 Sarah Al-Aqeel
Sarah Al-Aqeel Taewoo Ryu
Taewoo Ryu Huoming Zhang
Huoming Zhang Kondethimmanahalli H. Chandramouli
Kondethimmanahalli H. Chandramouli Timothy Ravasi
Timothy Ravasi- 1KAUST Environmental Epigenetics Program, Division of Biological and Environmental Sciences and Engineering, Division of Applied Mathematics and Computer Sciences, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
- 2APEC Climate Center, Busan, South Korea
- 3Bioscience Core Laboratories, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
The acorn barnacle, Balanus amphitrite, is the main biofouling organism in marine environments. In the present study we profiled the transcriptome and proteome of B. amphitrite at different life stages (nauplius II, nauplius VI, and cyprid) from the Red Sea, where the average water surface temperature is 34°C and the salinity reaches 41‰. We identified 65,784 expressed contigs, and a total of 1387 expressed proteins measured by quantitative proteomics. We found that osmotic stress, salt stress, hyperosmotic response and the Wnt signaling pathway were strongly up-regulated during the planktonic stage, while the MAPK pathway, lipid metabolism, and cuticle development genes were down-regulated. In the transition stage between the nauplius VI and the cyprid, genes that are involved in blood coagulation, cuticle development and eggshell formation were highly up-regulated, while the nitric oxide pathway, which stimulates the swimming and feeding response in marine invertebrates, was down-regulated. We are able to report for the first time that sound sensory system proteins are highly abundant in the nauplius VI stage, implying that these proteins are good targets for the development of new antifouling compounds. The results presented here together with the new genome-wide datasets for a non-model specie represent an important resource for the study of biofouling and development. Proteomics data are available via ProteomeXchange with identifier PXD004679.
Introduction
The barnacle Balanus amphitrite is one of the most studied biofouling organism, because of its wide distribution in the world's oceans (Wiegemann, 2005; Bacchetti De Gregoris et al., 2009), and its important role in costal ecosystems (Faimali et al., 2006). Barnacles are of interest in marine science because of their ecological and commercial impacts (Matsumura et al., 2000).
Few studies have described changes in the expression of genes and proteins important in larval development and attachment. Most of these have used 454 pyrosequencing or expressed sequence tag (EST) libraries to reveal changes between the later larval and cyprid stages (Bacchetti De Gregoris et al., 2009; Chen et al., 2011). A recent study of another barnacle species used Illumina next-generation sequencing technology to identify genes similar to those coding for cement proteins in B. amphitrite (Lin et al., 2014); these proteins are important for barnacle adhesion to a substrate. Other studies have used quantitative proteomics to identify proteins important in various developmental stages of barnacles (Zhang et al., 2010), and have identified the up-regulation of genes for several proteins responsible for cytoskeleton formation in juveniles (Chen et al., 2014).
In the present study we undertook the first integrated analysis of transcriptome and proteome data for free-swimming larval stages, generated using high-throughput techniques, with the aim of better understanding attachment mechanisms in B. amphitrite and identifying putative new targets for antifouling compounds. We focused primarily on the gene expression changes among different developmental stages. This was done by analyzing the transcriptome and proteome changes that occurred during three distinct pre-metamorphosis stages in B. amphitrite. We compared the first swimming nauplius II larvae, the last swimming nauplius VI larvae, and the pre-attachment cyprid stages of B. amphitrite (Figure 1). A total of 532,378 Illumina contigs were assembled, representing the most comprehensive expression dataset for B. Amphitrite or other biofouling organisms.
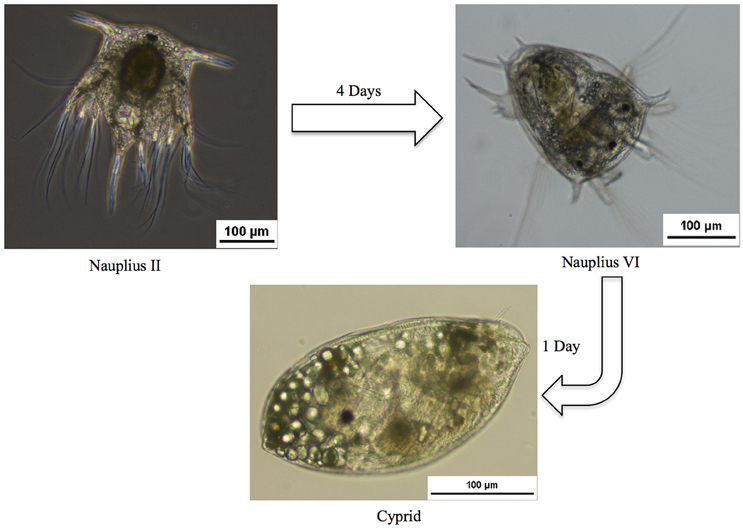
Figure 1. Developmental stages of B. Amphitrite.
To understand the molecular mechanism of B. amphitrite metamorphosis, we mapped differentially expressed genes and proteins into pathways important for each developmental stage. We identified stress response, hyperosmotic response, cell adhesion, Wnt signaling, MAPK cascade and blood coagulation cascade pathways as being the most dominant in driving B. amphitrite development prior to the attachment stage. Interestingly, we are able to report for the first time that sound sensory system proteins are strongly up-regulated in the later stage (nauplius VI), suggesting that these proteins are good candidate targets for the development of new antifouling compounds.
Materials and Methods
Water and Algal Feed Stock Preparation
Aged, purified seawater was prepared using a peristaltic pump to pass seawater through a Millipore filter (0.2 μm pore size, 142 mm diameter; Pall Corporation, Michigan, USA). The filtered water was autoclaved at 120°C for 20 min, and stored in a closed container without aeration for 7 days at room temperature (RT) (Qiu and Qian, 1997).
Chaetoceros calcitrans (obtained from the National Prawn Company, Al-Laith, Saudi Arabia) is a brown diatom having a 2.5 μm cell diameter (Laing and Britain, 1991). It is commonly used as a food source for filter feeding organisms including barnacles (Napolitano et al., 2007; Chotipuntu, 2011), and was used as food for B. amphitrite in this study. C. calcitrans was cultured in flasks containing f/2 medium (MKF220L, NCMA, New Haven, United States) at room temperature (24°C), 37 parts per thousand (ppt) salinity (PCSTestr 35 meter; Thermo Fisher Scientific, Waltham, MA, USA), continuous white fluorescent illumination (180–240.5 μmol m−2s−1; LI-250A light meter) (LI-Cor Bioscience, Lincoln, US), and good aeration. The diatom typically reached peak growth on day 5 (Huang and Madan, 1999). Serial dilution of the diatom cultures was performed before feeding to the barnacles.
Larval Collection and Culture
Adult B. amphitrite on mangroves were collected from the Red Sea at the King Abdullah University of Science and Technology, Kingdom of Saudi Arabia (22°18′15.8″ N, 039°06′9.6″ E), and maintained in fresh seawater. In the laboratory the mangroves having attached adult barnacles were washed at least 3 times with seawater and transferred to a small clean aquarium full with filtered seawater (FSW) at the same salinity as the collection site (37 ppt), and a thermostatic aquarium heater (Eheim Jager, Deizisau, Germany) was used to mimic the temperature at the barnacle collection site (33°C). The aquarium was covered with aluminum foil sheeting, which had a small gap to allow concentrated light to penetrate, to enhance larval release (Qiu and Qian, 1997; Thiyagarajan et al., 2002). Newly released nauplii (nauplius II) were attracted to the light source, and collected using a 1 ml pipette after 2 h. Approximately 5.4 × 104 larvae were collected and separated into three equal groups. The larvae in the first group were transferred to a 2-ml Eppendorf tube, excess water was removed, and the sample was snap frozen (liquid nitrogen) and stored at −80°C for later transcriptome and proteome analysis. The other two groups of nauplii were combined and cultured in a beaker (approximately 3 larvae/ml). The beaker were placed in a water bath to maintain the temperature in the beaker up to 33°C with continuous and proper filtered aeration (0.2 um) and illuminations. Cultured C. calcitrans cells (approximate concentration 246.5 × 106 cells/ml) were added to the beaker on a daily basis, and the water was changed when the bottom layer in the beaker become yellow. Nauplius VI larvae were collected on the 5th day of culture using 350 μm pore sized mesh (07-350/34; Elcofiltering, Miami, Florida, United States), and the collected sample was separated into two groups. The first group was washed with FSW to remove debris, and the larvae were transferred into a 2-ml Eppendorf tube, snap frozen, and stored into −80°C for further analysis.
The second group was returned to the aquarium to continue development to the cyprid stage, which was collected on the following 6th day using 120 μm mesh (07-120/34; Elcofiltering, Miami, Florida, United States). The harvested cyprids were prepared for analysis as described above.
RNA and Protein Extraction
Total RNA was extracted from each stage using the AllPrep RNA/protein kit (Qiagen, Hilden, Germany), following the manufacturer's instructions. In brief, the frozen larvae were transferred to a mortar and pestle, and homogenized to a fine powder in the presence of added liquid nitrogen. Buffer APL was added to the powder, incubated for 5 min, then pipetted into an AllPrep spin column and centrifuged for 1 min at ≥ 8000 × g. The resulting liquid was pipetted directly onto the center of the gel bed of a Protein Clean-up spin column, and centrifuged for 3 min at 240 × g. After centrifugation the resulting liquid contained the purified total protein in 1% SDS equilibration buffer. To isolate total RNA the AllPrep spin column was washed with Buffer RLT and centrifuged for 1 min at 8000 × g. Ethanol (70%, 1 × volume) was added to the centrifuged liquid, which was transferred to an RNeasy spin column. The spin column was washed three times with Buffer RW1 and Buffer RPE. The eluted RNA was kept in RNase free water. The reliability of the total RNA extraction was tested using a NanoDrop 1000 spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), and the integrity of RNA was checked using a 2100 Bioanalyzer (Agilent Technologies, CA, USA).
RNA Illumina Sequencing
The mRNA purification, double strand cDNA library construction, adaptor ligation, purification and library amplification were performed following the Illumina TruSeq™ protocol. Illumina sequencing (HiSeq2000) was used to produce short paired-end reads. The raw paired-end reads were submitted to the NCBI short read archive (SAR) (Bioproject no. SUB940958 and Biosample no. SAMN03650362-SAMN03650366).
Pre-processing, Assembly, and Functional Annotation of Data
Adaptors and low quality reads (Q-score ≤ 20) were trimmed using customized java scripts. Reads from different stages (in replicate) were pooled and assembled using ABySS v1.3.4 (Simpson et al., 2009) with parameters of 45, 55, 65, 75, and 85 k-mer, the scaffolding of the reads were using Trans-ABySS v1.4.4 (Robertson et al., 2010). Short reads (< 200 bp) were removed, and the pooled assembled data (total of 532,378 contigs) were submitted to the ORF prediction tool TransDecoder, integrated into the Trinity package (total of 120,000 predicted ORFs). The redundant contigs were clustered using CD-HIT-EST v4.6 if they were 95% similar; a total of 65,784 representative contigs resulted from this step. The translated sequences were subsequently submitted to BLASTp against a non-redundant (nr) database (Pruitt et al., 2005) and InterProScan (Pfam and Superfamily protein domains) for functional annotation of the representative contigs. BLASTp was used for searching for homologous proteins from other species, using a threshold of >50 bitscore. The gene ontology for homologous genes was obtained using the gene2go NCBI database.
Contigs Expression Analysis
The representative contigs were assigned to each stage by aligning the raw reads for each condition against the representative contigs using BWA aligner (Li and Durbin, 2009) against the original raw reads. The number of reads in each stage was assigned using SAMTools (Robertson et al., 2010). Because of the availability of only one replicate of nauplius II, normalization of the read count and analysis of differential gene expression was preformed using the generalized fold change algorithm “GFOLD” package V1.1.2 (Feng et al., 2012) which calculate the posterior distribution of fold change of the interest gene in each condition where the p-value is constant (0.01). If the GFOLD value is >0 then the gene of interest is up regulated whereas the GFOLD is < 0 means down regulation of the gene and finally if the GFOLD value is 0 then the expression level of this gene is not significantly changed.
Proteins Digestion and iTRAQ Labeling
The concentration of protein in each extract was measured using a 2-D Quant kit (GE Healthcare, United Kingdom). Protein (100 μg) from each stage was reduced by dithiothreitol (DTT), following the iTRAQ Reagents Multiplex Kit protocol (AB SCIEX, Foster City, CA), incubated for 30 min at 37°C, alkylated with iodoacetamide, incubated in the dark at room temperature for 1 h, then digested overnight using trypsin at 37°C. Following digestion, each sample was desalted using a SepPak C18 cartridge (Waters, Milford) and dried in a SpeedVac (Thermo Scientific, Waltham, MA, USA) for 2 h. Each sample was subsequently labeled using the iTRAQ Reagents Multiplex Kit (4 plex) (AB SCIEX, Foster City, CA), according to the manufacturer's protocol. In brief, the desalted dried protein digest was dissolved in 30 μL of dissolution buffer (0.5 M TEAB) and mixed with 70 μL of ethanol-suspended iTRAQ reagents. Thereafter, the sample was labeled with the appropriate reagent. For the first replicate, reagent 114 was labeled to the nauplius II sample, reagents 115 and 116 were labeled to the nauplius VI sample, and reagent 117 was labeled to the cyprid sample. For the second replicate, reagent 114 was labeled to the nauplius II sample, reagent 115 was labeled to the nauplius VI sample, and reagents 116 and 117 were labeled to the cyprid sample. The labeling reactions were carried out at room temperature for 60 min. The samples were then combined in a single tube, dried in a SpeedVac (Thermo Scientific), and fractionated using high performance liquid chromatography (HPLC).
Peptides Fractionation Using HPLC Coupled with LTQ-Orbitrap Mass Spectrometry
The dried iTRAQ labeled samples was dissolved in 85 μL strong cation exchange chromatography (SCX) buffer A (10 mM KH2PO4; pH 3.0; ACN/H2O 25/75, v/v). The sample was then centrifuged at 10,000 × g for 10 min at room temperature. The supernatant containing peptides was transferred to a PolySULFOETHYL A column (PolyLC, Columbia, MD, USA) and fractionated on an Accela HPLC system (Thermo Scientific). The sample was fractionated using a gradient of 100% buffer A, 5–30% buffer B (10 mM KH2PO4; pH 3.0; 500 mM KCl; ACN/H2O 25/75, v/v), 30–100% buffer B and 100% buffer B at a constant flow rate of 1 mL/min (total time 40 min). The fractions were collected at 1-min intervals, and consecutive fractions having low peak intensities were combined. A total of 15 fractions were obtained. Each fraction was dried in a SpeedVac for 3 h, dissolved in 0.1% trifluoroacetic acid (TFA), desalted using a Sep-Pak C18 cartridge, and dried in a SpeedVac for 2 h. For LTQ-Orbitrap mass spectrometry (MS) (Thermo Scientific), each fraction was resuspended in 20 μM of LC-MS Buffer A, vortexed, centrifuged at 10,000 × g for 5 min, then transferred to a LC-MS sample vial for mass spectrometry, which was conducted at the Bioscience Core Laboratory Facility at the King Abdullah University of Science and Technology, Thuwal, Saudi Arabia. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al., 2016) partner repository with the dataset identifier PXD004679 and 10.6019/PXD004679.
Proteins Identification
Raw MS data were processed as follows: higher-energy collisional dissociation (HCD) and collision-induced dissociation (CID) spectra were extracted independently using Proteome Discoverer V.1.2 software (Thermo Scientific), and subsequently processed using python script. The resulting Mascot generic format (MGF) files were submitted to Mascot v2.2 (Matrix Sciences Ltd, United Kingdom) for searching against the BLAST results in combine with its translated sequences from the transcriptomic data (120,000 sequence entries). The digestion enzyme was trypsin. The Mascot search had a fragment ion mass tolerance of 0.5 Da and the mass tolerance for the peptide was set to 10 ppm. A maximum of one missed cleavage was allowed. Variable modifications were set to 4-plex iTRAQ at tyrosine, and oxidation at methionine (M). The fixed modifications were set to carbamidomethylation at cysteine, 4-plex iTRAQ at N-terminal and lysine. The Mascot (.dat) files were processed using Scaffold v4.1.1 (Proteome Software Inc. Portland, OR, USA) software for validation, quantitation and identification of peptide and protein. The protein identifications were only accepted if the probability was >99.0% and they contained a minimum of 1 peptide with a probability greater than 95.0%. Using the Peptide and Protein Prophet algorithm with Scaffold delta-mass correction in the software; the false positive rates for peptide and protein were 1 and 0.1%, respectively. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters.
Proteins Quantitation
The iTRAQ label-based quantitation of the identified proteins was performed using the Scaffold Q+algorithm. The intensities of all labeled peptides were normalized across all runs. Individual quantitative data acquired in each run were normalized using the i-Tracker algorithm. Peptide intensity was normalized within the assigned protein. The reference channel (114 in this study) was normalized to produce a 1:1-fold change, and the iTRAQ ratios were then transformed to a log (2) scale. Kruskal–Wallis test was performed across all the three biological and three technical replicates. The differential expression was determined by a p-value of less than 0.05 and a |ratio|>0.584 (representing a 1.5 fold change). In addition, we reported differential expressed proteins that are consistently up or consistently down regulation in all technical and biological replicates. Up regulated proteins is having positive value and down regulated proteins is having negative value compared to the base proteins in each comparison.
cDNA Synthesis, Primer Design, and Real Time- qPCR Validation
Total RNA was extracted as previously described and top 1% of annotated genes were used for primer designing. cDNA was synthesized using SuperrScript III First strand Synthesis kit (Invetrogen). Quantitative polymerase chain reaction was performed after that on a 7900HT Fast Real-Time PCR system (Applied Biosystems) using RNA samples for all the stages, similar to those employed for the RNA-Seq experiments. PCR for the mentioned genes and the House keeping genes (actin) were designed using Pimer3.0 (Untergasser et al., 2012). The primers for all the genes are giving in the Table S8. SYBR Green Master Mix (Applied Biosystems) was used to study the gene expression. The Real-time PCR mixtures containing cDNA and each primer were heated at 95°C for 15 min and then subjected to 40 cycles consisting of denaturation at 95°C for 5 s, annealing at 60°C for 20 s, and extension at 72°C for 20 s, and finally for an extension of 10 min at 72°C. Each cDNA was analyzed in triplicate, after which the average threshold cycle (Ct) per sample was calculated, normilizing the expression levels of target genes based on the housekeeping gen (actin) was calculated as the following (ΔCt = Ct target gene − Ct reference gene). Since the total number of cycles in the Real-Time PCR was 40, for easier interpretation, the ΔCt was converted into 40-ΔCt (Czechowski et al., 2004) (Table S8).
Results
De novo Assembly of the B. amphitrite Transcriptome
Using Illumina paired-end sequencing we obtained 1,634,975 assembled pooled contigs from the nauplius II, nauplius VI, and cyprid stages. A total of 532,378 contigs, corresponding to 121,472 translated sequences, remained following removal of short reads (< 200 bp). Redundancy in the translated sequences was removed using CD-HIT-EST (≥90% sequence similarity), leaving a total of 65,784 representative expressed contigs. These representative contigs were used for the downstream analyses described below (Table 1).

Table 1. Summary of Illumina sequencing assembly of B. amphitrite.
Functional Annotation of the Expressed Contigs
To search for sequence similarities among other species, the predicted open reading frame (ORF) sequences were submitted to the NCBI nr database (BLASTp bit score >50; minimal match length) and InterProScan v5.4. The number of annotated hits in the NCBI nr database was 48,749, among which 31,945 contigs had at least one gene ontology (GO) annotation. In the InterProScan database, of the 120,093 hits we found 34,576 contigs having at least one protein domain (Pfam or Superfamily). A summary of the contig annotation is provided in Table 2.

Table 2. Summary of annotated contigs.
Differentially Expressed Contigs in Larval Developmental Stages
Of the 8344 contigs differentially expressed between the nauplius II and nauplius VI stages, 7295 were up-regulated and 1049 were down-regulated. Between the nauplius VI and the cyprid stages we found 200 differentially expressed contigs, 159 of which were up-regulated and 41 were down-regulated. The top 1% up-regulated and most down-regulated contigs were selected for further enrichment analysis.
Up- and Down-Regulated Genes between the Nauplius II and Nauplius VI Stages
Between the nauplius II and nauplius VI stages we found 7294 up-regulated genes having at least a factor of 2-fold change in expression. These included genes having an enriched GO term (P-value 0.01; hypergeometric test) and overall GO terms with significant enrichment to be response to cytokinin (GO:0009735); genes having developmental functions including reproductive structure development (GO:0048608), and nematode larval development (GO:0002119); and genes involved in metabolism including carbohydrate energy reserve formation (GO:0005975), hormone processes (GO:0042445), and regulation of cGMP processes (GO:0030823). Furthermore, there were GO terms related to stress responses including response to osmotic stress (GO:0006970), response to oxidative stress (GO:0006979), response to salt stress (GO:0009651), the hyperosmotic response (GO:0006972), and response to light intensity (GO:0009642). These are likely to be differentially expressed between these larval stages in order to adapt to the high salinity and temperature of the Red Sea (Thunell et al., 1988; Arz et al., 2003). Interestingly, in this phase there were also over-represented GO terms related to bone resorption (GO:0045453), and bone remodeling (GO:0046849).
In addition, innate immune response terms including activation of the immune response (GO:0002253), the immune response (GO:0006955), complement activation (GO:0006956), and the inflammatory response (GO:0006954) appeared to be important in this phase of larval development (Figure 2). On the other hand, we identified genes that were significantly up-regulated but had under-represented pathways, including the Wnt signaling pathway (GO:0016055), localization (GO:0051179), gland development (GO:0048732), nervous system development (GO:0007399), and compound eye photoreceptor cell differentiation (GO:0001751). Previous studies have shown the important role of the Wnt signaling pathway in B. amphitrite settlement (Chen et al., 2011), and we believe that these genes are important in the process of metamorphosis to the next developmental phase.
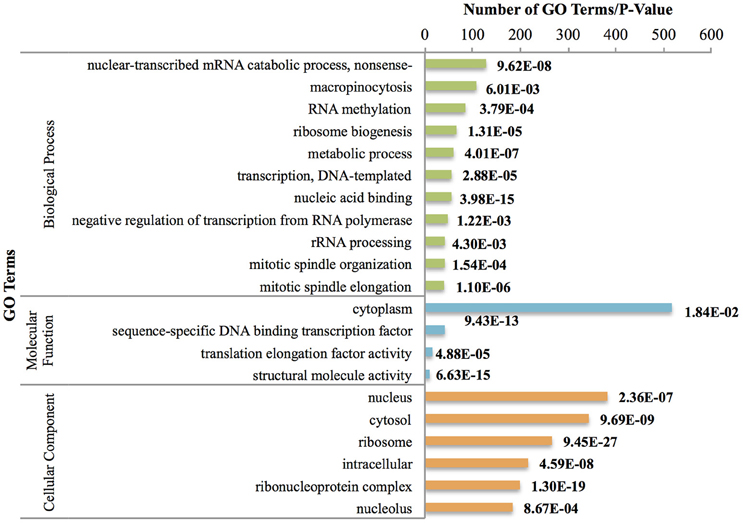
Figure 2. Most frequent up-regulated GO terms between Nauplius II and Nauplius VI. P-values are indicated on the right side. GO terms have been chosen based on hyper geometric test (P < 0.01) and most frequent terms.
The 1045 differentially expressed genes found to be down-regulated between the nauplius II and nauplius VI stages were for pathways involved in controlling organism shape, including chitin-based cuticle development (GO:0040003), replacement ossification (GO:0036075) and the molting cycle (GO:0042303). Genes for lipid metabolism, including positive regulation of lipid storage (GO:0010884), the response to lipid (GO:0033993), and cholesterol homeostasis (GO:0042632) were also down-regulated between these two larval stages. Few genes were down-regulated and enriched in relation to signaling, phosphorylation kinase activities and the regulation of translation and transcription pathways, including the MAPK cascade (GO:0000165), and Ral protein signal transduction (GO:0032484) (Figure S1). In a previous study it was reported that activation of the MAPK cascade, particularly at the aged cyprid stage, has a significant role in attachment mechanisms (He et al., 2012).
We investigated the transcription factors that are differentially expressed at different stages. We checked the transcription factor prediction database (DBD) to identify the transcription factors that were differentially expressed in our datasets. We found a total of 1411 putative transcription factors, of which 355 were up-regulated between the nauplius II and nauplius VI stages.
The main up-regulated transcription factors are predicted to be involved in cell adhesion (GO:0007155) and localization (GO:0051179), which suggests the need for an increase in the production of attachment proteins at the nauplius VI stage (Figure S2).
Up-Regulated Genes between the Nauplius VI and Cyprid Stages
In the short molting time between the nauplius VI and cyprid stages (approximately 1 day), only 156 genes were up-regulated. The most enriched up-regulated GO terms between these two stages were those involved in forming the outer layer of the cyprid (chitin-based cuticle development (GO:0040003); cuticle development (GO:0042335); and chorion-containing eggshell formation (GO:0007304)). Additionally, the main aim of the cyprid (non-feeding) stage is to find a place for settlement, which requires lipid metabolism to provide an energy source; this was evident in our data (prostanoid metabolic process (GO:0006692); fatty acid derivative metabolic process (GO:1901568)). Moreover, blood coagulation (GO:0007598), which is an extrinsic pathway, was enriched in the cyprid stage. This confirms the previous finding of a similarity between cement proteins in B. amphitrite and the mammalian blood clotting cascade (Dickinson et al., 2009; Figure S3).
We also found transcription factors that are involved in ganglion mother cell fate determination (GO:0007402), which may explain the formation of the nervous system in a later stage. The associated pathways include those involved in development of the larval body and neural fibers necessary to modify the antennules for settlement (nematode larval development (GO:0002119); ventral cord development (GO:0007419); and tracheal outgrowth, open tracheal system (GO:0007426)).
Genes Down-Regulated between the Nauplius VI and Cyprid Stages
There were 41 genes down-regulated between the nauplius VI and cyprid stages (Figure 3, Figure S4). Most notable was down-regulation of the nitric oxide (an intracellular signaling molecule) pathway at this stage [regulation of nitric-oxide synthase biosynthesis (GO:0051769)]. Previous studies have shown that nitric oxide plays important roles in marine invertebrates, including in the response to environmental stress, feeding behavior and swimming (Palumbo, 2005); these are functions that are not necessary at this sessile pre-attachment stage, and are therefore suppressed.
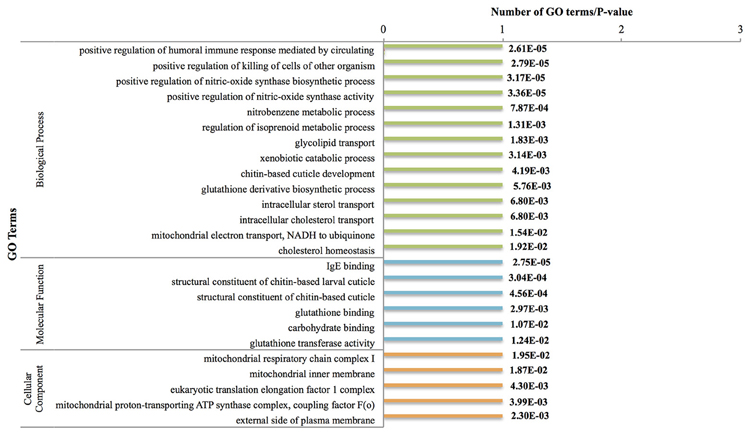
Figure 3. Most frequent down-regulated GO terms between Nauplius VI and Cyprid. P-values are indicated on the right side. GO terms have been chosen based on hyper geometric test (P < 0.01) and most frequent terms.
Up- and Down-Regulated Genes from Planktonic Stage to Sessile Stage
We assessed genes that were differentially expressed during the planktonic and sessile stages, and found 121 genes that were strongly up-regulated during the nauplius II, nauplius VI, and cyprid stages. The most up-regulated genes were those involved in the response to oxidative stress (GO:0006979), and in cuticle and outer layer development. The latter included chitin-based cuticle development (GO:0040003), cuticle development (GO:0042335), and chorion-containing eggshell formation (GO:0007304). This is consistent with the dramatic shape and physiological changes that occur between these two stages. We also found blood clotting cascade genes to be strongly up-regulated, including those believed to be involved in the formation of cement proteins (blood coagulation-extrinsic pathway (GO:0007598); blood coagulation-fibrin clot formation (GO:0072378).
Additionally, there were 46 transcription factors that are important in larval development, including nematode larval development (GO:0002119), and larval development (GO:0002164) (Table 3).
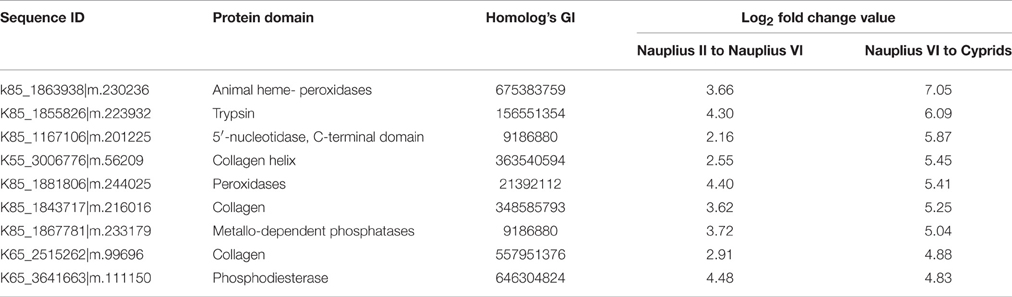
Table 3. Up-regulated genes from planktonic to sessile stage.
Seven transcription factors were down-regulated in the nauplius II, nauplius VI, and cyprid stages. These factors are responsible for nitrogen and aromatic biosynthetic processes (e.g., nitrogen compound metabolic process (GO:0006807); aromatic compound biosynthetic process (GO:0019438)). Furthermore, transcription factors related to heart development were substantially enriched, including sinus venosus morphogenesis (GO:0003236), growth involved in heart morphogenesis (GO:0003241), cardiac atrium formation (GO:0003210) and cardioblast migration (GO:0003260).
Genes Differentially Expressed between the Planktonic and Sessile Stages
All the cuticle development genes, including cuticle development (GO:0042335), and chitin-based cuticle development (GO:0040003) were up-regulated from the nauplius II to the nauplius VI stages, and were then sharply down-regulated from the nauplius VI to the cyprid stages (Figure 4). This can be explained by the metamorphosis that occurs from the larval to the juvenile stage, where the cuticle needs to be changed regularly. Furthermore, in the juvenile stage the calcium carbonate and calcium adhesive proteins are up-regulated to commence secretion of adhesive proteins, and to form the calcium shell at a later stage.
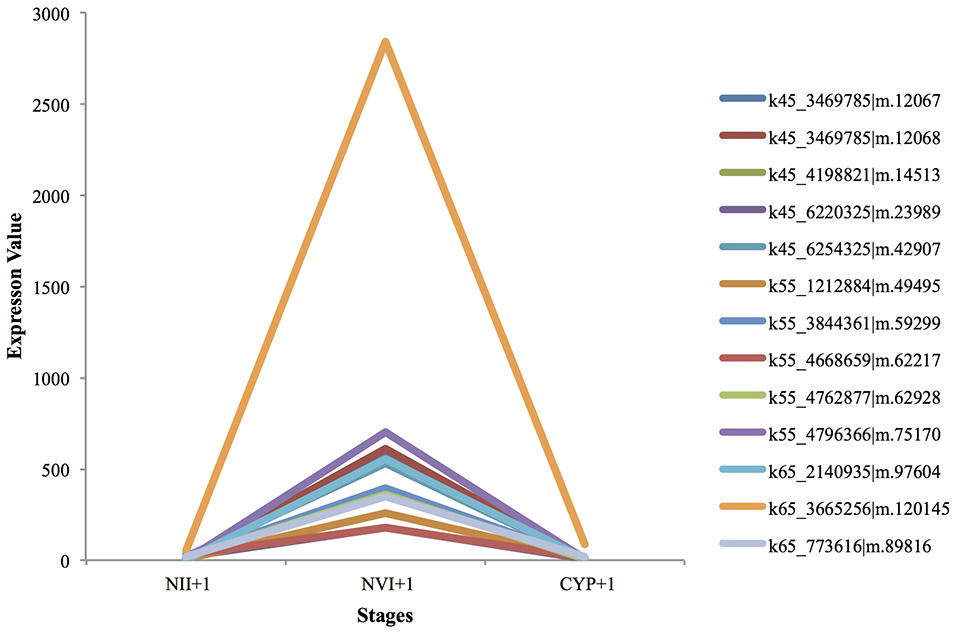
Figure 4. UP-regulated genes from Nauplius II to Nauplius VI following by down regulation in Cyprid.
We also found that several transcription factors were down-regulated in the first and last planktonic stages, before being strongly up-regulated in the cyprid stage. Amongst these were adhesion functions, including leukocyte cell-cell adhesion (GO:0007159), and mature structure development as notum development (GO:0007477) (Figure S5 and Table S1).
Quantitative Protein Expression Analysis between the Planktonic and Sessile Stages
To identify differentially expressed proteins between the three different stages of B. amphitrite and to compare the peptide sequence data with the sequences from the transcriptome analysis of the same organism, we used a linear ion trap (LTQ orbitrap) mass spectrometry technique that identified a total of 1387 proteins that were differentially expressed (at least a factor of 1.5-fold change) between the three stages.
Proteins Up-Regulated between the Nauplius II and Nauplius VI Stages
There were 43 differentially expressed proteins up-regulated in the nauplius II stage compared with the nauplius VI stage. These included vitellinogen, which provides the major egg yolk proteins that are a nutrient source during early development of vertebrates and invertebrates, and cuticular protein, which is important for cytoskeleton formation. Gene ontology annotation showed several enriched GO terms including protein autoprocessing (GO:0016540), which is important in the generation of mature proteins, and the oxidative stress response, which is important in immune defense and included the significant terms [detection of oxidative stress (GO:0070994) and response to oxidative stress (GO:0006979)]. Carbohydrate metabolism is important during the sessile phase, and is represented by the term canonical glycolysis (GO:0061621). GO categories involved in movement, secretion, enzyme production and gene expression were also up-regulated. Most importantly, a series of hormone signaling pathways, including the juvenile hormone mediated signaling pathway (GO:0035626), were significantly up-regulated. Cytokinesis formation, which is important in the assembly of actin, myosin and associated proteins, was also found to be expressed (Tables S1–S3).
Up- and Down-Regulation of Proteins between the Nauplius VI and Cyprid Stages
There were nine proteins up-regulated between the nauplius VI and cyprid stages. Most of these are hypothetical proteins, and included chitin-based cuticle development (GO:0040003), adult somatic muscle development (GO:0007527), and hematopoietic stem cell proliferation (GO:0071425) (Table S4).
The five proteins down-regulated between the nauplius VI and cyprid stages included myosin-9 and alpha-actinin proteins. Both these proteins play important roles in cytoskeletal formation and reorganization. Muscle contraction (GO:0006936), positive regulation of receptor activity (GO:2000273), focal adhesion assembly (GO:0048041), actin filament bundle assembly (GO:0051017), and positive regulation of potassium ion transport (GO:0043268) were also enriched and down-regulated in the cyprid stage (Table S5).
Protein Expression Change from the Planktonic to the Sessile Stage
The 37 proteins up-regulated during transition from nauplius II through nauplius VI to the cyprid stage included vitellogenin, cuticular protein, and enzymes involved in energy generation and protein aggregation, including AAA family ATPases, glyceraldehyde-3-phosphate dehydrogenase and NADP-specific isocitrate dehydrogenase. These proteins are also enriched in autoprocessing (GO:0016540), detection of oxidative stress (GO:0070994), the juvenile hormone mediated signaling pathway (GO:0035626), and response to oxidative stress (GO:0006979) (Table S6).
A total of 31 proteins were down-regulated during transition from the planktonic to the sessile stage. Chitin-based cuticle development (GO:0040003) was the most significant annotated GO term. Others included vinculin-like isoform 2 and myosin heavy chain kinase, which are cytoskeleton proteins involved in focal adhesion (Table S7).
Integration of the Transcriptome and Proteome Datasets
To identify robust pathways corroborated by both datasets, we integrated the differentially expressed transcripts and proteins for each pairwise stage comparison.
We found 81 differentially expressed contigs that encoded to proteins during transition from the nauplius II to the nauplius VI stage. Among the translated proteins, 14 were up-regulated and 10 were down-regulated (Table 4). The up-regulated genes and proteins included chitin-based cuticle attachment to epithelium (GO:0050741), lateral inhibition (GO:0059197), lipid transport (GO:0065842), signal transduction (GO:0065841), and chemotaxis (GO:0068511). The cuticle proteins play important roles in forming the exoskeleton layer in invertebrates, in addition to prevention of water loss. The lateral inhibition pathway was also up-regulated, and acts in increasing contrast and sharpness in the visual response; Matsumura and Qian (2014) suggested that compound eyes have an important role in larval settlement at later stages of larval life. Our results also show that lipid transport and vitellogenin were up-regulated; these play important roles as major energy sources. Lipid proteins are expressed in early larval stages, and increase in expression in the nauplius VI stage. Previous studies (Chen et al., 2011) have shown the important role of vitellogenin during the nauplius VI stage, but this is the first study to show the early expression of these proteins in the nauplius II stage. Signal transduction and chemotaxis were also up-regulated during this stage, highlighting the importance for organism movement and induction of the attachment response during this stage. These results confirm previous indications of the importance of larval vision and energy sources in the attachment process. Interestingly, many expressed genes were down-regulated and their protein products were up-regulated. Most of these gene products are hypothetical proteins involved in skeletal system development (GO:0001501), cell migration (GO:0016477), the G-protein coupled acetylcholine receptor signaling pathway (GO:0007213), and sensory perception of sound (GO:0007605). Decreased larval movement just prior to attachment could be explained by up-regulation of the skeletal development and cell migration pathways at the outer surface of the non-feeding stage. The G-protein coupled acetylcholine receptor plays an important role in larval attachment and metamorphosis, as explained by Okazaki and Shizuri (2000). Interestingly, we found sensory perception of sound was also up-regulated, perhaps explaining why Branscomb and Rittschof (1984) were able to use low frequency waves to inhibit the settlement of B. amphitrite cyprids.
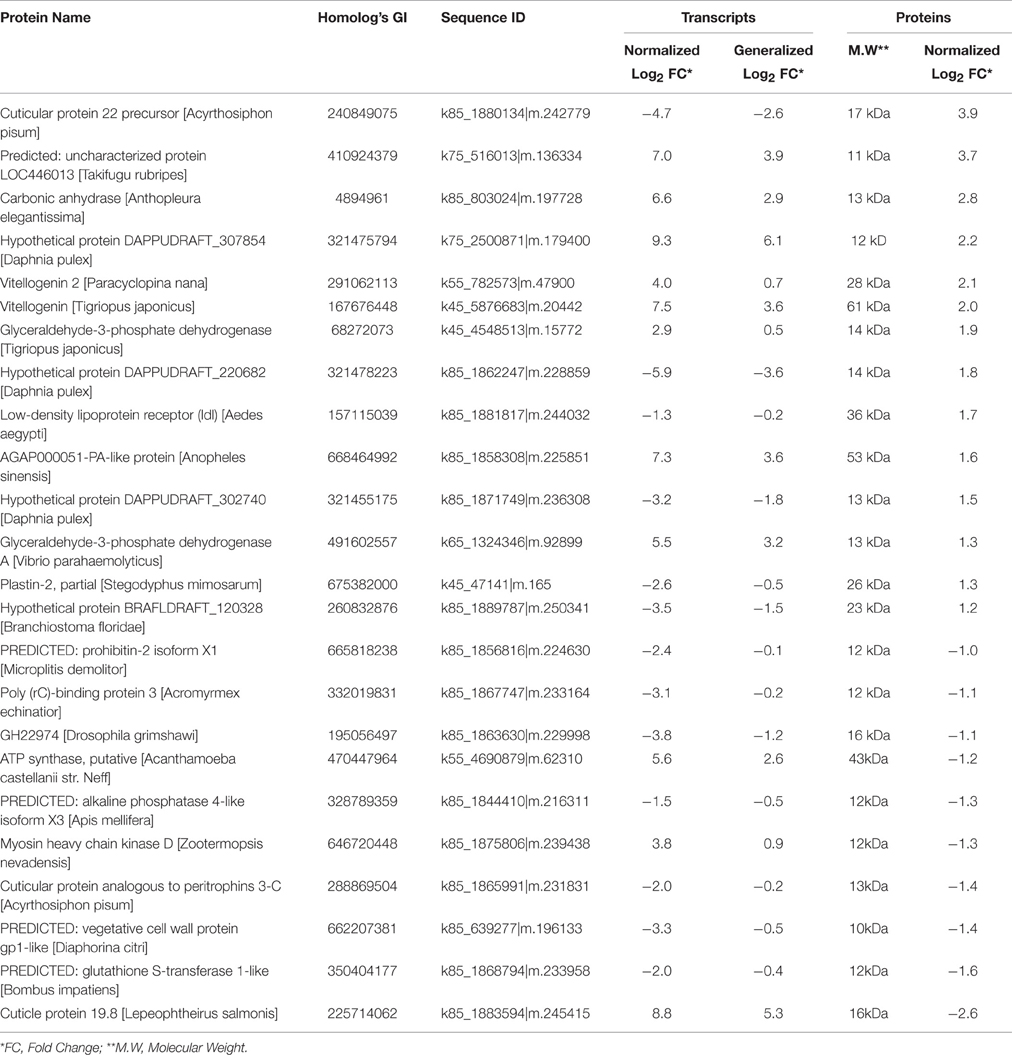
Table 4. Top up/down differentially integrative transcriptomic and proteomic data between Nauplius II and Nauplius VI.
We also found down-regulation of genes and their encoded proteins between the nauplius II and nauplius VI stages, including those involved in adult locomotor behavior (GO:0008344); at the cyprid stage locomotion activity decreases as attachment processes begin.
Three up-regulated contigs encoding proteins differentially expressed between the nauplius VI and cyprid stages (Table 5) are involved in cell proliferation (GO:0008283) and chitin-based cuticle development (GO:0040003) in the cyprid stage, where one of the most important processes is hardening and expansion of the cell population to enable attachment to the substrate.

Table 5. Top up differentially integrative transcriptomic and proteomic data between Nauplius VI and Cyprid.
Validation of the Expression Profiles
To further confirm the expression of genes in different developmental stages, eight selected highly up/down regulated genes together with housekeeping genes were selected to study their expression by mean of quantitative real-time PCR (RT-qPCR). Overall we were able to confirm the expression measured by the genome-wide transcriptome approach (Figure 5). We confirmed the up-regulatation of genes in naplius VI compared to nauplius II, between these Vitellogenin (Vit) “Figures 5A,B” and a gene with unknown function (ampb001) “Figures 5C,D.” Five genes, of unknown functions (ampb002 “Figures 5E,F,” ampb003 “Figures 5 G,H,” ampb004 “Figures 5I,J,” ampb005 “Figures 5K,L”) were down-regulated in the cyprid stage compared to napuis VI. Additionally we confirmed by RT-qPCR that ampb006 “Figures 5M,N,” and ampb007 “Figures 5O,P” genes are changing their expression pattern between planktonic and sessile stages. Overall this analysis had shown a good consistency between high-throughput transcriptomes and RT-qPCR results.
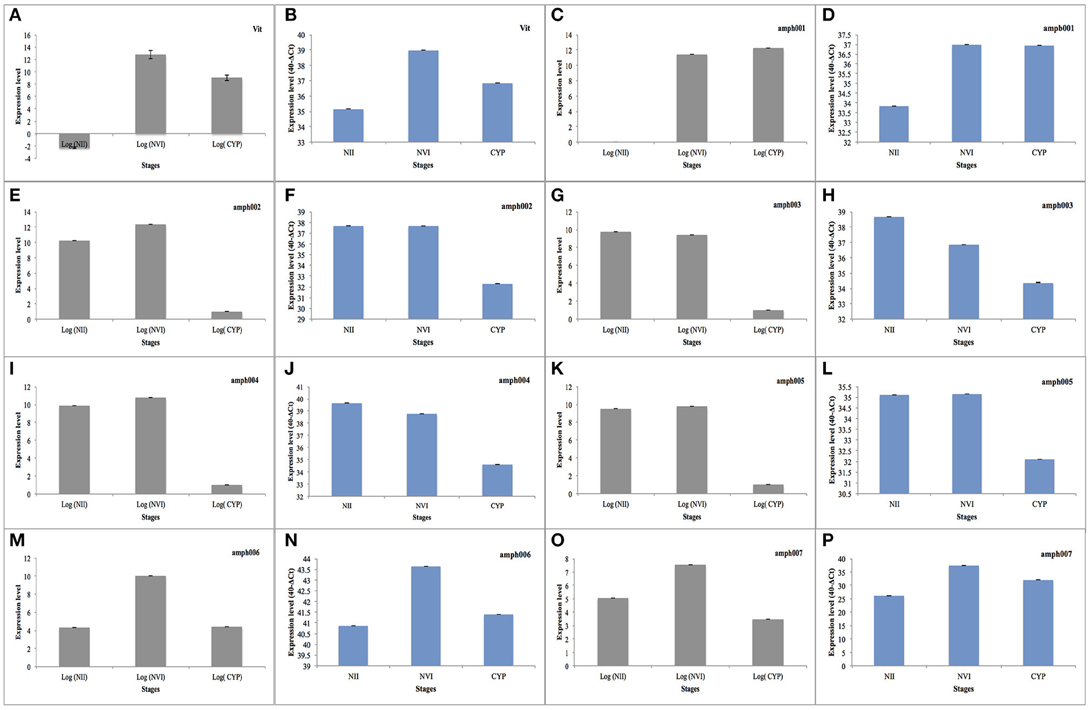
Figure 5. Bar plot showing the expression level of mRNA of highly expressed genes (in Gray) and the expression level of the same genes determined by RT-qPCR (in Blue). (NII, nauplius II; NVI, napluis VI; CYP, Cyprid). (A,B) Up regulation of Vitellogenin between naplius VI compared to naplius II. (C,D) Up regulation of unknown between naplius VI compared to naplius II. (E–L) Down regulation during Cyprid stage compared to napulis VI in five unknown genes ampb002, ampb003, ampb004, and ampb005, respectively. (M–P) Different expression pattern between planktonic and sessile stage in ampb006 and ampb007.
Discussion
In this study we identified a set of annotated gene products that are candidate factors in the settlement mechanism of B. amphitrite. To gain insight into the molecular mechanisms underlying larval attachment, we assembled a transcriptome dataset encompassing three stages of B. amphitrite development. We assembled 532,378 contigs, which were condensed into 65,784 representative contigs that were used in the downstream analysis.
We found osmotic stress, salt stress, hyperosmotic response and the Wnt signaling pathway to be strongly up-regulated during transition from the nauplius II to the nauplius VI stages. This was interpreted as a response to fluctuations in salinity and temperature, and adjustment between the early release larval stage and the later larval stage. Wnt signaling has an important role in early larval development. The MAPK cascade, lipid metabolism and cuticle development were down-regulated during this transition. The main target of the MAPK cascade is extracellular signaling, which is needed in the cyprid stages but not at the nauplius VI stage, and lipid metabolism was down-regulated because the main source of energy during the nauplius II is carbohydrate. In the transition from the nauplius VI to the cyprid stages we found blood coagulation proteins, cuticle development and eggshell formation to be extremely up-regulated (related to outer layer formation), while nitric oxide (important in the swimming and feeding responses in marine invertebrates) was down-regulated in the cyprid stage, as it is not needed during further stages. While most of the genes were equally distributed among stages, we found that some genes were up-regulated from the nauplius II to the nauplius VI stages and down-regulated from the nauplius VI to the cyprid stages. These included cuticle development and nitrogen biosynthetic process, both of which are needed during the first stage and consumed in the cyprid stage. We also identified that several adhesion proteins were down-regulated in the nauplius VI stage but up-regulated in the cyprid stage.
From the results of this study we conclude that the cyprid stage plays a critical role in the transition from the planktonic to the sessile phase of B. amphitrite, as there are many hypothetical genes that were translated to proteins having important roles in larvae development and attachment mechanism during later stages. These genes and proteins need to be further studied to fully clarify the attachment mechanism. By comparing gene and protein expression we were also able to confirm the importance of compound eyes at later larval stages and the cyprid stage, and their role in the settlement process during later life stages. Remarkably, our results show the importance of chitin-based cuticle development genes and proteins, which are involved in all life stages of B. amphitrite. Our study is also the first to show that the sound sensory system pathway is expressed during the last swimming larval stage before metamorphosis. This pathway is important, as we believe that sound is used for communication between the cyprid stage and later stage (juvenile and adult) during settlement to barnacle colonies on the substratum. Our study has provided a comprehensive insight into important pathways for the settlement of larvae of a major biofouling organism, and identified new putative targets for the development of antifouling compounds.
Author Contributions
Conceived the overall study: TRavasi, SA. Performed the culture and RNA and protein extraction: SA. iTRAQ labeling and mass spectrometry: SA, HZ. Transcriptome assembly and design of transcriptome assembly: TRyu. Functional annotation, homolog identification, differential gene expression analysis: SA. Drafting the manuscript: SA. Discussion and comment on writing: TRyu, TRavasi. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the King Abdullah University of Science and Technology (KAUST). We thank the National Prawn Company for providing algal samples; Sridharan Govindachary (SABIC-CRI KAUST) for help with culturing and comments; the Coastal and Marine Resources Core Laboratory for sample collection; Harris Mavromatis (KAUST), Tim Wong and Dineshram (HKUST) for laboratory assistance and comments; and Yanal Ghosheh and Allan Kamau for analysis comments and assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2016.00171
Table S1. Transcription factors in Down-regulated genes from planktonic to sessile stage.
Table S2. Top differentially up-regulated proteins between Nauplius II and Nauplius VI.
Table S3. Top differentially down regulated proteins between Nauplius II and Nauplius VI.
Table S4. Top differentially up regulated proteins between Nauplius VI and Cyprid.
Table S5. Top differentially down regulated proteins between Nauplius VI and Cyprid.
Table S6. Top differentially up regulated proteins between Nauplius II, Nauplius VI and Cyprid.
Table S7. Top differentially down regulated proteins between Nauplius II, Nauplius VI and Cyprid.
Table S8. Primers used to design the validation interested genes.
Figure S1. Most frequent down-regulated GO terms between Nauplius II and Nauplius VI. P-values are indicated on the right side. GO terms have been chosen based on hyper geometric test (P < 0.01) and most frequent terms.
Figure S2. Top frequent Pfam protein domains in up and down regulated genes between Nauplius II and Nauplius VI.
Figure S3. Most frequent up-regulated GO terms between Nauplius VI and Cyprids. P-values are indicated on the right side. GO terms have been chosen based on hyper geometric test (P < 0.01) and most frequent terms.
Figure S4. Top frequent Pfam protein domain in up and down regulated genes between Nauplius VI and Cyprid.
Figure S5. Down-regulated transcription factors from Nauplius II to Nauplius VI following by up regulation in Cyprid.
References
Arz, H. W., Lamy, F., Pätzold, J., Müller, P. J., and Prins, M. (2003). Mediterranean moisture source for an early-Holocene humid period in the northern Red Sea. Science 300, 118–121. doi: 10.1126/science.1080325
Bacchetti De Gregoris, T., Borra, M., Biffali, E., Bekel, T., Burgess, J. G., Kirby, R., et al. (2009). Construction of an adult barnacle (Balanus amphitrite) cDNA library and selection of reference genes for quantitative RT-PCR studies. BMC Mol. Biol. 10:62. doi: 10.1186/1471-2199-10-62
Branscomb, E. S., and Rittschof, D. (1984). An investigation of low frequency sound waves as a means of inhibiting barnacle settlement. J. Exp. Mar. Biol. Ecol. 79, 149–154. doi: 10.1016/0022-0981(84)90215-6
Chen, Z. F., Matsumura, K., Wang, H., Arellano, S. M., Yan, X., Alam, I., et al. (2011). Toward an understanding of the molecular mechanisms of barnacle larval settlement: a comparative transcriptomic approach. PLoS ONE 6:e22913. doi: 10.1371/journal.pone.0022913
Chen, Z.-F., Zhang, H., Wang, H., Matsumura, K., Wong, Y. H., Ravasi, T., et al. (2014). Quantitative proteomics study of larval settlement in the barnacle Balanus amphitrite. PLoS ONE 9:e88744. doi: 10.1371/journal.pone.0088744
Chotipuntu, P. (2011). Marine diatom (Chaetoceros calcitrans) as a monospecies diet for conditioning oyster (Crassostrea belcheri Sowerby) broodstock. Walailak J. Sci. Technol. 2, 201–207. doi: 10.2004/wjst.v2i2.164
Czechowski, T., Bari, R. P., Stitt, M., Scheible, W. R., and Udvardi, M. K. (2004). Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root-and shoot-specific genes. Plant J. 38, 366–379. doi: 10.1111/j.1365-313X.2004.02051.x
Dickinson, G. H., Vega, I. E., Wahl, K. J., Orihuela, B., Beyley, V., Rodriguez, E. N., et al. (2009). Barnacle cement: a polymerization model based on evolutionary concepts. J. Exp. Biol. 212, 3499–3510. doi: 10.1242/jeb.029884
Faimali, M., Garaventa, F., Piazza, V., Greco, G., Corrà, C., Magillo, F., et al. (2006). Swimming speed alteration of larvae of Balanus Amphitrite as a behavioural end-point for laboratory toxicological bioassays. Mar. Biol. 149, 87–96. doi: 10.1007/s00227-005-0209-9
Feng, J., Meyer, C. A., Wang, Q., Liu, J. S., Liu, X. S., and Zhang, Y. (2012). GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 28, 2782–2788. doi: 10.1093/bioinformatics/bts515
He, L.-S., Xu, Y., Matsumura, K., Zhang, Y., Zhang, G., Qi, S.-H., et al. (2012). Evidence for the involvement of p38 MAPK activation in barnacle larval settlement. PLoS ONE 7:e47195. doi: 10.1371/journal.pone.0047195
Huang, X., and Madan, A. (1999). CAP3: a DNA sequence assembly program. Genome Res. 9, 868–877. doi: 10.1101/gr.9.9.868
Laing, I., and Britain, G. (1991). Cultivation of Marine Unicellular Algae. Conwy: Ministry of Agriculture, Fisheries and Food.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Lin, H.-C., Wong, Y. H., Tsang, L. M., Chu, K. H., Qian, P.-Y., and Chan, B. K. (2014). First study on gene expression of cement proteins and potential adhesion-related genes of a membranous-based barnacle as revealed from Next-Generation Sequencing technology. Biofouling 30, 169–181. doi: 10.1080/08927014.2013.853051
Matsumura, K., Hills, J. M., Thomason, P. O., Thomason, J. C., and Clare, A. S. (2000). Discrimination at settlement in barnacles: laboratory and field experiments on settlement behaviour in response to settlement, Äêinducing protein complexes. Biofouling 16, 181–190. doi: 10.1080/08927010009378443
Matsumura, K., and Qian, P.-Y. (2014). Larval vision contributes to gregarious settlement in barnacles: adult red fluorescence as a possible visual signal. J. Exp. Biol. 217, 743–750. doi: 10.1242/jeb.096990
Napolitano, G. E., Ackman, R. G., and Ratnayake, W. (2007). Fatty acid composition of three cultured algal species (isochvysis galbana, chaetoceros gracilis and chaetoceros calcitrans) used as food for bivalve larvae. J. World Aquac. Soc. 21, 122–130. doi: 10.1111/j.1749-7345.1990.tb00532.x
Okazaki, Y., and Shizuri, Y. (2000). Effect of inducers and inhibitors on the expression of bcs genes involved in cypris larval attachment and metamorphosis of the barnacles Balanus amphitrite. Int. J. Dev. Biol. 44, 451–456. Available online at: http://www.ijdb.ehu.es/web/paper.php?doi=11032178
Palumbo, A. (2005). Nitric oxide in marine invertebrates: a comparative perspective. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 142, 241–248. doi: 10.1016/j.cbpb.2005.05.043
Pruitt, K. D., Tatusova, T., and Maglott, D. R. (2005). NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504. doi: 10.1093/nar/gki025
Qiu, J. W., and Qian, P. Y. (1997). Effects of food availability, larval source and culture method on larval development of Balanus amphitrite Darwin: implications for experimental design. J. Exp. Mar. Biol. Ecol. 217, 47–61. doi: 10.1016/S0022-0981(97)00037-3
Robertson, G., Schein, J., Chiu, R., Corbett, R., Field, M., Jackman, S. D., et al. (2010). De novo assembly and analysis of RNA-seq data. Nat. Methods 7, 909–912. doi: 10.1038/nmeth.1517
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Thiyagarajan, V., Harder, T., and Qian, P. Y. (2002). Relationship between cyprid energy reserves and metamorphosis in the barnacle Balanus amphitrite Darwin (cirripedia; thoracica). J. Exp. Mar. Biol. Ecol. 280, 79–93. doi: 10.1016/S0022-0981(02)00415-X
Thunell, R. C., Locke, S. M., and Williams, D. F. (1988). Glacio-eustatic sea-level control on Red Sea salinity. Nature 334, 601–604. doi: 10.1038/334601a0
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115–e115. doi: 10.1093/nar/gks596
Vizcaíno, J. A., Csordas, A., Del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., et al. (2016). 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 44, D447–D456. doi: 10.1093/nar/gkv1145
Wiegemann, M. (2005). Adhesion in blue mussels (Mytilus edulis) and barnacles (genus Balanus): mechanisms and technical applications. Aquat. Sci. 67, 166–176. doi: 10.1007/s00027-005-0758-5
Keywords: Amphibalanus amphitrite, biofouling, development, transcriptomics, proteomics, genomics
Citation: Al-Aqeel S, Ryu T, Zhang H, Chandramouli KH and Ravasi T (2016) Transcriptome and Proteome Studies Reveal Candidate Attachment Genes during the Development of the Barnacle Amphibalanus Amphitrite. Front. Mar. Sci. 3:171. doi: 10.3389/fmars.2016.00171
Received: 16 May 2016; Accepted: 30 August 2016;
Published: 21 September 2016.
Edited by:
Fengping Wang, Shanghai Jiao Tong University, ChinaCopyright © 2016 Al-Aqeel, Ryu, Zhang, Chandramouli and Ravasi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy Ravasi, dGltb3RoeS5yYXZhc2lAa2F1c3QuZWR1LnNh