 Shivang S. Shah
Shivang S. Shah Elizabeth F. Stone
Elizabeth F. Stone Richard O. Francis
Richard O. Francis Matthew S. Karafin3
Matthew S. Karafin3- 1Department of Pediatrics, Columbia University, New York, NY, United States
- 2Department of Pathology and Cell Biology, Columbia University, New York, NY, United States
- 3Department of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, NC, United States
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common enzymopathy in humans. G6PD is an essential enzyme in the pentose phosphate pathway (PPP), generating NADPH needed for cellular biosynthesis and reactive oxygen species (ROS) homeostasis, the latter especially key in red blood cells (RBCs). Beyond the RBC, there is emerging evidence that G6PD exerts an immunologic role by virtue of its functions in leukocyte oxidative metabolism and anabolic synthesis necessary for immune effector function. We review these here, and consider the global immunometabolic role of G6PD activity and G6PD deficiency in modulating inflammation and immunopathology.
1 Introduction
Glucose-6-phosphate dehydrogenase (G6PD) catalyzes the rate-limiting first step in the pentose phosphate pathway (PPP), converting nicotinamide adenine dinucleotide phosphate (NADP) to its reduced form: NADPH (Figure 1A). Levels of active G6PD are regulated at both the transcriptional level via various canonical signaling pathways (e.g., JAK-STAT, Wnt, mTOR), and at the post-translational level (e.g., via phosphorylation, de-acetylation), allowing tight coordination of G6PD activity to meet acute cellular demand in response to oxidative stress, metabolic demand, or systemic inflammation (Figure 1A-1). NADPH generation is critical to host antioxidant defense via glutathione reduction (Figure 1A-2), and is also essential for anabolic cellular metabolism, including synthesis of nucleotides, fatty acids, and amino acids (Figure 1A-3). Downstream production of ribulose-5-phosphate (R5P) is essential for formation of key nucleotides and cofactors (Figure 1A-3), in addition to acting as a glycolytic shunt intermediary (Figure 1A-4). Indeed, G6PD, by virtue of its importance in fundamental redox homeostasis and anabolic metabolism, plays a multifaceted, ubiquitous role in human physiology, including within immune responses.
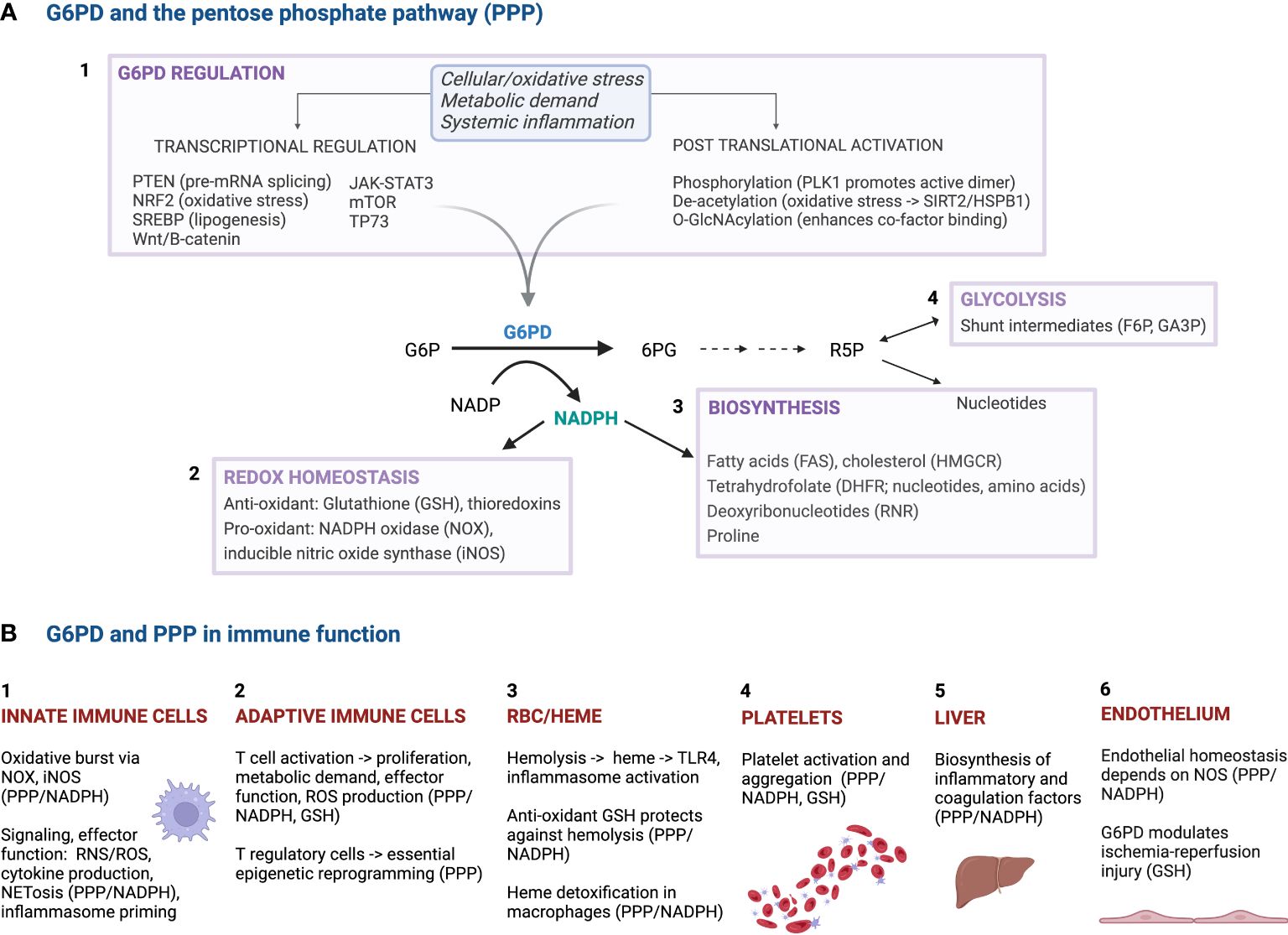
Figure 1 The roles of G6PD and the pentose phosphate pathway (PPP) in cellular and immune function. (A) The top panel highlights the multifaceted role of G6PD in cellular metabolism, including biosynthesis and the response to oxidative stress. In response to key triggers such as cellular/oxidative stress, metabolic demand and systemic inflammation, active G6PD production is upregulated by various factors/pathways at the transcriptional and post-transcriptional levels. The resultant NADPH produced is key in redox homeostasis and several biosynthetic functions, including lipid and nucleotide synthesis. (B) The bottom panel highlights the pleiotropic impact that G6PD and PPP activity have on immune function, through diverse impacts on biosynthesis and activation/effector phenotypes, redox and endothelial homeostasis, cell signaling and inflammasome priming, and mitigating RBC hemolytic stress. PTEN, phosphatase and tensin homolog; mRNA, messenger ribonucleic acid; NRF2, nuclear factor erythroid 2-related factor 2; SREBP, sterol regulatory element binding protein; JAK-STAT3, Janus kinase-signal transducer and activator of transcription 3; mTOR, mammalian target of rapamycin; TP73, tumor protein p73; PLK1, polo-like kinase 1; O-GlcNAc, O-linked β-N-acetylglucosamine; SIRT2, sirtuin 2; HSPB1, heat shock protein family B member 1; G6P, glucose 6-phosphate; G6PD, glucose 6-phosphate dehydrogenase; 6PG, 6-phosphogluconate; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of NADP; R5P, ribulose 5-phosphate; F6P, fructose 6-phosphate; GA3P, glyceraldehyde 3-phosphate; GSH, glutathione; TRX, thioredoxin reductase; FAS, fatty acid synthase; iNOS, inducible nitric oxide synthase; NOX, NADPH oxidase; HMGCR, 3-hydroxy-3-methylglutaryl coenzyme A reductase; RNR, ribonucleotide reductase; DHFR, dihydrofolate reductase; TRX, thioredoxin; PPP, pentose phosphate pathway; TLR4, Toll-like receptor 4; RNS, reactive nitrogen species; ROS, reactive oxygen species; NET, neutrophil extracellular trap.
Across human populations, basal G6PD enzyme activity is highly variable, with hundreds of missense genetic mutations described worldwide in the X-linked G6PD gene. G6PD mutations lead to a range of enzyme deficiency phenotypes as classified by the World Health Organization, characterized by decreased enzyme stability and/or reduced G6PD enzyme activity. Indeed, G6PD deficiency is the most common human enzymopathy, affecting >500 million people worldwide. It has been hypothesized that G6PD deficiency confers a selective advantage against malaria, as there is substantial overlap of G6PD deficiency prevalence in regions of historical malaria-endemicity (1), although experimental evidence supporting this malaria protection hypothesis remains mixed (2–6). In the red blood cell (RBC), where G6PD deficiency has best been studied, and where the trait is most prominently manifested, oxidant stressors—e.g., infection, diet, and medications—can trigger life-threatening hemolytic crises and anemia. Beyond RBC biology and malaria, investigators have also examined the function of G6PD in immunity. Here, we review the role(s) of G6PD activity and the effects of G6PD deficiency on immune system function, in health and disease.
2 Importance of G6PD for innate immunity
G6PD and the PPP are crucial for innate immune system function. Key phagocytes, such as neutrophils and macrophages, use reactive oxygen species (ROS) and reactive nitrogen species (RNS) to kill pathogens in phagolysosomes. To generate sufficient quantities of ROS and RNS quickly, these cells require rapid flux through the PPP to produce sufficient amounts of NADPH to enable superoxide formation from oxygen by the action of NADPH oxidase (NOX), and nitric oxide (NO) formation from arginine by inducible nitric oxide synthase (iNOS) (Figure 1A-2). The required rapid flux through the PPP necessitates sufficient G6PD enzyme activity capacity to allow this respiratory burst to occur. Given that NOX and NOS activity in neutrophils and macrophages depend upon G6PD-derived NADPH and reduced glutathione (GSH) (7), the role of G6PD and the PPP has been studied in these cell types, and roles for G6PD activity have been identified even beyond the oxidative burst.
2.1 G6PD and PPP function in neutrophils
As the most common circulating blood leukocyte, neutrophils are crucial for monitoring pathogens and tissue damage. Soon after stimulation by an appropriate trigger—e.g., chemokines/cytokines, pathogen components like lipopolysaccharide (LPS), and/or wounded tissue—neutrophils migrate to areas of injury and/or infection and mount an initial innate immune response which requires a rapid metabolic switch dependent on G6PD. Minutes after initial stimulation, neutrophils switch from primarily using glycolysis for basal metabolism, to the PPP, in order to allow for rapid generation of NADPH (8).
Brisk production of NADPH through the PPP is itself linked to sufficient basal and reserve capacity of G6PD enzyme activity (Figure 1A). The NADPH generated allows for biosynthesis of effector molecules (Figure 1A-3), as well as rapid generation of ROS within phagolysosomes (Figure 1A-2). This was first demonstrated by finding that chemical inhibition of the neutrophil oxidative burst exhibited PPP and G6PD dependence (9). Indeed, several distinct stimuli, each with different receptors and mechanisms of action, have been found to induce the neutrophil respiratory burst and pentose cycling ex vivo (8). This G6PD-dependent rapid metabolic switch allows several crucial neutrophil activities to occur (Figure 1B-1): phagocytosis, release of neutrophil extracellular traps (NETs), and degranulation accompanied by rapid production of large amounts of extracellular ROS. Indeed, neutrophil activation and degranulation, including extrusion of NETs via NETosis, are all PPP- and ROS-dependent processes, generated by NOX activity (8, 10, 11).
In particular, neutrophil NETosis is a critical neutrophil effector function, which releases chromatin and granule proteins into the extracellular space to entangle and kill bacteria (12). In activated neutrophils, NET formation occurs in response to pathogen stimuli and pro-inflammatory signaling and depends on superoxide generation by NADPH oxidase (Figure 1A-2). The rapid production of ROS in activated neutrophils, which requires increased pentose cycling, and thus increased G6PD activity, acts to promote NETosis via oxidative inhibition of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (13); this process can even be pathogenic when excessive, as in hyperinflammatory states such as severe acute respiratory syndrome coronavirus 2 (SARS−CoV−2) infection (14).
Overall, the uniquely rapid need for neutrophil effector functions, such as NETosis, phagocytosis and degranulation, to be mobilized quickly in the innate immune response necessitates brisk production of NADPH, which is chiefly controlled by the PPP rate-limiting activity of G6PD.
2.2 G6PD function in macrophages
G6PD activity is also critical in macrophage function. Similar to neutrophils, rapid production of NADPH is needed for phagocytosis, and the oxidative burst in activated macrophages. Sufficient reserve capacity for G6PD activity allows for rapid production of excess NADPH needed with macrophage activation. This G6PD-dependent production of NADPH is then used for biosynthesis of effector molecules (Figure 1A-3), as well as ROS/RNS production (via NOX, iNOS) needed for phagocytosis and the oxidative burst (Figure 1A-2).
Beyond the phagocytic respiratory burst, G6PD acts more generally as a key regulator of cellular redox homeostasis and provides substrate for NADPH oxidase by generating NADPH, thus exerting an innate immune modulatory role in these capacities. While some ROS generation, produced via NADPH-dependent NOX and iNOS, is advantageous and necessary for pathogen clearance, it must be carefully titrated by antioxidant GSH – which is itself regulated by NADPH-dependent glutathione reductase; as both processes are NADPH-dependent, both processes depend on G6PD activity (Figure 1A-2). With respect to redox homeostasis, ROS induction has numerous downstream effects. For instance, excess ROS leads to activation of nuclear factor erythroid 2-related factor 2 (NRF2) and upregulation of heme oxygenase (HMOX), which can dampen inflammation and protect against immunopathology more generally. As an example, immunopathologic vasculopathy in a hypoxic model of pulmonary hypertension was found to be driven by G6PD-induced pro-inflammatory epigenetic signaling — including upregulated expression of pro-inflammatory factors (e.g., tumor necrosis factor-alpha [TNF-α]), increased numbers of activated macrophages, and activated platelets — and conversely, was able to be mitigated with G6PD deficiency (15, 16).
G6PD-dependent ROS generation is also important in innate immune sensing by macrophages (Figure 1B-1). Nucleotide-binding oligomerization domain (Nod)-like receptor (NLR) signaling via pattern recognition receptors also depends on PPP flux, which can affect intracellular sensing by macrophages of pathogens, such as Aspergillus (17). Priming the canonical NLR family pyrin domain containing 3 (NLRP3) inflammasome also depends on ROS. Indeed, G6PD-deficient monocytes exhibit decreased inflammasome responsiveness to LPS, including poor induction of pro-interleukin-1β (IL-1β), which has been linked to insufficient NADPH oxidase production of ROS and impaired intracellular bacterial killing (18). This impaired inflammasome responsiveness in G6PD deficiency may also in part explain the decreased body temperatures seen in G6PD-deficient patients with malaria (19).
Furthermore, systemic inflammatory response signals and their downstream propagation is dependent on G6PD expression and activity in macrophages (Figure 1B-1). As an example, in response to gram-negative LPS, TNF-α induces upregulation of macrophage G6PD expression and activity (20), and increased G6PD expression has been visualized by ultrastructural techniques in activated macrophages (21). By contrast, G6PD-deficient mice have a diminished innate response to LPS, with impaired IL-6 production (22). Macrophage-derived G6PD activity also links the innate and acquired immune system, with ROS upregulating immunoresponsive gene 1 (IRG1) and major histocompatibility complex I (MHC I) expression in activated macrophages in a PPP- and NOX-dependent manner (23).
2.3 Effect of G6PD deficiency on innate immune system function and disease
Given the role of G6PD in phagocyte effector function, in the 1960s, it was theorized that G6PD deficiency might impact susceptibility to sepsis and meningitis (24). In the most severe variants of G6PD deficiency, e.g., those leading to chronic non-spherocytic hemolytic anemia and chronic granulomatous disease (CGD), it was observed that leukocyte G6PD activity and PPP flux were markedly reduced (25, 26), leading to diminished oxidative burst and impaired bacterial killing in phagolysosomes (27). Moreover, in these sporadic cases of severe G6PD deficiency with CGD, PPP flux is markedly diminished to the point of impaired NADPH oxidase activity and absent NETosis (28), and the impaired phagocytic respiratory burst was clinically linked to cases of mycobacterial infection (29). Beyond the severe deficiency variants, it was also noted that there was reduced G6PD activity in leukocytes isolated from individuals with moderate G6PD deficiency such as those with the common Mediterranean variant (30–34).
Clinically, there is much interest in understanding the role that G6PD deficiency and an associated aberrant innate immune effector response might play in childhood sepsis. For example, G6PD activity is markedly diminished in neutrophils isolated from neonates, with variable reports of deficient phagocytosis and chemotaxis and impaired bactericidal killing (35, 36). Indeed, a case-control study of 76 neonates with sepsis and 1214 healthy neonates identified a significantly increased prevalence of G6PD deficiency in those neonates specifically admitted for sepsis (37). In G6PD-deficient infants in Saudi Arabia, where the Mediterranean variant predominates, an increased risk of severe infection and sepsis was seen in childhood, especially with catalase-positive organisms (38, 39). In Kenya, where the milder G6PD A- deficiency variant predominates, large case-control studies in children reported an increased risk for bacteremia, especially with gram positive organisms such as Staphylococcus aureus and Streptococcus pneumoniae (40, 41). Increased risk has also been reported for other intracellular pathogens, including Salmonella typhi [typhoid fever (42, 43)], Rickettsia rickettsia [Rocky Mountain spotted fever (44, 45)], and Toxoplasma gondii [toxoplasmosis (46)]. The added impact of G6PD deficiency may be even more pronounced in settings of more severe immune compromise; for example, patients with acute myelogenous leukemia and concomitant G6PD deficiency exhibit increased risk for invasive fungal infections (47).
Apart from serious bacterial infection, other stressors can activate a brisk systemic innate immune response, including trauma and severe coronavirus disease 2019 (COVID-19). In one study of African-American trauma patients, those with G6PD A- deficiency were at increased risk for prolonged systemic inflammatory response syndrome, septicemia, and respiratory and wound infections (48). The hyperinflammation associated with severe COVID-19 infection has been linked to an overly exuberant innate immune phenotype with excessive macrophage activation (49) that could plausibly be impacted by G6PD deficiency. While genome-wide association studies of COVID-19 have not specifically linked G6PD polymorphic variants to severe illness (50), several retrospective studies have suggested that patients with G6PD deficiency were at increased risk of hospitalization and severe disease (51, 52), including acute respiratory distress syndrome with associated ventilator dependence (53).
3 G6PD in adaptive immunity
Adaptive immunity, specifically the effector arms of T and B cell activation, are also dependent on G6PD and PPP activity. Over 50 years ago, it was first noted that G6PD activity increases in lymphocytes activated by phytohemagglutinin [PHA] (54). More recently, several groups interested in cancer biology have detailed a variety of cell-type specific lymphocyte functions that are impacted by G6PD. In particular, these recent efforts have shown that activated T cells depend on G6PD and PPP activity for fundamental processes related to effector function, ranging from metabolic reprogramming and ROS homeostasis, to biosynthesis of effector molecules and signal transduction (Figure 1B-2).
In activated T cells, metabolic fitness and synthetic function has been directly linked to G6PD activity and ROS homeostasis by Ghergurovich and colleagues, who used a novel metabolite reporter and deuterium tracer assays to monitor cellular G6PD activity in the presence of a specific potent inhibitor of G6PD (55). In activated T cells, flux through the oxidative PPP increases nearly 10-fold, but chemical inhibition of G6PD can markedly decrease this and, as a result, impair T cell activation and decrease production of pro-inflammatory interferon-gamma (IFN-γ) (55). This dependence on G6PD activity in activated T cells occurs despite the partially redundant contribution of other enzymes like isocitrate dehydrogenase and malate dehydrogenase to the overall production of cellular NADPH, which is needed for biosynthesis of immune effector molecules (Figure 1A-3). In addition, G6PD has a critical role, along with NFKB-inducing kinase (NIK), in supporting metabolism in activated T cells (56). In particular, activated T cells maintain aerobic glycolytic flux by stabilizing hexokinase 2, which requires control of ROS via G6PD and the PPP (Figure 1A-2). G6PD-deficient activated T cells exhibit diminished glycolytic metabolism and reduced cytokine production (55), and cytokine-induced killer cells likewise depend on the PPP for effector function and proliferation (57).
Beyond metabolism, G6PD activity plays a role in T cell signal transduction and cytolytic function (Figure 1B-2). In early T cell receptor activation, intracellular calcium signaling is dependent on G6PD and dual oxidase 1 (DUOX1) enzyme activity, which work in concert to facilitate NADP/NADPH cycling (58). In cytotoxic T lymphocytes, G6PD activity has also been linked epigenetically to granzyme B production and cellular lytic function via acetyl coenzyme A synthesis and histone acetylation (59).
ROS homeostasis, which is highly dependent on G6PD activity (Figure 1A-2), is also critical for T cell function, including maturation and activation (60). In particular, ROS affects T cell receptor signaling through several downstream signaling pathways (57, 61, 62), including mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinases (PI3K), and Janus kinase-signal transducer and activator of transcription 3 (JAK-STAT3). Some amount of ROS is critical for normal T cell survival and maturation, and appropriate physiologic upregulation of ROS levels is needed for T cell receptor activation and downstream signaling that leads to T helper cell type 1 (Th1) differentiation and production of pro-inflammatory cytokines, such as IFN-γ and TNF-α (63). However, too much ROS can lead to excess Th1 activation and promote a pro-inflammatory state. Indeed, in high metabolic states, T cells depend on higher levels of G6PD activity to maintain homeostasis by mitigating ROS. For example, in T-lymphoblastic leukemia cells, inhibition of mammalian target of rapamycin (mTOR) signaling downregulates G6PD, resulting in excess ROS and cell death (64).
In B cells, basal G6PD expression and activity are low, suppressed by transcription factors paired box protein 5 (PAX5) and Ikaros family zinc finger protein 1 (IKZF1). However, in states of activation, including oncogenesis, B cell activation depends on G6PD and PPP activity to mitigate oxidative stress (65), and the hypermetabolic leukocytes of acute B-cell leukemia are likewise more sensitive to oxidant stressors, such as dihydroartemisinin (66).
4 The effect of G6PD in other cell types important for immunity
Beyond leukocytes, it is relevant to consider other cell types that contribute to innate immune activation and systemic inflammatory responses, including red blood cells, platelets, hepatocytes, and endothelial cells. These cell types are also impacted by G6PD and PPP activity with respect to ROS homeostasis and biosynthesis of effector molecules, and this can impact their downstream contribution to systemic inflammation.
4.1 Red blood cells
G6PD is a key regulator of oxidant stress in erythrocytes, critical to protection against membrane stress and hemolysis, which is a potent systemic inflammatory stimulus (Figure 1B-3). For example, free heme triggers macrophage Toll like receptor 4 (TLR4) activation and TNF-α production, as well as ROS-mediated NLRP3 inflammasome induction (67–70). Additionally, increased serum iron levels caused by frequent hemolysis may play a role in immune modulation, as several studies have linked hyperferremia with serious infection in humans, a concept termed “nutritional immunity” (71–74). Beyond hemolysis, recent evidence suggests a role for microvesicle formation and mitochondrial retention in oxidatively-stressed RBCs in potentiating immune activation (75, 76). Such oxidatively stressed RBCs then serve to facilitate macrophage activation by binding macrophage Fc receptors; indeed, with respect to malaria protection, it has been theorized that protection conferred by G6PD deficiency may be related to increased phagocytosis of parasitized oxidatively-damaged G6PD-deficient RBCs (77–79) via binding by macrophage Fcγ and lectin-like receptors (80), and that such parasites may be more susceptible to ROS (81).
4.2 Platelets
Platelets are another key participant in the innate immune response. They act as sensors, mediators, and direct effectors, and platelets promote endothelial activation, leukocyte recruitment, systemic inflammation, and pathogen neutralization. Platelets recruit and activate cells in both innate and adaptive immune responses and also secrete antimicrobial peptides (82, 83). Platelets directly sense pathogen- and damage-associated signals via TLRs and other receptors (84). Platelets also possess complement and cytokine/chemokine receptors, in addition to producing and releasing pro-inflammatory cytokines upon activation (85). Once activated, platelets secrete various immunomodulatory and microbicidal factors stored in their alpha-granules, including defensins, thrombocidins, kinocidins, and other antimicrobial peptides (85). Platelets can bind and be activated by damage and pathogen signals, including viral particles and bacterial surface proteins (86); in addition, platelet degranulation can promote direct bactericidal activity and enhance macrophage clearance (87). As a testament to their key roles in infection and systemic inflammation, it is noteworthy that pro-inflammatory cytokines, such as IL-6, regulate platelet production (88).
Thus, although platelets are less well studied in G6PD deficiency, they are also relevant for immune function (Figure 1B-4). As an example, platelets from G6PD-deficient individuals exhibit reduced G6PD enzymatic activity (30), affecting the availability of reduced glutathione required for platelet aggregation (89) and of NADPH needed for biosynthesis of fatty acids and phospholipids (90) (Figure 1A-3). Additionally, activated platelets require a switch to aerobic glycolysis, which increases PPP flux; a process that can be abrogated via chemical inhibition of G6PD (91).
4.3 Liver
G6PD function in the liver is relevant to systemic immunity, given the liver’s key role in producing relevant plasma proteins, including acute phase reactants, cytokines, and complement factors (Figure 1B-5). The liver also amplifies innate immune activation by producing mediators, such as IL-6 and C-reactive protein (CRP), and liver dysfunction results in a relative immunodeficient state (92). Hepatic G6PD enzymatic activity is diminished in G6PD-deficient individuals (93), can be altered in non-deficient individuals by exogenous factors, such as insulin and high fat diets (94, 95), and can be upregulated to meet increased demand in states of physiologic stress. For example, in G6PD-deficient individuals, residual hepatic enzyme activity can prove insufficient in stress states where increased G6PD activity is required, as in neonates with comorbid G6PD deficiency and Gilbert’s syndrome, who are at increased risk for hyperbilirubinemia (96, 97). As a corollary, in response to oxidant stress, G6PD-deficient hepatocytes exhibit transcriptomic network perturbations in redox pathways and immune response pathways (98). Clinically, G6PD deficiency has been linked to increased risk of Hepatitis A and B infections (99, 100), which may be related to deficient interferon responses in hepatocytes due to insufficient ROS generation by NADPH oxidase, as has been described for Dengue virus infection in G6PD-deficient monocytes (101).
4.4 Endothelium and epithelium
Tissue barrier cells, including epithelial and endothelial cells, also play key roles in host-pathogen interactions and immunopathology (Figure 1B-6). In G6PD-deficient individuals, epithelial and mesenchymal cells such as fibroblasts and lenses, exhibit decreased enzyme activity (102, 103). In the endothelium, G6PD activity plays a key role in mediating endothelial cell homeostasis and VEGF-mediated angiogenesis (104), as depletion of enzyme activity is associated with decreased endothelial NOS activity and decreased NO bioavailability. In response to systemic inflammatory factors (e.g., ROS, LPS-mediated TLR4 signaling, TNF-α), G6PD undergoes compensatory upregulation in the endothelium (20, 105). This is likely to be an issue in G6PD-deficient individuals because an inability to maintain endothelial homeostasis has been linked to both acute and chronic inflammatory endothelial activation and has relevance to pathologies as diverse as cardiovascular disease and sepsis. Less well studied, but equally intriguing, is the effect of G6PD deficiency on airway epithelial barrier function. For example, G6PD activity and PPP flux were found to be essential for innate immune sensing of Aspergillus infection by Nod-like receptors in airway epithelial cells (17).
5 Discussion
The G6PD story is one of variance: just as inherited levels of enzymatic activity vary across the geographic spectrum, so too do they vary across the spectrum of health and disease. G6PD is an acutely responsive sensor of cellular and metabolic stress, which is modulated by a myriad of factors, including diet and environment. In leukocytes, G6PD activity exhibits diurnal variation (106), is reduced in states of malnutrition (107), and is upregulated during bacterial infection (108). Beyond infection, G6PD may also play a role in the immunopathology of autoimmune disease. As examples, emerging evidence links increased G6PD activity and PPP flux to hyper-functional dendritic cells in aplastic anemia (109), as well as pro-inflammatory activated T cells in rheumatoid arthritis (110); still other reports link G6PD deficiency to a range of immune phenotypes (111), including celiac disease (112) and asthma (113). Finally, G6PD may play a role in immunosenescence, given that G6PD activity in lymphocytes declines with age (114, 115).
Indeed, given the ubiquity of G6PD in immunologically-relevant pathways and processes, it is clear that more research is needed to improve our understanding of the immunologic role of G6PD and G6PD deficiency in health and disease. At a macroscopic level, by considering the population genetics of the world’s most common enzymopathy, it is worth asking anew what the beneficial and harmful roles of G6PD deficiency are in immune function that might affect evolutionary fitness in the face of infectious and environmental stressors. Common functional variants at G6PD have independently arisen on diverse genetic backgrounds across a wide geographic range and exhibit variable penetrance with respect to enzyme activity; thus, the resulting immunologic phenotypes also likely vary considerably across populations. How might such variation differentially affect cellular and immune homeostasis and the body’s response to immunopathologic stressors, including innate and adaptive immune responses to severe infection? Could G6PD play a role in response to vaccination or in predispositions to autoimmunity? Ideally, answers to such questions will help paint a clearer picture of the role of this interesting enzyme in immune function and immunopathology, and might someday allow physicians to harness immunometabolic diagnostic and therapeutic tools to improve outcomes in severe sepsis and various immune disorders.
Author contributions
SS: Writing – original draft, Writing – review & editing. ES: Writing – original draft, Writing – review & editing. RF: Writing – original draft, Writing – review & editing. MK: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institutes of Health (5R01HL148151).
Acknowledgments
We thank Steven Spitalnik for his thoughtful critiques and suggestions to improve our manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tishkoff SA, Varkonyi R, Cahinhinan N, Abbes S, Argyropoulos G, Destro-Bisol G, et al. Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science. (2001) 293:455–62. doi: 10.1126/science.1061573
2. Bienzle U, Ayeni O, Lucas AO, Luzzatto L. Glucose-6-phosphate dehydrogenase and malaria. Greater resistance of females heterozygous for enzyme deficiency and of males with non-deficient variant. Lancet. (1972) 1:107–10. doi: 10.1016/S0140-6736(72)90676-9
3. Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. (1995) 376:246–9. doi: 10.1038/376246a0
4. Guindo A, Fairhurst RM, Doumbo OK, Wellems TE, Diallo DA. X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PloS Med. (2007) 4:e66. doi: 10.1371/journal.pmed.0040066
5. Malaria Genomic Epidemiology Network. Malaria Genomic Epidemiology Network. Reappraisal of known malaria resistance loci in a large multicenter study. Nat Genet. (2014) 46:1197–204. doi: 10.1038/ng.3107
6. Clarke GM, Rockett K, Kivinen K, Hubbart C, Jeffreys AE, Rowlands K, et al. Characterisation of the opposing effects of G6PD deficiency on cerebral malaria and severe malarial anaemia. Elife. (2017) 6:e15085. doi: 10.7554/eLife.15085
7. Vogt MT, Thomas C, Vassallo CL, Basford RE, Gee JB. Glutathione-dependent peroxidative metabolism in the alveolar macrophage. J Clin Invest. (1971) 50:401–10. doi: 10.1172/JCI106507
8. Britt EC, Lika J, Giese MA, Schoen TJ, Seim GL, Huang Z, et al. Switching to the cyclic pentose phosphate pathway powers the oxidative burst in activated neutrophils. Nat Metab. (2022) 4:389–403. doi: 10.1038/s42255-022-00550-8
9. Strauss RR, Paul BB, Sbarra AJ. Effect of phenylbutazone on phagocytosis and intracellular killing by guinea pig polymorphonuclear leukocytes. J Bacteriol. (1968) 96:1982–90. doi: 10.1128/jb.96.6.1982-1990.1968
10. Azevedo EP, Rochael NC, Guimarães-Costa AB, de Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A Metabolic Shift toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-induced Neutrophil Extracellular Trap (NET) Formation. J Biol Chem. (2015) 290:22174–83. doi: 10.1074/jbc.M115.640094
11. Leben R, Ostendorf L, van Koppen S, Rakhymzhan A, Hauser AE, Radbruch H, et al. Phasor-Based Endogenous NAD(P)H Fluorescence Lifetime Imaging Unravels Specific Enzymatic Activity of Neutrophil Granulocytes Preceding NETosis. Int J Mol Sci. (2018) 19:1018. doi: 10.3390/ijms19041018
12. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
13. Talwar D, Miller CG, Grossmann J, Szyrwiel L, Schwecke T, Demichev V, et al. The GAPDH redox switch safeguards reductive capacity and enables survival of stressed tumour cells. Nat Metab. (2023) 5:660–76. doi: 10.1038/s42255-023-00781-3
14. Li Y, Hook JS, Ding Q, Xiao X, Chung SS, Mettlen M, et al. Neutrophil metabolomics in severe COVID-19 reveal GAPDH as a suppressor of neutrophil extracellular trap formation. Nat Commun. (2023) 14:2610. doi: 10.1038/s41467-023-37567-w
15. Hashimoto R, Gupte SA. G6PD is a critical enabler of hypoxia-induced accumulation of macrophages and platelets in mice lungs and contributor to lung inflammation. Vascul Pharmacol. (2022) 144:106976. doi: 10.1016/j.vph.2022.106976
16. Joshi SR, Kitagawa A, Jacob C, Hashimoto R, Dhagia V, Ramesh A, et al. Hypoxic activation of glucose-6-phosphate dehydrogenase controls the expression of genes involved in the pathogenesis of pulmonary hypertension through the regulation of DNA methylation. Am J Physiol Lung Cell Mol Physiol. (2020) 318:L773–86. doi: 10.1152/ajplung.00001.2020
17. Kastelberg B, Ayubi T, Tubau-Juni N, Leber A, Hontecillas R, Bassaganya-Riera J, et al. Nlrx1-Regulated Defense and Metabolic Responses to Aspergillus fumigatus Are Morphotype and Cell Type Specific. Front Immunol. (2021) 12:749504. doi: 10.3389/fimmu.2021.749504
18. Yen WC, Wu YH, Wu CC, Lin HR, Stern A, Chen SH, et al. Impaired inflammasome activation and bacterial clearance in G6PD deficiency due to defective NOX/p38 MAPK/AP-1 redox signaling. Redox Biol. (2020) 28:101363. doi: 10.1016/j.redox.2019.101363
19. Kakande E, Greenhouse B, Bajunirwe F, Drakeley C, Nankabirwa JI, Walakira A, et al. Associations between red blood cell variants and malaria among children and adults from three areas of Uganda: a prospective cohort study. Malar J. (2020) 19:21. doi: 10.1186/s12936-020-3105-3
20. Spolarics Z, Wu JX. Tumor necrosis factor alpha augments the expression of glucose-6-phosphate dehydrogenase in rat hepatic endothelial and Kupffer cells. Life Sci. (1997) 60:565–71. doi: 10.1016/S0024-3205(96)00641-8
21. Matsubara S. Glucose-6-phosphate dehydrogenase and mouse Kupffer cell activation: an ultrastructural dual staining enzyme-cytochemical study. Histochem Cell Biol. (2002) 118:345–50. doi: 10.1007/s00418-002-0430-2
22. Chandra R, Villanueva E, Feketova E, Machiedo GW, Haskó G, Deitch EA, et al. Endotoxemia down-regulates bone marrow lymphopoiesis but stimulates myelopoiesis: the effect of G6PD deficiency. J Leukoc Biol. (2008) 83:1541–50. doi: 10.1189/jlb.1207838
23. Liu X, Wu XP, Zhu XL, Li T, Liu Y. IRG1 increases MHC class I level in macrophages through STAT-TAP1 axis depending on NADPH oxidase mediated reactive oxygen species. Int Immunopharmacol. (2017) 48:76–83. doi: 10.1016/j.intimp.2017.04.012
24. Schlegel RJ, Bellanti JA. Increased susceptibility of males to infection. Lancet. (1969) 2:826–7. doi: 10.1016/S0140-6736(69)92278-8
25. Cooper MR, DeChatelet LR, McCall CE, LaVia MF, Spurr CL, Baehner RL. Complete deficiency of leukocyte glucose-6-phosphate dehydrogenase with defective bactericidal activity. J Clin Invest. (1972) 51:769–78. doi: 10.1172/JCI106871
26. Gray GR, Stamatoyannopoulos G, Naiman SC, Kliman MR, Klebanoff SJ, Austin T, et al. Neutrophil dysfunction, chronic granulomatous disease, and non-spherocytic haemolytic anaemia caused by complete deficiency of glucose-6-phosphate dehydrogenase. Lancet. (1973) 2:530–4. doi: 10.1016/S0140-6736(73)92350-7
27. Cooper MR, DeChatelet LR, McCall CE, LaVia MF, Spurr CL, Baehner RL. Leucocyte G.-6-P.D. deficiency Lancet. (1970) 1:110. doi: 10.1016/S0140-6736(70)92693-0
28. Siler U, Romao S, Tejera E, Pastukhov O, Kuzmenko E, Valencia RG, et al. Severe glucose-6-phosphate dehydrogenase deficiency leads to susceptibility to infection and absent NETosis. J Allergy Clin Immunol. (2017) 139:212–9. doi: 10.1016/j.jaci.2016.04.041
29. Khan TA, Mazhar H, Nawaz M, Kalsoom K, Ishfaq M, Asif H, et al. Expanding the clinical and genetic spectrum of G6PD deficiency: The occurrence of BCGitis and novel missense mutation. Microb Pathog. (2017) 102:160–5. doi: 10.1016/j.micpath.2016.11.025
30. Ramot B, Szeinberg A, Adam A, Sheba C, Gafni D. A study of subjects with erythrocyte glucose-6-phosphate dehydrogenase deficiency: investigation of platelet enzymes. J Clin Invest. (1959) 38:1659–61. doi: 10.1172/JCI103943
31. Chan TK, Todd D, Wong CC. Tissue enzyme levels in erythrocyte glucose-6-phosphate dehydrogenase deficiency. J Lab Clin Med. (1965) 66:937–42.
32. Mordmüller B, Turrini F, Long H, Kremsner PG, Arese P. Neutrophils and monocytes from subjects with the Mediterranean G6PD variant: effect of Plasmodium falciparum hemozoin on G6PD activity, oxidative burst and cytokine production. Eur Cytokine Netw. (1998) 9:239–45.
33. Tzortzatou-Stathopoulou F, Zannos-Mariolea L, Karayiannis P, Constantsas N, Matsaniotis N. Leucocyte glucose-6-phosphate dehydrogenase (g-6-pd) activity in g-6-pd deficient subjects. Eur J Pediatr. (1980) 135:37–9. doi: 10.1007/BF00445890
34. Schilirò G, Russo A, Mauro L, Pizzarelli G, Marino S. Leukocyte function and characterization of leukocyte glucose-6-phosphate dehydrogenase in Sicilian mutants. Pediatr Res. (1976) 10:739–42. doi: 10.1203/00006450-197608000-00009
35. Holmes B, Day N, Haseman J, Good RA. Development of bactericidal capacity and phagocytosis-associated metabolism of fetal pig leukocytes. Infect Immun. (1972) 5:232–7. doi: 10.1128/iai.5.2.232-237.1972
36. Yeung CY, Wan TS, Tam AY. Neutrophil function in glucose-6-phosphate dehydrogenase deficient neonates. J Paediatr Child Health. (1999) 35:324–5. doi: 10.1046/j.1440-1754.1999.00376.x
37. Rostami-Far Z, Ghadiri K, Rostami-Far M, Shaveisi-Zadeh F, Amiri A, Rahimian Zarif B. Glucose-6-phosphate dehydrogenase deficiency (G6PD) as a risk factor of male neonatal sepsis. J Med Life. (2016) 9:34–8.
38. Mallouh AA, Abu-Osba YK. Bacterial infections in children with glucose-6-phosphate dehydrogenase deficiency. J Pediatr. (1987) 111:850–2. doi: 10.1016/S0022-3476(87)80202-0
39. Abu-Osba YK, Mallouh AA, Hann RW. Incidence and causes of sepsis in glucose-6-phosphate dehydrogenase-deficient newborn infants. J Pediatr. (1989) 114:748–52. doi: 10.1016/S0022-3476(89)80131-3
40. Gilchrist JJ, Uyoga S, Pirinen M, Rautanen A, Mwarumba S, Njuguna P, et al. Risk of pneumococcal bacteremia in Kenyan children with glucose-6-phosphate dehydrogenase deficiency. BMC Med. (2020) 18:148. doi: 10.1186/s12916-020-01604-y
41. Uyoga S, Macharia AW, Ndila CM, Nyutu G, Shebe M, Awuondo KO, et al. Glucose-6-phosphate dehydrogenase deficiency and susceptibility to childhood diseases in Kilifi, Kenya. Blood Adv. (2020) 4:5942–50. doi: 10.1182/bloodadvances.2020003015
42. Owusu SK, Foli AK, Konotey-Ahulu FI, Janosi M. Frequency of glucose-6-phosphate-dehydrogenase deficiency in typhoid fever in Ghana. Lancet. (1972) 1:320. doi: 10.1016/S0140-6736(72)90325-X
43. Lampe RM, Kirdpon S, Mansuwan P, Benenson MW. Glucose-6-phosphate dehydrogenase deficiency in Thai children with typhoid fever. J Pediatr. (1975) 87:576–8. doi: 10.1016/S0022-3476(75)80826-2
44. Walker DH, Kirkman HN. Rocky Mountain spotted fever and deficiency in glucose-6-phosphate dehydrogenase. J Infect Dis. (1980) 142:771. doi: 10.1093/infdis/142.5.771
45. Walker DH, Hawkins HK, Hudson P. Fulminant Rocky Mountain spotted fever. Its pathologic characteristics associated with glucose-6-phosphate dehydrogenase deficiency. Arch Pathol Lab Med. (1983) 107:121–5.
46. Tabbara KF, Sharara NA, Al-Momen AK. Toxoplasmosis in a group of glucose-6-phosphate dehydrogenase deficient patients. Saudi Med J. (2001) 22:330–2.
47. Sanna M, Caocci G, Ledda A, Orrù F, Fozza C, Deias P, et al. Glucose-6-phosphate dehydrogenase deficiency and risk of invasive fungal disease in patients with acute myeloid leukemia. Leuk Lymphoma. (2017) 58:2558–64. doi: 10.1080/10428194.2017.1312666
48. Spolarics Z, Siddiqi M, Siegel JH, Garcia ZC, Stein DS, Ong H, et al. Increased incidence of sepsis and altered monocyte functions in severely injured type A- glucose-6-phosphate dehydrogenase-deficient African American trauma patients. Crit Care Med. (2001) 29:728–36. doi: 10.1097/00003246-200104000-00005
49. Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature. (2022) 606:585–93. doi: 10.1038/s41586-022-04802-1
50. Pairo-Castineira E, Rawlik K, Bretherick AD, Qi T, Wu Y, Nassiri I, et al. GWAS and meta-analysis identifies 49 genetic variants underlying critical COVID-19. Nature. (2023) 617:764–8. doi: 10.1038/s41586-023-06034-3
51. Elsea SH, Razjouyan J, Lee KM, Lynch JA, Martini S, Pandit LM. Association of Glucose-6-Phosphate Dehydrogenase Deficiency With Outcomes in US Veterans With COVID-19. JAMA Netw Open. (2023) 6:e235626. doi: 10.1001/jamanetworkopen.2023.5626
52. Israel A, Berkovitch M, Merzon E, Golan-Cohen A, Green I, Ruppin E, et al. Glucose-6-Phosphate Dehydrogenase Deficiency and Coronavirus Disease 2019. Clin Infect Dis. (2023) 77(7):972-975. doi: 10.1093/cid/ciad348
53. Youssef JG, Zahiruddin F, Youssef G, Padmanabhan S, Ensor J, Pingali SR, et al. G6PD deficiency and severity of COVID19 pneumonia and acute respiratory distress syndrome: tip of the iceberg? Ann Hematol. (2021) 100:667–73. doi: 10.1007/s00277-021-04395-1
54. Nadler HL, Dowben RM, Hsia DY. Enzyme changes and polyribosome profiles in phytohemagglutinin stimulated lymphocytes. Blood. (1969) 34:52–62. doi: 10.1182/blood.V34.1.52.52
55. Ghergurovich JM, García-Cañaveras JC, Wang J, Schmidt E, Zhang Z, TeSlaa T, et al. A small molecule G6PD inhibitor reveals immune dependence on pentose phosphate pathway. Nat Chem Biol. (2020) 16:731–9. doi: 10.1038/s41589-020-0533-x
56. Gu M, Zhou X, Sohn JH, Zhu L, Jie Z, Yang JY, et al. NF-κB-inducing kinase maintains T cell metabolic fitness in antitumor immunity. Nat Immunol. (2021) 22:193–204. doi: 10.1038/s41590-020-00829-6
57. Zhang W, Huang H, Cai H, Tan WS. Enhanced metabolic activities for ATP production and elevated metabolic flux via pentose phosphate pathway contribute for better CIK cells expansion. Cell Prolif. (2019) 52:e12594. doi: 10.1111/cpr.12594
58. Gu F, Krüger A, Roggenkamp HG, Alpers R, Lodygin D, Jaquet V, et al. Dual NADPH oxidases DUOX1 and DUOX2 synthesize NAADP and are necessary for Ca2+ signaling during T cell activation. Sci Signal. (2021) 14:eabe3800. doi: 10.1126/scisignal.abe3800
59. Lu C, Yang D, Klement JD, Colson YL, Oberlies NH, Pearce CJ, et al. G6PD functions as a metabolic checkpoint to regulate granzyme B expression in tumor-specific cytotoxic T lymphocytes. J Immunother Cancer. (2022) 10:e003543. doi: 10.1136/jitc-2021-003543
60. Kesarwani P, Murali AK, Al-Khami AA, Mehrotra S. Redox regulation of T-cell function: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. (2013) 18:1497–534. doi: 10.1089/ars.2011.4073
61. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
62. Zhang Q, Yang Z, Ni Y, Bai H, Han Q, Yi Z, et al. NF-κB and pSTAT3 synergistically drive G6PD overexpression and facilitate sensitivity to G6PD inhibition in ccRCC. Cancer Cell Int. (2020) 20:483. doi: 10.1186/s12935-020-01576-2
63. Franchina DG, Dostert C, Brenner D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. (2018) 39:489–502. doi: 10.1016/j.it.2018.01.005
64. Silic-Benussi M, Sharova E, Ciccarese F, Cavallari I, Raimondi V, Urso L, et al. mTOR inhibition downregulates glucose-6-phosphate dehydrogenase and induces ROS-dependent death in T-cell acute lymphoblastic leukemia cells. Redox Biol. (2022) 51:102268. doi: 10.1016/j.redox.2022.102268
65. Xiao G, Chan LN, Klemm L, Braas D, Chen Z, Geng H, et al. B-Cell-Specific Diversion of Glucose Carbon Utilization Reveals a Unique Vulnerability in B Cell Malignancies. Cell. (2018) 173:470–484.e18. doi: 10.1016/j.cell.2018.02.048
66. Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. (2019) 131:356–69. doi: 10.1016/j.freeradbiomed.2018.12.011
67. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. (2007) 282:20221–9. doi: 10.1074/jbc.M610737200
68. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A. (2014) 111:E4110–4118. doi: 10.1073/pnas.1405023111
69. Bolívar BE, Brown-Suedel AN, Rohrman BA, Charendoff CI, Yazdani V, Belcher JD, et al. Noncanonical Roles of Caspase-4 and Caspase-5 in Heme-Driven IL-1β Release and Cell Death. J Immunol. (2021) 206:1878–89. doi: 10.4049/jimmunol.2000226
70. Salgar S, Bolívar BE, Flanagan JM, Anum SJ, Bouchier-Hayes L. The NLRP3 inflammasome fires up heme-induced inflammation in hemolytic conditions. Transl Res. (2023) 252:34–44. doi: 10.1016/j.trsl.2022.08.011
71. Levine RL, Lemons JA. Letter: Concentrations of serum iron in relation to infection in the neonate. J Pediatr. (1975) 87:331–2. doi: 10.1016/S0022-3476(75)80626-3
72. Weinberg ED. Iron and susceptibility to infectious disease. Science. (1974) 184:952–6. doi: 10.1126/science.184.4140.952
73. Barry DM, Reeve AW. Increased incidence of gram-negative neonatal sepsis with intramuscula iron administration. Pediatrics. (1977) 60:908–12. doi: 10.1542/peds.60.6.908
74. Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol. (2012) 10:525–37. doi: 10.1038/nrmicro2836
75. Moriconi C, Dzieciatkowska M, Roy M, D’Alessandro A, Roingeard P, Lee JY, et al. Retention of functional mitochondria in mature red blood cells from patients with sickle cell disease. Br J Haematol. (2022) 198:574–86. doi: 10.1111/bjh.18287
76. Zecher D, Cumpelik A, Schifferli JA. Erythrocyte-derived microvesicles amplify systemic inflammation by thrombin-dependent activation of complement. Arterioscler Thromb Vasc Biol. (2014) 34:313–20. doi: 10.1161/ATVBAHA.113.302378
77. Allison AC, Eugui EM. A radical interpretation of immunity to malaria parasites. Lancet. (1982) 2:1431–3. doi: 10.1016/S0140-6736(82)91330-7
78. Cappadoro M, Giribaldi G, O’Brien E, Turrini F, Mannu F, Ulliers D, et al. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. (1998) 92:2527–34. doi: 10.1182/blood.V92.7.2527
79. Bussolino F, Turrini F, Arese P. Measurement of phagocytosis utilizing [14C]cyanate-labelled human red cells and monocytes. Br J Haematol. (1987) 66:271–4. doi: 10.1111/j.1365-2141.1987.tb01311.x
80. Horn S, Bashan N, Gopas J. Phagocytosis of phenylhydrazine oxidized and G-6-PD-deficient red blood cells: the role of cell-bound immunoglobulins. Blood. (1991) 78:1818–25. doi: 10.1182/blood.V78.7.1818.bloodjournal7871818
81. Yuthavong Y, Bunyaratvej A, Kamchonwongpaisan S. Increased susceptibility of malaria-infected variant erythrocytes to the mononuclear phagocyte system. Blood Cells. (1990) 16:591–7.
82. Scherlinger M, Richez C, Tsokos GC, Boilard E, Blanco P. The role of platelets in immune-mediated inflammatory diseases. Nat Rev Immunol. (2023) 23:495–510. doi: 10.1038/s41577-023-00834-4
83. Gaertner F, Massberg S. Patrolling the vascular borders: platelets in immunity to infection and cancer. Nat Rev Immunol. (2019) 19:747–60. doi: 10.1038/s41577-019-0202-z
84. Aslam R, Speck ER, Kim M, Crow AR, Bang KWA, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. (2006) 107:637–41. doi: 10.1182/blood-2005-06-2202
85. Yeaman MR. Platelets: at the nexus of antimicrobial defence. Nat Rev Microbiol. (2014) 12:426–37. doi: 10.1038/nrmicro3269
86. Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, et al. Migrating Platelets Are Mechano-scavengers that Collect and Bundle Bacteria. Cell. (2017) 171:1368–82. doi: 10.1016/j.cell.2017.11.001
87. Ali RA, Wuescher LM, Dona KR, Worth RG. Platelets Mediate Host Defense against Staphylococcus aureus through Direct Bactericidal Activity and by Enhancing Macrophage Activities. J Immunol. (2017) 198:344–51. doi: 10.4049/jimmunol.1601178
88. Hill RJ, Warren MK, Levin J. Stimulation of thrombopoiesis in mice by human recombinant interleukin 6. J Clin Invest. (1990) 85:1242–7. doi: 10.1172/JCI114559
89. Bosia A, Spangenberg P, Lösche W, Arese P, Till U. The role of the GSH-disulfide status in the reversible and irreversible aggregation of human platelets. Thromb Res. (1983) 30:137–42. doi: 10.1016/0049-3848(83)90236-0
90. Schwartz JP, Cooperberg AA, Rosenberg A. Platelet-function studies in patients with glucose-6-phosphate dehydrogenase deficiency. Br J Haematol. (1974) 27:273–80. doi: 10.1111/j.1365-2141.1974.tb06793.x
91. Kulkarni PP, Tiwari A, Singh N, Gautam D, Sonkar VK, Agarwal V, et al. Aerobic glycolysis fuels platelet activation: small-molecule modulators of platelet metabolism as anti-thrombotic agents. Haematologica. (2019) 104:806–18. doi: 10.3324/haematol.2018.205724
92. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
93. Oluboyede OA, Esan GJ, Francis TI, Luzzatto L. Genetically determined deficiency of glucose 6-phosphate dehydrogenase (type-A-) is expressed in the liver. J Lab Clin Med. (1979) 93:783–9.
94. Gozukara EM, Frolich M, Holten D. The effect of unsaturated fatty acids on the rate of synthesis of rat liver glucose-6-phosphate dehydrogenase. Biochim Biophys Acta. (1972) 286:155–63. doi: 10.1016/0304-4165(72)90101-8
95. Nakamura T, Yoshimoto K, Aoyama K, Ichihara A. Hormonal regulations of glucose-6-phosphate dehydrogenase and lipogenesis in primary cultures of rat hepatocytes. J Biochem. (1982) 91:681–93. doi: 10.1093/oxfordjournals.jbchem.a133741
96. Kaplan M, Renbaum P, Levy-Lahad E, Hammerman C, Lahad A, Beutler E. Gilbert syndrome and glucose-6-phosphate dehydrogenase deficiency: a dose-dependent genetic interaction crucial to neonatal hyperbilirubinemia. Proc Natl Acad Sci U S A. (1997) 94:12128–32. doi: 10.1073/pnas.94.22.12128
97. Kaplan M, Muraca M, Vreman HJ, Hammerman C, Vilei MT, Rubaltelli FF, et al. Neonatal bilirubin production-conjugation imbalance: effect of glucose-6-phosphate dehydrogenase deficiency and borderline prematurity. Arch Dis Child Fetal Neonatal Ed. (2005) 90:F123–127. doi: 10.1136/adc.2004.058313
98. Du G, Xiao M, Wei X, Zhou C, Li S, Cai W. Hepatic transcriptional profiling response to fava bean-induced oxidative stress in glucose-6-phosphate dehydrogenase-deficient mice. Gene. (2018) 652:66–77. doi: 10.1016/j.gene.2018.02.014
99. Zhao J, Zhang X, Guan T, Dai Q, He W, Zhang H, et al. The association between low glucose-6-phosphate dehydrogenase activity level and hepatitis B virus infection among pre-pregnant reproductive-age Chinese females. Sci Rep. (2019) 9:3865. doi: 10.1038/s41598-019-40354-7
100. Gotsman I, Muszkat M. Glucose-6-phosphate dehydrogenase deficiency is associated with increased initial clinical severity of acute viral hepatitis A. J Gastroenterol Hepatol. (2001) 16:1239–43. doi: 10.1046/j.1440-1746.2001.02611.x
101. Al-Alimi AA, Ali SA, Al-Hassan FM, Idris FM, Teow SY, Mohd Yusoff N. Dengue virus type 2 (DENV2)-induced oxidative responses in monocytes from glucose-6-phosphate dehydrogenase (G6PD)-deficient and G6PD normal subjects. PloS Negl Trop Dis. (2014) 8:e2711. doi: 10.1371/journal.pntd.0002711
102. Gartler SM, Gandini E, Ceppellini R. Glucose-6-phosphate dehydrogenase deficient mutant in human cell culture. Nature. (1962) 193:602–3. doi: 10.1038/193602a0
103. Orzalesi N, Sorcinelli R, Binaghi B. Glucose-6-Phosphate Dehydrogenase in Cataracts of Subjects Suffering from Favism. Ophthalmic Res. (1976) 8:192–4. doi: 10.1159/000264816
104. Leopold JA, Walker J, Scribner AW, Voetsch B, Zhang YY, Loscalzo AJ, et al. Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. J Biol Chem. (2003) 278:32100–6. doi: 10.1074/jbc.M301293200
105. Spolarics Z. Endotoxemia, pentose cycle, and the oxidant/antioxidant balance in the hepatic sinusoid. J Leukoc Biol. (1998) 63:534–41. doi: 10.1002/jlb.63.5.534
106. Wolach B, Ashkenazi M, Grossmann R, Gavrieli R, Friedman Z, Bashan N, et al. Diurnal fluctuation of leukocyte G6PD activity. A possible explanation for the normal neutrophil bactericidal activity and the low incidence of pyogenic infections in patients with severe G6PD deficiency in Israel. Pediatr Res. (2004) 55:807–13. doi: 10.1203/01.PDR.0000120680.47846.47
107. Selvaraj RJ, Bhat KS. Phagocytosis and leucocyte enzymes in protein-calorie malnutrition. Biochem J. (1972) 127:255–9. doi: 10.1042/bj1270255
108. Okuda K, Tadokoro I, Noguchi Y. The metabolic and phagocytic activities of leukocytes from patients receiving corticosteroid and radiation therapy, and patients with bacterial infections. Jpn J Microbiol. (1975) 19:105–13. doi: 10.1111/j.1348-0421.1975.tb00856.x
109. Liu C, Sheng W, Fu R, Wang H, Li L, Liu H, et al. Differential expression of the proteome of myeloid dendritic cells in severe aplastic anemia. Cell Immunol. (2013) 285:141–8. doi: 10.1016/j.cellimm.2013.09.007
110. Weyand CM, Goronzy JJ. Immunometabolism in the development of rheumatoid arthritis. Immunol Rev. (2020) 294:177–87. doi: 10.1111/imr.12838
111. Israel A, Schäffer AA, Berkovitch M, Ozeri DJ, Merzon E, Green I, et al. Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency and Long-Term Risk of Immune-Related diseases. medRxiv. (2023) 2023:23287616. doi: 10.1101/2023.03.23.23287616
112. Dore MP, Errigo A, Bibbò S, Manca A, Pes GM. High Frequency of Glucose-6-Phosphate Dehydrogenase Deficiency in Patients Diagnosed with Celiac Disease. Nutrients. (2022) 14:1815. doi: 10.3390/nu14091815
113. Fois A, Dore MP, Manca A, Scano V, Pirina P, Pes GM. Association between Glucose-6-Phosphate Dehydrogenase Deficiency and Asthma. J Clin Med. (2021) 10:5639. doi: 10.3390/jcm10235639
114. Rosa LF, De Almeida AF, Safi DA, Curi R. Metabolic and functional changes in lymphocytes and macrophages as induced by ageing. Physiol Behav. (1993) 53:651–6. doi: 10.1016/0031-9384(93)90169-G
Keywords: glucose-6-phosphate dehydrogenase, G6PD, immunity, immunometabolism, G6PD deficiency
Citation: Shah SS, Stone EF, Francis RO and Karafin MS (2024) The global role of G6PD in infection and immunity. Front. Immunol. 15:1393213. doi: 10.3389/fimmu.2024.1393213
Received: 04 March 2024; Accepted: 24 May 2024;
Published: 13 June 2024.
Edited by:
Giovanni Mario Pes, University of Sassari, ItalyReviewed by:
Ariel Israel, Leumit Health Care Services, IsraelRoberto Rosales-Reyes, National Autonomous University of Mexico, Mexico
Copyright © 2024 Shah, Stone, Francis and Karafin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shivang S. Shah, ss5235@columbia.edu