 Camille A. Daniels1
Camille A. Daniels1 Sebastian Baumgarten1Lauren K. Yum1Craig T. Michell1
Sebastian Baumgarten1Lauren K. Yum1Craig T. Michell1 Till Bayer1,2Chatchanit Arif1
Till Bayer1,2Chatchanit Arif1 Cornelia Roder1
Cornelia Roder1 Ernesto Weil3
Ernesto Weil3 Christian R. Voolstra1*
Christian R. Voolstra1*- 1Division of Biological and Environmental Science and Engineering, Red Sea Research Center, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
- 2GEOMAR Department: Evolutionary Ecology of Marine Fishes, GEOMAR Helmholtz Centre for Ocean Research, Kiel, Germany
- 3Department of Marine Sciences, University of Puerto Rico, Mayaguez, Puerto Rico
White Plague Disease (WPD) is implicated in coral reef decline in the Caribbean and is characterized by microbial community shifts in coral mucus and tissue. Studies thus far have focused on assessing microbial communities or the identification of specific pathogens, yet few have addressed holobiont response across metaorganism compartments in coral disease. Here, we report on the first metatranscriptomic assessment of the coral host, algal symbiont, and microbial compartment in order to survey holobiont structure and function in healthy and diseased samples from Orbicella faveolata collected at reef sites off Puerto Rico. Our data indicate holobiont-wide as well as compartment-specific responses to WPD. Gene expression changes in the diseased coral host involved proteins playing a role in innate immunity, cytoskeletal integrity, cell adhesion, oxidative stress, chemical defense, and retroelements. In contrast, the algal symbiont showed comparatively few expression changes, but of large magnitude, of genes related to stress, photosynthesis, and metal transport. Concordant with the coral host response, the bacterial compartment showed increased abundance of heat shock proteins, genes related to oxidative stress, DNA repair, and potential retroelement activity. Importantly, analysis of the expressed bacterial gene functions establishes the participation of multiple bacterial families in WPD pathogenesis and also suggests a possible involvement of viruses and/or phages in structuring the bacterial assemblage. In this study, we implement an experimental approach to partition the coral holobiont and resolve compartment- and taxa-specific responses in order to understand metaorganism function in coral disease.
Introduction
Coral disease is a major threat to coral reef ecosystems that has increased and contributed to the decline of reef ecosystems globally over the last 40 years (Weil and Rogers, 2011). While the number of diseases reported worldwide ranges from 18 to 29 (Pollock et al., 2011), many are yet to be characterized and few etiological agents have been identified. Coral diseases that affect multiple species, such as White Plague Disease (WPD), are of particular interest, since reef diversity and structure is adversely impacted on large scales.
WPD is described as a bacterial infection (Dustan, 1977; Richardson et al., 1998) reported to potentially afflict over 40 coral species (Weil et al., 2006) and is responsible for several local and widespread epizootic events that have caused significant reef degradation in the Caribbean (Miller et al., 2009; Weil and Croquer, 2009). Reports of three WPD types (I, II, and III) in the Caribbean are distinguished by disease progression rates (Dustan, 1977; Richardson et al., 1998), which range from mm to cm of tissue loss per day.
Aurantimonas coralicida and Thalassomonas loyana were independently confirmed as WPD pathogens in different coral species and geographic locations (Denner et al., 2003; Thompson et al., 2006). However, recent surveys of colonies showing WPD signs failed to identify either bacterial pathogen (Pantos et al., 2003; Barash et al., 2005; Sunagawa et al., 2009; Cárdenas et al., 2012; Roder et al., 2014a). Roder et al. (2014a) recently described similar bacterial profiles in two coral genera (Porites and Pavona) that exhibited WPD-like signs in Thailand. Subsequent comparisons to Caribbean samples (Orbicella faveolata and Orbicella franksi) suggest a conserved disease microbiome across oceans (Roder et al., 2014b), supporting a view that bacterial community changes are likely a result of opportunistic bacteria induced by environmental stressors that compromise coral immunity (Lesser, 2007). In addition to ongoing studies targeting bacterial community changes during WPD progression (Sunagawa et al., 2009; Cárdenas et al., 2012; Roder et al., 2014a,b), a recent metagenomic survey of Orbicella annularis found distinct viral communities associated with healthy, WPD-affected, and bleached samples (Soffer et al., 2014). But causation still remains elusive and few studies have addressed coral holobiont response to disease. Using a combination of 16S and cDNA microarrays, Closek et al. (2014) found an increase in microbial diversity in Yellow Band Disease (YBD)-infected colonies and reduced expression of defense- and metabolism-related genes in the coral host Orbicella faveolata [formerly Monstastraea faveolata (Budd et al., 2012)].
To further understand the response of the coral holobiont to coral disease, we employed a metatranscriptomic sequencing approach to target the coral host, algal symbiont, and the associated microbial community through comprehensive sequencing of the prokaryotic and eukaryotic RNA fractions from healthy and WPD-infected colonies of the Caribbean coral O. faveolata. To do this, we partitioned total RNA into polyA+ and polyA− fractions. PolyA+ RNA was sequenced to analyze expression of coral host and algal symbiont genes. Gene expression in the bacterial community was assayed by sequencing of rRNA-reduced polyA− RNA (Stewart et al., 2010). Our aim was to establish a metatranscriptomic framework that allows examination of functional patterns across holobiont members to dissect how coral holobiont compartments respond. We were further interested in elucidating whether responses occur between holobiont members that would suggest a coordinated interplay.
Materials and Methods
Sample Collection
O. faveolata specimens were collected by SCUBA at Weimberg reef off La Parguera, southwest coast of Puerto Rico (between N 17°53′17.40/W 66°59′52.90 and N 17°53′25.40/W 66°59′19.00) on September 5 and 6, 2011. Samples were collected at 20 ± 2 m depth with hammer and chisel from healthy (HH) colonies and those displaying phenotypic signs of White Plague Disease (DD) (Weil and Croquer, 2009). Diseased colonies displayed an abrupt, white lesion line that spanned the colony surface, and separated living tissue from algal-colonized dead coral skeleton. No bleaching was observed on specimens. DD samples were taken at the disease margin interface of healthy and diseased tissues sensu Roder et al. (2014b), whereas samples from HH colonies were chiseled off the uppermost part of the colonies. A total of 2 DD and 2 HH samples were collected that represent distinct specimens from the identical coral colonies as those analyzed in Roder et al. (2014b). All samples were handled wearing gloves and directly transferred into sterile Whirl-Pak sampling bags upon collection. On board, specimens were rinsed with filtered seawater (0.22 μm). All samples were kept on ice during transportation and were subsequently flash frozen in liquid nitrogen before being stored at −80°C.
Metatranscriptome and RNAseq Library Generation
For generation of the metatranscriptomes, we followed a protocol by Stewart et al. (2010) with the addition of a polyA+ separation step. Briefly, after separation of polyA+ eukaryotic mRNAs via magnetic bead-labeled oligo(dT)s, rRNAs were reduced in the polyA− prokaryotic mRNA compartment via differential hybridization of the polyA− fraction to biotinylated rRNA probes generated from 16S-, 23S-, 18S-, 28S-specific PCRs on DNA extracted from the same samples. For DNA isolation, coral fragments were crushed to powder using an autoclaved CryoCup (BioSpec Products, Inc., Bartlesville, OK) and pestle. DNA was extracted from 100 mg of coral powder with the DNEasy Plant kit (Qiagen, Hilden, Germany). DNAs were pooled by condition and 16S, 23S, 18S, 28S PCRs were run with T7-amended rRNA primer sets using ~20–25 ng DNA (Stewart et al., 2010). Total RNA extractions with on-column DNase digestions as per manufacturer's instructions were performed on 100 mg of coral powder using the Qiagen RNeasy Kit. Samples were purified with the MinElute kit (Qiagen), eluted in 14 μL RNase free water, and total RNA quality was assayed via Bioanalyzer. Eukaryotic (i.e., coral host, algal symbiont, remaining eukaryotes) polyA+ mRNA was isolated using the DynaBeads mRNA purification kit (Invitrogen, Carlsbad, CA) sensu Moitinho-Silva et al. (2014) on 0.35–1 μg of total RNA. This yielded the following samples: 1_HH_polyA+, 2_HH_polyA+, 1_DD_polyA+, 2_DD_polyA+. The remaining polyA− mRNA fraction contained bacterial mRNAs, rRNAs, and other RNAs. rRNAs were reduced via subtractive hybridization of biotinylated sample-specific rRNA probes following the protocol by Stewart et al. (2010). This yielded the following samples: 1_HH_polyA−, 2_HH_polyA−, 1_DD_polyA−, 2_DD_polyA−. Each polyA+ fraction was amplified with the MessageAmp Kit II aRNA kit (Ambion, Austin, TX) according to manufacturer's instructions.
Overlapping paired-end libraries with 180 bp insert lengths were generated from all samples (i.e., 1_HH_polyA+, 2_HH _polyA+, 1_DD_polyA+, 2_DD_polyA+, 1_HH_polyA−, 2_HH_polyA−, 1_DD_polyA−, and 2_DD_polyA−) with the TruSeq v1 kit (llumina, San Diego, CA). The libraries were multiplexed at a 5–10 nM range and 8 pmol were sequenced on a single HiSeq2000 lane at the KAUST BioSicence Core facility (Thuwal, Saudi Arabia).
Data Processing, Transcriptome Assembly, and Gene Expression Analysis
An overview of the analytical pipeline devised in this study for assessing holotranscriptome response of O. faveolata to WPD is outlined in the Supplemental Material (Supplementary Data 1). FASTQ files for all paired-end libraries were imported into FastQC software (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc) to inform downstream processing and filtering. Adapter removal, read and quality trimming were completed in Trimmomatic-0.22 (Bolger et al., 2014), where a 4 bp sliding window was applied to retain bases with quality scores > 20, and only complete read pairs were retained. Trimmed paired-end reads from the polyA+ libraries were error-corrected using the ALLPATHS-LG v.47609 standalone module (Gnerre et al., 2011) and assembled with the Trinity pipeline (version: r20131110) (Haas et al., 2013) to generate a de novo reference transcriptome (67,593 genes > 200 bp, mean length: 541 bp, N50: 656 bp) that contained assembled gene loci from coral, Symbiodinium, and other eukaryotes (e.g., endolithic algae, fungi, etc.) (Supplementary Data 2). Reference assembly annotations were determined by subsequent BLASTX queries against SwissProt and TrEMBL (Altschul et al., 1990; Boeckmann et al., 2003; Bayer et al., 2012). 20,638 of 67,593 genes (30.5%) were annotated at < 1e−5 (Supplementary Data 3). Gene ontology annotations were determined by mapping SwissProt/TrEMBL IDs to the UniProt-GOA database (Dimmer et al., 2012). Protein domains were assigned by InterProScan implemented in UniProt KnowledgeBase (Hunter et al., 2009; Magrane and Consortium, 2011). polyA+ libraries were mapped to the reference transcriptome using Bowtie2-2.1.0 (Langmead and Salzberg, 2012) and loaded into SAMtools-0.1.18 (Li et al., 2009) to create sorted BAM files. These were analyzed in eXpress-1.3.0 (Roberts and Pachter, 2013) to quantify transcript abundances for each gene (read counts and FPKM), imported into edgeR (Robinson et al., 2010), and filtered according to gene loci >0-counts-per-million present in both samples. Subsequently, TMM library normalization and dispersion estimation were conducted. An exact test was performed on negative binomial fitted data to determine differentially expressed genes between HH and DD samples at an FDR cutoff of < 0.1 (Benjamini and Hochberg, 1995). Expression data for eukaryotic genes are reported as log2 fold changes (Supplementary Data 4).
To sort the reference transcriptome into holobiont compartments (i.e., coral, Symbiodinium, other), all 67,593 genes were queried (BLASTN) against a custom database (492,343 sequences; Supplementary Data 5) containing four coral (Acropora digitifera, Acropora hyacinthus, O. faveolata, Stylophora pistillata) and three cnidarian (Aiptasia pallida, Hydra vulgaris, Nematostella vectensis) transcriptomes. The assembly was also screened against four Symbiodinium transcriptomes (199,199 sequences; clade A, B, C3, D) (Schwarz et al., 2008; Shinzato et al., 2011; Bayer et al., 2012; Barshis et al., 2013, 2014; Liew et al., 2014). For each gene, the top hit (< 1e−5) was compared between coral and Symbiodinium database searches, and assigned to the compartment with the smaller e-value. If a gene had identical e-values for both queries, it was assigned to the compartment with the larger bitscore. Remaining contigs (>1e−5) were classified as “other” and represent taxa other than coral or symbiont (e.g., endolithic algae, fungi, etc.) that also comprise the coral holobiont.
Taxonomic and Functional Annotation of Microbial Compartment
Given the complexity of microbial metatranscriptomes, we conducted annotation-based gene abundance analyses, irrespective of species of origin. To do this, paired-end reads for each polyA− library were merged via perl script (merge_fastq.pl, Supplementary Data 6) and imported into MG-RAST 3.3.2.1 (Meyer et al., 2008) to receive taxonomic and functional annotations. The libraries 1_HH_polyA−, 2_HH_polyA−, 1_DD_polyA−, and 2_DD_polyA− correspond to the following MG-RAST sample names mfav_3-_HH, mfav_33-_HH, mfav_1 -_DD, and mfav_31-_DD, respectively. Bacterial polyA− reads were queried against the MG-RAST Subsystems database (≤1e−4, ≥33% identity, min alignment length 15 aa) and assigned Subsystem Level 4 annotations (“gene functions”). Resulting data were downloaded to yield gene function abundance estimates for bacterial reads from each polyA− library. Gene function abundances were scaled to the library with the highest read counts and log2(x + 1) transformed. Significant differences between HH and DD were assessed via two-sided t-tests on these transformed abundance estimates using an FDR cutoff of < 0.1 (Storey, 2015). Fold changes were derived for significantly different genes using 2(AV DD−AV HH). Given the limited replication (n = 2), only genes with a fold-change of two or greater were considered (Supplementary Data 7).
In order to determine taxonomic composition in healthy and diseased microbiomes, bacterial mRNAs from the polyA− fractions were queried against the M5NR database in MG-RAST (≤1e−4, ≥33% identity, min alignment length 15 aa) using best-hit phylogenetic annotation. Read counts were averaged for each condition to compare family level distributions between HH and DD libraries. To identify which taxa are contributing to the expression of highly abundant gene functions in diseased corals, top 10 contributing bacterial families were calculated as the percentage of total sequence abundance via phylogenetic assignment of associated reads (Supplementary Data 8).
Algal Symbiont (Symbiodinium) Typing
Denaturing Gradient Gel Electrophoresis (DGGE) was performed to identify Symbiodinium types associated with healthy and WPD affected O. faveolata specimens. The intergenic spacer ITS2 was amplified from ~20 ng DNA per sample following LaJeunesse et al. (2003) with the following modifications: annealing temperature was maintained at 52°C for 27 cycles after a 20 cycle touchdown. ITS2 DGGE-PCR products were cleaned using an Illustra ExoStar One Step kit (GE Life Sciences, Piscataway, NJ) and sent for Sanger sequencing to the KAUST Bioscience Core Facility (Thuwal, Saudi Arabia). Sequences were quality-trimmed with Codon Code Aligner (Codon Code, Centerville, MA) and aligned against a reference Symbiodinium ITS2 database (Arif et al., 2014) containing representatives from six different Symbiodinium clades found in corals. All O. faveolata samples produced identical DGGE banding patterns and were composed of Symbiodinium sp. C7 and C12 (Supplementary Data 9).
Results
Holotranscriptome Sequencing of the Coral Metaorganism
In order to survey coral holobiont response to WPD, we conducted metatranscriptomic sequencing of the coral host, algal symbiont, and microbial compartment of healthy and diseased samples from O. faveolata (Supplementary Data 1). To analyze expression of coral host and symbiont genes, Illumina sequencing of the polyA+ fractions for healthy and diseased samples yielded a total of 116.6 million paired-end (PE) reads. Subsequent quality filtering and adapter trimming yielded 90.2 million PE reads (Table 1), which were assembled into a reference transcriptome containing 67,593 genes and used for subsequent read mapping and differential expression analysis.
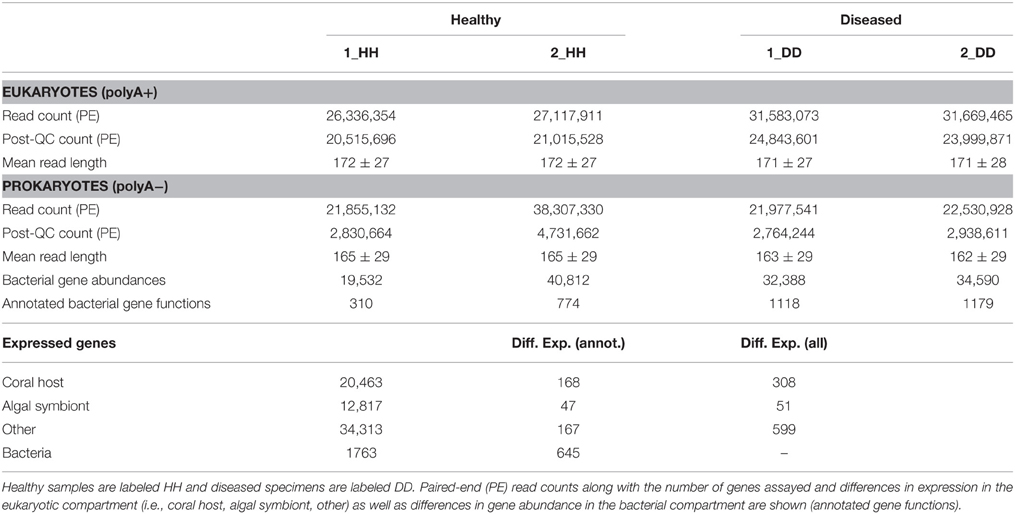
Table 1. Sequence overview, statistics, and gene expression in healthy and WPD affected samples of Orbicella faveolata.
Of the 67,593 genes, 20,458 could be assigned to coral, 12,817 to the algal symbiont, and 34,313 were assigned to “other” (i.e., non-coral and non-symbiont eukaryotic genes). For a homology-based assessment of bacterial function, polyA− fractions of the same healthy and diseased samples were sequenced to produce a total of 104.5 million PE reads. Subsequent quality assessment and trimming resulted in 13.1 million PE reads that were merged before annotation using the MG-RAST (Meyer et al., 2008) pipeline.
Functional Response of the Coral Host
Expression across the 20,458 coral genes showed differences in 308 genes (1.5%) between healthy and WPD samples [10% false-discovery rate (FDR) correction] (Supplementary Data 4). These genes had an average log2-fold change of 7.5 (range 3.25–14.8 fold) for upregulated and −7.9 (range −3.43 to −14.4 fold) for downregulated genes. 168 (55%) of these coral genes had annotations and could be assigned to the following processes in diseased samples: innate immunity, response to reactive oxygen/nitrogen species (ROS/RNS), mitochondrial/ER stress, chemical defense, ciliary/flagellar motility, cell cycle, transcription/chromatin remodeling, translation, cytoskeletal organization, and retroelement activity (Figure 1). More specifically, pattern recognition receptors for viral and bacterial agents were differentially expressed, e.g., upregulation of putative RNA helicase DDX60 along with a membrane attack complex pore-forming (MACPF) homolog. Decreased expression (−7.75 fold) for a C-type lectin, an innate immunity gene that recognizes bacteria and the coral symbiont, was noted. Two Ras homologs involved in phagocytosis of apoptotic cells were highly expressed (~10–11 fold increase). Host RNS/ROS response was marked by increased expression (3.7–9.8 fold) of five antioxidant homologs: peroxiredoxin-5, monothiol glutaredoxin-S15, cytochrome 450 3A13, and two cyan fluorescence-encoding genes in diseased samples. Mitochondrial stress was noted by cytochrome c upregulation (5.3 fold) and reduced DNA repair (DNA2) expression. Unfolded protein response (UPR) genes were upregulated, which included a mono [ADP-ribose] polymerase PARP16 (3.8 fold) and a viral susceptibility factor, protein isomerase 5-1A (9.8 fold), which suggests endoplasmic reticulum (ER) stress. Golgi-associated plant pathogenesis-related protein 1 (GAPR-1), an autophagy inhibitor, was also upregulated (3.7 fold). Chemical defense was altered as demonstrated by repression of six of seven protease inhibitors (−7 to −9 fold). Toxin AvTX-60A, a membrane attacking perforin, was the only upregulated gene in this group (5.5 fold). Mitotic arrest was signified by consistent downregulation of cell cycle genes, and we also observed a 4–14.3 fold reduction of four male meiosis homologs, which implies spermatogenic impairment. Ciliary/flagellar dysfunction was revealed by lower expression of assembly and motility genes, including three sperm-specific genes (4.0–8.8 fold), and epithelial encoding genes (hydrocephalus-inducing and putative flagellar-associated proteins, 3.4–9.2 fold). All transcription-related genes were downregulated except for p53 repressor and N-lysine methyltransferase SETD8 (4.3 fold). Moreover, p53 transcriptional regulators (peregrin and histone acetyltransferase KAT2B) had 9–13 fold lower expression in diseased samples, along with multiple histones (i.e., H1, H2B.1/H2B.2, H4). Conversely, genes associated with translation were upregulated (5–11 fold), which comprised two eukaryotic initiation factors (EIF4G3 and EIF5-1A) and 40S/60S ribosomal proteins (Supplementary Data 4). Cell adhesion genes were reduced by 9–12 fold, except for spondin-1 (9.5 fold upregulation). Host cytoskeletal homologs had decreased expression (−3.5 to −9.1 fold), including tubulin-β, actin-3, and collagen, and alludes to loss of cytoskeletal integrity. Finally, 2-epi-5-epi-valiolone synthase, a mycosporin amino acid synthesis homolog, was downregulated (−4.3 fold), and also an RNA helicase homolog TDRD-9 (−9.1 fold) involved in retrotransposon silencing.
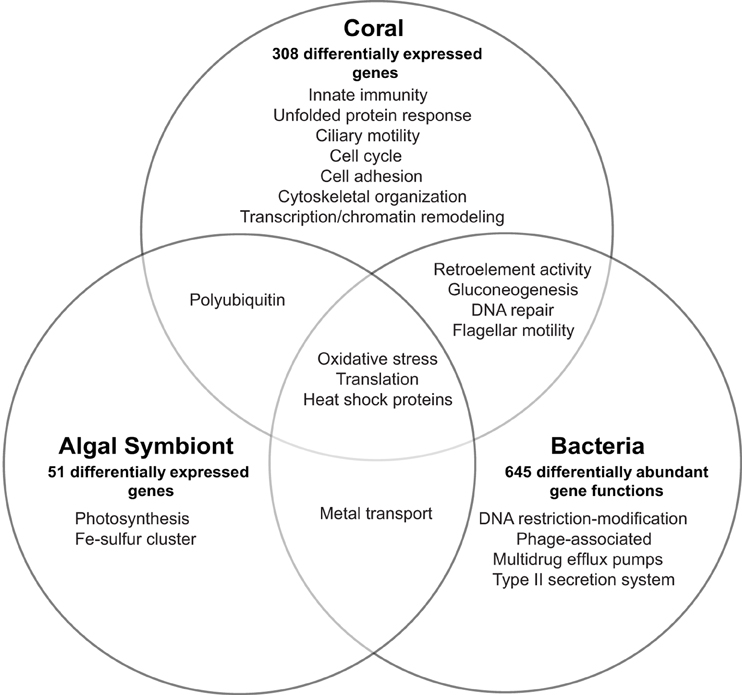
Figure 1. Overview of transcriptional response and affected biological processes in the Orbicella faveolata coral holobiont in White Plague Disease. The number of differentially expressed eukaryotic and differentially abundant bacterial genes are shown for each holobiont compartment.
Functional Response of the Algal Symbiont
Relative to the coral expression profile, the Symbiodinium compartment produced few genes that responded to WPD, but with substantial fold-changes. Expression was followed across 12,817 genes and only 51 (0.003%) showed a differential response, of which 47 genes were annotated (Supplementary Data 4, Figure 1). All genes were upregulated showing an average log2-fold change of 8.1 (range 3.9–12.9 fold). Affected genes could be assigned to photosynthesis, ROS and stress response, translation, iron-sulfur cluster assembly, and metal transport. Eleven photosynthesis-related homologs were upregulated (range 4.4–12.3 fold) that included electron carriers, ferredoxin, and cytochrome b 6-f complex subunits. Oxidative stress was noted by a 6.4 fold increase of superoxide dismutase in diseased samples. Further, two heat shock proteins (Hsp90 family; 6.2 and 10 fold), and three peptidyl-prolyl cis-trans isomerases (PPIases) that bind cyclophorins to induce protein folding were upregulated (5.5–11.1 fold). Translation-associated genes were composed of only 40S, 50S, and 60S ribosomal proteins (5.2–12.9 fold increase). Increased expression of ZupT transporter and vacuolar proton ATPase a2 (both 4.6 fold) suggested increased zinc transport in the Symbiodinium compartment. Symbiont identification using DGGE determined Symbiodinium sp. type C7 and C12 as the dominant symbiont types across all samples (Supplementary Data 9).
Functional Response of Coral-associated Bacteria
To assess response of the bacterial compartment to coral disease, we applied a homology-based annotation approach and analyzed differences in overall abundance for expressed gene functions. Expression of 1763 annotated functions (representing 848 genes) was assayed, of which 645 annotated functions representing 281 genes (~37%) were differentially abundant between healthy and diseased corals (fold-change ≥ 2, 10% FDR correction, Supplementary Data 7, Figure 1). Fold-changes ranged from −29.0 to 25.3 fold (lowest FC: DNA helicase, restriction/modification system component YeeB; highest FC: pyruvate phosphate dikinase). Bacterial gene expression revealed stress signatures reflected by an increased abundance of multiple DNA repair and heat shock dnaK cluster homologs (2 to 9 fold), choline-sulfatase (6.2 fold), an osmoprotectant precursor, as well as a RNA polymerase factor and two genes encoding for oxidative stress proteins (3.5–8.5 fold). In contrast, DNA restriction modification system components (type II helicase and type III methylation subunit) were highly decreased (−29.0 and −16.2 fold) in WPD. Furthermore, higher abundances of tyrosine recombinase XerC (2.2 fold), transcription factor for phage regulation of gene expression (8.5 fold) and two Staphylococcus pathogenicity island associated (SaPI) homologs were detected (3.2 and 17.2 fold). Other virulence and antibiotic resistance-associated genes were also enriched and encoded for flagellar motility (4.6 to 21.2 fold), type II bacterial secretion proteins (4.8 to 13.4 fold), beta lactamase C (7.1 fold), and multidrug efflux pumps (9.6 to 10.2 fold). Genes associated with multiple iron acquisition systems increased in the microbial compartment as a consequence of disease, where a TonB receptor showed the highest upregulation (8.28 fold). Other genes encoding for metal transport proteins were differentially expressed, including a molybdenum permase homolog and a GTPase-associated zinc chaperone (17.0 and 21.2 fold). Interestingly, metabolic shifts to gluconeogenesis (25 fold increase for pyruvate phosphate dikinase), ammonia assimilation (~10 to 20 fold increase for glutamate synthase and 2 to 4 fold reduction of three nitrogenases), and organic sulfur assimilation (5 to 8 fold decrease for inorganic sulfur assimilation enzymes) became evident in response to WPD. Additionally, tRNA aminoacylation and translation homologs increased 10.5 to 25 fold. Also, we found an about 5 fold increase for retron-type reverse transcriptases (RRTs).
Linking Microbial Functional Response to Bacterial Taxa
To further understand the microbial landscape in WPD, we assigned bacterial mRNAs to cognate species. Overall, expression of genes from a total of 258 bacterial families was identified in healthy and diseased corals. Of these, 232 bacterial families (89.9%) were detected in both conditions, but at different relative abundances. Further, the amount of expressed gene functions increased in diseased corals. For instance, the overall number of expressed functions for the 25 most abundant families increased from an average of 41 in healthy corals to 79 in diseased corals (Table 2). Higher abundances of mRNA sequences from Enterobacteriaceae, Vibrionaceae, Rhodobacteraceae, Flavobacteriaceae, Campylobacteraceae, and Burkholderiaceae were observed in diseased samples. Interestingly, the diseased corals also indicated a less even distribution of bacterial families than healthy corals. To test whether distinct bacterial taxa were dominant in highly expressed functions in diseased corals, we determined the 10 most contributing bacterial families of 35 differentially abundant genes based on mRNA read abundance and associated taxonomic annotations (Figure 2). Three genes were exclusively represented by a single bacterial family: RNA polymerase sigma factor for flagellar operon was mapped to Planctomycetaceae, probable RND efflux system membrane fusion protein to Alteromonadaceae, and ribokinase to the bacterial order Chroococcales. However, the majority of expressed functions contained sequences that mapped to multiple bacterial families. RRTs represented only 17.9% (5387 sequence reads) of bacterial genes in healthy corals, but were by far the most abundant expressed gene in diseased samples (71.9%; 24,217 sequence reads) (Supplementary Data 7). Interestingly, RRTs were expressed by only six bacterial families and dominated by Clostridiaceae (Figure 2). We found multiple stress and virulence genes to be associated with Alteromonadaceae, Rhodobacteraceae, Flavobacteriaceae, Enterobacteriaceae, and Vibrionaceae, which were also consistently found in WPD-affected coral species by Roder et al. (2014a) and suggested to be opportunistic bacteria (Supplementary Data 7). Intriguingly, these same bacterial families also constituted the top 10 families for nutrient acquisition- and translation-related functions (Figure 2). Further, RNA polymerase sigma factors for phage gene regulation (58% sequence contribution), oxidative stress (57% sequence contribution), and flagellar operon (100% sequence contribution) were functions predominantly expressed by Planctomycetaceae. Alteromonadaceae dominated sequences for type II secretion pathway, RND efflux membrane, and heat DnaK genes (20–100% overall contribution). TonB receptor (iron acquisition) sequences were primarily represented by Pseudoalteromonadaceae, Pseudomonadaceae, and Alteromonadaceae, while bacteria from the family Colwelliaceae dominated the putative zinc chaperone (COG 0523) sequence pool. Lastly, the most abundant families associated with translation-related sequences were Enterobacteriaceae and Flavobacteriaceae (Supplementary Data 7).
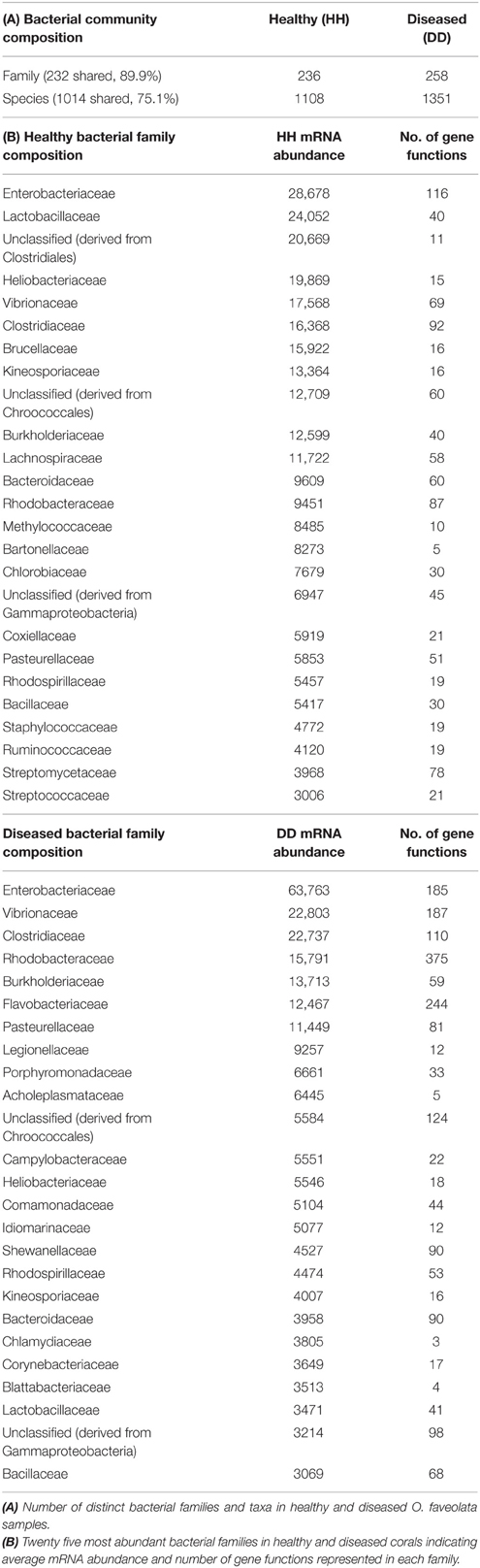
Table 2. Bacterial community and family composition of Orbicella faveolata.
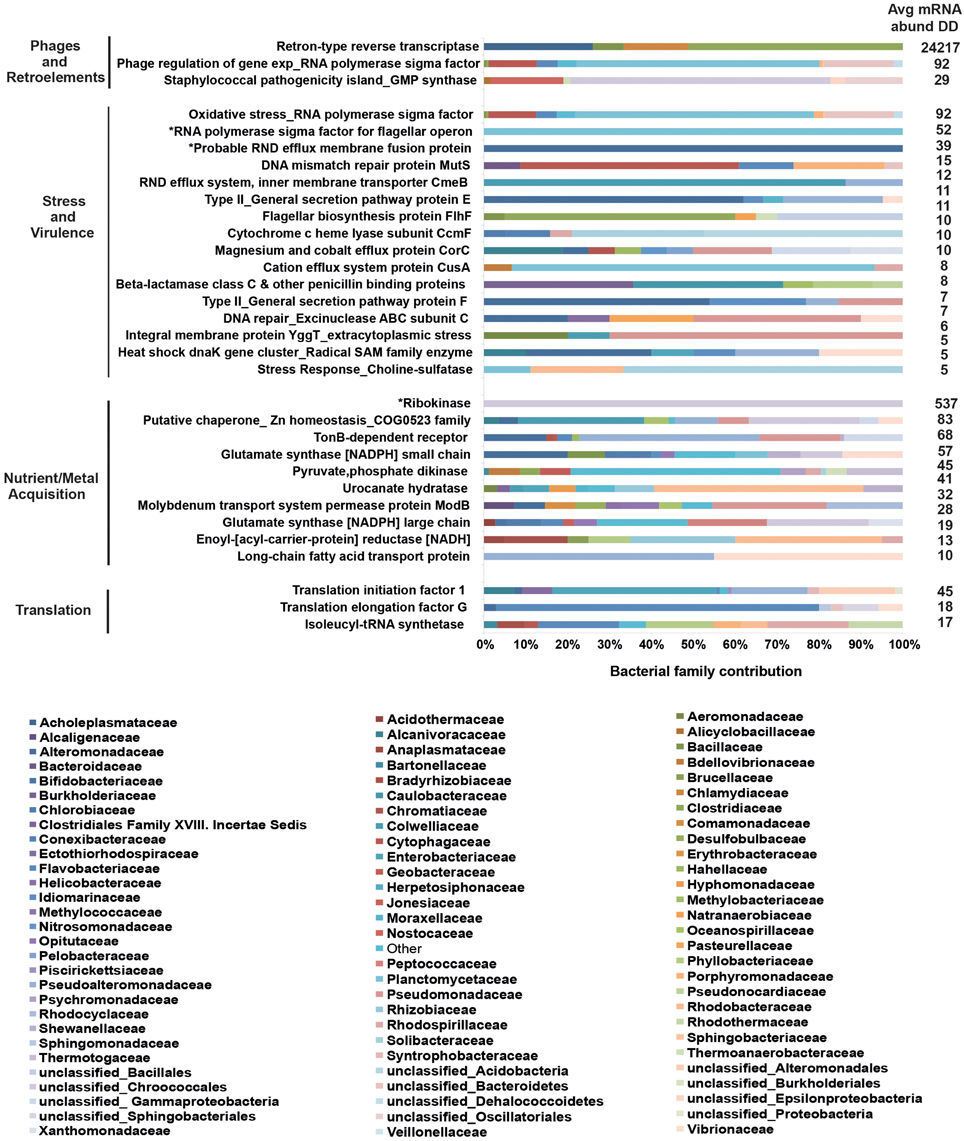
Figure 2. Top 10 bacterial family distributions for highly abundant genes in coral disease. Bacterial mRNA sequences were taxonomically annotated to obtain family composition for each gene function. Categories at the left designate higher order processes for highly abundant genes. Asterisks (*) indicate gene functions represented by a single bacterial family. Values on the right depict average mRNA sequence abundance in diseased samples (DD). Bacterial families are listed below the graph, and their contributing percentages are detailed in Supplementary Data 8.
Discussion
All Holobiont Compartments Respond to Coral Disease
By comparing active gene functions across the coral host, algal symbiont, and associated microbes of the coral holobiont, our data show that all compartments respond to WPD infection and indicate that studying the bacterial community in isolation may bias interpretation of etiopathology. Although our data are only based on a limited number of samples, a multi-compartment response to coral disease has been suggested previously by Closek et al. (2014). Our study implements an experimental approach to sequence expressed genes across the entire coral holobiont in order to understand metaorganism function in coral disease. To substantiate the findings discussed in the following, future studies should employ highly replicated designs of the here-developed pipeline to further resolve compartment- and taxa-specific responses. In addition, the analysis of healthy tissue samples from diseased coral colonies (Closek et al., 2014; Wright et al., 2015) promises to yield further insight in regard to separating causes and consequences of coral disease.
We found that about 1.5% of coral host genes showed differential expression in the diseased state, which is within range of recent coral RNASeq studies (0.05–4%) investigating heat or microbial stress (Barshis et al., 2013; Burge et al., 2013; Libro et al., 2013; Wright et al., 2015). Differentially expressed genes encoded for innate immunity, oxidative stress, translation, and regulation of retroelement activity indicating that the coral host is responding to the WPD infection. A recent study by Wright et al. (2015) also found differential expression of genes related to oxidative stress and translation in diseased tissues. In comparison, transcriptomic adjustment in Symbiodinium was marginal, i.e., only few genes were regulated but with pronounced fold-changes, which aligns with previous stress studies (Leggat et al., 2011; Baumgarten et al., 2013; Libro et al., 2013; Barshis et al., 2014). WPD microbiomes have been shown to reflect increased bacterial diversity compared to healthy consortia in 16S based surveys (Sunagawa et al., 2009; Roder et al., 2014a,b). Since our metatranscriptomics approach allowed for assessment of both, phylogenetic and functional profiles of bacteria, we could show that the vast majority (90%) of bacterial families were shared between healthy and diseased coral samples. However, diseased corals displayed a less even distribution of bacterial family abundances compared to healthy corals and an overall increase in expressed gene functions (Table 2). These findings support the notion that bacterial families already associated with the coral colony increase in abundance and have access to a more diverse functional repertoire under stress, further demonstrating that opportunistic bacteria might be a hallmark of coral disease (Lesser, 2007; Roder et al., 2014a,b).
We detected a large functional response to WPD in the microbial compartment where 37% of measured genes were differentially abundant between healthy and diseased coral samples. The microbiome of diseased corals reflected a functional profile of virulence, stress response, and metabolic adjustment for acquisition of host nutrients/metals. Genes associated with these processes were expressed by multiple bacterial families, but the family contribution was distinct for each gene. RRTs were most commonly expressed, and we found an overall compositional dominance of Proteobacteria and bacterial families representing known animal/coral pathogens previously identified in diseased coral, e.g., Altermonadaceae, Vibrionaceae, Campylobacteraceae, Enterobacteriaceae, Flavobacteriaceae, and Rhodobacteraceae (Sunagawa et al., 2009; Roder et al., 2014a,b). The contribution of multiple bacterial families to the functional expression landscape in diseased samples (Figure 2) provides further support for WPD as an opportunistic, polymicrobial disease.
Although WPD induced distinct transcriptomic responses in the different holobiont compartments, we could categorize a portion of the differentially expressed genes into related or shared processes (Figure 1). All compartments showed signs of increased translation. Further, oxidative stress was detected in all holobiont compartments as indicated by upregulation of genes associated with ROS scavenging. Symbiodinium and bacterial profiles had increased expression of heat shock proteins and metal transporters, which allude to stress response and acquisition/transport of metals that may be released as a result of host tissue damage. DNA repair and flagellar genes had contrasting profiles (Supplementary Data 4, 7): both were downregulated in the coral host, but significantly higher abundances were observed in the microbial compartments, while gluconeogenesis and retroelement-activity related genes were either upregulated or significantly increased in both compartments.
Potential Indication for a Role of Viruses and/or Phages
In addition to the overlapping expressed functions, our metatranscriptomic survey identified a common theme between the coral host and associated bacteria: response to viruses and/or phages giving way to the hypothesis that WPD might be a viral-associated disease that promotes secondary bacterial infections. This is a compelling idea as, at present, it is not possible to attribute causation to a single pathogen or consortia for WPD (Pollock et al., 2011). Rather, WPD studies continue to produce conserved diseased microbiomes in different species (Roder et al., 2014a) and across oceans (Roder et al., 2014b). Viruses have recently been implicated in a metagenomic survey by Soffer et al. (2014), where distinct DNA viromes between healthy, WPD affected, and bleached M. annularis samples were reported. Further, Soffer et al. (2014) found higher viral diversity and dominance of ssDNA viruses in diseased samples, which they suggested to be involved in WPD pathogenesis. In this study, we detected upregulation of putative RNA helicase DDX60 in diseased coral tissue. DDX60 is an innate immunity factor that binds ss/dsRNA or dsDNA viruses and interacts with RIG-I-like signaling receptors to mount an antiviral response (Vabret and Blander, 2013). Moreover, our WPD specimens were sampled from the same coral genus (Orbicella) and region (Caribbean) described in the survey by Soffer et al. (2014). We also observed increased expression of eukaryotic initiation factors and 40S/60S ribosomal activity in the coral compartment, which could be producing viral proteins, rather than host proteins. Additionally, upregulation of the autophagy inhibitor GAPR-1 (Shoji-Kawata et al., 2013) may be a strategy employed by the host to prevent further viral replication. Alternatively, GAPR-1 might also be one of the genes that viruses manipulate to evade lysosomal elimination. To unequivocally establish a link between WPD and viruses, future studies should include analyzing samples across a time series, potentially in combination with a viral enrichment approach (Soffer et al., 2014; Weynberg et al., 2014), to characterize viral communities associated with the coral holobiont before, during, and after a WPD outbreak. The reduced expression of a C-type lectin that recognizes bacterial pathogens and Symbiodinium (Vidal-Dupiol et al., 2009) detected in WPD affected corals, as well as a microbiome shift to opportunistic/pathogenic taxa and functional diversification (Table 2) suggest weakened antibacterial defense and possible symbiont loss.
Along with the virus-specific immune sensing observed in the coral compartment, we also find evidence for a response to phages in the coral microbiome. While phage-derived immunity has been suggested for mucosal defense in healthy corals (Barr et al., 2013), we hypothesize that phages may also be involved in promoting bacterial virulence during WPD infection. This is supported by an increased abundance of transcription factors for phage regulation of gene expression and staphylococcal associated pathogenicity island (SaPI) genes in the diseased bacterial compartment. Phage-directed intergenic transfer of SaPI toxin genes between bacteria has been demonstrated (Chen and Novick, 2009) and might be a strategy for various bacterial taxa to acquire virulence. Bacterial pathogens are known to use phase variation [a process that employs type III DNA restriction modification system (T3DRMS) methyltransferases] to turn on/off expression of multiple genes (Srikhanta et al., 2010), which results in phenotypic diversity of bacteria. Given that a T3DRMS methylation subunit was highly repressed in diseased bacterial mRNA libraries, this could suggest epigenetic modulation of expression by bacteria in WPD-affected samples.
Increased Retroelement Activity
In conjunction with transcriptomic alterations implicated in antiviral defense and phage interaction in WPD, we also observed expression of genes associated with retroelement (i.e., retrotransposons and retrons) activity across multiple compartments. Although previously detected in heat stressed corals and healthy/WPD metagenomes (Desalvo et al., 2008; Garcia et al., 2013), we resolved differential expression of genes involved in retroelement regulation to associations with coral, fungal, and bacterial members of the Orbicella holobiont. TDRD9 is an ATPase/DExH-type helicase involved in germline defense in Hydra and higher eukaryotes (Shoji et al., 2009; Lim et al., 2014) and its downregulation has been shown to increase LINE-1 retrotransposon expression and demethylation, leading to male sterility (Shoji et al., 2009). Our data corroborate these findings, where repression of a TDRD9 homolog in the coral compartment was noted along with meiotic arrest and spermatogenic impairment signatures in response to WPD (Supplementary Data 4) suggesting deleterious impacts on coral development and reproductive capacity of diseased coral colonies. Additionally, increased LINE-1 expression induces innate antiviral responses in several human autoimmune diseases (Madigan et al., 2012; Volkman and Stetson, 2014). Given the upregulation of DDX60 in diseased corals, we cannot rule out that WPD may inadvertently promote coral immune detection of these endogenous retroelements. Another possibility is that DDX60 is sensing complementary viral DNA, which is produced by some RNA viruses that harness retrotransposon reverse transcriptase during LINE-1 expression in infected human cells (Shimizu et al., 2014), but further inquiry is necessary to confirm this mechanism in an invertebrate system.
Oxidative stress is known to stimulate Tf2 retrotransposon activity in yeast (Chen et al., 2003) and was subsequently shown to regulate oxygen-dependent expression of adjacent genes (Sehgal et al., 2007). Interestingly, we observed upregulation of fungal homologs for Ty3 Gal-Pol polyprotein and a Tf2 retrotransposon, in addition to antioxidant genes in the compartment, which contained non-coral and non-symbiont eukaryotic genes (“other”) (Supplementary Data 4). These results imply the potential for transposition-induced gene modulation in the coral holobiont and may be a mechanism for adjusting the transcriptional response to stress. Impacts on neighboring gene expression that occur post-transposition require further data to determine whether these modifications are a silent, beneficial, or harmful holobiont response to WPD infection.
Unlike retrotransposons in eukaryotes, retrons are reverse transcriptase-encoding genes unique to bacteria, which are non-mobile and used to produce satellite DNA (msDNA) composed of an ssDNA- and ssRNA-complex (Dhundale et al., 1987). Retrons are associated with prophages, plasmids, integrons, and are suggested to promote genomic variation via mutation, duplication, or by other means (Lampson et al., 2005). The overwhelming majority of expressed genes in the bacterial compartment represented retron-type reverse transcriptases (RRTs, 71.9%) in response to WPD, but we can only speculate about their definite function. RRT expression might play a role in introducing genomic variation (Lampson et al., 2005) that influences bacterial gene expression, which would provide an avenue for microbiome adaptation under changing environmental conditions. At the same time, misannotations of rRNAs as proteins seems to be a widespread phenomenon associated with metatranscriptomics (Tripp et al., 2011). In particular, RRTs seem to be commonly misannotated 23S rRNA genes (Tripp et al., 2011), which would explain their abundance in our data. Hence, the bacterial RRT signature in our data should be treated with caution, and it remains to be determined to what extend increased retroelement activity is a shared characteristic between bacteria and the eukaryotic compartment.
Conclusions
Coral diseases are an ongoing and pervasive threat to corals and the reef ecosystems they build. Studies thus far have focused on assessing microbial communities or the identification of specific pathogens, but the functional response of the coral metaorganism is uncertain. Metatranscriptome analysis of the reef-building coral O. faveolata in this study indicates that the entire coral holobiont responds to the disease. Our data support a potential role or involvement of viruses and/or phages in WPD, but further experiments are needed to unequivocally resolve this observation. In line with other recent studies, we could confirm participation of multiple bacterial families contributing to the disease state. The incorporation of compartment-based approaches to capture holobiont patterns as devised in this study will aid in understanding the contribution of individual taxa to metaorganism function in health and disease.
Author Contributions
CRV conceived and designed the experiments. EW, CA, and CR collected samples. CD, SB, LY, CRV, CM, TB, CA, CR, and EW generated data and provided materials. CD and CRV analyzed and interpreted data. CRV and CD wrote the manuscript.
Data Accessibility
Sequence data determined in this study have been deposited in the NCBI Sequence Read Archive under PRJNA289876 (http://www.ncbi.nlm.nih.gov/bioproject/?term=289876). The polyA− sequence data were deposited in the MG-RAST Metagenomics Analysis Server and are accessible at http://metagenomics.anl.gov/linkin.cgi?project=11266.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CD is supported by an AEA KAUST Grant. We acknowledge Shajahan Ali and the KAUST Bioscience Core facility. This project was partially funded by NSF IOS # 1017510 and OCE -1105143 to EW. Financial support was provided by KAUST baseline funds to CRV. Logistical support and lab space in Puerto Rico provided by the Department of Marine Sciences, University of Puerto Rico.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2015.00062
Supplementary Data 1. Workflow of metatranscriptome data analysis pipeline.
Supplementary Data 2. Orbicella faveolata holobiont polyA+ transcriptome assembly (67,593 genes, min: 200 bp, max: 5212 bp, N50: 656 bp).
Supplementary Data 3. Orbicella faveolata holobiont polyA+ transcriptome annotation (67,593 genes). BLASTX matches (<1e−5) in SwissProt (14,887) and TrEMBL (additional 5751) databases annotated 20,638 genes (30.5%).
Supplementary Data 4. Eukaryotic differentially expressed genes (FDR < 0.1) between healthy and diseased Orbicella faveolata samples. Fungal retroelement- and antioxidant-associated genes in the “other” compartment are indicated in bold.
Supplementary Data 5. Cnidarian and Symbiodinium BLASTN database overview for assignment of transcriptomic genes to eukaryotic coral holobiont compartments.
Supplementary Data 6. merge_fastq.pl script.
Supplementary Data 7. Differentially abundant bacterial gene functions (FDR < 0.1) between healthy and diseased Orbicella samples.
Supplementary Data 8. Top 10 bacterial family distributions for highly abundant gene functions in coral disease. List of bacterial families and percent abundances as depicted in Figure 2. Average read abundance in diseased samples for a given gene function are denoted in the Avg_mRNA_abund_DD column.
Supplementary Data 9. DGGE-PCR gel image of Symbiodinium ITS2 profiles for healthy (HH) and WPD-affected (DD) Orbicella faveolata coral specimens. Diagnostic bands are marked with the Symbiodinium clade type and indicate the algal symbionts for these samples.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Arif, C., Daniels, C., Bayer, T., Banguera-Hinestroza, E., Barbrook, A., Howe, C. J., et al. (2014). Assessing Symbiodinium diversity in scleractinian corals via next-generation sequencing-based genotyping of the ITS2 rDNA region. Mol. Ecol. 23, 4418–4433. doi: 10.1111/mec.12869
Barash, Y., Sulam, R., Loya, Y., and Rosenberg, E. (2005). Bacterial Strain BA-3 and a filterable factor cause a White Plague-like disease in corals from the Eilat coral reef. Aquat. Microb. Ecol. 40, 183–189. doi: 10.3354/ame040183
Barr, J. J., Auro, R., Furlan, M., Whiteson, K. L., Erb, M. L., Pogliano, J., et al. (2013). Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. U.S.A. 110, 10771–10776. doi: 10.1073/pnas.1305923110
Barshis, D. J., Ladner, J. T., Oliver, T. A., and Palumbi, S. R. (2014). Lineage-specific transcriptional profiles of Symbiodinium spp. unaltered by heat stress in a coral host. Mol. Biol. Evol. 31, 1343–1352. doi: 10.1093/molbev/msu107
Barshis, D. J., Ladner, J. T., Oliver, T. A., Seneca, F. O., Traylor-Knowles, N., and Palumbi, S. R. (2013). Genomic basis for coral resilience to climate change. Proc. Natl. Acad. Sci. U.S.A. 110, 1387–1392. doi: 10.1073/pnas.1210224110
Baumgarten, S., Bayer, T., Aranda, M., Liew, Y. J., Carr, A., Micklem, G., et al. (2013). Integrating microRNA and mRNA expression profiling in Symbiodinium microadriaticum, a dinoflagellate symbiont of reef-building corals. BMC Genomics 14:704. doi: 10.1186/1471-2164-14-704
Bayer, T., Aranda, M., Sunagawa, S., Yum, L. K., Desalvo, M. K., Lindquist, E., et al. (2012). Symbiodinium transcriptomes: genome insights into the dinoflagellate symbionts of reef-building corals. PLoS ONE 7:e35269. doi: 10.1371/journal.pone.0035269
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300.
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Budd, A. F., Fukami, H., Smith, N. D., and Knowlton, N. (2012). Taxonomic classification of the reef coral family Mussidae (Cnidaria: Anthozoa: Scleractinia). Zool. J. Linn. Soc. 166, 465–529. doi: 10.1111/j.1096-3642.2012.00855.x
Burge, C. A., Mouchka, M. E., Harvell, C. D., and Roberts, S. (2013). Immune response of the Caribbean sea fan, Gorgonia ventalina, exposed to an Aplanochytrium parasite as revealed by transcriptome sequencing. Front. Physiol. 4:180. doi: 10.3389/fphys.2013.00180
Cárdenas, A., Rodriguez-R, L. M., Pizarro, V., Cadavid, L. F., and Arévalo-Ferro, C. (2012). Shifts in bacterial communities of two caribbean reef-building coral species affected by White Plague Disease. ISME J. 6, 502–512. doi: 10.1038/ismej.2011.123
Chen, D., Toone, W. M., Mata, J., Lyne, R., Burns, G., Kivinen, K., et al. (2003). Global transcriptional responses of fission yeast to environmental stress. Mol. Biol. Cell 14, 214–229. doi: 10.1091/mbc.E02-08-0499
Chen, J., and Novick, R. P. (2009). Phage-mediated intergeneric transfer of toxin genes. Science 323, 139–141. doi: 10.1126/science.1164783
Closek, C. J., Sunagawa, S., Desalvo, M. K., Piceno, Y. M., Desantis, T. Z., Brodie, E. L., et al. (2014). Coral transcriptome and bacterial community profiles reveal distinct Yellow Band Disease states in Orbicella faveolata. ISME J. 8, 2411–2422. doi: 10.1038/ismej.2014.85
Denner, E. B. M., Smith, G. W., Busse, H. J., Schumann, P., Narzt, T., Polson, S. W., et al. (2003). Aurantimonas coralicida gen. nov., sp nov., the causative agent of White Plague Type II on Caribbean scleractinian corals. Int. J. Syst. Evol. Microbiol. 53, 1115–1122. doi: 10.1099/ijs.0.02359-0
Desalvo, M. K., Voolstra, C. R., Sunagawa, S., Schwarz, J. A., Stillman, J. H., Coffroth, M. A., et al. (2008). Differential gene expression during thermal stress and bleaching in the Caribbean coral Montastraea faveolata. Mol. Ecol. 17, 3952–3971. doi: 10.1111/j.1365-294X.2008.03879.x
Dhundale, A., Lampson, B., Furuichi, T., Inouye, M., and Inouye, S. (1987). Structure of msDNA from Myxococcus xanthus: evidence for a long, self-annealing RNA precursor for the covalently linked, branched RNA. Cell 51, 1105–1112. doi: 10.1016/0092-8674(87)90596-4
Dimmer, E. C., Huntley, R. P., Alam-Faruque, Y., Sawford, T., O'donovan, C., Martin, M. J., et al. (2012). The UniProt-GO Annotation database in 2011. Nucleic Acids Res. 40, D565–D570. doi: 10.1093/nar/gkr1048
Dustan, P. (1977). Vitality of reef coral populations off Key Largo, Florida - recruitment and mortality. Environ. Geol. 2, 51–58. doi: 10.1007/BF02430665
Garcia, G. D., Gregoracci, G. B., Santos, E. D., Meirelles, P. M., Silva, G. G. Z., Edwards, R., et al. (2013). Metagenomic analysis of healthy and White Plague-Affected Mussismilia braziliensis corals. Microb. Ecol. 65, 1076–1086. doi: 10.1007/s00248-012-0161-4
Gnerre, S., Maccallum, I., Przybylski, D., Ribeiro, F. J., Burton, J. N., Walker, B. J., et al. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 108, 1513–1518. doi: 10.1073/pnas.1017351108
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hunter, S., Apweiler, R., Attwood, T. K., Bairoch, A., Bateman, A., Binns, D., et al. (2009). InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215. doi: 10.1093/nar/gkn785
LaJeunesse, T. C., Loh, W. K. W., van Woesik, R., Hoegh-Guldberg, O., Schmidt, G. W., and Fitt, W. K. (2003). Low symbiont diversity in southern Great Barrier Reef corals, relative to those of the Caribbean. Limnol. Oceanogr. 48, 2046–2054. doi: 10.4319/lo.2003.48.5.2046
Lampson, B. C., Inouye, M., and Inouye, S. (2005). Retrons, msDNA, and the bacterial genome. Cytogenet. Genome Res. 110, 491–499. doi: 10.1159/000084982
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leggat, W., Seneca, F., Wasmund, K., Ukani, L., Yellowlees, D., and Ainsworth, T. D. (2011). Differential responses of the coral host and their algal symbiont to thermal stress. PLoS ONE 6:e26687. doi: 10.1371/journal.pone.0026687
Lesser, M. P. (2007). Coral reef bleaching and global climate change: can corals survive the next century? Proc. Natl. Acad. Sci. U.S.A. 104, 5259–5260. doi: 10.1073/pnas.0700910104
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Libro, S., Kaluziak, S. T., and Vollmer, S. V. (2013). RNA-seq profiles of immune related genes in the staghorn coral Acropora cervicornis infected with white band disease. PLoS ONE 8:e81821. doi: 10.1371/journal.pone.0081821
Liew, Y. J., Aranda, M., Carr, A., Baumgarten, S., Zoccola, D., Tambutte, S., et al. (2014). Identification of microRNAs in the coral Stylophora pistillata. PLoS ONE 9:e91101. doi: 10.1371/journal.pone.0091101
Lim, R. S., Anand, A., Nishimiya-Fujisawa, C., Kobayashi, S., and Kai, T. (2014). Analysis of Hydra PIWI proteins and piRNAs uncover early evolutionary origins of the piRNA pathway. Dev. Biol. 386, 237–251. doi: 10.1016/j.ydbio.2013.12.007
Madigan, A. A., Sobek, K. M., Cummings, J. L., Green, W. R., Bacich, D. J., and O'keefe, D. S. (2012). Activation of innate anti-viral immune response genes in symptomatic benign prostatic hyperplasia. Genes Immun. 13, 566–572. doi: 10.1038/gene.2012.40
Magrane, M., and Consortium, U. (2011). UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford). 2011:bar009. doi: 10.1093/database/bar009
Meyer, F., Paarmann, D., D'souza, M., Olson, R., Glass, E. M., Kubal, M., et al. (2008). The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Miller, J., Muller, E., Rogers, C., Waara, R., Atkinson, A., Whelan, K. R. T., et al. (2009). Coral disease following massive bleaching in 2005 causes 60% decline in coral cover on reefs in the US Virgin Islands. Coral Reefs 28, 925–937. doi: 10.1007/s00338-009-0531-7
Moitinho-Silva, L., Seridi, L., Ryu, T., Voolstra, C. R., Ravasi, T., and Hentschel, U. (2014). Revealing microbial functional activities in the Red Sea sponge Stylissa carteri by metatranscriptomics. Environ. Microbiol. 16, 3683–3698. doi: 10.1111/1462-2920.12533
Pantos, O., Cooney, R. P., Le Tissier, M. D. A., Barer, M. R., O'donnell, A. G., and Bythell, J. C. (2003). The bacterial ecology of a plague-like disease affecting the Caribbean coral Montastrea annularis. Environ. Microbiol. 5, 370–382. doi: 10.1046/j.1462-2920.2003.00427.x
Pollock, F. J., Morris, P. J., Willis, B. L., and Bourne, D. G. (2011). The urgent need for robust coral disease diagnostics. PLoS Pathog. 7:e1002183. doi: 10.1371/journal.ppat.1002183
Richardson, L., Goldberg, W. M., Carlton, G., and Halas, J. C. (1998). Coral disease outbreak in the Florida keys: plague type II. Rev. Biol. Tropical 46, 187–198.
Roberts, A., and Pachter, L. (2013). Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 10, 71–73. doi: 10.1038/nmeth.2251
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Roder, C., Arif, C., Bayer, T., Aranda, M., Daniels, C., Shibl, A., et al. (2014a). Bacterial profiling of White Plague disease in a comparative coral species framework. ISME J. 8, 31–39. doi: 10.1038/ismej.2013.127
Roder, C., Arif, C., Daniels, C., Weil, E., and Voolstra, C. R. (2014b). Bacterial profiling of White Plague disease across corals and oceans indicates a conserved and distinct disease microbiome. Mol. Ecol. 23, 965–974. doi: 10.1111/mec.12638
Schwarz, J. A., Brokstein, P. B., Voolstra, C., Terry, A. Y., Manohar, C. F., Miller, D. J., et al. (2008). Coral life history and symbiosis: functional genomic resources for two reef building Caribbean corals, Acropora palmata and Montastraea faveolata. BMC Genomics 9:97. doi: 10.1186/1471-2164-9-97
Sehgal, A., Lee, C. Y., and Espenshade, P. J. (2007). SREBP controls oxygen-dependent mobilization of retrotransposons in fission yeast. PLoS Genet. 3:e131. doi: 10.1371/journal.pgen.0030131
Shimizu, A., Nakatani, Y., Nakamura, T., Jinno-Oue, A., Ishikawa, O., Boeke, J. D., et al. (2014). Characterisation of cytoplasmic DNA complementary to non-retroviral RNA viruses in human cells. Sci. Rep. 4:5074. doi: 10.1038/srep05074
Shinzato, C., Shoguchi, E., Kawashima, T., Hamada, M., Hisata, K., Tanaka, M., et al. (2011). Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476, 320–323. doi: 10.1038/nature10249
Shoji, M., Tanaka, T., Hosokawa, M., Reuter, M., Stark, A., Kato, Y., et al. (2009). The TDRD9-MIWI2 complex is essential for piRNA-mediated retrotransposon silencing in the mouse male germline. Dev. Cell 17, 775–787. doi: 10.1016/j.devcel.2009.10.012
Shoji-Kawata, S., Sumpter, R., Leveno, M., Campbell, G. R., Zou, Z., Kinch, L., et al. (2013). Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494, 201–206. doi: 10.1038/nature11866
Soffer, N., Brandt, M. E., Correa, A. M. S., Smith, T. B., and Thurber, R. V. (2014). Potential role of viruses in White Plague Coral disease. ISME J. 8, 271–283. doi: 10.1038/ismej.2013.137
Srikhanta, Y. N., Fox, K. L., and Jennings, M. P. (2010). The phasevarion: phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat. Rev. Microbiol. 8, 196–206. doi: 10.1038/nrmicro2283
Stewart, F. J., Ottesen, E. A., and Delong, E. F. (2010). Development and quantitative analyses of a universal rRNA-subtraction protocol for microbial metatranscriptomics. ISME J. 4, 896–907. doi: 10.1038/ismej.2010.18
Storey, J. (2015). qvalue: Q-value Estimation for False Discovery Rate Control. R package version 2.0.0. Available online at: http://qvalue.princeton.edu/; http://github.com/jdstorey/qvalue
Sunagawa, S., Desantis, T. Z., Piceno, Y. M., Brodie, E. L., Desalvo, M. K., Voolstra, C. R., et al. (2009). Bacterial diversity and White Plague Disease-associated community changes in the Caribbean coral Montastraea faveolata. ISME J. 3, 512–521. doi: 10.1038/ismej.2008.131
Thompson, F. L., Barash, Y., Sawabe, T., Sharon, G., Swings, J., and Rosenberg, E. (2006). Thalassomonas loyana sp. nov., a causative agent of the White Plague-like disease of corals on the Eilat coral reef. Int. J. Syst. Evol. Microbiol. 56, 365–368. doi: 10.1099/ijs.0.63800-0
Tripp, H. J., Hewson, I., Boyarsky, S., Stuart, J. M., and Zehr, J. P. (2011). Misannotations of rRNA can now generate 90% false positive protein matches in metatranscriptomic studies. Nucleic Acids Res. 39, 8792–8802. doi: 10.1093/nar/gkr576
Vabret, N., and Blander, J. M. (2013). Sensing microbial RNA in the cytosol. Front. Immunol. 4:468. doi: 10.3389/fimmu.2013.00468
Vidal-Dupiol, J., Adjeroud, M., Roger, E., Foure, L., Duval, D., Mone, Y., et al. (2009). Coral bleaching under thermal stress: putative involvement of host/symbiont recognition mechanisms. BMC Physiol. 9:14. doi: 10.1186/1472-6793-9-14
Volkman, H. E., and Stetson, D. B. (2014). The enemy within: endogenous retroelements and autoimmune disease. Nat. Immunol. 15, 415–422. doi: 10.1038/ni.2872
Weil, E., and Croquer, A. (2009). Spatial variability in distribution and prevalence of Caribbean scleractinian coral and octocoral diseases. I. Community-level analysis. Dis. Aquat. Org. 83, 195–208. doi: 10.3354/dao02011
Weil, E., and Rogers, C. S. (2011). “Coral reef disease in the Atlantic-Caribbean,” in Coral Reefs: An Ecosystem in Transition eds Z. Dubinski and N. Stambler (Dordrecht; Heidelberg; London; New York, NY: Springer-Verlag), 465–492. doi: 10.1007/978-94-007-0114-4_27
Weil, E., Smith, G., and Gil-Agudelo, D. L. (2006). Status and progress in coral reef disease research. Dis. Aquat. Org. 69, 1–7. doi: 10.3354/dao069001
Weynberg, K. D., Wood-Charlson, E. M., Suttle, C. A., and van Oppen, M. J. (2014). Generating viral metagenomes from the coral holobiont. Front. Microbiol. 5:206. doi: 10.3389/fmicb.2014.00206
Keywords: coral reef, coral disease, metatranscriptomics, Symbiodinium, metaorganism
Citation: Daniels CA, Baumgarten S, Yum LK, Michell CT, Bayer T, Arif C, Roder C, Weil E and Voolstra CR (2015) Metatranscriptome analysis of the reef-building coral Orbicella faveolata indicates holobiont response to coral disease. Front. Mar. Sci. 2:62. doi: 10.3389/fmars.2015.00062
Received: 14 May 2015; Accepted: 18 August 2015;
Published: 11 September 2015.
Edited by:
Andrew Stanley Mount, Clemson University, USAReviewed by:
Jian-Wen Qiu, Hong Kong Baptist University, Hong KongMikhail V. Matz, The University of Texas at Austin, USA
Copyright © 2015 Daniels, Baumgarten, Yum, Michell, Bayer, Arif, Roder, Weil and Voolstra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian R. Voolstra, Reef Genomics Lab, Red Sea Research Center, King Abdullah University of Science and Technology, Bldg 2, Room 2226, 23955 Thuwal, Saudi Arabia,Y2hyaXN0aWFuLnZvb2xzdHJhQGthdXN0LmVkdS5zYQ==