 Rebecca A. Fieth
Rebecca A. Fieth Marie-Emilie A. Gauthier
Marie-Emilie A. Gauthier Joanne Bayes1
Joanne Bayes1 Kathryn M. Green
Kathryn M. Green Sandie M. Degnan
Sandie M. Degnan- 1Marine Genomics Lab, School of Biological Sciences, The University of Queensland, Brisbane, QLD, Australia
- 2Centre for Microscopy and Microanalysis, The University of Queensland, Brisbane, QLD, Australia
Vertical transmission of bacterial symbionts, which is known in many species of sponge (Porifera), is expected to promote strong fidelity between the partners. Combining 16S rRNA gene amplicon sequencing and electron microscopy, we have assayed the relative abundance of vertically-inherited bacterial symbionts in several stages of the life cycle of Amphimedon queenslandica, a tropical coral reef sponge. We reveal that adult A. queenslandica house a low diversity microbiome dominated by just three proteobacterial OTUs, with a single gammaprotebacterium clearly dominant through much of the life cycle. This ontogenetic perspective has revealed that, although vertical transmission occurs very early in development, the inherited symbionts do not maintain proportional dominance of the bacterial community at every developmental stage. A reproductive bottleneck in the A. queenslandica life cycle is larval settlement, when a free-swimming pelagic larva settles out of the water column onto the benthos and completes metamorphoses into the sessile body plan within just 3–4 days. During this dramatic life cycle transition, an influx of environmentally-derived bacteria leads to a major reorganization of the microbiome, potentially challenging the fidelity and persistence of the vertically-inherited symbiotic relationships. However, dominance of the primary, vertically-inherited symbionts is restored in adult sponges. The mechanisms underlying ontogenetic changes in the bacterial community are unknown, including how the dominance of the primary symbionts is restored in the adult sponge—does the host or symbiont regulate this process? Using high-resolution transcriptional profiling in multiple stages of the A. queenslandica life cycle combined with this natural perturbation of the microbiome immediately following larval settlement, we are beginning to identify candidate host genes associated with animal-bacterial crosstalk. Among the sponge host genes upregulated during the times of active microbiome assembly, there is an enrichment of genes potentially involved in innate immunity, including scavenger receptors, and of genes containing eukaryote-like domains, which have elsewhere been implicated in host-symbiont interactions. Intriguingly, we also see an enrichment of sponge genes arising from ancient horizontal transfer events from bacteria, which raises the possibility that host-bacterial associations in the evolutionary past may help to regulate host-bacterial associations in the ecological present.
Introduction
Animal-bacterial symbioses are both ubiquitous and simultaneously exclusive because, although most organisms are involved in such relationships, most species do not participate in most symbioses (Douglas, 2014). This strongly suggests that host and symbionts carefully choose their partners, and thus that mechanisms exist for specific selection and persistence of suitable partners (McFall-Ngai et al., 2013; Douglas, 2014). One way in which selection and persistence can be regulated is via direct vertical inheritance of the symbionts by the animal host, usually from mother to offspring. This aligns the interests of both partners because the maintenance and spread of the symbiont depends directly on the reproduction of the host (Frank, 1996; Bright and Bulgheresi, 2010). This in turn should promote fidelity and specificity between the partners (Russell and Moran, 2005), so it not surprising that vertical transmission is a widespread feature of animal-bacterial symbioses (Bright and Bulgheresi, 2010). Vertical transmission also provides a mechanism for hosts to sanction against costly “cheater” symbionts by restricting their rate of transmission, but this depends on a hosts' capacity to maintain a suitable abundance and distribution of chosen partners throughout its life cycle (Bright and Bulgheresi, 2010; Douglas, 2014). Determining modes of symbiont transmission are thus important for understanding the ecology and fidelity of the symbiosis (Bulgheresi, 2011), and it becomes particularly important to identify whether critical points for transmission exist in the life cycle. These critical points can inform predictions of which environmental factors are most likely to affect transmission dynamics and microbiome composition (Gundel et al., 2011; Ebert, 2013), and can provide natural perturbations for identifying genes that may mediate host-symbiont interactions.
Marine sponges are well-known for harboring phylogenetically-diverse bacterial communities as adults, often with a high degree of host-symbiont fidelity (Webster et al., 2001, 2010; Hentschel et al., 2002, 2003, 2012; Taylor et al., 2004, 2007, 2013; Thomas et al., 2016). They provide compelling examples of how an animal's ecology comprises not only its external environment, but also its internally-housed microbes. In particular, the bacterial symbionts that coexist with sponges provide their hosts with an extended multitude of metabolic functions that include nitrogen and phosphorous cycling, sulfur oxidation, carbon fixation, conversion of dissolved organic matter, and the production of secondary metabolites that serve the sponge in defense against predation (Taylor et al., 2007; Fan et al., 2012; Hentschel et al., 2012; de Goeij et al., 2013; Wilson et al., 2014; Zhang et al., 2015). The nature of bacterial community-specificity has been explored for multiple sponge species across a variety of phylogenetic, spatial, and temporal scales, revealing that adult sponge microbiomes generally are dominated by core symbionts that comprise a mix of both specialists and generalists (Webster et al., 2001, 2010; Schmitt et al., 2011; Erwin et al., 2012, 2015; Simister et al., 2012; Blanquer et al., 2013; Taylor et al., 2013; Easson and Thacker, 2014; Burgsdorf et al., 2015a; Thomas et al., 2016).
These observations raise question about the relative importance of animal hosts and the core symbionts themselves in regulating the assembly and maintenance of the total microbiome through the host life cycle, and in homeostasis of host functionality (Thomas et al., 2016). In an ontogenetic context, numerous studies have demonstrated vertical inheritance of putatively core symbionts during development of embryos and larvae, based on evidence from electron microscopy (see for example Ereskovsky et al., 2005; Oren et al., 2005; Maldonado, 2007; Schmitt et al., 2008) or sequencing of prokaryotic 16S rRNA gene amplicons (see for example Oren et al., 2005; Enticknap et al., 2006; Schmitt et al., 2007, 2008; Sharp et al., 2007; Steger et al., 2008; Lee et al., 2009; Webster et al., 2010; Gloeckner et al., 2012). Horizontal transmission, characterized by de novo symbiont acquisition from the environment with each new generation, has also been identified in several sponges (de Caralt et al., 2007; Maldonado, 2007; Schmitt et al., 2007; Gloeckner et al., 2013) and could include the introduction into the microbiome of opportunistic, non-core bacterial species. In the absence of complete ontogenetic perspectives, however, the nature and extent of microbiome compositional changes through the host life cycle are unknown, including whether fidelity of core, vertically-inherited symbionts is challenged at any stage.
The demosponge Amphimedon queenslandica (Hooper and Van Soest, 2006) lives on the intertidal reef flat of tropical Pacific coral reefs, and has a well-characterized life cycle that comprises typical pelagic larva and benthic adult stages (reviewed in Degnan et al., 2008, 2015). It is particularly tractable for investigating host-symbiont interactions through the entire life cycle, as it houses asynchronously developing embryos and larvae through much of the year, in easily accessible brood chambers (Leys and Degnan, 2002; Degnan et al., 2005, 2015; Leys et al., 2008). It also is genome-enabled (Srivastava et al., 2010), facilitating investigations of molecular crosstalk between host and symbionts (Hentschel et al., 2012). Here we combine 16S rRNA gene amplicon sequencing and electron microscopy to identify the community composition and modes of inheritance of prokaryote symbionts in A. queenslandica, and to provide a novel perspective on post-transmission stability of symbionts in this phylum. We then explore the changes in host transcriptional activity at life cycle transitions that coincide with microbiome composition shifts that we observe, as a starting point for identifying candidate genes that may be involved in regulating interactions between the sponge host and its bacterial partners.
Materials and Methods
Characterization of the Prokaryote Community Using 16S rRNA Gene Amplicon Sequencing
Collection of Biological Material
Three healthy adult specimens (no bleaching or tissue necrosis) of A. queenslandica were removed intact from the reef flat at Shark Bay, Heron Island, southern Great Barrier Reef, Australia (23°27′ S, 151°55′ E) in each of January, May and October 2014, and immediately transported in cold seawater to the Heron Island Research Station (HIRS). From each adult, 0.5 g biopsies were frozen at −80°C for subsequent genomic DNA (gDNA) extraction. Because microbial symbiont communities have been shown to vary spatially between cortex and endosome regions in at least some sponges (Yang and Li, 2012), we sampled from four different sites within each sponge, and pooled all samples to comprise our adult sample from each season.
Fifteen additional adults collected in January 2014 were used for dissection of brood chambers as a source of embryos to document ontogenetic changes in microbial community composition and to identify microbes acquired through vertical transmission. Twenty embryos at each of four developmental stages (white, beige, spot and ring stages, which cover early to late embryogenesis) were extracted from brood chambers as previously described (Leys and Degnan, 2002; Leys et al., 2008) and immediately frozen in a minimal volume of 0.22 μm-filtered seawater (FSW) at −80°C for subsequent gDNA extraction.
For larval collection, another 15 adults collected in January 2014 were housed at HIRS in flow-through seawater aquaria drawing water from the reef flat. Plankton mesh traps attached to aquaria outflow were used to passively collect all larvae that emerged naturally from adult sponges between 12 noon and 3 p.m. (Degnan et al., 2015). Immediately upon emergence, ~40 larvae were frozen in minimal volume FSW at −80°C; these represented 0–3 h post-emergence (hpe) pre-competent larvae (see Degnan and Degnan, 2010). Remaining larvae were maintained in ambient seawater for a further 4 h, at which 40 individuals were collected to represent the 4–7 hpe competent larvae.
Finally, as a source of postlarval sponge material, all remaining competent larvae (4–7 hpe) were induced to settle onto the crustose coralline algae (CCA) Mesophyllum sp., which is common at the sponge collection site. Mesophyllum sp. was removed from rocks using hammer and chisel and broken into shards from which epi- and in-faunal animals were removed before shards were used to induce larval settlement. At 48 h post-induction (hpi), 40 postlarvae were carefully removed from the CCA using a sterile scalpel and immediately frozen in minimal volume FSW at −80°C for subsequent gDNA extraction.
To distinguish vertically-inherited prokaryotes from those environmentally acquired, we also profiled the microbial community of both the settlement substrate and the seawater of the native sponge habitat at Shark Bay. For the settlement substrate, shards of Mesophyllum sp. were cleaned to remove epi- and in-faunal animals, and stored at −80°C for subsequent gDNA extraction. For the seawater, sterile Millepore filter papers of two pore sizes (0.22 and 0.45 μm) were used to vacuum-filter ~70 L of seawater collected at each of two different tidal states across 3 consecutive days to maximize microbial coverage; filters were replaced every 2 L to prevent loss of filtering capacity due to blocking of pores. Used filters were transferred aseptically to individual 15 ml falcon tubes and stored at −80°C for subsequent gDNA extraction.
Extraction of Genomic DNA
Prokaryote DNA was extracted from batches of ~40 frozen sponge embryos, larvae or postlarvae, and ~0.25 g of frozen adult sponge tissue. We used the Experienced Users Protocol from the PowerSoil DNA Isolation kit (MO BIO Laboratories) following the manufacturer's protocol, except at step 3 where 20 μl of lysozyme from chicken egg white (Sigma-Aldrich; 20 mg/ml) was added with solution C1 in order to increase microbial lysis. Purified gDNA was eluted in a total volume of 50 μl of MOBIO PowerSoil C6 buffer and stored at −20°C.
To isolate total gDNA from the CCA, the frozen shards of Mesophyllum sp. were ground into a paste with 60 μl of the MOBIO PowerSoil kit (MOBIO) C1 solution using a sterilized ceramic pestle and mortar before being added to the Powerbead tube supplied with the kit. The remainder of the extraction procedure followed the manufacturer's instructions, with the exception of eluting the gDNA in a final volume of 50 μl of C6 buffer before storing at −20°C.
To isolate total gDNA from the seawater, the frozen collection filters were processed using a modified version of the Isolation of Genomic DNA from Tissues protocol of the Qiagen QIAamp® DNA Micro Kit Handbook. Filters were sliced into strips using sterile scalpels and then placed aseptically into 1.7 ml microcentrifuge tubes (maximum 2 filters per tube). DNA was extracted by addition of 180 μl of ATL buffer and 20 μl of lysozyme (20 mg/ml) per filter, followed by an incubation for 1 h at 56°C. After transferring lysates to new microcentrifuge tubes, this first step was repeated two more times for each sample. Cell lysis and DNA precipitation steps were performed as recommended by the manufacturer, and the purified gDNA was dissolved in 50 μl of C6 buffer and stored at −20°C.
Extracted gDNA from all samples was quantified using a Qubit® 2.0 Fluorometer (Invitrogen) and quality was assessed using a 2% TBE agarose gel.
Prokaryote 16S rRNA Gene Amplicon Sequencing
Twenty nanogram of each sample was used to generate PCR amplicons for 454 pyrosequencing with the broad specificity bacterial and archaeal forward (pyroL803F mix CCT ATC CCC TGT GTG CCT TGG CAG TCc tpp TCA Gtt aga Kac ccB Ngt agtc) and reverse (pyroL1392R CCA TCT CAT CCC TGC GTG TCT CCG Acct ppT CAG ACA GCa cgg gcg gtg tgtRc) primers as per the protocol used at The Australian Centre for Ecogenomics (ACE) at The University of Queensland. Due to sequence similarity between the A. queenslandica 16S mtDNA (GenBank accession DQ915601; (Erpenbeck et al., 2007) and bacterial 16S rRNA gene sequences, we performed a two-step PCR protocol. The initial A. queenslandica PCR product was ~100 base pairs smaller than the expected prokaryote product and thus could be separated from it by 1% TAE agarose gel electrophoresis; the excised prokaryote product was purified from gel matrix using a silica-based cleanup and dissolved in 15 μl of ultrapure water. At ACE, a second 10 cycle PCR was performed using pyrotagged reverse primers (Berry et al., 2011), and sequencing was performed on a Roche Life Sciences 454 GS-FLX system.
In silico Analysis of Prokaryote 16S Gene Amplicon Sequences
Raw 16S rRNA gene amplicon sequences were processed using QIIME version 1.8.0 (Caporaso et al., 2010b). For the filtration step, we discarded sequences that were <300 nucleotides in length, had >1 ambiguous base, had homopolymer runs longer than six nucleotides, had up to two mismatch in the adapter sequence, or did not pass a sliding window test of quality scores (window size = 300 bp; average quality score 25). Remaining sequences were trimmed to 300 nucleotides in length. Any residual sequences matching to mitochondrial DNA of the sponge host were identified using Bowtie2 (Langmead and Salzberg, 2012) and excluded from all downstream analyses. All sequences with more than 97% similarity to each other were grouped into Operational Taxonomic Units (OTUs) using UCLUST (Edgar, 2010), with the cluster seed used as a representative sequence for each OTU. Chimeric OTUs identified by ChimeraSlayer (Haas et al., 2011) were removed. Of the remaining OTUs, only those for which at least five reads were recovered across all samples were retained for further analyses.
We assigned taxonomies to these OTUs using UCLUST, with the 2013 Greengenes database as a reference. Using the OTU abundance matrix table created by QIIME, the relative abundance of OTUs for each sample was obtained by normalizing the abundances to the total number of reads in each sample. To assess the number of shared and unique OTUs among samples, the data sets were pooled according to their source (i.e., sponge or seawater). We derived heatmaps depicting OTU abundance using the R package pheatmap. To assess microbial community structure within our samples, ecological indices were measured using QIIME on data sets rarefied to the smallest sampling effort, as recommended by Gihring et al. (2012); because we recovered very low number of reads for the coralline algae sample, it was excluded from the ecological indices analyses. Alpha diversity was estimated by Chao1, Simpson and Shannon indices implemented within QIIME.
Phylogenetic trees were built for putative vertically-inherited bacterial OTUs, as defined by having a relative abundance of 5% or greater in at least one sponge developmental sample and being present in more than half of the developmental stages sampled. Nearest related and described sequences were retrieved from the 2013 Greengenes and NCBI databases. Sequence alignments were derived with PyNAST (Caporaso et al., 2010a) using the template alignment file “core_set_aligned.fasta.imputed,” filtered for highly variable regions using QIIME, and manually adjusted using Mesquite (Maddison and Maddison, 2014). A maximum-likelihood tree was generated with RAxML v. 8 (Stamatakis, 2014) using the rapid bootstrapping algorithm and 1000 bootstrap resamples, and visualized using FigTree v. 1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/).
Characterization of the Prokaryote Community Using Microscopy
Transmission Electron Microscopy (TEM)
TEM was conducted both on embryos and larvae collected from brood chambers as previously described (Leys et al., 2008; Degnan et al., 2015) and fixed as described in Leys and Degnan (2002, 2001). In addition, free-swimming larvae were collected and prepared for TEM by cryo-immobilizing live specimens using a Leica EM PACT2 high pressure freezer (HPF), followed by freeze-substitution in a Leica AFS2 in a fixative of 1% OsO4 and 0.5% uranyl acetate in acetone with 5% water (adapted from McDonald et al., 2007). Post-larval sponge samples obtained as previously described (Leys et al., 2008; Degnan et al., 2015) were fixed in 3% glutaraldehyde in 0.1 M sodium cacodylate buffer in FSW for 3 h at RT and osmicated in 1% OsO4 in 0.1M sodium cacodylate. Samples were then treated with 4% (v/v) hydrofluoric acid for 2 h at RT to remove siliceous spicules, followed by dehydration through a graded ethanol series. Samples were embedded in EPON 812 resin and polymerized in a 60°C oven for 2 days.
All sections were cut on a UC6 ultramicrotome (Leica), mounted on Formvar coated copper grids, and contrasted with uranyl acetate and lead citrate. Grids were imaged on a JEOL JEM 1011 transmission electron microscope at 80 kV, equipped with an Olympus Morada digital camera.
Enrichment of Prokaryote Cells from Adult Sponge Tissue
Three healthy adult A. queenslandica were collected from the Shark Bay, Heron Reef field site in March 2014 and immediately transported back to the Heron Island Research Station; they were subsequently maintained at 4°C or on ice at all times during the enrichment process. For each individual separately, sponge tissue was cut into ca. 1 cm3 pieces using sterile scalpel blades, and placed in Ca/Mg-free seawater (CMF-ASW). Samples were agitated on a shaker table for 5 min at 200 rpm, and the supernatant poured to waste. Agitation was repeated with fresh CMF-ASW, and the supernatant poured to waste. A sterile 25 μm mesh cell squeeze was performed on the sponge tissue, and the cell squeeze centrifuged at 100 g for 15 min at 4°C. Supernatant was kept and centrifuged further at 200 g for 15 min at 4°C. Supernatant was kept and centrifuged further at 300 g for 15 min at 4°C. The supernatant was removed and aliquoted into 1.7 mL Eppendorf tubes, and centrifuged at 8800 g for 20 min at 4°C to pellet cells. The resultant bacterial cell enrichments were fixed via two methods, for different downstream analyses: (i) for in situ hybridization, bacterial cells were fixed in 0.05% glutaraldehyde, 1x MOPS buffer, and 4% paraformaldehyde in Milli-Q water for 1 h, and stepped into 70% ethanol for storage at −20 °C; or (ii) for electron microscopy, bacterial cells were snap frozen in 15% glycerol at −80 °C.
Scanning Electron Microscopy (SEM)
Bacterial cell enrichments frozen in 15% glycerol (see above) were defrosted on ice and centrifuged at 10,000 g for 20 min to pellet the cells, and the supernatant was removed. Samples were fixed and embedded using a Pelco Biowave (Ted Pella), using a protocol adapted from Wendt et al. (2004). Cells were fixed in 3% glutaraldehyde in 0.1 M sodium cacodylate buffer in filtered seawater (FSW), and washed in fresh 0.1 M sodium cacodylate buffer before centrifugation at 10,000 g for 10 min. The supernatant was removed and an equal part Milli-Q water was added for cell resuspension. Fifty microliters of the resuspended cells were aliquoted onto poly-l-sine coated 12 mm coverslips, air dried, and dehydrated through a graded ethanol series. Sections of adult sponge were fixed following this same method. Both cells on coverslips and adult tissue samples were critically point dried (Autosamdri-815, Tousimis), mounted onto a stub, and gold coated (SPI-MODULE). SEM images were captured using a Jeol JCM-5000 NeoScope or HITACHI SU3500 SEM.
Confirmation of the Dominant Symbiont Using Fluorescence In situ Hybridization (FISH)
An oligonucleotide probe specific to the most numerically dominant OTU (OTU 3C 5′-CCCCAGAGTTCCCGGCCGAA-3′) was designed using Geneious software (version R7, Biomatters) and confirmed as unique by BLAST against all other 16S rRNA gene sequences in our data set. A reverse complement negative control (OTU 3NC, 5′- TTCGGCCGGGAACTCTGGGG -3′) was used to test for non-specific binding. Probes were 5′ end-labeled with the indocarbocyanine fluorochrome Cy3 during synthesis by Sigma-Aldrich, Australia and dissolved in Ultrapure distilled water (Invitrogen, Life Technologies) to a working concentration of 50 ng μL−1. Bacterial cell enrichments, fixed as described above, were used for FISH following the method of Webster and Hill (2001) with minor adjustments. Specifically, bacterial cells were dehydrated in 100% ethanol to facilitate their adherence to slides, and a 20 μL aliquot transferred to poly-1-sine-coated 10-well-slides to air dry.
Hybridization solution [8 μL 0.9 M NaCl, 20 mM Tris-Cl (pH 7.2), 0.01% SDS, 17% (v/v) formamide] was mixed with 1 μL of the fluorescently-labeled oligonucleotide probe and applied to wells. Slides were incubated in a dark chamber at 46°C for 3 h. Post-hybridization, slides were washed in a prewarmed wash buffer [20 mM Tris-Cl (pH 7.2), 0.01% SDS, 0.225 M NaCl] at 48°C for 15 min in the dark. Slides were dipped in ice cold Milli-Q water to remove excess salts, air dried, and mounted in ProLong Gold antifade reagent (Life Technologies) with DAPI (4′,6-diamidino-2-phenylindole, Sigma-Aldrich; 1:1000 μL dilution) used as a counter stain. Labeled bacteria on slides were visualized using a 63x or 100x oil immersion objective with Nikon filter sets on a Nikon TiE inverted microscope, and imaged using a Nikon DS-U3 camera.
Differential Expression Analysis of Sponge Host Genes
To identify candidate host genes that may be involved in regulating or responding to the bacterial symbiont community, we asked which genes show significant changes in expression across times in the life cycle where we identify changes in the bacterial community. The transcriptomes used in this analysis were generated using a Cel-Seq protocol (Hashimshony et al., 2012) for multiple, replicated stages distributed throughout the life cycle of A. queenslandica, as described elsewhere (Anavy et al., 2014; Levin et al., 2016). We retrieved raw transcript read counts for three individual samples each of larvae (2–6 hpe), chamber-stage postlarvae (~48 hpi), and adults, selected because they correspond to the life cycle stages between which we observed the most dynamic changes in microbiome composition. From these raw reads, we retained only those genes for which counts were detected in one out of three replicates, in at least two of the three stages studied; the final dataset represented 32% of all sponge gene models presented in Fernandez-Valverde et al. (2015). We used the R software package DeSeq2 to perform differential gene expression analysis (Love et al., 2014). Following normalization by the variance-stabilizing transformation implemented within DeSeq2, genes with mean gene counts less than the 40% quantile were filtered out of the dataset. We then performed pair-wise comparisons between successive life cycle stages (i.e., larva-postlarva and postlarva-adult) and retained subsets of genes that had a Benjamini and Hochberg's false discovery rate (adj p) of <0.05. These significantly differentially expressed genes were then separated into eight discrete clusters (Cluster I–VIII) according to their profiles of expression in the life cycle stages of interest.
For each profile, we used Gene Ontology (GO) annotations derived by Blast2GO (Conesa et al., 2005), followed by the tool Bingo (Maere et al., 2005) to test for statistical over-representation of molecular function categories. We applied Benjamini and Hochberg's (BH) false discovery rate (FDR) correction, using a significance level of 0.05. Redundant terms were removed from the enriched GO list with the online tool REViGO (Supek et al., 2011), using the enriched corrected p-value derived with Bingo as a guide to choose representatives for each GO annotation group, the EBI GOA database (which annotates all UniProt entries with GO terms) as reference, and SimRel as semantic similarity measure with a cut-off value of 0.7. Here we assumed that the relative frequencies of gene functions in the A. queenslandica genome were not far from the overall trends in the genomic databases.
For each cluster, PFAM domains within each gene were also identified with the HMMER package using the Pfam-A database (e-value cutoff of 1e3). PFAM domain enrichment was calculated using an R script as per (Chandran et al., 2009); p-values were computed using a hypergeometric distribution, and adjusted for multiple hypothesis testing by applying a BH FDR correction with a significance level of 0.05.
Data Deposition
16S rRNA gene amplicon sequences generated in this study (Table S1) and further detailed below are deposited in GenBank under Accession Numbers SUB1383828: KX375419–KX375763.
The Cel-Seq transcriptome data analyzed for this study were extracted from a larger dataset, spanning the complete life cycle of A. queenslandica. Raw read data for the nine samples analyzed herein can be accessed as part of the complete dataset deposited in the NCBI Gene Expression Omnibus database under accession GSE60483. Normalized count data for just the nine samples analyzed in this paper are presented as Table S2.
Results
16S rRNA Gene Amplicon Sequencing Reveals a Low Complexity Microbiome Dominated by a Small Number of Proteobacterial OTUs That Appear to Be Vertically Inherited
In total, we identified 422 OTUs based on more than 97% sequence similarity (Table 1). Of these, 169 OTUs were identified in the combined life cycle sponge samples, 364 OTUs derived from the combined seawater samples, and 31 OTUs derived from the coralline algal samples (Table 1). Close inspection of these coralline algal sequences revealed they were dominated by cyanobacteria-like chloroplast DNA, which likely originate from chloroplasts of the algae itself; these reads were thus removed from all downstream analyses. Also of note, although the primers that we used target both bacterial and archaeal 16S rRNA gene sequences, we recovered only four archaeal OTUs in the seawater samples and none in A. queenslandica (Table S1). We combined the 16S rRNA gene sequence reads derived from the separate non-rarefied normalized sponge developmental datasets and seawater seasonal datasets, respectively, to estimate OTU overlap between these sources. Most of the OTUs were recovered in only one source, with 56 restricted to A. queenslandica and 251 restricted to seawater. However, these represent only 6.5 and 13.5% of the total number of reads, respectively. The remaining OTUs were shared between the sponge and seawater pooled samples, representing a total of 113 OTUs and a large proportion (79.7%) of the total dataset sequences (Table 1).
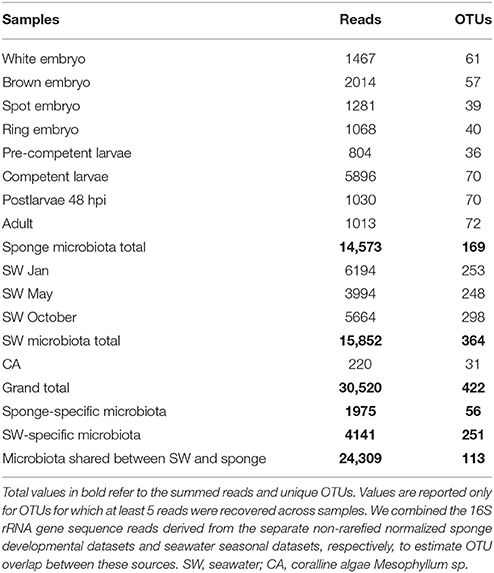
Table 1. 16S rRNA gene amplicon sequence summary for sponge and environmental samples, based on non-rarefied, normalized datasets.
Rarefaction curves based on total sequence counts do not display asymptomatic behavior for some of the data sets (Figure S1), suggesting that additional OTUs might have been discovered with deeper sequencing. The OTU richness and diversity estimations were obtained from rarefied data sets to the smallest sampling effort (i.e., 800 sequences in each data set), excluding the coralline algal sample. Overall, the seawater samples had the highest species richness and diversity, followed by the sponge postlarval (48 hpi) and adult stages, with little variation among the other sponge developmental stages (Table 2).
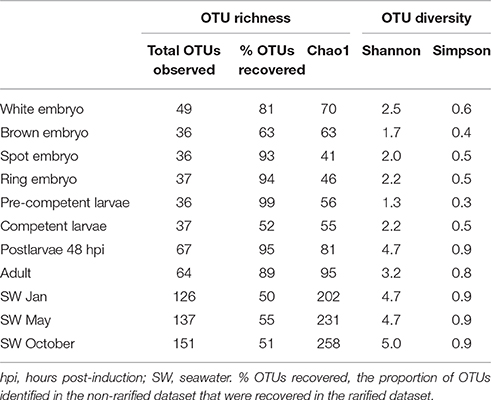
Table 2. Observed number of operational taxonomic units (OTUs) and estimation of richness (Chao1) and diversity (Shannon, Simpson) obtained from rarefied datasets.
We combined the 16S rRNA gene sequence reads derived from the separate non-rarefied normalized sponge developmental datasets to determine the taxon diversity of the total microbiome present in Amphimedon (with taxonomic representation >1%). The sponge microbiota was clearly dominated by Proteobacteria (71.6%), followed by Bacteroidetes (11.2%), Firmicutes (6.5%), Actinobacteria (4.1%), Planctomycetes (1.8%), and Cyanobacteria (1.8%; Figure 1). No Poribacteria OTUs were recovered from A. queenslandica (Table S1). Based upon OTU relative abundance within each developmental sample, the Proteobacteria, and specifically the Gammaproteobacteria, clearly dominated the bacterial community throughout the entire sponge ontogeny (Figures 2A,B). The relative abundance of Proteobacteria dropped below 90% only in the 48 hpi postlarval sample, at which stage other phyla (Actinobacteria, Bacteroidetes, and Firmicutes) were more prominent than observed in other life cycle stages. Similarly, within the Proteobacteria, the relative abundance of Gammaproteobacteria was lowest in postlarvae, followed by the adults, with other classes of Proteobacteria being more numerically dominant at these stages (Figures 2A,B). These results are consistent with the highest species richness and diversity observed in the postlarval stage of the sponge ontogeny (Table 2).
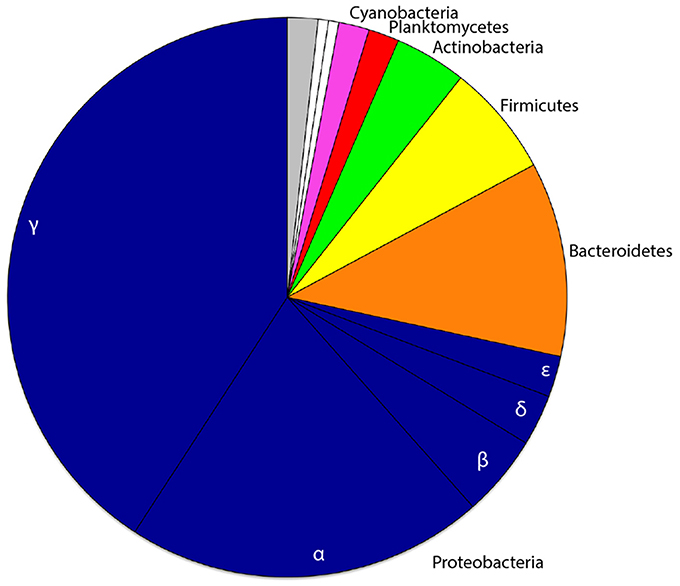
Figure 1. Proportional phyletic representation of bacterial OTUs recovered from one or more developmental stages of the sponge host Amphimedon queenslandica. Common phyla (taxonomic representation ≥1% across pooled developmental samples) are depicted in color, rare phyla (taxonomic representation ≤1% across pooled developmental samples) in white, and OTUs unclassified at the phylum level in gray. The class level divisions of proteobacterial OTUs are also indicated.
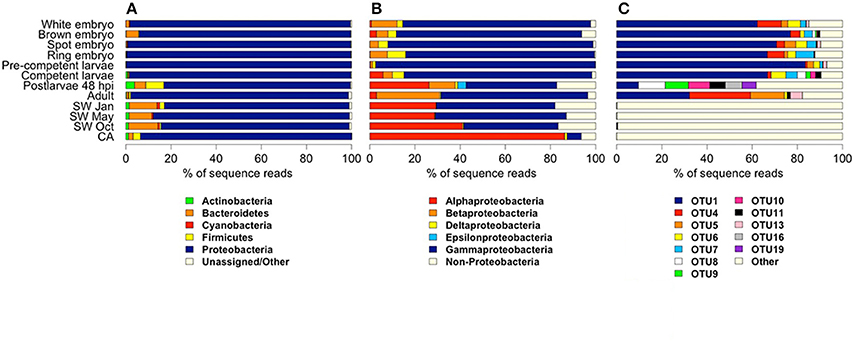
Figure 2. Relative abundance of bacterial and archaeal 16S rRNA gene amplicon reads derived from samples representing different stages in the life cycle of the sponge Amphimedon queenslandica, seasonal seawater (SW) from the sponge native habitat at Shark Bay, and the coralline algae (CA) settlement substrate Mesophyllum sp. Abundances are shown at (A) phylum level, (B) class level within Proteobacteria, and (C) OTU level. In (A) and (B), common taxa (displaying ≥ 1% read abundance in at least one sample) are coloured. For the CA sample, OTUs matching cyanobacteria-like chloroplast DNA were removed before deriving relative abundance estimates. In (C), bacterial OTUs shown are those that displayed a relative abundance ≥ 5% in at least one sponge life cycle stage.
We recovered 12 bacterial OTUs with a relative abundance of 5% or greater in at least one sponge developmental stage; six of these (OTU 1, 4, 5, 6, 7, 13) were identified in several stages of the lifecycle (Figures 2C, 3A). Four of these six belong to three orders within the class Gammaproteobacteria (OTU 1, order Chromatiales; OTU 5 and 6, order Oceanospirillales, but see below; OTU 13, order Alteromonadales), while the remaining two belong to unknown orders of the Beta- and Delta-proteobacteria (OTU 4 and 7, respectively; Figure 3B). OTU 1 was by far the most abundant OTU in the A. queenslandica microbiome; it made up 63.5% of the total bacterial community across all stages of the lifecycle, and dominated the microbiome of all embryonic and larval stages (Figures 2C, 3A). Three other OTUs (OTU 4, 6, and 7) also were represented in every lifecycle stage except for the postlarval stage, and OTU 5 and 13 were detected in the majority of stages. Of the six major sponge OTUs, three were also detected in the seawater community but only in very low numbers and never in all seasons (OTU 1, 4, and 6); OTU 5, 7 and 13 were not detected at all in the seawater samples (Figure 3A; Table S1). In contrast, the three main OTUs in the seawater samples (OTU 3, 2, and 12 in order of abundance, assigned, respectively to the orders Alteromonadales, Rhodobacterales and Sphingomonadales) represent 11.8% of the total normalized dataset sequences, and 64.7% of the total seawater community, but are either absent or recovered at low abundance (<1%) in the sponge samples. OTU 2 is also abundant in the coralline algal sample (Figure 3A; Table S1).
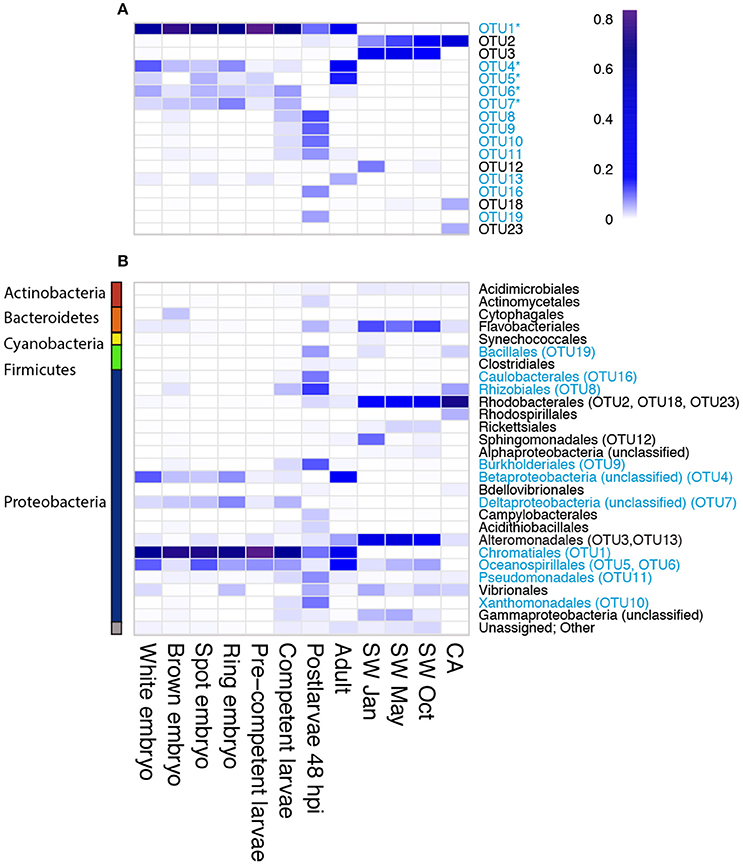
Figure 3. Heatmap depicting relative OTU abundance at (A) OTU level for the most abundant OTUs (relative abundance ≥5% within any sample), and (B) order level for all OTUs. The dominant sponge microbionts (relative abundance ≥5% within at least one developmental stage) are indicated in blue. Amongst these, the putative vertically-inherited symbionts (defined as being present in more than half of the developmental stages sampled) are indicated with an asterisk. The order with which each of the most abundant OTUs was affiliated by the program QIIME is indicated in (B). The color scale (0–0.8) indicates percent of total reads assigned to a particular bacterial OTU (panel A) or bacterial order (panel B).
Because OTU 1, 4, 5, 6, 7, and 13 were found generally throughout the sponge life cycle, but were barely, or not at all, detected in the seawater (Figures 2C, 3A), we consider them putative vertically-inherited bacterial symbionts. Together, they dominated the sponge microbiome through embryonic and larval development in the maternal brood chamber, and on into free-swimming larvae. However, the composition of the sponge bacterial community changed substantially in the postlarval 48 hpi sample, at which stage OTU 1 reduced to its lowest observed abundance and OTUs 4, 5, 6, 7, and 13 became undetectable (Figures 2C, 3A). Instead in this stage, we observed the numerical dominance of several other OTUs that were detectable only sporadically, or not at all, in earlier development stages (Figures 2, 3; Table S1). These include most predominantly OTU 16 [Proteobacteria (alpha), Caulobacterales] and OTU 19 (Firmicutes, Bacillales; Figures 2C, 3A), but also several others assigned to diverse orders within the Proteobacteria (see Figure 3, Table S1 for details). At least some of these appeared to be environmentally-derived from either the benthic settlement substrate (the coralline algae) or from the seawater, given their presence in one or more of those samples (Figures 3A,B; Table S1). There was a low level of retention of some of these horizontally-acquired bacteria into the adult stage, but generally the microbiome composition of the adult sponges again mirrored that observed in early development. Specifically, in adult sponges, OTU 1, 4, 5, 6, 7, and 13 were all again detected, and the first three together comprised 75% of the bacterial community (OTU 1, 30%; OTU 4, 30%; OTU 5, 15%; Figures 2C, 3A).
OTU 1 shows highest similarity (95%) to other Ectothiorodospiraceae bacteria within the order Chromatiales in the NCBI database. In our maximum-likelihood (ML) phylogenetic analyses, it forms a strongly-supported clade with other bacterial clones originating from other demosponges, within the family Ectothiorodospiraceae (Figure S2). OTU 4 also forms a well-supported clade with other uncultured demosponge-associated bacterial clones, which is quite divergent from other clades within the phylum Betaproteobacteria, and thus cannot be assigned at this time to any particular order (Figure S3). OTU 5 clusters with other clones derived from both marine and terrestrial environments in a clade within the genus Halomona (order Oceanospirillales; Figure S4). OTU 6 is a proteobacterium that shows highest similarity to other genera in the orders Chromatiales (94% identity) and Oceanospirillales representatives (88.3% identity) in the Greengenes and NCBI databases, while the closest match in the Greengene reference collection used by QIIME is an uncultured bacterium belonging to the Halomonadaceae family. Thus, it is identified as Oceanospirillales in the QIIME analyses reported above (Table S1; Figure 3B). Our ML phylogenetic analyses do not permit confident placement of this OTU, but they do suggest instead that it might belong to the order Chromatiales (Figure S2).
Electron Microscopy (EM) and Fluorescence In situ Hybridization (FISH) Reveal the Morphologies of the Most Dominant OTUs, and Changes in Their Abundance through the Life Cycle
Scanning electron microscopy (SEM) of the cell preparation used for the FISH experiment revealed that the dominant bacterial symbiont is characterized by a rod-shape morphology with a spiral membrane (Figure 4A). Using a fluorescent probe specific to OTU 1, in conjunction with 4′,6-diamidino-2-phenylindole (DAPI) as a counterstain, we confirmed that this Chromatiales bacterium was the morphotype that numerically dominated the microbe-enriched cell preparations obtained from adult A. queenslandica (Figures 4B,C). The same morphotype is clearly visible also in intact adult tissue prepared observed under SEM (Figure 5A).
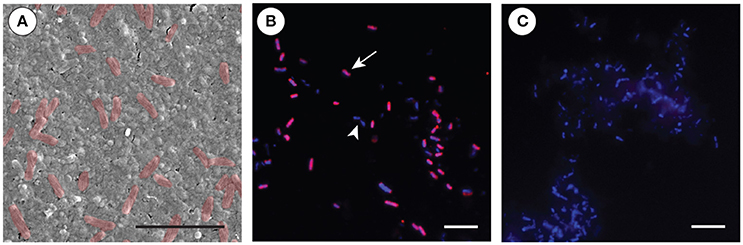
Figure 4. Fluorescent in situ hybridization (FISH) of the primary Amphimedon queenslandica symbiont (OTU 1) in microbe-enriched cell preparations from adult sponges. (A) SEM of the cell preparation used for the FISH experiments, showing the abundance of the OTU 1 bacterial morphology (false colored in red). Scale bar 5 μm. (B) FISH on bacterial-enriched cells using Cy3-labeled-OTU 1-specific probe (red; arrow), complemented by DAPI that labels all bacterial cells (blue; non-OTU 1-labeled cells, arrowhead). Scale bar 20 μm. (C) Negative control using reverse complement OTU 1-specific probe, with DAPI labeling all bacteria (blue). Scale bar 20 μm.

Figure 5. Electron microscopy (EM) images of the two bacterial symbionts that are numerically the most dominant through all Amphimedon queenslandica life stages. (A) Scanning EM of adult tissue showing OTU 1 (arrow) and putative OTU 4 (arrowhead) morphotypes. Scale bar 1 μm. (B) Transmission EM section of juvenile tissue with OTU 1 shown in cross- and longitudinal-section between sponge cells (SC). The distinctive 9 equidistant “arms” of this bacterium are formed by the spiral cell wall and outer cell membrane. Scale bar 1 μm. (C) TEM section showing a direct comparison of the morphologies of OTU 1 and putative OTU 4; clearly visible are the acidocalcisome-like structures (ac) often found in OTU 1, situated toward the poles of the bacterium. Scale bar 1 μm.
The OTU 1 morphotype is also clearly distinguishable by transmission electron microscopy (TEM) in thin sections of A. queenslandica and was the most abundant morphotype observed in all stages sampled for EM; it was mostly observed extracellularly. The OTU 1 morphotype is up to 1.5–2 μm in length and 0.5–0.8 μm in diameter, and has a spiral envelope formed by the cytoplasmic membrane and the cell wall, which are 20 nm apart. This envelope seems to be coated in a glycol-protein membrane and equipped with longitudinal ridges, giving cross-sectioned bacteria the appearance of a 9-armed star (Figures 5B,C). These protuberances are uniform in height (50–80 nm from tip to valley) and arranged equidistantly around the bacterial cell. There is a continuous riboplasm throughout the intracytoplasmic membrane (0.8–1 μm long, 0.3–0.5 μm wide). The OTU 1 morphotype lacks any noticeable photosynthetic structures, flagella or pili, but pseudo-spherical, membrane-bound vacuoles are visible (Figures 5B,C). These resemble commonly-found bacterial organelles that contain elemental granules either of phosphorus, sulfur or polyhydroxybutyrate (Lawry et al., 1981; Robertson and Kuenen, 2006; Docampo et al., 2010).
We also frequently observed a second morphotype, albeit in lower abundance generally across the developmental stages than OTU 1. Based on its proportional abundance, we infer the second most common morphotype is likely to be OTU 4, the Betaproteobacteria of currently unknown order, although we do not yet have direct experimental evidence of this. Under SEM, the putative OTU 4 morphotype resembles a slightly curved pointed rod with small external ridges (Figure 5A). Under TEM, we observed that the putative OTU 4 morphotype is 1.3–2.7 μm long and 0.3–0.5 μm wide, with a slim curved intracellular membrane. In longitudinal-section, irregular 10–40 nm peaks give the cell envelope a ribbed appearance, and in cross-section, the cell membrane appears 10 nm wide and circular (Figure 5C). These betaproteobacteria have a uniform riboplasm visible in the high-pressure frozen larval samples, and 50–250 nm long fimbriae coating their cell wall (Figure 5C).
By using TEM on multiple ontogenetic stages of A. queenslandica, we were able to visually confirm the vertical inheritance of OTU 1, which our 16S rRNA gene amplicon sequencing data suggests comprises at least 60% of the microbiome in developing embryos (Figure 2C). Specifically, we observed maternal nurse cells (trophocytes) laden with these bacteria in the region of brood chambers immediately adjacent to early developing embryos (Figure 6A). Trophocytes were easily distinguishable from other sponge cells by the presence of lipid inclusions and yolk granules within their cytoplasm; they housed a moderate number of bacteria, usually 10–20 cells (Figure 6A). We also observed bacteria-laden trophocytes, either intact or in the process of rupturing and releasing their bacterial contents, in the extracellular space of cleavage-stage (white) embryos (Figure 6B), but never inside embryos of later developmental stages. In blastula-stage (brown) embryos, we observed OTU 1 morphotypes that were no longer associated with nurse cells, but were now freely and randomly distributed in the extracellular space of the embryos (Figure 6C). Phagocytes, containing a visually distinct Golgi apparatus and partially-digested bacteria, were also observed between the inner and outer embryonic follicle layer of blastulas (Figure 6D). This is the earliest stage of the life cycle in which we observed evidence of sponge cells phagocytosing the bacterial symbionts.
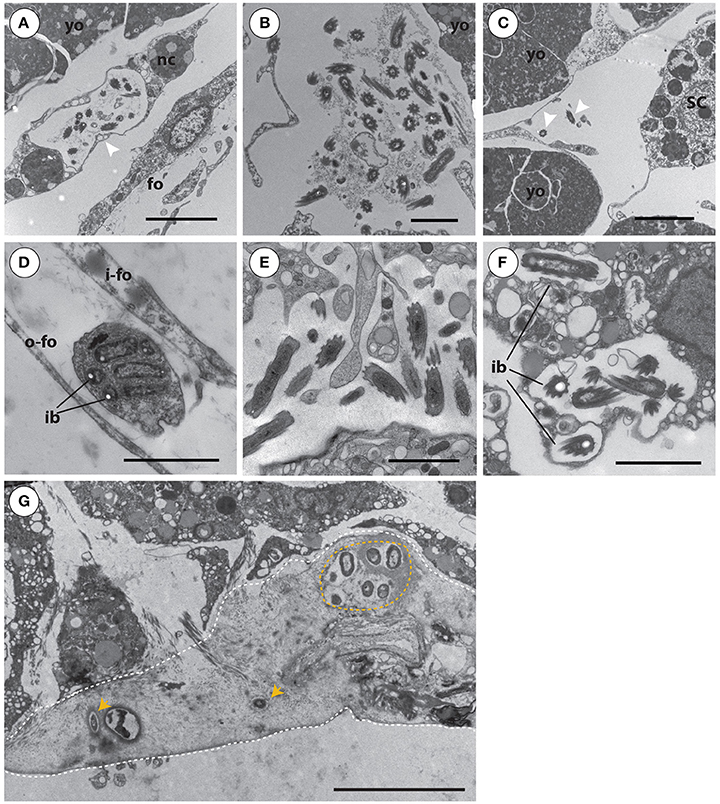
Figure 6. Transmission electron microscopy images of Amphimedon queenslandica symbionts in multiple different life cycle stages that span cleavage-stage embryos to juveniles. (A) Bacterial symbionts in large pocket vesicles are transported into cleavage-stage (white) embryos by maternal nurse cells (arrowhead). Scale bar 5 μm. (B) Upon entering cleavage-stage embryos, the nurse cells rupture and release their bacterial cargo into the extracellular space. Scale bar 2 μm. (C) In blastula stage (brown) embryos, bacterial symbionts (arrow heads) are randomly distributed at low density through the extracellular space. Scale bar 2 μm. (D) In blastula stage (brown) embryos, between the inner and outer embryonic follicle layer, sponge cells were observed phagocytosing symbiont bacteria. Scale bar 2 μm. (E) By larval stages, individual symbionts are observed in highest density in the inner cell mass. Scale bar 2 μm. (F) During metamorphosis of larvae into postlarvae (shown at 48 h post-induction of metamorphosis), bacterial symbionts are predominantly observed toward the endosome, including often intracellularly, being phagocytosed by sponge cells. (G) In postlarvae by 48 hpi, a collagen- and spongin-rich layer has been excreted to adhere the postlarva to the settlement substrate. Through this extracellular spongin- and collagen-rich basal layer (outlined by dashed white line), foreign bacteria (orange arrowheads; orange circle) enter the sponge body from the environment. The most basal edge of the postlarva, where it has been detached from the substrate, is the lower dashed line of the image. Scale bar (A–F), 2 μm; (G), 5 μm. Yolk masses (yo), nurse cells (nc), inner follicle (i-fo) and outer follicle (o-fo) membrane, cilia (ci), and intracellular bacteria (ib) in sponge vesicles.
Although active transport of symbionts by maternal cells into the sponge embryos appeared to occur only up to blastula stage, and some phagocytosis was observed from this time on, the abundance of the OTU 1 morphotype remained high through the rest of embryogenesis, with small clusters consistently found between sponge cells throughout the embryo, particularly in the inner cell mass (ICM; not shown). Embryogenesis ends with mature larvae released from the brood chamber into the water column. By this stage, symbionts were observed in low abundance along the dense columnar epithelial cell (CEC) layer, and in much higher abundance throughout the ICM, in which they occurred unevenly in spaces between collagen fibers (Figure 6E). After larvae settled onto the benthic substrate and began metamorphosis (48 hpi postlarvae), we observed these vertically-inherited core bacterial symbionts predominantly toward the endosome; at this stage, we occasionally observed these core symbionts intracellularly, which we interpret as possible phagocytosis by sponge cells (Figure 6F). Also in the 48 hpi postlarvae, we observe previously-unseen bacterial morphotypes now present in the basal layer of the young sponge (Figure 6G), and spatially separated from the vertically-inherited bacteria in the endosome. At this stage, it is apparent that the basal pinacoderm has not yet grown across to seal the young sponge from the environment, and thus bacteria are able to move freely from the environment into the extracellular space in this spongin-containing layer. Our 16S rRNA gene amplicon results (see above and Figure 3, Table S1) suggest that these new bacterial types may be horizontally acquired from the coralline algae settlement substrate itself or, more often, from the surrounding seawater. Some of these appear to persist into adulthood (Figure 3, Table S1), while others become undetectable in adults either because they were only transiently present in postlarvae or because they persist into adulthood but at undetectable abundances.
Differential Expression Analysis of Sponge Genes Reveals Possible Candidates for Animal-Bacterial Interactions
To identify genes that may be involved in regulating microbiome assembly, we performed pairwise comparisons of normalized gene expression between successive sponge life cycle stages across the metamorphic transition that encompasses the dramatic shift in microbiome composition (i.e., larva to 48 hpi postlarva, and postlarva to adult). From this analysis, we identified 4480 genes that exhibited differential expression in one or more of these pairwise comparisons. As larvae settled and began metamorphosis into the postlarval sessile body plan, 1999 genes were significantly upregulated and 1620 were significantly downregulated (Table S3; Figure 7A). Maturation from the postlarval to the adult stage was accompanied by the significant upregulation of 978 genes and downregulation of 394 genes (Table S3; Figure 7A). To follow the expression trajectory of each gene across the three life cycle stages tested, we categorized the genes that showed a significant shift in expression into eight profiles (Cluster I–VIII; Table S3; Figure 7B). The expression of a large proportion of genes (69.4%) significantly changed during the larval to postlarval transition period but then remained constant in the adult stage (Cluster I and II). We recovered fewer differentially expressed genes (19.2%) across the transition from the postlarval to the adult stage (Cluster III and IV), while 11% genes displayed either a sustained or reversed expression profile across the successive life cycle stages tested (Cluster V–VIII; Table S3; Figure 7B). Based on Blast2GO annotations (Table S3), we identified among the differentially expressed genes (DEGs) several candidates worthy of future investigation. In particular, we discuss the 1689 Cluster I DEGs—those upregulated from free-swimming larvae to 48 hpi postlarvae—because we consider these especially likely to include genes involved in monitoring and regulating the microbiome around the time of the compositional shift. Accordingly, we focus on discussing those genes from Cluster I that have Blast2GO annotations suggesting putative functions in bacterial recognition and response. Future experimental validation of these potential candidates will require functional analyses that are beyond the scope of the current study.
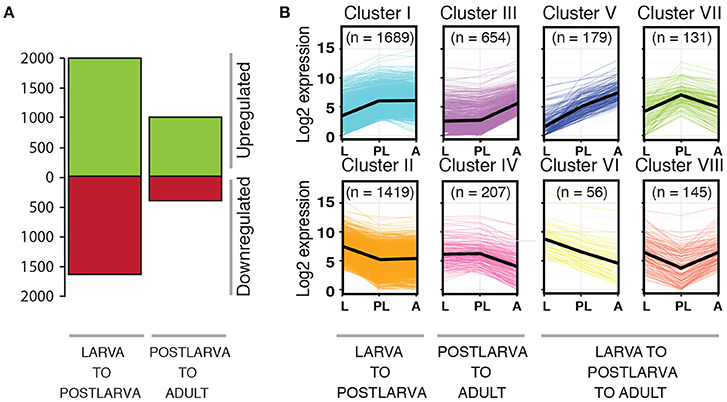
Figure 7. Differential gene expression across the metamorphic transition in Amphimedon queenslandica. (A) Number of genes that are differentially expressed across two major life cycle transitions (p < 0.05), whether upregulated (green) or downregulated (red). (B) Gene clusters (I–VIII) that share common expression profiles across the life cycle. The black line represents the average trend for each life cycle stage. The number in brackets indicates the number of genes recovered in each gene cluster.
The Cluster 1 gene list (Table S3) includes 14 genes containing ankyrin repeat domains (ANKs) of various types, and 24 genes containing tetratricopeptide repeats (TPRs); one of the latter (Aqu2.1.21073, a glycosyltransferase/tetratricopeptide repeat domain-containing protein) has been identified with high confidence as a gene horizontally transferred from bacteria into the A. queenslandica genome (Conaco et al., 2016). The next most abundant genes families of interest are 19 Deleted in Malignant Brain Tumor 1 (DMBT1) genes, each of which contain scavenger receptor cysteine-rich (SRCR) domains (Rosenstiel et al., 2007), followed by six each of tnf receptor-associated factor (TRAF) genes with putative roles in activation of the immune response (Wiens and Glenney, 2011), and tripartite motif-containing protein family (TRIM) genes that are hypothetically involved in pathogen recognition and host defense (Ozato et al., 2008). We also identified several single genes with putative functions of interest, including an AIG1 family protein, an anthrax toxin receptor 2 gene, a gram-negative bacteria binding protein, and a macrophage mannose receptor 1-like gene (Hibino et al., 2006). Interestingly, the Cluster 1 list also includes several genes involved in autophagy (e.g., cdgsh iron-sulfur domain-containing protein and autophagy-related proteins; Virgin and Levine, 2009), as well as several kinds of extracellular matrix proteins, such as fibropellins, genes involved in modulation of cell shape and migration, such as filamins and Rho GTPase-activating proteins, and genes encoding transmembrane receptors that bridge cell-extracellular matrix, such as integrins (Hook et al., 1990).
To search for gene families that were overrepresented in the clusters of DEGs, we performed both a GO annotation enrichment analysis (Table S4A) and a PFAM domain enrichment analysis (Table S4B) on each gene cluster. Among genes that showed a sustained upregulation between the successive stages (Cluster V), notable is a significant enrichment of scavenger receptor-type genes. Despite identifying many genes containing either ANK repeat or TPR domains among multiple clusters, we did not find a significant enrichment of either domain in any particular cluster (Table S4).
We were interested in specifically identifying genes in the A. queenslandica genome that are putatively derived from horizontal gene transfer (HGT; Conaco et al., 2016) and significantly upregulated from the larval to the postlarval stage. Among these, we recovered several associated with hydrolase activity, oxidoreductase activity, carbohydrate metabolism and glycogenesis activity (Table S3). We also note that HGTs significantly upregulated in the postlarval to adult stage included those annotated as metalloproteases (e.g., Aspzincin-like peptidase m35, extracellular protease), glycogenesis, hydrolase and sulfatase activity, while those encoding extracellular proteases and metalloproteases showed a sustained upregulation across all three of the successive stages. Several of these horizontally-transferred genes contained domains that were recovered in our GO and PFAM enrichment analyses (Table S4).
Discussion
Amphimedon queenslandica Adults House a Low Complexity Microbiome Dominated by a Small Number of Proteobacterial OTUs Whose Closest Relatives Are Other Sponge Symbionts
As measured by the number of distinct OTUs identified by 16S rRNA gene amplicon sequencing, A. queenslandica supports a microbiome of low complexity. We recovered a total of 169 OTUs for the combined sponge samples, and just 72 OTUs in adults alone, which is toward the lower end of the range (50–3820) currently known from adult marine sponge microbiomes (Thomas et al., 2016). We do note, however, that our study had relatively low sequencing depth, which could have compromised the recovery of rarer OTUs. Of the OTUs associated with A. queenslandica, 56 appear to be sponge-enriched in that they are not also found in the surrounding seawater. Consistent with observations in other sponge taxa (Thomas et al., 2016 and references therein), the A. queenslandica microbiome is heavily dominated by the phylum Proteobacteria, distantly followed by Bacteroidetes and Firmicutes, and complemented by at least 9 other phyla. We found no evidence of phyla that are typical of many other sponges (Simister et al., 2012; Thomas et al., 2016), such as Chloroflexi and Acidobacteria. The proteobacterial OTUs are dominated in adult A. queenslandica by classes Beta- and Gamma-proteobacteria, as observed in several species of low microbial abundance (LMA) sponge (Hentschel et al., 2003; Giles et al., 2012), rather than the more generally observed dominance by Gamma- and Alpha-classes (Thomas et al., 2016). Together, these observations indicate that A. queenslandica is appropriately considered as a LMA sponge species, as has been reported for congeners (Gloeckner et al., 2014; Poppell et al., 2014).
Indeed, at least 75% of the adult microbiome is comprised of just three OTUs. Two of these (OTU 1, 4) phylogenetically cluster into clades formed by sponge-derived sequences, as commonly observed for core sponge symbionts (Erwin and Thacker, 2008; Simister et al., 2012; Taylor et al., 2013; Thomas et al., 2016). Based on our phylogenetic analyses, OTU 1 is a gammaproteobacterium in the order Chromatiales (Family Ectothiorhodospiraceae) and clusters with other uncultivated clones found in both LMA and HMA sponges, including a chemoautotrophic sulfur-oxidizing bacterium associated with the demosponge Haliclona cymaeformis (Tian et al., 2014). We used FISH with an OTU 1-specific probe to confirm that the very distinctive 9-armed rod-shape bacterium clearly visible in all stages of the life cycle is most likely this Chromatiales bacterium, which morphologically resembles that reported from larvae of another demosponge, Haliclona (Gellius) caerulea (Maldonado, 2007). Its unusual star-shaped morphology may provide it with an increased surface area to volume ratio, and thus a possible advantage over competitors for nutrient acquisition (Wanger et al., 2008). The OTU 1 morphotype lacks photosynthetic structures, but does contain globules inside the cell membrane, which we hypothesize to be sulfur, consistent with it belonging to the purple-sulfur bacteria. Chemoautotrophic Chromatiales are found relatively frequently in association with marine sponge species, implying that they play an important functional role in the partnership. The second most abundant symbiont, OTU 4, is a betaproteobacterium that is quite divergent from known orders within that phylum, and thus cannot be assigned to a lower taxonomic level at this time. In a companion paper (Gauthier et al., 2016), we report the draft genomes for these two OTUs, which we name therein as AqS1 (OTU 1; A. queenslandica Symbiont 1) and AqS2 (OTU 4), and discuss their potential functional contributions to extending the metabolic capabilities of the A. queenslandica holobiont. Here, we confine our discussion to the dynamic contribution of these OTUs to the microbiome in different stages of the sponge life cycle.
Together, OTUs 1 and 4 comprise the majority of the adult microbiome, proportionally at least 30% each according to our 16S rRNA gene amplicon data, although EM observations suggest that OTU 1 in particular may be even higher. In SEM of adult sponges, we see irregular distribution of two common morphotypes, either aggregating near choanocytes or distributed randomly and sparsely throughout brood chamber, mesohyl tissue, and aquiferous system. We have personally examined hundreds of A. queenslandica adults from Heron Island Reef across multiple years, and have observed these two putative OTU 1 and 4 morphotypes in every single healthy individual. We consider both to be core members of the A. queenslandica microbiome, as defined by Thomas et al. (2016) as an OTU present in at least 85% of the replicates for a single host species. A third bacterium, OTU 5, which is a gammaproteobacterium in the order Oceanospirillales, comprises another 15% of the adult microbiome as indicated by the 16S rRNA gene amplicon sequencing, but this translates to a low enough abundance that we rarely encounter it by electron microscopy. Species with such low complexity adult microbiomes dominated by just a few proteobacterial species, as reported for a handful of other sponge species (Giles et al., 2012; Blanquer et al., 2013; Croué et al., 2013), can be considered as very tractable models for studying sponge-bacteria interactions. In A. queenslandica, this value is further enhanced by the availability of draft genomes for both the sponge host (Srivastava et al., 2010) and two of the core bacterial symbionts (Gauthier et al., 2016).
Several OTUs Are Present Throughout the Life Cycle, and There is Evidence for Both Vertical Inheritance and Phagocytosis of Core Symbionts during Development of This LMA Sponge
In A. queenslandica, both our EM and 16S rRNA gene data unequivocally revealed the presence of several OTUs in all developmental stages. Together with OTUs 1, 4, and 5, three other proteobacterial OTUs had a relative abundance of 5% or greater in at least one sponge developmental sample (OTU 6, 7, 13). All six were identified in several stages of the lifecycle but were barely, or not at all, detected in the seawater. We thus consider these six OTUs to be putative vertically-inherited core bacterial symbionts. These findings contrast those of Schmitt et al. (2008), who reported the reproductive stages of LMA sponges to generally be bacteria-free, although core symbionts have been observed at least in larvae of the LMA sponge Crambe crambe (Croué et al., 2013), and certainly vertical inheritance is commonplace in HMA sponges (Schmitt:2008bm; Enticknap et al., 2006; see for example Ereskovsky et al., 2005; Maldonado, 2007; Schmitt et al., 2007, 2008; Sharp et al., 2007; Steger et al., 2008; Lee et al., 2009; Webster et al., 2010; Gloeckner et al., 2012).
OTU 1 is by far the most abundant in the A. queenslandica microbiome, alone comprising more than 60% of the total bacterial community in all embryonic and larval stages that we assessed by 16S rRNA gene amplicon sequencing. We directly observe its vertical inheritance in very early development, and its apparent phagocytosis by sponge cells in blastula stage embryos and then throughout the remainder of development in the brood chamber. Despite the phagocytosis, abundance of the OTU 1 morphotype increases substantially from embryonic to larval stages, and we continue to see evidence of phagocytosis in the free-swimming larvae. Other studies have also reported phagocytosis of symbionts during the larval stage, and the use of symbiotic bacteria as a nutrition source in the non-feeding sponge larva has been suggested to contribute to lengthening the planktonic phase or even to increasing settlement success of competent larvae (Ereskovsky and Tokina, 2004; de Caralt et al., 2007). In the context of the latter proposition, it is interesting that, in A. queenslandica, OTU 1 constitutes as much as 85% of the total microbiome of larvae that have developmentally matured such that they are competent to settle. Notably, OTU 1 is at its highest proportion of the microbiome at this competent larva stage, while proportion of the other vertically-inherited symbionts remains unchanged, or even decreases. We hypothesize that OTU 1 may have a critical functional role in facilitating larval settlement and metamorphosis, by supplying the amino acid L-arginine required for the sponge larvae to produce the gaseous signaling molecule, nitric oxide (NO), via nitric oxide synthase. We have shown that NO is required by these larvae to successfully settle and metamorphose (Ueda et al., in press), but its production requires the presence of L-arginine as a substrate. A. queenslandica itself is unable to synthesize this amino acid, but OTU 1 can do so (Gauthier et al., 2016).
The Composition of the Core Microbiome is Stable through the Life Cycle, Except for a Dramatic Shift at the Time of Larval Settlement and Metamorphosis
Abundance estimates based on 16S rRNA gene amplicon sequencing at phylum, class and OTU level, as well as the richness and diversity indices, all indicate that the A. queenslandica microbiome is relatively stable throughout much of the life cycle. The six, putative vertically-inherited symbionts (OTUs 1, 4, 5, 6, 7, and 13) together dominate the sponge microbiome through embryonic and larval development in the maternal brood chamber, and on into free-swimming larvae. However, there is a substantial re-organization of the bacterial community composition within the first 48 h after larvae settle onto the benthos and initiate metamorphosis into the sessile body plan. In particular, the proportional abundance of OTUs 1 and 4, otherwise so dominant in all developmental stages, has declined dramatically in 48 h post-induction (hpi) postlarvae. At this time, OTU 1 was reduced to by far its lowest abundance (<10%) and OTUs 4, 5, 6, 7, and 13 were undetectable at the sequencing depth applied in this study. Instead, in 48 hpi larvae, we observed the emergent dominance of several other OTUs that were barely, or not at all, detectable in earlier development stages. Indeed, even the relative abundance of Proteobacteria dropped below 90% only in the 48 hpi postlarval sample, at which stage other phyla (Actinobacteria, Bacteroidetes, and Firmicutes) became more prominent than observed at any other stage of the life cycle. As a result, we saw the highest species richness and diversity in the postlarval stage of the sponge ontogeny.
We propose that two distinct factors contribute to this shift in microbiome composition. First, we continue to observe phagocytosis particularly of the OTU 1 morphotype during these transitional stages, such that its decline might be caused at least in part by its increased use as a source of nutrition by the early postlarval sponge that cannot yet filter feed. It is unclear whether the sponge preferentially ingests the OTU 1 morphotype over all others, or whether we simply are more likely to observe this morphotype being phagocytosed because it remains the single most abundant morphotype even though its proportional abundance has declined at this stage. At 48 hpi, the aquiferous system composed of canals lined by choanocytes and endopinacocytes is clearly developing, but there is no osculum and thus there is not yet a flow-through water current for filter feeding (Degnan et al., 2015). The aquiferous system required for feeding does not fully develop until 3–5 days post-settlement, and we observe the major microbiome shift through the first 48 h post-settlement. Second, at least some of the novel OTUs that appeared at this stage—having not being detected in earlier developmental stages—seem to be environmentally-derived from either the benthic settlement substrate (the coralline algae) or from the seawater, based on their presence in one or more of those samples. This raises interesting questions as to whether the exact composition of the post-larval microbiome may vary according to the specific settlement site chosen by individual larvae. We currently are investigating this question in more detail, in a higher temporal resolution analysis of the composition of both vertically- and horizontally-acquired bacterial communities, from the time of larval settlement through metamorphosis and establishment of the feeding juvenile body plan.
The influence of specific settlement substrate may be particularly important because we saw a low level retention of some horizontally-acquired bacteria into the adult stage, although generally the microbiome composition of the adult sponges again mirrored that observed in early development. Specifically, in adult sponges, all six of the vertically-inherited core symbionts were again detected, with OTUs 1, 4, and 5 together comprising 75% of the bacterial community. It is worth noting that 16S rRNA gene sequences specific to four of these symbionts were identified in seawater samples taken from the sponge habitat, albeit in very low abundance. This suggests that horizontal acquisition of microbes present in the surrounding environment as inactive dispersal stages could complement vertical transmission. These could thus act as “seed banks,” permitting juveniles and adults to recover symbionts that might have been lost during their life cycle, and thus help to ensure their persistence, as has been proposed elsewhere (Schmitt et al., 2008; Webster et al., 2010; Gloeckner et al., 2013; Taylor et al., 2013).
This is, to the best of our knowledge, the first report of microbiome composition through multiple different stages of a sponge life cycle, including across the metamorphic transition. The singular upheaval that we observe when postlarvae take up novel OTUs from their settlement environment, followed by a recovery to a long-term stable adult state, strongly supports the existence of mechanisms for specific selection and persistence of suitable, core partners in a high-fidelity symbiosis (McFall-Ngai et al., 2013; Douglas, 2014). The identity of the selection mechanisms at both molecular and cellular levels, and the relative roles of the various partners in making the selections and maintaining the homeostasis, both remain to be determined, but we propose that the A. queenslandica holobiont can provide a valuable model system for such investigations.
Differential Expression Analysis of Sponge Genes Indicates Some Potential Players in the Molecular Crosstalk between Host and Symbionts
In particular, this naturally-occurring perturbation of the microbiome through settlement and metamorphosis can be exploited as a means to identify specific genes that might be involved in mediating animal-bacterial interactions, including both symbiont and opportunistic bacteria. Many of the genes differentially expressed across this life cycle transition will be associated with the extensive morphological, ecological, and physiological changes that constitute metamorphosis of the sponge body plan, rather than specifically with sponge-bacterial interactions. We thus used Blast2GO annotations of differentially expressed genes (DEGs) to search for candidates that, based on their demonstrated function in other organisms, could be involved in animal-bacterial crosstalk. To identify candidates particularly worthy of further investigation, we focus here on those identified in Cluster I—genes significantly upregulated from free-swimming larvae to 48 hpi postlarvae—because we consider these especially likely to include genes involved in monitoring and regulating the microbiome around the time of the compositional shift revealed by our 16S rRNA gene amplicon sequencing and EM data.
It is of particular interest that the Cluster I gene list includes multiple genes containing ankyrin repeat domains (ANKs; 14 genes) or tetratricopeptide repeats (TPRs; 24 genes), although neither showed significant GO or PFAM domain enrichment in any cluster. Both ANKs and TPRs mediate protein-protein interactions and are known as eukaryote-like proteins (ELPs), in reference to their identification first in eukaryote genomes, but more recently also in many prokaryote genomes. TPR-containing proteins are known to play critical roles as bacterial virulence factors (Cerveny et al., 2013), and their presence in the genome of a sponge Pseudovibrio sp. symbiont has been postulated to aid in attachment to the eukaryotic host and in avoidance of the host's immune response (Alex and Antunes, 2015). ANK-repeat proteins (ARPs) have been identified in abundance in the genomes of multiple sponge-associated bacteria (Liu et al., 2010; Thomas et al., 2010; Siegl et al., 2011; Fan et al., 2012; Hentschel et al., 2012; Tian et al., 2014; Alex and Antunes, 2015; Burgsdorf et al., 2015b; Britstein et al., 2016), including in the most dominant core symbiont of A. queenslandica, OTU 1, although not in OTU 4 (Gauthier et al., 2016). ARPs from bacterial symbionts have been experimentally shown to influence sponge-bacterial interactions, specifically via interfering with phagocytosis as a possible mechanism to escape digestion in a sponge host (Nguyen et al., 2014). Together, these findings lead us to suggest that the ANK- and TPR-containing proteins upregulated in Cluster 1 might well be important in mediating sponge-bacterial crosstalk through larval settlement and metamorphosis because they could directly interact with symbiont proteins containing the same domains. It is further notable that one of the Cluster I genes encoding a TPR has been putatively identified as a gene horizontally transferred from bacteria into the A. queenslandica genome (Conaco et al., 2016).
Among these genes differentially upregulated from pelagic larvae to settled 48 hpi postlarvae, and then through to maturation into a filter-feeding adult (Cluster I), we also identified several genes belonging to each of three families that include genes with demonstrated roles in innate immunity in other animals. Innate immune genes are of interest because of their hypothesized roles in invertebrates in establishing and maintaining bacterial symbiont communities (Nyholm and Graf, 2012). As discussed above, both our histological data and that of other studies report that phagocytosis of symbionts can occur at least during the larval stage in some sponge species (Ereskovsky and Tokina, 2004; de Caralt et al., 2007). It is unknown whether this occurs because larvae do not yet possess a well-developed immune system to select for or against specific types of bacteria, or because the bacteria themselves lack mechanisms to evade detection and consumption. The fact that phagocytosis of the dominant bacterial symbiont is not observed in Amphimedon adults gives some support to the former, although it could also be that the presence of ARPs in the genome of this symbiont (OTU 1; see Gauthier et al., 2016) protects it from detection. Our transcriptome data further suggests that the larvae might lack means to differentiate between generic microbes and symbionts because it is only as larvae metamorphose into the benthic, sessile body plan that we see the upregulation of several genes expected to equip postlarval and adult individuals with the means to mediate microbial recognition and removal of invaders, as discussed below.
First, we found 19 genes containing Scavenger Receptor Cysteine-Rich (SRCR) domains, which constitute a group of conserved receptors whose function is associated with non-self-recognition (Buckley and Rast, 2015). Scavenger receptor type genes also were enriched in our GO and PFAM domain analyses in Cluster V, which comprises genes that show sustained upregulation from larvae to postlarvae to adults. Significant expansions of multi-domain SRCR gene families have occurred in sea urchins, some of which were upregulated in response to immune challenge (Pancer, 2000, 2001; Hibino et al., 2006; Rast et al., 2006). An expanded repertoire of SRCR domain-containing proteins is also encoded by the A. queenslandica genome (Srivastava et al., 2010), and these include conserved pattern recognition receptors that could interact directly with bacterial-derived signals (Hentschel et al., 2012). Evidence for the involvement of SRCR domain-containing proteins in immunity in poriferans includes the expression of membrane-associated SRCR proteins on spherule cells that appear to be involved in cell aggregation in Geodia cydonium (Blumbach et al., 1998). Perhaps even more compelling, an SRCR domain-containing protein is highly upregulated when the sponge Petrosia ficiformis is colonized by symbiotic cyanobacteria (Steindler et al., 2007), and the LMA sponge S. carteri very interestingly has an expanded complement of SRCR-like domains compared to the HMA sponge X. testudinaria (Ryu et al., 2016). Notably, the SRCR factors upregulated in A. queenslandica postlarvae are composed of SRCR repeats that show structural similarity to those of vertebrate DMBT1, which binds directly to bacterial proteins and is linked with immune-related NLR and TLR signaling (Rosenstiel et al., 2007). These multiple lines of evidence from divergent phyla suggest that SRCR proteins could play critical roles in host-microbiome interactions.
Two further immune gene families for which we identify multiple DEG Cluster I genes are the Tnf Receptor-Associated Factor (TRAF) family (6 DEGs), with putative roles in cytokine signaling and activation of the immune response to stress (Arch et al., 1998), and tripartite motif-containing protein family (TRIM) genes (6 DEGs) that are hypothetically involved in pathogen recognition and host defense (Ozato et al., 2008). We also identified several single genes with putative functions of interest, including an AIG1 family protein, orthologs of which confer resistance to bacteria in plants, an anthrax toxin receptor 2 gene, a gram-negative bacteria binding protein that is hypothesized to function as a pattern recognition receptor for LPS from gram-negative bacteria, and a macrophage mannose receptor 1-like that may act as a phagocytic receptor for bacteria (Christophides et al., 2002). Interestingly, the Cluster 1 list also includes several genes involved in autophagy (e.g., cdgsh iron-sulfur domain-containing protein and autophagy-related proteins), which is often upscaled during times of cellular stress (Kroemer et al., 2010).
Not surprisingly, given the major cellular-level body plan reorganization occurring at this time in the sponge life cycle (Degnan et al., 2015), we also see in Cluster 1 the upregulation of: (i) several kinds of extracellular matrix proteins, such as fibropellins; (ii) genes involved in modulation of cell shape and migration, such as filamins and Rho GTPase-activating proteins; and (iii) genes encoding transmembrane receptors that bridge cell-extracellular matrix, such as integrins. These are of interest because a stable symbiosis relies at least in part on the hosts' ability to maintain a suitable abundance and distribution of chosen partners throughout its life cycle (Bright and Bulgheresi, 2010; Douglas, 2014). One way in which animal hosts appear to control their symbiont abundance is by restricting the habitat available to them (Davy et al., 2012; Douglas, 2014), and the substantial cell-level reorganization in postlarvae could provide a means to achieve this.
The A. queenslandica genome encodes other genes whose transcriptional upregulation is expected to help the host recognize and/or respond to diverse bacteria that themselves may have genome-encoded means to shape a host immune system and thus its microbiota (Comstock and Kasper, 2006). In particular, the expression of various catabolic enzymes increases either at the larva to 48 hpi postlarva, or postlarva to adult, transition. These include sulfuric ester hydrolases (e.g., iduronate 2-sulfatase, arylsulfatase) and fucosidases (e.g., alpha-L-fucosidase) that play a role in the break down of glycoproteins, glycolipids, and sulfated molecules, all recognized components of at least some bacteria, as well as cysteine-type endopeptidases (e.g., cathepsins) that degrade proteins. Another upregulated catabolytic enzyme, which is associated with hydrolase activity, is a gram-negative bacteria binding protein that shows most similarity to a protein that displayed (1,3)-beta-d-glucan recognition in Suberites domuncula (Perovic-Ottstadt et al., 2004). Together with other upregulated polysaccharide binding molecules (e.g., jacalin lectin family protein, novel protein vertebrate galectin, macrophage mannose receptor 1-like), these could equip the postlarval sponge with means to recognize both wanted and unwanted microbes, and act accordingly.
Among putatively horizontally-transferred genes in the A. queenslandica (Conaco et al., 2016) that show significant upregulation from larvae to postlarvae, we identified several associated with hydrolase activity, oxidoreductase activity, carbohydrate metabolism and glycogenesis activity; some of these have putative functions in breaking down cellulose, which is interesting given their upregulation at the time that non-feeding larvae, and subsequently postlarvae, are settling and reorganizing their body plan on coralline algae substrates. We also note that HGTs significantly upregulated later, from the 48 hpi postlarval to adult stage, included those annotated as metalloproteases (e.g., Aspzincin-like peptidase m35, extracellular protease), glycogenesis, hydrolase and sulfatase activity, while those encoding extracellular proteases and metalloproteases showed a sustained upregulation across all three of the successive stages. This presence of these HGTs among our potential candidates for animal-bacterial crosstalk raises the possibility that host-bacterial associations in the evolutionary past may help to regulate host-bacterial associations in the ecological present (Degnan, 2014) and calls for molecular biological and experimental approaches to help determine the specific functions of these genes in the A. queenslandica holobiont.
Conclusions and Future Directions
Vertically-inherited symbionts have the potential to affect their hosts during all developmental stages, and their contributions to the ecology and fitness of the host may vary across the life cycle (Yule et al., 2013). The discovery in A. queenslandica of distinct life cycle stages with distinct microbiome compositions provides a framework for a demographic approach that quantifies the effects of symbionts on growth, survival, and even stage-specific behaviors, to characterize the ecological dynamics of symbioses across the host ontogeny. Despite the fidelity and stability that is expected to accompany vertical transmission of core symbionts, we observe an influx of potentially opportunistic bacteria at larval settlement, followed by a return to a more stable state that is accompanied by increased activity of genes encoding immune recognition and response proteins and lysosomal enzymes. Animal hosts may regulate their symbionts via this kind of genome-encoded molecular crosstalk, by restricting habitat space available to symbionts, and perhaps also by restricting rates of symbiont proliferation to match host demand for symbiotic services (Davy et al., 2012; Douglas, 2014). The perturbation of the microbiome that we observe immediately following larval settlement provides a natural experimental system, without need for artificial manipulation, to investigate host-symbiont interactions in regulating microbiome composition and stability. It also provides a means to address whether the specific nature of a larval settlement sites influences microbiome composition, and, if so, are there consequences of this for local adaptation of the holobiont.
Author Contributions
SD, RF, and JB designed the research. RF, KG, and JB prepared the biological material. JB assisted in early stages of analysis of 454 sequence data. MG conducted the 454 sequence data and the CelSeq transcriptome data analyses. RF and KG conducted the TEM and SEM work; RF conducted the FISH experiments. SD, MG, and RF wrote the paper. All authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the facilities, and scientific and technical assistance, of the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland. Thanks to Jabin Watson for assistance with adult sponge collection and microbial cell enrichments. This work was supported by the Australian Research Council under DP110104601 and DP0985995 to SD, and an Australian Postgraduate Award to RF. We are sincerely grateful to Laura Steindler and Lucas Moitinho-Silva, whose thoughtful comments greatly improved this article.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2016.00228/full#supplementary-material
References
Alex, A., and Antunes, A. (2015). Whole genome sequencing of the Symbiont Pseudovibriosp. from the intertidal marine sponge polymastia penicillusrevealed a gene repertoire for host-switching permissive lifestyle. Genome Biol. Evol. 7, 3022–3032. doi: 10.1093/gbe/evv199
Anavy, L., Levin, M., Khair, S., Nakanishi, N., Fernandez-Valverde, S. L., Degnan, B. M., et al. (2014). BLIND ordering of large-scale transcriptomic developmental timecourses. Development 141, 1161–1166. doi: 10.1242/dev.105288
Arch, R. H., Gedrich, R. W., and Thompson, C. B. (1998). Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes Dev. 12, 2821–2830. doi: 10.1101/gad.12.18.2821
Berry, D., Ben Mahfoudh, K., Wagner, M., and Loy, A. (2011). Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl. Environ. Microbiol. 77, 7846–7849. doi: 10.1128/AEM.05220-11
Blanquer, A., Uriz, M. J., and Galand, P. E. (2013). Removing environmental sources of variation to gain insight on symbionts vs. transient microbes in high and low microbial abundance sponges. Environ. Microbiol. 15, 3008–3019. doi: 10.1111/1462-2920.12261
Blumbach, B., Pancer, Z., Pancer, Z., Diehl-Seifert, B., Diehl-Seifert, B., Müller, I., et al. (1998). The putative sponge aggregation receptor. Isolation and characterization of a molecule composed of scavenger receptor cysteine-rich domains and short consensus repeats. J. Cell. Sci. 111(Pt 17), 2635–2644.
Bright, M., and Bulgheresi, S. (2010). A complex journey: transmission of microbial symbionts. Nat. Rev. Micro. 8, 218–230. doi: 10.1038/nrmicro2262
Britstein, M., Devescovi, G., Handley, K. M., Malik, A., Haber, M., Saurav, K., et al. (2016). A new N-acyl homoserine lactone synthase in an uncultured symbiont of the red sea sponge Theonella swinhoei. Appl. Environ. Microbiol. 82, 1274–1285. doi: 10.1128/AEM.03111-15
Buckley, K. M., and Rast, J. P. (2015). Diversity of animal immune receptors and the origins of recognition complexity in the deuterostomes. Dev. Comp. Immunol. 49, 179–189. doi: 10.1016/j.dci.2014.10.013
Bulgheresi, S. (2011). “Microbial symbiont transmission: basic principles and dark sides,” in Beneficial Microorganisms in Multicellular Life Forms, eds E. Rosenberg and U. Gophna (Berlin; Heidelberg: Springer-Verlag), 299–311. doi: 10.1007/978-3-642-21680-0_22
Burgsdorf, I., Erwin, P. M., and López-Legentil, S. (2015a). Biogeography rather than association with cyanobacteria structures symbiotic microbial communities in the marine sponge Petrosia ficiformis. Front. Microbiol. 5:529. doi: 10.3389/fmicb.2014.00529
Burgsdorf, I., Slaby, B. M., Handley, K. M., Haber, M., Blom, J., Marshall, C. W., et al. (2015b). Lifestyle evolution in cyanobacterial symbionts of sponges. MBio 6, e00391–e00315. doi: 10.1128/mBio.00391-15
Caporaso, J. G., Bittinger, K., Bushman, F. D., DeSantis, T. Z., Andersen, G. L., and Knight, R. (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cerveny, L., Straskova, A., Dankova, V., Hartlova, A., Ceckova, M., Staud, F., et al. (2013). Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect. Immun. 81, 629–635. doi: 10.1128/IAI.01035-12
Chandran, D., Tai, Y. C., Hather, G., Dewdney, J., Denoux, C., Burgess, D. G., et al. (2009). Temporal global expression data reveal known and novel salicylate-impacted processes and regulators mediating powdery mildew growth and reproduction on Arabidopsis. Plant Physiology 149, 1435–1451. doi: 10.1104/pp.108.132985
Christophides, G. K., Zdobnov, E., Barillas-Mury, C., Birney, E., Blandin, S., Blass, C., et al. (2002). Immunity-related genes and gene families in anopheles gambiae. Science 298, 159–165. doi: 10.1126/science.1077136
Comstock, L. E., and Kasper, D. L. (2006). Bacterial glycans: key mediators of diverse host immune responses. Cell 126, 847–850. doi: 10.1016/j.cell.2006.08.021
Conaco, C., Tsoulfas, P., Sakarya, O., Dolan, A., Werren, J., and Kosik, K. S. (2016). Detection of prokaryotic genes in the Amphimedon queenslandica genome. PLoS ONE 11:e0151092. doi: 10.1371/journal.pone.0151092
Conesa, A., Götz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Croué, J., West, N. J., Escande, M.-L., Intertaglia, L., Lebaron, P., and Suzuki, M. T. (2013). A single betaproteobacterium dominates the microbial community of the crambescidine-containing sponge Crambe crambe. Sci. Rep. 3:2583. doi: 10.1038/srep02583
Davy, S. K., Allemand, D., and Weis, V. M. (2012). Cell biology of cnidarian-dinoflagellate symbiosis. Microbiol. Mol. Biol. Rev. 76, 229–261. doi: 10.1128/MMBR.05014-11
de Caralt, S., Uriz, M. J., and Wijffels, R. H. (2007). Vertical transmission and successive location of symbiotic bacteria during embryo development and larva formation in Corticium candelabrum (Porifera: Demospongiae). J. Mar. Biol. Ass. U.K. 87, 1693–1699. doi: 10.1017/S0025315407056846
Degnan, B. M., Adamska, M., Craigie, A., Degnan, S. M., Fahey, B., Gauthier, M., et al. (2008). The demosponge Amphimedon queenslandica: reconstructing the ancestral metazoan genome and deciphering the origin of animal multicellularity. CSH Protoc. 2008:pdb.emo108. doi: 10.1101/pdb.emo108
Degnan, B. M., Adamska, M., Richards, G. S., Larroux, C., Leininger, S., Bergum, B., et al. (2015). “Porifera,” in Evolutionary Developmental Biology of Invertebrates 1, ed A. Wanninger (Vienna: Springer Vienna), 65–106.
Degnan, B. M., Leys, S. P., and Larroux, C. (2005). Sponge development and antiquity of animal pattern formation. Integr. Comp. Biol. 45, 335–341. doi: 10.1093/icb/45.2.335
Degnan, S. M. (2014). Think laterally: horizontal gene transfer from symbiotic microbes may extend the phenotype of marine sessile hosts. Front. Microbiol. 5:638. doi: 10.3389/fmicb.2014.00638
Degnan, S. M., and Degnan, B. M. (2010). The initiation of metamorphosis as an ancient polyphenic trait and its role in metazoan life-cycle evolution. Philos. Trans. R. Soc. B Biol. Sci. 365, 641–651. doi: 10.1098/rstb.2009.0248
de Goeij, J. M., van Oevelen, D., Vermeij, M. J. A., Osinga, R., Middelburg, J. J., de Goeij, A. F. P. M., et al. (2013). Surviving in a marine desert: the sponge loop retains resources within coral reefs. Science 342, 108–110. doi: 10.1126/science.1241981
Docampo, R., Ulrich, P., and Moreno, S. N. J. (2010). Evolution of acidocalcisomes and their role in polyphosphate storage and osmoregulation in eukaryotic microbes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 365, 775–784. doi: 10.1098/rstb.2009.0179
Douglas, A. E. (2014). Symbiosis as a general principle in eukaryotic evolution. Cold Spring Harb. Perspect. Biol. 6:a016113. doi: 10.1101/cshperspect.a016113
Easson, C. G., and Thacker, R. W. (2014). Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Front. Microbiol. 5:532. doi: 10.3389/fmicb.2014.00532
Ebert, D. (2013). The epidemiology and evolution of symbionts with mixed-mode transmission. Annu. Rev. Ecol. Evol. Syst. 44, 623–643. doi: 10.1146/annurev-ecolsys-032513-100555
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Enticknap, J. J., Kelly, M., Peraud, O., and Hill, R. T. (2006). Characterization of a culturable alphaproteobacterial symbiont common to many marine sponges and evidence for vertical transmission via sponge larvae. Appl. Environ. Microbiol. 72, 3724–3732. doi: 10.1128/AEM.72.5.3724-3732.2006
Ereskovsky, A., Gonobobleva, E., and Vishnyakov, A. (2005). Morphological evidence for vertical transmission of symbiotic bacteria in the viviparous sponge Halisarca dujardini Johnston (Porifera, Demospongiae, Halisarcida). Mar. Biol. 146, 869–875. doi: 10.1007/s00227-004-1489-1
Ereskovsky, A. V., and Tokina, D. B. (2004). Morphology and fine structure of the swimming larvae of Ircinia oros (Porifera, Demospongiae, Dictyoceratida). Invertebr. Reprod. Dev. 45, 137–150. doi: 10.1080/07924259.2004.9652583
Erpenbeck, D., Voigt, O., Adamski, M., Adamska, M., Hooper, J., Wörheide, G., et al. (2007). Mitochondrial diversity of early-branching metazoa is revealed by the complete mt genome of a haplosclerid demosponge. Mol. Biol. Evol. 24, 19–22. doi: 10.1093/molbev/msl154
Erwin, P. M., Coma, R., López-Sendino, P., Serrano, E., and Ribes, M. (2015). Stable symbionts across the HMA-LMA dichotomy: low seasonal and interannual variation in sponge-associated bacteria from taxonomically diverse hosts. FEMS Microbiol. Ecol. 91, fiv115. doi: 10.1093/femsec/fiv115
Erwin, P. M., Pita, L., López-Legentil, S., and Turon, X. (2012). Stability of sponge-associated bacteria over large seasonal shifts in temperature and irradiance. Appl. Environ. Microbiol. 78, 7358–7368. doi: 10.1128/AEM.02035-12
Erwin, P. M., and Thacker, R. W. (2008). Cryptic diversity of the symbiotic cyanobacterium Synechococcus spongiarum among sponge hosts. Mol. Ecol. 17, 2937–2947. doi: 10.1111/j.1365-294X.2008.03808.x
Fan, L., Reynolds, D., Liu, M., Stark, M., Kjelleberg, S., Webster, N. S., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. 109, E1878–E1887. doi: 10.1073/pnas.1203287109
Fernandez-Valverde, S. L., Calcino, A. D., and Degnan, B. M. (2015). Deep developmental transcriptome sequencing uncovers numerous new genes and enhances gene annotation in the sponge Amphimedon queenslandica. BMC Genomics 16, 720–711. doi: 10.1186/s12864-015-1588-z
Frank, S. A. (1996). Host control of symbiont transmission: the separation of symbionts into germ and soma. Am. Nat. 148, 1113–1124. doi: 10.1086/285974
Gauthier, M. E. A., Watson, J. R., and Degnan, S. M. (2016). Draft genomes shed light on the dual bacterial symbiosis that dominates the microbiome of the coral reef sponge Amphimedon queenslandica. Front. Mar. Sci. 3:196. doi: 10.3389/fmars.2016.00196
Gihring, T. M., Green, S. J., and Schadt, C. W. (2012). Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ. Microbiol. 14, 285–290. doi: 10.1111/j.1462-2920.2011.02550.x
Giles, E. C., Kamke, J., Moitinho-Silva, L., Taylor, M. W., Hentschel, U., Ravasi, T., et al. (2012). Bacterial community profiles in low microbial abundance sponges. FEMS Microbiol. Ecol. 83, 232–241. doi: 10.1111/j.1574-6941.2012.01467.x
Gloeckner, V., Hentschel, U., Ereskovsky, A., and Schmitt, S. (2013). Unique and species-specific microbial communities in Oscarella lobularis and other Mediterranean Oscarella species (Porifera: Homoscleromorpha). Mar. Biol. 160, 781–791. doi: 10.1007/s00227-012-2133-0
Gloeckner, V., Lindquist, N., Schmitt, S., and Hentschel, U. (2012). Ectyoplasia ferox, an experimentally tractable model for vertical microbial transmission in marine sponges. Microb. Ecol. 65, 462–474. doi: 10.1007/s00248-012-0142-7
Gloeckner, V., Wehrl, M., Moitinho-Silva, L., Gernert, C., Schupp, P., Pawlik, J. R., et al. (2014). The HMA-LMA dichotomy revisited: an electron microscopical survey of 56 sponge species. Biol. Bull. 227, 78–88. doi: 10.1086/BBLv227n1p78
Gundel, P. E., Rudgers, J. A., and Ghersa, C. M. (2011). Incorporating the process of vertical transmission into understanding of host—symbiont dynamics. Oikos 120, 1121–1128. doi: 10.1111/j.1600-0706.2011.19299.x
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., et al. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504. doi: 10.1101/gr.112730.110
Hashimshony, T., Wagner, F., Sher, N., and Yanai, I. (2012). CEL-Seq: single-Cell RNA-seq by multiplexed linear Amplification. 2, 666–673. doi: 10.1016/j.celrep.2012.08.003
Hentschel, U., Fieseler, L., Wehrl, M., Gernert, C., Steinert, M., Hacker, J., et al. (2003). Microbial diversity of marine sponges. Prog. Mol. Subcell. Biol. 37, 59–88. doi: 10.1007/978-3-642-55519-0_3
Hentschel, U., Hopke, J., Horn, M., Friedrich, A. B., Wagner, M., Hacker, J., et al. (2002). Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 68, 4431–4440. doi: 10.1128/AEM.68.9.4431-4440.2002
Hentschel, U., Piel, J., Degnan, S. M., and Taylor, M. W. (2012). Genomic insights into the marine sponge microbiome. Nat. Rev. Micro. 10, 641–654. doi: 10.1038/nrmicro2839
Hibino, T., Loza-Coll, M., Messier, C., Majeske, A. J., Cohen, A. H., Terwilliger, D. P., et al. (2006). The immune gene repertoire encoded in the purple sea urchin genome. Dev. Biol. 300, 349–365. doi: 10.1016/j.ydbio.2006.08.065
Hook, M., McGavin, M. J., Switalski, L. M., Raja, R., Raucci, G., Lindgren, P. E., et al. (1990). Interactions of bacteria with extracellular matrix proteins. Cell Differ. Dev. 32, 433–438. doi: 10.1016/0922-3371(90)90060-A
Hooper, J., and Van Soest, R. (2006). A new species of Amphimedon (Porifera, Demospongiae, Haplosclerida, Niphatidae) from the Capricorn-Bunker Group of Islands, Great Barrier Reef, Australia: target species for the ‘sponge genome project’. Zootaxa 1314, 31–39.
Kroemer, G., Mariño, G., and Levine, B. (2010). Autophagy and the integrated stress response. Mol. Cell 40, 280–293. doi: 10.1016/j.molcel.2010.09.023
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lawry, N., Jani, V., and Jensen, T. (1981). Identification of the sulfur inclusion body in Beggiatoa alba B18LD by energy-dispersive X-ray microanalysis. Curr. Microbiol. 6, 71–74. doi: 10.1007/BF01569006
Lee, O. O., Chui, P. Y., Wong, Y. H., Pawlik, J. R., and Qian, P. Y. (2009). Evidence for vertical transmission of bacterial symbionts from adult to embryo in the Caribbean sponge svenzea zeai. Appl. Environ. Microbiol. 75, 6147–6156. doi: 10.1128/AEM.00023-09
Levin, M., Anavy, L., Cole, A. G., Winter, E., Mostov, N., Khair, S., et al. (2016). The mid-developmental transition and the evolution of animal body plans. Nature 531, 637–641. doi: 10.1038/nature16994
Leys, S. P., and Degnan, B. M. (2002). Embryogenesis and metamorphosis in a Haplosclerid Demosponge: gastrulation and transdifferentiation of larval ciliated cells to choanocytes. Invertebr. Biol. 121, 171–189. doi: 10.1111/j.1744-7410.2002.tb00058.x
Leys, S. P., Larroux, C., Gauthier, M., Adamska, M., Fahey, B., Richards, G. S., et al. (2008). Isolation of amphimedon developmental material. Cold Spring Harb. Protoc. 2008:pdb.prot5095. doi: 10.1101/pdb.prot5095
Leys, S. P., and Degnan, B. M. B. (2001). Cytological basis of photoresponsive behavior in a sponge larva. Biol. Bull 201, 323–338. doi: 10.2307/1543611
Liu, M. Y., Kjelleberg, S., and Thomas, T. (2010). Functional genomic analysis of an uncultured δ-proteobacterium in the sponge Cymbastela concentrica. ISME J. 5, 427–435. doi: 10.1038/ismej.2010.139
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. bioRxiv. doi: 10.1101/002832
Maddison, W. P., and Maddison, D. R. (2014). Mesquite: A Modular System for Evolutionary Analysis. Version 3.01. Available online at: mesquiteproject.org
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. doi: 10.1093/bioinformatics/bti551
Maldonado, M. (2007). Intergenerational transmission of symbiotic bacteria in oviparous and viviparous demosponges, with emphasis on intracytoplasmically-compartmented bacterial types. J. Mar. Biol. Ass. 87, 1701–1713. doi: 10.1017/S0025315407058080
McDonald, K., Morphew, M., Verkade, P., and Müller-Reichert, T. (2007). “Recent advances in high-pressure freezing,” in Electron Microscopy Methods in Molecular Biology, ed J. Kuo (Totowa, NJ: Humana Press), 143–173.
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C. G., Carey, H. V., Domazet-Loso, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. 110, 3229–3236. doi: 10.1073/pnas.1218525110
Nguyen, M. T. H. D., Liu, M., and Thomas, T. (2014). Ankyrin-repeat proteins from sponge symbionts modulate amoebal phagocytosis. Mol. Ecol. 23, 1635–1645. doi: 10.1111/mec.12384
Nyholm, S. V., and Graf, J. (2012). Knowing your friends: invertebrate innate immunity fosters beneficial bacterial symbioses. Nat. Rev. Micro. 10, 815–827. doi: 10.1038/nrmicro2894
Oren, M., Steindler, L., and Ilan, M. (2005). Transmission, plasticity and the molecular identification of cyanobacterial symbionts in the Red Sea sponge Diacarnus erythraenus. Mar. Biol. 148, 35–41. doi: 10.1007/s00227-005-0064-8
Ozato, K., Shin, D.-M., Chang, T.-H., and Morse, H. C. (2008). TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860. doi: 10.1038/nri2413
Pancer, Z. (2000). Dynamic expression of multiple scavenger receptor cysteine-rich genes in coelomocytes of the purple sea urchin. Proc. Natl. Acad. Sci. U.S.A. 97, 13156–13161. doi: 10.1073/pnas.230096397
Pancer, Z. (2001). Individual-specific repertoires of immune cells SRCR receptors in the purple sea urchin (S. Purpuratus). Adv. Exp. Med. Biol. 484, 31–40. doi: 10.1007/978-1-4615-1291-2_3
Perovic-Ottstadt, S., Adell, T., Proksch, P., Wiens, M., Korzhev, M., Gamulin, V., et al. (2004). A (1 → 3)-β-d-glucan recognition protein from the sponge Suberites domuncula. Eur. J. Biochem. 271, 1924–1937. doi: 10.1111/j.1432-1033.2004.04102.x
Poppell, E., Weisz, J., Spicer, L., Massaro, A., Hill, A., and Hill, M. (2014). Sponge heterotrophic capacity and bacterial community structure in high- and low-microbial abundance sponges. Mar. Ecol. 35, 414–424. doi: 10.1111/maec.12098
Rast, J. P., Smith, L. C., Loza-Coll, M., Hibino, T., and Litman, G. W. (2006). Genomic insights into the immune system of the Sea Urchin. Science 314, 952–956. doi: 10.1126/science.1134301
Robertson, L., and Kuenen, J. G. (2006). “The colorless sulfur bacteria,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 985–1011.
Rosenstiel, P., Sina, C., End, C., Renner, M., Lyer, S., Till, A., et al. (2007). Regulation of DMBT1 via NOD2 and TLR4 in intestinal epithelial cells modulates bacterial recognition and invasion. J. Immunol. 178, 8203–8211. doi: 10.4049/jimmunol.178.12.8203
Russell, J. A., and Moran, N. A. (2005). Horizontal transfer of bacterial symbionts: heritability and fitness effects in a novel aphid host. Appl. Environ. Microbiol. 71, 7987–7994. doi: 10.1128/AEM.71.12.7987-7994.2005
Ryu, T., Seridi, L., Moitinho-Silva, L., Oates, M., Liew, Y. J., Mavromatis, C., et al. (2016). Hologenome analysis of two marine sponges with different microbiomes. BMC Genomics 17:158. doi: 10.1186/s12864-016-2501-0
Schmitt, S., Angermeier, H., Schiller, R., Lindquist, N., and Hentschel, U. (2008). Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Appl. Environ. Microbiol. 74, 7694–7708. doi: 10.1128/AEM.00878-08
Schmitt, S., Tsai, P., Bell, J., Fromont, J., Ilan, M., Lindquist, N., et al. (2011). Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6, 564–576. doi: 10.1038/ismej.2011.116
Schmitt, S., Wehrl, M., Lindquist, N., and Weisz, J. B. (2007). Morphological and Molecular Analyses of Microorganisms in Caribbean Reef Adult Sponges and in Corresponding Reproductive Material. Faculty Publications. Published Version. Submission 1. Available online at: http://digitalcommons.linfield.edu/biolfac_pubs/1
Sharp, K. H., Eam, B., Faulkner, D. J., and Haygood, M. G. (2007). Vertical transmission of diverse microbes in the tropical sponge Corticium sp. Appl. Environ. Microbiol. 73, 622–629. doi: 10.1128/AEM.01493-06
Siegl, A., Kamke, J., Hochmuth, T., Piel, J., Richter, M., Liang, C., et al. (2011). Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5, 61–70. doi: 10.1038/ismej.2010.95
Simister, R. L., Deines, P., Botté, E. S., Webster, N. S., and Taylor, M. W. (2012). Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ. Microbiol. 14, 517–524. doi: 10.1111/j.1462-2920.2011.02664.x
Srivastava, M., Simakov, O., Chapman, J., Fahey, B., Gauthier, M. E. A., Mitros, T., et al. (2010). The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466, 720–726. doi: 10.1038/nature09201
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Steger, D., Ettinger-Epstein, P., Whalan, S., Hentschel, U., de Nys, R., Wagner, M., et al. (2008). Diversity and mode of transmission of ammonia-oxidizing archaea in marine sponges. Environ. Microbiol. 10, 1087–1094. doi: 10.1111/j.1462-2920.2007.01515.x
Steindler, L., Schuster, S., Ilan, M., Avni, A., Cerrano, C., and Beer, S. (2007). Differential gene expression in a marine sponge in relation to its symbiotic state. Mar. Biotechnol. 9, 543–549. doi: 10.1007/s10126-007-9024-2
Supek, F., Bosnjak, M., Škunca, N., and Šmuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6:e21800. doi: 10.1371/journal.pone.0021800
Taylor, M. W., Hill, R. T., Piel, J., Thacker, R. W., and Hentschel, U. (2007). Soaking it up: the complex lives of marine sponges and their microbial associates. ISME J. 1, 187–190. doi: 10.1038/ismej.2007.32
Taylor, M. W., Schupp, P. J., Dahllöf, I., Kjelleberg, S., and Steinberg, P. D. (2004). Host specificity in marine sponge-associated bacteria, and potential implications for marine microbial diversity. Environ. Microbiol. 6, 121–130. doi: 10.1046/j.1462-2920.2003.00545.x
Taylor, M. W., Tsai, P., Simister, R. L., Deines, P., Botte, E., Ericson, G., et al. (2013). ‘Sponge-specific’ bacteria are widespread (but rare) in diverse marine environments. ISME J. 7, 438–443. doi: 10.1038/ismej.2012.111
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7:11870. doi: 10.1038/ncomms11870
Thomas, T., Rusch, D., DeMaere, M. Z., Yung, P. Y., Lewis, M., Halpern, A., et al. (2010). Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 4, 1557–1567. doi: 10.1038/ismej.2010.74
Tian, R.-M., Wang, Y., Bougouffa, S., Gao, Z.-M., Cai, L., Bajic, V., et al. (2014). Genomic analysis reveals versatile heterotrophic capacity of a potentially symbiotic sulfur-oxidizing bacterium in sponge. Environ. Microbiol. 16, 3548–3561. doi: 10.1111/1462-2920.12586
Ueda, N., Richards, G. S., Degnan, B. M., Kranz, A., Adamska, M., and Croll, R. (in press). An ancient role for nitric oxide in regulating the animal pelagobenthic life cycle: evidence from a marine sponge. Sci. Rep.
Virgin, H. W., and Levine, B. (2009). Autophagy genes in immunity. Nat. Immunol. 10, 461–470. doi: 10.1038/ni.1726
Wanger, G., Onstott, T. C., and Southam, G. (2008). Stars of the terrestrial deep subsurface: a novel “star-shaped” bacterial morphotype from a South African platinum mine. Geobiology 6, 325–330. doi: 10.1111/j.1472-4669.2008.00163.x
Webster, N. S., and Hill, R. T. (2001). The culturable microbial community of the Great Barrier Reef sponge Rhopaloeides odorabile is dominated by an α-Proteobacterium. Mar. Biol. 138, 843–851. doi: 10.1007/s002270000503
Webster, N. S., Taylor, M. W., Behnam, F., Lücker, S., Rattei, T., Whalan, S., et al. (2010). Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070–2082. doi: 10.1111/j.1462-2920.2009.02065.x
Webster, N. S., Wilson, K. J., Blackall, L. L., and Hill, R. T. (2001). Phylogenetic diversity of bacteria associated with the marine sponge Rhopaloeides odorabile. Appl. Environ. Microbiol. 67, 434–444. doi: 10.1128/AEM.67.1.434-444.2001
Wendt, K. D., Jensen, C. A., Tindall, R., and Katz, M. L. (2004). Comparison of conventional and microwave-assisted processing of mouse retinas for transmission electron microscopy. J. Microsc. 214, 80–88. doi: 10.1111/j.0022-2720.2004.01310.x
Wiens, G. D., and Glenney, G. W. (2011). Origin and evolution of TNF and TNF receptor superfamilies. Dev. Comp. Immunol. 35, 1324–1335. doi: 10.1016/j.dci.2011.03.031
Wilson, M. C., Mori, T., Rückert, C., Uria, A. R., Helf, M. J., Takada, K., et al. (2014). An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506, 58–62. doi: 10.1038/nature12959
Yang, Z., and Li, Z. (2012). Spatial distribution of prokaryotic symbionts and ammoxidation, denitrifier bacteria in marine sponge Astrosclera willeyana. Sci. Rep. 2:528. doi: 10.1038/srep00528
Yule, K. M., Miller, T., and Rudgers, J. A. (2013). Costs, benefits, and loss of vertically transmitted symbionts affect host population dynamics. Oikos 122, 1512–1520. doi: 10.1111/j.1600-0706.2012.00229.x
Keywords: marine invertebrate microbiome, larval settlement, Porifera, Chromatiales, vertical inheritance
Citation: Fieth RA, Gauthier M-EA, Bayes J, Green KM and Degnan SM (2016) Ontogenetic Changes in the Bacterial Symbiont Community of the Tropical Demosponge Amphimedon queenslandica: Metamorphosis Is a New Beginning. Front. Mar. Sci. 3:228. doi: 10.3389/fmars.2016.00228
Received: 28 June 2016; Accepted: 31 October 2016;
Published: 15 November 2016.
Edited by:
Ute Hentschel Humeida, GEOMAR Helmholtz Centre for Ocean Research Kiel, GermanyReviewed by:
Laura Steindler, University of Haifa, IsraelLucas Moitinho-Silva, University of New South Wales, Australia
Copyright © 2016 Fieth, Gauthier, Bayes, Green and Degnan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandie M. Degnan, cy5kZWduYW5AdXEuZWR1LmF1