 Yanli Zhao
Yanli Zhao Zujuan Huang1†
Zujuan Huang1†- 1Department of Cardiovascular Medicine, Shenzhen Longhua District Central Hospital, Shenzhen, China
- 2School of Clinical Medicine, Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan, China
Vascular calcification (VC) is considered a common pathological process in various vascular diseases. Accumulating studies have confirmed that VC is involved in the inflammatory response in heart disease, and SPP1+ macrophages play an important role in this process. In VC, studies have focused on the physiological and pathological functions of macrophages, such as pro-inflammatory or anti-inflammatory cytokines and pro-fibrotic vesicles. Additionally, macrophages and activated lymphocytes highly express SPP1 in atherosclerotic plaques, which promote the formation of fatty streaks and plaque development, and SPP1 is also involved in the calcification process of atherosclerotic plaques that results in heart failure, but the crosstalk between SPP1-mediated immune cells and VC has not been adequately addressed. In this review, we summarize the regulatory effect of SPP1 on VC in T cells, macrophages, and dendritic cells in different organs’ VC, which could be a potential therapeutic target for VC.
Introduction
Vascular diseases, particularly cardiovascular and brain diseases, are the leading causes of human disease mortality. Vascular calcification (VC) is considered a common pathological process in various vascular diseases, such as diabetes (1), atherosclerosis (2, 3), vascular injury (4, 5), chronic kidney disease (CKD) (6), liver fibrosis (7), and aging (8), and is closely associated with chronic calcium-phosphate deposition in blood vessels (9, 10). Intimal calcification and medial calcification are two types of VC; the former is closely related to the infiltration of inflammatory cells, vascular inflammation, lipid deposits, hyperlipidemia, and hypertension, and the latter is associated with aging, diabetes, CKD, and arterial stiffness (11, 12). The inflammatory response plays a vital role in different VCs. Pro-inflammatory macrophages contribute to microcalcification though intimal extracellular matrix (ECM) degradation and mineralization, which are closely related to the production of IL-1β, TNFα, and IL-18. Microcalcification induces an osteoblast-like phenotype in vascular smooth muscle cells (VSMCs) and then enhances the production of inflammatory cytokines, such as IL-1β, IL-6, and osteopontin (OPN), resulting in more dense calcification in atherosclerotic plaques (13).
SPP1 (Secreted Phosphoprotein 1 or OPN), a macrophage-derived OPN, plays an important regulatory role in cardiac repair after myocardial injury and pathological cardiac hypertrophy (14). After myocardial infarction, infiltrated macrophages are found in the myocardial infarction site, and the expression of SPP1, which is derived from macrophages, increases, but SPP1 is not expressed in normal cardiac tissue (15). The data in the SPP1 knockout mouse model revealed that loss of the SPP1 gene does not affect cardiac function but results in a decrease in collagen (16). In addition, the expression of the SPP1 gene is positively correlated with cardiac hypertrophy, which causes cardiomyocyte apoptosis and fibrosis (17). Studies in vascular endothelial cells have shown that SPP1 promotes angiogenesis and endothelial migration (18, 19). Excessive SPP1 is involved in the proliferation and migration of VSMCs, leading to vascular hyperplasia (20, 21). In addition, macrophages and activated lymphocytes highly express SPP1 in atherosclerotic plaques, which promote the formation of fatty streaks and plaque development, and SPP1 is also involved in the calcification process of atherosclerotic plaques and results in heart failure (22).
Single-cell transcriptomics analysis revealed that macrophages, natural killer T cells, and T and B lymphocytes are major immune cell subsets in calcified atheromatous plaques in asymptomatic patients (23). Additionally, an increasing number of studies have suggested that SPP1 participates in different inflammatory responses to VC by regulating immune cells. TREM2hi (triggered receptor expressed on myeloid cells 2) macrophages display a unique gene signature with expression of SPP1 in mouse atherosclerotic lesions (24). Compared with those in the normal group, SPP1 is positively correlated with dendritic cells (DCs) and regulatory T cells in the calcific aortic valve disease (CAVD), but there is a negative correlation between SPP1 and M2 phenotype macrophage (25). SPP1 mediates immune cell adhesion, migration, activation, anti-apoptosis, and other biological functions, suggesting that it is a potential mediator of VC (26). Thus, this review summarized the potential function of SPP1 in VC through the regulation of immune cells.
SPP1 signaling and immune cells
Immune cell infiltration
SPP1 is regarded as a key mediator of cell adhesion and migration, and SPP1 binding to OPN receptors (integrins and CD44) promotes cell migration and effector functions (27). A recent study showed that CD4+ T cells generating SPP1 exerted a beneficial effect on controlling acute graft-versus-host disease (aGVHD) through limiting gastrointestinal pathology (a major target organ of aGVHD) in a mouse model of aGVHD (28). In addition, immune correlation analysis revealed that SPP1 expression was higher in resting CD4 memory T cells and lower in regulatory T cells in the diagnosis of biliary atresia, suggesting that SPP1 mediated CD4+ T-cell and regulatory T-cell infiltration (29) (Figure 1A). Additionally, integrated analysis of bulk and single-cell RNA sequencing data showed that SPP1 was positively associated with myeloid cell infiltration but negatively associated with CD4/CD8 cell infiltration in the prognosis of patients with hepatocellular carcinoma (30) (Figure 1A).
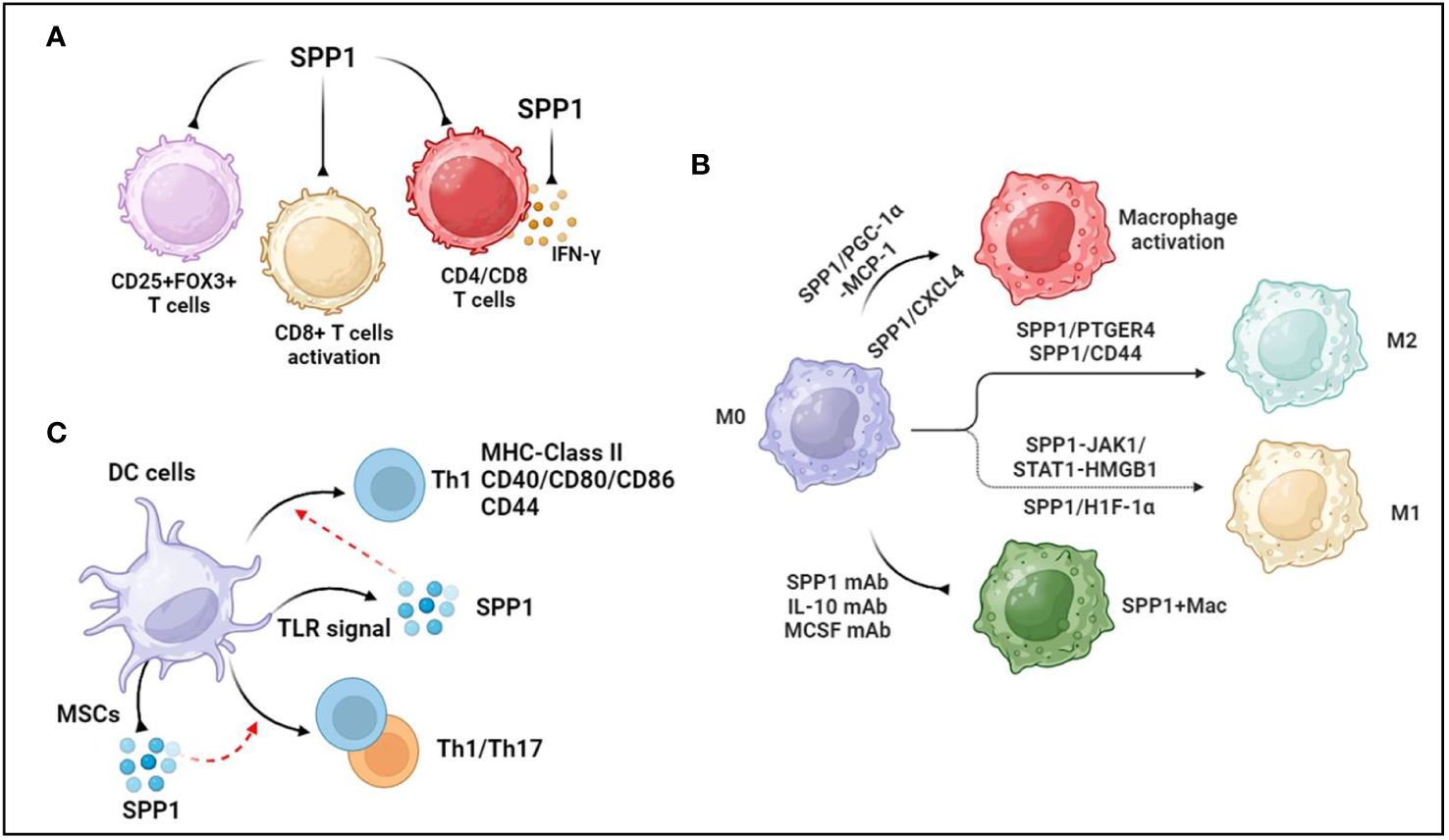
Figure 1 Association between SPP1 and immune cells. (A) SPP1 reduces regulatory T cells, inhibits CD8+T cells activation and suppresses CD4+ and CD8+ T-cell infiltration. Additionally, SPP1 inhibits IFN-γ secretion. (B) PGC-1α increases SPP1 secretion from monocytes, mediating macrophage activation and recruitment through MCP-1 expression. Platelet-derived CXC chemokine ligand 4 (CXCL4) is required for SPP1+ macrophage activation. Anti-SPP1, anti-IL-10, and anti-MCSF antibodies reduce the number of SPP1+ macrophages. During macrophage polarization, SPP1 induces the polarization of macrophages to M2-like TAMs through SPP1/CD44 and SPP1-PTGER4 signaling. SPP1 stimulates Janus kinase 1/signal transducers and activators of transcription 1 (JAK1/STAT1) signaling in hepatocytes to produce high-mobility group box 1 (HMGB1), which facilitates macrophage polarization toward the M1 phenotype. Upregulated SPP1 promotes M1 macrophage polarization through the overexpression of hypoxia-inducible factor 1α (HIF-1α). (C) TLR signaling promotes the production of SPP1 in dendritic cells. SPP1 induces DC differentiation toward the T helper 1 (Th1) phenotype, accompanied by increased MHC class II, costimulatory (CD40, CD80, and CD86), and adhesion molecule (CD44) levels. Activated mesenchymal stromal cells (MSCs) reduce the level of SPP1 generation by DCs cocultured with IL-1β, IL-6, and TNFα, but increased levels of SPP1 in CD103-DCs induce Th1 and Th17 immune cell responses during experimental colitis. The figures were generated with BioRender (https://biorender.com/).
Similarly, SPP1 is positively correlated with CD8 + cells, CD4 + cells, macrophages, neutrophils, and DCs in ovarian cancer (31). In the context of intrahepatic cholangiocarcinoma (iCCA), there were more SPP1+ macrophages that infiltrated the peripheral small duct type of S100P-SPP1 + iCCA (32). A study in OPN/SPP1 knockout mice showed that there was disorganized wound remodeling and defective macrophage infiltration after injury or infection (33). Furthermore, OPN-deficient DCs transduced with SPP1 could rescue Th17 cell generation in vivo and in vitro (34). On the other hand, intracellular OPN (iOPN) decreases the population size of myeloid progenitor cells and myeloid cells, and secreted OPN (sOPN) increases the population size of lymphoid cells (35). Supernatants of CD153+PD-1+CD4+ T cells remarkably promoted macrophage migration in a cell migration assay, which was inhibited in the presence of an anti-SPP1 antibody (36). Interestingly, OPN treatment during C. neoformans infection sharply increased the number of pulmonary eosinophils but decreased the total number of neutrophils without affecting the number of CD4+ T cells, DCs, or alveolar macrophages (37). These data demonstrate that OPN/SPP1 not only regulates immune cell infiltration but also controls immune cell differentiation.
SPP1 and T-cells
OPN inhibits the activity of cytotoxic CD8+T lymphocytes (CTLs), contributing to the progression of malignant disease (38). OPN is expressed in myeloid regulatory cells (MRCs) and malignant cells, which are two major components of the tumor microenvironment. SPP1 suppressed IFN-γ secretion by binding to CD44, which was more highly expressed in activated T lymphocytes (38) (Figure 1A). Another study showed that SPP1 decreased CD69+CD8+ T cells, CD25−CD8+ T cells, and PD-1+CD8+ T cells, suggesting that the SPP1 protein likely suppressed CD8+ T-cell activation (39) (Figure 1A). Knocking out SPP1 in colon tumor cells increased the CTL lytic activity of T cells in vitro and inhibited tumor growth in vivo, whereas the protein level of OPN increased in the peripheral blood of tumor-bearing mice (40). A recent study showed that macrophage-specific deletion of SPP1 strengthened the efficacy of anti-PD-1 treatment in liver cancer and reduced cancer-associated fibroblasts (CAFs) infiltration and increased cytotoxic T-cell infiltration (41). Additionally, SPP1 knockout expanded granulocyte-oriented myeloid-derived suppressor cells (MDSCs), which was associated with the inhibition of lung metastases (42). Further data suggested that SPP1 deletion decreased the amount of regulatory T-cell accumulation at the metastatic site.
SPP1 and macrophages
Given that SPP1 is predominantly secreted from macrophages, SPP1+ macrophages are widely recognized to regulate various diseases, such as cancers (31), cardiovascular diseases (43), tissue fibrosis (44, 45), and nonalcoholic steatohepatitis (46). Another study demonstrated that PGC-1α increased SPP1 secretion from monocytes, mediating macrophage activation and recruitment through MCP-1 expression (47) (Figure 1B). Moreover, secreted SPP1 regulates monocyte/macrophage biology (48, 49). As a case in point, upregulated SPP1 promoted macrophage polarization toward the M1 phenotype but not the M2 phenotype through the overexpression of hypoxia-inducible factor 1α (HIF-1α) (50) (Figure 1B). In colorectal cancer, SPP1+ macrophages highly express complement component 1C chain (C1QC), mannose receptor C type 1 (MRC1), signal transducer and activator of transcription 1 (STAT1), and peroxisome proliferator-activated receptor gamma (PPARG), which are associated with macrophage polarization (51). Furthermore, single-cell RNA-seq analysis demonstrated that SPP1 controlled the interaction between HCC cells and macrophages through SPP1/CD44 and SPP1-PTGER4 signaling, and in vitro data also showed that SPP1 induced the polarization of macrophages to M2-like tumor-associated macrophages (TAMs) (52) (Figure 1B). However, SPP1 stimulated Janus kinase 1/signal transducers and activators of transcription 1 (JAK1/STAT1) signaling in hepatocytes to secrete high-mobility group box 1 (HMGB1), which facilitated macrophage polarization toward the M1 phenotype (53) (Figure 1B). Nevertheless, a recent study suggested that compared with monocytes and MARK4+ macrophages, macrophages expressing both SPP1High and CXCL9High TAMs exhibit upregulated M0, M1, and M2 phenotype markers in human cancers, but the expression levels of M2 markers were higher than those of M1 markers in SPP1High TAMs (54). Thus, the roles of SPP1 in macrophage polarization should be further investigated.
In terms of tissue-resident macrophages, SPP1 is known as a key mediator of IL-10–STAT3–Galectin-3 axis signaling in cardiac macrophages after MI (15). Further study revealed a reduction in the number of SPP1-producing macrophages following the administration of the anti-IL-10 antibody alone or the coadministration of the anti-IL-10 antibody plus the anti-MCSF antibody after MI (55) (Figure 1B). Senescent fibro-adipogenic progenitors actively increased macrophage recruitment in vitro, which inhibited the recruitment of macrophages after anti-SPP1 antibody treatment (56) (Figure 1B). The data showed that SPP1 regulated macrophage recruitment during the senescence of fibro-adipogenic progenitors. Additionally, platelet-derived CXC chemokine ligand 4 (CXCL4) is required for SPP1+ macrophage activation and organ fibrosis, and an expansion in SPP1+ macrophages of patients with CKD and those with heart failure has also been found (57) (Figure 1B). IL-6 secreted from tumor enteric glial cells (EGCs) facilitates monocyte differentiation toward SPP1+TAMs (58). There was an increase in the expression level of SPP1 in silica-treated RAW264.7 macrophages (59). These data suggested that SPP1 expression was associated with cytokines and materials, thus potentially offering solutions to human diseases by regulating SPP1+ macrophages.
SPP1 and dendritic cells
DCs, known as a source of SPP1, are central mediators of the induction of T-cell immunity. SPP1 regulates the migration, differentiation, maturation, and survival of DCs (60–62). In addition, TLR signaling modulated the production of SPP1 in DCs (63) (Figure 1C). There was a significant association between SPP1 and DC markers in ovarian cancer, where SPP1 regulated DC infiltration (31). The cytokine milieu mediates the production of SPP1 in DCs, including IL-27, IFN-I, IL-1β, IL-6, and TNFα (34, 64). SPP1 induced DC differentiation toward the T helper 1 (Th1) phenotype, accompanied by increased MHC class II, costimulatory (CD40, CD80, and CD86), and adhesion molecule (CD44) levels (60) (Figure 1C). There was an increase in the level of SPP1 in CD103-DCs during experimental colitis, which induced Th1 and Th17 immune cell responses (65) (Figure 1C). Activated mesenchymal stromal cells (MSCs) reduced the level of SPP1 generation by DCs cocultured with IL-1β, IL-6, and TNFα (64) (Figure 1C). These data demonstrated that SPP1 mediated the biological functions of DCs, which promoted DC-induced immune responses in various diseases.
The association between SPP1 and immune cells in vascular calcification
Cardiovascular calcification
Valve interstitial cells undergo myofibrogenesis, osteogenic differentiation, calcification, and mineralization in CAVD, which contributes to cardiac outflow obstruction (66). In addition to common risks (aging, obesity, diabetes, hypertension, smoking, etc.), plasma lipids, inflammation, mineralization, and fibrosis contribute to CAVD (67). More interestingly, immune cells play a vital role in CAVD, and the interaction between SPP1 and these immune cells may mediate the progression of CAVD (68). There was an increase in the level of SPP1 in the heart with age, and SPP1 is known as one of the hub genes in CAVD (69). Additionally, highly expressed SPP1 was found in the cardiac valve of adult sheep and was associated with the progression of CAVD during aging (70). An increasing number of studies have demonstrated that different T-cell subsets, such as T helper cells, CTLs, regulatory T cells, memory effector T cells, and natural killer T cells, are present in the aortic valve during the development of CAVD (71, 72). Another study demonstrated that SPP1 expression in the calcific aortic valve resulted in CD4+ and CD8+ T-cell infiltration, which, in turn, accelerated CAVD (73) (Figure 2A). Cytokines such as IL-22 and chemokines (CXCL9) are derived from T cells and promote CAVD (74, 75). Interestingly, IL-22 promoted mineral deposition and osteoblastic differentiation via IL-22R1 during mineralization in human CAVD (74) (Figure 2A). Furthermore, T cells predominantly secreted OPN (28) (Figure 2A). In diabetic mice, increased SPP1 production was detected in senescent CD153+PD-1+CD44hiCD4+ T cells (76). SPP1 also induced IFN-γ and IL-17 expression in T cells but inhibited IL-10 expression in T cells and B cells (77) (Figure 2A). Consequently, there was a reduction in valve calcium deposition accompanied by reduced RUNX2 expression in response to IL-17A-neutralizing antibody treatment (78) (Figure 2A). Therefore, there was an association between T cells and SPP1 in mediating the progression of CAVD. Additionally, a recent study showed that there has been a significant increase in the expression levels of SPP1, HMOX1, and CD28 in the CAVD group (25). Further data suggested that lymphocyte counts were correlated with CAVD. However, there is little evidence to support the mechanisms by which SPP1 affects T cells in CAVD.
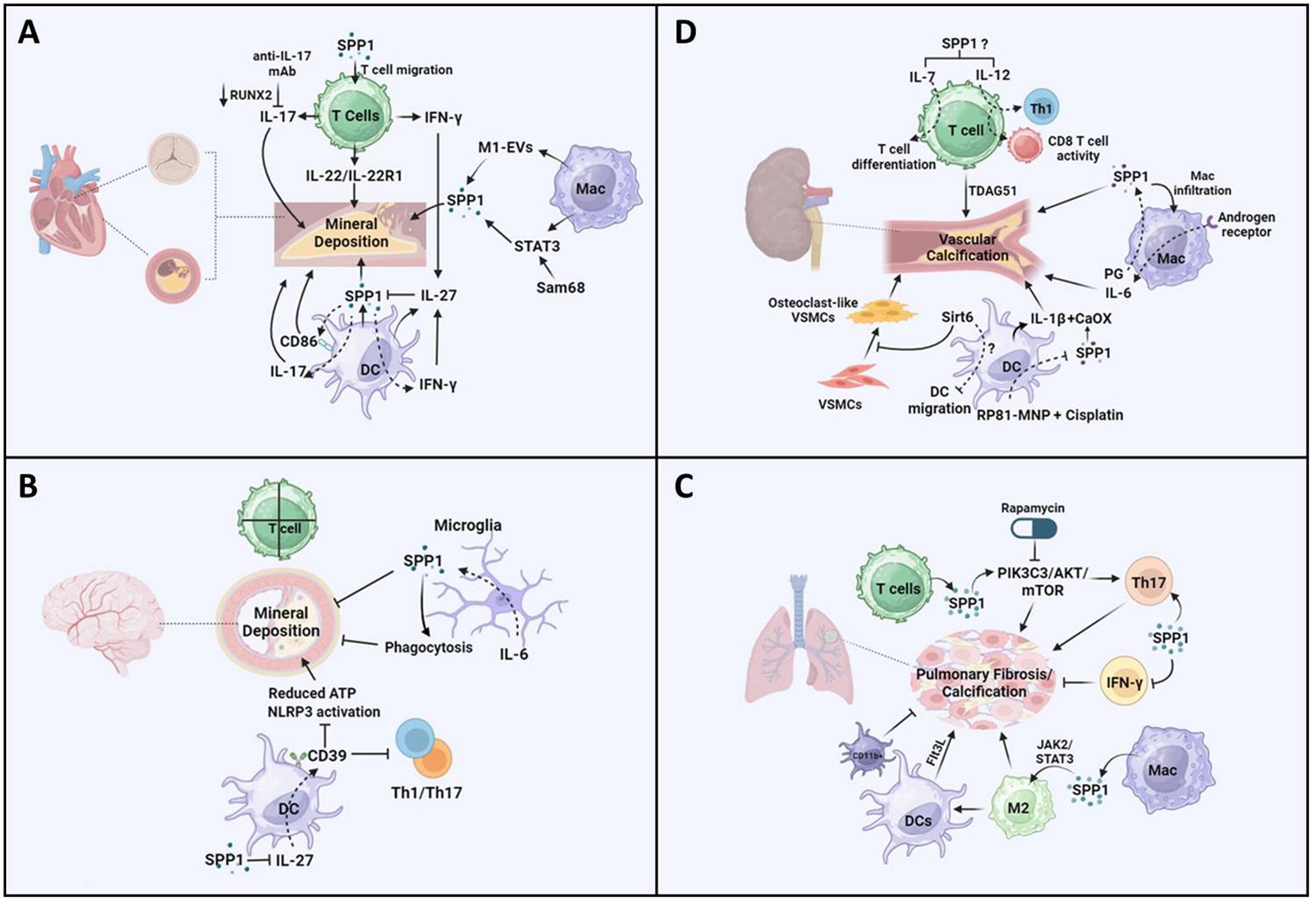
Figure 2 Potential pathways of SPP1-mediated T cells, macrophages, and dendritic cells in the progression of vascular calcification. (A) Increased production of SPP1 is detected in senescent CD153+PD-1+CD44hiCD4+ T cells in diabetic mice and consequently may induce IFN-γ/IL-17. SPP1 resulted in CD4+ and CD8+ T-cell infiltration, IL-22 promotes mineral deposition and osteoblastic differentiation via IL-22R1. There was a reduction in valve calcium deposition accompanied by reduced RUNX2 expression in response to IL-17A-neutralizing antibody treatment. Extracellular vesicles (EVs) derived from M1-polarized macrophages (M1-EVs) enhanced the expression of osteogenesis-related genes such as SPP1 and calcium nodule formation. Increased macrophage infiltration regulates STAT3 to promote osteogenic calcification. Src-associated in mitosis 68-KD (Sam68) coordinated with STAT3 in human CAV, resulting in mineral deposition via SPP1 signaling. CD86 expression has been found in DCs in coronary artery disease, and SPP1 expression is positively related to CD86. IFN-γ increased IL-27 expression and decreased SPP1 expression in DCs, and the inhibitory effect of IL-27 on SPP1 was detected. (B) There is no evidence showing that T cells induce vessel calcification in the brain. Upregulated SPP1 is detected in cerebellar microglia from the CNS-targeted production of IL-6 (GFAP-IL6 mice). Moreover, SPP1-producing microglia inhibit the progression of peripheral ectopic calcification in vivo. SPP1 suppressed the expression of IL-27 in DCs. IL-27 expression is detected in astrocytes in multiple sclerosis (MS) brains. IL-27 increases the expression of CD39 on DCs, which enhances tolerance by inhibiting Th1- and Th17-induced immune responses. There was a reduction in the concentration of ATP and activation of the NLRP3 inflammasome pathway. (C) Calcified fibrotic nodules that resorbed non-active lesions contained fewer CD3+ T cells, producing SPP1. SPP1 stimulates the PIK3C3-AKT-mTOR pathway, promoting chronic inflammation and Th17 differentiation. Rapamycin inhibits the progression of mesenchymal stromal cell calcification. Additionally, SPP1 increased the ratio of IL-17-producing T cells to IFN-γ-producing T cells, resulting in lung fibrosis. SPP1 regulates macrophage polarization toward the anti-inflammatory M2 phenotype via upregulation of the Janus kinase 2 (JAK2)/STAT3 signaling pathway, resulting in pulmonary fibrosis. Additionally, a higher expression level of SPP1 regulates macrophage polarization toward the M2 phenotype, resulting in the activation of dendritic cell infiltration in patients with lung adenocarcinoma with EGFR mutations. The expression level of FMS-like tyrosine kinase-3 ligand (Flt3L) and the number of lung DCs increased significantly during the progression of pulmonary fibrosis, but the accumulation of CD11b-positive cells inhibited lung fibrosis in a mouse model. (D) TDAG51 (T-cell death-associated gene 51) is a key predictor of vascular calcification in patients with chronic kidney disease. There is an increase in the expression of IL-7 and IL-12 in chronic kidney disease, which promotes T-cell differentiation and CD8+ cytotoxic T cells. In macrophages, there was a reduction in macrophage infiltration and kidney fibrosis in SPP1 KO mice following ischemia-reperfusion injury. Targeting the androgen receptor suppresses phosphate-induced vascular smooth muscle cell calcification by decreasing IL-6 expression. PG treatment inhibits the reduced H3K4me3 modification of SPP1. IL-1β-producing dendritic cells are related to kidney stone formation, which is associated with CaOx crystals inducing an inflammatory response. SPP1 has consistently been recognized as one of the inner cores of CaOx deposits. There was a reduction in SPP1 expression in all kidney cells treated with MNP-encapsulated RP81 (RP81-MNPs) after cisplatin activation. Furthermore, Sirt6 alleviated vascular calcification in CKD by inhibiting the osteogenic transdifferentiation of VSMCs and that Sirt6 inhibited the progression of experimental autoimmune encephalomyelitis through a reduction in DC migration. The figures were generated with BioRender (https://biorender.com/).
Macrophages mainly participate in the progression of cardiovascular diseases, including atherosclerosis (79), myocardial infarction (80), cardiac repair (81), and CAVD (82). A recent study showed that SPP1+ macrophages promoted atrial fibrillation and that the level of SPP1 in macrophages increased during atrial fibrillation (43). Furthermore, studies in CAVD have suggested that macrophages secrete SPP1, which is associated with CAVD progression (25, 83). Aortic valve interstitial cells (AVICs) internalized DiI-labeled extracellular vesicles (EVs) derived from M1-polarized macrophages (M1-EVs), which promoted the expression of osteogenesis-related genes such as SPP1 and calcium nodule formation (84) (Figure 2A). On the other hand, SPP1 mediated IL-10–STAT3–Galectin-3 axis signaling in cardiac macrophages after MI (15) (Figure 2A). A study in Notch1+/− aortic valve disease demonstrated that macrophage infiltration increased and macrophage phenotype shifted toward pro-inflammatory macrophage, resulting in decreased STAT3β, which inhibited the expression of STAT3α and RUNX2 (82). Consequently, changes in these genes led to osteogenic calcification (82). Additionally, upregulated Src-associated in mitosis 68-KD (Sam68) expression was found in human CAV, which coordinated with STAT3, resulting in mineral deposition and osteogenic differentiation (85) (Figure 2A). These results showed that SPP1-Sam68-STAT3 signaling could mediate macrophages in CAVD.
DCs are one of the main leukocyte populations in heart valve leukocytes (86). A recent integrated bioinformatics analysis revealed that CD86 was upregulated in the aortic valve stenosis (87). In fact, the expression of CD86 has been found in DCs in coronary artery disease (88) (Figure 2A). Additionally, SPP1 expression is positively related to CD86, which is associated with M2-type macrophages in colorectal cancer (41). These data suggested that SPP1-CD86 signaling could modulate DC function to affect the progression of CAVD. Furthermore, SPP1 increased IFN-γ expression and the Th17/Treg ratio in different models (50, 89), but IFN-γ and IL-17 mediated DC migration and T-cell activation through DCs (90, 91). In turn, IFN-γ alleviated IL-17-induced autoimmune inflammation by increasing IL-27 expression and decreasing SPP1 expression in DCs, and a further study indicated that IL-27 inhibited the expression of SPP1 in DCs (92) (Figure 2A). Another study discovered that IL-27R knockout contributed to the accumulation of myeloid cells and T cells, resulting in severe atherosclerosis in mice (93). However, there are few concerns in CAVD through the differential regulation of SPP1 in DCs.
Brain vascular calcification
Brain VC is largely related to aging and neurodegenerative and neuroinflammatory diseases (94). Recent studies have shown that stroke and myocardial infarction result in intracranial arterial calcifications, which are not associated with cognitive outcomes (95). Additionally, large artery stiffness causes brain vascular dysfunction, which is linked to an inflammatory response and increased oxidative stress (96). Accumulating studies have demonstrated that the expression level of SPP1 increases in various CNS disease models, which exerted an association between SPP1 and immune cells (27). Another study discovered that platelet-derived growth factor BB (PDGF-BB) phosphorylates its receptors PDGFRβ and ERK, resulting in RUNX2 activation (94). Additionally, SPP1 expression increased significantly in a platelet-derived growth factor BB (PDGF-BB)-induced brain VC model (94). SPP1 is expressed in the brain vasculature, while SPP1 is expressed in pericytes and fibroblast-like cells in the brain (97). In the context of immune cells, CD3+ T cells have been found in the surroundings of calcifications in the brain parenchyma, but there is no evidence showing that T cells induce vessel calcification in the brain (98) (Figure 2B). However, the expression of SPP1 and integrin subunit alpha X (ITGAX) is associated with cognitive decline and neuropathologies through the regulation of microglial subsets (99). Thus, further studies should investigate whether SPP1 expression during VC affects T-cell function or recruitment to promote mineral deposition during brain VC.
SPP1-producing macrophages play an important role in calcification. Monocytes differentiate into SPP1-producing macrophages via calcium, which contributes to the pro-inflammatory macrophage response (100). VCs are positively correlated with Alzheimer’s disease in elderly individuals (101). Additionally, a study in Alzheimer’s disease models suggested that SPP1 expression was required for microglial synaptic phagocytosis (102, 103) (Figure 2B). Thus, SPP1+ macrophages could modulate brain VC, thus representing a potential target for treating brain diseases in individuals of different ages. On the other hand, microglia limited calcification induced by IL-6- and IFN-α-mediated neuroinflammation, and upregulated SPP1 was detected in cerebellar microglia from CNS-targeted production of IL-6 (GFAP-IL6 mice) (104) (Figure 2B). Moreover, SPP1, which is produced in microglia, inhibited the progression of peripheral ectopic calcification in vivo (105, 106) (Figure 2B). Therefore, SPP1 may restrict calcium deposition through microglia, which could connect with the IL-6 signaling pathway.
Additionally, recent data have shown that different types of DCs have been found in cerebral ischemia, such as conventional type 1 DCs (cDC1s), conventional type 2 DCs (cDC2s), monocyte-derived DCs, migratory DCs, and plasmacytoid (103). Additional data also showed that there was an increased expression of SPP1 in four clusters of DCs after experimental stroke (103). Additionally, a previous study illustrated that SPP1 (iOPN) suppressed the expression of IL-27 in DCs but promoted that of Th17 cells (107) (Figure 2B). The expression level of IL-27 was detected in astrocytes in multiple sclerosis (MS) brains (108). Furthermore, IL-27 increased the expression of CD39 on DCs, which enhanced tolerance by inhibiting Th1- and Th17-induced immune responses (109) (Figure 2B). Further data showed that there was a reduction in the concentration of ATP and activation of the NLRP3 inflammasome pathway (109–111) (Figure 2B). These data demonstrated that secretion of SPP1 from DCs could regulate the progression of brain VC through IL-27 and CD39. Thus, more mechanisms by which SPP1 regulates DCs should be investigated in the context of brain VCs.
Pulmonary vascular calcification
Pulmonary arterial hypertension (PAH) contributes to pulmonary VC, which is associated with impaired vascular stiffness, pulmonary artery atherosclerosis, and inflammation (4, 112–114). In the pulmonary artery, there was a reduction in SPP1 expression with age (70). A recent meta-analysis study showed that there was a higher increase in the expression level of SPP1 in patients with idiopathic pulmonary fibrosis (IPF) (70). Moreover, small calcified lung nodules frequently contribute to dystrophic calcification in injured lungs (115). Calcified fibrotic nodules that resorbed non-active lesions contained fewer CD3+ T cells (116). A great deal of CD3+ and CD4+ T cells have been found in other sites, such as closed necrotic, non-necrotic cellular granulomas, and central cavities of open granulomas (116). However, in terms of the cavity surface, there were no T cells in the necrotic zone or on the cavity surface, which contributed to preventing the interaction between macrophages and T cells (116). Furthermore, the role of SPP1 in T-cell migration, adhesion, and activation has been well investigated. Thus, SPP1 could regulate T cells to control the progression of calcified fibrotic nodules. Interestingly, an increase in the expression of SPP1 was found in acid fast bacilli (AFB)-scarce lesions (116). Additionally, increasing levels of IL-15 and SPP1 contribute to decreasing the number of bacteria in AFB-scarce lesions (116). These data showed that SPP1 plays an anti-microbial role in lung diseases (116). Another study demonstrated that inhibition of SPP1 blocked the PIK3C3-AKT-mTOR pathway, resulting in the alleviation of chronic inflammation and mediating Th17/Treg differentiation in chronic obstructive pulmonary disease (COPD) (117) (Figure 2C). Furthermore, rapamycin, an mTOR inhibitor, suppressed the progression of MSC calcification (Figure 2C) (118). Additionally, SPP1 increased the ratio of IL-17-producing T cells to IFN-γ-producing T cells, resulting in lung fibrosis (119, 120) (Figure 2C). These data suggest that T cells modulate lung inflammation and calcification via SPP1 signaling pathways.
Macrophages also affected lung calcifications, which are divided into two types of tissue-resident macrophages [alveolar macrophages (AMs) or interstitial macrophages (IMs)] (121). There was an increase in the number of SPP1high macrophages in fibrotic lungs rather than FABP4high and FCN1high (122). Another study also showed that SPP1-expressing monocytes/macrophages were mainly found around microliths in patients with pulmonary alveolar microlithiasis (PAM) (123). Interestingly, SPP1 regulates macrophage polarization toward the anti-inflammatory M2 phenotype via upregulation of the Janus kinase 2 (JAK2)/STAT3 signaling pathway, resulting in pulmonary fibrosis (Figure 2C) (124). On the other hand, the expression level of MERTK is highly increased in SPP1high macrophages, which could be a potential treatment for IPF (125). MERTK was highly elevated in macrophages, promoting profibrotic effects in pulmonary fibrosis (126). These data showed that MERTKhigh SPP1high macrophages could regulate pulmonary fibrosis and consequently result in lung calcification.
Additionally, increased SPP1 expression regulated macrophage polarization toward the M2 phenotype, which resulted in the activation of DC infiltration in patients with lung adenocarcinoma with EGFR mutations (127) (Figure 2C). Additionally, the expression level of FMS-like tyrosine kinase-3 ligand (Flt3L) and the number of lung DCs increased significantly during the progression of pulmonary fibrosis in both mice and humans, but the accumulation of CD11b-positive cells inhibited lung fibrosis in a mouse model (Figure 2C) (128). Another study suggested that there was a direct association between SPP1 and KRAS mutation in lung cancer and that SPP1 deficiency had a protective effect on patients with KRAS mutation in lung cancer (129). An increased number of DC1s and eosinophils was detected in the advanced-IPF group compared with the control group or the early-IPF group, demonstrating that these immune cells may modulate the late stage of IPF (130). However, the effects of SPP1-mediated DCs on lung calcification have been less studied.
Chronic kidney diseases
VC, which injures endothelial cells and vascular smooth muscle, is an important factor that induces morbidity and mortality in patients with CKD (131). Compared with those in the normal group, calcium salts are prone to pathological deposition in the arterial wall of patients with CKD (132). Additionally, elevated extracellular phosphate, severe inflammation, and cellular senescence lead to VC in CKD patients (132). In the context of the inflammatory response, a recent study demonstrated that TDAG51 (T-cell death-associated gene 51) was a key predictor of VC in patients with CKD (133) (Figure 2D). IL-7 is a key cytokine involved in T- and B-cell development and regulates the immune response in various diseases (134) (Figure 2D). Additionally, there was an increase in the expression level of IL-7 in CKD (Figure 2D). The level of IL-12, another cytokine, was also increased in CKD, which differentiated naive T cells into Th1 subsets and promotes CD8+ cytotoxic T-cell and NK cell activity (135, 136) (Figure 2D). However, no association between SPP1 and VC via the IL-7 or IL-12 signaling pathway has been reported in CKD. In addition, microglia treated with SPP1 exhibited a decrease in IL-6 expression. sIL-6 is associated with VC and chronic inflammation in CKD (137). Thus, the effects of SPP1/IL-6 on T cells to regulate kidney VC should be investigated.
Furthermore, there was a reduction in macrophage infiltration and kidney fibrosis in SPP1 KO mice following ischemia-reperfusion injury (138) (Figure 2D). Targeting macrophage androgen receptor decreased IL-6 expression, which suppressed phosphate-induced VSMC calcification (139) (Figure 2D). Epigenetic regulation is involved in inflammation and CAD progression, and histone methyltransferase inhibitors alleviate the progression of the pro-inflammatory response by switching macrophages to foam cells (140, 141). Additionally, a study of E3 ligase von Hippel–Lindau protein (VHL)-deficient macrophages showed that 3-phosphoglyceric acid (PG) treatment inhibited the reduced H3K4me3 modification of SPP1, resulting in increased SPP1 expression (142, 143) (Figure 2D). Thus, epigenetic regulation of SPP1+ macrophages could be an effective target in VC. In the context of kidney stone disease (KSD), SPP1 was extensively associated with other hub genes among the 30 hub genes, which may be an interaction with macrophages to regulate KSD (144). Additionally, a study in OPN knockout mice with glyxylate administration suggested that OPN contributed to the formation of kidney stones, which was also associated with macrophage activity (145). During mineral deposition, SPP1 expression was detected in surrounding mineralized tissues in patients with KSD. More interestingly, large numbers of M1 and M1/M2 macrophages as well as T cells were found in the same area. However, this study did not confirm the correlation between SPP1 and immune cells related to mineral deposition or whether SPP1 recruited immune cells to plaques or SPP1+-producing macrophages promoted mineral deposition during the development of fibrosis (146). Another study demonstrated that calcium promoted monocyte differentiation into SPP1+ macrophages (100). Thus, targeting SPP1+ macrophages could be a potential therapy for the formation of kidney VC, and other regulatory mechanisms of SPP1+ macrophages should be studied in the future.
A recent study also revealed four subclusters of DC populations among kidney immune cells (147). IL-1β-producing DCs are also related to kidney stone formation, which is associated with CaOx crystals inducing an inflammatory response (148–151) (Figure 2D). Furthermore, SPP1 has consistently been recognized as one of the inner cores of CaOx deposits (152) (Figure 2D). In terms of cisplatin-induced CKD, the inflammatory response was enhanced in the RNLS-KO model, especially for those DC populations increased to fivefold compared with that in the wild type (153). Moreover, further data showed that there was a reduction in SPP1 expression in all kidney cells treated with MNP-encapsulated RP81 (RP81-MNPs) after cisplatin activation (153) (Figure 2D). However, there was no evidence that SPP1 was associated with DCs in this kind of model. Sirt6 inhibited the progression of experimental autoimmune encephalomyelitis through a reduction in DC migration (154) (Figure 2D). Furthermore, a study in SIRT6-transgenic (SIRT6-Tg) mice demonstrated that Sirt6 alleviated VC in CKD by inhibiting the osteogenic transdifferentiation of VSMCs (155) (Figure 2D). Therefore, the association between SPP1 and other signaling pathways (IL-1β and Sirt6) in DCs should be addressed more specifically in the future, which could provide an effective target for VCs.
Conclusions and future perspectives
Vascular diseases are the main cause of mortality in humans, and accumulating studies have confirmed that VC is involved in the inflammatory response in human diseases and that SPP1-mediated immune cells play an important role in the progression of VC. The specific mechanisms by which SPP1 mediates immune cells and VC should be adequately addressed, as SPP1 could be a potential therapeutic target for VC.
In recent decades, the role of the inflammatory response in VC has been well reported. However, the regulatory effects of SPP1 on immune cells in VC remain unclear. In this review, we summarize the effects of SPP1 on T cells, macrophages, and DCs to decipher the association between SPP1 and these immune cells in VC. Identifying specific regulatory mechanisms between SPP1 and immune cells is essential because these mechanisms contribute to VC in different diseases. The SPP1–cytokine–T-cell axis might be an important mediator in the progression of VC in different organs. Cytokine-neutralizing antibodies, T-cell antibodies, or mTOR inhibitors could offer potential therapeutic strategies to alleviate VC by regulating mineral deposition in patients. Moreover, SPP1+ macrophages have been identified as significant immune cells in VC. SPP1/IL-6 signaling can regulate macrophages to affect VC progression, and targeting SPP1/IL-6 signaling provides a unique method to decipher the different mechanisms involved in the formation of VC. Additionally, targeting SPP1/STAT3 pathway signaling in macrophages plays a vital role in VC development, which could mediate the progression of mineral deposition and osteogenic differentiation. On the other hand, the effects of SPP1 on DCs should be further investigated in the context of VC, given that interactions between SPP1 and DCs have been detected in diseased tissues.
Author contributions
YZ: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. ZH: Conceptualization, Data curation, Formal analysis, Methodology, Validation, Visualization, Writing – original draft. LG: Conceptualization, Data curation, Formal analysis, Methodology, Validation, Visualization, Writing – original draft. HM: Formal analysis, Validation, Visualization, Writing – original draft. RC: Project administration, Resources, Supervision, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Guangdong Medical Research Foundation, China (Grant number A2024454); the Basic and Applied Basic Research Foundation of Guangdong Province, China (Grant number 2023A1515220216); and the Scientific Research Projects of Medical and Health Institutions of Longhua District, Shenzhen, China (Grant number 2023003).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ghosh S, Luo D, He W, Chen J, Su X, Huang H. Diabetes and calcification: The potential role of anti-diabetic drugs on vascular calcification regression. Pharmacol Res. (2020) 158:104861. doi: 10.1016/j.phrs.2020.104861
2. Shioi A, Ikari Y. Plaque calcification during atherosclerosis progression and regression. J Atheroscl Thrombosis. (2018) 25:294–303. doi: 10.5551/jat.RV17020
3. Onnis C, Virmani R, Kawai K, Nardi V, Lerman A, Cademartiri F, et al. Coronary artery calcification: current concepts and clinical implications. Circulation. (2024) 149:251–66. doi: 10.1161/CIRCULATIONAHA.123.065657
4. Yuan C, Ni L, Zhang C, Hu X, Wu X. Vascular calcification: New insights into endothelial cells. Microvascular Res. (2021) 134:104105. doi: 10.1016/j.mvr.2020.104105
5. Greenberg HZE, Zhao G, Shah AM, Zhang M. Role of oxidative stress in calcific aortic valve disease and its therapeutic implications. Cardiovasc Res. (2022) 118:1433–51. doi: 10.1093/cvr/cvab142
6. Amaya-Garrido A, Brunet M, Buffin-Meyer B, Piedrafita A, Grzesiak L, Agbegbo E, et al. Calprotectin is a contributor to and potential therapeutic target for vascular calcification in chronic kidney disease. Sci Trans Med. (2023) 15:eabn5939. doi: 10.1126/scitranslmed.abn5939
7. Chang Y, Ryu S, Sung KC, Cho YK, Sung E, Kim HN, et al. Alcoholic and non-alcoholic fatty liver disease and associations with coronary artery calcification: evidence from the Kangbuk Samsung Health Study. Gut. (2019) 68:1667–75. doi: 10.1136/gutjnl-2018-317666
8. Pescatore LA, Gamarra LF, Liberman M. Multifaceted mechanisms of vascular calcification in aging. Arteriosclerosis Thrombosis Vasc Biol. (2019) 39:1307–16. doi: 10.1161/ATVBAHA.118.311576
9. Leopold JA. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med. (2015) 25:267–74. doi: 10.1016/j.tcm.2014.10.021
10. Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. (2006) 99:1044–59. doi: 10.1161/01.RES.0000249379.55535.21
11. Mori H, Torii S, Kutyna M, Sakamoto A, Finn AV, Virmani R. Coronary Artery Calcification and its Progression: What Does it Really Mean? JACC Cardiovasc Imaging. (2018) 11:127–42. doi: 10.1016/j.jcmg.2017.10.012
12. Petho AG, Tapolyai M, Browne M, Fulop T. Hypomagnesemia as a risk factor and accelerator for vascular aging in diabetes mellitus and chronic kidney disease. Metabolites. (2023) 13:306. doi: 10.3390/metabo13020306
13. Waring OJ, Skenteris NT, Biessen EAL, Donners M. Two-faced Janus: the dual role of macrophages in atherosclerotic calcification. Cardiovasc Res. (2022) 118:2768–77. doi: 10.1093/cvr/cvab301
14. Shirakawa K, Sano M. Osteopontin in cardiovascular diseases. Biomolecules. (2021) 11:1047. doi: 10.3390/biom11071047
15. Shirakawa K, Endo J, Kataoka M, Katsumata Y, Yoshida N, Yamamoto T, et al. IL (Interleukin)-10-STAT3-galectin-3 axis is essential for osteopontin-producing reparative macrophage polarization after myocardial infarction. Circulation. (2018) 138:2021–35. doi: 10.1161/CIRCULATIONAHA.118.035047
16. Trueblood NA, Xie Z, Communal C, Sam F, Ngoy S, Liaw L, et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res. (2001) 88:1080–7. doi: 10.1161/hh1001.090842
17. Subramanian V, Krishnamurthy P, Singh K, Singh M. Lack of osteopontin improves cardiac function in streptozotocin-induced diabetic mice. Am J Physiol Heart Circulatory Physiol. (2007) 292:H673–83. doi: 10.1152/ajpheart.00569.2006
18. Wang C, Ying J, Nie X, Zhou T, Xiao D, Swarnkar G, et al. Targeting angiogenesis for fracture nonunion treatment in inflammatory disease. Bone Res. (2021) 9:29. doi: 10.1038/s41413-021-00150-4
19. Wein M, Huelter-Hassler D, Nelson K, Fretwurst T, Nahles S, Finkenzeller G, et al. Differential osteopontin expression in human osteoblasts derived from iliac crest and alveolar bone and its role in early stages of angiogenesis. J Bone Mineral Metab. (2019) 37:105–17. doi: 10.1007/s00774-017-0900-1
20. Cheng J, Wu H, Xie C, He Y, Mou R, Zhang H, et al. Single cell mapping of large and small arteries during hypertensive aging. J Gerontol A Biol Sci Med Sci. (2023) 79:glad188. doi: 10.1093/gerona/glad188
21. Voelkl J, Lang F, Eckardt KU, Amann K, Kuro OM, Pasch A, et al. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci CMLS. (2019) 76:2077–91. doi: 10.1007/s00018-019-03054-z
22. Sama IE, Woolley RJ, Nauta JF, Romaine SPR, Tromp J, Ter Maaten JM, et al. A network analysis to identify pathophysiological pathways distinguishing ischaemic from non-ischaemic heart failure. Eur J Heart Failure. (2020) 22:821–33. doi: 10.1002/ejhf.1811
23. Alsaigh T, Evans D, Frankel D, Torkamani A. Decoding the transcriptome of calcified atherosclerotic plaque at single-cell resolution. Commun Biol. (2022) 5:1084. doi: 10.1038/s42003-022-04056-7
24. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
25. Zheng Y, Wen S, Jiang S, He S, Qiao W, Liu Y, et al. CircRNA/lncRNA-miRNA-mRNA network and gene landscape in calcific aortic valve disease. BMC Genomics. (2023) 24:419. doi: 10.1186/s12864-023-09441-y
26. Icer MA, Gezmen-Karadag M. The multiple functions and mechanisms of osteopontin. Clin Biochem. (2018) 59:17–24. doi: 10.1016/j.clinbiochem.2018.07.003
27. Lin EY, Xi W, Aggarwal N, Shinohara ML. Osteopontin (OPN)/SPP1: from its biochemistry to biological functions in the innate immune system and the central nervous system (CNS). Int Immunol. (2023) 35:171–80. doi: 10.1093/intimm/dxac060
28. Aggarwal N, Deerhake ME, DiPalma D, Shahi SK, Gaggioli MR, Mangalam AK, et al. Secreted osteopontin from CD4(+) T cells limits acute graft-versus-host disease. Cell Rep. (2021) 37:110170. doi: 10.1016/j.celrep.2021.110170
29. Kong M, Ma T, Xiang B. ANKRD1 and SPP1 as diagnostic markers and correlated with immune infiltration in biliary atresia. Medicine. (2021) 100:e28197. doi: 10.1097/MD.0000000000028197
30. Zheng SS, Wu YF, Zhang BH, Huang C, Xue TC. A novel myeloid cell marker genes related signature can indicate immune infiltration and predict prognosis of hepatocellular carcinoma: Integrated analysis of bulk and single-cell RNA sequencing. Front Mol Biosci. (2023) 10:1118377. doi: 10.3389/fmolb.2023.1118377
31. Gao W, Liu D, Sun H, Shao Z, Shi P, Li T, et al. SPP1 is a prognostic related biomarker and correlated with tumor-infiltrating immune cells in ovarian cancer. BMC Cancer. (2022) 22:1367. doi: 10.1186/s12885-022-10485-8
32. Song G, Shi Y, Meng L, Ma J, Huang S, Zhang J, et al. Publisher Correction: Single-cell transcriptomic analysis suggests two molecularly distinct subtypes of intrahepatic cholangiocarcinoma. Nat Commun. (2022) 13:2848. doi: 10.1038/s41467-022-30599-8
33. Rittling SR, Matsumoto HN, McKee MD, Nanci A, An XR, Novick KE, et al. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Mineral Res Off J Am Soc Bone Mineral Res. (1998) 13:1101–11. doi: 10.1359/jbmr.1998.13.7.1101
34. Shinohara ML, Kim JH, Garcia VA, Cantor H. Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: role of intracellular osteopontin. Immunity. (2008) 29:68–78. doi: 10.1016/j.immuni.2008.05.008
35. Kanayama M, Xu S, Danzaki K, Gibson JR, Inoue M, Gregory SG, et al. Skewing of the population balance of lymphoid and myeloid cells by secreted and intracellular osteopontin. Nat Immunol. (2017) 18:973–84. doi: 10.1038/ni.3791
36. Shirakawa K, Yan X, Shinmura K, Endo J, Kataoka M, Katsumata Y, et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Invest. (2016) 126:4626–39. doi: 10.1172/JCI88606
37. Hansakon A, Png CW, Zhang Y, Angkasekwinai P. Macrophage-derived osteopontin influences the amplification of cryptococcus neoformans-promoting type 2 immune response. J Immunol. (2021) 207:2107–17. doi: 10.4049/jimmunol.2100202
38. Shurin MR. Osteopontin controls immunosuppression in the tumor microenvironment. J Clin Invest. (2018) 128:5209–12. doi: 10.1172/JCI124918
39. Klement JD, Paschall AV, Redd PS, Ibrahim ML, Lu C, Yang D, et al. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J Clin Invest. (2018) 128:5549–60. doi: 10.1172/JCI123360
40. Klement JD, Poschel DB, Lu C, Merting AD, Yang D, Redd PS, et al. Osteopontin blockade immunotherapy increases cytotoxic T lymphocyte lytic activity and suppresses colon tumor progression. Cancers (Basel). (2021) 13:1006. doi: 10.3390/cancers13051006
41. Liu Y, Xun Z, Ma K, Liang S, Li X, Zhou S, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. (2023) 78:770–82. doi: 10.1016/j.jhep.2023.01.011
42. Sangaletti S, Tripodo C, Sandri S, Torselli I, Vitali C, Ratti C, et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. (2014) 74:4706–19. doi: 10.1158/0008-5472.CAN-13-3334
43. Hulsmans M, Schloss MJ, Lee IH, Bapat A, Iwamoto Y, Vinegoni C, et al. Recruited macrophages elicit atrial fibrillation. Science. (2023) 381:231–9. doi: 10.1126/science.abq3061
44. De Muynck K, Heyerick L, De Ponti FF, Vanderborght B, Meese T, Van Campenhout S, et al. Osteopontin characterizes bile duct-associated macrophages and correlates with liver fibrosis severity in primary sclerosing cholangitis. Hepatology. (2023) 79:269–88. doi: 10.1097/HEP.0000000000000557
45. Ouyang JF, Mishra K, Xie Y, Park H, Huang KY, Petretto E, et al. Systems level identification of a matrisome-associated macrophage polarisation state in multi-organ fibrosis. eLife. (2023) 12:e85530. doi: 10.7554/eLife.85530
46. Han H, Ge X, Komakula SSB, Desert R, Das S, Song Z, et al. Macrophage-derived osteopontin (SPP1) protects from nonalcoholic steatohepatitis. Gastroenterology. (2023) 165:201–17. doi: 10.1053/j.gastro.2023.03.228
47. Rowe GC, Raghuram S, Jang C, Nagy JA, Patten IS, Goyal A, et al. PGC-1alpha induces SPP1 to activate macrophages and orchestrate functional angiogenesis in skeletal muscle. Circ Res. (2014) 115:504–17. doi: 10.1161/CIRCRESAHA.115.303829
48. Duvall CL, Weiss D, Robinson ST, Alameddine FM, Guldberg RE, Taylor WR. The role of osteopontin in recovery from hind limb ischemia. Arteriosclerosis Thrombosis Vasc Biol. (2008) 28:290–5. doi: 10.1161/ATVBAHA.107.158485
49. Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. (2007) 117:2877–88. doi: 10.1172/JCI31986
50. Chen L, Yang J, Zhang M, Fu D, Luo H, Yang X. SPP1 exacerbates ARDS via elevating Th17/Treg and M1/M2 ratios through suppression of ubiquitination-dependent HIF-1alpha degradation. Cytokine. (2023) 164:156107. doi: 10.1016/j.cyto.2022.156107
51. Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z, et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat Commun. (2022) 13:1742. doi: 10.1038/s41467-022-29366-6
52. Liu L, Zhang R, Deng J, Dai X, Zhu X, Fu Q, et al. Construction of TME and Identification of crosstalk between Malignant cells and macrophages by SPP1 in hepatocellular carcinoma. Cancer Immunol Immunother CII. (2022) 71:121–36. doi: 10.1007/s00262-021-02967-8
53. Xu Z, Xi F, Deng X, Ni Y, Pu C, Wang D, et al. Osteopontin promotes macrophage M1 polarization by activation of the JAK1/STAT1/HMGB1 signaling pathway in nonalcoholic fatty liver disease. J Clin Transl Hepatol. (2023) 11:273–83. doi: 10.14218/JCTH.2021.00474
54. Bill R, Wirapati P, Messemaker M, Roh W, Zitti B, Duval F, et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science. (2023) 381:515–24. doi: 10.1126/science.ade2292
55. Shirakawa K, Endo J, Kataoka M, Katsumata Y, Anzai A, Moriyama H, et al. MerTK expression and ERK activation are essential for the functional maturation of osteopontin-producing reparative macrophages after myocardial infarction. J Am Heart Assoc. (2020) 9:e017071. doi: 10.1161/JAHA.120.017071
56. Zhang X, Ng YE, Chini LCS, Heeren AA, White TA, Li H, et al. Senescent skeletal muscle fibroadipogenic progenitors recruit and promote M2 polarization of macrophages. Aging Cell. (2023) 23:e14069. doi: 10.1111/acel.14069
57. Hoeft K, Schaefer GJL, Kim H, Schumacher D, Bleckwehl T, Long Q, et al. Platelet-instructed SPP1(+) macrophages drive myofibroblast activation in fibrosis in a CXCL4-dependent manner. Cell Rep. (2023) 42:112131. doi: 10.1016/j.celrep.2023.112131
58. Baarle Lv, Simone VD, Schneider L, Santhosh S, Abdurahiman S, Biscu F, et al. IL-1R signaling drives enteric glia-macrophage interactions in colorectal cancer. bioRxiv. (2023) 2023:06. doi: 10.1101/2023.06.01.543246
59. Huang R, Hao C, Wang D, Zhao Q, Li C, Wang C, et al. SPP1 derived from silica-exposed macrophage exosomes triggers fibroblast transdifferentiation. Toxicol Appl Pharmacol. (2021) 422:115559. doi: 10.1016/j.taap.2021.115559
60. Renkl AC, Wussler J, Ahrens T, Thoma K, Kon S, Uede T, et al. Osteopontin functionally activates dendritic cells and induces their differentiation toward a Th1-polarizing phenotype. Blood. (2005) 106:946–55. doi: 10.1182/blood-2004-08-3228
61. Schulz G, Renkl AC, Seier A, Liaw L, Weiss JM. Regulated osteopontin expression by dendritic cells decisively affects their migratory capacity. J Invest Dermatol. (2008) 128:2541–4. doi: 10.1038/jid.2008.112
62. Kawamura K, Iyonaga K, Ichiyasu H, Nagano J, Suga M, Sasaki Y. Differentiation, maturation, and survival of dendritic cells by osteopontin regulation. Clin Diagn Lab Immunol. (2005) 12:206–12. doi: 10.1128/CDLI.12.1.206-212.2005
63. Salvi V, Scutera S, Rossi S, Zucca M, Alessandria M, Greco D, et al. Dual regulation of osteopontin production by TLR stimulation in dendritic cells. J Leukocyte Biol. (2013) 94:147–58. doi: 10.1189/jlb.0412194
64. Scutera S, Salvi V, Lorenzi L, Piersigilli G, Lonardi S, Alotto D, et al. Adaptive regulation of osteopontin production by dendritic cells through the bidirectional interaction with mesenchymal stromal cells. Front Immunol. (2018) 9:1207. doi: 10.3389/fimmu.2018.01207
65. Kourepini E, Aggelakopoulou M, Alissafi T, Paschalidis N, Simoes DC, Panoutsakopoulou V. Osteopontin expression by CD103- dendritic cells drives intestinal inflammation. Proc Natl Acad Sci United States America. (2014) 111:E856–65. doi: 10.1073/pnas.1316447111
66. Blaser MC, Kraler S, Luscher TF, Aikawa E. Multi-omics approaches to define calcific aortic valve disease pathogenesis. Circ Res. (2021) 128:1371–97. doi: 10.1161/CIRCRESAHA.120.317979
67. Moncla LM, Briend M, Bosse Y, Mathieu P. Calcific aortic valve disease: mechanisms, prevention and treatment. Nat Rev Cardiol. (2023) 20:546–59. doi: 10.1038/s41569-023-00845-7
68. Broeders W, Bekkering S, El Messaoudi S, Joosten LAB, van Royen N, Riksen NP. Innate immune cells in the pathophysiology of calcific aortic valve disease: lessons to be learned from atherosclerotic cardiovascular disease? Basic Res Cardiol. (2022) 117:28. doi: 10.1007/s00395-022-00935-6
69. Sun JY, Hua Y, Shen H, Qu Q, Kan JY, Kong XQ, et al. Identification of key genes in calcific aortic valve disease via weighted gene co-expression network analysis. BMC Med Genomics. (2021) 14:135. doi: 10.1186/s12920-021-00989-w
70. Tsang HG, Clark EL, Markby GR, Bush SJ, Hume DA, Corcoran BM, et al. Expression of calcification and extracellular matrix genes in the cardiovascular system of the healthy domestic sheep (Ovis aries). Front Genet. (2020) 11:919. doi: 10.3389/fgene.2020.00919
71. Shimoni S, Bar I, Meledin V, Gandelman G, George J. Circulating regulatory T cells in patients with aortic valve stenosis: Association with disease progression and aortic valve intervention. Int J Cardiol. (2016) 218:181–7. doi: 10.1016/j.ijcard.2016.05.039
72. Raddatz MA, Madhur MS, Merryman WD. Adaptive immune cells in calcific aortic valve disease. Am J Physiol Heart Circulatory Physiol. (2019) 317:H141–H55. doi: 10.1152/ajpheart.00100.2019
73. Gong S, Xiang K, Chen L, Zhuang H, Song Y, Chen J. Integrated bioinformatics analysis identified leucine rich repeat containing 15 and secreted phosphoprotein 1 as hub genes for calcific aortic valve disease and osteoarthritis. IET Syst Biol. (2024). doi: 10.1049/syb2.12091
74. Deng H, Li H, Liu Z, Shen N, Dong N, Deng C, et al. Pro-osteogenic role of interleukin-22 in calcific aortic valve disease. Atherosclerosis. (2023) 388:117424. doi: 10.1016/j.atherosclerosis.2023.117424
75. Schlotter F, Halu A, Goto S, Blaser MC, Body SC, Lee LH, et al. Spatiotemporal multi-omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation. (2018) 138:377–93. doi: 10.1161/CIRCULATIONAHA.117.032291
76. Shirakawa K, Sano M. T cell immunosenescence in aging, obesity, and cardiovascular disease. Cells. (2021) 10:2435. doi: 10.3390/cells10092435
77. Stromnes IM, Goverman JM. Osteopontin-induced survival of T cells. Nat Immunol. (2007) 8:19–20. doi: 10.1038/ni0107-19
78. Yang Z, Zhang J, Zhu Y, Zhang C, Li G, Liu S, et al. IL-17A induces valvular endothelial inflammation and aggravates calcific aortic valve disease. Biochem Biophys Res Commun. (2023) 672:145–53. doi: 10.1016/j.bbrc.2023.04.079
79. Barrett TJ. Macrophages in atherosclerosis regression. Arteriosclerosis Thrombosis Vasc Biol. (2020) 40:20–33. doi: 10.1161/ATVBAHA.119.312802
80. Yap J, Irei J, Lozano-Gerona J, Vanapruks S, Bishop T, Boisvert WA. Macrophages in cardiac remodelling after myocardial infarction. Nat Rev Cardiol. (2023) 20:373–85. doi: 10.1038/s41569-022-00823-5
81. Rizzo G, Gropper J, Piollet M, Vafadarnejad E, Rizakou A, Bandi SR, et al. Dynamics of monocyte-derived macrophage diversity in experimental myocardial infarction. Cardiovasc Res. (2023) 119:772–85. doi: 10.1093/cvr/cvac113
82. Raddatz MA, Huffstater T, Bersi MR, Reinfeld BI, Madden MZ, Booton SE, et al. Macrophages promote aortic valve cell calcification and alter STAT3 splicing. Arteriosclerosis Thrombosis Vasc Biol. (2020) 40:e153–e65. doi: 10.1161/ATVBAHA.120.314360
83. Monzack EL, Masters KS. Can valvular interstitial cells become true osteoblasts? A side-by-side comparison. J Heart Valve Dis. (2011) 20:449–63.
84. Xia H, Gao M, Chen J, Huang G, Xiang X, Wang Y, et al. M1 macrophage-derived extracellular vesicle containing tsRNA-5006c promotes osteogenic differentiation of aortic valve interstitial cells through regulating mitophagy. PeerJ. (2022) 10:e14307. doi: 10.7717/peerj.14307
85. Liu X, Zheng Q, Wang K, Luo J, Wang Z, Li H, et al. Sam68 promotes osteogenic differentiation of aortic valvular interstitial cells by TNF-alpha/STAT3/autophagy axis. J Cell Commun Signal. (2023) 17:863–79. doi: 10.1007/s12079-023-00733-2
86. Hulin A, Anstine LJ, Kim AJ, Potter SJ, DeFalco T, Lincoln J, et al. Macrophage transitions in heart valve development and myxomatous valve disease. Arteriosclerosis Thrombosis Vasc Biol. (2018) 38:636–44. doi: 10.1161/ATVBAHA.117.310667
87. Liu YH, Liu Y, Xin YF, Zhang Q, Ding ML. Identification of key genes involved in calcific aortic valve disease based on integrated bioinformatics analysis. Exp Biol Med. (2023) 248:52–60. doi: 10.1177/15353702221118088
88. Dopheide JF, Sester U, Schlitt A, Horstick G, Rupprecht HJ, Munzel T, et al. Monocyte-derived dendritic cells of patients with coronary artery disease show an increased expression of costimulatory molecules CD40, CD80 and CD86 in vitro. Coronary Artery Dis. (2007) 18:523–31. doi: 10.1097/MCA.0b013e3282eff1ad
89. Zhao K, Zhang M, Zhang L, Wang P, Song G, Liu B, et al. Intracellular osteopontin stabilizes TRAF3 to positively regulate innate antiviral response. Sci Rep. (2016) 6:23771. doi: 10.1038/srep23771
90. Kamath AT, Sheasby CE, Tough DF. Dendritic cells and NK cells stimulate bystander T cell activation in response to TLR agonists through secretion of IFN-alpha beta and IFN-gamma. J Immunol. (2005) 174:767–76. doi: 10.4049/jimmunol.174.2.767
91. Jirmo AC, Busse M, Happle C, Skuljec J, Daluge K, Habener A, et al. IL-17 regulates DC migration to the peribronchial LNs and allergen presentation in experimental allergic asthma. Eur J Immunol. (2020) 50:1019–33. doi: 10.1002/eji.201948409
92. Murugaiyan G, Mittal A, Weiner HL. Identification of an IL-27/osteopontin axis in dendritic cells and its modulation by IFN-gamma limits IL-17-mediated autoimmune inflammation. Proc Natl Acad Sci United States America. (2010) 107:11495–500. doi: 10.1073/pnas.1002099107
93. Peshkova IO, Fatkhullina AR, Mikulski Z, Ley K, Koltsova EK. IL-27R signaling controls myeloid cells accumulation and antigen-presentation in atherosclerosis. Sci Rep. (2017) 7:2255. doi: 10.1038/s41598-017-01828-8
94. Wang J, Fang CL, Noller K, Wei Z, Liu G, Shen K, et al. Bone-derived PDGF-BB drives brain vascular calcification in male mice. J Clin Invest. (2023) 133:e168447. doi: 10.1172/JCI168447
95. Goluke NMS, de Brouwer EJM, de Jonghe A, Claus JJ, Staekenborg SS, Emmelot-Vonk MH, et al. Intracranial artery calcifications: Risk factors and association with cardiovascular disease and cognitive function. J Neuroradiol. (2022) 49:281–7. doi: 10.1016/j.neurad.2020.08.001
96. Winder NR, Reeve EH, Walker AE. Large artery stiffness and brain health: insights from animal models. Am J Physiol Heart Circulatory Physiol. (2021) 320:H424–H31. doi: 10.1152/ajpheart.00696.2020
97. Nahar K, Lebouvier T, Andaloussi Mae M, Konzer A, Bergquist J, Zarb Y, et al. Astrocyte-microglial association and matrix composition are common events in the natural history of primary familial brain calcification. Brain Pathol. (2020) 30:446–64. doi: 10.1111/bpa.12787
98. Zarb Y, Sridhar S, Nassiri S, Utz SG, Schaffenrath J, Maheshwari U, et al. Microglia control small vessel calcification via TREM2. Sci Adv. (2021) 7:eabc4898. doi: 10.1126/sciadv.abc4898
99. Lopes KP, Yu L, Shen X, Qiu Y, Tasaki S, Iatrou A, et al. Associations of cortical SPP1 and ITGAX with cognition and common neuropathologies in older adults. Alzheimers Dement. (2023) 20:525–37. doi: 10.1002/alz.13474
100. Murthy S, Karkossa I, Schmidt C, Hoffmann A, Hagemann T, Rothe K, et al. Danger signal extracellular calcium initiates differentiation of monocytes into SPP1/osteopontin-producing macrophages. Cell Death Dis. (2022) 13:53. doi: 10.1038/s41419-022-04507-3
101. Kuller LH, Lopez OL, Mackey RH, Rosano C, Edmundowicz D, Becker JT, et al. Subclinical cardiovascular disease and death, dementia, and coronary heart disease in patients 80+ Years. J Am Coll Cardiol. (2016) 67:1013–22. doi: 10.1016/j.jacc.2015.12.034
102. De Schepper S, Ge JZ, Crowley G, Ferreira LSS, Garceau D, Toomey CE, et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer's disease. Nat Neurosci. (2023) 26:406–15. doi: 10.1038/s41593-023-01257-z
103. Garcia-Bonilla L, Shahanoor Z, Sciortino R, Nazarzoda O, Racchumi G, Iadecola C, et al. Analysis of brain and blood single-cell transcriptomics in acute and subacute phases after experimental stroke. Nat Immunol. (2024) 25:357–70. doi: 10.1038/s41590-023-01711-x
104. West PK, Viengkhou B, Campbell IL, Hofer MJ. Microglia shield the murine brain from damage mediated by the cytokines IL-6 and IFN-alpha. Front Immunol. (2022) 13:1036799. doi: 10.3389/fimmu.2022.1036799
105. Steitz SA, Speer MY, McKee MD, Liaw L, Almeida M, Yang H, et al. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol. (2002) 161:2035–46. doi: 10.1016/S0002-9440(10)64482-3
106. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci Off J Soc Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
107. Clemente N, Raineri D, Cappellano G, Boggio E, Favero F, Soluri MF, et al. Osteopontin bridging innate and adaptive immunity in autoimmune diseases. J Immunol Res. (2016) 2016:7675437. doi: 10.1155/2016/7675437
108. Lalive PH, Kreutzfeldt M, Devergne O, Metz I, Bruck W, Merkler D, et al. Increased interleukin-27 cytokine expression in the central nervous system of multiple sclerosis patients. J Neuroinflamm. (2017) 14:144. doi: 10.1186/s12974-017-0919-1
109. Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. (2013) 14:1054–63. doi: 10.1038/ni.2695
110. Wang P, Zhang N, Wu B, Wu S, Zhang Y, Sun Y. The role of mitochondria in vascular calcification. J Transl Int Med. (2020) 8:80–90. doi: 10.2478/jtim-2020-0013
111. Zheng Y, Xu L, Dong N, Li F. NLRP3 inflammasome: The rising star in cardiovascular diseases. Front Cardiovasc Med. (2022) 9:927061. doi: 10.3389/fcvm.2022.927061
112. Liu SF, Nambiar Veetil N, Li Q, Kucherenko MM, Knosalla C, Kuebler WM. Pulmonary hypertension: Linking inflammation and pulmonary arterial stiffening. Front Immunol. (2022) 13:959209. doi: 10.3389/fimmu.2022.959209
113. Garg PK, Guan W, Karger AB, Steffen BT, Budoff M, Tsai MY. Lipoprotein (a) and risk for calcification of the coronary arteries, mitral valve, and thoracic aorta: The Multi-Ethnic Study of Atherosclerosis. J Cardiovasc Comput Tomogr. (2021) 15:154–60. doi: 10.1016/j.jcct.2020.06.002
114. Liu S, Kucherenko MM, Sang P, Li Q, Yao J, Veetil NN, et al. Pulmonary vascular calcification as A pathomechanism in pulmonary hypertension due to left heart disease. Eur Respir J. (2022) 60:2466. doi: 10.1164/ajrccm-conference.2022.C105
115. Brown K, Mund DF, Aberle DR, Batra P, Young DA. Intrathoracic calcifications: radiographic features and differential diagnoses. Radiographics. (1994) 14:1247–61. doi: 10.1148/radiographics.14.6.7855339
116. Subbian S, Tsenova L, Kim MJ, Wainwright HC, Visser A, Bandyopadhyay N, et al. Lesion-specific immune response in granulomas of patients with pulmonary tuberculosis: A pilot study. PloS One. (2015) 10:e0132249. doi: 10.1371/journal.pone.0132249
117. Lin H, Cheng S, Yang S, Zhang Q, Wang L, Li J, et al. Isoforskolin modulates AQP4-SPP1-PIK3C3 related pathway for chronic obstructive pulmonary disease via cAMP signaling. Chin Med. (2023) 18:128. doi: 10.1186/s13020-023-00778-w
118. Babicheva A, Makino A, Yuan JX. mTOR signaling in pulmonary vascular disease: pathogenic role and therapeutic target. Int J Mol Sci. (2021) 22:2144. doi: 10.3390/ijms22042144
119. Oh K, Seo MW, Kim YW, Lee DS. Osteopontin potentiates pulmonary inflammation and fibrosis by modulating IL-17/IFN-gamma-secreting T-cell ratios in bleomycin-treated mice. Immune Network. (2015) 15:142–9. doi: 10.4110/in.2015.15.3.142
120. Deng L, Huang T, Zhang L. Correction to: T cells in idiopathic pulmonary fibrosis: crucial but controversial. Cell Death Discovery. (2023) 9:74. doi: 10.1038/s41420-023-01375-4
121. Ogawa T, Shichino S, Ueha S, Matsushima K. Macrophages in lung fibrosis. Int Immunol. (2021) 33:665–71. doi: 10.1093/intimm/dxab040
122. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. (2019) 54:1802441. doi: 10.1183/13993003.02441-2018
123. Uehara Y, Tanaka Y, Zhao S, Nikolaidis NM, Pitstick LB, Wu H, et al. Insights into pulmonary phosphate homeostasis and osteoclastogenesis emerge from the study of pulmonary alveolar microlithiasis. Nat Commun. (2023) 14:1205. doi: 10.1038/s41467-023-36810-8
124. Hou J, Ji J, Chen X, Cao H, Tan Y, Cui Y, et al. Alveolar epithelial cell-derived Sonic hedgehog promotes pulmonary fibrosis through OPN-dependent alternative macrophage activation. FEBS J. (2021) 288:3530–46. doi: 10.1111/febs.15669
125. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. (2020) 6:eaba1983. doi: 10.1126/sciadv.aba1983
126. She Y, Xu X, Yu Q, Yang X, He J, Tang XX. Elevated expression of macrophage MERTK exhibits profibrotic effects and results in defective regulation of efferocytosis function in pulmonary fibrosis. Respir Res. (2023) 24:118. doi: 10.1186/s12931-023-02424-3
127. Zheng Y, Hao S, Xiang C, Han Y, Shang Y, Zhen Q, et al. The correlation between SPP1 and immune escape of EGFR mutant lung adenocarcinoma was explored by bioinformatics analysis. Front Oncol. (2021) 11:592854. doi: 10.3389/fonc.2021.592854
128. Tort Tarres M, Aschenbrenner F, Maus R, Stolper J, Schuette L, Knudsen L, et al. The FMS-like tyrosine kinase-3 ligand/lung dendritic cell axis contributes to regulation of pulmonary fibrosis. Thorax. (2019) 74:947–57. doi: 10.1136/thoraxjnl-2018-212603
129. Giopanou I, Kanellakis NI, Giannou AD, Lilis I, Marazioti A, Spella M, et al. Osteopontin drives KRAS-mutant lung adenocarcinoma. Carcinogenesis. (2020) 41:1134–44. doi: 10.1093/carcin/bgz190
130. Yu J, Thomas J, Haub J, Pizarro C, Zhang M, Biener L, et al. Early-stage idiopathic pulmonary fibrosis is characterized by bronchoalveolar accumulation of SPP1+ macrophages. bioRxiv. (2023) 2023:12. doi: 10.1101/2023.12.06.569201
131. Zhang YX, Tang RN, Wang LT, Liu BC. Role of crosstalk between endothelial cells and smooth muscle cells in vascular calcification in chronic kidney disease. Cell Proliferation. (2021) 54:e12980. doi: 10.1111/cpr.12980
132. Nelson AJ, Raggi P, Wolf M, Gold AM, Chertow GM, Roe MT. Targeting vascular calcification in chronic kidney disease. JACC Basic to Trans Sci. (2020) 5:398–412. doi: 10.1016/j.jacbts.2020.02.002
133. Platko K, Lebeau PF, Gyulay G, Lhotak S, MacDonald ME, Pacher G, et al. TDAG51 (T-cell death-associated gene 51) is a key modulator of vascular calcification and osteogenic transdifferentiation of arterial smooth muscle cells. Arteriosclerosis Thrombosis Vasc Biol. (2020) 40:1664–79. doi: 10.1161/ATVBAHA.119.313779
134. Perna AF, Russo L, D'Esposito V, Formisano P, Bruzzese D, Vigorito C, et al. Lanthionine, a novel uremic toxin, in the vascular calcification of chronic kidney disease: the role of proinflammatory cytokines. Int J Mol Sci. (2021) 22:6875. doi: 10.3390/ijms22136875
135. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. (2019) 176:1248–64. doi: 10.1016/j.cell.2019.01.021
136. Anderson CE, Hamm LL, Batuman G, Kumbala DR, Chen CS, Kallu SG, et al. The association of angiogenic factors and chronic kidney disease. BMC Nephrol. (2018) 19:117. doi: 10.1186/s12882-018-0909-2
137. Lopez-Mejias R, Gonzalez-Gay MA. IL-6: linking chronic inflammation and vascular calcification. Nat Rev Rheumatol. (2019) 15:457–9. doi: 10.1038/s41584-019-0259-x
138. Persy VP, Verhulst A, Ysebaert DK, De Greef KE, De Broe ME. Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int. (2003) 63:543–53. doi: 10.1046/j.1523-1755.2003.00767.x
139. Pang H, Xiao L, Lu Z, Chen H, Shang Z, Jiang N, et al. Targeting androgen receptor in macrophages inhibits phosphate-induced vascular smooth muscle cell calcification by decreasing IL-6 expression. Vasc Pharmacol. (2020) 130:106681. doi: 10.1016/j.vph.2020.106681
140. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arteriosclerosis Thrombosis Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
141. Chao CT, Kuo FC, Lin SH. Epigenetically regulated inflammation in vascular senescence and renal progression of chronic kidney disease. Semin Cell Dev Biol. (2024) 154:305–15. doi: 10.1016/j.semcdb.2022.09.012
142. Zhang W, Li Q, Li D, Li J, Aki D, Liu YC. The E3 ligase VHL controls alveolar macrophage function via metabolic-epigenetic regulation. J Exp Med. (2018) 215:3180–93. doi: 10.1084/jem.20181211
143. Cai M, Bompada P, Atac D, Laakso M, Groop L, De Marinis Y. Epigenetic regulation of glucose-stimulated osteopontin (OPN) expression in diabetic kidney. Biochem Biophys Res Commun. (2016) 469:108–13. doi: 10.1016/j.bbrc.2015.11.079
144. Hong SY, Xia QD, Xu JZ, Liu CQ, Sun JX, Xun Y, et al. Identification of the pivotal role of SPP1 in kidney stone disease based on multiple bioinformatics analysis. BMC Med Genomics. (2022) 15:7. doi: 10.1186/s12920-022-01157-4
145. Okada A, Yasui T, Hamamoto S, Hirose M, Kubota Y, Itoh Y, et al. Genome-wide analysis of genes related to kidney stone formation and elimination in the calcium oxalate nephrolithiasis model mouse: detection of stone-preventive factors and involvement of macrophage activity. J Bone Mineral Res Off J Am Soc Bone Mineral Res. (2009) 24:908–24. doi: 10.1359/jbmr.081245
146. Canela VH, Bowen WS, Ferreira RM, Syed F, Lingeman JE, Sabo AR, et al. A spatially anchored transcriptomic atlas of the human kidney papilla identifies significant immune injury in patients with stone disease. Nat Commun. (2023) 14:4140. doi: 10.1038/s41467-023-38975-8
147. Fu J, Sun Z, Wang X, Zhang T, Yuan W, Salem F, et al. The single-cell landscape of kidney immune cells reveals transcriptional heterogeneity in early diabetic kidney disease. Kidney Int. (2022) 102:1291–304. doi: 10.1016/j.kint.2022.08.026
148. Canales BK, Anderson L, Higgins L, Ensrud-Bowlin K, Roberts KP, Wu B, et al. Proteome of human calcium kidney stones. Urology. (2010) 76:1017 e13–20. doi: 10.1016/j.urology.2010.05.005
149. Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1beta secretion. J Clin Invest. (2013) 123:236–46. doi: 10.1172/JCI63679
150. Rhee E, Santiago L, Park E, Lad P, Bellman GC. Urinary IL-6 is elevated in patients with urolithiasis. J Urol. (1998) 160:2284–8. doi: 10.1097/00005392-199812010-00101
151. Suen JL, Liu CC, Lin YS, Tsai YF, Juo SH, Chou YH. Urinary chemokines/cytokines are elevated in patients with urolithiasis. Urol Res. (2010) 38:81–7. doi: 10.1007/s00240-010-0260-y
152. Mushtaq S, Siddiqui AA, Naqvi ZA, Rattani A, Talati J, Palmberg C, et al. Identification of myeloperoxidase, alpha-defensin and calgranulin in calcium oxalate renal stones. Clin Chim Acta; Int J Clin Chem. (2007) 384:41–7. doi: 10.1016/j.cca.2007.05.015
153. Guo X, Xu L, Velazquez H, Chen TM, Williams RM, Heller DA, et al. Kidney-targeted renalase agonist prevents cisplatin-induced chronic kidney disease by inhibiting regulated necrosis and inflammation. J Am Soc Nephrol JASN. (2022) 33:342–56. doi: 10.1681/ASN.2021040439
154. Ferrara G, Benzi A, Sturla L, Marubbi D, Frumento D, Spinelli S, et al. Sirt6 inhibition delays the onset of experimental autoimmune encephalomyelitis by reducing dendritic cell migration. J Neuroinflamm. (2020) 17:228. doi: 10.1186/s12974-020-01906-1
Keywords: OPN, SPP1, immune cells, vascular calcification, vascular diseases
Citation: Zhao Y, Huang Z, Gao L, Ma H and Chang R (2024) Osteopontin/SPP1: a potential mediator between immune cells and vascular calcification. Front. Immunol. 15:1395596. doi: 10.3389/fimmu.2024.1395596
Received: 04 March 2024; Accepted: 22 May 2024;
Published: 11 June 2024.
Edited by:
Liwu Li, Virginia Tech, United StatesReviewed by:
Marta Scatena, University of Washington, United StatesCopyright © 2024 Zhao, Huang, Gao, Ma and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanli Zhao, yanlizhao2015@126.com; Rong Chang, qhschangrong@126.com
†These authors have contributed equally to this work and share first authorship