 Andreas J. Andersson
Andreas J. Andersson- Scripps Institution of Oceanography, University of California San Diego, La Jolla, CA, USA
The long-term success of coral reefs depends on a positive balance of calcium carbonate production exceeding dissolution, erosion, and material export. As a result of ocean acidification, coral reefs could transition from net accretion to net erosion owing to decreasing rates of calcification and increasing rates of chemical dissolution and bioerosion. Here, I present a fundamental paradigm that aims to explain the main driver of carbonate sediment dissolution on coral reefs based on theory and a new empirical dataset of pore water carbonate chemistry from the Bermuda coral reef platform. The paradigm shows that carbonate sediment dissolution is most strongly controlled by the extent of organic matter decomposition in the sediments, but that the magnitude of dissolution is influenced by how much decomposition is required to reach pore water undersaturation with respect to the most soluble bulk carbonate mineral phase present in the sediments, a condition defined as the Carbonate Critical Threshold (CCT). Decomposition of organic matter beyond the CCT under aerobic conditions results in stoichiometric proportional dissolution of carbonate sediments. As ocean acidification proceeds over the next several decades, the extent of organic matter decomposition required to reach the CCT will decrease, carbonate dissolution will increase, and subsequently the accumulation of carbonate sediments will decrease. Since drastic reductions in anthropogenic CO2 emission are unlikely in the foreseeable future, the paradigm shows that active controls and reduction of organic matter input to coral reefs at the local scale might be an effective mitigation strategy to prevent or delay coral reefs transitioning to a state of net dissolution.
Ocean Acidification and Increasing Coral Reef Dissolution
Oceanic uptake of anthropogenic CO2 has resulted in significant changes in the surface seawater dissolved inorganic carbon system and acid-base balance. Since the onset of the industrial revolution, the average surface seawater pH has decreased by 0.1 units, which corresponds to an increase in the hydrogen ion concentration and acidity by 25% (e.g., Orr et al., 2005; Doney et al., 2009). Future predictions suggest that surface seawater pH will decrease by another 0.3–0.4 units by the end of this century (Orr et al., 2005). These major changes in ocean chemistry combined with climate change have raised major concerns on the potential consequences to corals and coral reef ecosystems (Kleypas et al., 1999; Hoegh-Guldberg et al., 2007). A number of studies indicate that corals and other marine calcifiers will have an increasingly difficult time depositing their skeletons and shells made of CaCO3 under lower seawater pH conditions (Kroeker et al., 2013). Furthermore, bioerosion and dissolution of CaCO3 substrates, sediments, and structures are likely to increase (Andersson et al., 2009; Tribollet et al., 2009; Wisshak et al., 2012; Dove et al., 2013). It has also been proposed that these processes will be more sensitive to ocean acidification than the process of calcification (Andersson and Gledhill, 2013; Eyre et al., 2014). Models and mesocosm experiments suggest that coral reefs in general will transition from a condition of net accretion to net erosion as a result of the projected decreasing rates of calcification and increasing rates of chemical dissolution and bioerosion, but the timing of when, how fast, and at what seawater CO2 conditions this will occur varies (Andersson et al., 2009; Silverman et al., 2009; Dove et al., 2013). Despite these dire projections with potential devastating consequences to coral reef ecosystems and the millions of people dependent on these systems for nutrition, financial revenue, and protection from storms, the effect of ocean acidification on CaCO3 dissolution has received relatively little attention from both the scientific research community and the general public (Eyre et al., 2014).
Here, I present a theoretical paradigm based on fundamental biogeochemical principles and empirical data from the Bermuda coral reef platform describing the mechanisms and controls of coral reef carbonate sediment dissolution and how this will change under future anthropogenic ocean acidification. Note that throughout this manuscript the term CaCO3 refers to the general group of carbonate mineral phases (i.e., aragonite, Mg-calcite, and calcite) that make up contemporary coral reef carbonate sediments.
Materials and Methods
The empirical data for this study were based on surface seawater and pore water inorganic carbon chemistry measurements from the Bermuda coral reef platform collected between 2007–2012 (Figure 1). The surface seawater data have previously been reported in Andersson et al. (2014). Surface seawater samples were collected at four stations across the Bermuda coral reef platform at 0.5–1 m depth using a Niskin bottle every month between 2007 and 2012. On occasion, high-resolution spatial surveys were also conducted. The TA and DIC were sampled according to standard protocols (Dickson et al., 2007) using 250 ml Kimax brand glass sample bottles. Samples were immediately poisoned with 100 μL saturated solution of HgCl2. Temperature and salinity were measured in situ using YSI 556 multi-parameter sonde. On occasion samples for salinity were also analyzed using an auto-salinometer (Guildline Instruments). Bottom water and sediment pore water samples were collected every other month for 1 year starting in June 2011 from pore water wells (Falter and Sansone, 2000) permanently installed at 5 locations across the reef platform (Figure 1). Eight wells were installed at each site within an approximate area of 0.5×0.5 m with the capability to extract pore water from 2, 4, 6, 8, 10, 12, 16, and 20 cm sediment depth. Each 25 ml sample roughly represented the volume of a sphere with a radius of 2–3 cm depending on the porosity of the sediments at each site. The wells were staggered to minimize overlap and interference between sampling depths. Pore water was sampled by a diver during daytime using a 25 ml luer-lok syringe (no nighttime samples were collected). Back on the boat, temperature (±0.1°C), pH-NBS (±0.02), and salinity (±0.1) were immediately measured on a subsample using an Accumet AP72 portable pH/T/S meter. The remainder of the sample was carefully passed through a 0.45 μm Millipore filter into a 20 ml glass vial, treated with a 20 μl saturated solution of mercuric chloride (HgCl2), fitted with a rubber stopper, and crimped with an aluminum seal.
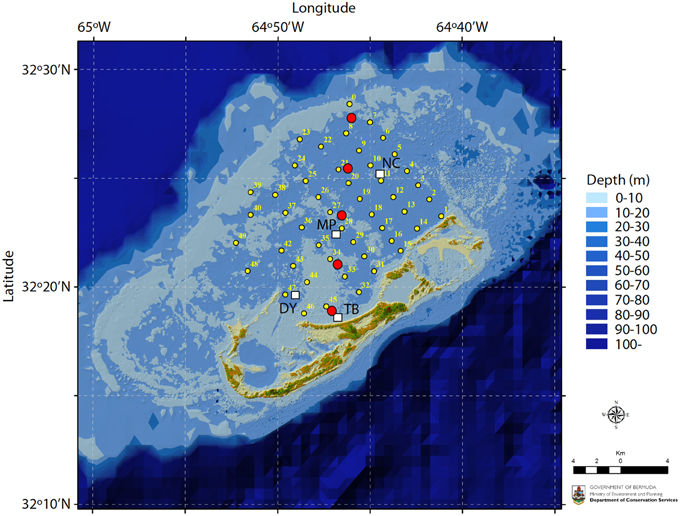
Figure 1. Map of the Bermuda coral reef platform. Surface seawater samples were collected monthly at stations TB, DY, MP, and NC between 2007–2012 (white squares) and in the winter and summer of 2010 at stations 1–49 (small yellow circles). Pore water samples were collected every other month between June 2011 and June 2012 at five locations traversing the platform (large red circles). Map credit: M. Shailer, Dept. of Conservation Services, Government of Bermuda.
DIC samples were determined by coulometry or infrared detection whereas TA samples were analyzed by closed cell acidimetric titration (e.g., Dickson et al., 2007; Dickson, 2010). Surface seawater DIC samples were analyzed using a UIC 5011 coulometer coupled with a VINDTA3C, VINDTA3D or a SOMMA system while pore waters DIC were analyzed using a LiCOR 7000 based AIRICA system. Surface seawater TA was analyzed using a VINDTA3S titration system. All instruments were manufactured or modified by Marianda Inc. (www.marianda.com). Performance and precision of the instruments were regularly checked using certified reference material (CRM) prepared by A. Dickson at Scripps Institution of Oceanography. The accuracy and precision of replicate CRMs analyzed in conjunction with surface seawater samples was on any given day of analysis typically in the range of ±2–4 μmol kg−1 for both TA and DIC. The mean accuracy and precision of replicate CRM injections analyzed in conjunction with pore water samples was ±4.6 μmol kg−1 (n = 118). Pore water TA was calculated based on pH and DIC measurements using CO2sys_v2.1 (Lewis and Wallace, 1998) at in situ temperature and salinity conditions based on stoichiometric constants defined by Peng et al. (1987) on the pH-NBS scale with K1 and K2 from Mehrbach et al. (1973).
Present Day Biogeochemical Function of Coral Reefs
When open ocean seawater flushes on to a coral reef, it has a certain composition of major ions, inorganic and organic carbon, and nutrients that reef organisms will utilize to produce organic material and calcium carbonate rock (Figure 2A). The ultimate energy that fuels these processes comes from the sun, but it is from these basic ingredients corals and other marine calcifiers build immense reef structures that provide a habitat for thousands of marine organisms.
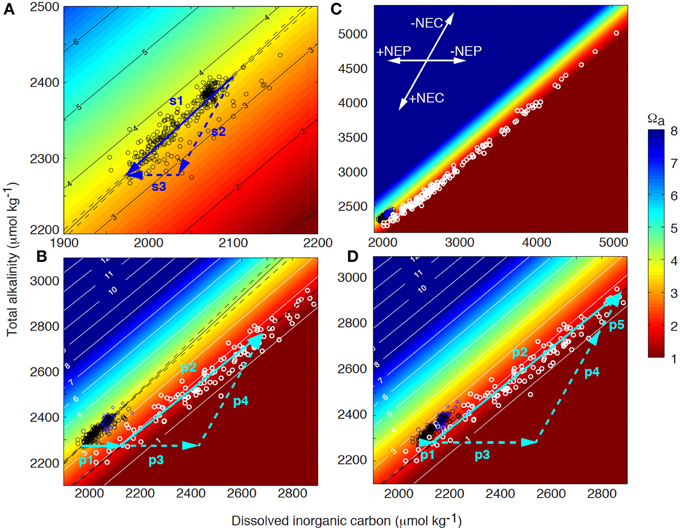
Figure 2. Coral reef surface seawater (black circles) and pore water (white circles) total alkalinity (TA) as a function of dissolved inorganic carbon (DIC) superimposed on contours of aragonite saturation state (Ωa) at 25°C and salinity of 36.5. (A) Net drawdown of surface seawater TA and DIC (vector s1) owing to net ecosystem organic carbon production (NEP, vector s3) and net ecosystem calcification (NEC, vector s2) as seawater transits the Bermuda coral reef platform (also see inset in panel C). (B) Surface seawater acts as the starting composition for pore water. Initially, DIC increases and Ωa decreases as a result of organic matter decomposition in the sediments (vector p1). The pore water Ωa will decrease until it becomes undersaturated with respect to the most soluble bulk carbonate mineral phase present in the sediments, which will start to dissolve at a condition referred to as the Carbonate Critical Threshold (CCT). Further, organic matter decomposition (vector p3) will drive dissolution (vector p4) with the sum of these processes illustrated by vector p2 (i.e., metabolic dissolution). The extent of dissolution is quantitatively controlled by the amount of organic matter decomposed beyond the CCT, and the resulting change in pore water TA and DIC, (C) is a function of the reaction rates and flushing with overlying surface seawater. (D) As a result of ocean acidification, surface seawater DIC will increase and Ωa decrease. Consequently, decomposition of less organic material will be required to reach the CCT (vector p1) and more organic material (vector p3) can fuel additional dissolution (vector p5) compared to present day conditions, i.e., decomposition of the same amount of organic matter (vectors p1+p3) will result in increasing dissolution under future high CO2 conditions (vectors p4 compared to p4+p5).
As seawater transits across a coral reef, different types of benthic algae and symbiotic algae living within corals utilize dissolved inorganic carbon () from the water column via the process of photosynthesis to generate organic matter and oxygen:
The organic matter is subsequently utilized by heterotrophic organisms on the reef as a source of food and energy. Concurrently, some organisms that could be either autotrophic or heterotrophic, manufacture skeletons and shells made of CaCO3 by combining calcium ions with DIC in the process of calcification:
The reason why many marine organisms deposit CaCO3 and how this process evolved is somewhat debated, but it provides a number of advantages including protection, a rigid structure, increasing surface area, and a method to keep up with sea level rise. The removal of calcium from the water column as a result of calcification alters seawater total alkalinity (TA = Σ[Na+ + 2Mg2+ + 2Ca2+ + 2Sr2+ + K+] − Σ[Cl− + ]), which somewhat simplified is a measure of the seawater buffer or acid neutralization capacity (e.g., Zeebe and Wolf-Gladrow, 2001). The greater the seawater TA, the greater the amount of DIC the seawater can hold. Equation (2) illustrates that calcification decrease TA by 2 moles and DIC by 1 mole for every mole of CaCO3 produced while organic carbon production mainly affects DIC (Equation 1; see inset in Figure 2). It is frequently assumed that production and decomposition of organic material have no effect on the seawater TA in coral reefs. Strictly speaking, this is not true as the associated uptake and release of nutrients actually do have an effect on the TA, but for coral reefs this effect is typically less than 5% of the change in DIC associated with this process (Atkinson and Smith, 1983; Watanabe et al., 2006) and can be neglected in most instances.
The net effect of the ecosystem organic carbon production (NEP = photosynthesis—autotrophic and heterotrophic respiration) and ecosystem calcification (NEC = calcification – CaCO3 dissolution) on the seawater TA-DIC balance can be illustrated in a Deffeyes diagram (Figure 2A; Deffeyes, 1965). Vector s1 in Figure 2A indicates the modification occurring to the seawater DIC and TA owing to net positive primary production and calcification as seawater transits across the Bermuda coral reef. The slope of this vector (TA vs. DIC) depends on the relative importance of the net inorganic and organic carbon cycles (i.e., NEC vs. NEP), with observed slopes from different coral reefs ranging from 0.3 to 2 (Suzuki and Kawahata, 2003; Watanabe et al., 2006; Andersson et al., 2014; Lantz et al., 2014). In a system where NEC and NEP are strongly coupled and balanced, this slope is close to 1, which can be inferred by combining Equations 1 and 2:
Furthermore, depending on the slope of the reaction pathway owing to the net reef metabolism, the seawater pH and saturation state with respect to CaCO3 (Ω) will be modified such that a slope <0.95 will increase Ω whereas a slope >0.95 will decrease Ω at a temperature of 25°C and salinity of 36.5 (Figure 2; Andersson and Gledhill, 2013). Thus, it is obvious that coral reefs have the ability to modify the seawater pH and Ω depending on the community composition and the relative importance of NEC and NEP (Anthony et al., 2011; Kleypas et al., 2011; Andersson et al., 2014). The seawater Ω is a thermodynamic index on the favorability of precipitation (Ω > 1) and CaCO3 dissolution (Ω < 1), and is for aragonite, the mineral deposited by corals, defined as: Ωa = [Ca2+][]/Ksp*, where brackets refers to the concentrations of the ions, and Ksp* is the ion concentration product at equilibrium. In addition to aragonite, coral reef sediments contain significant proportions biogenic Mg-calcites of varying Mg content, many which are more soluble than aragonite (Morse et al., 2006).
If net positive organic carbon production and calcification were continuously taking place, both particulate organic matter and inorganic CaCO3 would accumulate and constitute the bulk composition of coral reefs. However, except for the standing stock of organic material in living reef organisms, none to very little organic material (<1%) is found in the sediments of most coral reef environments (Yamamoto et al., 2001; Atkinson, 2011). Thus, most of the organic material that is produced locally or imported from the open ocean or adjacent terrestrial ecosystems is either exported or decomposed within the reef (i.e., Equation 1 reversed). Net respiration occurs in the dark at night, in sediment pore waters, and typically also in coral reef lagoons where organic material is deposited and relatively little benthic primary production occurs. The majority of decomposition takes place in the upper few centimeters of the sediments and is producing DIC, which causes a decrease in the pore water Ω (Figure 2B). Because of the restricted vertical space (mm to cm) and seawater volume over which this decomposition occurs, the seawater Ω and pH can be driven to very low values in the sediment pore waters (Morse et al., 1985; Morse and Mackenzie, 1990).
Based on the pore water carbonate chemistry data collected across the Bermuda coral reef platform, the pore water Ω will decrease until it becomes undersaturated with the most soluble bulk carbonate mineral phase present in the sediments, which at this point starts to dissolve (Figure 2B; Equation 2 reversed), a condition I refer to as the Carbonate Critical Threshold (CCT). In the Bermuda dataset, this occurs at an aragonite saturation state close to 1.75 ± 0.4, which corresponds to a Mg-calcite mineral phase at metastable equilibrium with the pore water containing 11–20 mol% MgCO3 (depending on the solubility curve used for the calculation; Morse et al., 2006). Mg-calcite mineral phases constitute 20–35% of Bermuda surface sediments with an average Mg-calcite composition of 13.3 ± 0.5 mol% for grain sizes <63 μm and a broader range of 8–16 mol% for grain sizes >63 μm (Andersson, unpublished data). In contrast to the Bermuda data, Yamamoto et al. (2015) observed the CCT at seawater Ωa of ~3 in sediments at Ishigaki Island, Japan.
Continued decomposition of organic material and production of CO2 beyond the CCT under aerobic conditions will drive stoichiometrically proportional CaCO3 sediment dissolution, which is evident based on the observed close to proportional increase in DIC and TA in the pore water (i.e., metabolic dissolution, Figures 2B,C; Emerson and Bender, 1981; Moulin et al., 1985; Hales and Emerson, 1997; Burdige et al., 2008):
The extent of DIC and TA increase (Figure 2C) is mainly a function of the rates of decomposition and CaCO3 dissolution, and the rate of seawater flushing, which is controlled by a number of factors including sediment grain size, depth, waves, and currents. Regardless of the resulting change in pore water chemistry, under aerobic conditions the total flux and the amount of CaCO3 dissolution is directly proportional and approximately equal to the amount of organic material decomposed beyond the CCT. For suboxic and anoxic conditions, this is not the case as anaerobic processes such as denitrification and sulfate reduction generate alkalinity that will counteract the reduction in pore water carbonate mineral saturation state caused by CO2 production (e.g., Morse and Mackenzie, 1990; Burdige, 2006). However, for denitrification, excess CO2 is still produced that can drive CaCO3 dissolution, but quantitatively less CaCO3 is dissolved for every mole of organic material that is remineralized compared to aerobic conditions (Emerson et al., 1982; Burdige, 2006). Nevertheless, nitrate/nitrite concentrations generated from organic matter decomposition in coral reef sediments are in general low (Atkinson and Smith, 1983), and the influence of denitrification on carbonate sediment dissolution is probably small. Similar to denitrification, sulfate reduction initially produce excess H+ that can drive CaCO3 dissolution, but as the process proceeds, production of excess alkalinity will increase the pore water carbonate mineral saturation state and promote precipitation of CaCO3 (Morse and Mackenzie, 1990; Walter and Burton, 1990; Burdige, 2006). This process is mostly important in areas of extensive organic matter deposition, in small grained sediments controlled by diffusional processes, and in the presence of Fe, which may form iron sulfides that will cause an increase in pore water alkalinity and pH (Walter and Burton, 1990). In permeable or intensely bioturbed coral reef carbonate sediments void of Fe, the influence of sulfate reduction on carbonate precipitation is probably small since oxygen is relatively rapidly resupplied in these sediments. Reoxidation of sulfide generated from sulfate reduction will produce acid that could drive carbonate dissolution (Aller, 1982).
For the purpose of the present paper, which mainly considers permeable carbonate sediments that make up the largest proportion of CaCO3 stored in coral reefs (Eyre et al., 2014), the relative contributions from decomposition of organic matter under suboxic and anoxic condition, and the influence on CaCO3 dissolution, were assumed to be minor relative to the influence of aerobic decomposition (Moulin et al., 1985). Hence, if this assumption is valid, in a coral reef system where organic carbon production and calcification are strongly coupled and roughly equal (Suzuki et al., 1995), and the trophic status is balanced (NEP = 0), the amount of decomposed organic material required to drive pore water Ω to undersaturation with respect to the most soluble bulk carbonate mineral phase, roughly corresponds to the amount of CaCO3 accumulation (ignoring the influence of bioerosion, transport processes, and suboxic/anoxic CaCO3 precipitation/dissolution). If this statement is correct, it has a number of implications:
(1) Higher surface seawater Ω promotes greater accumulation of CaCO3 because decomposition of more organic matter is required to reach the CCT.
(2) Lower surface seawater Ω promotes less accumulation of CaCO3 because decomposition of less organic matter is required to reach the CCT.
(3) External inputs of organic matter from terrestrial and open ocean ecosystems promote less accumulation of CaCO3 as decomposition of this extra material drive additional dissolution beyond the CCT.
(4) Less external organic matter and decomposition promotes greater accumulation of CaCO3 because less “fuel” is available to drive additional CaCO3 dissolution.
(5) Coral reef systems dominated by organic carbon cycling (TA vs. DIC slope <1), accumulate less CaCO3 than those dominated by inorganic carbon cycling (TA vs. DIC slope >1) because of higher relative levels of metabolic dissolution. Consequently, these systems exist closer to a condition of net dissolution.
It is important to note that the foundation for these hypotheses and predictions is based on the condition that no or little organic material accumulates in the sediments of coral reefs (Yamamoto et al., 2001; Atkinson, 2011) and that aerobic decomposition of organic matter is the dominant metabolic pathway. Should significant amounts of organic material be permanently buried or exported, additional CaCO3 would accumulate. However, because coral reefs and CaCO3 accumulation have significantly expanded and increased since the Last Glacial Maximum (LGM) (Vecsei and Berger, 2004), it can be inferred that the amount of organic matter deposited and decomposed within coral reefs during this time period has been insufficient to overcome both the CCT and the amount required to drive reefs to a conditions of net dissolution.
Future Biogeochemical Function of Coral Reefs
How will the observed biogeochemical function of coral reefs and CaCO3 accumulation described here be affected by ocean acidification? As a result of oceanic uptake of anthropogenic CO2, the surface seawater DIC will gradually increase in the open ocean, and thus, the DIC of the source water for coral reefs will increase while the seawater pH and Ω will decrease (Figure 2D). Consequently, significantly less organic matter decomposition will be required to reach the CCT (vector p1), more organic matter will be available to drive dissolution (vector p3), and consequently there will be less accumulation of CaCO3 compared to present day conditions. Vector p5 in Figure 2D illustrates the additional amount of TA and DIC generated from dissolution under a future scenario of ocean acidification compared to a present day scenario, assuming decomposition of the same amount of organic matter in both scenarios (vectors p1+p3). These quantitative vector illustrations demonstrate that smaller amounts of external input of organic material and decomposition will be required to push the reef to a condition of net dissolution in a high CO2 world. However, as the most soluble bulk carbonate mineral phase in the sediments change as a result of dissolution, the CCT will also gradually change toward lower Ωa, potentially partly counteracting the change in surface seawater chemistry. Nonetheless, the rate of change in surface seawater chemistry owing to rising atmospheric CO2 is much faster than current estimates of carbonate dissolution rates and the resulting changes in sediment bulk mineral composition (Andersson et al., 2005; Morse et al., 2006).
In addition to lower seawater Ω arising from ocean acidification, changes in reef biogeochemical processes may also influence the amount of organic matter decomposition required to reach the CCT. The projected future changes in seawater chemistry in conjunction with warming and overfishing could change the community structure of coral reefs, and thus, the relative importance of organic and inorganic carbon cycling (i.e., NEP vs. NEC). Specifically, it has been hypothesized that reefs could transition from domination by calcifiers to non-calcifying macroalgae (Hoegh-Guldberg et al., 2007). Such changes could potentially alter the reaction pathway and consequently the influence by reef metabolism on seawater chemistry as seawater transits across reefs (Suzuki and Kawahata, 2003; Watanabe et al., 2006; Andersson et al., 2014). This could alter the starting chemical composition of the pore water depending on the relative change in NEP and NEC, and could either increase or decrease the amount of organic matter decomposition required to reach the CCT relative to a non-altered reaction pathway.
For many coral reefs significantly influenced by human activities, increasing input of organic material and nutrients combined with warmer sea surface temperature and faster decomposition of organic matter are likely to experience additional CaCO3 dissolution on top of the increase resulting from ocean acidification (Cai et al., 2011). Furthermore, decreasing rates of calcification, increasing bioerosion, and increasing frequency and intensity of storms will also contribute to accelerate net erosion of coral reefs (Andersson and Gledhill, 2013). The threshold when coral reefs will undergo net dissolution will be different between different locations depending on local factors and processes, but given the rate of increasing CO2 emissions, and preliminary empirical results from a handful of studies (Andersson et al., 2009; Silverman et al., 2009; Fabricius et al., 2011), there is little doubt that most reefs will experience this transition during this or the next century.
What are the implications of a net dissolving coral reef? One can easily deduce that a reef that is losing more CaCO3 than it can produce will not be able to sustain itself indefinitely. Similarly, it is well known that coral reef biodiversity is strongly linked to the structural complexity of the reef (Fabricius et al., 2011), and without this complexity, a reef cannot support as high and diverse number of species. The question though is how fast will coral reefs dissolve and break down once this threshold has been surpassed? At this time, we do not know the answer to this critical question, but evidence from severely bleached coral reefs resulting in mass coral mortality, suggest that complex reef structures that have been built up over hundreds of years could erode away in relatively short time periods with small chances of rapid recovery (Eakin, 1996; Manzello et al., 2014). A process facilitated by high rates of bioerosion by sea urchins, sponges, and other critters. However, assuming that the majority of reefs will not experience such catastrophic erosion resulting from the combination of coral bleaching, mortality, bioerosion, and dissolution, it is the organic matter input and decomposition that are the major controlling factors of carbonate sediment dissolution on these reefs. In a future high CO2 world, this will still be the case, but the amount of organic matter required to reach the CCT will be less compared to present conditions. Consequently, in coral reefs strongly influenced by external organic matter inputs, an effective mitigation strategy might be to attempt to control these inputs and the subsequent organic matter decomposition since it is unlikely CO2 emissions to the atmosphere will be reduced in the foreseeable future. This strategy can be managed locally and conducted over a range of scales and costs, but further investigations are needed to evaluate the efficiency and the practicality of this strategy. Combined with other local actions aimed at minimizing sedimentation, nutrification, and maximizing healthy fish populations, these actions are likely to promote healthier coral reefs and greater resistance against ocean acidification. Additional studies are also needed investigating the possibility to promote actively coral reef growth by manipulating seawater chemistry at the local scale by for example seaweed farming, alkalinity additions, or feeding of corals.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author gratefully acknowledges support from NSF Grants OCE 09-28406 and OCE 12-55042 and NOAA grant NA10AR4310094. I am also thankful to Tim Noyes, Andrew Collins and Kyra Freeman for assistance with sample collection and analyses, and Professor Fred Mackenzie at the University of Hawaii for providing comments on an early draft of this manuscript. Constructive comments from the two reviewers also significantly improved this manuscript.
References
Aller, R. C. (1982). Carbonate dissolution in nearshore terrigenous muds: the role of physical and biological reworking. J. Geol. 90, 98–154.
Andersson, A. J., and Gledhill, D. (2013). Ocean acidification and coral reefs: effects on breakdown, dissolution and net ecosystem calcification. Ann. Rev. 5, 321–348. doi: 10.1146/annurev-marine-121211-172241
Andersson, A. J., Kuffner, I. B., Mackenzie, F. T., Jokiel, P. L., Rodgers, K. S., and Tan, A. (2009). Net loss of CaCO3 from a subtropical calcifying community due to seawater acidification: mesocosm-scale experimental evidence. Biogeosciences 6, 1811–1823. doi: 10.5194/bg-6-1811-2009
Andersson, A. J., Mackenzie, F. T., and Lerman, A. (2005). Coastal ocean and carbonate systems in the high CO2 world of the Anthropocene. Am. J. Sci. 305, 875–918. doi: 10.2475/ajs.305.9.875
Andersson, A. J., Yeakel, K., Bates, N. R., and de Putron, S. (2014). Partial offsets in ocean acidification from changing coral reef biogeochemistry. Nat. Clim. Change 4, 56–61. doi: 10.1038/nclimate2050
Anthony, K. R. N., Kleypas, J. A., and Gattuso, J.-P. (2011). Coral reefs modify their seawater carbon chemistry – implications for impacts of ocean acidification. Glob. Change Biol. 17, 3655–3666. doi: 10.1111/j.1365-2486.2011.02510.x
Atkinson, M. (2011). “Carbon fluxes in coral reefs,” in Encyclopedia of modern coral reefs: Structure, Form and Process, ed D. Hopley (Dordrecht: Springer), 181–185.
Atkinson, M. J., and Smith, S. V. (1983). C:N:P ratios of benthic marine plants. Limnol. Oceanogr. 28, 568–574. doi: 10.4319/lo.1983.28.3.0568
Burdige, D. J., Zimmerman, R. C., and Hu, X. (2008). Rates of carbonate dissolution in permeable sediments from pore-water profiles: the role of sea grasses. Limnol. Oceanogr. 53, 549–565. doi: 10.4319/lo.2008.53.2.0549
Cai, W.-J., Hu, X., Huang, W.-J., Murell, M. C., Lehrter, J. C., Lohrenz, S. E., et al. (2011). Acidification of subsurface coastal waters enhanced by eutrophication. Nat. Geosci. 4, 766–770, doi: 10.1038/ngeo1297
Deffeyes, K. S. (1965). Carbonate equilibria: a graphic and algebraic approach. Limnol. Oceanogr. 10, 412–426.
Dickson, A. G. (2010). “The carbon dioxide system in seawater: equilibrium chemistry and measurements,” in Guide to Best Practices for Ocean Acidification Research and Data Reporting, eds U. Riebesell, V. J. Fabry, L. Hansson, and J.-P. Gattuso (Brussels: European Commission, Directorate-General for Research), 17–40.
Dickson, A. G., Sabine, C. L., and Christian, J. R. (eds.). (2007). Guide to Best Practices for Ocean CO2 Measurements. PICES special publication 3, IOCCP.
Doney, S. C., Fabry, V. J., Feely, R. A., and Kleypas, J. A. (2009). Ocean acidification: the other CO2 problem. Ann. Rev. Mar. Sci. 1, 169–192. doi: 10.1146/annurev.marine.010908.163834
Dove, S. G., Kline, D. I., Pantos, O., Angly, F. E., Tyson, G. W., and Hoegh-Guldberg, O. (2013). Future reef decalcification under a business-as-usual CO2 emission scenario. Proc. Natl. Acad. Sci. U.S.A. 110, 15342–15347. doi: 10.1073/pnas.1302701110
Eakin, C. M. (1996). Where have all the carbonates gone? A model comparison of calcium carbonate budgets before and after the 1982-1983 El Nino at Uva Island in the eastern Pacific. Coral Reefs 15, 109–119.
Emerson, S., Grundmanis, V., and Graham, D. (1982). Carbonate chemistry in marine pore waters: MANOP sites C and S. Earth Planet. Sci. Lett. 61, 220–232.
Emerson, S. R., and Bender, M. L. (1981). Carbon fluxes at the sediment-water interface in the deep-sea: calcium carbonate preservation. J. Mar. Res. 39, 139–162.
Eyre, B., Andersson, A. J., and Cyronak, T. (2014). Benthic coral reef calcium carbonate dissolution in an acidifying ocean. Nat. Clim. Change 4, 969–976, doi: 10.1038/nclimate2380
Fabricius, K. E., Langdon, C., Uthicke, S., Humphrey, C., Noonan, S., De'ath, G., et al. (2011). Losers and winners in coral reefs acclimatized to elevated carbon dioxide concentrations. Nat. Clim. Change 1, 165–169. doi: 10.1038/nclimate1122
Falter, J. L., and Sansone, F. J. (2000). Shallow pore water sampling in reef sediments. Coral Reefs 19, 93–97. doi: 10.1007/s003380050233
Hales, B., and Emerson, S. (1997). Calcite dissolution in sediments of the Ceara Rise: in situ measurements of porewater O2, pH, and CO2(aq). Geochim. Cosmochim. Acta 61, 501–514.
Hoegh-Guldberg, O., Mumby, P. J., Hooten, A. J., Steneck, R. S., Greenfield, P., Gomez, E., et al. (2007). Coral reefs under rapid climate change and ocean acidification. Science 318, 1737–1742. doi: 10.1126/science.1152509
Kleypas, J. A., Anthony, K. R. N., and Gattuso, J.-P. (2011). Coral reefs modify their seawater carbon chemistry – case study from a barrier reef (Moorea, French Polynesia). Glob. Change Biol. 17, 3667–3678. doi: 10.1111/j.1365-2486.2011.02530.x
Kleypas, J. A., Buddemeier, R. W., Archer, D., Gattuso, J.-P., Langdon, C., and Opdyke, B. N. (1999). Geochemical consequences of increased atmospheric carbon dioxide on coral reefs. Science 284, 118–120. doi: 10.1126/science.284.5411.118
Kroeker, K. J., Kordas, R. L., Crim, R., Hendriks, I. E., Ramajo, L., Singh, G. S., et al. (2013). Impacts of ocean acidification on marine organisms: quantifying sensitivities and interaction with warming. Glob. Change Biol. 19, 1884–1896. doi: 10.1111/gcb.12179
Lantz, C. A., Atkinson, M. J., Winn, C. W., and Kahng, S. E. (2014). Dissolved inorganic carbon and total alkalinity of a Hawaiian fringing reef: chemical techniques for monitoring the effects of ocean acidification on coral reefs. Coral Reefs 33, 105–115. doi: 10.1007/s00338-013-1082-5
Lewis, E., and Wallace, D. W. R. (1998). Program developed for CO2 system calculations, ORNL/CDIAC-105, Carbon Dioxide Inf. Anal. Cent., Oak Ridge Natl. Lab. OakRidge, TN: U.S. Department of Energy.
Manzello, D. P., Enochs, I. C., Bruckner, A., Renaud, P. G., Kolodziej, G., Budd, D. A., et al. (2014). Galapagos coral reef persistence after ENSO warming across an acidification gradient. Geophys. Res. Lett. 41, 9001–9008. doi: 10.1002/2014GL062501
Mehrbach, C., Culberson, C.H., Hawley, J.E., and Pytkowicz, R.N. (1973). Measurement of the apparent dissociation constants of carbonic acid in seawater at atmospheric pressure. Limnol. Oceanogr. 18, 897–907.
Morse, J. W., Andersson, A. J., and Mackenzie, F. T. (2006). Initial responses of carbonate-rich shelf sediments to rising atmospheric pCO2 and “ocean acidification”: role of high Mg-calcite. Geochim. Cosmochim. Acta 70, 5814–5830, doi: 10.1016/j.gca.2006.08.017
Morse, J. W., and Mackenzie, F. T. (1990). Geochemistry of Sedimentary Carbonates. Amsterdam: Elsevier Science and Technology.
Morse, J. W., Zullig, J. J., Bernstein, L. D., Millero, F. J., Milne, P., Mucci, A., et al. (1985). Chemistry of calcium carbonate-rich shallow water sediments in the Bahamas. Am. J. Sci. 285, 147–185.
Moulin, E., Jordens, A., and Wollast, R. (1985). “Influence of the aerobic bacterial respiration on the early dissolution of carbonates in coastal sediments,” in Progress in Belgian Oceanographic Research: Proceedings of a Symposium Held at the Palace of Academies Brussels, R. Van Griekenand R. Wollast Brussels, 196–208.
Orr, J. C., Fabry, V. J., Aumont, O., Bopp, L., Doney, S. C., Feely, R. A., et al. (2005). Anthropogenic ocean acidification over the twenty-first century and its impacts on calcifying organisms. Nature 437, 681–686. doi: 10.1038/nature04095
Peng, T. H., Takahashi, T., and Broecker, W. S. (1987). Seasonal variability of carbon dioxide, nutrients and oxygen in the northern North Atlantic surface water: observations and a model. Tellus B 39, 439–458.
Silverman, J., Lazar, B., Cao, L., Caldeira, K., and Erez, J. (2009). Coral reefs may start dissolving when atmospheric CO2 doubles. Geophys. Res. Lett. 36, L05606. doi: 10.1029/2008GL036282
Suzuki, A., and Kawahata, H. (2003). Carbon budget of coral reef systems: an overview of observations in fringing reefs, barrier reefs and atolls in the Indo-Pacific regions. Tellus B 55, 428–444. doi: 10.1034/j.1600-0889.2003.01442.x
Suzuki, A., Nakamori, T., and Kayanne, H. (1995). The mechanism of production enhancement in coral reef carbonate systems: model and empirical results. Sediment. Geol. 99, 259–280.
Tribollet, A., Godinot, C., Atkinson, M., and Langdon, C. (2009). Effects of elevated pCO2 on dissolution of coral carbonates by microbial euendoliths. Glob. Biogeochem. Cycles 23, GB3008. doi: 10.1029/2008GB003286
Vecsei, A., and Berger, W. H. (2004). Increase of atmospheric CO2 during deglaciation: constraints on the coral reef hypothesis from patterns of deposition. Glob. Biogeochem. Cycles 18, GB1035. doi: 10.1029/2003GB002147
Walter, L. M., and Burton, E. A. (1990). Dissolution of recent platform carbonate sediments in marine pore fluids. Am. J. Sci. 290, 601–643.
Watanabe, A., Kayanne, H., Hata, H., Kudo, S., Nozaki, K., Kato, K., et al. (2006). Analysis of the seawater CO2 system in the barrier reef-lagoon system of Palau using total alkalinity-dissolved inorganic carbon diagrams. Limnol. Oceanogr. 51, 1614–1628. doi: 10.4319/lo.2006.51.4.1614
Wisshak, M., Schönberg, C. H. L., Form, A., and Freiwald, A. (2012). Ocean acidification accelerates reef bioerosion. PLoS ONE 7:e45124. doi: 10.1371/journal.pone.0045124
Yamamoto, M., Kayanne, H., and Yamamuro, M. (2001). Characteristic of organic matter in lagoonal sediments from the Great Barrier Reef. Geochem. J. 35, 385–401. doi: 10.2343/geochemj.35.385
Yamamoto, S., Kayanne, H., Tokoro, T., Kuwae, T., and Watanabe, A. (2015). Total alkalinity flux in coral reefs estimated from eddy covariance and sediment pore-water profiles. Limnol. Oceanogr. 60, 229–241. doi: 10.1002/lno.10018
Keywords: CaCO3, dissolution, ocean acidification, coral reef, carbonate sediments
Citation: Andersson AJ (2015) A fundamental paradigm for coral reef carbonate sediment dissolution. Front. Mar. Sci. 2:52. doi: 10.3389/fmars.2015.00052
Received: 04 May 2015; Accepted: 03 July 2015;
Published: 21 July 2015.
Edited by:
Hajime Kayanne, The University of Tokyo, JapanReviewed by:
Shoji Yamamoto, The University of Tokyo, JapanAtsushi Watanabe, Tokyo Institute of Technology, Japan
Copyright © 2015 Andersson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas J. Andersson, Scripps Institution of Oceanography, University of California, San Diego, 9500 Gilman Dr. #0244 La Jolla, CA 92093-0244, USA,YWFuZGVyc3NvbkB1Y3NkLmVkdQ==