 Christopher J. Miller
Christopher J. Miller Andrew L. Rose
Andrew L. Rose T. David Waite
T. David Waite- 1School of Civil and Environmental Engineering, UNSW Australia, Sydney, NSW, Australia
- 2Southern Cross GeoScience, Southern Cross University, Lismore, NSW, Australia
The reaction between Fe(II) and H2O2 to yield hydroxyl radicals (HO•), the Fenton reaction, is of interest due to its role in trace metal and natural organic matter biogeochemistry, its utility in water treatment and its role in oxidative cell degradation and associated human disease. There is significant dispute over whether HO•, the most reactive of the so-called reactive oxygen species (ROS), is formed in this reaction, particularly under circumneutral conditions relevant to natural systems. In this work we have studied the oxidation kinetics of Fe(II) complexed by L = citrate, ethylenediaminetetraacetic acid (EDTA) and diethylenetriaminepentaacetic acid (DTPA) and also measured HO• production using phthalhydrazide as a probe compound at pH 8.2. A kinetic model has been developed and utilized to confirm that HO• is the sole product of the Fe(II)L-H2O2 reaction for L = EDTA and DTPA. Quantitative HO• production also appears likely for L = citrate, although uncertainties with the speciation of Fe(II)-citrate complexes as well as difficulties in modeling the oxidation kinetics of these complexes has prevented a definitive conclusion. In the absence of ligands at circumneutral pH, inorganic Fe(II) reacts with H2O2 to yield a species other than HO•, contrary to the well-established production of HO• from inorganic Fe(II) at low pH. Our results suggest that at high pH Fe(II) must be complexed for HO• production to occur.
Introduction
The reaction between Fe(II) and H2O2 (the Fenton reaction) occurs naturally in aquatic environments (White et al., 2003), biological systems (Backa et al., 1993; Winterbourn, 1995; Jomova et al., 2012; Lee et al., 2012), and is also employed in water treatment applications (von Sonntag, 2008). Although ubiquitous and an important source of reactive oxygen species (ROS), if HO• is indeed produced, the identity of the intermediate formed in this reaction has long been the subject of debate (Goldstein et al., 1993; MacFaul et al., 1998; Walling, 1998; Goldstein and Meyerstein, 1999; Dunford, 2002; Rachmilovich-Calis et al., 2009a; Remucal Keenan and Sedlak, 2011). It is generally accepted that HO• is formed under acidic conditions and in the absence of ligands. However, an alternative mechanism invoking higher valent Fe complexes such as Fe(IV), first proposed by Bray and Gorin (1932), is also consistent with many findings (Kremer, 1999). Under circumneutral conditions the mechanism is particularly controversial (Remucal Keenan and Sedlak, 2011), with recent studies now mostly in agreement that at higher pH the product of the Fenton reaction with inorganic Fe(II) shifts to a species other than HO•, possibly to a high-valent Fe species such as Fe(IV) (Hug and Leupin, 2003; Keenan and Sedlak, 2008; Bataineh et al., 2012; Lee et al., 2013). The underlying cause of this is not clear. The presence of organic ligands that are important to the speciation of Fe(II) in natural waters (Roy and Wells, 2011; Hopwood et al., 2015), as well as those employed to solubilize Fe under circumneutral conditions in industrial practices, complicates this further.
For reasons which will be discussed later, we have examined the ligands citrate, ethylenediaminetetraacetic acid (EDTA), and diethylenetriaminepentaacetic acid (DTPA) in this study. Despite extensive study, it is still not clear whether or not HO• is formed when the Fe(II) complexes of these ligands are oxidized by H2O2. Previous investigations have used a wide array of probe compounds to examine the nature of the oxidative intermediate formed in this reaction. By consideration of these methodologies used in this earlier work, particularly their limitations, it has been possible to design a study to avoid these issues and more-effectively isolate the Fe(II)L-H2O2 reaction. For example, Rahhal and Richter (1988, 1989) compared the impact of t-BuOH and MeOH upon the reaction stoichiometry and kinetics when Fe(II)/Fe(III)DTPA solutions reacted with H2O2 or were exposed to radiolytically produced HO• and concluded that HO• could not be the reactive intermediate. Similar work however by Rush and Koppenol (1987) (with L = EDTA and DTPA) employing a wider range of scavengers was generally consistent with HO• formation (although t-BuOH and benzoate scavengers yielded contradictory results), in direct contrast to their own earlier work which suggested that HO• was not formed by peroxidation of Fe(II)EDTA (Rush and Koppenol, 1986). Luzzatto et al. (1995) later examined the Fe(II)EDTA-H2O2 system using a β-elimination based assay with alcohol probe compounds, concluding that Fe(II)EDTA reacts with H2O2 to yield HO•. All these studies were undertaken in deoxygenated solutions at relatively high Fe concentrations (10−5–10−3 M), under which conditions the relatively poorly studied reactions between organic radical intermediates (whatever their mode of formation) and FeL species potentially become important. Although •CH2COH(CH3)CH3 radicals formed from the reaction of HO• and t-BuOH were shown to be unreactive toward Fe-DTPA complexes (Rahhal and Richter, 1989) and unable to reduce Fe(III)EDTA (Rush and Koppenol, 1987), Croft et al. (1992) suggested that they could oxidize Fe(II)EDTA. Fe(III)EDTA has also been shown to oxidize radicals (Rush and Koppenol, 1987). Most carbon-centered organic radicals also typically react rapidly with O2. It is possible that incomplete O2-exclusion further complicated the analysis of the earlier work, as even very small amounts of O2 will lead to competition between O2 and Fe species for the organic radicals (Rush and Koppenol, 1987). It would therefore seem likely that the complications introduced from the poorly-studied reactions between Fe and the carbon-centered radicals may be the reason for the discrepancies in these previous studies.
The nature of the intermediate formed has also been proposed to be dependent upon the initial reactant concentrations. Yamazaki and Piette (1990, 1991) studied the Fe(II)L-H2O2 (L = EDTA, DTPA) reaction under oxic conditions by DMPO spin trapping concluding that Fe(II)DTPA produces only HO•. They demonstrated that HO• formation was quantitative at low Fe(II) concentrations (<10−6 M). However, competitive scavenging with t-BuOH was not consistent with HO• formation, in agreement with similar observations by Rahhal and Richter (1988). Based upon observation of an increase in the total trapped spin when ethanol (EtOH) was added to the system, they also suggested that Fe(II)EDTA peroxidation forms another oxidative intermediate in addition to HO•. This concentration dependence however could also conceivably be caused by competition between DMPO and Fe-L complexes for the oxidant.
Croft et al. (1992) undertook a detailed ESR study of the Fe(II)EDTA/H2O2 system by monitoring organic radicals formed from substrate oxidation. With carefully controlled studies they demonstrated that anomalous results from scavenging experiments could be explained if oxidation/reduction of organic radicals by Fe(III)L/Fe(II)L and H2O2 was incorporated into the modeling. Previous anomalous results in the Fe(II)EDTA/H2O2 system with t-BuOH and EtOH as scavengers were thus reconciled when reduction of t-BuOH and β-EtOH radicals by Fe(II)EDTA and oxidation of α-EtOH radicals by Fe(III)EDTA was invoked. Often contradictory results have been rationalized by insufficient consideration of intermediate organic radical species, which may further oxidize Fe(II) or reduce Fe(III), depending upon the nature of the radical thus formed (Croft et al., 1992; Kosaka et al., 1992; Yurkova et al., 1999).
In comparison to the Fe(II)-EDTA/DTPA system, the Fe(II)-citrate system has been studied comparatively little. Gutteridge (1991) provided evidence that citrate is able to reduce Fe(III), with the Fe(II)-citrate thus formed able to produce HO•, although the mechanism was not examined nor was quantitative production confirmed (note that the reduction process is not significant at the concentrations and time scales used in this work). Studies on the photo-reduction of Fe(III)-citrate in the presence of H2O2 also support the formation of HO•, although details of reaction rates were not examined (Zepp et al., 1992). Vile et al. (1987) directly studied the reaction between H2O2 and Fe(II)-citrate (Fe(II)cit) and suggested that an oxidant whose properties are consistent with HO• is formed based upon formation of similar thiobarbituric acid reactive substances (TBARS) to those formed during deoxyribose degradation with Fe(II)EDTA, which was considered to produce HO• based upon the impact of various scavengers. Separate experiments measuring CO2 formation during formate oxidation demonstrated that Fe(II)cit peroxidation produces 60% less HO• than Fe(II)EDTA peroxidation. The extent of deoxyribose degradation by Fe(II)cit was similar to that observed with Fe(II)EDTA, whereas inorganic Fe(II) produced ~3 to 4-fold more TBARS, suggesting that inorganic Fe(II) may produce HO•. Results from this work should be considered with some caution in light of more recent evidence regarding the inherent complexity of interpreting results from the TBARS method (Rachmilovich-Calis et al., 2009b).
From these previous studies it is clear that there is significant disagreement as to the reactive intermediate formed in these systems and that detailed consideration of the fate of any organic radicals formed must be considered in any quantitative analysis, as highlighted by Masarwa et al. (2005). By consideration of the fate of these radical species it is conceivable that a kinetic model may be constructed that could adequately describe the concentration profiles of Fe(II)/Fe(III), as well as any production of HO•, should it indeed be formed. The main drawback with such an approach is the paucity of studies into the reactivity of carbon-centered radicals with Fe, especially when complexed by organic ligands. However, most carbon-centered radicals react at near diffusion-controlled rates with O2 to yield organoperoxyl radicals, with some of these organoperoxyl radicals subsequently rapidly eliminating (von Sonntag et al., 1997), with the reactions of and Fe-complexes thoroughly investigated. Therefore, provided Fe concentrations are significantly lower than O2 concentrations, and the organoperoxyl radicals that are formed are of the type that rapidly eliminate , any potential reaction between Fe and these carbon-centered radicals will be insignificant. Under such conditions organic radicals are effectively converted to , a species with well-known reactivity with Fe and Fe-complexes. This study has been designed around these requirements to allow for a definitive conclusion to be reached, and to avoid the complications that have plagued earlier work in this area.
In the present study the oxidation of inorganic Fe(II) and Fe(II)L by H2O2 was examined under oxic conditions at low micromolar concentrations of Fe(II), combining measurements of Fe(II) oxidation kinetics, HO• production kinetics, and competition between well-defined competitive HO• scavengers and the HO• trapping agent phthalhydrazide (Phth). Kinetic modeling was then used to quantitatively examine whether HO• was the key intermediate formed. By employing oxic conditions with relatively low concentrations of FeL and H2O2, the fate of intermediate organic radicals was determined by well-defined O2-mediated pathways, resulting in minimal interference to the Fe(II)L-H2O2 system under study. This approach, combined with quantitative kinetic modeling incorporating the fate and further interaction of intermediate organic radical species, has allowed the Fe(II)L-H2O2 reaction to be effectively isolated and studied. A critical aspect of this approach is being able to reasonably predict the actual fate of any oxidizing radical formed, as well as the kinetics and mechanism of Fe(II) oxidation [and potentially Fe(III) reduction] in the presence of the ligand. For this reason, well-defined synthetic ligands (L = citrate, EDTA and DTPA) for which the relevant chemistry is reasonably well-understood have been employed in place of the poorly characterized ligands that are typical of marine, estuarine and freshwater systems. A low ionic strength has been used in this study to prevent introduction of redox-active contaminants from the salt solutions, as well as to simplify the kinetic and thermodynamic treatment of the Fe-ligand interaction which can be greatly complicated by high concentrations of cations such as Mg2+ and Ca2+ (Fujii et al., 2008; Miller et al., 2012).
Materials and Methods
Chemicals
Solutions were prepared in 18 MΩ.cm Milli-Q water (MQ) obtained from a Millipore Milli-Q system. Glassware and plasticware were soaked for 3 days in 0.5 M HCl. Reagents were of analytical reagent grade unless noted otherwise. EDTA and DTPA were added from stock solutions adjusted to pH ≈ 7.2 while citrate was added to solutions as solid disodium hydrogen citrate sesquihydrate (C6H6Na2O7·1.5H2O). H2O2 stock solutions were prepared by dilution of a 30.7% w/v H2O2 solution (Riedel-de Haën, stabilized with 40 mg/L dipicolinic acid) and standardized spectrophotometrically using ε250 = 22.7 M−1·cm−1 (Morgan et al., 1988). Fe(II) stock solutions were prepared daily from Fe(II)SO4·7H2O (4 mM) and adjusted to pH 4 with HCl. 1 M Na2CO3 buffer was prepared by dissolving Na2CO3 in MQ and allowing it to stand for several days. It was then filtered to remove precipitates (0.22 μm Millipore Millex-GV PVDF membrane) and then adjusted to pH 11 with 32% HCl. K5Cu(HIO6)2 (Cu(III)) was synthesized as described previously (Miller et al., 2011). For scavenging experiments, appropriate amounts of sodium formate (Sigma-Aldrich, 99+% ACS Reagent grade) or 2-methyl-propan-2-ol (t-BuOH, Ajax Analytical Reagent grade) were added to the standard reaction matrix, which consisted of 10 mM NaCl and 2 mM NaHCO3. When performing HO• production experiments, 0.55 mM phthalhydrazide (Phth, 99%, Aldrich) was also added to the standard matrix. In all experiments the pH of the matrix was adjusted to 8.20 ± 0.05 with HCl and/or NaOH, which remained relatively constant (±0.05 pH units) during an experiment. All reactions were studied under conditions minimizing exposure to room lighting through use of brown glass vessels and shielding with aluminum foil.
Fe(II) Oxidation Experiments
The kinetics of Fe(II)cit oxidation by O2 and H2O2 were examined by monitoring the loss of Fe(II) using luminol chemiluminescence (Rose and Waite, 2001; Miller et al., 2009) with [Fe(II)]0 = 0.2, 0.6, 1.2, and 2 μM, where the subscript 0 denotes initial concentration. Fe(II)L oxidation kinetics (L = EDTA or DTPA; [Fe(II)]0 = 2 μM) were investigated by following Fe(III)L formation spectrophotometrically at 260 nm [ε ≈ 7600 M−1·cm−1 for both complexes, background corrected at 500 nm, with the molar absorbance coefficient determined here consistent with the values determined by Hill-Cottingham (1955) after accounting for the pH dependence]. In all cases, inorganic Fe(II) was added to a matrix containing an oxidant and ligand. Although explicitly considered during kinetic modeling, initial competition between Fe(II)-complex formation and oxidation of inorganic Fe(II) was expected to be minimal. This was confirmed by calculations using known rate constants for complex formation and inorganic Fe(II) oxidation for L = EDTA or citrate, and validated experimentally for L = DTPA as described in Supplementary Material Section 1. In all cases, no Fe(III) precipitation was expected to occur in the final solution on short timescales as the solubility product for amorphous ferric oxide (AFO) was not exceeded (see Supplementary Material Section 2 for calculations; in the absence of ligand AFO precipitation occurs rapidly at 2 μM Fe(III) at pH 8.2).
Phth was not present when monitoring Fe(II) oxidation as both Phth and Phth-OH absorb strongly in the UV (interfering with the Fe(III)EDTA and Fe(III)DTPA absorbance) and also due to the formation of chemiluminescent Phth-OH (which interferes with luminol chemiluminescence).
Fe(II) Oxidation in the Presence of Formate
The oxidation of formate by HO• initially yields , which reacts with O2 with a rate constant of 2 × 109 M−1·s−1 to yield and CO2 (Buxton et al., 1976). As such, adding an excess of formate quantitatively converts all HO• that is produced to and CO2. Under the standard reaction conditions used here, excess ligand effectively competes with Fe(II) for HO•, forming ligand-derived organic radicals that are potentially able to influence Fe(II) oxidation kinetics. As the reactivity of these organic radicals is unknown, Fe(II) oxidation experiments were conducted with addition of sodium formate ([formate]0 = 50 mM for L = EDTA and DTPA and [formate]0 = 3 mM for L = citrate) at formate concentrations sufficiently high for quantitative scavenging of HO•, i.e., such that the reaction of HO• with excess ligand is rendered insignificant. When formate is present at these concentrations, all HO• will be converted to ; if addition of formate does not change the reaction kinetics then this implies that excess ligand, the primary sink for HO• in the absence of formate, also reacts with HO• in a similar fashion as formate to ultimately yield , validating the assumption that the reaction of ligand with HO• can be simplified to a reaction that produces along with some form of oxidized ligand.
HO• Production Experiments
Experiments were performed at 22 ± 2°C in vessels shielded from light. Experiments were initiated by addition of Fe(II) (2 μM) from an acidic stock (4 mM FeIISO4·7H2O in 0.1 mM HCl) to a vigorously stirred solution containing H2O2 (0–50 μM), Phth (0.55 mM), and either no ligand, 1 mM citrate, 10 μM EDTA or 10 μM DTPA. HO• formation was monitored using the phthalhydrazide method (Miller et al., 2011). Briefly, HO• reacts with Phth to form a hydroxy-derivative (Phth-OH) that accumulates in solution. Phth-OH is subsequently quantified using chemiluminescence detection during oxidation in an FeLume flowcell (Waterville Analytical) employing Cu(III)/H2O2 as an oxidant in 0.5 M Na2CO3 buffer at pH 11. The term “Phth-OH” is used to represent the total amount of HO• that has reacted with Phth, with the actual analyte, 5-hydroxy-2,3-dihydrophthalazine-1,4-dione (5-HO-Phth), formed in approximately 20% yield (Miller et al., 2011).
Impact of t-BuOH Upon Phth-Trappable HO• Production
HO• production experiments were repeated in the presence of a range of t-BuOH concentrations (2, 5, 20, or 100 mM) with 0.55 mM Phth present in each case to examine whether competition between Phth and t-BuOH was consistent with the known behavior of HO•, thus supporting or refuting HO• formation. Experiments were performed with 5 μM H2O2 for L = EDTA, 50 μM H2O2 for L = DTPA, and 10 μM H2O2 for L = citrate.
Kinetic Modeling
Experimental results were initially evaluated in the context of a basic kinetic model for Fe(II) oxidation in the presence of L, O2, and H2O2, which we denote as the “Fe(II)L model,” that was constructed using known reactions (Table 1). All rate constants were either taken directly from the literature or calculated from literature values, except for rate constants for the oxidation of Fe(II)L by O2 and the oxidation of Fe(II)L by H2O2, which were treated as variable parameters to be determined by fitting of the model to the experimental data. It was assumed in the Fe(II)L model that the reaction of H2O2 with Fe(II) (either inorganic or organically complexed) yielded HO•; this was treated as a null hypothesis that was tested by attempting to fit the model to the experimental data. It was also assumed that, under the oxygenated experimental conditions employed, the reaction of HO• with L resulted in the oxidation of L to a compound Lox that did not undergo further reaction, with concomitant formation of ; this is expected for L = EDTA based on the mechanism established by Höbel and von Sonntag (1998) and hypothesized to also be the case for L = DTPA or citrate. This assumption was tested by examining Fe(II)L oxidation in the presence of formate as described earlier. For experiments conducted in the presence of formate, the additional reactions in Table 2 were also included in the kinetic model.
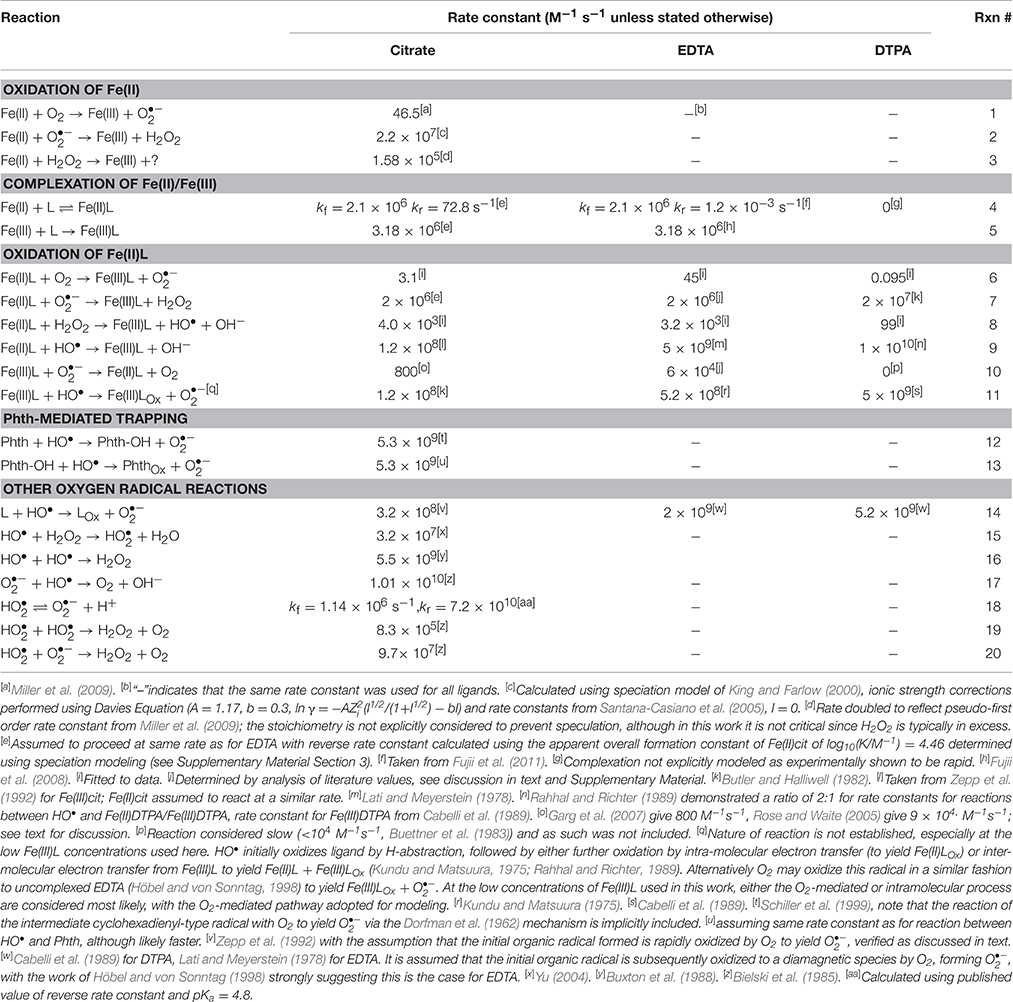
Table 1. “Fe(II)L” kinetic model for Fe(II)L oxidation.

Table 2. Additional reactions in the presence of formate.
Kinetic model simulations were performed using Presto-Kinetics V 3.28.3 (Wulkow, 2004) and Kintecus V 4.1 (Ianni, 2003). All model fitting was performed using the routines available in Presto unless specified otherwise, in which case parameters were manually adjusted and the quality of fit assessed qualitatively using Kintecus simulations.
Results and Discussion
HO• Production by Inorganic Fe(II)
When Fe(II) was oxidized by H2O2 in the absence of ligands, Phth-OH concentrations were always below the detection limit of 15 nM, implying no significant production of HO• occurred. This is despite H2O2 being the dominant oxidant of Fe(II) under these conditions (King and Farlow, 2000; González-Dávila et al., 2005; Santana-Casiano et al., 2006; Pham and Waite, 2008; Miller et al., 2009), with ≈1 μM Phth-OH expected to form in this process if HO• was indeed the product of the Fe(II)-H2O2 reaction. Control experiments confirmed that inorganic Fe(III) did not impact the recovery of 5-HO-Phth nor interfere with any other aspect of the method, confirming the result and suggesting that under the conditions of this study the Fe(II)-H2O2 reaction, although the dominant oxidation path for Fe(II), does not yield HO•. This result further supports the notion that a pH threshold exists above which the peroxidation of Fe(II) produces a species other than HO•, the nature of which cannot be determined from this work, although likely to be some form of Fe(IV) compound (Hug and Leupin, 2003; Keenan and Sedlak, 2008; Bataineh et al., 2012; Lee et al., 2013).
Kinetics of Fe(II) Oxidation and HO• Production by Fe(II)EDTA and Fe(II)DTPA
The oxidation kinetics of Fe(II)EDTA and Fe(II)DTPA were both well-described by the Fe(II)L model in Table 1 (see Figure 1). The kinetics of Fe(II)L oxidation (L = EDTA and DTPA) were unchanged in the presence of formate, supporting the formation of during ligand oxidation by HO•, as anticipated (Höbel and von Sonntag, 1998). It should be noted that bridged dimeric species of the form such as Fe(III)L()Fe(III)L, proposed to occur during O2-mediated oxidation by Seibig and Van Eldik (1997) for L = EDTA are not considered in the kinetic model as they are unimportant at the low Fe concentrations used here despite their demonstrated importance at millimolar concentrations of Fe(II) (Zang and Van Eldik, 1990b). The rate constants for O2- and H2O2-mediated oxidation of Fe(II)L (L = EDTA, DTPA) found here are generally consistent with previously determined values under similar conditions (see Supplementary Material Sections 4 and 5 for details). For both oxidants, the oxidation of Fe(II)DTPA was an order of magnitude slower than that of Fe(II)EDTA, despite the similar nature of these ligands, which is consistent with previous results (Graf et al., 1984). The absence of a readily-exchangeable bound water molecule in the Fe(II)DTPA complex (Zang and Van Eldik, 1990a) could explain the much slower oxidation kinetics observed for this complex if oxidation of Fe(II)L (L = EDTA, DTPA) by O2 and H2O2 was an inner sphere process; in this case, with the initial binding of the oxidant to Fe(II)L being the rate-limiting step, exchange of a bound H2O molecule for an oxidant molecule should presumably proceed at a much greater rate and with less steric hindrance than would be observed when oxidant binding first requires breaking of an Fe(II)-chelate bond (Graf et al., 1984). Summers et al. (2008) have demonstrated the importance of the water-loss rate to the related superoxide-mediated reduction of Fe(III)EDTA and Fe(III)DTPA.
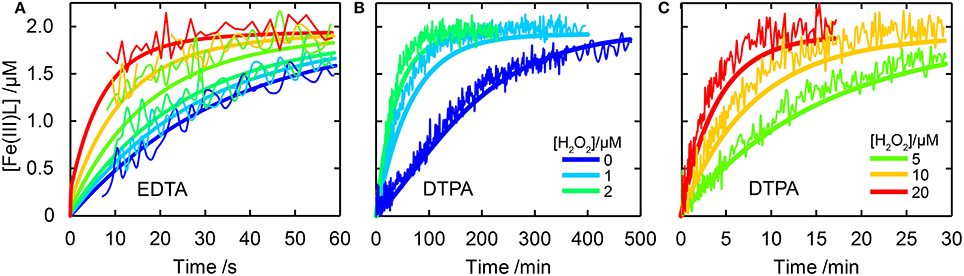
Figure 1. Comparison of experimental data to model results for Fe(II)EDTA and Fe(III)EDTA oxidation studies. In all cases thin lines are experimental data and thick lines model output. Experimental conditions were [Fe(II)]0 = 2 μM and [EDTA or DTPA] = 10 μM. Panel (A) shows results for EDTA for [H2O2] = 0 (blue), 1, 2, 5, 10, and 20 μM (red). Panel (B,C) show results for DTPA with [H2O2] as labeled.
For both Fe(II)EDTA and Fe(II)DTPA, significant Phth-trappable HO• production was observed. However, the interaction of the Fe-EDTA and Fe-DTPA complexes with (which is formed from the reaction of O2 with Fe(II)L and also during oxidation of the hydroxycyclohexadienyl-radical resulting from the reaction of HO• and Phth) complicates the kinetic modeling analysis. Although EDTA and DTPA are structurally quite similar, their Fe complexes exhibit markedly different reactivity with , likely due to the presence (EDTA) or absence (DTPA) of a readily exchangeable bound water molecule (Summers et al., 2008). Fe(III)DTPA is essentially unreactive with , with reported rate constants of <104 M−1s−1 (Buettner et al., 1983) and ≈103 M−1s−1 (Bolann et al., 1992), whereas Fe(II)DTPA is readily oxidized, with k7 = 2 × 107 M−1s−1 (Butler and Halliwell, 1982), such that acts only as an oxidant and Fe redox cycling does not occur. In contrast, Fe(II)EDTA is readily oxidized by , with k7 ≈2–8 × 106 M−1s−1 (Ilan and Czapski, 1977; Bull et al., 1982, 1983), with k7 = 2 × 106 M−1s−1 appropriate under the conditions of this study (see Supplementary Material Section 6 for further discussion). Unlike DTPA, Fe(III) complexed by EDTA is readily reduced by , k10 = 103–107 M−1s−1 (Bull et al., 1983). The wide range of reported values for the reduction rate constant is due to significant media/ionic strength effects (due to changes in the thermodynamics of the speciation and because the reaction involves two like-charged ions with z = −1) and also a pH effect as only the [FeIII(EDTA)(H2O)]− form is reactive with ; the hydrolyzed complex [FeIII(EDTA)(OH)]2− is essentially unreactive, with the pKa for [FeIII(EDTA)(H2O)]− ≈7.6 (Bull et al., 1983). As the current work was conducted at pH 8.2, near the pKa of Fe(III)EDTA, it is difficult to obtain a reliable literature estimate for the reduction rate constant. From a compilation of literature rate constants (discussed further in the Supplementary Material Section 6), a value for k10 of between 2 × 104 and 6 × 105 M−1s−1 is considered appropriate. Using the kinetic model it was shown that significant redox cycling of Fe occurs when k10 is at the upper end of this range, but not at the lower end. This Fe redox cycling results in elevated Fe(II) concentrations at later stages of the reaction; although this has negligible impact upon the kinetics of Fe(II) oxidation, when H2O2 is present in excess of Fe, this continual flux of Fe(II)EDTA results in ongoing HO• production even after the oxidation of Fe(II)EDTA appears to be complete (see Supplementary Figures 4, 5 in the Supplementary Material). Such an ongoing production of HO• is not supported by the data, suggesting the value for k10 appropriate to the conditions of study is at the lower end of the possible range.
It is clear that the behavior of the system, especially with regards to the kinetics and magnitude of HO• production, is strongly influenced by the interaction of Fe-EDTA complexes with . The sensitivity of the system to the rate constant for reduction of Fe(III)EDTA by is not evident when examining Fe(II) oxidation kinetics alone, and highlights the need to carefully consider the fate of the reactive intermediates. Due to the sensitivity of the model to the magnitude of k10, several values of this rate constant were trialed within the plausible bounds determined from previous studies, and the magnitude of the O2- and H2O2-mediated oxidation rate constants then fitted to the data. From this process a value for k10 of 6 × 104 M−1s−1 was found to both adequately describe the data and lie within the range expected from previous studies. After accounting for the complications introduced by reactions involving , both the Fe-EDTA and Fe-DTPA systems were well described by a kinetic model that proposed HO• as the sole-product of the reaction between H2O2 and Fe(II)L (L = EDTA, DTPA), as can be seen in Figures 2, 3. The ability to describe all the experimental results using the kinetic model developed demonstrates that there is no need to invoke higher-valent Fe species when L = EDTA or DTPA, and the hypothesis that quantitative formation of HO• occurs in this system is not inconsistent with the observed behavior.
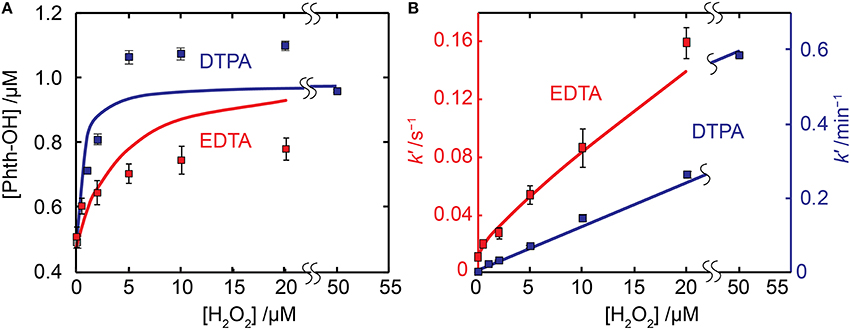
Figure 2. Comparison of experimental and model results for the magnitude (A) and pseudo-first order rate constant (B) of Phth-OH production for L = EDTA and DTPA. Experimental data are shown as symbols in all panels with the model results shown as solid lines. In all cases [Fe(II)]0 = 2 μM and [L] = 10 μM.
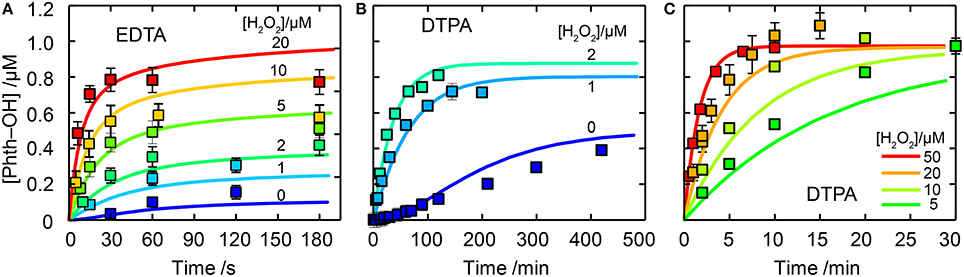
Figure 3. Phth-OH production by Fe(II)EDTA (A) and Fe(II)DTPA (B,C). Experimental data are shown as symbols with error bars the standard deviation of three replicate experiments. Kinetic model results are shown as solid lines (with k10 = 6 × 104 M−1s−1 for EDTA). In all cases [Fe(II)] = 2 μM, [L] = 10 μM and [H2O2] is as labeled.
Experiments with the addition of t-BuOH (0, 2, 5, 20, 100 mM), which competes with Phth (0.55 mM) for HO• via a well-described mechanism (Rxn 12 in Table 1; see Supplementary Material Section 7 for the reactions of t-BuOH), were used to validate that HO• was the species trapped by Phth. The final Phth-OH concentration was determined from both the experimental data and model predictions, then the impact of t-BuOH assessed by fitting a standard competition equation to the data:
where [Phth-OH]∞ is the final Phth-OH concentration in the absence of t-BuOH and R is the ratio of the rate constants for reaction of HO• with t-BuOH and Phth, i.e., k(HO•, t-BuOH)/k(HO•, Phth). The value of R was calculated for both the experimental data and also the kinetic model prediction; agreement between both values provides additional evidence for HO• production. For L = EDTA, the experimental data yielded R = 0.117 ± 0.003, in good agreement with R = 0.107 ± 0.001 obtained from fitting the competition equation to kinetic model output. For L = DTPA the experimental data yielded R = 0.073 ± 0.007 which is also in good agreement with R = 0.089 ± 0.002 obtained from the kinetic model output (in all cases R is given as best fit value ±one standard error from the curve fitting procedure, which likely underestimates the true uncertainty). For each ligand, the kinetic model output value of R differed from the value of 0.11 predicted from literature constants (Buxton et al., 1988; Schiller et al., 1999) due to the more complicated interactions present in the kinetic models compared to the assumptions made in the simplified competition equation; however, in both cases, the results are consistent with HO• as the only source of Phth-OH in this system. While Rahhal and Richter (1988) have previously asserted that the product of the Fe(II)DTPA-H2O2 reaction is completely unreactive with t-BuOH, our results clearly demonstrate that the product of this reaction does react with t-BuOH in a manner quantitatively consistent with the product being HO•.
Kinetics of Fe(II) Oxidation and HO• Production by Fe(II)Citrate
The apparent rate of H2O2-mediated Fe(II)-citrate oxidation varied with Fe(II) concentration. This dependence could not be reproduced by the Fe(II)L model in Table 1, which fitted the data poorly (Figure 4). Attempts to fit this model simultaneously to the Fe(II) oxidation and HO• production data were also unsuccessful. Due to the inability of the Fe(II)L model to reproduce the Fe(II) concentration dependence, and given that HO• production experiments were only conducted with [Fe(II)]0 = 2 μM, a second model scenario was examined in which the kinetic model in Table 1 was fitted to the HO• production data (which was conducted with [Fe(II)]0 = 2 μM) and also only to the oxidation data where [Fe(II)]0 = 2 μM. This fitting scenario, which we denote as the “Fe(II)L-2 μM” model, also yielded poor results. The failure of the Fe(II)L model, which only considers one form of Fe(II)-cit species, is not surprising, as the most likely reason for a concentration dependence of the oxidation rate is the formation of higher-order complexes, which will lead to speciation being a function of the Fe(II) concentration as well as the Fe(II):cit ratio.
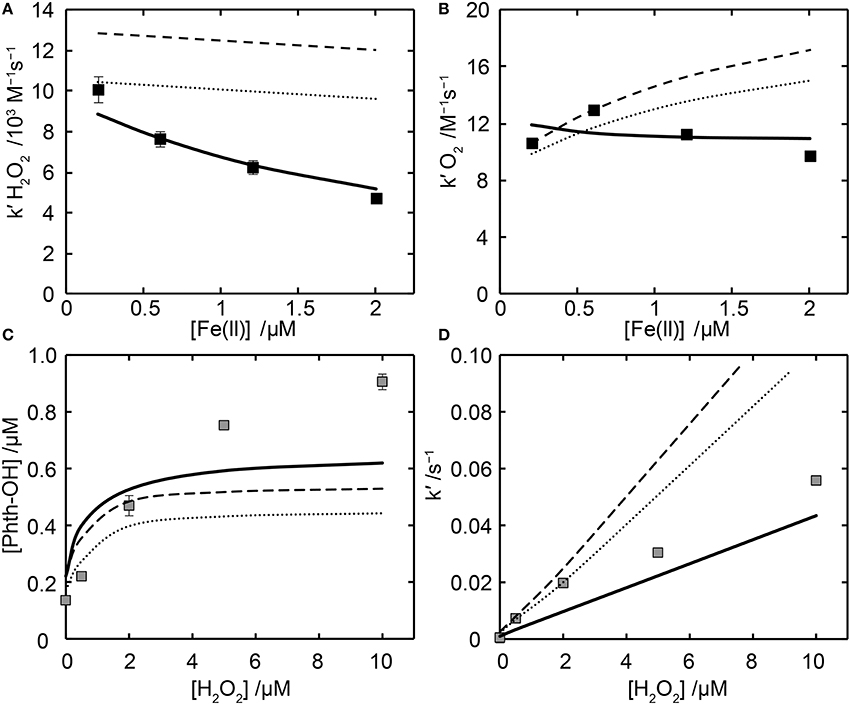
Figure 4. Kinetics of Fe(II) oxidation and resulting Phth-OH production in the presence of citrate. Panels (A,B) show the apparent rate constants for Fe(II)-citrate oxidation by (A) H2O2 and (B) O2 as a function of initial Fe(II) concentration ([cit] = 1 mM in all cases). Symbols are derived from linear fits of experimental pseudo-first order oxidation rate constants as a function of [H2O2] (A) or from the pseudo-first order rate constant divided by [O2] (B). Panels (C,D) show HO• production, with panel (C) showing the magnitude of total Phth-OH production when [Fe(II)]0 = 2 μM and panel (D) showing the kinetics of Phth-OH formation (the pseudo-first order rate constant of Phth-OH production). In all panels model predictions are indicated by lines, with the Fe(II)L model shown as a dashed line, the Fe(II)L-2 μM model as a dotted line and the Fe(II)2L model as a solid line.
None-the-less, several possible explanations were considered for the poor performance of this model. The possibility of organic radicals reducing Fe(III)L was discarded since the rate constant for reaction between carbon-centered radicals and O2, which is typically near 2 × 109 M−1s−1 (von Sonntag et al., 1997), is likely much greater than that for the reaction of carbon centered radicals with Fe(III)cit; reported rate constants for reduction of inorganic Fe(III) by hydroxyalkyl radicals are (1–6) × 108 M−1s−1 (Neta et al., 1996). Formate addition experiments further supported the assumption that O2 was the main sink for carbon-centered radicals, as its presence did not alter the Fe(II) oxidation results. The reaction of organic radicals with O2 is likely to ultimately yield , which may oxidize or reduce iron. Garg et al. (2007) reported a rate constant for -mediated reduction of Fe(III)cit of 800 M−1s−1, which is likely to be applicable under the current conditions based upon the kinetic model presented in their work. Assuming that the rate constant for -mediated oxidation of Fe(II)cit is similar to that of Fe(II)EDTA (2 × 106 M−1s−1), will be a net oxidant. This reasoning was supported by the HO• trapping experiments, in which a limiting Phth-OH concentration of ≈1 μM (which is half the initial Fe(II) concentration) was obtained at high [H2O2]. This limit occurs because formed during the HO•-Phth reaction oxidizes Fe(II)cit, hence limiting the amount of Fe(II)cit that is able to react with H2O2. If was also able to effectively reduce Fe(III)cit complexes, such an effect would not be observed, and enhanced Phth-OH production would be expected as a result of this additional flux of Fe(II)cit. This conclusion is consistent with the results of Sutton (1985) who employed similar logic to formate degradation studies. Even if a higher rate constant for -mediated reduction was used in the modeling [e.g., 9 × 104 M−1s−1 from Rose and Waite (2005) which was determined at lower citrate:Fe ratios than those used here] it was still not possible to replicate the trends observed. As such, it can be deduced that reactions of are unable to account for the apparent Fe(II)cit concentration dependence.
The trends observed can be reproduced if the existence of a di-iron citrate species of the form Fe(II)2L is proposed with differing oxidation kinetics to Fe(II)L [termed the “Fe(II)2L” model, which includes the additional reactions shown in Table 3]. The oxidation data could only poorly constrain parameters in the Fe(II)2L model, with Presto unable to converge to a unique solution. As such, an analytical expression was used to fit the Fe(II)2L complexation constants (K4 and K24) to the data (see Supplementary Material Section 8 for details; note that the related formation constants, βM, L are also referred to, with β1, 1 = K4 and β2, 1 = K4K24). Provided that log K4 > 4 and log K24 ≈ log K4 + 3.1, it was possible to find values for the Fe(II)L oxidation rate constant that fitted the data well. The physical basis for this lower bound on K4 being that small amounts of inorganic Fe(II) have low impact, so as long as the majority of Fe(II) is in complexed form, the results will be similar. In all cases a good fit could only be achieved if it was assumed that Fe(II)2L was comparatively unreactive (i.e., apparent H2O2-mediated oxidation rate constant <103 M−1s−1, with a value of 1 M−1s−1 adopted for modeling). A value of log K4 = 4.75 was determined from fitting to the HO• production data, with log K24 then fixed from the analytical modeling process. As is apparent in Figures 4A,B, only the Fe(II)2L model was able to provide a good fit to the Fe(II)cit oxidation data under all conditions. The Fe(II)2L species is speculative and does not accord with the speciation model of Königsberger et al. (2000), with published formation constants of similar species from other proposed models e.g., Fe(II)2(cit)2 from Amico et al. (1979), also inconsistent with those required to reproduce the data here. Inclusion of the proposed species is considered justified however due to the general uncertainty surrounding Fe(II)-citrate speciation at the high pH of this work, and that inclusion of this species was able to describe the data well, while all other possibilities considered failed to do so. This issue is explored further in Supplementary Material Section 9.
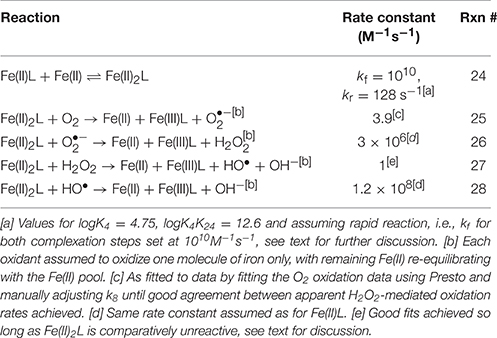
Table 3. Additional reactions used in the Fe(II)2L model.
Peroxidation of Fe(II) in the presence of citrate resulted in substantial HO• production. The apparent rate constant for reaction with H2O2 of (4.9 ± 0.4) × 103 M−1s−1 was determined from the Phth-OH production data (assuming quantitative formation of HO•); this compares to an apparent rate constant of (4.7 ± 0.2) × 103 M−1s−1 for the H2O2-mediated oxidation of Fe(II)cit when [Fe(II)]0 = 2 μM (i.e., under comparable conditions to the HO• production experiments). The close correspondence between these rate constants provides evidence that the production of HO• is closely coupled to the reaction of H2O2 with Fe(II)cit, and that it is likely that HO• is the sole product of this reaction. Competition experiments with t-BuOH supported the conclusion that HO• is the only species trapped by Phth in the Fe(II)-citrate system, with experiments yielding a value of R = 0.088 ± 0.004, in good agreement with predicted values of R = 0.078, 0.085, and 0.088 for the Fe(II)2L model, the Fe(II)L model, and the Fe(II)L-2 μM model, respectively. However, the Fe(II)L, Fe(II)L-2μM, and the Fe(II)2L models all provided a poor fit to the experimental HO• production data (see Figures 4C,D, 5). Although this may suggest that HO• is not the sole product of the Fe(II)cit-H2O2 interaction, it is far more likely that the poor fit is due to inadequacies in all the models described and that HO• is still likely the dominant product. As shown above for L = EDTA, without a thorough understanding of the oxidation and reduction kinetics of Fe(II)L/Fe(III)L by O2, , and H2O2 it is difficult to quantitatively determine whether HO• is the exclusive intermediate from the reaction of Fe(II)L and H2O2. It is clear that existing models of Fe(II)-citrate speciation and oxidation are not sufficiently refined to definitively rule out the presence of additional oxidative species, with additional work needed to better understand the Fe(II)-citrate-H2O2 system. However, this work strongly supports the notion that HO•, although potentially not the sole oxidative intermediate present, is certainly the dominant species formed.
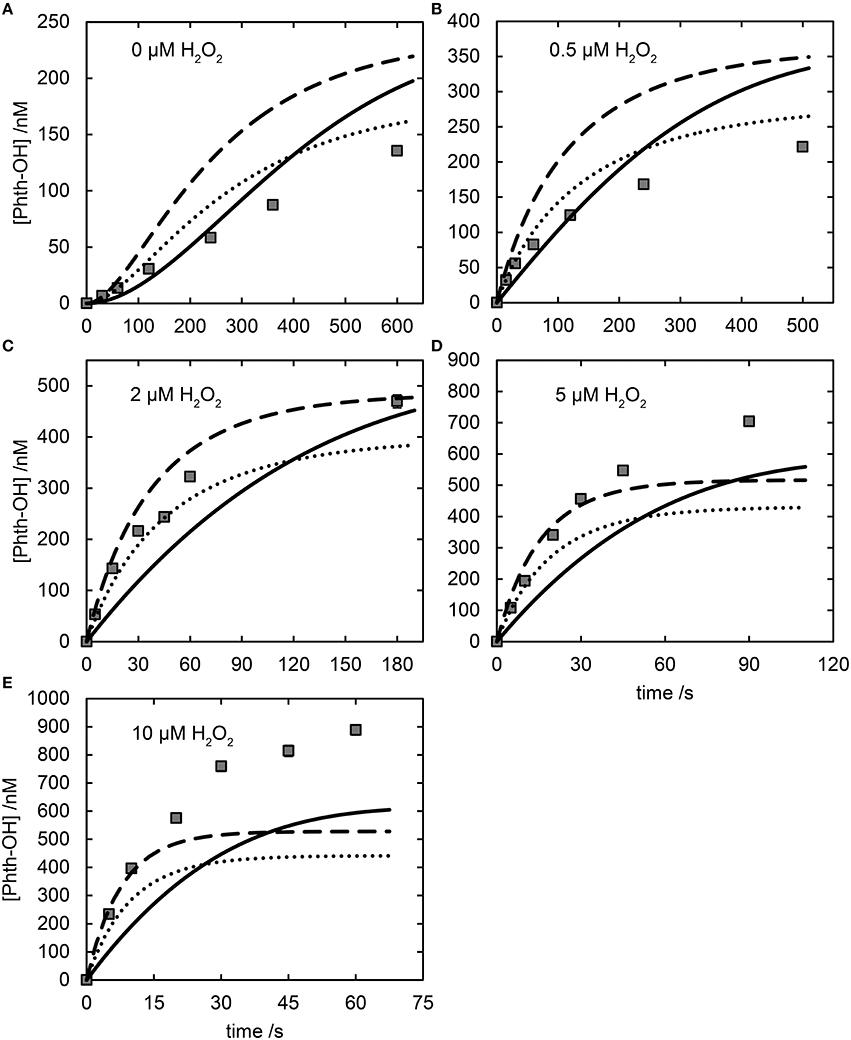
Figure 5. Comparison of experimental data and model results for HO• production by Fe(II)cit when oxidized by H2O2 (0–10 μM, panels A–E respectively). In all panels [Fe(II)] = 2 μM, [cit] = 1 mM, experimental data are shown as symbols, the Fe(II)L model as a dashed line, the Fe(II)L-2 μM model as a dotted line and the Fe(II)2L model as a solid line.
Marine Implications
In this study we have tested simple model ligands as a proxy for the far more complex natural materials that bind Fe(II) in marine, estuarine and freshwater systems. This simplification is necessary due to the importance of thorough treatment of all potential radical reactions before any quantitative assessment of the yield of HO• production can be made. Even a ligand as simple as citrate introduces significant challenges in the interpretation of the data, with further work to constrain both the nature of the complexes formed as well as their individual ROS reactivity needed before a definitive conclusion could be drawn.
Although this study has further confirmed that at pH ~ 8, inorganic Fe(II) does not react with H2O2 to give HO•, it is still not clear why the nature of the oxidant formed on oxidation of inorganic Fe(II) by H2O2 is pH dependent, nor why the organic ligands that we have examined here are able to overcome this limitation. While it has been demonstrated that this same logic seems to apply to terrigeneous NOM (Miller et al., 2013), which would appear to be a good model of estuarine Fe(II) ligands (Hopwood et al., 2015), it is not clear if this is of relevance to oceanic Fe-ligands, which are known to be important controls on Fe(II) oxidation rates in the open ocean (Roy and Wells, 2011). It is also unknown how colloidal Fe(II) associated with organic material may behave (von der Heyden et al., 2014). It is also still unclear indeed what properties a complexing agent must have to promote formation of HO•, however it would seem quite likely that complexed Fe(II) in marine, estuarine and freshwater environments would react with H2O2 to yield HO•.
Despite the minimal role of cycling of Fe between the +2 and +3 oxidation states under the conditions of this work, in natural systems this process is potentially more important. As superoxide is now recognized to be produced in a wide variety of natural contexts (Shaked and Rose, 2013), a steady flux of superoxide may be sufficient to continually reduce Fe(III) complexes to the Fe(II) form, which combined with observed H2O2 concentrations on the order of tens to hundreds of nanomolar (Kieber and Helz, 1995; Avery et al., 2005) may lead to a continual, albeit slow, production pathway for HO•.
Conclusions
Under circumneutral conditions and in the presence of organic ligands, Fe(II) is oxidized by H2O2 to yield hydroxyl radicals. Competition experiments with t-BuOH confirmed that HO• was indeed the species being measured. Kinetic modeling indicates that the difference in magnitude of HO• production between ligands can be adequately explained by consideration of all relevant reactions between ROS and Fe(II)L/Fe(III)L without the need to invoke different HO•-production efficiencies for various Fe(II)L complexes. Of particular importance are the reactions of Fe(II)L with O2, , and H2O2 and the reaction of Fe(III)L with . For the well-studied ligands EDTA and DTPA there is excellent agreement between model predictions and experimental results, with the poor HO• production predictions for Fe(II)citrate likely due to an incomplete description of the speciation and oxidation mechanism rather than the production of an alternative oxidant by Fe(II)-citrate complexes. In the absence of organic ligands, no HO• production could be measured under conditions where H2O2 was the dominant oxidant of Fe(II). As such, it is concluded that, under the conditions of this work, inorganic Fe(II) and H2O2 react to form an intermediate that is not HO•, the nature of which was unable to be examined. Overall, it is clear that under environmentally relevant conditions the Fenton reaction will only result in HO• production if Fe(II) is complexed by an organic ligand.
Author Contributions
TW initiated the study which was undertaken by CM and supervised by TW and AR. All authors contributed to manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding provided through the Australian Research Council's Discovery Scheme (Project No. DP0987188) is gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2016.00134
References
Amico, P., Daniele, P. G., Cucinotta, V., Rizzarelli, E., and Sammartano, S. (1979). Equilibrium study of iron(II) and manganese(II) complexes with citrate ion in aqueous solution: relevance to coordination of citrate to the active site of aconitase and to gastrointestinal absorption of some essential metal ions. Inorganica Chim. Acta 36, 1–7. doi: 10.1016/S0020-1693(00)89363-9
Avery, J. G. B., Cooper, W. J., Kieber, R. J., and Willey, J. D. (2005). Hydrogen peroxide at the Bermuda Atlantic Time Series Station: temporal variability of seawater hydrogen peroxide. Mar. Chem. 97, 236–244. doi: 10.1016/j.marchem.2005.03.006
Backa, S., Gierer, J., Reitberger, T., and Nilsson, T. (1993). Hydroxyl radical activity associated with the growth of white-rot fungi. Holzforschung 47, 181–187. doi: 10.1515/hfsg.1993.47.3.181
Bataineh, H., Pestovsky, O., and Bakac, A. (2012). pH-induced mechanistic changeover from hydroxyl radicals to iron(IV) in the Fenton reaction. Chem. Sci. 3, 1594–1599. doi: 10.1039/c2sc20099f
Bielski, B. H. J., Cabelli, D. E., Arudi, R. L., and Ross, A. B. (1985). Reactivity of HO2/ radicals in aqueous solution. J. Phys. Chem. Ref. Data 14, 1041–1100. doi: 10.1063/1.555739
Bolann, B. J., Henriksen, H., and Ulvik, R. J. (1992). Decay kinetics of studied by direct spectrophotometry. Interaction with catalytic and non-catalytic substances. Biochim. Biophys. Acta Gen. Subj. 1156, 27–33. doi: 10.1016/0304-4165(92)90091-8
Bray, W. C., and Gorin, M. H. (1932). Ferryl ion, a compound of tetravalent iron. J. Am. Chem. Soc. 54, 2124–2125. doi: 10.1021/ja01344a505
Buettner, G. R., Doherty, T. P., and Patterson, L. K. (1983). The kinetics of the reaction of superoxide radical with Fe(III) complexes of EDTA, DETAPAC and HEDTA. FEBS Lett. 158, 143–146. doi: 10.1016/0014-5793(83)80695-4
Bull, C., Fee, J. A., O'neill, P., and Fielden, E. M. (1982). Iron-ethylenediaminetetraacetic acid (EDTA)-catalyzed superoxide dismutation revisited: an explanation of why the dismutase activity of Fe-EDTA cannot be detected in the cytochrome c/xanthine oxidase assay system. Arch. Biochem. Biophys. 215, 551–555. doi: 10.1016/0003-9861(82)90115-1
Bull, C., McClune, G. J., and Fee, J. A. (1983). The mechanism of iron EDTA catalyzed superoxide dismutation. J. Am. Chem. Soc. 105, 5290–5300. doi: 10.1021/ja00354a019
Butler, J., and Halliwell, B. (1982). Reaction of iron-EDTA chelates with the superoxide radical. Arch. Biochem. Biophys. 218, 174–178. doi: 10.1016/0003-9861(82)90333-2
Buxton, G. V., Greenstock, C. L., Helman, W. P., and Ross, A. B. (1988). Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O−) in aqueous solution. J. Phys. Chem. Ref. Data 17, 513–886. doi: 10.1063/1.555805
Buxton, G. V., Sellers, R. M., and McCracken, D. R. (1976). Pulse radiolysis study of monovalent cadmium, cobalt, nickel and zinc in aqueous solution. Part 2.-Reactions of the monovalent ions. J. Chem. Soc. Faraday Trans. 1: Phys. Chem. Condens. Phases 72, 1464–1476. doi: 10.1039/f19767201464
Cabelli, D. E., Rush, J. D., Thomas, M. J., and Bielski, B. H. J. (1989). Kinetics of the free-radical-induced reduction of (diethylenetriaminepentaacetato)iron(III) to (diethylenetriaminepentaacetato)iron(II): a pulse radiolysis study. J. Phys. Chem. 93, 3579–3586. doi: 10.1021/j100346a042
Croft, S., Gilbert, B. C., Smith, J. R. L., and Whitwood, A. C. (1992). An E.S.R. investigation of the reactive intermediate generated in the reaction between FeII and H2O2 in aqueous solution. Direct evidence for the formation of the hydroxyl radical. Free Radic. Res. 17, 21–39. doi: 10.3109/10715769209061086
Dorfman, L. M., Taub, I. A., and Buhler, R. E. (1962). Pulse radiolysis studies. I. Transient spectra and reaction-rate constants in irradiated aqueous solutions of benzene. J. Chem. Phys. 36, 3051–3061. doi: 10.1063/1.1732425
Duesterberg, C. K., Cooper, W. J., and Waite, T. D. (2005). Fenton-mediated oxidation in the presence and absence of oxygen. Environ. Sci. Technol. 39, 5052–5058. doi: 10.1021/es048378a
Dunford, H. B. (2002). Oxidations of iron(II)/(III) by hydrogen peroxide: from aquo to enzyme. Coord. Chem. Rev. 233–234, 311–318. doi: 10.1016/S0010-8545(02)00024-3
Fujii, M., Dang, T. C., Rose, A. L., Omura, T., and Waite, T. D. (2011). Effect of light on iron uptake by the freshwater cyanobacterium Microcystis aeruginosa. Environ. Sci. Technol. 45, 1391–1398. doi: 10.1021/es103311h
Fujii, M., Rose, A. L., Waite, T. D., and Omura, T. (2008). Effect of divalent cations on the kinetics of Fe(III) complexation by organic ligands in natural waters. Geochim. Cosmochim. Acta 72, 1335–1349. doi: 10.1016/j.gca.2007.12.017
Garg, S., Rose, A. L., and Waite, T. D. (2007). Superoxide mediated reduction of organically complexed iron(III): comparison of non-dissociative and dissociative reduction pathways. Environ. Sci. Technol. 41, 3205–3212. doi: 10.1021/es0617892
Goldstein, S., and Meyerstein, D. (1999). Comments on the mechanism of the “Fenton-like” reaction. Acc. Chem. Res. 32, 547–550. doi: 10.1021/ar9800789
Goldstein, S., Meyerstein, D., and Czapski, G. (1993). The Fenton reagents. Free Radic. Biol. Med. 15, 435–445. doi: 10.1016/0891-5849(93)90043-T
González-Dávila, M., Santana-Casiano, J. M., and Millero, F. J. (2005). Oxidation of iron(II) nanomolar with H2O2 in seawater. Geochim. Cosmochim. Acta 69, 83–93. doi: 10.1016/j.gca.2004.05.043
Graf, E., Mahoney, J. R., Bryant, R. G., and Eaton, J. W. (1984). Iron-catalyzed hydroxyl radical formation. Stringent requirement for free iron coordination site. J. Biol. Chem. 259, 3620–3624.
Gutteridge, J. M. C. (1991). Hydroxyl radical formation from the auto-reduction of a ferric citrate complex. Free Radic. Biol. Med. 11, 401–406. doi: 10.1016/0891-5849(91)90157-X
Hill-Cottingham, D. G. (1955). Spectrophotometric determination of iron chelates. Analyst 80, 906–908.
Höbel, B., and von Sonntag, C. (1998). OH-Radical induced degradation of ethylenediaminetetraacetic acid (EDTA) in aqueous solution: a pulse radiolysis study. J. Chem. Soc. Perkin Trans. 2, 509–514. doi: 10.1039/a708167g
Hopwood, M. J., Statham, P. J., Skrabal, S. A., and Willey, J. D. (2015). Dissolved iron(II) ligands in river and estuarine water. Mar. Chem. 173, 173–182. doi: 10.1016/j.marchem.2014.11.004
Hug, S. J., and Leupin, O. (2003). Iron-catalyzed oxidation of arsenic(III) by oxygen and by hydrogen peroxide: pH-Dependent formation of oxidants in the Fenton reaction. Environ. Sci. Technol. 37, 2734–2742. doi: 10.1021/es026208x
Ianni, J. C. (2003). “A comparison of the Bader-Deuflhard and the Cash-Karp Runge-Kutta integrators for the GRI-MECH 3.0 model based on the chemical kinetics code Kintecus,” in Computational Fluid and Solid Mechanics, ed K. J. Bathe (Oxford: Elsevier Science Ltd.), 1368–1372.
Ilan, Y. A., and Czapski, G. (1977). The reaction of superoxide radical with iron complexes of EDTA studied by pulse radiolysis. Biochim. Biophys. Acta Gen. Subj. 498, 386–394. doi: 10.1016/0304-4165(77)90277-X
Jomova, K., Baros, S., and Valko, M. (2012). Redox active metal-induced oxidative stress in biological systems. Transit. Metal Chem. 37, 127–134. doi: 10.1007/s11243-012-9583-6
Keenan, C. R., and Sedlak, D. L. (2008). Factors affecting the yield of oxidants from the reaction of nanoparticulate zero-valent iron and oxygen. Environ. Sci. Technol. 42, 1262–1267. doi: 10.1021/es7025664
Kieber, R. J., and Helz, G. R. (1995). Temporal and seasonal variations of hydrogen peroxide levels in estuarine waters. Estuar. Coast. Shelf Sci. 40, 495–503. doi: 10.1006/ecss.1995.0034
King, D. W., and Farlow, R. (2000). Role of carbonate speciation on the oxidation of Fe(II) by H2O2. Mar. Chem. 70, 201–209. doi: 10.1016/S0304-4203(00)00026-8
Königsberger, L. C., Königsberger, E., May, P. M., and Hefter, G. T. (2000). Complexation of iron(III) and iron(II) by citrate. Implications for iron speciation in blood plasma. J. Inorg. Biochem. 78, 175–184. doi: 10.1016/S0162-0134(99)00222-6
Kosaka, H., Katsuki, Y., and Shiga, T. (1992). Spin trapping study on the kinetics of Fe2+ autoxidation: formation of spin adducts and their destruction by superoxide. Arch. Biochem. Biophys. 293, 401–408. doi: 10.1016/0003-9861(92)90412-P
Kremer, M. L. (1999). Mechanism of the Fenton reaction. Evidence for a new intermediate. Phys. Chem. Chem. Phys. 1, 3595–3605. doi: 10.1039/a903915e
Kundu, K. P., and Matsuura, N. (1975). Gamma-radiolysis of ferric ethylene diamine tetra-acetate in neutral aqueous solution. Int. J. Radiat. Phys. Chem. 7, 565–571. doi: 10.1016/0020-7055(75)90021-2
Kwan, W. P., and Voelker, B. M. (2002). Decomposition of hydrogen peroxide and organic compounds in the presence of dissolved iron and ferrihydrite. Environ. Sci. Technol. 36, 1467–1476. doi: 10.1021/es011109p
Lati, J., and Meyerstein, D. (1978). Oxidation of first-row bivalent transition-metal complexes containing ethylenediaminetetra-acetate and nitrilotriacetate ligands by free radicals: a pulse-radiolysis study. J. Chem. Soc. Dalton Trans. 1978, 1105–1118. doi: 10.1039/dt9780001105
Lee, H., Lee, H.-J., Sedlak, D. L., and Lee, C. (2013). pH-Dependent reactivity of oxidants formed by iron and copper-catalyzed decomposition of hydrogen peroxide. Chemosphere 92, 652–658. doi: 10.1016/j.chemosphere.2013.01.073
Lee, J.-C., Son, Y.-O., Pratheeshkumar, P., and Shi, X. (2012). Oxidative stress and metal carcinogenesis. Free Radic. Biol. Med. 53, 742–757. doi: 10.1016/j.freeradbiomed.2012.06.002
Luzzatto, E., Cohen, H., Stockheim, C., Wieghardt, K., and Meyerstein, D. (1995). Reactions of low valent transition metal complexes with hydrogen peroxide. Are they “Fenton-like” or not? 4. The case of Fe(II)L, L = EDTA; HEDTA and TCMA. Free Radic. Res. 23, 453–463. doi: 10.3109/10715769509065266
MacFaul, P. A., Wayner, D. D. M., and Ingold, K. U. (1998). A radical account of “Oxygenated Fenton Chemistry.” Acc. Chem. Res. 31, 159–162. doi: 10.1021/ar970057z
Masarwa, A., Rachmilovich-Calis, S., Meyerstein, N., and Meyerstein, D. (2005). Oxidation of organic substrates in aerated aqueous solutions by the Fenton reagent. Coord. Chem. Rev. 249, 1937–1943. doi: 10.1016/j.ccr.2005.01.003
Miller, C. J., Lee, S. M. V., Rose, A. L., and Waite, T. D. (2012). Impact of natural organic matter on H2O2-mediated oxidation of Fe(II) in coastal seawaters. Environ. Sci. Technol. 46, 11078–11085. doi: 10.1021/es3022792
Miller, C. J., Rose, A. L., and Waite, T. D. (2009). Impact of natural organic matter on H2O2-mediated oxidation of Fe(II) in a simulated freshwater system. Geochim. Cosmochim. Acta 73, 2758–2768. doi: 10.1016/j.gca.2009.02.027
Miller, C. J., Rose, A. L., and Waite, T. D. (2011). Phthalhydrazide chemiluminescence method for determination of hydroxyl radical production: modifications and adaptations for use in natural systems. Anal. Chem. 83, 261–268. doi: 10.1021/ac1022748
Miller, C. J., Rose, A. L., and Waite, T. D. (2013). Hydroxyl radical production by H2O2-mediated oxidation of Fe(II) complexed by Suwannee River fulvic acid under circumneutral freshwater conditions. Environ. Sci. Technol. 47, 829–835. doi: 10.1021/es303876h
Morgan, M. S., Van Trieste, P. F., Garlick, S. M., Mahon, M. J., and Smith, A. L. (1988). Ultraviolet molar absorptivities of aqueous hydrogen peroxide and hydroperoxyl ion. Anal. Chim. Acta 215, 325–329. doi: 10.1016/S0003-2670(00)85294-0
Neta, P., Grodkowski, J., and Ross, A. B. (1996). Rate constants for reactions of aliphatic carbon-centred radicals in aqueous solutions. J. Phys. Chem. Ref. Data 25, 709–1050. doi: 10.1063/1.555978
Pham, A. N., and Waite, T. D. (2008). Oxygenation of Fe(II) in natural waters revisited: kinetic modeling approaches, rate constant estimation and the importance of various reaction pathways. Geochim. Cosmochim. Acta 72, 3616–3630. doi: 10.1016/j.gca.2008.05.032
Rachmilovich-Calis, S., Masarwa, A., Meyerstein, N., Meyerstein, D., and Van Eldik, R. (2009a). New mechanistic aspects of the Fenton reaction. Chem. A Eur. J. 15, 8303–8309. doi: 10.1002/chem.200802572
Rachmilovich-Calis, S., Meyerstein, N., and Meyerstein, D. (2009b). A mechanistic study of the effects of antioxidants on the formation of malondialdehyde-like products in the reaction of hydroxyl radicals with deoxyribose. Chem. A Eur. J. 15, 7717–7723. doi: 10.1002/chem.200802272
Rahhal, S., and Richter, H. W. (1988). Reduction of hydrogen peroxide by the ferrous iron chelate of diethylenetriamine-N,N,N',N”,N”-pentaacetate. J. Am. Chem. Soc. 110, 3126–3133. doi: 10.1021/ja00218a022
Rahhal, S., and Richter, H. W. (1989). Reaction of hydroxyl radicals with the ferrous and ferric iron chelates of diethylenetriamine-N,N,N',N”,N”-pentaacetate. Free Radic. Res. 6, 369–377. doi: 10.3109/10715768909087920
Remucal Keenan, C., and Sedlak, D. L. (2011). “The role of iron coordination in the production of reactive oxidants from ferrous iron oxidation by oxygen and hydrogen peroxide,” in Aquatic Redox Chemistry, eds P. G. Tratnyek, T. J. Grundl, and S. B. Haderlein (Washington, DC: American Chemical Society), 177–197.
Rose, A. L., and Waite, T. D. (2001). Chemiluminescence of luminol in the presence of iron(II) and oxygen: oxidation mechanism and implications for its analytical use. Anal. Chem. 73, 5909–5920. doi: 10.1021/ac015547q
Rose, A. L., and Waite, T. D. (2005). Reduction of organically complexed ferric iron by superoxide in a simulated natural water. Environ. Sci. Technol. 39, 2645–2650. doi: 10.1021/es048765k
Roy, E. G., and Wells, M. L. (2011). Evidence for regulation of Fe(II) oxidation by organic complexing ligands in the Eastern Subarctic Pacific. Mar. Chem. 127, 115–122. doi: 10.1016/j.marchem.2011.08.006
Rush, J. D., and Koppenol, W. H. (1986). Oxidizing intermediates in the reaction of ferrous EDTA with hydrogen peroxide. Reactions with organic molecules and ferrocytochrome c. J. Biol. Chem. 261, 6730–6733.
Rush, J. D., and Koppenol, W. H. (1987). The reaction between ferrous polyaminocarboxylate complexes and hydrogen peroxide: an investigation of the reaction intermediates by stopped flow spectrophotometry. J. Inorg. Biochem. 29, 199–215. doi: 10.1016/0162-0134(87)80027-2
Santana-Casiano, J. M., González-Dávila, M., and Millero, F. J. (2005). Oxidation of nanomolar levels of Fe(II) with oxygen in natural waters. Environ. Sci. Technol. 39, 2073–2079. doi: 10.1021/es049748y
Santana-Casiano, J. M., González-Dávila, M., and Millero, F. J. (2006). The role of Fe(II) species on the oxidation of Fe(II) in natural waters in the presence of O2 and H2O2. Mar. Chem. 99, 70–82. doi: 10.1016/j.marchem.2005.03.010
Schiller, J., Arnhold, J., Schwinn, J., Sprinz, H., Brede, O., and Arnold, K. (1999). Differences in the reactivity of phthalic hydrazide and luminol with hydroxyl radicals. Free Radic. Res. 30, 45–57. doi: 10.1080/10715769900300061
Seibig, S., and Van Eldik, R. (1997). Kinetics of [FeII(edta)] oxidation by molecular oxygen revisited. New evidence for a multistep mechanism. Inorg. Chem. 36, 4115–4120. doi: 10.1021/ic970158t
Shaked, Y., and Rose, A. L. (2013). Seas of superoxide. Science 340, 1176–1177. doi: 10.1126/science.1240195
Summers, J. S., Baker, J. B., Meyerstein, D., Mizrahi, A., Zilbermann, I., Cohen, H., et al. (2008). Measured rates of fluoride/metal association correlate with rates of superoxide/metal reactions for FeIIIEDTA(H2O)− and related complexes. J. Am. Chem. Soc. 130, 1727–1734. doi: 10.1021/ja077193b
Sutton, H. C. (1985). Efficiency of chelated iron compounds as catalysts for the Haber-Weiss reaction. J. Free Radic. Biol. Med. 1, 195–202. doi: 10.1016/0748-5514(85)90118-7
Vile, G. F., Winterbourn, C. C., and Sutton, H. C. (1987). Radical-driven fenton reactions: studies with paraquat, adriamycin, and anthraquinone 6-sulfonate and citrate, ATP, ADP, and pyrophosphate iron chelates. Arch. Biochem. Biophys. 259, 616–626. doi: 10.1016/0003-9861(87)90528-5
von der Heyden, B. P., Hauser, E. J., Mishra, B., Martinez, G. A., Bowie, A. R., Tyliszczak, T., et al. (2014). Ubiquitous presence of Fe(II) in aquatic colloids and its association with organic carbon. Environ. Sci. Technol. Lett. 1, 387–392. doi: 10.1021/ez500164v
von Sonntag, C. (2008). Advanced oxidation processes: mechanistic aspects. Water Sci. Technol. 58, 1015–1021. doi: 10.2166/wst.2008.467
von Sonntag, C., Dowideit, P., Xingwang, F., Mertens, R., Xianming, P., Schuchmann, M. N., et al. (1997). The fate of peroxyl radicals in aqueous solution. Water Sci. Technol. 35, 9–15. doi: 10.1016/S0273-1223(97)00003-6
Walling, C. (1998). Intermediates in the reactions of Fenton type reagents. Acc. Chem. Res. 31, 155–157. doi: 10.1021/ar9700567
White, E. M., Vaughan, P. P., and Zepp, R. G. (2003). Role of the photo-Fenton reaction in the production of hydroxyl radicals and photobleaching of colored dissolved organic matter in a coastal river of the southeastern United States. Aquat. Sci. 65, 402–414. doi: 10.1007/s00027-003-0675-4
Winterbourn, C. C. (1995). Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol. Lett. 82–83, 969–974. doi: 10.1016/0378-4274(95)03532-X
Yamazaki, I., and Piette, L. H. (1990). ESR spin-trapping studies on the reaction of Fe2+ ions with H2O2-reactive species in oxygen toxicity in biology. J. Biol. Chem. 265, 13589–13594.
Yamazaki, I., and Piette, L. H. (1991). ESR spin-trapping study on the oxidizing species formed in the reaction of the ferrous ion with hydrogen peroxide. J. Am. Chem. Soc. 113, 7588–7593. doi: 10.1021/ja00020a021
Yu, X.-Y. (2004). Critical evaluation of rate constants and equilibrium constants of hydrogen peroxide photolysis in acidic aqueous solutions containing chloride ions. J. Phys. Chem. Ref. Data 33, 747–763. doi: 10.1063/1.1695414
Yurkova, I. L., Schuchmann, H.-P., and Von Sonntag, C. (1999). Production of OH radicals in the autoxidation of the Fe(II)-EDTA system. J. Chem. Soc. Perkin Trans. 2: Phys. Org. Chem. 10, 2049–2052. doi: 10.1039/a904739e
Zang, V., and Van Eldik, R. (1990a). Influence of the polyamino carboxylate chelating ligand (L) on the kinetics and mechanism of the formation of FeII(L)NO in the system FeII(L)/NO/HONO/ in aqueous solution. Inorg. Chem. 29, 4462–4468. doi: 10.1021/ic00347a026
Zang, V., and Van Eldik, R. (1990b). Kinetics and mechanism of the autoxidation of iron(II) induced through chelation by ethylenediaminetetraacetate and related ligands. Inorg. Chem. 29, 1705–1711. doi: 10.1021/ic00334a023
Keywords: reactive oxygen species, hydroxyl radical, Fenton, EDTA, DTPA, citrate
Citation: Miller CJ, Rose AL and Waite TD (2016) Importance of Iron Complexation for Fenton-Mediated Hydroxyl Radical Production at Circumneutral pH. Front. Mar. Sci. 3:134. doi: 10.3389/fmars.2016.00134
Received: 15 March 2016; Accepted: 19 July 2016;
Published: 04 August 2016.
Edited by:
Colleen Hansel, Woods Hole Oceanographic Institution, USAReviewed by:
William P. Inskeep, Montana State University, USAWilliam Sunda, National Oceanic and Atmospheric Administration, USA
Copyright © 2016 Miller, Rose and Waite. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: T. David Waite, ZC53YWl0ZUB1bnN3LmVkdS5hdQ==