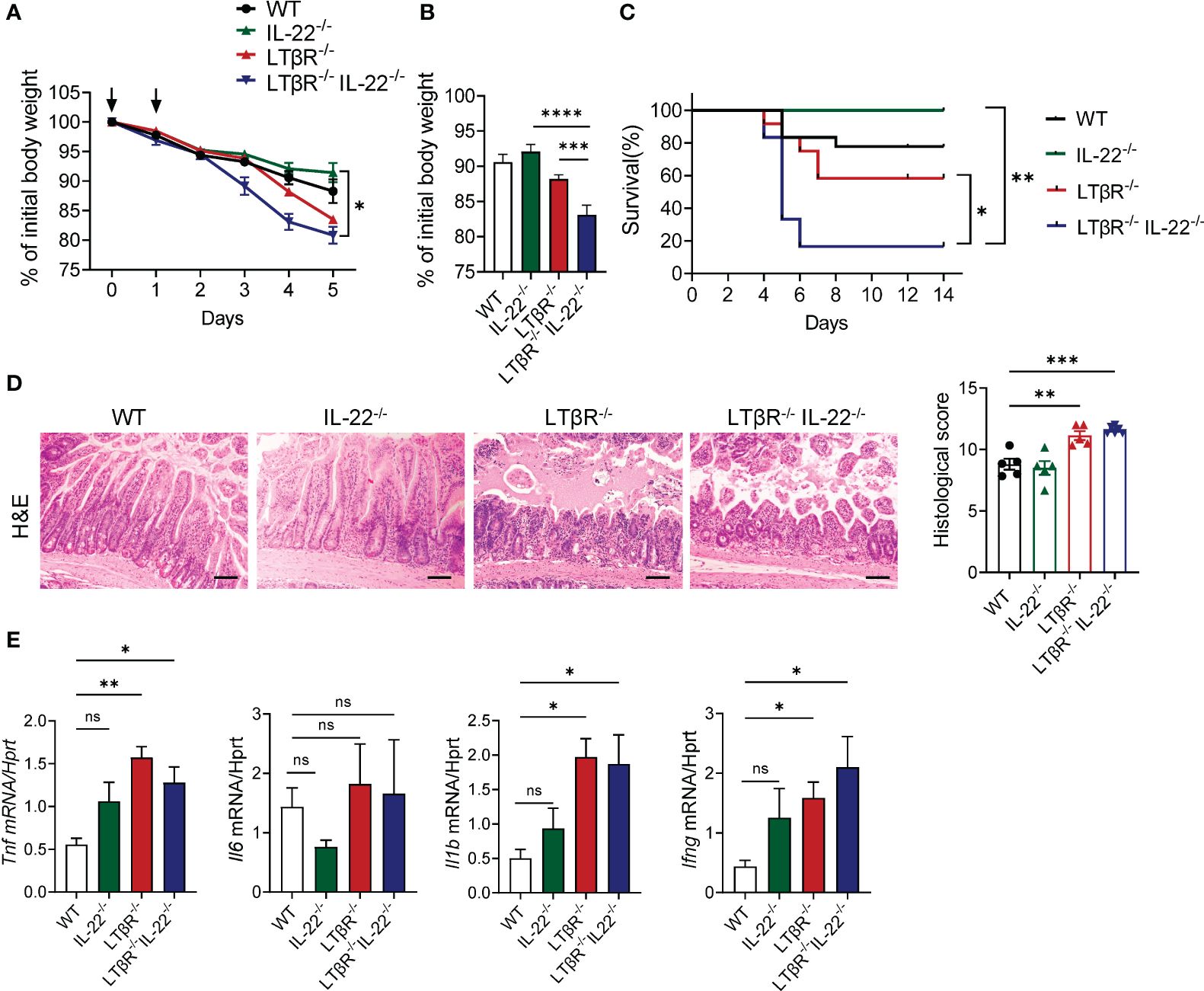
Figure 4 LTβR signaling cooperates with IL-22 for mucosal protection. LTβR-/- mice were intercrossed with IL-22-/- mice. Mice were treated with MTX as in Figure 1A. (A) Kinetics of body weight loss. n=8–18 mice per group. (B) Body weight loss at day 4 after MTX treatment. (C) Survival, n=6–18 mice per group. (D) Representative H&E images (scale bars, 100μm) and histopathology scores. (E) Expression of proinflammatory cytokines in the ileum on day 5. n=6–8 mice per group. Data was combined from 4–7 experiments with similar results. Statistics were determined using two-way ANOVA with Geisser-Greenhouse correction (A), unpaired t test (B), log-rank (Mantel-Cox) test (C), ordinary one-way ANOVA (D), Kruskal-Wallis test (E). ns, not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
LTβR signaling in epithelial cells is essential for mucosal repair following MTX-induced damage
Next, we sought to determine which LTβR-expressing cells are important for protection against MTX-induced epithelial injury. Since previous studies highlighted the role of LTβR signaling in intestinal epithelial cells for protection against epithelial injury caused by bacterial infection or by chemical agent (43, 45, 59), we tested whether LTβR signaling in epithelial cells is essential for mucosal repair during MTX-induced damage. Therefore, we generated mice with specific inactivation of LTβR in intestinal epithelial cells (Vil-LTβR-/- mice) by crossing LTβR floxed mice (45) with Villin-Cre (51). Vil-LTβR-/- mice demonstrated an accelerated body weight loss and increased mortality after MTX treatment, compared to littermate Cre-negative control mice (Figures 5A, F). Histological analysis and analysis of Ki-67 expression revealed increased tissue damage and reduced epithelial cell proliferation in the ileum of Vil-LTβR-/- mice compared to control mice (Figures 5B, C). Expression of proinflammatory cytokines TNF, IL-6, IL-1β and IFNγ was increased in the ileum of Vil-LTβR-/- mice on Day 5 (Figure 5D). Additionally, we found increased expression of CXCL1, CXCL2, CXCL9, CXCL10, CXCL13 and CCL2 chemokines in the ileum of Vil-LTβR-/- mice (Figure 5E). These results demonstrate that LTβR signaling in intestinal epithelial cells is essential for protection against MTX-induced injury.
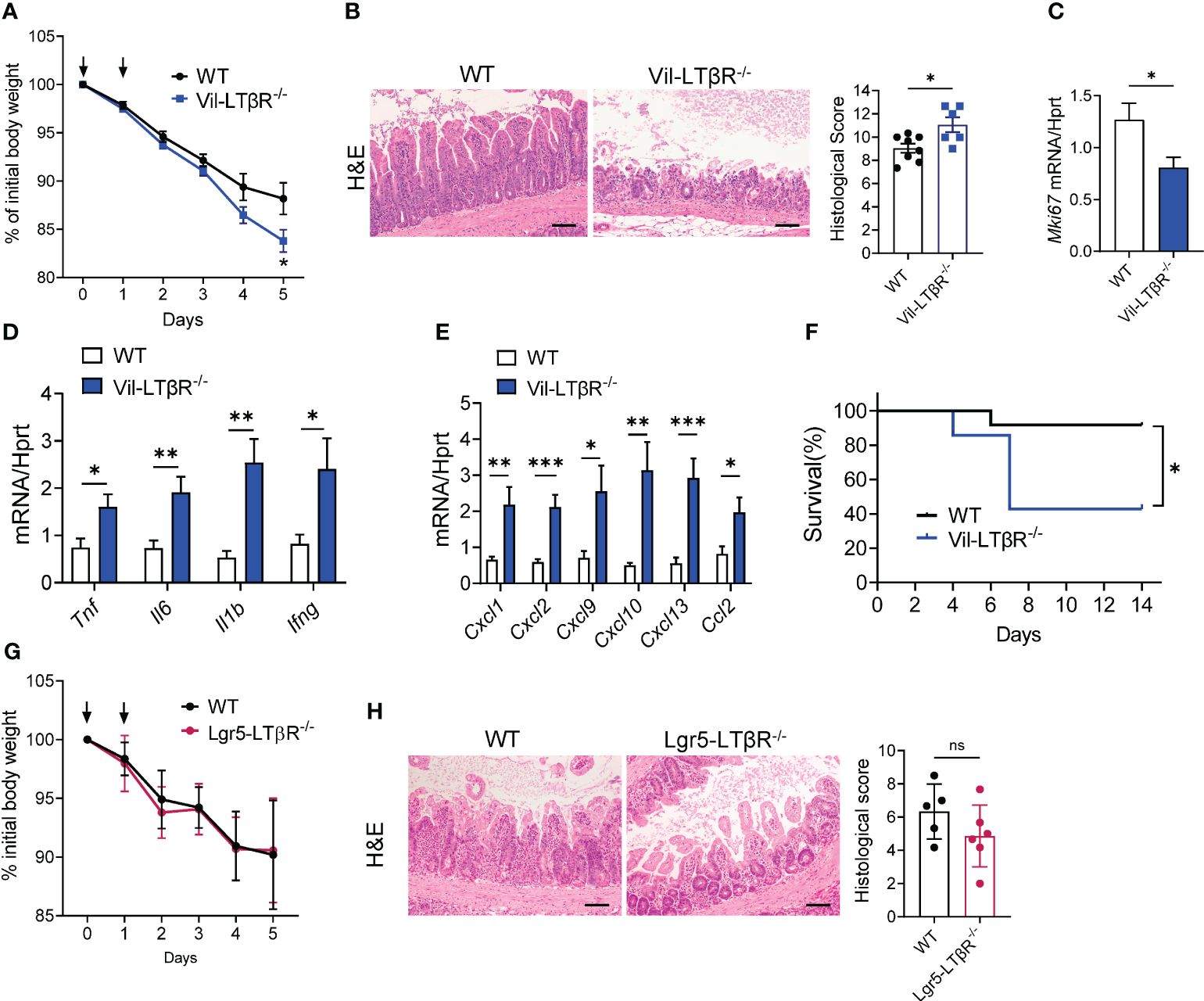
Figure 5 LTβR signaling in intestinal epithelial cells is required for protection against MTX-induced intestinal damage. (A–F) WT and Vil-LTβR-/- mice were treated with MTX, as in Figure 1A. (A) Body weight loss; n=19–22 mice per group. (B) Representative H&E images (scale bars, 100μm) and histopathology scores; Expression of (C) Ki-67, (D) cytokines and (E) chemokines in the ileum on day 5 after MTX treatment. n= 6–8 mice per group. (F) Survival; n=7–12 mice per group. (G, H) LTβR expression by Lgr5+ cells is dispensable for protection. WT and Lgr5-LTβR-/- mice were treated with MTX, as in Figure 1A, Lgr5-Cre expression was induced by tamoxifen administration. Mice were euthanized on day 5 for analysis. (G) Body weight change, n=11–14 mice per group (H) representative H&E images (scale bars, 100μm) and histopathology scores. Data combined from 2–5 independent experiments with similar results. Data shown as mean ± SEM. Statistics were determined using multiple unpaired t test (A), Mann-Whitney test (B), unpaired t test (C–E, H) or Log-rank (Mantel-Cox) test (F). ns, not significant; *p<0.05, **p<0.01, ***p<0.001.
Regeneration of intestinal epithelium after damage depends on continuous differentiation of epithelial cells from ISCs (70). Lgr5+ ISCs have the ability to give rise to all intestinal epithelial cells (71). The maintenance of ISCs after intestinal damage is dependent on IL-22 production by ILC3s (26, 30). Since LTβR signaling controls IL-22 production by ILC3s in several models of intestinal inflammation (45, 47), we sought to determine whether LTβR signaling in Lgr5+ ISCs directly contributes to epithelium regeneration after MTX-induced injury. Therefore, we generated Lgr5-LTβR-/- mice by crossing LTβR floxed mice (45) with Lgr5-EGFP-IRES-CreERT2 mice (54), and treated them with MTX. However, Lgr5-LTβR-/- mice did not show increased weight loss or aggravated intestinal pathology, compared to littermate Cre- control mice (Figures 5G, H). Moreover, analysis of publicly available single-cell RNA-sequence survey of the small intestine epithelium in naïve WT mice (72) revealed that while LTβR was highly expressed in goblet cells and enterocytes, LTβR expression was low-to moderate in Lgr5hi ISC, TA.G2 or Paneth cells (Supplementary Figure S4A). Furthermore, to test whether global LTβR deficiency affects maintenance and/or proliferation of ISCs after mucosal damage, we intercrossed LTβR-/- mice with Lgr5-EGFP-IRES-CreERT2 reporter mice and analyzed epithelial cell populations in the ileum on day 5 after MTX treatment. We did not find significant difference in the ratio of Lgr5+ ISCs, Paneth cells, tuft cells, epithelial cells, goblet cells between control and LTβR-/- mice (Supplementary Figures S4B–G). Collectively, these data suggest that LTβR signaling is dispensable for ISC maintenance and proliferation after MTX-induced injury.
Previous studies have implicated the role of LTβR signaling in CD11c+ DCs for IL-22 production and mucosal protection against intestinal bacterial infection (47). In addition, LTβR expression in neutrophils contributes to mucosal repair in DSS-induced colitis (46). To define whether expression of LTβR in DCs and macrophages/monocytes contributes to protection from MTX-induced injury, we treated mice with CD11c+ DC-specific deficiency of LTβR (CD11c-LTβR-/-mice) (45) and macrophage/neutrophil-specific LTβR deficiency (LysM-LTβR-/- mice) (43), as well as mice with combined deficiency (CD11c, LysM-LTβR-/-) with MTX, and then analyzed body weight loss and pathology on day 5 after MTX administration. We did not find difference in body weight loss or intestinal pathology in any of these strains, compared to Cre- littermate controls (Supplementary Figures S5A, B). Interestingly, IL-22 expression was decreased in the ileum of CD11c-LTβR-/- mice (Supplementary Figure S5C). This decrease suggests that while LTβR signaling in CD11c+ cells is not critical for control of intestinal injury after MTX treatment, it may contribute to the IL-22-dependent maintenance of ISCs. Collectively, our data suggest that LTβR signaling in epithelial cells, but not immune cells is essential for protection from MTX-induced intestinal damage.
Non-canonical NF-κB signaling in intestinal epithelial cells protects from MTX-induced intestinal damage
As our experiments with LTβR-/-IL-22-/- mice (Figure 4) implied that LTβR-dependent IL-22- independent signaling could contribute to protection from MTX-induced damage, we next tested whether LTβR-dependent regulation of the NF-κB pathway is important for mucosal healing. LTβR signaling can activate both canonical and non-canonical NF-κB signaling pathways to produce various proinflammatory cytokines and chemokines in response to the inflammatory stimuli (39, 42, 73). NF-κB signaling in intestinal epithelial cells can contribute to protection from intestinal inflammation in several animal models of disease (74). Recent studies demonstrated the important role of non-canonical NF-κB signaling in intestinal epithelial cells for protection from gut bacterial infections and intestinal inflammation (59, 75). We found that treatment of CMT-93 intestinal epithelial cells in vitro with MTX or with an agonistic αLTβR antibody induced expression of NF-κB2 (Figure 6A). Moreover, NF-κB2 was upregulated in the ileum after MTX treatment in vivo (Figure 6B). To test whether non-canonical NF-κB signaling in intestinal epithelial cells protects from intestinal inflammation caused by MTX treatment, we generated mice with specific inactivation of RelB in intestinal epithelial cells (Vil-RelB-/- mice) by crossing RelB floxed mice (55) with Villin-Cre mice (51). Vil-RelB-/- mice treated with MTX demonstrated aggravated weight loss and increased intestinal pathology, compared to littermate Cre- control mice (Figures 6C, D). Whereas proliferation of intestinal epithelial cells in these mice was decreased (Figure 6E), expression of proinflammatory cytokines TNF and IL-1β was elevated (Figures 6F). In contrast to Vil-RelB-/-, mice with inactivation of RelB in CD11c+ DCs (CD11c-RelB-/-) did not display an increased body weight loss post MTX treatment (Supplementary Figure S5D), consistent with results in CD11c-LTβR-/- mice (Supplementary Figures S5A, B). Together, these results suggest that LTβR on intestinal epithelial cells activates non-canonical NF-κB signaling to promote recovery after MTX-induced injury (Figure 7).
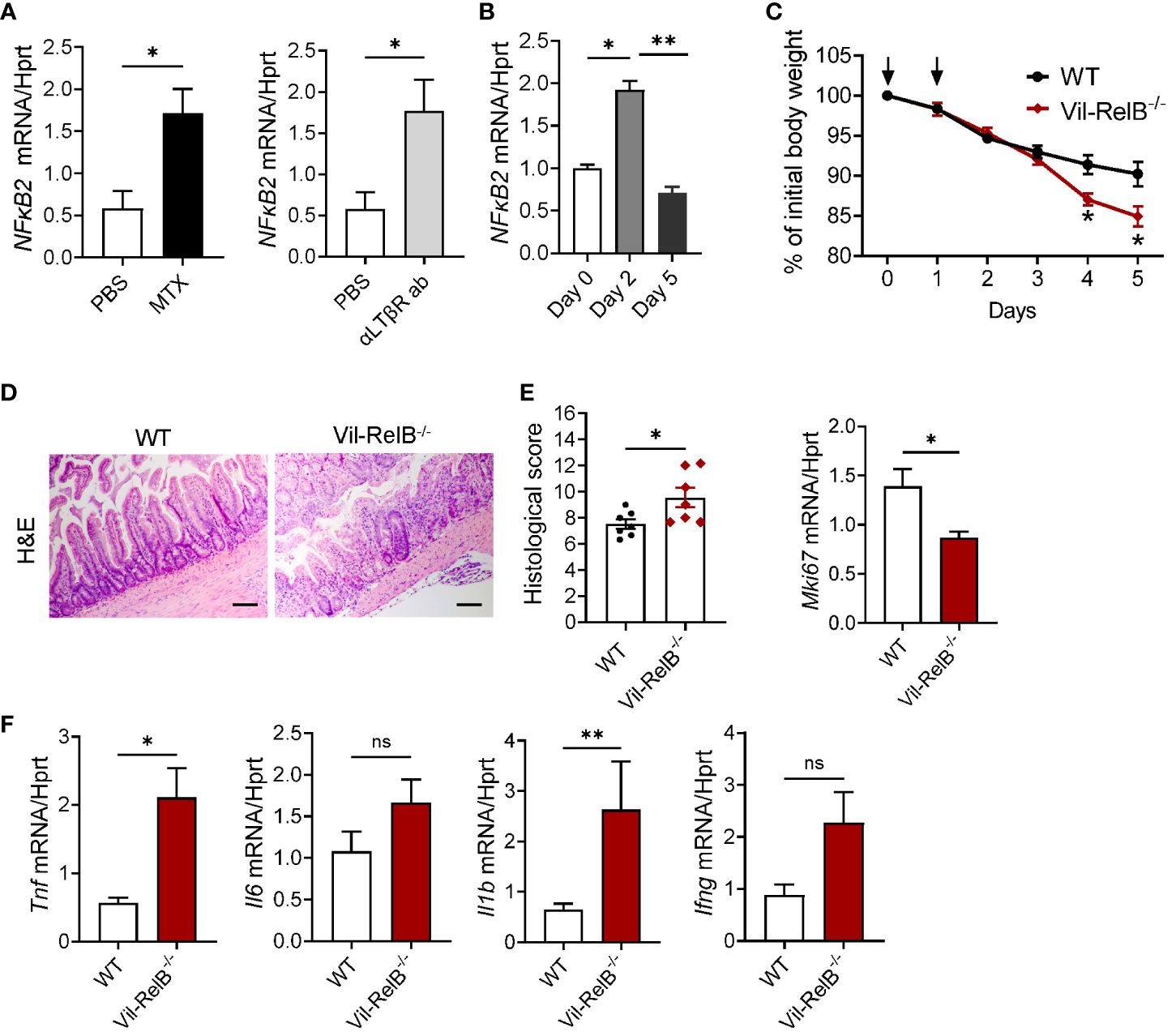
Figure 6 Non-canonical NF-κB signaling in intestinal epithelial cells protects from MTX-induced intestinal damage. (A) CMT-93 epithelial cells were treated with MTX (5 μmol/L) or agonistic αLTβR antibody (ACH6, 0.5μg/ml) for 24 hours. Nfκb2 expression was measured by real-time PCR. (B) Nfκb2 expression in the ileum of WT mice treated with MTX was measured by real-time PCR. n= 4–7 mice per group. (C–F) WT and Vil-RelB-/- mice were treated with MTX as on Figure 1A. (C) Body weight loss; n=14–30 per group. (D) representative H&E images (scale bars, 100μm) and histopathology scores; Expression of (E) Ki-67, and (F) proinflammatory cytokines in the ileum on day 5 after treatment. n= 5 mice per group. Data is combined from 3–4 independent experiments with similar results. Data shown as mean ± SEM. Statistics were determined using unpaired t test (A, D), Mann-Whitney test (A, B), two-way ANOVA with Geisser-Greenhouse correction (C), or Kruskal-Wallis test (E, F). ns, not significant; *p<0.05; **p<0.01.
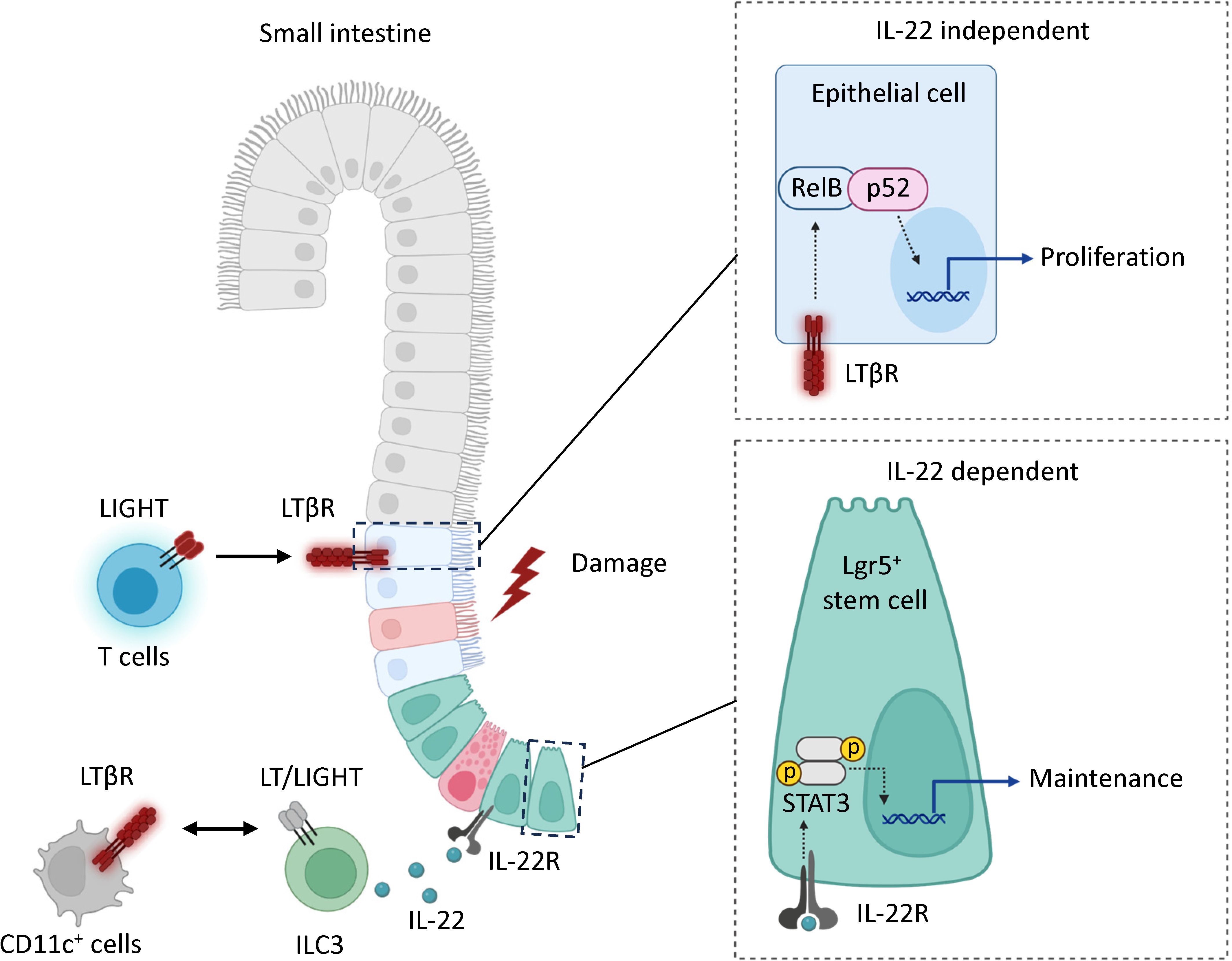
Figure 7 Model. LTβR signaling promotes mucosal healing following MTX-induced injury by controlling IL-22 dependent and IL-22 independent pathways. Mucosal damage promotes expression of LIGHT and LTβ in the intestine. In the IL-22 independent pathway, the interaction of LIGHT expressing T cells with LTβR in intestinal epithelial cells activates non-canonical RelB/p52 NF-kB signaling to promote proliferation of epithelial cells after injury. In the IL-22 dependent pathway, interaction of LIGHT/LT expressing ILC3s with CD11c+ LTβR-expressing cells promotes secretion of IL-22 which interacts with IL-22R to support the maintenance of Lgr5+ intestinal stem cells. LTβR expression in Lgr5+ stem cells is dispensable for protection.
Discussion
Accumulating evidence suggests that immune mechanisms may either exacerbate or ameliorate intestinal damage caused by chemotherapeutic drugs. Recent studies implicated the role of IL-22 and ILC3s in mucosal repair following MTX-induced intestinal damage (30, 31), however the role of other immune components and cytokines remains less defined. In this study we revealed the critical role of LTβR in protection from chemotherapy-induced intestinal damage. As previous studies demonstrated the role of LTβR in regulation of IL-22 production by ILC3s (45, 47), we hypothesized that LTβR-dependent regulation of ILC3s and IL-22 mediates protection against chemotherapy-induced intestinal damage. However, our results suggest that although LT expression in ILC3s is critical for control of IL-22 production, it is dispensable for protection from MTX-induced injury. Instead, another LTβR ligand, LIGHT, produced by T cells was critical for protection. Moreover, LTβR and IL-22 pathways jointly participate in mucosal protection. Furthermore, we demonstrate that LTβR-dependent non-canonical NF-kB signaling in intestinal epithelial cells is required for mucosal repair.
Although the role of LTβR signaling in the development and maintenance of lymphoid tissues and inflammatory diseases is well established (41, 76–78), accumulating evidence suggests that LTβR regulates intestinal inflammation (43, 45–47, 64, 79, 80). However, the role of LTβR in chemotherapy-induced epithelial injury has not been investigated. Our data demonstrate that LTβR-deficient mice display increased body weight loss, severe pathology, reduced epithelial cell proliferation and increased mortality post MTX administration. This phenotype was associated with increased expression of proinflammatory cytokines TNF, IL-1β, IFNγ and chemokines CXCL1, CXC2, CXCL9, CXCL10, and CCL2 in the small intestine at day 5 post MTX administration, whereas IL-22 and IL-22 dependent expression of antibacterial proteins were reduced. These results are consistent with previous studies supporting the role of LTβR in regulation of colonic IL-22 production and protection against C. rodentium infection (44, 47). As increased expression of proinflammatory cytokines at day 5 can be a result of impaired epithelial cell proliferation, we next analyzed immune cell populations and cytokines at day 2 post MTX administration, during the disease induction phase. Our results show that expression of CXCL2 and IL-22 was reduced in the ileum of LTβR-/- mice, whereas IFNγ, CXCL9, CXCL10 were increased at day 2 post MTX treatment. This is consistent with LTβR function in controlling neutrophil recruiting chemokines in response to mucosal bacterial pathogen C. rodentium (43). Flow cytometry revealed an increased frequency of CD8αα+ IELs whereas proportion of CD4+ T cells was reduced in the IEL and LP of LTβR-/- mice. CD8αα+ IELs are known to play regulatory role in intestinal inflammation (81, 82). How LTβR signaling controls CD8αα+ IELs recruitment and the role of these cells in chemotherapy-induced epithelial damage remains to be determined.
Both LTβR ligands LT and LIGHT have been implicated in the regulation of inflammatory responses in the gut (43, 69, 79). Surprisingly, LIGHT but not LTβ, was essential for protection from MTX-induced intestinal damage, as LIGHT-/- mice displayed increased intestinal pathology post MTX treatment whereas LTβ-/- mice did not exhibit an exacerbated pathology. Furthermore, inactivation of LTβ in ILC3s did not result in increased intestinal pathology, despite reduced IL-22 levels in the ileum of RORγt-LTβ-/- mice. These results highlight distinct roles of LIGHT and LT in different models of intestinal damage. Thus, LTβ expressed by RORγt+ ILC3s is critical for protections against C. rodentium, while LIGHT is dispensable in this model (47). In contrast, LIGHT, rather than LTβ, was critical for protection against DSS-induced intestinal damage (69, 79). Interestingly, LIGHT-/- mice displayed reduced levels of IL-22 in the ileum post MTX treatment, suggesting that LIGHT signaling can also control IL-22 production in this model of intestinal damage. In contrast, in the C. rodentium colitis model, LTβ, but not LIGHT, was critical for IL-22 production (47). The distinct role of LIGHT and LTβ in these models of intestinal damage could be attributed to different LIGHT and LTβ producing cell types. Our data revealed that T cells are the major contributors to mucosal protection against MTX induced damage, because TCRβδ-/- mice displayed an exacerbated intestinal pathology compared to RORγt-/- mice. As LIGHT expression was rapidly increased in the intestine at day 2 post MTX treatment, but was ablated in TCRβδ-/- mice, this data suggest that LIGHT provided by T cells contribute to mucosal protection. It is also possible that LIGHT expression by other immune or stromal cells contribute to protection. Our results are in line with a previous study suggesting the role of LIGHT in regulation of intestinal stem cell gene signatures (83). The kinetics and level of LIGHT expression may explain protective versus pathogenic LIGHT-mediated responses in the gut. Consistent with this hypothesis, we detected only a transient induction of LIGHT expression in the MTX-induced injury model. In contrast, sustained overexpression of LIGHT on T cells can break down the immunosuppressive state mediated by Tregs and induce T cell- mediated intestinal inflammation (84, 85).
Previous studies demonstrated the critical role of IL-22 in promoting ISC proliferation after injury (19, 26, 29, 30). However, a recent study demonstrated that IL-22 deficient mice do not display increased intestinal pathology after MTX treatment, implicating IL-22 independent pathways, such as Hippo-Yap, in promoting intestinal epithelial cell proliferation after injury (31). Consistently, our study also did not detect an increased intestinal pathology in IL-22-/- mice post MTX treatment. Although IL-22 expression was impaired in the ileum of RORγt-LTβ-/- mice, these mice did not exhibit increased intestinal pathology. However, we revealed that genetic inactivation of IL-22 further exacerbated MTX-induced intestinal pathology in LTβR-/- mice. These results suggest that LTβR and IL-22 jointly promote mucosal repair after MTX-induced intestinal damage. Interestingly, LTβR stimulation may suppress YAP/TAZ activity in fibroblastic reticular cells in lymph nodes (86). However, the connection between LTβR and Yap signaling in intestinal epithelial cells remains to be determined.
LTβR is expressed on a variety of epithelial, stromal, and myeloid cells in the gut, thereby participating in regulation of mucosal immune homeostasis (43, 45, 46, 59, 64, 87). Therefore, we wanted to determine which LTβR expressing cells are critical for protection against MTX-induced damage. Our results suggest that LTβR expression in intestinal epithelial cells is essential for protection, whereas LTβR expression on macrophages and dendritic cells is dispensable. The protective role of LTβR on intestinal epithelial cells was previously demonstrated in C. rodentium infection and DSS-induced colitis models (43, 45). However, genetic inactivation of LTβR in ISCs did not exacerbate intestinal disease, consistent with low expression of LTβR on Lgr5+ stem cells (72). These results suggest that although LTβR on intestinal epithelial cells is critical for mucosal repair after MTX-induced damage, LTβR signaling in ISCs is dispensable for protection. The role of specific subsets of LTβR-expressing intestinal epithelial cells in mucosal repair after MTX-induced damage will be further defined in future studies.
LTβR stimulation leads to non-canonical NF-kB signaling, which involves NF-κB-inducing kinase (NIK) and IKKα, processing of p100 precursor and nuclear translocation of the non-canonical NF-κB complex p52/RelB (39, 42, 88). Additionally, LTβR stimulation can lead to activation of the canonical NF-κB pathway operating via NFκB1 (p50/RelA) transcription, which usually occurs within minutes and does not require novel gene expression, in contrast to the non-canonical pathway (73, 88). Non-canonical NF-kB signaling is thought to play a central role in induction of proinflammatory cytokines TNF, IL-6, IL-18, IL-1β early during chemotherapy-induced intestinal injury, thereby promoting inflammation (2, 3). In contrast, non-canonical NF-κB signaling in intestinal epithelial cells is important for protection from gut bacterial infections and intestinal inflammation (59, 74, 75). Our results are consistent with these studies and identify a previously unrecognized role for epithelial cell-intrinsic RelB expression in regulating mucosal repair after chemotherapy-induced intestinal damage.
Based on our results, we propose a model for a LTβR-dependent mechanism for mucosal healing after MTX-induced intestinal damage (Figure 7). MTX injury results in early upregulation of chemokines and increased recruitment of T cells to the epithelial layer. LIGHT, presumably produced by T cells interacts with LTβR on intestinal epithelial cells to activate non-canonical RelB signaling thereby promoting proliferation of epithelial cells after injury. Interactions between LIGHT/LT expressing RORγt+ ILC3s and LTβR expressing CD11c+ cells can also contribute to IL-22-dependent maintenance of ISCs after injury. Our data suggest that LTβR also promotes mucosal healing in 5-FU induced intestinal mucositis. The critical LTβR expressing cells and LTβR ligands in 5-FU induced intestinal injury remain to be determined.
Gaining insight into the immune regulation of mucosal healing post-cytotoxic drug exposure holds crucial implications for developing targeted therapeutic interventions. In summary, our study revealed a previously unrecognized role for the LTβR-RelB pathway in intestinal epithelial cells which promotes mucosal repair after chemotherapy-induced intestinal damage. These findings provide valuable insights into the immune mechanisms orchestrating mucosal healing after chemotherapy-induced intestinal injury, paving the way for potential therapeutic interventions.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://portals.broadinstitute.org/single_cell/study/small-intestinal-epithelium.
Ethics statement
The animal study was approved by The University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AVT: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. QC: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. AM: Conceptualization, Formal analysis, Investigation, Supervision, Writing – review & editing. AK: Data curation, Formal analysis, Investigation, Visualization, Writing – review & editing. YS: Investigation, Writing – original draft. JV: Investigation, Writing – original draft. AWT: Investigation, Writing – review & editing. SS: Data curation, Formal analysis, Investigation, Writing – review & editing. EK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by grants from the National Institutes of Health (NIH) NS112263, DE029187, the Cancer Prevention and Research Institute of Texas (CPRIT) RP210105, RP220470, and by the Max and Minnie Tomerlin Voelcker Fund. AM was supported by K12 GM111726 San Antonio Biomedical Education and Research-Institutional Research and Academic Career Development Award (SABER-IRACDA). The Flow Cytometry Shared Resource at UT Health San Antonio is supported by a grant from the National Cancer Institute (P30CA054174) to the Mays Cancer Center, a grant from the Cancer Prevention and Research Institute of Texas (CPRIT) (RP210126), a grant from the National Institutes of Health (1S10OD030432), and support from the Office of the Vice President for Research at UT Health San Antonio.
Acknowledgments
We thank Dr. Michael Croft and Dr. Mitchell Kronenberg (La Jolla Institute for Immunology) for providing LIGHT-/- mice. We are grateful to Biogen Idec for providing anti-LTβR agonistic antibody. We thank Anna Tumanova for editing the manuscript. Figure 7 was created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1388496/full#supplementary-material
Supplementary Figure 1 | LTβR signaling protects against 5-FU induced intestinal inflammation. WT and LTβR-/- mice were treated with 5-Fluorouracil (5-FU, 50 mg/kg, i.p.) daily for 4 consecutive days, and analyzed at day 5. (A) Body weight change. Black arrows: days of 5-FU treatment. n=14–17 mice per group. (B) Disease score. (C) Representative photographs of colons and colon length. (D) Representative H&E images and histological scores. Scale bars, 100μm. (E) Cytokine expression in the colon. n= 7 mice per group. Data represents 1 of 3 independent experiments with similar results. Data shown as mean ± SEM. Statistics were determined using two-way ANOVA with Geisser-Greenhouse correction (A), unpaired t test (B–E). ns, not significant; * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Supplementary Figure 2 | Gating strategy of immune cell populations in SI. (A) Gating strategy of immune cell populations in IEL. Lin+(Lineage+): B220, Ly6G. Neutrophils were defined as CD45+Lin+MHCII-CD11b+; B cells were defined as CD45+Lin+CD11b-MHCII+; T cells were defined as CD45+CD3+ (B) Gating strategy of immune cell populations in LP. Neutrophils, CD45+Ly6G+CD11b+; B cells, CD45+Ly6G-B220+; Eosinophils, CD45+Ly6G-B220-CD11b+Siglec F+; T cells, CD45+Ly6G-B220-SiglecF-TCRβ+; ILC1s, CD45+Ly6G-B220-SiglecF-TCRβ-CD64-NK1.1+; Dendritic cells (DCs), CD45+Ly6G-B220-SiglecF-TCRβ-CD64-MHCII+CD11c+; Macrophages (Mph), CD45+Ly6G-B220-SiglecF-TCRβ-CD64+MHCII+CD11b+; Monocytes (Mo), CD45+Ly6G-B220-SiglecF-TCRβ-CD64+MHCII-CD11b+CCR2+.
Supplementary Figure 3 | Analysis of immune cell populations and cytokines in WT SI after MTX treatment. (A–G) WT mice were treated with MTX as in Figure 1A. Mice were euthanized on day 2 and small intestines were collected for analysis. (A) Representative flow cytometry plots and frequency of T cell populations in SI IEL. Frequency is calculated in live CD45+ cells. (B, C) Representative flow cytometry plots and frequency of cell populations in LP. Tregs (B220-CD3+CD4+CD25+FoxP3+); ILC1s (CD45+Ly6G-B220-SiglecF-TCRβ- CD64-NK1.1+); CD4+ T cells; CD3+ T cells; Macrophages (Mph, CD11c-Ly6G-SiglecF-CD11b+MHCII+CD64+); Neutrophils (Nph, Ly6G+ CD11b+); B cells (B220+); DCs (CD45+Ly6G-B220-SiglecF-TCRβ-CD64-MHCII+CD11c+). Expression of (D) cytokines and (E) chemokines in the ileum at day 0, 2 and 5 post MTX treatment. (F, G) LTα expression after MTX was measured by Real-Time PCR in WT (F) ileum, jejunum, and duodenum as well as (G) LP and IEL from small intestine. (D–G) Data are representative of two experiments (n=3–7 per group). Data shown as mean ± SEM. Statistics were determined using unpaired t test (A–C, F, G), Mann-Whitney test (D, E), Kruskal-Wallis test (D–F). ns, not significant, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Supplementary Figure 4 | Analysis of epithelial cell populations in the ileum of WT and LTβR-/- mice. (A) Expression of Ltbr and Lgr5 in various small intestine derived cell types was determined by single-cell RNA-seq. Data was obtained from the study conducted by Haber et al. (72), using the Broad Institute Single-Cell Portal for data analysis (https://portals.broadinstitute.org/single_cell/study/small-intestinal-epithelium). (B) WT and LTβR-/- mice were crossed with Lgr5-GFP reporter mice. GFP expression was induced by tamoxifen administration and mice were treated with MTX as in Figure 1A. Mice were euthanized on day 5 and ileum epithelial cells analyzed by flow cytometry. Gating strategy. Tuft cells: EpCAM+CD45+; Lgr5+ cells: EpCAM+Lgr5+CD31-Ter119-CD45-; Paneth cells: EpCAM+c-Kit+CD31-Ter119-CD45-; Epithelial cells: EpCAM+CD31-Ter119-CD45-. (C–F) Representative flow plot and frequency of cell populations. (G) Goblet cells analysis by Alcian Blue staining in small intestine. Scale bars, 100μm. Data show 1 of 2 independent experiments with similar results (n=3–5 per group). Data shown as mean ± SEM. Statistics were determined using unpaired t test. ns, not significant.
Supplementary Figure 5 | LTβR signaling in macrophages and DCs is not essential for the protection from MTX induced intestinal injury. (A–D). WT, LysM-LTβR-/-, CD11c-LTβR-/-, CD11c, LysM-LTβR-/- and CD11c-RelB-/- mice were treated with MTX as in Figure 1A. (A, D) Body weight loss (n=9–14 mice per group) and (B) Representative H&E images (scale bars, 100μm) with histopathology scores. (C) IL-22 expression in the ileum on day 5 after MTX treatment. n= 5–8 mice per group. Data are combined from 3–5 independent experiments with similar results. Data shown as mean ± SEM. Statistics were determined using two-way ANOVA with Geisser-Greenhouse correction (A, D), Kruskal-Wallis test (B), unpaired t test (C). ns, not significant, * p<0.05.
Abbreviations
LTβR, lymphotoxin beta receptor; LT, lymphotoxin; LIGHT, lymphotoxin-like inducible protein that competes with glycoprotein D for herpes virus entry on T cells, MTX, methotrexate; 5-FU, 5-Fluorouracil; SI, small intestine; WT, wild type; ISC, intestinal stem cells.
References
1. Dahlgren D, Sjoblom M, Hellstrom PM, Lennernas H. Chemotherapeutics-induced intestinal mucositis: pathophysiology and potential treatment strategies. Front Pharmacol. (2021) 12:681417. doi: 10.3389/fphar.2021.681417
2. Sougiannis AT, VanderVeen BN, Davis JM, Fan D, Murphy EA. Understanding chemotherapy-induced intestinal mucositis and strategies to improve gut resilience. Am J Physiol Gastrointest Liver Physiol. (2021) 320:G712–G9. doi: 10.1152/ajpgi.00380.2020
3. Basile D, Di Nardo P, Corvaja C, Garattini SK, Pelizzari G, Lisanti C, et al. Mucosal injury during anti-cancer treatment: from pathobiology to bedside. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11060857
4. Villa A, Sonis ST. Mucositis: pathobiology and management. Curr Opin Oncol. (2015) 27:159–64. doi: 10.1097/CCO.0000000000000180
6. Elting LS, Cooksley CD, Chambers MS, Garden AS. Risk, outcomes, and costs of radiation-induced oral mucositis among patients with head-and-neck Malignancies. Int J Radiat Oncol Biol Phys. (2007) 68:1110–20. doi: 10.1016/j.ijrobp.2007.01.053
7. O'Brien ME, Borthwick A, Rigg A, Leary A, Assersohn L, Last K, et al. Mortality within 30 days of chemotherapy: A clinical governance benchmarking issue for oncology patients. Br J Cancer. (2006) 95:1632–6. doi: 10.1038/sj.bjc.6603498
8. Iacovelli R, Pietrantonio F, Maggi C, de Braud F, Di Bartolomeo M. Combination or single-agent chemotherapy as adjuvant treatment of gastric cancer: A systematic review and meta-analysis of published trials. Crit Rev Oncol Hematol. (2016) 98:24–8. doi: 10.1016/j.critrevonc.2015.09.002
9. Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, et al. Folfiri plus cetuximab versus folfiri plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (Fire-3): A randomised, open-label, phase 3 trial. Lancet Oncol. (2014) 15:1065–75. doi: 10.1016/S1470–2045(14)70330–4
10. Becerra CR, Frenkel EP, Ashfaq R, Gaynor RB. Increased toxicity and lack of efficacy of rofecoxib in combination with chemotherapy for treatment of metastatic colorectal cancer: A phase II study. Int J Cancer. (2003) 105:868–72. doi: 10.1002/ijc.11164
11. Mego M, Chovanec J, Vochyanova-Andrezalova I, Konkolovsky P, Mikulova M, Reckova M, et al. Prevention of irinotecan induced diarrhea by probiotics: A randomized double blind, placebo controlled pilot study. Complement Ther Med. (2015) 23:356–62. doi: 10.1016/j.ctim.2015.03.008
12. Lee JY, Chu SH, Jeon JY, Lee MK, Park JH, Lee DC, et al. Effects of 12 weeks of probiotic supplementation on quality of life in colorectal cancer survivors: A double-blind, randomized, placebo-controlled trial. Dig Liver Dis. (2014) 46:1126–32. doi: 10.1016/j.dld.2014.09.004
13. Lu H, Liu H, Wang J, Shen J, Weng S, Han L, et al. The chemokine CXCL9 exacerbates chemotherapy-induced acute intestinal damage through inhibition of mucosal restitution. J Cancer Res Clin Oncol. (2015) 141:983–92. doi: 10.1007/s00432-014-1869-y
14. Zhou B, Xia X, Wang P, Chen S, Yu C, Huang R, et al. Induction and amelioration of methotrexate-induced gastrointestinal toxicity are related to immune response and gut microbiota. EBioMedicine. (2018) 33:122–33. doi: 10.1016/j.ebiom.2018.06.029
15. Sethy C, Kundu CN. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: implication of DNA repair inhibition. BioMed Pharmacother. (2021) 137:111285. doi: 10.1016/j.biopha.2021.111285
16. Sangild PT, Shen RL, Pontoppidan P, Rathe M. Animal models of chemotherapy-induced mucositis: translational relevance and challenges. Am J Physiol Gastrointest Liver Physiol. (2018) 314:G231–G46. doi: 10.1152/ajpgi.00204.2017
17. Sakai H, Sagara A, Matsumoto K, Hasegawa S, Sato K, Nishizaki M, et al. 5-fluorouracil induces diarrhea with changes in the expression of inflammatory cytokines and aquaporins in mouse intestines. PloS One. (2013) 8:e54788. doi: 10.1371/journal.pone.0054788
18. Sultani M, Stringer AM, Bowen JM, Gibson RJ. Anti-inflammatory cytokines: important immunoregulatory factors contributing to chemotherapy-induced gastrointestinal mucositis. Chemother Res Pract. (2012) 2012:490804. doi: 10.1155/2012/490804
19. Hanash AM, Dudakov JA, Hua G, O'Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. (2012) 37:339–50. doi: 10.1016/j.immuni.2012.05.028
20. Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. (2011) 12:21–7. doi: 10.1038/ni.1962
21. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414–112123
22. Keir M, Yi Y, Lu T, Ghilardi N. The role of il-22 in intestinal health and disease. J Exp Med. (2020) 217:e20192195. doi: 10.1084/jem.20192195
23. Renauld JC. Class ii cytokine receptors and their ligands: key antiviral and inflammatory modulators. Nat Rev Immunol. (2003) 3:667–76. doi: 10.1038/nri1153
24. Saxton RA, Henneberg LT, Calafiore M, Su L, Jude KM, Hanash AM, et al. The tissue protective functions of interleukin-22 can be decoupled from pro-inflammatory actions through structure-based design. Immunity. (2021) 54:660–72.e9. doi: 10.1016/j.immuni.2021.03.008
25. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. (2004) 21:241–54. doi: 10.1016/j.immuni.2004.07.007
26. Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. (2015) 528:560–4. doi: 10.1038/nature16460
27. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. (2012) 36:276–87. doi: 10.1016/j.immuni.2011.12.011
28. Lorchner H, Poling J, Gajawada P, Hou Y, Polyakova V, Kostin S, et al. Myocardial healing requires reg3beta-dependent accumulation of macrophages in the ischemic heart. Nat Med. (2015) 21:353–62. doi: 10.1038/nm.3816
29. Gronke K, Hernandez PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature. (2019) 566:249–53. doi: 10.1038/s41586–019-0899–7
30. Aparicio-Domingo P, Romera-Hernandez M, Karrich JJ, Cornelissen F, Papazian N, Lindenbergh-Kortleve DJ, et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J Exp Med. (2015) 212:1783–91. doi: 10.1084/jem.20150318
31. Romera-Hernandez M, Aparicio-Domingo P, Papazian N, Karrich JJ, Cornelissen F, Hoogenboezem RM, et al. Yap1-driven intestinal repair is controlled by group 3 innate lymphoid cells. Cell Rep. (2020) 30:37–45.e3. doi: 10.1016/j.celrep.2019.11.115
32. Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur J Immunol. (2006) 36:1309–23. doi: 10.1002/eji.200535503
33. Eken A, Singh AK, Treuting PM, Oukka M. IL-23R+ Innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. (2014) 7:143–54. doi: 10.1038/mi.2013.33
34. Munoz M, Heimesaat MM, Danker K, Struck D, Lohmann U, Plickert R, et al. Interleukin (IL)-23 mediates toxoplasma gondii-induced immunopathology in the gut via matrixmetalloproteinase-2 and IL-22 but independent of IL-17. J Exp Med. (2009) 206:3047–59. doi: 10.1084/jem.20090900
35. Gunasekera DC, Ma J, Vacharathit V, Shah P, Ramakrishnan A, Uprety P, et al. The development of colitis in IL10(-/-) mice is dependent on IL-22. Mucosal Immunol. (2020) 13:493–506. doi: 10.1038/s41385–019-0252–3
36. Kamanaka M, Huber S, Zenewicz LA, Gagliani N, Rathinam C, O'Connor W Jr., et al. Memory/effector (Cd45rb(Lo)) cd4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J Exp Med. (2011) 208:1027–40. doi: 10.1084/jem.20102149
37. Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. (2005) 23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719
38. Upadhyay V, Fu YX. Lymphotoxin signalling in immune homeostasis and the control of microorganisms. Nat Rev Immunol. (2013) 13:270–9. doi: 10.1038/nri3406
39. Albarbar B, Dunnill C, Georgopoulos NT. Regulation of cell fate by lymphotoxin (LT) receptor signalling: functional differences and similarities of the LT system to other TNF superfamily (TNFSF) members. Cytokine Growth Factor Rev. (2015) 26:659–71. doi: 10.1016/j.cytogfr.2015.05.001
40. Ware CF, Croft M, Neil GA. Realigning the light signaling network to control dysregulated inflammation. J Exp Med. (2022) 219. doi: 10.1084/jem.20220236
41. Koroleva EP, Fu YX, Tumanov AV. Lymphotoxin in physiology of lymphoid tissues - implication for antiviral defense. Cytokine. (2018) 101:39–47. doi: 10.1016/j.cyto.2016.08.018
42. Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. (2002) 17:525–35. doi: 10.1016/s1074–7613(02)00423–5
43. Wang Y, Koroleva EP, Kruglov AA, Kuprash DV, Nedospasov SA, Fu YX, et al. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity. (2010) 32:403–13. doi: 10.1016/j.immuni.2010.02.011
44. Ota N, Wong K, Valdez PA, Zheng Y, Crellin NK, Diehl L, et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with citrobacter rodentium. Nat Immunol. (2011) 12:941–8. doi: 10.1038/ni.2089
45. Macho-Fernandez E, Koroleva EP, Spencer CM, Tighe M, Torrado E, Cooper AM, et al. Lymphotoxin beta receptor signaling limits mucosal damage through driving IL-23 production by epithelial cells. Mucosal Immunol. (2015) 8:403–13. doi: 10.1038/mi.2014.78
46. Riffelmacher T, Giles DA, Zahner S, Dicker M, Andreyev AY, McArdle S, et al. Metabolic activation and colitis pathogenesis is prevented by lymphotoxin beta receptor expression in neutrophils. Mucosal Immunol. (2021) 14:679–90. doi: 10.1038/s41385–021-00378–7
47. Tumanov AV, Koroleva EP, Guo X, Wang Y, Kruglov A, Nedospasov S, et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe. (2011) 10:44–53. doi: 10.1016/j.chom.2011.06.002
48. Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor rorgamma(T) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. (2004) 5:64–73. doi: 10.1038/ni1022
49. Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, et al. Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature. (1992) 360:225–31. doi: 10.1038/360225a0
50. Ahlfors H, Morrison PJ, Duarte JH, Li Y, Biro J, Tolaini M, et al. IL-22 fate reporter reveals origin and control of IL-22 production in homeostasis and infection. J Immunol. (2014) 193:4602–13. doi: 10.4049/jimmunol.1401244
51. Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (Crypt) and horizontal (Duodenum, cecum) axes of the intestine. J Biol Chem. (2002) 277:33275–83. doi: 10.1074/jbc.M204935200
52. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using lysmcre mice. Transgenic Res. (1999) 8:265–77. doi: 10.1023/a:1008942828960
53. Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, et al. Elimination of antigen-presenting cells and autoreactive T cells by fas contributes to prevention of autoimmunity. Immunity. (2007) 26:629–41. doi: 10.1016/j.immuni.2007.03.016
54. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. (2007) 449:1003–7. doi: 10.1038/nature06196
55. Powolny-Budnicka I, Riemann M, Tanzer S, Schmid RM, Hehlgans T, Weih F. Rela and relb transcription factors in distinct thymocyte populations control lymphotoxin-dependent interleukin-17 production in gammadelta T cells. Immunity. (2011) 34:364–74. doi: 10.1016/j.immuni.2011.02.019
56. Alimzhanov MB, Kuprash DV, Kosco-Vilbois MH, Luz A, Turetskaya RL, Tarakhovsky A, et al. Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc Natl Acad Sci U.S.A. (1997) 94:9302–7. doi: 10.1073/pnas.94.17.9302
57. Scheu S, Alferink J, Potzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of light causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med. (2002) 195:1613–24. doi: 10.1084/jem.20020215
58. Tumanov A, Kuprash D, Lagarkova M, Grivennikov S, Abe K, Shakhov A, et al. Distinct role of surface lymphotoxin expressed by B cells in the organization of secondary lymphoid tissues. Immunity. (2002) 17:239–50. doi: 10.1016/S1074-7613(02)00397-7
59. Pian Y, Chai Q, Ren B, Wang Y, Lv M, Qiu J, et al. Type 3 innate lymphoid cells direct goblet cell differentiation via the lt-ltbetar pathway during listeria infection. J Immunol. (2020) 205:853–63. doi: 10.4049/jimmunol.2000197
60. de Koning BA, van Dieren JM, Lindenbergh-Kortleve DJ, van der Sluis M, Matsumoto T, Yamaguchi K, et al. Contributions of mucosal immune cells to methotrexate-induced mucositis. Int Immunol. (2006) 18:941–9. doi: 10.1093/intimm/dxl030
61. Koelink PJ, Wildenberg ME, Stitt LW, Feagan BG, Koldijk M, van 't Wout AB, et al. Development of reliable, valid and responsive scoring systems for endoscopy and histology in animal models for inflammatory bowel disease. J Crohns Colitis. (2018) 12:794–803. doi: 10.1093/ecco-jcc/jjy035
62. Koroleva EP, Halperin S, Gubernatorova EO, Macho-Fernandez E, Spencer CM, Tumanov AV. Citrobacter rodentium-induced colitis: A robust model to study mucosal immune responses in the gut. J Immunol Methods. (2015) 421:61–72. doi: 10.1016/j.jim.2015.02.003
63. Mackay F, Browning JL, Lawton P, Shah SA, Comiskey M, Bhan AK, et al. Both the lymphotoxin and tumor necrosis factor pathways are involved in experimental murine models of colitis. Gastroenterology. (1998) 115:1464–75. doi: 10.1016/s0016–5085(98)70025–3
64. Gubernatorova EO, Tumanov AV. Tumor necrosis factor and lymphotoxin in regulation of intestinal inflammation. Biochem (Mosc). (2016) 81:1309–25. doi: 10.1134/S0006297916110092
65. Li HL, Lu L, Wang XS, Qin LY, Wang P, Qiu SP, et al. Alteration of gut microbiota and inflammatory cytokine/chemokine profiles in 5-fluorouracil induced intestinal mucositis. Front Cell Infect Microbiol. (2017) 7:455. doi: 10.3389/fcimb.2017.00455
66. Badr AM, Alkharashi LA, Sherif IO, Alanteet AA, Alotaibi HN, Mahran YF. Il-17/notch1/stat3 pathway contributes to 5-fluorouracil-induced intestinal mucositis in rats: amelioration by thymol treatment. Pharm (Basel). (2022) 15. doi: 10.3390/ph15111412
67. Metzemaekers M, Vanheule V, Janssens R, Struyf S, Proost P. Overview of the mechanisms that may contribute to the non-redundant activities of interferon-inducible cxc chemokine receptor 3 ligands. Front Immunol. (2017) 8:1970. doi: 10.3389/fimmu.2017.01970
68. Sawant KV, Sepuru KM, Lowry E, Penaranda B, Frevert CW, Garofalo RP, et al. Neutrophil recruitment by chemokines Cxcl1/KC and Cxcl2/MIP2: Role of Cxcr2 activation and glycosaminoglycan interactions. J Leukoc Biol. (2021) 109:777–91. doi: 10.1002/JLB.3A0820-207R
69. Krause P, Zahner SP, Kim G, Shaikh RB, Steinberg MW, Kronenberg M. The tumor necrosis factor family member TNFSF14 (LIGHT) is required for resolution of intestinal inflammation in mice. Gastroenterology. (2014) 146:1752–62.e4. doi: 10.1053/j.gastro.2014.02.010
70. Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. (2014) 15:19–33. doi: 10.1038/nrm3721
71. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. (2009) 459:262–5. doi: 10.1038/nature07935
72. Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, et al. A single-cell survey of the small intestinal epithelium. Nature. (2017) 551:333–9. doi: 10.1038/nature24489
73. Piao W, Kasinath V, Saxena V, Lakhan R, Iyyathurai J, Bromberg JS. Ltbetar signaling controls lymphatic migration of immune cells. Cells. (2021) 10. doi: 10.3390/cells10040747
74. Wullaert A, Bonnet MC, Pasparakis M. Nf-kappab in the regulation of epithelial homeostasis and inflammation. Cell Res. (2011) 21:146–58. doi: 10.1038/cr.2010.175
75. Giacomin PR, Moy RH, Noti M, Osborne LC, Siracusa MC, Alenghat T, et al. Epithelial-intrinsic ikkalpha expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J Exp Med. (2015) 212:1513–28. doi: 10.1084/jem.20141831
76. Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. (1998) 9:59–70. doi: 10.1016/s1074-7613(00)80588-9
77. Browning JL. Inhibition of the lymphotoxin pathway as a therapy for autoimmune disease. Immunol Rev. (2008) 223:202–20. doi: 10.1111/j.1600-065X.2008.00633.x
78. Shou Y, Koroleva E, Spencer CM, Shein SA, Korchagina AA, Yusoof KA, et al. Redefining the role of lymphotoxin beta receptor in the maintenance of lymphoid organs and immune cell homeostasis in adulthood. Front Immunol. (2021) 12:712632. doi: 10.3389/fimmu.2021.712632
79. Giles DA, Zahner S, Krause P, van der Gracht E, Riffelmacher T, Morris V, et al. The tumor necrosis factor superfamily members TNFSF14 (LIGHT), lymphotoxin beta and lymphotoxin beta receptor interact to regulate intestinal inflammation. Front Immunol. (2018) 9:2585. doi: 10.3389/fimmu.2018.02585
80. Guendel F, Kofoed-Branzk M, Gronke K, Tizian C, Witkowski M, Cheng HW, et al. Group 3 innate lymphoid cells program a distinct subset of IL-22BP-producing dendritic cells demarcating solitary intestinal lymphoid tissues. Immunity. (2020) 53:1015–32.e8. doi: 10.1016/j.immuni.2020.10.012
81. Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat Rev Immunol. (2011) 11:445–56. doi: 10.1038/nri3007
82. Ma H, Qiu Y, Yang H. Intestinal intraepithelial lymphocytes: maintainers of intestinal immune tolerance and regulators of intestinal immunity. J Leukoc Biol. (2021) 109:339–47. doi: 10.1002/JLB.3RU0220–111
83. Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. (2018) 175:372–86.e17. doi: 10.1016/j.cell.2018.08.067
84. Wang J, Lo JC, Foster A, Yu P, Chen HM, Wang Y, et al. The regulation of T cell homeostasis and autoimmunity by T cell-derived light. J Clin Invest. (2001) 108:1771–80. doi: 10.1172/JCI13827
85. Shaikh RB, Santee S, Granger SW, Butrovich K, Cheung T, Kronenberg M, et al. Constitutive expression of light on T cells leads to lymphocyte activation, inflammation, and tissue destruction. J Immunol. (2001) 167:6330–7. doi: 10.4049/jimmunol.167.11.6330
86. Choi SY, Bae H, Jeong SH, Park I, Cho H, Hong SP, et al. YAP/TAZ direct commitment and maturation of lymph node fibroblastic reticular cells. Nat Commun. (2020) 11:519. doi: 10.1038/s41467–020-14293–1
87. Cheng HW, Morbe U, Lutge M, Engetschwiler C, Onder L, Novkovic M, et al. Intestinal fibroblastic reticular cell niches control innate lymphoid cell homeostasis and function. Nat Commun. (2022) 13:2027. doi: 10.1038/s41467–022-29734–2
Keywords: LTβR, LIGHT, lymphotoxin, RelB, IL-22, methotrexate, intestinal damage
Citation: Chen Q, Muñoz AR, Korchagina AA, Shou Y, Vallecer J, Todd AW, Shein SA, Tumanov AV and Koroleva E (2024) LTβR-RelB signaling in intestinal epithelial cells protects from chemotherapy-induced mucosal damage. Front. Immunol. 15:1388496. doi: 10.3389/fimmu.2024.1388496
Received: 19 February 2024; Accepted: 01 May 2024;
Published: 30 May 2024.
Edited by:
Jian Zheng, University of Louisville, United StatesReviewed by:
Huihong Zeng, Nanchang University, ChinaShahan Mamoor, Independent Researcher, East Islip, NY, United States
Copyright © 2024 Chen, Muñoz, Korchagina, Shou, Vallecer, Todd, Shein, Tumanov and Koroleva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexei V. Tumanov, tumanov@uthscsa.edu; Ekaterina Koroleva, koroleva@uthscsa.edu
†Present address: Amanda R. Muñoz, Department of Biology & Chemistry, Texas A&M International University, Laredo, TX, United States
‡These authors have contributed equally to this work