 Alejandro A. Murillo
Alejandro A. Murillo Salvador Ramírez-Flandes
Salvador Ramírez-Flandes Edward F. DeLong
Edward F. DeLong Osvaldo Ulloa
Osvaldo Ulloa- 1Laboratorio de Oceanografía Microbiana, Departmento de Oceanografía & Instituto Milenio de Oceanografía, Universidad de Concepción, Concepción, Chile
- 2Programa de Doctorado en Ingeniería de Sistemas Complejos, Universidad Adolfo Ibáñez, Santiago, Chile
- 3Department of Biological Engineering and Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
Sulfur-oxidizing γ-proteobacteria are abundant in marine oxygen-deficient waters, and appear to play a key role in a previously unrecognized cryptic sulfur cycle. Metagenomic analyses of members of the uncultured SUP05 lineage in the Canadian seasonally anoxic fjord Saanich Inlet (SI), hydrothermal plumes in the Guaymas Basin (GB) and single cell genomics analysis of two ARCTIC96BD-19 representatives from the South Atlantic Sub-Tropical Gyre (SASG) have shown them to be metabolically versatile. However, SI and GB SUP05 bacteria seem to be obligate chemolithoautotrophs, whereas ARCTIC96BD-19 has the genetic potential for aerobic respiration. Here, we present results of a metagenomic analysis of sulfur-oxidizing γ-proteobacteria (GSO), closely related to the SUP05/ARCTIC96BD-19 clade, from a coastal ecosystem in the eastern South Pacific (ESP). This ecosystem experiences seasonal anoxia and accumulation of nitrite and ammonium at depth, with a corresponding increase in the abundance of GSO representatives. The ESP-GSOs appear to have a significantly different gene complement than those from SI, GB, and SASG. Genomic analyses of de novo assembled contigs indicate the presence of a complete aerobic respiratory complex based on the cytochrome bc1 oxidase. Furthermore, they appear to encode a complete TCA cycle and several transporters for dissolved organic carbon species, suggesting a mixotrophic lifestyle. Thus, the success of sulfur-oxidizing γ-proteobacteria in oxygen-deficient marine ecosystems appears due not only to their previously recognized anaerobic metabolic versatility, but also to their capacity to function under aerobic conditions using different carbon sources. Finally, members of ESP-GSO cluster also have the genetic potential for reducing nitrate to ammonium based on the nirBD genes, and may therefore facilitate a tighter coupling of the nitrogen and sulfur cycles in oxygen-deficient waters.
Introduction
Oxygen-deficient marine ecosystems support a high diversity of pelagic microorganisms that perform a variety of anaerobic metabolisms, particularly those associated with the nitrogen cycle (Stevens and Ulloa, 2008; Lam and Kuypers, 2011; Wright et al., 2012). Molecular taxonomic surveys in oceanic, coastal, and inland seas oxygen-deficient systems have revealed a high representation of pelagic sulfur-oxidizing γ-proteobacteria (Fuchs et al., 2005; Stevens and Ulloa, 2008; Lavik et al., 2009; Walsh et al., 2009; Wright et al., 2012; Glaubitz et al., 2013; Marshall and Morris, 2013). Phylogenetic analyses of this bacterial group have indicated that it is formed by two closely related lineages: ARCTIC96BD-19 (Bano and Hollibaugh, 2002) and SUP05 (Sunamura et al., 2004). The ARCTIC96BD-19 cluster comprises autotrophic sulfur-oxidizing bacteria capable of aerobic respiration (Swan et al., 2011; Marshall and Morris, 2013), while members of the SUP05 cluster have the genetic potential to oxidize sulfur compounds using nitrate as the terminal electron acceptor (Walsh et al., 2009). The latter have been implicated in a cryptic sulfur cycle in the permanent anoxic marine zone (AMZ) off northern Chile, where the production of sulfide is masked by its immediate reoxidation to sulfate coupled to nitrate reduction, suggesting an important role of sulfur cycling in this nitrate-rich anoxic environment (Canfield et al., 2010; Ulloa et al., 2012). The importance of SUP05 members has also been highlighted by a metatranscriptomic study in the core of the permanent AMZ off northern Chile (Stewart et al., 2012), where abundant transcripts related to sulfur-oxidation and nitrate reduction by relatives of SUP05 and sulfur-oxidizing endosymbionts of deep-sea clams (i.e., Candidatus Ruthia magnifica and Candidatus Vesicomyosocius okutanii) were found.
Walsh et al. (2009) reported a draft metagenome assembly of members of the SUP05 lineage from the seasonally anoxic fjord Saanich Inlet (SI) in British Columbia, and described the gene clusters required for sulfur oxidation to sulfate, carbon fixation, nitrate reduction and N2O production. According to this analysis, SI SUP05 bacteria apparently lack a complete TCA cycle and an aerobic respiratory electron transport chain, restricting them to an obligatory anaerobic and chemolithoautotrophic lifestyle. Recently, Anantharaman et al. (2013) showed that metagenomes and metatranscriptomes from a hydrothermal plume and deep-sea background at the Guaymas Basin (GB) contain a high representation of sequences matching SUP05. These chemolithoautotrophic GB sulfur-oxidizers have the genetic potential for H2 oxidation, stressing the metabolic plasticity of this group of bacteria.
Here, we analyzed assembled genome fragments of planktonic γ-proteobacteria, associated with the SUP05/ARCTIC96BD-19 lineage, recovered from the seasonal oxygen-deficient waters over the continental shelf off central Chile in the eastern South Pacific (ESP-GSO). Comparative analyses with respect to gene content, similarity and synteny were performed to understand the similarities and differences of this group compared to SUP05 representatives reported from the SI and GB, and to ARCTIC96BD-19 representatives reported from the South Atlantic Sub-Tropical Gyre (SASG). Our analyses suggest that the genomes of the ESP-GSO ecotypes encode genes for different metabolic capabilities than have been reported for their close relatives from SI and GB.
Materials and Methods
Area of Sampling and Data Collection
Water samples for metagenomic and ancillary data were collected at the COPAS time-series station, Station 18 (36°27′ S, 73°00′ W; water depth: 93 m), located on the continental shelf off central Chile (Figure 1A). Profiles for the physical variables and dissolved oxygen concentration were registered with a SeaBird 25 conductivity-temperature-depth (CTD) profiler and a SeaBird SB43 oxygen sensor. Additionally, on several occasions in situ oxygen concentrations were measured with the ultra-sensitive STOX sensor (Revsbech et al., 2009). NO−3, NO−2, and NH+4 concentrations were determined as described in Galán et al. (2012).
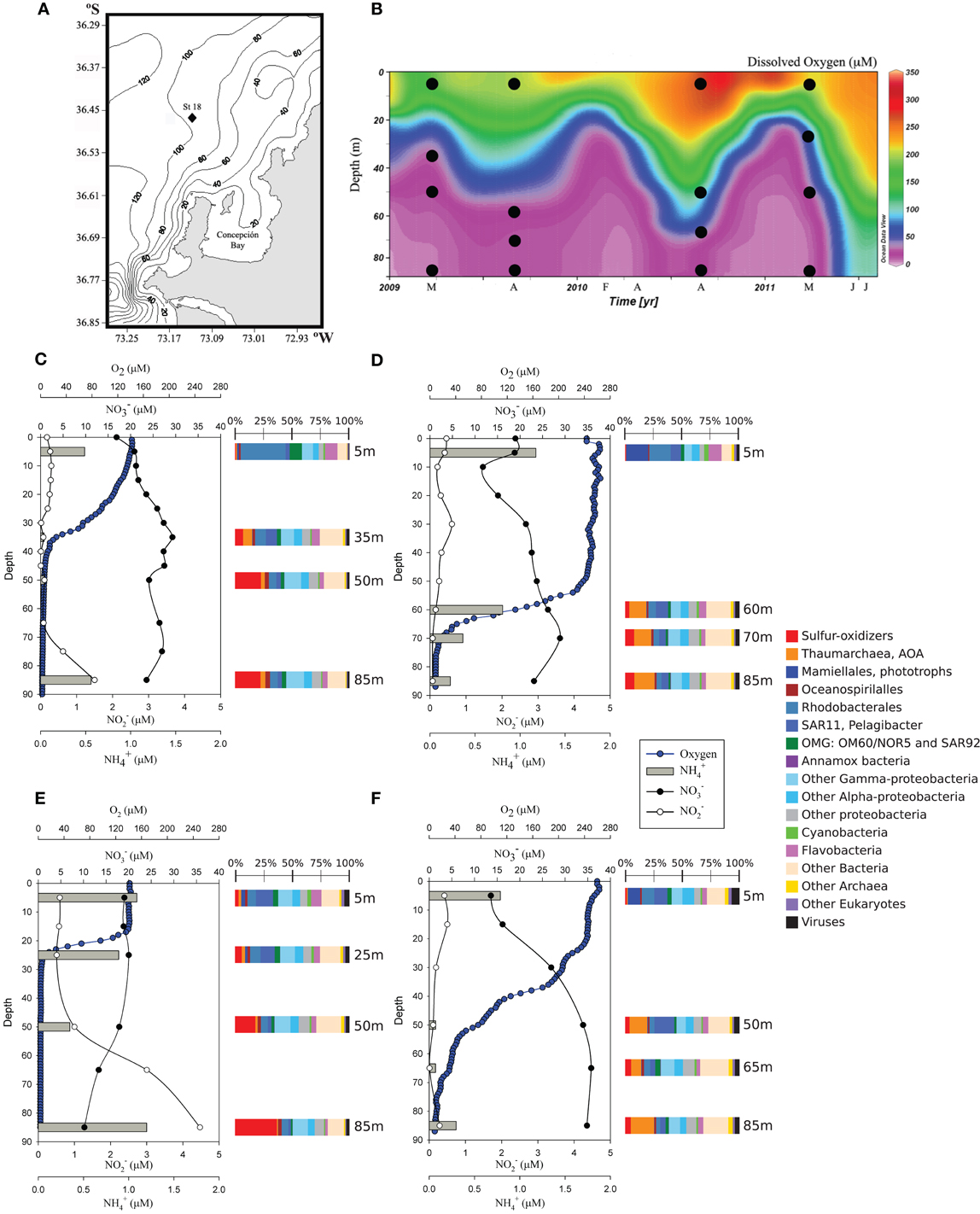
Figure 1. Area of sampling and nutrient profiles in the water column. (A) Location of the time-series Station 18 off Concepción Bay. (B) Contour map for dissolved oxygen concentration in the water column at Station 18 during the 3-year period of the study. Depths sampled for metagenomics in March 2009 and 2011 (upwelling) and in August 2009 and September 2010 (non-upwelling) are shown as black dots. (C,E) Nutrient profiles for the upwelling periods sampled in 2009 and 2011, respectively. (D,F) Nutrient profiles for the non-upwelling periods sampled in 2009 and 2010, respectively. For each metagenome analyzed, the relative abundance of the inferred major taxonomic groups derived from the protein-coding sequences are shown as horizontal stacked bars.
DNA samples for metagenomic analyses were collected from four discrete depths during the upwelling period on March 24, 2009 and March 30, 2011, as well as during the non-upwelling period in August 31, 2009 and September 01, 2010. An additional sample obtained from 85 m depth, on January 12, 2009 was also incorporated in the analysis. The sampling depths covered the surface photic layer (5 m), the mid-waters (35–60 m) and the deeper waters (70–87 m) over the shelf (Figures 1B–F). For each depth, a volume of 10 L was pre-filtered through a 20-μ m mesh, then a 1.6-μ m glass-fiber prefilter (APFA04700, Millipore) in a 47-mm Swinex, and the cells collected on a 0.2-μ m Sterivex filter (SVGP01050, Millipore). The Sterivex filters were filled with 2 mL of lysis buffer (50 mM Tris•HCl, 40 mM EDTA, and 0.73 M sucrose) and fast frozen in liquid nitrogen. Genomic DNA was extracted using a modified protocol from Galán et al. (2012). Briefly, 2 mg of lysozyme in 40 μ L lysis buffer was added to each filter and cells were lysed for 45 min at 37°C. Following the lysis, 100 μ L of proteinase K and 100 μ L SDS 20% were added to each filter and the samples were digested at 50°C for 2 h. The lysate was removed, and each filter was incubated with 1 mL of lysis buffer at 55°C for an additional 15 min. The total pooled lysates were then extracted, once with phenol-chloroform-isoamyl alcohol (25:24:1; pH 8) and once with chloroform-isoamyl alcohol (24:1), and samples were concentrated by isopropanol precipitation. The purified genomic DNA was used for the generation of single-stranded DNA libraries and emulsion PCR according to manufacturers protocols using the 454 Genome Sequencer (Roche Applied Science, Indianapolis, IN, USA). Clonally amplified library fragments were sequenced using FLX chemistry for the 2009 samples and Titanium chemistry for remaining samples on a Roche Genome Sequencer FLX instrument, yielding 3.23 × 105–1.33 × 106 reads per sample (Table S1), according to the manufacturer's protocols. De novo contig sequences have been submitted to the NCBI under GenBank accession numbers KJ783402-KJ783431, and raw sequences of the metagenomes have been deposited in CAMERA under accession number CAM_P_0000692.
Bioinformatics Analysis
Taxonomic characterization of the metagenomes according to 16S rRNA sequences (Figures S2–S7) was carried out by comparing individual reads against a customized version of the NCBI RefSeq-rRNA database (BLASTN bitscore cutoff at 50) and filtered (by annotation) to count only the 16S-rRNA-associated sequences. This customized database consisted in the RefSeq rRNA as of January 2014 plus 16S rRNA sequences from SUP05 and ARCTIC96BD-19, not included in the original RefSeq rRNA database.
The raw sequences for each community DNA sample were analyzed against the NCBI-nr database using the BLASTX algorithm, with a bit-score threshold of 50, obtaining a taxonomic classification of the protein-coding sequences and an estimation of the community composition of each sample. For the contig assembly, samples enriched in sequences assigned to sulfur-oxidizing organisms were selected, corresponding to those obtained in the upwelling period from 85 m in January (Figure S1) and 35, 50, and 85 m in March 2009, which constitute the contig data set for 2009. The contig data set for 2011 was assembled with sequences from 50 and 85 m collected in March 2011 (Table S1). Both data sets were processed and analyzed separately, due to the differences in sequencing technologies and the greater number of sequences of the 2011 dataset, allowing us to compare the robustness of contig assembling and the possibility of chimeras generation between both data collections. Assembly of metagenomic reads was performed using the Newbler/454/Roche assembler, with a minimum overlap length of 40 bp and a minimum overlap identity of 97%. These values were more stringent than the default Newbler parameters (40 bp and 90%, respectively) in order to reduce the possibility of chimeric contigs. With these parameters, 299 and 1497 contigs were assembled from the March 2009 and 2011 datasets, respectively (Table S1). The largest contigs obtained for each data set were of 76,353 and 76,610 bp, respectively. The largest contigs that specifically associated with GSOs were of 56,914 bp (formed by 7083 sequences) for 2009 and 35,253 bp (formed by 1011 sequences) for 2011. However, the largest contigs reported in this work are of 51,536 bp for 2009 and 15,603 bp for 2011, since the other ones did not contain genes associated with the metabolisms of interest (i.e., for the carbon, nitrogen, and sulfur cycles). The assembled contigs were annotated using Metagenemark (Zhu et al., 2010) and genes were preliminarily identified by the best BLASTX hit against NCBI-nr. Those contigs associated with sulfur-oxidizing γ-proteobacteria and containing open reading frames (ORFs) related to carbon, nitrogen and sulfur metabolisms were manually re-annotated using the Geneious Pro software version 5.4.6 (Drummond et al., 2011), utilizing BLASTX against the NCBI-nr database with a Max E-value of 1e−5, word size of 3, the BLOSUM62 matrix, and an open and extend gap penalty of 11/1, respectively. The contigs were also compared for synteny analysis against the genomic scaffolds of the uncultured SUP05 cluster bacterium from SI (Accession ACSG00000000), the genomic scaffolds of the uncultured SUP05 cluster bacterium from hydrothermal vent plumes in the GB of the deep Gulf of California (Accession AJXC00000000), the genomic scaffolds of the ARCTIC96BD-19 cluster (AAA0001, accession AFHZ00000000 and AAA0007, accession AFHY00000000), and the complete genomic sequences of the reference symbionts Candidatus Vesicomyosocius okutanii HA (Accession AP009247) and Candidatus Ruthia magnifica str. Cm (Accession CP000488), using the “Trebol” software for prokaryotic comparative genomics (http://bioinf.udec.cl).
Taxonomic binning and classification of this subset of contigs was performed using BLASTN for each contig against a custom nucleotide database that consisted of all available genomic DNA sequences from SUP05 (accession numbers GG729945-GG730036), Candidatus Vesicomyosocius okutanii HA, Candidatus Ruthia magnifica str. Cm, ARTIC96BD-19 (AAA0001, accession AFHZ00000000) and ARTIC96BD-19 (AAA0007, accession AFHY00000000), this with the objective of differentiating inside of the GSO cluster if the contigs belonged to the SUP05 or the ARCTIC96BD-19 subclade. We retrieved the contigs whose best hit corresponded to SI SUP05, C. Ruthia magnifica and Candidatus Vesicomyosocius okutanii (contig00465, contig05874, contig07238, contig10131, contig13071, contig13139, contig13504, contig17720, contig28161, contig29238, contig31001, contig31297), assigned to sulfur-oxidizing γ-proteobacteria. Although these contigs contained some homologous regions to ARCTIC96BD-19, all of them had better sequence identity to SI SUP05 (Tables S2–S15). An in-house program was also used to compute the overall frequencies of di, tri and tetranucleotides for the (meta)genomes of SUP05 and ARCTIC96BD-19. These frequencies were normalized by the total number of base pairs in each dataset. Our contigs were processed in the same way. Spearman rank order correlation was then used for the binning of the contigs. However, it was impossible to assign unequivocally our contigs to SUP05 or to ARCTIC96BD-19 through tetra- tri- or di-nucleotide frequency analysis, most likely because of the nature of the reference data available. To corroborate this ambiguity in the binning process, global di-, tri-, and tetranucleotide frequencies were calculated for the SUP05 metagenome and an ARCTIC96BD-19 single-cell partial genome (SUP05: 96 contigs from http://www.ncbi.nlm.nih.gov/nuccore?term=34785[BioProject] and ARCTIC: gamma proteobacterium SCGC AAA007-O20). Then, for each of the 96 original contigs of SUP05 we performed the composition-based analysis as above. Many of the SUP05 contigs were better correlated with the ARCTIC96BD-19 global frequencies than with those from their own metagenome, independent of the size of the contig. As an example, a dinucleotide analysis for a >20 kb SUP05 contig (gi|264676130|gb|ACSG01000005.1| Uncultured SUP05 cluster bacterium Contig34357) gives a better Spearman rank order RHO against ARCTIC19BD-19 than against SUP05 (0.9058 vs. 0.9029). Similar results are obtained for tri and tetranucleotides analysis. Therefore, we could not unambiguously bin sequences against SUP05 vs. ARCTIC96BD-19 with the available data using the composition-based method. A further analysis was performed by using average coverage for each analyzed contig. These values were calculated as the sum of all the reads lengths (only the segment involved in the alignment that contribute to the consensus sequence of the contig) divided by the total contig length. The results of this analysis are presented in Tables S19–S30 and discussed in the following sections.
For those key genes that were not found assembled in contigs, we analyzed the possibility of recovering homologous sequences from single reads (singlets). For this, the singlets were compared against a BLAST database composed of selected proteins involved in key steps of the metabolic pathways reconstructed, namely Sqr, FccAB, NirK, NorBC, NosZ (Table S16).
Results and Discussion
Environmental and Microbial Context
The coastal area of central Chile is characterized by the presence of strong wind-driven upwelling during the spring and summer seasons. This process brings nutrient-rich and oxygen-poor Equatorial Subsurface Water (ESSW) over the continental shelf, enhancing phytoplankton primary production in the euphotic zone (Montero et al., 2007; Sobarzo et al., 2007). The respiration of the sinking organic matter produced by photosynthesis enhances the oxygen deficiency at depth. Indeed, measurements with the ultra-sensitive STOX sensor (Revsbech et al., 2009) carried out in January 2010, during the upwelling period, revealed that waters below 75 m had less than 13-nM of oxygen, the detection limit of the sensor used. These essentially anoxic waters present during the upwelling season showed accumulation of nitrite (NO−2) and ammonium (NH+4) (Figures 1C,E), reflecting an active nitrogen cycle. Conversely, during the non-upwelling season (austral autumn and winter) the wind shifts to the northerly direction, favoring downwelling and the presence of waters with higher oxygen concentrations over the shelf than in spring-summer (Figures 1D,F; Sobarzo et al., 2007; Galán et al., 2012).
The planktonic microbial community reaches maximal cell densities (~1.5–2.0 × 106) in the upwelling season, and includes anammox bacteria of the Candidatus Scalindua clade (Galán et al., 2012) and ammonia-oxidizing thaumarchaea (Molina et al., 2010). However, the sulfur-oxidizing γ-proteobacteria related to the SUP05/ARCTIC96-BD19 clade are by far the most abundant organisms at the anoxic depths in the water column of this shelf ecosystem (Figures S2–S7). The 16S rRNA gene sequences recovered from the metagenomic analysis showed a clear dominance of the SUP05/ARCTIC96BD-19 clade, being 17% of the total bacterial 16S rRNA sequences at 35 m in March 2009, and up to the 44% at 85 m in March 2011 (Table S17). The protein-coding sequences that associated with γ-proteobacteria sulfur oxidizers related to the SUP05 and ARCTIC96-BD19 clades and sulfur-oxidizing endosymbionts of deep-sea clams Candidatus Ruthia magnifica (Roeselers et al., 2010) and Candidatus Vesicomyosocius okutanii (Kuwahara et al., 2007) also dominated the metagenomes of the anoxic waters, with more than 25% of the total sequences in the upwelling period of 2011 (Figure 1E). Based on these results, we assembled and analyzed genome fragments that could be assigned to the GSO cluster, i.e., that were most closely associated with the SUP05/ARCTIC96BD-19 lineage (see Materials and Methods), and that encoded genes associated with carbon, nitrogen, and sulfur metabolisms.
Carbon Metabolism
The metagenomic data for members of the eastern South Pacific (ESP) GSO from the seasonal anoxic waters showed the presence of genes related to the Calvin-Benson-Bassham (CBB) cycle, encoding the large subunit form II (LII) of the ribulose-1,5-bisphosphate carboxylase-oxygenase (RuBisCO) enzyme CbbM, the RuBisCO operon transcriptional regulator belonging to the LysR family of proteins CbbR, and the regulatory proteins CbbQ and CbbO (Figure 2 and Figure S8; Table 1 and Table S18). This operon was retrieved from de novo assembled contigs obtained during the two independent upwelling seasons considered, March 2009 and 2011, and is in synteny with the RuBisCO operon described in the SI and GB SUP05 metagenomes (Walsh et al., 2009; Anantharaman et al., 2013). However, the ESP RuBisCO operon contains an extra ORF between cbbM and cbbQ, related to a putative membrane protein (Figure S8). The cbbM gene in ARCTIC96BD-19 has been recovered as just partial single sequences in the single-cell genomic (SCGC) projects (Swan et al., 2011), which prevented the comparison for synteny against the ARCTIC96BD-19 sub-clade. On the other hand, only the operon for the large subunit form I of RuBisCO has been found in contigs from ARCTIC96BD-19 (Swan et al., 2011).
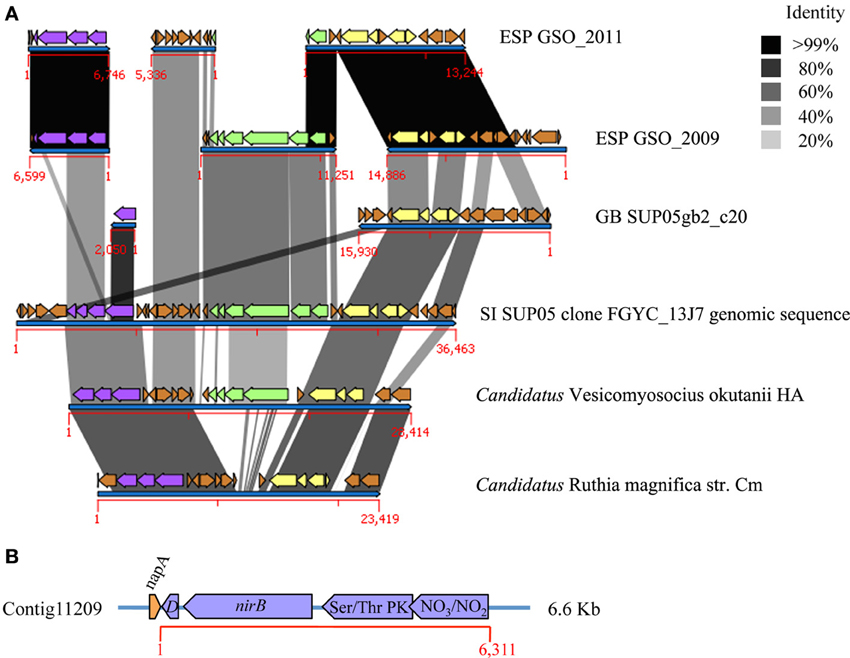
Figure 2. Genetic context of the nirBD genes, nar, and RuBisCO operons. (A) Comparison of de novo contigs constructed with metagenomic sequences from 2009 and 2011 sampling (≥99% of identity is represented by the black color of the synteny comparisons). The nirBD operon is shown in purple and is located physically near the nitrate reductase (nar, light green) and RuBisCO operons (yellow) in the Saanich Inlet SUP05 clone FGYC_13J7, and in the genome of Candidatus Vesicomyosocius okutanii. In the Candidatus Ruthia magnifica genome the nar operon is absent, but the region maintains some similarity to the narG gene. In the case of the GB-SUP05 metagenome, the RuBisCO operon was found in synteny with that in SI- and ESP-GSO. However, the nar operon was not found in the comparative analysis, and the nirB gene was retrieved as a single gene in a short contig (2050 bp), with no identity to the ESP-GSO nirB gene. (B) Specific annotation of the nirBD genes flanked by a truncated form of the nitrate reductase napA gene at the 5′ end and by a bifunctional serine/threonine kinase/phosphatase and a nitrate/nitrite transporter in the 3′ end, both sequences related to the regulation and sensing of the cellular level of nitrate and nitrite, and therefore considered as part of the operon.
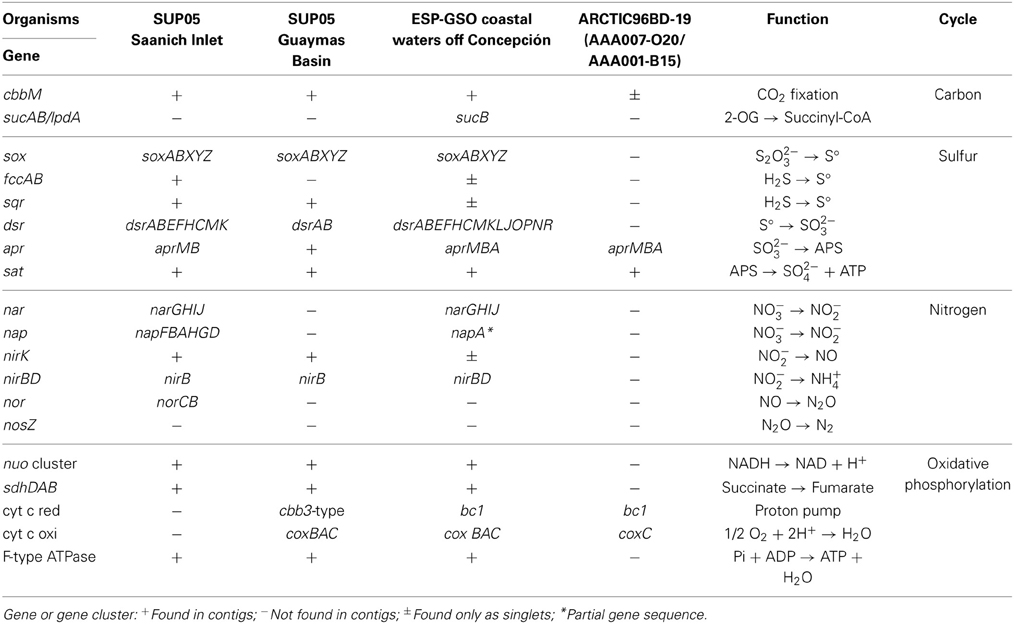
Table 1. Summary of genes and gene clusters reconstructed for GSO cluster from the oxygen-deficient coastal waters off Concepción (ESP), compared with those from Saanich Inlet (SI), the hydrothermal vent plumes in Guaymas Basin (GB) and two ARCTIC96BD-19 representatives from the South Atlantic Sub-Tropical Gyre.
The cbbM-cbbQ-cbbO found in our data presents the basic genetic arrangement described for the form II of the enzyme in obligate autotrophs, with no clustering with regulatory genes found in facultative ones. Such arrangement suggests that the CBB cycle in ESP-GSOs is constitutively expressed (Badger and Bek, 2008). However, we cannot totally exclude the possibility that they have another form of RuBisCO outside the form II operon, although no such RuBisCO was found in our dataset. The three γ-proteobacteria described so far as obligate chemolithoautotrophs that encode the form II type of RuBisCO also have at least one extra form of this enzyme. Halothiobacillus neapolitanus, a saline aquatic/soil bacterium, has forms IAc and II of RuBisCO, while Hydrogenovibrio marinus, a marine bacterium, and Thiomicrospira crunogena XCL-2, a marine bacterium inhabiting hydrothermal vents, both have forms IAc, IAq, and II. The reason for having several RuBisCO forms has been explained by their different kinetic properties (Badger and Bek, 2008). Moreover, bacteria with a single form of RuBisCO have been classified as specialists, with CO2 fixation occurring under constant levels of CO2 and O2. In the case of form II, such conditions are low O2 and high CO2, due to a low discrimination against O2 as an alternative substrate, a poor affinity for CO2, and a relatively high transformation rate (kcat = K2). The recovery of just one type of RuBisCO operon from the metagenomic data of the ESP-GSO bacteria, together with the apparent absence of carbon-concentrating mechanisms, such as carbonic anhydrase enzymes, bicarbonate transporters and carboxysome related proteins, suggest there may be an alternative source of carbon for these microorganisms when oxygen is present.
Historically, the absence of the α-ketoglutarate dehydrogenase complex (KDH) has been interpreted as being diagnostic for obligate autotrophy and methanotrophy (Wood et al., 2004). The biosynthetic form of the incomplete TCA cycle has been described for the SI and GB SUP05 metagenomes (Walsh et al., 2009; Anantharaman et al., 2013), and no genetic evidence for the KDH gene was found in the ARCTIC96BD-19 single amplified genomes (SAGs). These observations suggested an obligate autotrophic lifestyle, fueled by CO2 fixation through the form II of RuBisCO. In a fully functional TCA cycle, the α-ketoglutarate dehydrogenase complex converts α-ketoglutarate to succinyl coenzyme A, which is an essential precursor for porphyrin synthesis (Wood et al., 2004). In the case of obligate autotrophs and methanotrophs, the incomplete TCA cycle acts as a biosynthetic “horseshoe,” channeling carbon from carbon dioxide or methane into central biosynthetic pathways (Wood et al., 2004). In this context, our data show the presence of the gene sucB encoding a dihydrolipoamide acyltransferase, part of the KDH complex, and consistent with the reactions in Figure 3A, necessary for a complete TCA cycle (Figure 3A inset, Table 1 and Table S19). The sucB gene has not been described for the closely related symbionts Candidatus Ruthia magnifica and Candidatus Vesicomyosocius okutanii, but a phylogenetic analysis of SucB amino acid sequences showed a closer relationship to other sulfur-oxidizing γ-proteobacteria symbionts (Figure S9). Furthermore, synteny analysis showed the homology of flanking genes at the 3′ end of this GSO gene cluster to those of GB and SI SUP05, but a clearly a different genetic context for ESP-GSO at the 5′ end, where the sucB gene was encoded (Figure 3B).
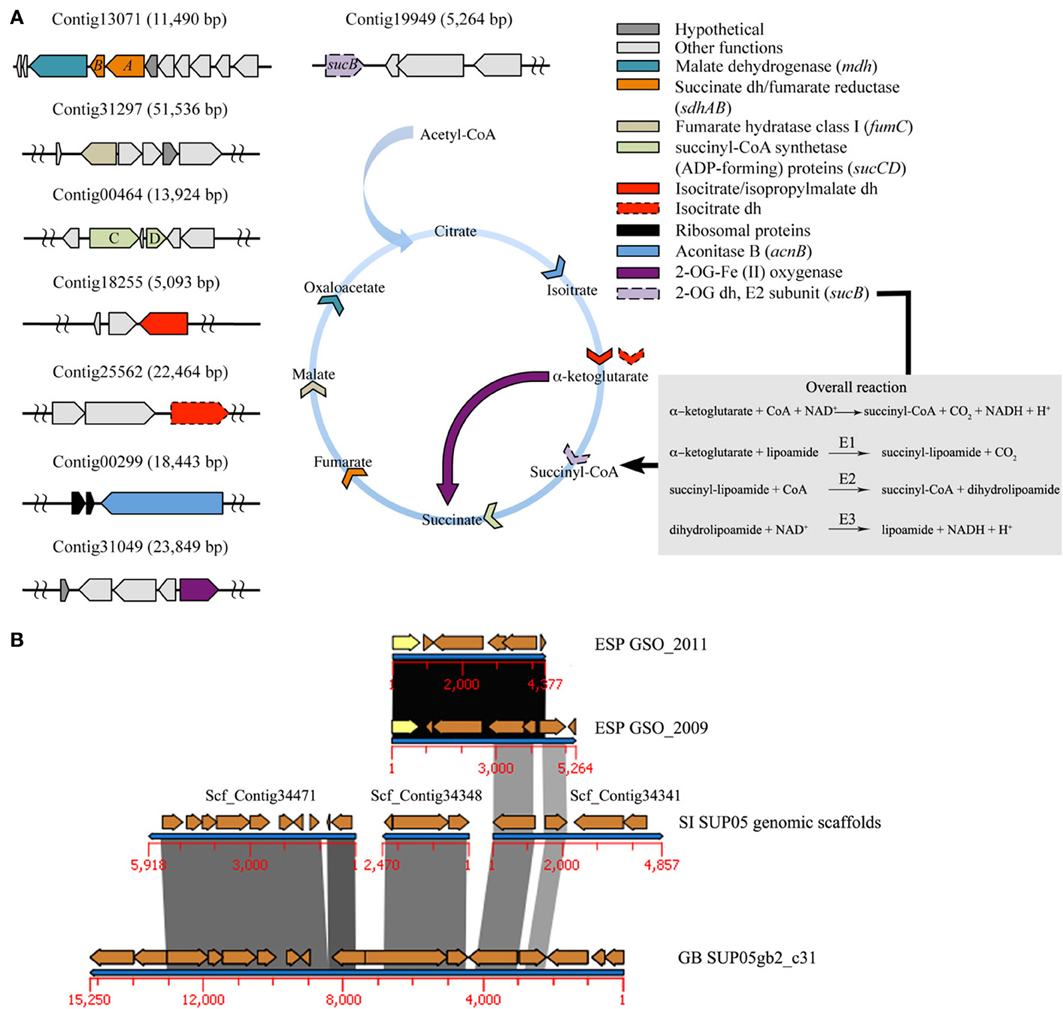
Figure 3. Complete TCA cycle. (A) Sequences for each step of the TCA were retrieved from the metagenomic data associated with the ESP-GSO lineage bacterium. The color arrowheads show the specific function in the cycle for each enzyme. The overall reaction and semi-reactions for the α-ketoglutarate dehydrogenase complex are shown in the inset; the E2 subunit (sucB gene) of this complex was retrieved from the data. Also, an alternative 2OG-Fe[II] oxygenase gene was recovered from the data and its function is shown as a dark purple arrow. (B) Synteny analysis of de novo contigs for 2009 and 2011 datasets, coding for sucB gene (yellow), compared to SI SUP05 genomic scaffolds and GB SUP05 subclade gb2 contig31.
In addition to the α-ketoglutarate dehydrogenase subunit E2 (sucB gene), we retrieved two genes related to alternative pathways that could close the TCA cycle. Firstly, we retrieved the 2-oxoglutarate-iron (II) (2OG-Fe[II]) oxygenase gene (Figure 3), which encodes the enzymatic function to converts 2-oxoglutarate to succinate bypassing the succinyl-CoA molecule, consuming O2 in the reaction and producing CO2 as by-product (EC 1.14.11.2) (Hutton et al., 1967; Kivirikko et al., 1972; Berg and Prockop, 1973). This enzymatic function, in turns, belongs to the 2OG-Fe(II) dioxygenase superfamily, which is widely represented in bacteria and eukaryotes, but apparently not in archaea. While the function of some members of this family has been characterized in H. sapiens (Chen et al., 1994; Wei et al., 1996), C. elegans (Darby et al., 1999) and E. coli (Kondo et al., 1986), an extensive list of homologous proteins in eukaryotes and bacteria, including the sequences retrieved from our metagenomic data, still awaits for biochemical characterization (Aravind and Koonin, 2001). Secondly, we recovered the gene that encodes for succinic semialdehyde dehydrogenase (SSADH), recently described by Zhang and Bryant (2011), which work in coordination with the enzyme 2-oxoglutarate decarboxylase (2-OGDC) that transforms 2-OG into succinic semialdehyde (SSA). Subsequently, SSA is oxidized to succinate by SSADH, closing the TCA cycle in cyanobacteria (Zhang and Bryant, 2011). These results imply the potential use of the full TCA cycle in members of the ESP-GSO cluster.
Supporting the above results, and based on dissolved organic carbon (DOC)-related transporters expressed by coastal bacterioplankton (Poretsky et al., 2010), we recovered several genes related to ABC and TRAP transport systems for general and branched-chain amino acids (BCAA), carbohydrates, polyamines, compatible solutes such as glycine-betaine-proline, and cations antiporters in several contigs (Table S20). Our results thus indicate that members of the ESP-GSO cluster off central Chile have the metabolic potential of utilizing sources of DOC heterotrophically, and suggested a facultative autotrophic or even a mixotrophic lifestyle for these bacteria, which appear capable of utilizing both inorganic and organic substrates.
Oxidative Phosphorylation
Consistent with a fully functional TCA cycle, the electrons released during the oxidation of TCA intermediates are transported to NAD+ or FAD+. In the case of aerobic respiration, the NADH is oxidized and the electrons are transported to oxygen via an electron transport chain. However, published data on the SI SUP05 metagenome describe a facultative or strictly anaerobic lifestyle, due to the absence of the aerobic respiratory complexes (Walsh et al., 2009).
We found two ESP-GSO contigs from the anoxic depths that encoded ORFs related to the cytochrome bc1 reductase and cytochrome c oxidase (complexes III and IV, respectively) (Figures 4, 5). Both complexes can be found in contigs from the ARCTIC96BD-19 whole genome shotgun dataset (Swan et al., 2011) and in the GB SUP05 metagenome (Anantharaman et al., 2013). They are also in synteny with the genomes of the symbionts Candidatus Vesicomyosocius okutanii and Candidatus Ruthia magnifica (Figure S10). The contig for complex IV (contig 17720), however, presented a lower coverage than the average of the other contigs analyzed (9.50 vs. 33.48×), which may indicate that it is found in a highly variable region, reflecting genetic variability within the GSOs, given that all their component genes were consistently affiliated to these organisms. Additionally, the GB metagenome and the genomes of symbionts have genes for the cbb3-type terminal cytochrome c oxidase, which are apparently absent in the SI and ESP-GSO metagenomes and show a different genomic context in the synteny analysis (Figure 5). The detailed identity for each ORF is presented in Tables S21–S25. These complexes complete the oxidative phosphorylation pathway for ATP production through the aerobic respiration process, well characterized in model organisms (Table 1). The complexes I, II, and V are complete and in synteny with the SI and GB SUP05 metagenomes and with the mentioned symbiont genomes (Figures S11–S13). These findings support the metabolic potential for both GB and ESP-GSO lineages to utilize oxygen as a terminal electron acceptor, apparently with different affinities, giving them the ability to more widely populate the water column.
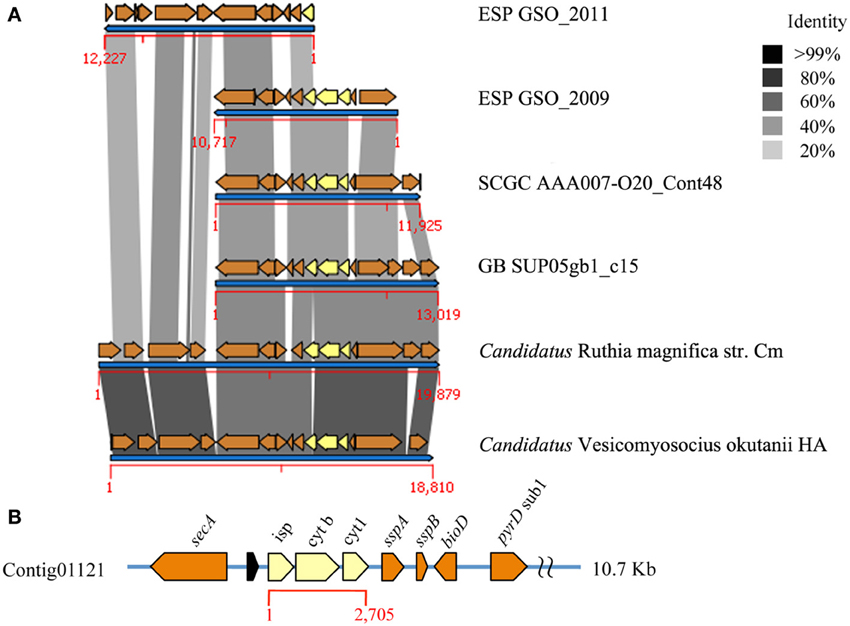
Figure 4. Complex III of the oxidative phosphorylation pathway of the ESP-GSO lineage off the coast of Concepción. (A) Comparison of de novo contigs constructed with metagenomic sequences from 2009 and 2011 sampling. Cytochrome bc1 complex, previously undescribed for Saanich Inlet SUP05, is shown in yellow and the synteny of genetic background was analyzed against ARCTIC96BD-19 strain AAA007-O20, GB SUP05 and the reference genomes of endosymbionts Candidatus Ruthia magnifica and Candidatus Vesicomyosocius okutanii. No genomic scaffolds were recovered for the complex III or any flanking gene from the SI SUP05 data. (B) Specific annotation of the cytochrome bc1 complex and surrounding genes from the 2009 data.
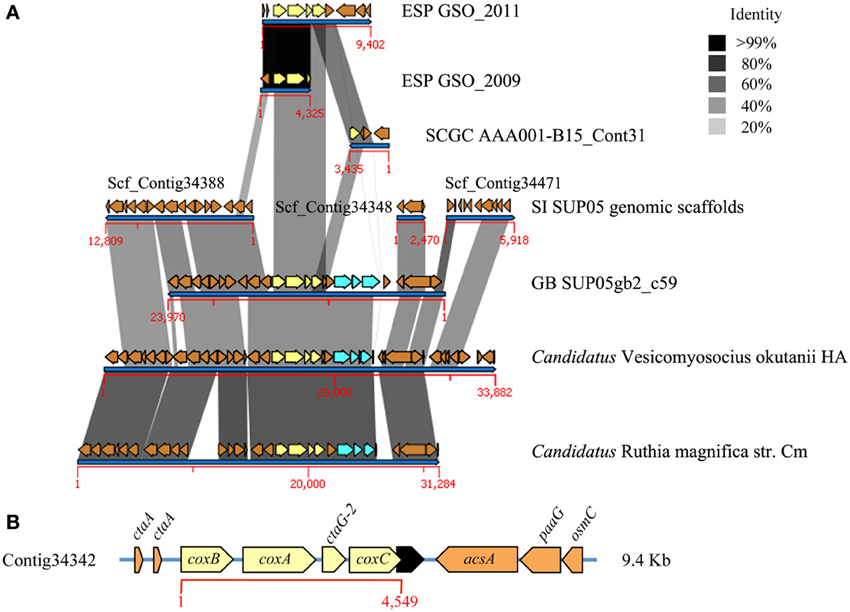
Figure 5. Complexes IV of the oxidative phosphorylation pathway in the ESP-GSO lineage off coast of Concepcion. In (A) are shown de novo contigs constructed with metagenomic sequences from 2009 and 2011 sampling, compared against contigs from the uncultured SI SUP05 and GB SUP05 cluster bacterium, ARCTIC96BD-19 strain AAA001-B15 and the reference symbionts genomes Candidatus Ruthia magnifica and Candidatus Vesicomyosocius okutanii. The cytochrome c oxidase complex is shown in yellow and the high affinity cytochrome c oxidase cbb3 in cyan. We were not able to find any contig of SUP05 from Saanich Inlet that have identity with complex IV, but the genomic scaffolds coding for flanking genes are shown. (B) Specific annotation of the cytochrome c oxidase complex and surrounding genes for the 2011 data.
Sulfur and Nitrogen Dissimilatory Pathways
From an energetic point of view, the complete oxidation of sulfide or thiosulfate to sulfate, with a net flow of eight electrons, is one of the most energetically favorable processes developed by chemolithoautotrophs for energy production (Shao et al., 2010). In the anoxic zone, the flow of electrons, utilized for the anaerobic respiration, must be deposited in an alternative electron acceptor. Energetically, nitrate (NO−3) is the next preferred electron acceptor for respiration after oxygen, and the energy produced in the reductive transformation of NO−3 into N2 is as high as that from oxic respiration of organic matter (Lam and Kuypers, 2011). This coupling of dissimilatory pathways is consistent with the high percentage of sequences associated with sulfur-oxidizing γ-proteobacteria in the nitrate-rich anoxic environment (Figures 1C,E). In this context, we recovered an incomplete form of the sulfur oxidation (Sox) system, involved in the oxidation of thiosulfate, and similar to those described for obligate photolithotrophs, which are distinguished by the formation of sulfur globules during the oxidation of thiosulfate to sulfate (Dahl et al., 2005; Grimm et al., 2008). The soxXAYZ/soxB, retrieved (Table 1; Figures S14, S15), hypothetically should bind thiosulfate through the SoxXA heterodimer, which couples the sulfone sulfur to a SoxY-cysteine-sulfhydryl group of the SoxYZ complex, from which the terminal sulfone group is released by the activity of SoxB (Frigaard and Bryant, 2008). However, the apparent lack of the sulfur dehydrogenase SoxCD complex, which regenerates the SoxYZ complex, prevents the direct oxidation of sulfone sulfur atoms to sulfate, and instead accumulates sulfur that must be further oxidized to sulfate (Ghosh and Dam, 2009). This truncated Sox system, a common feature of the SUP05/ARCTIC96BD-19 lineage (Walsh et al., 2009; Anantharaman et al., 2013; Marshall and Morris, 2013), represents an inefficient energy source, considering that the complete oxidation of one mole of thiosulfate by the complete Sox system generates eight moles of electrons. In contrast, the truncated form only generates two moles, not allowing lithotrophic growth with thiosulfate as the solely source of energy, but still being able to oxidize thiosulfate at low rates (Friedrich et al., 2001). The BLASTX identity and affiliation for each sox gene are shown in Table S26.
The coupling of an incomplete Sox system for the oxidation of sulfur to sulfite is due to the presence of a reverse dissimilatory sulfite reductase (dsr) pathway in this cluster of organisms (Figure S16). We retrieved a complete version of dsr cluster encoded in a 12.4 Kb region in contig31001 with 34,585 bp of total length (Figure S16 and Table S27). This cluster shares synteny with the Allochromatium vinosum cluster (Pott and Dahl, 1998; Dahl et al., 2005; Sander et al., 2006) and only lacks the gene dsrS, which along with dsrR are soluble cytoplasmic proteins of unknown function (Grimm et al., 2008). Also, the retrieved cluster shares a high degree of synteny with those in the SI- and GB-SUP05 metagenomes and the symbionts Candidatus Ruthia magnifica and Candidatus Vesicomyosocius okutanii genomes (Figure S16).
Consistent with a complete and functional version of the dsr cluster, sulfite has to be transformed into sulfate; we retrieved sequences related to the aprMBA-sat genes, involved in this transformation (Figure 6 and Figure S17; Table 1 and Table S28). This system takes the sulfite generated and transforms it into adenosine 5′-phosphosulfate (APS) and then into ATP plus sulfate through substrate-level phosphorylation, a process that could represent an important source of energy for sulfur-oxidizing bacteria, such as is the case for Ach. vinosum (Sánchez et al., 2001).
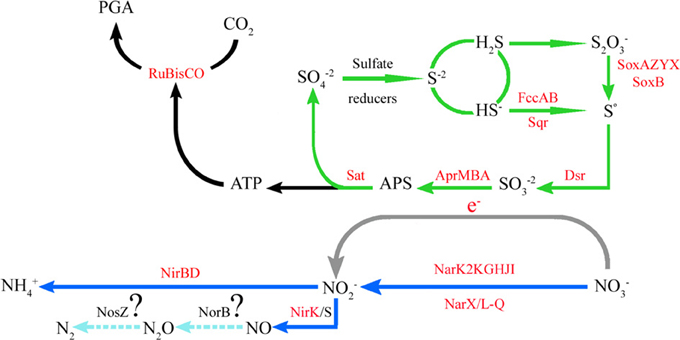
Figure 6. Predicted coupled sulfur and nitrogen cycles in the ESP-GSO of the seasonal oxygen-deficient upwelling waters off the coast of Concepcion, Chile. The functions in red were retrieved and reconstructed from the metagenomic data: the incomplete Sox pathway for the oxidation of thiosulfate to sulfur, the reverse dissimilatory sulfite reductase (dsr) pathway to oxidize sulfur to sulfite, sulfite is sequentially transformed in APS and sulfate plus ATP by the Sat-Apr system, and the ATP generated could be utilized to fix inorganic carbon through the CBB cycle. Coupled to sulfur oxidation, nitrate is reduced as final electron acceptor through the nitrate reductase NarG. The nitrite produced could be transformed into ammonium in this system, given the presence of nitrite reductase NirBD, which is related to the ammonification process in several organisms.
Although we did not find contigs encoding genes for the sulfide:quinone reductase (sqr) or flavocytochrome c (fccAB), involved in sulfide oxidation, we found un-assembled sequences associated with the SUP05 linage, Candidatus Ruthia magnifica str. Cm and Candidatus Vesicomyosocius okutanii HA with significant identity to sqr and fccAB (Table S16). The utilization of reduced sulfur species seems to be the central metabolic pathway for electron flow in the ESP-GSO lineage, due to the absence of membrane-bound group 1 Ni/Fe hydrogenase genes, described for GB SUP05 lineage (Anantharaman et al., 2013), coherent with the different ecological niche of these groups. None of the 18 genes of the hydrogenase cluster were recovered from the ESP metagenomic data.
Chemolithoautotrophic denitrification, fueled by the described thiosulfate oxidation, leads to the utilization of NO−3/NO−2 as electron acceptors and releases gaseous N2O and/or N2, utilizing the energy generated in this process to fix inorganic carbon. However, a complete conventional denitrification process consists of four reaction steps via the metaloenzymes nitrate reductase (Nar), nitrite reductase (Nir), nitric oxide reductase (Nor), and nitrous oxide reductase (Nos), acting sequentially under anaerobic conditions (Philippot, 2002). We found genes for membrane bound dissimilatory nitrate reductase (nar) pathway (Figure 6 and Figure S18A; Table 1 and Table S29), which in turn has been shown to be directly involved in ATP formation through the generation of a transmembrane proton motive force (Jormakka et al., 2003). The narK2KGHJI operon recovered from the metagenomic data (Figure S18A) was associated and shares synteny with the operon described for the SUP05 metagenome from SI (Walsh et al., 2009), but that was absent from the GB metagenomes (Figure 2). In a different contig, we identified a two-component regulatory system nitrate/nitrite response regulator, related to NarX/L and a signal transduction histidine kinase nitrate/nitrite-specific, related to NarQ (Figure S18B and Table S29); both genes have been shown to be associated with the regulation of the narGHIJ operon expression in Pseudomonas stutzeri and E. coli (Chiang et al., 1992; Dong et al., 1992; Walker and DeMoss, 1994). The narX/L gene is annotated as encoding a hypothetical protein, Sup05_1368, in the metagenome of the uncultured SI SUP05 (Walsh et al., 2009).
The next step in denitrification is the reduction of nitrite to nitric oxide, the key step in the dissimilative process. This reaction can be catalyzed by the copper-containing NirK or cytochrome cd1-containing NirS nitrite reductases. However, none of these genes (or known accessory genes) were retrieved in the analyzed contigs, although two unassembled sequences with putative homology to NirK that were taxonomically associated to the SI SUP05 lineage were found (Table S16). Nonetheless, one of the analyzed contigs contained the nirBD genes, related to the anaerobic nitrite reductase (NirBD, Table 1; Figure 2B and Table S30). In this contig we found a case similar to the one for the cytochrome complex IV described above, which had an average coverage lower than the majority of the analyzed contigs. However, in this case we also found that all the constituent genes affiliated consistently to the sulfur-oxidizing symbionts, and shared a coherent synteny with their genomes, supporting the idea of a genuine genomic region within a highly related community of sulfur-oxidizing γ-proteobacteria thriving in this ecosystem. On the other hand, only nirB was found in the GB and SI metagenomes, with high identity between themselves, but with low identity to the ESP nirB gene (Figure 2).
NirBD appears to be synthesized only during anaerobiosis, presumably to regenerate NAD+ and detoxify nitrite that accumulates as result of nitrate respiration (Gennis and Stewart, 1996). The detoxification function has been described for E. coli, whereas in various bacteria and fungi this enzyme typically mediates nitrate assimilation (Lin and Stewart, 1998). The assimilation process has been tested experimentally in Mycobacterium tuberculosis, where the presence of both narGHJI and nirBD genes are related to an assimilatory nitrate and nitrite reduction (Malm et al., 2009). Therefore, it is probable that the ESP sulfur-oxidizing community utilizes the excess of nitrate as an electron acceptor for energy generation through the sulfur-oxidizing pathway, but does not go further with the dissimilatory pathway, but reduces nitrite to ammonium (Figure 6); this “ammonification” process would not generate proton motive force and would only serve as an electron sink (Stewart, 1988). This was supported by the apparent absence of genes related to the subsequent steps in the dissimilatory denitrification pathway, the norCB and nosRZDFYL operons, of which no related sequences were detected in the contigs generated, nor in the singlets associated with the ESP SUP05 lineage. Also these genes were not found in the metagenomes of GB SUP05. The absence of a dissimilatory denitrification pathway seems evident than in both GB and ESP-GSO populations.
The ESP-GSO ammonification process fueled by sulfur oxidation would be analogous to the process occurring in the top sediment underlying the anoxic waters, where the trichome-forming sulfur-oxidizing bacteria Thioploca spp. chemolithotrophically reduce nitrate to ammonium (Otte et al., 1999). In sediments, the high input of biomass from the water column fuels microbial sulfate reduction at high yields, but the sulfide produced is rapidly removed in a stepwise manner by Thioploca mats, avoiding sulfide poisoning (Teske, 2010), coupling this to nitrate reduction to ammonium and using the energy generated to fix CO2 (Otte et al., 1999). These two spatially separated but parallel processes could represent an important source of ammonium for the anaerobic ammonia-oxidizing (anammox) community in the water column (Galán et al., 2012) or sediments, leading indirectly to nitrogen loss from the system in the form of N2. The coupling between dissimilatory nitrate reduction to ammonium and anammox has already been proposed for oxygen deficient waters off Peru (Lam et al., 2007) and over the Omani shelf in the Arabian Sea (Jensen et al., 2011). However, in both cases the heterotrophic DNRA process based on the NrfA nitrite reductase has been invoked, while in the case of the ESP-GSO sulfur-oxidizing bacteria, nitrate reduction to ammonium appears to be mediated by the NirBD encoded nitrite reductase, and coupled to sulfur oxidation and autotrophic carbon fixation (Figure 6).
Conclusion
Coastal upwelling ecosystems are important sites of biogeochemical cycling of nutrients, trace elements and greenhouse gases, cycling that is mediated by microorganisms. The loss of dissolved oxygen in marine waters, either through natural or anthropogenic causes, significantly alters the microbial community structure and the spatial structure and functions of associated biogeochemical cycles. Here, we have described the partial gene complement of the SUP05/ARCTIC96BD-19 clade found in the ESP, that suggests an extended metabolic potential and versatility for this group, that thrives in waters having annual cycles of widely varying oxygen concentrations. These bacteria have the potential to oxidize the sulfur oxyanion thiosulfate (Figure 6). This oxidation, in turn, may be coupled to nitrate reduction to nitrite, but instead of continuing with the dissimilative nitrite reduction to N2O (Walsh et al., 2009), the group appears capable of the reducing nitrogen to ammonia, thereby restoring instead of removing nitrogen from the system. The energy produced through the thiosulfate oxidation pathway with nitrate can be utilized for carbon fixation via RuBisCO (Figure 6). But additionally, there appears to be a complete TCA cycle in these microorganisms, suggesting their potential to sustain a mixotrophic lifestyle. Consistent with their potential to use DOC and their distribution in waters with highly variable oxygen concentrations, we found a complete oxidative phosphorylation complex, which may confer them the ability to thrive in micromolar concentrations of oxygen. Overall, these results emphasize the central role that this group of γ-proteobacteria may play in the coupling of carbon, nitrogen and sulfur cycles in oxygen-deficient marine waters, and highlight the occurrence of a metabolic diversity that has allowed the success a different ecotype of the SUP05/ARCTIC96BD-19 clade in a variety of environmental contexts.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Lucy Belmar for help in the field, collecting the samples that allowed the development of this study, and to the captain and crew of the L/C Kay-Kay II. This work was supported by the Gordon and Betty Moore Foundation through the Mi_Loco Project, grant MMI1661 to Universidad de Concepción, and MMI Investigator award to Edward F. DeLong, as well as by the Chilean Research Council (CONICYT) through the FONDECYT #3120047 grant to Alejandro A. Murillo. Additional support was provided by the Millennium Scientific Initiative, grant IC 120019 to Osvaldo Ulloa.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmars.2014.00018/abstract
References
Anantharaman, K., Breier, J. A., Sheik, C. S., and Dick, G. J. (2013). Evidence for hydrogen oxidation and metabolic plasticity in widespread deep-sea sulfur-oxidizing bacteria. Proc. Natl. Acad. Sci. U.S.A. 110, 330–335. doi: 10.1073/pnas.1215340110
Aravind, L., and Koonin, E. V. (2001). The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol. 2, 0007.1–0007.8. doi: 10.1186/gb-2001-2-3-research0007
Badger, M. R., and Bek, E. J. (2008). Multiple Rubisco forms in proteobacteria: their functional significance in relation to CO2 acquisition by the CBB cycle. J. Exp. Bot. 59, 1525–1541. doi: 10.1093/jxb/erm297
Bano, N., and Hollibaugh, J. (2002). Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean. Appl. Environ. Microbiol. 68, 505–518. doi: 10.1128/AEM.68.2.505-518.2002
Berg, R. A., and Prockop, D. J. (1973). Affinity column purification of protocollagen proline hydroxylase from chick embryos and further characterization of the enzyme. J. Biol. Chem. 248, 1175–1182.
Canfield, D. E., Stewart, F. J., Thamdrup, B., De Brabandere, L., Dalsgaard, T., Delong, E. F., et al. (2010). A cryptic sulfur cycle in oxygen-minimum-zone waters off the Chilean coast. Science 330, 1375–1378. doi: 10.1126/science.1196889
Chen, B. J., Carroll, P., and Samson, L. (1994). The Escherichia coli AlkB protein protects human cells against alkylation-induced toxicity. J. Bacteriol. 176, 6255–6261.
Chiang, R. C., Cavicchioli, R., and Gunsalus, R. P. (1992). Identification and characterization of narQ, a second nitrate sensor for nitrate-dependent gene regulation in Escherichia coli. Mol. Microbiol. 6, 1913–1923. doi: 10.1111/j.1365-2958.1992.tb01364.x
Dahl, C., Engels, S., Pott-Sperling, A. S., Schulte, A., Sander, J., Lübbe, Y., et al. (2005). Novel genes of the dsr gene cluster and evidence for close interaction of Dsr proteins during sulfur oxidation in the phototrophic sulfur bacterium Allochromatium vinosum. J. Bacteriol. 187, 1392–1404. doi: 10.1128/JB.187.4.1392-1404.2005
Darby, C., Cosma, C. L., Thomas, J. H., and Manoil, C. (1999). Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 96, 15202–15207. doi: 10.1073/pnas.96.26.15202
Dong, X. R., Li, S. F., and DeMoss, J. A. (1992). Upstream sequence elements required for NarL-mediated activation of transcription from the narGHJI promoter of Escherichia coli. J. Biol. Chem. 267, 14122–14128.
Drummond, A., Ashton, B., Buxton, S., Cheung, M., Cooper, A., Duran, C., et al. (2011). Geneious. Available online at: http://www.geneious.com/
Friedrich, C. G., Rother, D., Bardischewsky, F., Quentmeier, A., and Fischer, J. (2001). Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microbiol. 67, 2873–2882. doi: 10.1128/AEM.67.7.2873-2882.2001
Frigaard, N.-U., and Bryant, D. A. (2008). “Genomic and evolutionary perspectives on sulfur metabolism in green sulfur bacteria,” in Microbial Sulfur Metabolism, eds C. Dahl and C. G. Friedrich (New York, NY; Berlin; Heidelberg: Springer-Verlag), 60–73.
Fuchs, B. M., Woebken, D., Zubkov, M. V., Burkill, P., and Amann, R. (2005). Molecular identification of picoplankton populations in contrasting waters of the Arabian Sea. Aquat. Microb. Ecol. 39, 145–157. doi: 10.3354/ame039145
Galán, A., Molina, V., Belmar, L., and Ulloa, O. (2012). Temporal variability and phylogenetic characterization of planktonic anammox bacteria in the coastal upwelling ecosystem off central Chile. Prog. Oceanogr. 95, 110–120. doi: 10.1016/j.pocean.2011.07.007
Gennis, R. B., and Stewart, V. (1996). “Respiration,” in Escherchia coli and Salmonella: Cellular and Molecular Biology, eds F. C. Neidhardt, R. Curtis III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (Washington DC: ASM Press), 217–261.
Ghosh, W., and Dam, B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33, 999–1043. doi: 10.1111/j.1574-6976.2009.00187.x
Glaubitz, S., Kießlich, K., Meeske, C., Labrenz, M., and Jürgens, K. (2013). SUP05 dominates the Gammaproteobacterial sulfur oxidizer assemblages in pelagic redoxclines of the central Baltic and Black seas. Appl. Environ. Microbiol. 79, 2767–2776. doi: 10.1128/AEM.03777-12
Grimm, F., Franz, B., and Dahl, C. (2008). “Thiosulfate and sulfur oxidation in purple sulfur bacteria, microbial sulfur metabolism,” in Microbial Sulfur Metabolism, eds C. Dahl and C. G. Friedrich (New York, NY; Berlin; Heidelberg: Springer-Verlag), 101–116.
Hutton, J. J., Tappel, A. L., and Udenfriend, S. (1967). Cofactor and substrate requirements of collagen proline hydroxylase. Arch. Biochem. Biophys. 118, 231–240. doi: 10.1016/0003-9861(67)90302-5
Jensen, M. M., Lam, P., Revsbech, N. P., Nagel, B., Gaye, B., Jetten, M. S., et al. (2011). Intensive nitrogen loss over the Omani Shelf due to anammox coupled with dissimilatory nitrite reduction to ammonium. ISME J. 5, 1660–1670. doi: 10.1038/ismej.2011.44
Jormakka, M., Byrne, B., and Iwata, S. (2003). Proton motive force generation by a redox loop mechanism. FEBS Lett. 545, 25–30. doi: 10.1016/S0014-5793(03)00389-2
Kivirikko, K. I., Kishida, Y., Sakakibara, S., and Prockop, J. (1972). Hydroxylation of (X-Pro-Gly)n by protocollagen proline hydroxylase. Effect of chain length, helical conformation and amino acid sequence in the substrate. Biochim. Biophys. Acta 271, 347–356. doi: 10.1016/0005-2795(72)90209-7
Kondo, H., Nakabeppu, Y., Kataoka, H., Kuhara, S., Kawabata, S., and Sekiguchi, M. (1986). Structure and expression of the alkB gene of Escherichia coli related to the repair of alkylated DNA. J. Biol. Chem. 261, 15772–15777.
Kuwahara, H., Yoshida, T., Takaki, Y., Shimamura, S., Nishi, S., Harada, M., et al. (2007). Reduced genome of the thioautotrophic intracellular symbiont in a deep-sea clam, Calyptogena okutanii. Curr. Biol. 17, 881–886. doi: 10.1016/j.cub.2007.04.039
Lam, P., Jensen, M. M., Lavik, G., McGinnis, D. F., Müller, B., Schubert, C. J., et al. (2007). Linking crenarchaeal and bacterial nitrification to anammox in the Black Sea. Proc. Natl. Acad. Sci. U.S.A. 104, 7104–7109. doi: 10.1073/pnas.0611081104
Lam, P., and Kuypers, M. M. M. (2011). Microbial nitrogen cycling processes in oxygen minimum zones. Ann. Rev. Mar. Sci. 3, 317–345. doi: 10.1146/annurev-marine-120709-142814
Lavik, G., Stührmann, T., Brüchert, V., Van der Plas, A., Mohrholz, V., Lam, P., et al. (2009). Detoxification of sulphidic African shelf waters by blooming chemolithotrophs. Nature 457, 581–584. doi: 10.1038/nature07588
Lin, J. T., and Stewart, V. (1998). Nitrate assimilation by bacteria. Adv. Microb. Physiol. 39, 1–30, 379.
Malm, S., Tiffert, Y., Micklinghoff, J., Schultze, S., Joost, I., Weber, I., et al. (2009). The roles of the nitrate reductase NarGHJI, the nitrite reductase NirBD and the response regulator GlnR in nitrate assimilation of Mycobacterium tuberculosis. Microbiology 155, 1332–1339. doi: 10.1099/mic.0.023275-0
Marshall, K. T., and Morris, R. M. (2013). Isolation of an aerobic sulfur oxidizer from the SUP05/Arctic96BD-19 clade. ISME J. 7, 452–455. doi: 10.1038/ismej.2012.78
Molina, V., Belmar, L., and Ulloa, O. (2010). High diversity of ammonia-oxidizing archaea in permanent and seasonal oxygen-deficient waters of the eastern South Pacific. Environ. Microbiol. 12, 2450–2465. doi: 10.1111/j.1462-2920.2010.02218.x
Montero, P., Daneri, G., Cuevas, L. A., González, H. E., Jacob, B., Lizárraga, L., et al. (2007). Productivity cycles in the coastal upwelling area off Concepción: the importance of diatoms and bacterioplankton in the organic carbon flux. Prog. Oceanogr. 75, 518–530. doi: 10.1016/j.pocean.2007.08.013
Otte, S., Kuenen, J., Nielsen, L., Paerl, H., Zopfi, J., Schulz, H., et al. (1999). Nitrogen, carbon, and sulfur metabolism in natural thioploca samples. Appl. Environ. Microbiol. 65, 3148–3157.
Philippot, L. (2002). Denitrifying genes in bacterial and archaeal genomes. Biochim. Biophys. Acta 1577, 355–376. doi: 10.1016/S0167-4781(02)00420-7
Poretsky, R. S., Sun, S., Mou, X., and Moran, M. A. (2010). Transporter genes expressed by coastal bacterioplankton in response to dissolved organic carbon. Environ. Microbiol. 12, 616–627. doi: 10.1111/j.1462-2920.2009.02102.x
Pott, A. S., and Dahl, C. (1998). Sirohaem sulfite reductase and other proteins encoded by genes at the dsr locus of Chromatium vinosum are involved in the oxidation of intracellular sulfur. Microbiology 144, 1881–1894. doi: 10.1099/00221287-144-7-1881
Revsbech, N., Larsen, L., Gundersen, J., Dalsgaard, T., Ulloa, O., and Thamdrup, B. (2009). Determination of ultra-low oxygen concentrations in oxygen minimum zones by the STOX sensor. Limnol. Oceanogr. Methods 7, 371–381. doi: 10.4319/lom.2009.7.371
Roeselers, G., Newton, I. L. G., Woyke, T., Auchtung, T. A., Dilly, G. F., Dutton, R. J., et al. (2010). Complete genome sequence of Candidatus Ruthia magnifica. Stand. Genomic Sci. 3, 163–173. doi: 10.4056/sigs.1103048
Sánchez, O., Ferrera, I., Dahl, C., and Mas, J. (2001). In vivo role of adenosine-5′-phosphosulfate reductase in the purple sulfur bacterium Allochromatium vinosum. Arch. Microbiol. 176, 301–305. doi: 10.1007/s002030100327
Sander, J., Engels-Schwarzlose, S., and Dahl, C. (2006). Importance of the DsrMKJOP complex for sulfur oxidation in Allochromatium vinosum and phylogenetic analysis of related complexes in other prokaryotes. Arch. Microbiol. 186, 357–366. doi: 10.1007/s00203-006-0156-y
Shao, M.-F., Zhang, T., and Fang, H. H.-P. (2010). Sulfur-driven autotrophic denitrification: diversity, biochemistry, and engineering applications. Appl. Microbiol. Biotechnol. 88, 1027–1042. doi: 10.1007/s00253-010-2847-1
Sobarzo, M., Bravo, L., Donoso, D., Garcés-Vargas, J., and Schneider, W. (2007). Coastal upwelling and seasonal cycles that influence the water column over the continental shelf off central Chile. Prog. Oceanogr. 75, 363–382. doi: 10.1016/j.pocean.2007.08.022
Stevens, H., and Ulloa, O. (2008). Bacterial diversity in the oxygen minimum zone of the eastern tropical South Pacific. Environ. Microbiol. 10, 1244–1259. doi: 10.1111/j.1462-2920.2007.01539.x
Stewart, F. J., Ulloa, O., and DeLong, E. F. (2012). Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ. Microbiol. 14, 23–40. doi: 10.1111/j.1462-2920.2010.02400.x
Stewart, V. (1988). Nitrate respiration in relation to facultative metabolism in enterobacteria. Microbiol. Rev. 52, 190–232.
Sunamura, M., Higashi, Y., Miyako, C., Ishibashi, J., and Maruyama, A. (2004). Two bacteria phylotypes are predominant in the Suiyo Seamount hydrothermal plume. Appl. Environ. Microbiol. 70, 1190. doi: 10.1128/AEM.70.2.1190-1198.2004
Swan, B. K., Martinez-Garcia, M., Preston, C. M., Sczyrba, A., Woyke, T., Lamy, D., et al. (2011). Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science 333, 1296–1300. doi: 10.1126/science.1203690
Teske, A. (2010). Oceans. Cryptic links in the ocean. Science 330, 1326–1327. doi: 10.1126/science.1198400
Ulloa, O., Canfield, D. E., DeLong, E. F., Letelier, R. M., and Stewart, F. J. (2012). Microbial oceanography of anoxic oxygen minimum zones. Proc. Natl. Acad. Sci. U.S.A. 109, 15996–16003. doi: 10.1073/pnas.1205009109
Walker, M. S., and DeMoss, J. A. (1994). NarL-phosphate must bind to multiple upstream sites to activate transcription from the narG promoter of Escherichia coli. Mol. Microbiol. 14, 633–641. doi: 10.1111/j.1365-2958.1994.tb01302.x
Walsh, D. A., Zaikova, E., Howes, C. G., Song, Y. C., Wright, J. J., Tringe, S. G., et al. (2009). Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones. Science 326, 578–582. doi: 10.1126/science.1175309
Wei, Y. F., Carter, K. C., Wang, R. P., and Shell, B. K. (1996). Molecular cloning and functional analysis of a human cDNA encoding an Escherichia coli AlkB homolog, a protein involved in DNA alkylation damage repair. Nucleic Acids Res. 24, 931–937. doi: 10.1093/nar/24.5.931
Wood, A. P., Aurikko, J. P., and Kelly, D. P. (2004). A challenge for 21st century molecular biology and biochemistry: what are the causes of obligate autotrophy and methanotrophy? FEMS Microbiol. Rev. 28, 335–352. doi.: 10.1016/j.femsre.2003.12.001
Wright, J. J., Konwar, K. M., and Hallam, S. J. (2012). Microbial ecology of expanding oxygen minimum zones. Nat. Rev. Microbiol. 10, 381–394. doi: 10.1038/nrmicro2778
Zhang, S., and Bryant, D. A. (2011). The tricarboxylic acid cycle in cyanobacteria. Science 334, 1551–1553. doi: 10.1126/science.1210858
Keywords: ESP-GSO, metagenomic analysis, OMZ, SUP05, sulfur oxidizers
Citation: Murillo AA, Ramírez-Flandes S, DeLong EF and Ulloa O (2014) Enhanced metabolic versatility of planktonic sulfur-oxidizing γ-proteobacteria in an oxygen-deficient coastal ecosystem. Front. Mar. Sci. 1:18. doi: 10.3389/fmars.2014.00018
Received: 12 March 2014; Accepted: 16 June 2014;
Published online: 07 July 2014.
Edited by:
James Cotner, University of Minnesota, USAReviewed by:
Susannah Green Tringe, DOE Joint Genome Institute, USAEdward Hall, United States Geological Survey, USA
Julie A. Huber, Marine Biological Laboratory, USA
Copyright © 2014 Murillo, Ramírez-Flandes, DeLong and Ulloa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Osvaldo Ulloa, Departmento de Oceanografía & Instituto Milenio de Oceanografía, Universidad de Concepción, Casilla 160-C, 4070386 Concepción, Chile e-mail:b3VsbG9hQHVkZWMuY2w=