 Rachel E. Boschen
Rachel E. Boschen Ashley A. Rowden1
Ashley A. Rowden1 Malcolm R. Clark
Malcolm R. Clark Jonathan P. A. Gardner
Jonathan P. A. Gardner- 1National Institute for Water and Atmospheric Research, Wellington, New Zealand
- 2School of Biological Sciences, Faculty of Science, Victoria University of Wellington, Wellington, New Zealand
Genetic connectivity studies can inform the design of mitigation strategies used in environmental management. However, the expense of developing species-specific molecular markers and collecting samples at appropriate spatial and temporal scales can be prohibitive. Using archived material and existing molecular markers may provide a cost-effective way to assess population connectivity. Genetic connectivity studies are increasingly in demand in the deep sea in response to mounting anthropogenic pressures, including seafloor massive sulfide (SMS) mining. The feasibility of using archived material was assessed using the New Zealand-endemic vent mussel Gigantidas gladius, which inhabits areas licensed for the prospecting phase of SMS mining. Four molecular markers were tested, but only one (mitochondrial COI) provided suitable sequences. Of 942 specimens, only 150 individuals were informative, largely due to poor tissue quality of archived samples. Seven populations spanning the distributional range of G. gladius were assessed. The results indicate that G. gladius has high levels of gene flow among sites 10 to 100 s km apart and limited genetic structure. Haplotypic diversity was not equally distributed among populations, with lower diversity for the Macauley Volcano population at the northern extent of the species distribution and greater diversity within central populations. Migrant exchange was also greatest between central populations, with one population at Rumble V Seamount appearing important in terms of maintaining genetic diversity within the Kermadec Volcanic Arc region. However, interpretation of the results should be viewed with caution as small sample sizes may have limited the ability to detect genetic structure. Despite these limitations, mitigation strategies that protect areas of seabed from mining activities should consider the genetic vulnerability of the population at the northern edge of the species' distribution and the significance of certain central populations.
Introduction
The deep sea is subject to an increasing number of anthropogenic pressures (Ramirez-Llodra et al., 2011). These pressures include deep-sea mining for resources such as cobalt-rich crusts, polymetallic nodules, and seafloor massive sulfide (SMS). Of these, SMS deposits are expected to undergo exploitation before 2020 (Baker and Beaudoin, 2013). SMS deposits form through hydrothermal activity and exhibit a patchy distribution along seafloor tectonic margins, with deposits occurring on average every 100 km along the oceanic plate boundaries (Hannington et al., 2011). Active deposits support chemosynthetic communities restricted to hydrothermally active areas; many of these hydrothermal fauna have limited distributions and are endemic to one region, such as the crab Kiwa tyleri, endemic to the East Scotia Ridge in the Southern Ocean (Marsh et al., 2012, 2015) and the gastropod Ifremeria nautilei, restricted to three basins in the southwest Pacific (Bouchet and Warén, 1991; Thaler et al., 2011). Such limited distributions make these species particularly vulnerable to disturbance. However, there are many unknowns associated with the ecology of seabed communities found at SMS deposits, including the population connectivity of key species.
SMS mining activity is expected to remove all organisms inhabiting the area to be mined (Van Dover, 2011, 2014), potentially influencing the persistence of vent species in the region. To ensure vent populations remain sufficiently connected to sustain regional genetic diversity and to facilitate any repopulation of the mine site, it is important to assess the genetic structure and connectivity of populations of species vulnerable to mining disturbance. Connectivity patterns can provide important information on the sources and sinks of genetic diversity within the region and can be used to inform decisions on suitable preservation or “set-aside” areas to mitigate the effects of mining. For example, connectivity patterns of I. nautilei at SMS deposits in the Manus Basin identified the proposed set-aside site as an important source population in the region (Thaler et al., 2011), whilst in the deep sea, connectivity patterns of the quill worm Hyalinoecia longibranchiata within the New Zealand Exclusive Economic Zone (EEZ) raised questions on the suitability of existing Benthic Protection Areas (Bors et al., 2012).
To conduct genetic connectivity studies, many different marker types are available, based on an assessment of variation in allozymes, nuclear DNA, and organelle DNA, such as mitochondria (Hellberg et al., 2002; Liu and Cordes, 2004; Freeland, 2005). Each marker type has advantages and disadvantages, such as the relative cost, time and expertise required for development. Some markers are “generic” and can be applied to multiple taxa. For DNA sequencing, these include the mitochondrial cytochrome c oxidase subunit I (COI) and the nuclear internally transcribed spacer region (ITS), which are amplified with generic primers (White et al., 1990; Folmer et al., 1994). For DNA fragment analysis they include primers for amplified fragment length polymorphisms (AFLPs), randomly amplified polymorphic DNA (RAPDs) and sequence-related amplified polymorphisms (SRAPs). Other species-specific markers require more investment in development and characterization, such as microsatellites and single nucleotide polymorphisms (SNPs). Typically, suitable markers for population genetic connectivity studies must demonstrate intraspecific variation, be neutral (i.e., not subject to selection pressures), and have a known means of inheritance (Hellberg et al., 2002).
For assessments to be informative, sufficient individuals must be sampled from multiple populations spanning the geographic range of interest. However, obtaining sufficient fresh samples can be challenging, particularly within the marine environment, where the cost of sampling generally increases with depth and distance from shore. This challenge can be problematic in the deep sea, where financial and technical demands (such as vessel costs and the need for specialized sampling equipment) can inhibit collection of fresh material, restricting the spatial, and temporal coverage of samples. In the case of SMS mining, the first wave of exploitation is expected to occur within the EEZs of Pacific Island nations. Most of these nations have limited resources and the cost of collecting new samples at a range of spatial scales and developing new molecular markers could restrict the use of genetic connectivity studies. Although mining contractors would be expected to cover the cost of studies at prospective mine sites, connectivity studies outside of the mining lease area would not be sponsored. Regional-scale studies are essential for understanding the connectivity of vent organisms at meaningful biological spatial scales; cost-effective methods could enable large-scale studies to be conducted in resource-limited situations.
An alternative to obtaining fresh samples is to use archived material collected over many years and curated in national biological collections. Although this material has potential drawbacks, such as incomplete specimen records, small sample sizes, and occasionally low quality DNA (Wandeler et al., 2007), it is less expensive than obtaining fresh material and could provide a collection of samples with suitable temporal and spatial coverage for genetic connectivity studies. Using generic molecular markers that can be applied to multiple taxa would overcome the expense and expertise required to develop new species-specific markers, which could be beneficial for scientists in nations where resources and expertise are limited.
Multiple areas within the New Zealand EEZ have been licensed for the prospecting phase of SMS mining1 (Figure 1), but there is currently limited information on the population connectivity of species and assemblages which could be impacted. The mineral worth of New Zealand SMS deposits, which are rich in copper, zinc, gold, and silver (De Ronde et al., 2011) is balanced by their biological value, with the deposits and the surrounding seabed supporting a wide range of benthic assemblages (Boschen et al., 2015). These assemblages include chemosynthetic species (Clark and O'shea, 2001; Rowden et al., 2003) such as the vent mussel Gigantidas gladius, which is often very abundant (Figure 2) but is potentially vulnerable to disturbance from mining activity. G. gladius was chosen for this study because it inhabits multiple sites of active hydrothermal venting where licenses for the prospecting phase of SMS mining have been issued. As a vent obligate species endemic to the New Zealand EEZ (Von Cosel and Marshall, 2003), any disturbance from mining activities could be particularly detrimental to the persistence of the species. Assessing the population genetic connectivity of species that are representative of vent systems is essential for designing management strategies to mitigate the impacts of mining on biodiversity within the Kermadec Volcanic Arc region.
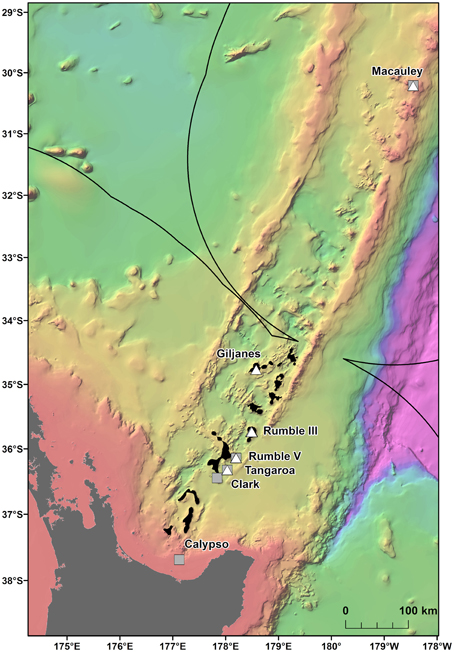
Figure 1. Gigantidas gladius sample locations along the Kermadec Volcanic Arc. Solid line indicates the New Zealand EEZ. Gray squares are samples used in this study, white triangles are excluded samples and black areas indicate where prospecting licenses have been issued for SMS mining.

Figure 2. Gigantidas gladius adults and juveniles in situ at Rumble V Seamount, on the periphery of an active hydrothermal vent (A), in clumps on the seabed (B) and within the sediment, predated upon by the asteroid Sclerasterius eructans (C). Images are from the joint New Zealand-USA 2005 NOAA “Ring of Fire Expedition.” Image credit NIWA.
Materials and Methods
Rationale
This study assesses the feasibility of using generic markers and archived material in genetic connectivity studies through populations of the hydrothermal vent endemic mussel G. gladius from within the New Zealand EEZ. G. gladius was chosen for this study because it is well represented as archived material in the NIWA Invertebrate Collection (NIC) and samples span the entire known species' distribution (Figure 1) from Macauley Volcano in the north to Calypso Vents in the south (830 km distance and ~7.5° of latitude), enabling the connectivity of all currently known populations to be assessed. The collections encompass more than a decade (November 2000–October 2012) of sampling across a substantial depth range (191–884 m), enabling the investigation of connectivity over time and depth, as well as geographical space (Table 1, Table S1). Off-the-shelf extraction kits were used in the study as low-tech cost-effective options that would be suitable for use by scientists in nations where resources and expertise are limited. Generic markers were chosen as alternatives to the development of species-specific markers, enabling the feasibility of their use in low-cost connectivity studies to be assessed.
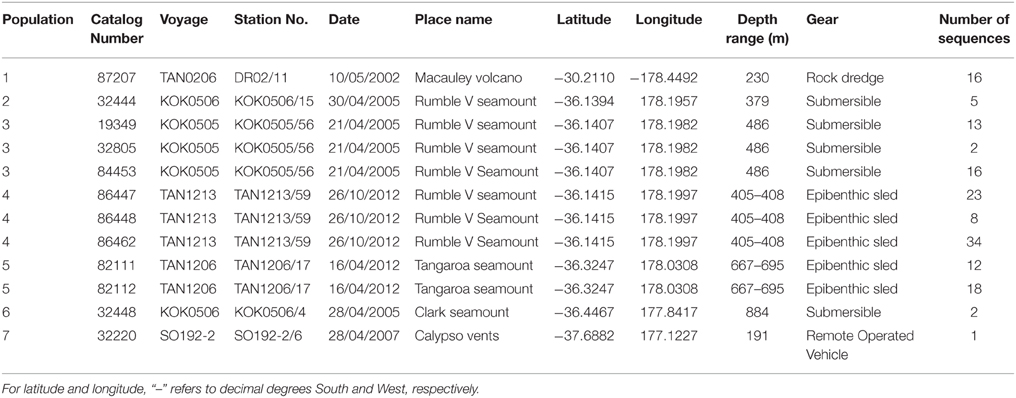
Table 1. Gigantidas gladius specimens used for analysis from the NIWA Invertebrate Collection.
Sampling and DNA Extraction
In total, 942 specimens of G. gladius were cataloged in the NIC as available for tissue sub-sampling. Where shells were available, the shell length of each individual was measured for cohort analysis. In expectation that different preservation methods and the length of time since sample collection would mean not all individuals had tissue of sufficient quality for genetic analysis (Wandeler et al., 2007), an initial analysis (DNA extraction, PCR, and sequencing) of six individuals per catalog lot number (sample) from the NIC was used to screen collections of specimens and identify samples with DNA of sufficient quality and quantity for analysis. In total, 792 individuals (84% of those cataloged as available) were unsuitable for genetic analysis (Table S1). These included 53 paratypes, 75 preserved in formalin, ~20 that were too damaged to differentiate individually, 36 too small to dissect successfully, 669 with DNA of too low quality to sequence, as determined by NanoDrop™ ND-1000 (Thermo Scientific) quantification of DNA concentrations and the A260/A280 ratios, and 16 with sequences too poor to be used in analysis. As valuable taxonomic vouchers, the paratypes were unavailable to the study. A subset of formalin-preserved individuals was initially trialed but DNA extraction using a Genomic DNA Mini Kit (Tissue; Geneaid Biotech Ltd., Taiwan) failed to obtain usable sequences, so formalin samples were excluded from further genetic analysis. Individuals that were too small to dissect (~5 mm long) had insufficient DNA for successful extraction and so were discarded. As a result, only 150 sequences (16% of the available individuals) from seven locations along the Kermadec Volcanic Arc were available for genetic analysis (Table 1). The individuals used for analysis were collected on multiple research cruises using different sampling gear between January 2000 and October 2012 (Table 1). All of the 150 sequences were from individuals fixed and stored in ethanol. A population consisted of all the individuals collected from a discrete sampling location (Table 1) and the seven populations used for analysis span the known distributional range of G. gladius; population 1, Macauley Volcano; populations 2, 3 and 4, Rumble V Seamount; population 5, Tangaroa Seamount; population 6, Clark Seamount; and population 7, Calypso Vents (Figure 1).
DNA was extracted using a Genomic DNA Mini Kit (Tissue; Geneaid Biotech Ltd., Taiwan) from mantle edge tissue (~5 mm). Extraction followed the manufacturer's instructions, except that the Proteinase K step was increased from 30 min to 3 h and the GBT buffer step was increased from 20 min to 1 h to increase DNA yield, thus obtaining DNA of sufficient quantity and quality for sequencing. Two alternative DNA extraction kits (ISOLATE II Genomic DNA Kit, Bioline; DNeasy Blood & Tissue Kit, Qiagen) were trialed but as the DNA yield and quality, as measured by NanoDrop™ quantification of DNA concentrations and the A260/A280 ratios, was similar among kits, the Genomic DNA Mini Kit was selected for all further extractions as the most cost effective option.
DNA Sequencing—PCR Conditions and Primers
Four DNA regions were investigated; mitochondrial COI, mitochondrial NADH4, nuclear ITS, and nuclear 28S. Primers used in this study were predominantly “generic,” being both widely available and applicable to multiple taxa. Primers and PCR cycle regimes are detailed in Table 2. PCRs were undertaken using a MultiGene™ gradient thermocycler (Labnet International Inc., USA). The PCR mixture consisted of 2 μl of 20–50 ng/μl DNA, 0.45 μl each of 10 μM forward and reverse primers (Integrated DNA Technologies, Singapore), 0.10 μl of 5.5 U μl−1 Fisher BioReagents™ Taq DNA polymerase (Thermo Fisher Scientific Inc., USA), 1.50 μl of the supplied 10x buffer, 0.6 μl of 5 mM dNTP, 1.2 μl of 25 mM MgCl2, and 0.60 μl 10 mg ml−1 bovine serum albumin (BSA). Total reaction volume was made up to 15 μl using 8.10 μl of double distilled H2O. PCR products were visualized on a 1% agarose gel prepared with 0.5 g agarose and 50 ml 0.5x TBE buffer. Gel band size was calculated using the molecular ladder Hyperladder™ 100 bp (Bioline, UK). PCR products were loaded onto the gel using 5x loading buffer (Bioline, UK). Gels were imaged using an Essential V2 UV transilluminator (Uvitec, Cambridge). All PCR products were cleaned prior to sequencing using ExoSAP-IT® (Affymetrix, USA) following manufacturer's instructions, except that only half the recommended volume of ExoSAP-IT® was required to produce sufficiently clean PCR products. Sanger ABI sequencing of cleaned PCR products was undertaken by Massey Genome Service (Massey University, New Zealand) on an ABI 3730 (Applied Biosystems Inc., USA). All individuals were sequenced using the forward primer. For quality control purposes, mussel-specific haplotype designations were checked by repeat sequencing of 12 individuals.
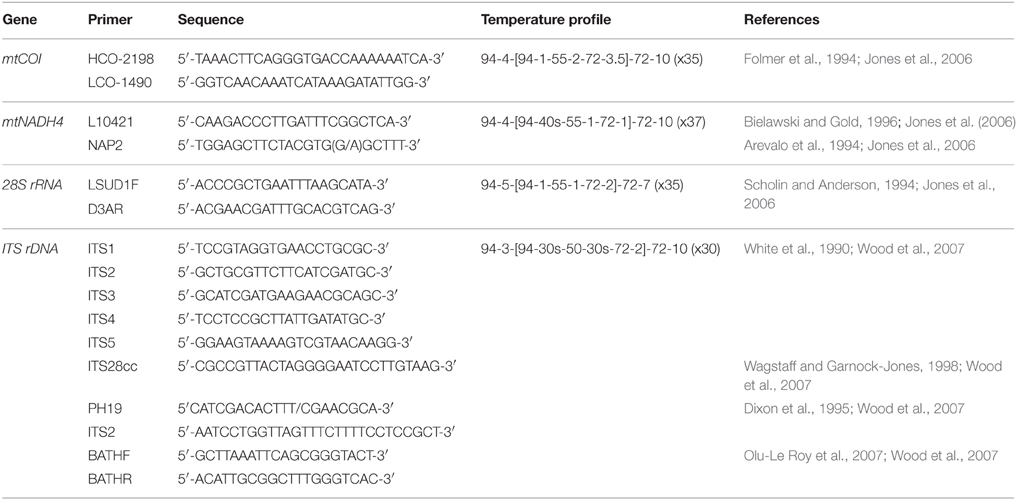
Table 2. Primers and PCR cycle regimes.
Data Analysis
Sequences were aligned and edited using Geneious 6.06 software2 with default settings for Geneious alignment; 65% similarity (5.0/-4.0) cost matrix, gap open penalty 12, gap extension penalty 3, global alignment with free end gaps. Sequence ends were trimmed to obtain sequences of 586 bp length and miscalls were manually replaced by visually assessing peak height; ambiguous bases were replaced with the base that had the highest peak at that site. The base(s) characterizing each haplotype were checked for adequate peak height and differentiation. Haplotype validity was further checked by pairwise alignment in Geneious of the 12 duplicate sequences.
The Geneious alignment in fasta format was used as an input file for FaBox (Villesen, 2007). Basic haplotypic diversity statistics for the seven populations were generated in FaBox and the project file was used as the input file for analyses using Arlequin v3.5.1.2 (Excoffier et al., 2005). To assess whether sufficient individuals were sampled to capture the haplotypic diversity of the seven populations, rarefaction curves (per population and overall) were calculated in RarefactWin3 and plotted in Excel 2013. To visualize the connectivity of and relationships among haplotypes, a minimum spanning haplotype network (Bandelt et al., 1999) was generated using Popart4, with the default epsilon value (0). The network was generated using the fasta file of the Geneious alignment, converted to nex format in DnaSPv5 (Librado and Rozas, 2009). Haplotype pie charts for each population were generated in Excel 2013 and plotted over a base map in ArcMap 10.1 (Environmental Systems Research Institute, USA) to display the haplotype distribution along the Kermadec Volcanic Arc. Analysis of Molecular variance (AMOVA) tests (Excoffier et al., 1992) in Arlequin were used to investigate the effect of metapopulation (all seven populations combined), location (North, Central or South Kermadec Volcanic Arc), year, and depth of collection on genetic connectivity among populations (Table 3). AMOVAs were performed using standard AMOVA computations (haplotypic format) and conventional F-statistics (Φ-statistics for DNA sequence data). Additional AMOVAs were conducted on population 4 (Station 59, Rumble V Seamount), population 5 (Station 17, Tangaroa Seamount) and combined population 4 and 5 to investigate the effect of age cohort (as determined by individual shell length) on genetic connectivity among individuals sampled from neighboring seamounts in the same year. To assess the effect of geographic distance on genetic connectivity, a Mantel test (Mantel, 1967; Smouse et al., 1986) was performed in Arlequin. The Mantel test was performed with 1000 permutations, the distance matrix was a log of the distance between populations in meters and the ΦST matrix was based on slatkinlinearfst (Slatkin, 1995). The number of migrants between populations per generation was calculated using the formula M = (1 – ΦST)/(2*ΦST) (Excoffier et al., 2005).
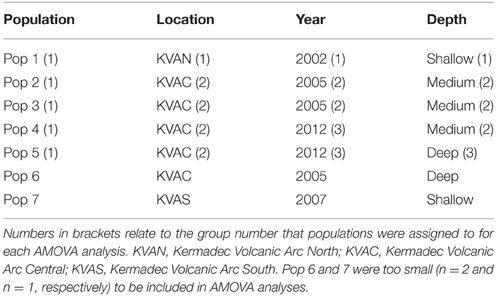
Table 3. Group structure used for AMOVA tests.
The mitochondrial haplotype sequences from this study have been submitted to GenBank. The accession numbers are as following: KU180249, KU180250, KU180251, KU180252, KU180253, KU180254, KU180255, KU180256, KU180257, KU180258, KU180259, KU180260, KU180261, KU180262, KU180263, KU180264, KU180265, KU180266, KU180267, KU180268, KU180269, KU180270, KU180271, KU180272, KU180273, KU180274, KU180275, KU180276, KU180277, KU180278, KU180279, KU180280, KU180281, KU180282, KU180283.
Results
Use of Generic Primers
PCR amplification of the NADH4 gene was unsuccessful using the selected primers; no bands (DNA products from the PCR) were visible on a 1% agarose gel. For the 28S gene, the region was successfully amplified but there was no sequence variation among the 20 test individuals, so the gene was not investigated further. The multiple primer pairs used to amplify the ITS region produced sequences of the expected length but the presence of multiple base peaks suggested that duplicate copies of ITS were present within the same individual. One short (200 bp) region of single copy ITS2 produced clean and usable sequences, but there was no variation within the region among the 20 test individuals and so the region was not used for analysis. The universal primers used for COI resulted in 586 bp of good quality sequence for 150 G. gladius individuals distributed across the seven populations. All analyses of population genetic connectivity were performed using these COI sequences.
Quality Control Tests
To check the observed high levels of haplotypic diversity, 12 individuals were repeat sequenced. Pairwise alignment of duplicate sequences demonstrated 100% identity of mussel-specific DNA sequences after ambiguous bases were resolved, indicating that the observed haplotypic diversity was real and not a PCR or sequencing artifact. Repeat sequencing indicated that sequencing using the forward primer was sufficient to consistently produce accurate sequences of the appropriate length and that duplicate sequencing using the reverse primer was not necessary.
Rarefaction curves of haplotypic diversity were used to assess sampling effort to capture genetic diversity within populations. Curves for all individuals regardless of population (Figure 3A) did not approach an asymptote, suggesting that insufficient individuals were sampled to capture the full haplotypic diversity of the metapopulation. Similarly, rarefaction curves for individual populations (Figure 3B) also did not approach an asymptote. The rarefaction curves for populations 4 and 5 had similar gradients, but the slope for population 3 was steeper and for populations 1 and 2 was shallower than for the other populations. Populations 6 and 7 were too small for rarefaction analysis.
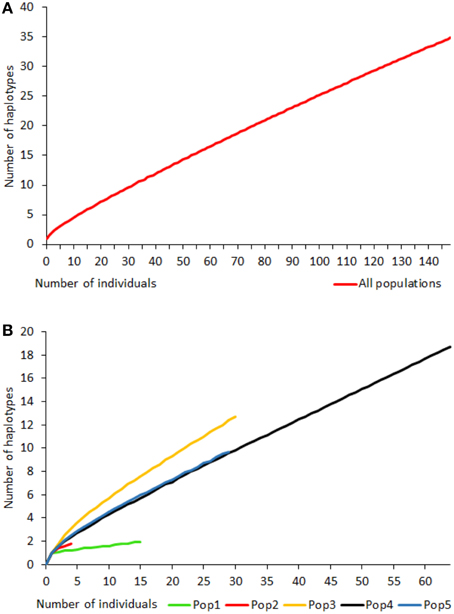
Figure 3. Rarefaction curves for haplotype diversity for all Gigantidas gladius individuals (A) and per population (B); populations 6 and 7 were too small for rarefaction analysis.
Patterns in Genetic Diversity
In total, 35 mitochondrial DNA COI haplotypes were identified from the 150 individuals across seven populations. This diversity was not evenly distributed (Table 4), with population 4 having the greatest number of haplotypes, populations 3 and 5 exhibiting intermediate diversity and populations 1, 2, 6, and 7 having the smallest number of haplotypes. In general, COI haplotypic diversity was a function of population sample size, with the notable exception of population 1. Haplotypic diversity was higher in the central part of the Kermadec Volcanic Arc (populations 3, 4, and 5; 13, 19, and 10 haplotypes, respectively) and lower at the northern and southern ends of the sampling range (population 1 and population 5; 2 and 1 haplotypes, respectively) (Figure 4).
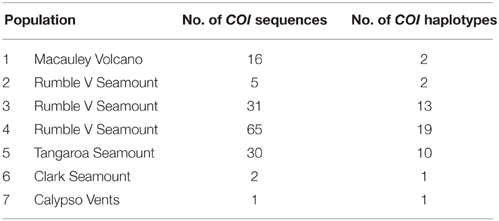
Table 4. Basic population statistics summary information.
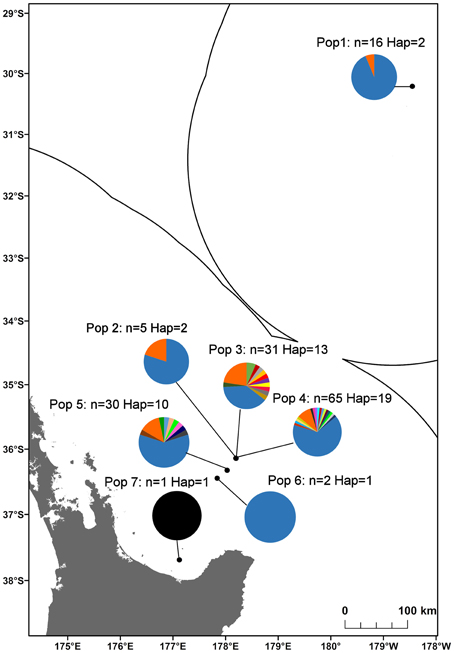
Figure 4. Haplotype diversity along the Kermadec Volcanic Arc for the seven populations of Gigantidas gladius. Each haplotype is represented by a different colored slice of the pie chart. “n” is the number of individuals sequenced per population, “Hap” is the number of haplotypes.
Many haplotypes were only observed in one individual (n = 29 or 83% of all haplotypes) with the occurrence of these “private” haplotypes increasing with sample size, again with the exception of population 1 (Figure 5). Six haplotypes were shared among populations; haplotypes 3 and 5 were found in all populations, haplotypes 1 and 11 were found in populations 3 and 4, and haplotypes 12 and 20 were found in populations 4 and 5 (Figure 5). The majority of haplotypes in the network are the result of a single mutational step from the most common haplotype, haplotype 3, or in some cases haplotype 5. Five intermediate haplotypes (represented by a double mutational step) were not sampled from the metapopulation.
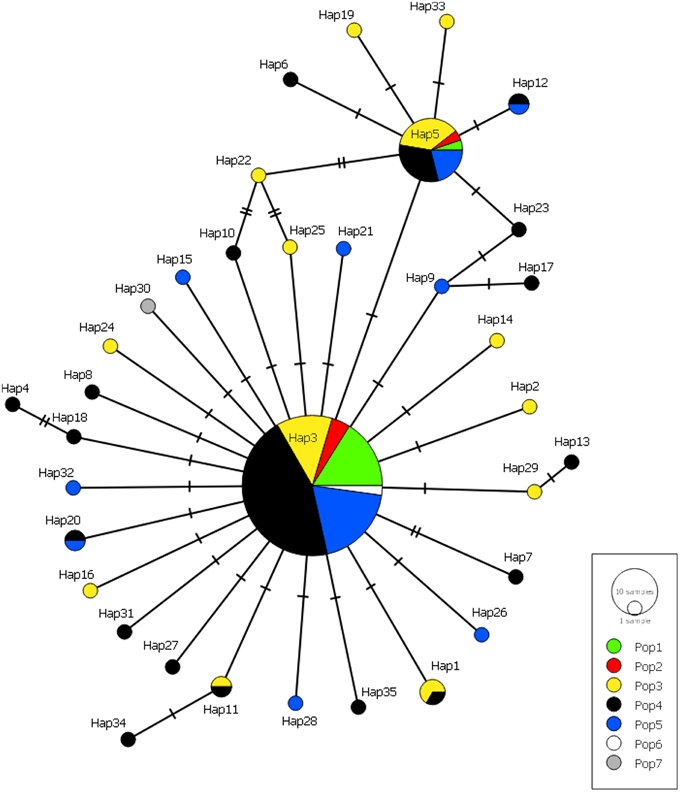
Figure 5. Minimum-spanning haplotype network for all Gigantidas gladius individuals from the seven populations. Circle size is proportional to the number of individuals with that haplotype, lines indicate relatedness of haplotypes. Line breaks indicate mutational steps in the connectivity between haplotypes.
There has been extensive migrant exchange among populations along the Kermadec Volcanic Arc. The number of migrants per generation between pairs of populations (Table 5) was greatest between populations 2 and 3 (both on Rumble V Seamount) and also high between populations 3 and 5 and populations 3 and 6. Populations 5 and 6 occurred on Tangaroa and Clark Seamounts respectively, the two seamounts closest to population 3, on Rumble V. Each population exchanged migrants with at least one other population, with population 3 the only population to exchange migrants with all other populations.
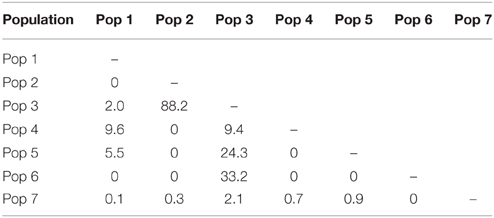
Table 5. The number of migrants per generation between pairs of populations.
There was limited genetic structure among populations. The AMOVA test of metapopulation identified significant haplotypic variation among populations (Table 6A), with pairwise testing between populations identifying population 1 (on Macauley Volcano, the most isolated seamount) as being different from populations 3 and 5. Populations 3 and 4 (both on Rumble V Seamount) were also significantly different from each other (Table 6B).

Table 6A. Results for AMOVA grouped as metapopulation.
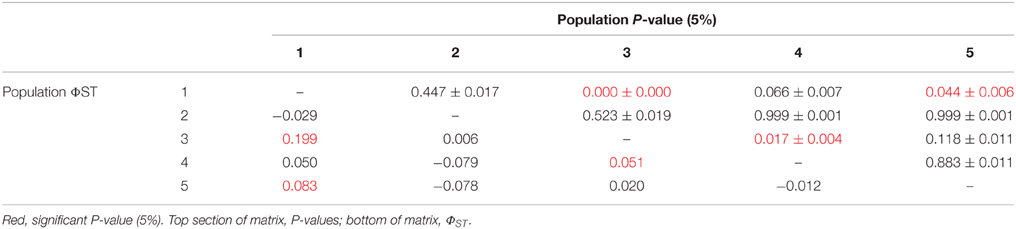
Table 6B. Pairwise testing between populations.
Factors Influencing Genetic Diversity
There was no effect of geographical distance on genetic differentiation among the five populations (Mantel test results, p = 0.446). Small samples sizes at population 6 and 7 (n = 2 and n = 1, respectively) meant that these populations were excluded from AMOVA tests. These tests demonstrated there was no significant influence of location, year sampled or depth sampled on genetic structure. Focused AMOVA tests of the effect of cohort (shell length) within populations 4, 5, and 4 and 5 combined revealed no effect of individual size on genetic connectivity between populations.
Discussion
The present study assessed the feasibility of using archived G. gladius samples to investigate patterns of genetic connectivity. Emphasis was placed on the use of standard, cost effective laboratory, and analytical approaches, because many countries that will experience vent mining activities in the near future may not have the highly developed infrastructure or experience necessary for in depth genetic connectivity research. Below, the implications of connectivity patterns for G. gladius are considered, particularly in light of proposed SMS mining activities within the New Zealand EEZ. The practicalities and limitations of the current approach are also discussed, with particular consideration given to the ability to provide robust recommendations for environmental management.
Population Genetic Connectivity of Gigantidas gladius
G. gladius exhibited high haplotypic diversity along the Kermadec Volcanic Arc; 35 mitochondrial haplotypes within 150 individuals. This diversity was comparable to the closely related hydrothermal vent mussel Bathymodiolus thermophilus, where 10 mitochondrial haplotypes occurred within 58 individuals in the eastern Pacific (Craddock et al., 1995) and to both Bathymodiolus platifrons and Bathymodiolus japonicus off Japan, where there were 15 mitochondrial haplotypes in 40 individuals and 20 haplotypes in 41 individuals, respectively (Kyuno et al., 2009; Miyazaki et al., 2013).
The star-like pattern of the haplotype network suggests that a recent expansion event has occurred, which could result from rapid colonization of new hydrothermal habitat. Hydrothermal vent systems are ephemeral, with tectonic and volcanic activity creating new habitat that is rapidly colonized by vent organisms (Lutz et al., 1994; Tunnicliffe et al., 1997). Many of the individuals used in this study were juveniles, which points to the possibility of a recent recruitment event. Star-like mitochondrial networks, implying recent population expansion, exist for many vent organisms (Vrijenhoek, 2010), including B. platifrons and B. japonicus (Kyuno et al., 2009; Miyazaki et al., 2013).
There was limited genetic structure among G. gladius populations, but no significant structuring effect was evident (i.e., no effect of location/distance, depth, year or individual size). A previous study on G. gladius (then called Bathymodiolus new species NZ-1) sampled from two seamounts (Rumble V and Rumble III Seamounts; 50 km apart) also found no population structure using COI, although using allozymes, pooled populations from one seamount were different to those at the other seamount (Smith et al., 2004). There was also no significant effect of individual size on genetic structure and no difference between populations on the same seamount (Smith et al., 2004). These earlier results agree in general with our findings using COI from a considerably larger collection of G. gladius samples, namely that there is high gene flow between populations along the Kermadec Volcanic Arc, although limited genetic structure was identified in the current study through pairwise testing.
A lack of geographic signal in patterns of genetic diversity was also found for B. thermophilus, with no evidence for barriers to gene flow across the known species' range of 2370 km (Craddock et al., 1995), whilst there was no effect of depth on connectivity among populations of the chemosynthetic seep mussel Bathymodiolus childressi (Carney et al., 2006). Another study found no differentiation among populations of B. platifrons and B. japonicus inhabiting both hydrothermal vent and cold seep habitats, with extensive gene flow between populations 1500 km apart (Kyuno et al., 2009; Miyazaki et al., 2013). Although B. platifrons and B. japonicus populations occur at both vents and seeps, which could improve dispersal through stepping-stones of reducing habitats (Smith, 1989), to date G. gladius has only been positively identified at vent habitats. Apparent high gene flow among Bathymodiolus populations is thought to result from them having a planktotrophic larval stage, which is able to spend approximately a year in the plankton (Arellano and Young, 2009) and so potentially disperse over large distances. Although the reproductive mode of G. gladius has not been determined, it is thought to have planktotrophic larvae from the shape of the prodissoconch (Von Cosel and Marshall, 2003) and so could have long-lived larvae with high dispersal potential. Such larvae could be sufficiently long-lived to populate sites as far north as Macauley Volcano, facilitating genetic exchange among populations.
The large number of migrants between G. gladius populations supports the concept of panmixia, with high levels of genetic connectivity among populations. However, the degree of exchange between populations is not equal, as indicated by the differences in migration rates and relative differences in haplotypic diversity. In particular, population 3 has the highest number of migrants and it also exchanges migrants with all other populations, suggesting it could be a key population for maintaining genetic diversity within the Kermadec Volcanic Arc region. The relatively lower haplotypic diversity of population 1 suggests it could be more isolated than other populations along the Arc and so is more vulnerable to disturbance. Population 1 is also genetically distinct from populations 3 and 5 (Rumble V and Tangaroa Seamounts, respectively), which supports this concept of relative isolation. Long-lived planktotrophic larvae should be capable of reaching population 1 at Macauley Volcano, but an absence of suitable habitat between central populations and Macauley could preclude a “stepping-stone” mode of connectivity between central and northern populations. Sampling has occurred on hydrothermally active seamounts of the Arc both south and north of Macauley, but there are no records of additional G. gladius populations from sampling efforts 440 km to the north or between Macauley and Giljanes Seamount to the south, a distance of 580 km (Figure 1). G. gladius can colonize a suite of hydrothermal habitats, including hard and soft substrata and both high and diffuse flow vent sites (Figure 2), across a depth range of 191–884 m. Whilst G. gladius is thought to host chemoautotrophic bacteria (Von Cosel and Marshall, 2003) it may also be an opportunistic suspension feeder in the same way as B. thermophilus (Page et al., 1991). By inhabiting multiple hydrothermal habitat types over a 900 m depth range and exhibiting a flexible feeding strategy, G. gladius increases the potential for colonization success. As a result, a lack of suitable hydrothermal habitat between Macauley and central populations seems unlikely to be the reason for the relative genetic isolation of population 1. Instead, larval dispersal distance could be limited by prevalent deep-sea currents. The East Cape Eddy may influence circulation down to 1000 m depth (Chiswell and Sutton, 2015) and so constrain the majority of larvae within the central Kermadec Volcanic Arc. The mean flow along the Arc at 1000 m is thought to be southwards from Macauley toward the central populations (Chiswell et al., 2015), which could explain the lower haplotypic diversity of population 1, as larval dispersal to Macauley could be restricted to occasions when breeding events coincide with shifts in the dominant current flow. These patterns in current flow could explain the restriction of G. gladius to the Kermadec Volcanic Arc Region. However, greater resolution on deep-sea currents along the Arc would be needed before current flow could be confirmed as the reason for the lower haplotypic diversity of population 1.
Limitations Associated with the Use of Archived Material and Generic Molecular Markers
There are known drawbacks to the use of archived material in population genetic connectivity assessments, including small sample sizes and the likelihood of low quality DNA (Wandeler et al., 2007). However, archived material also has the potential to provide valuable samples. The G. gladius material archived in the NIC represents all of the available samples for this species, spanning the known distributional range of 830 km and nearly 8° of latitude, thus providing an opportunity to characterize the genetic connectivity across an entire species' range. It is also a large collection in terms of number of individuals, and is therefore an appropriate target for investigation. However, of the 942 individuals available, ultimately only 150 could be used for this study. A high rate of sample loss also occurred in similar studies using archived material, such as investigations using deep-sea coral tissue (Miller et al., 2010).
Small samples sizes can fail to capture the total haplotypic diversity, leave gaps in coverage across space or time, and limit the power of statistical tests (e.g., G. gladius AMOVA tests for the effect of location, depth, year, individual size) with the problem that such limitations complicate interpretation of the results. In the case of G. gladius, although panmixia is biologically feasible, the apparent lack of genetic structuring factors could also result from low statistical power to detect differences among populations. Although larger sample sizes would enable more robust statistical testing, it is often not possible and many deep-sea connectivity studies have to make do with far fewer specimens, in some cases less than 10 individuals per population (Miller et al., 2010; Bors et al., 2012). As such, although the small numbers of individuals used for the present study are far from ideal, they are within the range commonly seen in deep-sea studies.
The majority of specimens omitted from this study were excluded due to low quality DNA, an acknowledged risk of using archived material (Wandeler et al., 2007). Low quality DNA also has the potential to overestimate genetic diversity due to errors during amplification and sequencing (Sefc et al., 2007). As DNA quality of archived material tends to decrease with time, the risk of such errors increases with specimen age. As far it was possible to ascertain, there was no evidence for a bias in genetic diversity associated with sample age, and hence presumptive DNA quality, of G. gladius. In particular, the highest genetic diversity was actually observed in the freshest sample (population 4: n = 65, haplotype = 19, sampled 2012), suggesting that the high haplotypic diversity observed for G. gladius is real and not due to preservation artifacts.
Four universal markers were trialed in this study, but only one was able to provide sequences suitable for population genetic connectivity assessment. This apparently low level of success highlights a problem with so-called universal markers—they are not necessarily applicable to all species or samples. Investment in further troubleshooting and testing is always possible but adds significant time and cost to a project, and also requires a higher level of technical expertise, which may not be available in all cases. For G. gladius, it was not feasible to troubleshoot for NADH4 or to resolve the issue of duplicate ITS sequences. Although ITS sequences have been successfully used in phylogenetic studies of other mussels, such as shallow water Perna spp. (Wood et al., 2007), studies using ITS from Bathymodiolus spp. required an additional cloning step as part of the amplification process (Jollivet et al., 1998; Olu-Le Roy et al., 2007), presumably to overcome the issue of multiple copies. Ideally, studies should incorporate both mitochondrial (e.g., COI) and nuclear (e.g., ITS) markers to test for patterns of genetic connectivity, but as in the case for this study, only one marker may be informative.
For G. gladius, the only successful marker was mitochondrial COI, which although commonly used to investigate interspecific genetic variation, demonstrated sufficient intraspecific variation to be used in this study. COI has been used in other deep-sea studies to assess population connectivity of the squat lobster Munida gracilis and quill worm H. longibranchiata (Bors et al., 2012) and the hydrothermal vent gastropod I. nautilei (Thaler et al., 2011). Using mitochondrial DNA from bivalves can be complicated by separate male and female mitochondrial genomes being transmitted independently to offspring (Fisher and Skibinski, 1990). However, there is no evidence to date for gender-biased mitochondria in G. gladius' closest relatives, Bathymodiolus spp. (Won et al., 2003; Jones et al., 2006), and mitochondrial sequences are commonly used in phylogenetic studies of this group (Smith et al., 2004; Jones et al., 2006; Fontanez and Cavanaugh, 2013). As such, we consider mitochondrial COI to be a suitable marker for connectivity assessment of G. gladius.
The limitations encountered in this study suggest that, using the cost-effective techniques described, archived material alone may be insufficient to assess genetic connectivity. As genetic techniques, such as formalin extraction, advance and become more affordable, samples that have historically been problematic to sequence may become available for use in connectivity studies. Although, at the present, archived material alone may be insufficient for conducting relatively low-tech, cost-effective connectivity studies, it may be used as a valuable starting point to identify spatial or temporal “gaps” in existing material. This could permit the use of available resources to target gaps as priorities for the collection of fresh samples. Once suitable supplementary fresh material has been collected and sequences obtained, more robust environmental management recommendations can be made. A suggested work flow to obtain sequences from appropriate archived material, resulting in recommendations for management, is detailed in Figure 6.
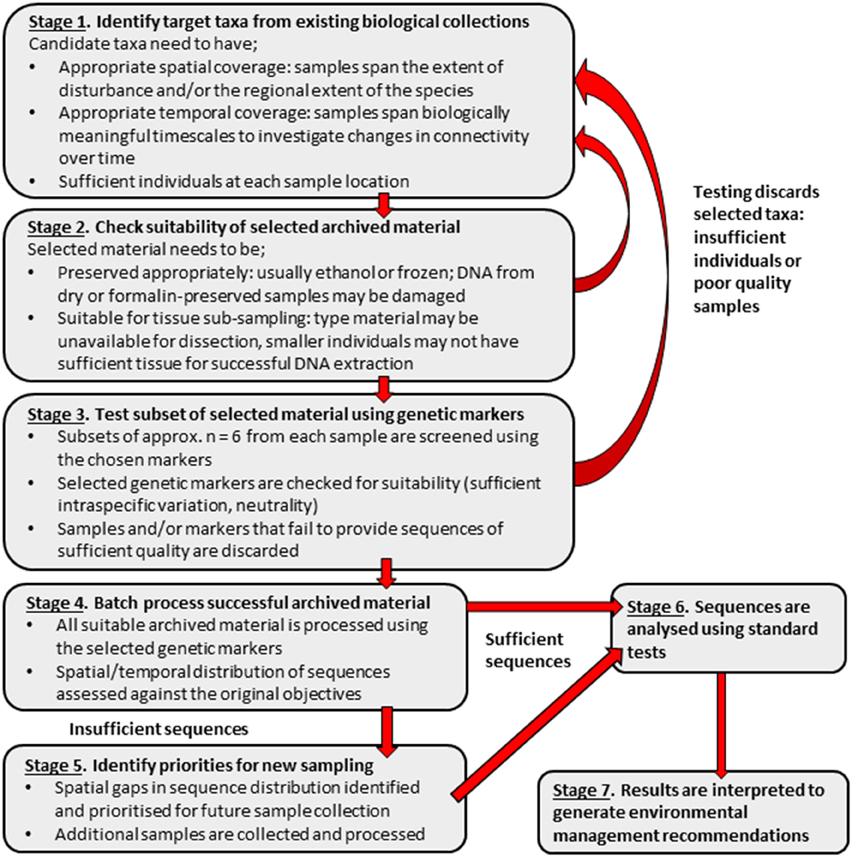
Figure 6. Suggested stepwise plan for using archived biological material in population genetic connectivity assessments with the aim to inform environmental management decisions.
Implications for the Environmental Management of SMS Mining
SMS mining activities have the potential to impact benthic organisms, such as G. gladius, mainly through habitat removal, altered hydrothermal flow or smothering with suspended sediment (Coffey Natural Systems, 2008; Van Dover, 2011, 2014; Boschen et al., 2013). One of the proposed mitigation strategies for SMS mining is the designation of “set-aside” areas to preserve similar habitat and biodiversity within the region (International Seabed Authority, 2010; Collins et al., 2013a,b). This includes the conservation of genetic diversity, with patterns in population connectivity providing important information on the sources and sinks of diversity and the potential for recolonization of impacted habitat.
A number of populations of G. gladius inhabit areas within the New Zealand EEZ that have been licensed for the prospecting phase of SMS mining and are potentially at risk from mining activities. The results from our study suggest that although there is generally high genetic diversity within and high gene flow among populations for G. gladius, this is not distributed equally among populations and any set-aside area or network of set-aside sites would need to take this into consideration. For example, detrimental impacts to population 1 from mining activity could result in the loss of the northern extent of the species' distribution, contracting the known species range by 4.5° of latitude. Equally, the high haplotypic diversity of population 3 and high migrant exchange between population 3 and all other populations suggests that it may be a source population. As such, population 3 is of particular importance in maintaining genetic diversity at the metapopulation level and may require protection, because disturbance to this population from mining activities has the potential to impact genetic diversity at a regional scale.
SMS mining is expected to occur on a relatively small spatial scale, with the impacts of mining predicted to be localized. For example at Solwara 1, an SMS deposit in the Manus Basin, the area of seabed licensed for exploitation is only 0.112 km2, whilst the majority of sedimentation impacts are expected to occur within 1 km of the discharge site (Coffey Natural Systems, 2008). The nature of exploitation, removing relatively small mineral-rich patches within a wider area, reflects the distribution of deposits, which is determined by hydrothermal activity. Many organisms that rely on hydrothermal activity and so inhabit these deposits subsequently have a patchy spatial distribution, with the potential for small-scale genetic variation among patches that could be lost through mining activity. For example, along the East Pacific Rise, genetic differentiation was discovered between patches of the vent tubeworm Riftia pachyptila just 400 m apart (Shank and Halanych, 2007). Although vents undergo natural habitat loss through volcanic and tectonic events, mining activities could compound this effect (Van Dover, 2011), potentially enhancing the loss of localized-genetic structure. To investigate small spatial scale genetic differentiation, it is important to assess genetic connectivity not just over the 100 or 1000 s of km that may encompass a species' range, but also at smaller spatial scales appropriate to mining disturbance. Such a nested design was used in a study of I. nautilei in the Manus Basin and demonstrated panmixia across all spatial scales, with no barriers to connectivity between individuals among patches within an SMS deposit-mound, among SMS mounds within a site, or among sites within a vent field (Thaler et al., 2011). In the one instance where archived samples of G. gladius occurred at multiple locations within the same seamount, Rumble V, there was evidence of genetic structure between two of the three populations. However, these populations were sampled seven years apart, so whether the difference in population structure relates to genetic patchiness within seamounts or temporal variation is unknown.
Although this study demonstrates that archived material can provide important information for environmental management, this material may be insufficient on its own for assessing the genetic connectivity of populations at SMS deposits. However, archived material can be used to inform targeted sampling (Figure 6). Combining archived and fresh material may be a cost effective strategy to provide samples with sufficient spatial and temporal coverage for connectivity studies at SMS deposits. Although sampling is necessary to characterize and assess the suitability of potential set-aside areas, collecting from these sites also has the potential to impact resident populations. Conducting connectivity assessments with both archived and fresh material may reduce the impact of sampling at potential set-aside sites and would be a strategy in-line with guidelines for responsible sampling at hydrothermal vents (Devey et al., 2007; Interridge, 2009).
Conclusion
The use of archived material in genetic connectivity studies is known to have limitations, many of which were encountered in this study. In particular, largely due to issues with sample preservation and DNA quality, only 16% of the cataloged G. gladius individuals yielded suitable DNA sequences for analysis. Although the connectivity results for G. gladius suggest high gene flow between populations with limited genetic structuring, small sample sizes reduced the power of statistical tests and ultimately mean results must be interpreted with caution. This study suggests that the use solely of archived material may be insufficient to generate robust results to inform management decisions. However, in the absence of fresh samples, using archived material can provide information that would otherwise be unavailable for making such decisions. Connectivity assessments using archived material may also be used to identify gaps in existing material, informing targeted sampling to provide the additional material required for robust statistical testing. Strategic sampling to fill gaps in archived sample coverage would be a practical option for studies where obtaining fresh samples is particularly challenging, such as within the deep sea. Establishing open-access reference collections for deep-sea samples, as well as the development of mechanisms whereby global genetic expertise can utilize such samples, would further facilitate the use of archived material in connectivity studies. Maximizing the use of archived material would also serve to reduce the expense and environmental disturbance of sampling for fresh material.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the NIWA Invertebrate Collection staff (Kareen Schnabel, Sadie Mills, and Dean Stotter) for their assistance in locating G. gladius material. We also thank the scientists and crew aboard the research voyages used to collect this material and acknowledge the support of the institutions involved with these voyages. The NIWA Invertebrate Collection provided specimens collected under the following projects: “Seamounts: their importance to fisheries and marine ecosystems,” funded by the former New Zealand Foundation for Research, Science and Technology; “Impact of resource use on vulnerable deep-sea communities” project (DSC) (CO1X0906), funded by the Ministry of Business, Innovation & Employment (MBIE); “Nascent Inter-Ridge Volcanic And Neotectonic Activity,” funded by the Ministry for Primary Industries (MPI), in collaboration with Auckland University, GNS Science (NZ) and the University of New Hampshire (USA) and with funding from the DSC program; the joint New Zealand-USA 2005 NOAA “Ring of Fire Expedition”; “Marine Geoscientific Research on Input and Output in the Tonga-Kermadec Subduction Zone,” a joint German-Canadian-New Zealand expedition led by the University of Kiel and the Federal Institute for Geosciences and Natural Resources; “Scientific Observer Program” funded by MPI. RB would like to thank Catarina Silva for assistance in the laboratory work involved in this study. RB is supported by PhD scholarship funding from NIWA and Victoria University of Wellington. The research reported here is part of the NIWA project “Deep-sea mining of the Kermadec Arc—Geophysical prospectivity and environmental impacts” funded by the New Zealand Ministry of Business, Innovation and Employment (formally the Foundation for Research Science and Technology) (contract CO1X0702). We would also like to thank two anonymous reviewers for their constructive comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2015.00105
Footnotes
1. ^http://www.nzpam.govt.nz/cms/online-services/current-permits/.
References
Arellano, S. M., and Young, C. M. (2009). Spawning, development, and the duration of larval life in a deep-sea cold-seep mussel. Biol. Bull. 216, 149–162.
Arevalo, E., Davis, S. K., and Sites, J. W. Jr. (1994). Mitochondrial DNA sequnce divergence and phylogenetic relationships among eight chromosome races of the Scleroporus grammicus complex (Phrynosomatidae) in central Mexico. Syst. Biol. 43, 387–418. doi: 10.1093/sysbio/43.3.387
Baker, E., and Beaudoin, Y. (eds.). (2013). Deep Sea Minerals: Sea Floor Massive Sulphides, a Physical, Biological, Environmental, and Technical review. SPC.
Bandelt, H. J., Forster, P., and Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Bielawski, J. P., and Gold, J. R. (1996). Unequal synonymous substitution rates within and between two protein-coding mitochondrial genes. Mol. Biol. Evol. 13, 889–892. doi: 10.1093/oxfordjournals.molbev.a025649
Bors, E. K., Rowden, A. A., Maas, E. W., Clark, M. R., and Shank, T. M. (2012). Patterns of deep-sea genetic connectivity in the New Zealand region: implications for management of benthic ecosystems. PLoS ONE 7:e49474. doi: 10.1371/journal.pone.0049474
Boschen, R. E., Rowden, A. A., Clark, M. R., Barton, S. J., Pallentin, A., and Gardner, J. P. A. (2015). Megabenthic assemblage structure on three New Zealand seamounts: implications for seafloor massive sulfide mining. Mar. Ecol. Prog. Ser. 523, 1–14. doi: 10.3354/meps11239
Boschen, R. E., Rowden, A. A., Clark, M. R., and Gardner, J. P. A. (2013). Mining of deep-sea seafloor massive sulfides: a review of the deposits, their benthic communities, impacts from mining, regulatory frameworks and management strategies. Ocean Coast. Manage. 84, 54–67. doi: 10.1016/j.ocecoaman.2013.07.005
Bouchet, P., and Warén, A. (1991). Ifremeria nautilei, a new gastropod from hydrothermal vents, probably associated with symbiotic bacteria. C. R. Acad. Sci. Paris Ser. III 312, 495–501.
Carney, S. L., Formica, M. I., Divatia, H., Nelson, K., Fisher, C. R., and Schaeffer, S. W. (2006). Population structure of the mussel “Bathymodiolus” childressi from Gulf of Mexico hydrocarbon seeps. Deep Sea Res. I 53, 1061–1072. doi: 10.1016/j.dsr.2006.03.002
Chiswell, S. M., Bostock, H. C., Sutton, P. J. H., and Williams, M. J. M. (2015). Physical oceanography of the deep seas around New Zealand: a review. N.Z. J. Mar. Freshw. 49, 286–317. doi: 10.1080/00288330.2014.992918
Chiswell, S. M., and Sutton, P. J. H. (2015). Drifter- and float-derived mean circulation at the surface and 1000 m in the New Zealand region. N.Z. J. Mar. Freshw. 49, 259–277. doi: 10.1080/00288330.2015.1008522
Clark, M. R., and O'shea, S. (2001). Hydrothermal vent and seamount fauna from the southern Kermadec ridge, New Zealand. InterRidge News 10, 14–17.
Coffey Natural Systems (2008). Environmental Impact Statement, Solwara 1 project, Nautilus Minerals Niugini Limited, Main Report. Brisbane, QLD: Coffey Natural Systems.
Collins, P. C., Croot, P., Carlsson, C., Colaco, A., Grehan, A., Hyeong, K., et al. (2013a). A primer for the 631 environmental impact assessment of mining at seafloor massive sulfide deposits. Mar. Pol. 42, 198–209. doi: 10.1016/j.marpol.2013.01.020
Collins, P. C., Kennedy, R., Copley, J., Boschen, R., Fleming, N., Forde, J., et al. (2013b). VentBase: developing a consensus among stakeholders in the deep-sea regarding environmental impact assessment for deep-sea mining. Mar. Pol. 42, 334–336. doi: 10.1016/j.marpol.2013.03.002
Craddock, C., Hoeh, W. R., Lutz, R. A., and Vrijenhoek, R. C. (1995). Extensive gene flow among mytilid (Bathymodiolus thermophilus) populations from hydrothermal vents of the eastern Pacific. Mar. Biol. 124, 137–146. doi: 10.1007/BF00349155
De Ronde, C. E. J., Massoth, G. J., Butterfield, D. A., Christenson, B. W., Ishibashi, J., Ditchburn, R. G., et al. (2011). Submarine hydrothermal activity and gold-rich mineralization at Brothers Volcano, Kermadec Arc, New Zealand. Miner Deposita 46, 541–584. doi: 10.1007/s00126-011-0345-8
Devey, C. W., Fisher, C. R., and Scott, S. (2007). Responsible science at hydrothermal vents. Oceanography 20, 162–171. doi: 10.5670/oceanog.2007.90
Dixon, D. R., Sole-Cava, A. M., Pascoe, P. L., and Holland, P. W. H. (1995). Periostracal adventitious hairs on spat of the mussel Mytilus edulis. J. Mar. Biol. Assoc. U.K. 75, 363–372. doi: 10.1017/S0025315400018233
Excoffier, L., Laval, G., and Schneider, S. (2005). Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol. Bioinform. 1, 47–50.
Excoffier, L., Smouse, P. E., and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491.
Fisher, C., and Skibinski, D. (1990). Sex-biased mitochondrial DNA heteroplasmy in the marine mussel Mytilus. Proc. R. Soc. Lond. B 242, 149–156. doi: 10.1098/rspb.1990.0118
Folmer, O., Black, M., Hoeh, W., Lutz, R., and Vrijenhoek, R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotech. 3, 294–299.
Fontanez, K. M., and Cavanaugh, C. M. (2013). Phylogenetic relationships of hydrothermal vent mussels (Bathymodiolinae) and their symbionts. Mar. Ecol. Prog. Ser. 474, 147–154. doi: 10.3354/meps10086
Freeland, J. (2005). “Molecular markers in ecology,” in Molecular Ecology, ed H. Kirk (Chichester: John Wiley and Sons), 31–62.
Hannington, M., Jamieson, J., Monecke, T., Petersen, S., and Beaulieu, S. (2011). The abundance of seafloor massive sulfide deposits. Geology 39, 1155–1158. doi: 10.1130/G32468.1
Hellberg, M. E., Burton, R. S., Neigel, J. E., and Palumbi, S. R. (2002). Genetic assessment of connectivity among marine populations. Bull. Mar. Sci. 70, 273–290.
International Seabed Authority (2010). Regulations on Prospecting and Exploration for Polymetallic Sulphides in the Area. ISBA/16/A/12/Rev.1. Kingston: International Seabed Authority.
Interridge (2009). InterRidge Statement of Commitment to Responsible Research Practices at Deep-Sea Hydrothermal Vents Available online at: http://www.interridge.org/IRStatement (Accessed March 27, 2015).
Jollivet, D., Comtet, T., Chevaldonné, P., Hourdez, S., Desbruyeres, D., and Dixon, D. R. (1998). Unexpected relationship between dispersal strategies and speciation within the association Bathymodiolus (Bivalvia)-Branchipolynoe (Polychaeta) inferred from the rDNA neutral ITS2 marker. Cah. Biol. Mar. 39, 359–362.
Jones, W. J., Won, Y. J., Maas, P. A. Y., Smith, P. J., Lutz, R. A., and Vrijenhoek, R. C. (2006). Evolution of habitat use by deep-sea mussels. Mar. Biol. 148, 841–851. doi: 10.1007/s00227-005-0115-1
Kyuno, A., Shintaku, M., Fujita, Y., Matsumoto, H., Utsumi, M., Watanabe, H., et al. (2009). Dispersal and differentiation of deep-sea mussels of the genus Bathymodiolus (Mytilidae, Bathymodiolinae). J. Mar. Biol. 2009, 1–15. doi: 10.1155/2009/625672
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 11, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, Z. J., and Cordes, J. F. (2004). DNA marker technologies and their applications in aquaculture genetics. Aquaculture 238, 1–37. doi: 10.1016/j.aquaculture.2004.05.027
Lutz, R. A., Shank, T. M., Fornari, D. J., Haymon, R. M., Lilley, M. D., Vondamm, K. L., et al. (1994). Rapid growth at deep-sea vents. Nature 371, 663–664. doi: 10.1038/371663a0
Mantel, N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220.
Marsh, L., Copley, J. T., Huvenne, V. A. I., Linse, K., Reid, W. D. K., Rogers, A. D., et al. (2012). Microdistribution of faunal assemblages at deep-sea hydrothermal vents in the southern ocean. PLoS ONE 7:e48348. doi: 10.1371/journal.pone.0048348
Marsh, L., Copley, J. T., Tyler, P. A., and Thatje, S. (2015). In hot and cold water: differential life-history traits are key to success in contrasting thermal deep-sea environments. J. Anim. Ecol. 2015, 1–15. doi: 10.1111/1365-2656.12337
Miller, K., Williams, A., Rowden, A. A., Knowles, C., and Dunshea, G. (2010). Conflicting estimates of connectivity among deep-sea coral populations. Mar. Ecol. 31, 144–157. doi: 10.1111/j.1439-0485.2010.00380.x
Miyazaki, J. I., Beppu, S., Kajio, S., Dobashi, A., Kawato, M., Fujiwara, Y., et al. (2013). Dispersal ability and environmental adaptability of deep-sea mussels Bathymodiolus (Mytilidae: Bathymodiolinae). OJMS 3, 31–39. doi: 10.4236/ojms.2013.31003
Olu-Le Roy, K., von Cosel, R., Hourdez, S., Carney, S. L., and Jollivet, D. (2007). Amphi-Atlantic cold-seep Bathymodiolus species complexes across the equatorial belt. Deep Sea Res. 54, 1890–1911. doi: 10.1016/j.dsr.2007.07.004
Page, H. M., Faila-Médioni, A., Fisher, C. R., and Childress, J. J. (1991). Experimental evidence for filter-feeding by the hydrothermal vent mussel, Bathymodiolus thermophilus. Deep Sea Res. 38, 1455–1461. doi: 10.1016/0198-0149(91)90084-S
Ramirez-Llodra, E., Tyler, P. A., Baker, M. C., Bergstad, O. A., Clark, M. R., Escobar, E., et al. (2011). Man and the last great wilderness: human impact on the deep sea. PLoS ONE 6:e22588. doi: 10.1371/journal.pone.0022588
Rowden, A. A., Clark, M. R., O'shea, S., and Mcknight, D. G. (2003). Benthic Biodiversity of Seamounts on the Southern Kermadec Volcanic arc. Marine Biodiversity Biodescurity Report 3, 1–23. National Institute of Water and Atmospheric Research, Wellington.
Scholin, C. A., and Anderson, D. M. (1994). Identification of group-and strain-specific generic markers for globally distributed Alexandrium (Dinophyceae). I. RFLP analysis of SSU rRNA genes. J. Phycol. 30, 44–54.
Sefc, K., Payne, R., and Sorenson, M. (2007). Single base errors in PCR products from avian museum specimens and their effect on estimates of historical genetic diversity. Conserv. Genet. 8, 879–884. doi: 10.1007/s10592-006-9240-8
Shank, T. M., and Halanych, K. M. (2007). Toward a mechanistic understanding of larval dispersal: insights from genomic fingerprinting of the deep-sea hydrothermal vent tubeworm Riftia pachyptila. Mar. Ecol. 28, 25–35. doi: 10.1111/j.1439-0485.2007.00146.x
Slatkin, M. (1995). A measure of population subdivision based on microsatellite allele frequencies. Genetics 139, 457–462.
Smith, P. J., Mcveagh, S. M., Won, Y., and Vrijenhoek, R. C. (2004). Genetic heterogeneity among New Zealand species of hydrothermal vent mussels (Mytilidae: Bathymodiolus). Mar. Biol. 144, 537–545. doi: 10.1007/s00227-003-1207-4
Smouse, P. E., Long, J. C., and Sokal, R. R. (1986). Multiple regression and correlation extensions of the Mantel Test of matrix correspondence. Syst. Zool. 35, 627–632. doi: 10.2307/2413122
Thaler, A. D., Zelnio, K., Saleu, W., Schutltz, T. F., Carlsson, J., Cunningham, C., et al. (2011). The spatial scale of genetic subdivision in populations of Ifremeria nautilei, a hydrothermalvent gastropod from the southwest Pacific. BMC Evol. Biol. 11:372. doi: 10.1186/1471-2148-11-372
Tunnicliffe, V., Embley, R. W., Holden, J. F., Butterfield, D. A., Massoth, G. J., and Juniper, S. K. (1997). Biological colonization of new hydrothermal vents following an eruption on Juan de Fuca Ridge. Deep Sea Res. I 44, 1627–1644. doi: 10.1016/S0967-0637(97)00041-1
Van Dover, C. L. (2011). Mining seafloor massive sulphides and biodiversity: what is at risk? ICES J. Mar. Sci. 68, 341–348. doi: 10.1093/icesjms/fsq086
Van Dover, C. L. (2014). Impacts of anthropogenic disturbances at deep-sea hydrothermal vent ecosystems: a review. Mar. Environ. Res. 102, 59–72. doi: 10.1016/j.marenvres.2014.03.008
Villesen, P. (2007). FaBox: an online toolbox for fasta sequences. Mol. Ecol. Notes 7, 965–968. doi: 10.1111/j.1471-8286.2007.01821.x
Von Cosel, R. M., and Marshall, B. A. (2003). Two new species of large mussels (Bivalvia: Mytilidae) from active submarine volcanoes and a cold seep off the eastern North Island of New Zealand, with description of a new genus. Nautilus 117, 31–46.
Vrijenhoek, R. C. (2010). Genetic diversity and connectivity of deep-sea hydrothermal vent metapopulations. Mol. Ecol. 19, 4391–4411. doi: 10.1111/j.1365-294X.2010.04789.x
Wagstaff, S. J., and Garnock-Jones, P. J. (1998). Evolution and bigeography of the Hebe compex (Scrophulariaceae) inferred from ITS sequences. N.Z. J. Bot. 36, 425–437. doi: 10.1080/0028825X.1998.9512581
Wandeler, P., Hoeck, P. E. A., and Keller, L. F. (2007). Back to the future: museum specimens in population genetics. Trends Ecol. Evol. 22, 634–642. doi: 10.1016/j.tree.2007.08.017
White, T. J., Bruns, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J.J. Sninsky, and T. J. White. (San Diego, CA: Academic Press), 315–322.
Won, Y., Young, C. R., Lutz, R. A., and Vrijenhoek, R. C. (2003). Dispersal barriers and isolation among deep-sea mussel populations (Mytilidae: Bathymodiolus) from eastern Pacific hydrothermal vents. Mol. Ecol. 12, 169–184. doi: 10.1046/j.1365-294X.2003.01726.x
Keywords: population genetic connectivity, deep-sea mining, archived samples, environmental management, molecular markers, seafloor massive sulfides
Citation: Boschen RE, Rowden AA, Clark MR and Gardner JPA (2015) Limitations in the Use of Archived Vent Mussel Samples to Assess Genetic Connectivity Among Seafloor Massive Sulfide Deposits: A Case Study with Implications for Environmental Management. Front. Mar. Sci. 2:105. doi: 10.3389/fmars.2015.00105
Received: 29 March 2015; Accepted: 17 November 2015;
Published: 08 December 2015.
Edited by:
Anthony Grehan, National University of Ireland, Galway, IrelandReviewed by:
Daniela Zeppilli, Institut Français de Recherche pour l'Exploitation de la MER, FranceRaul Bettencourt, IMAR Center, University of the Azores, Portugal
Copyright © 2015 Boschen, Rowden, Clark and Gardner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel E. Boschen, cmFjaGVsLmJvc2NoZW5AdnV3LmFjLm56