 Agnes K. M. Weiner1,2*
Agnes K. M. Weiner1,2* Raphael Morard1
Raphael Morard1 Manuel F. G. Weinkauf1,3
Manuel F. G. Weinkauf1,3 Kate F. Darling4,5Aurore André6Frédéric Quillévéré7
Kate F. Darling4,5Aurore André6Frédéric Quillévéré7 Yurika Ujiie8
Yurika Ujiie8 Christophe J. Douady9,10Colomban de Vargas11,12
Christophe J. Douady9,10Colomban de Vargas11,12 Michal Kucera1
Michal Kucera1- 1MARUM Center for Marine Environmental Sciences, University of Bremen, Bremen, Germany
- 2Department of Marine Biodiversity Research, Japan Agency for Marine Earth Science and Technology (JAMSTEC), Yokosuka, Japan
- 3Department of Earth Sciences, University of Geneva, Geneva, Switzerland
- 4School of GeoSciences, University of Edinburgh, Edinburgh, UK
- 5School of Geography and GeoSciences, University of St. Andrews, St. Andrews, UK
- 6UFR Sciences Exactes et Naturelles, Université de Reims-Champagne-Ardenne, Reims, France
- 7Centre Nationnal de la Recherche Scientifique (CNRS), École Normale Supérieure (ENS) de Lyon, University of Lyon, Villeurbanne, France
- 8Center for Advanced Marine Core Research, Kochi University, Kochi, Japan
- 9Laboratoire d'Ecologie des Hydrosystémes Naturels et Anthropisés, Univ Lyon, Université Lyon 1, CNRS UMR 5023, ENTPE, Villeurbanne, France
- 10Institut Universitaire de France, Paris, France
- 11Centre Nationnal de la Recherche Scientifique (CNRS), UMR 7144, Station Biologique de Roscoff, Roscoff, France
- 12Station Biologique de Roscoff, Sorbonne Universités, UPMC Univ Paris 06, UMR 7144, Roscoff, France
Single-cell genetic analysis is an essential method to investigate the biodiversity and evolutionary ecology of marine protists. In protist groups that do not reproduce under laboratory conditions, this approach provides the only means to directly associate molecular sequences with cell morphology. The resulting unambiguous taxonomic identification of the DNA sequences is a prerequisite for barcoding and analyses of environmental metagenomic data. Extensive single-cell genetic studies have been carried out on planktonic foraminifera over the past 20 years to elucidate their phylogeny, cryptic diversity, biogeography, and the relationship between genetic and morphological variability. In the course of these investigations, it has become evident that genetic analysis at the individual specimen level is confronted by innumerable challenges ranging from the negligible amount of DNA present in the single cell to the substantial amount of DNA contamination introduced by endosymbionts or food particles. Consequently, a range of methods has been developed and applied throughout the years for the genetic analysis of planktonic foraminifera in order to enhance DNA amplification success rates. Yet, the description of these methods in the literature rarely occurred with equivalent levels of detail and the different approaches have never been compared in terms of their efficiency and reproducibility. Here, aiming at a standardization of methods, we provide a comprehensive review of all methods that have been employed for the single-cell genetic analysis of planktonic foraminifera. We compile data on success rates of DNA amplification and use these to evaluate the effects of key parameters associated with the methods of sample collection, storage and extraction of single-cell DNA. We show that the chosen methods influence the success rates of single-cell genetic studies, but the differences between them are not sufficient to hinder comparisons between studies carried out by different methods. The review thus not only provides a comprehensive reference with guidelines for future genetic studies on foraminifera, but it also establishes an important benchmark for investigations using existing single-cell datasets. The methods are widely applicable and the review may help to establish similar standard principles for their utilization in other protist groups.
Introduction
Molecular characterization of marine unicellular eukaryotes using a single-cell approach has provided invaluable insights into evolutionary and ecological processes at play within their populations. This includes studies in phylogenetics, (cryptic) diversity, speciation, organismal interactions, biogeography, and environmental adaptations (e.g., Heywood et al., 2011; Yoon et al., 2011). The term “single-cell approach” refers to a method, which involves the extraction, amplification and sequencing of DNA from an individual living unicellular organism. Using this approach in marine protists with skeletal frameworks, DNA sequences can be directly associated with morphology, allowing the revision of species limits (e.g., Harper et al., 2009) and discovering the extent of cryptic diversity within morphologically defined species (e.g., de Vargas et al., 1999, 2004). The application of a single-cell approach is a necessity in all groups of protists that cannot be cultured to the stage of reproduction in the laboratory. Likewise, it has to be applied for protists that only reproduce sexually, since this precludes establishing clonal strains for obtaining generous quantities of DNA for genetic analysis.
Planktonic foraminifera represent an excellent model group to study the evolution and ecology of marine microplankton. These exclusively marine protists are globally distributed in the world ocean and their calcite shells are well preserved in deep-sea sediments. Their continuous fossil record allows the tracking of speciation and extinction events and the evolution and phylogenetic relationships of the group are therefore well known (Aze et al., 2011). Due to the high number of morphological features of their calcite shells, a comprehensive morpho-taxonomy was established, which resolves the entire morphological diversity within the group (e.g., Hemleben et al., 1989).
The first molecular genetic studies were carried out on planktonic foraminifera in the 1990s, with the aim to validate their phylogenetic relationships at the molecular level (Darling et al., 1996a; de Vargas et al., 1997). Applying a single-cell approach proved particularly valuable, since planktonic foraminifera are thought to reproduce only sexually (Hemleben et al., 1989) and so far have never been observed to reproduce in culture. Using the small subunit ribosomal RNA gene (SSU rDNA) as a marker, hidden genetic diversity was discovered within established morphospecies (Huber et al., 1997; de Vargas et al., 1999). This triggered a series of subsequent studies that screened morphospecies for their cryptic diversity (e.g., Darling et al., 1999; de Vargas et al., 1999; Aurahs et al., 2009a; Morard et al., 2009, 2011; Weiner et al., 2012, 2014; André et al., 2013). Planktonic foraminifera are thus a prime example of a group with fully resolved morphological taxonomy that could be tested by genetic information (Aurahs et al., 2009b) and where the extent of hidden genetic diversity could be established in great detail (Darling and Wade, 2008). Since the development of DNA extraction methods which leave the calcite shell intact, the DNA sequences can be directly associated with the morphology of the same individual, establishing a definitive link between genetic diversity and subtle morphological variability (e.g., Morard et al., 2009, 2011; Quillévéré et al., 2013). In some cases, the combination of genetics and morphology has even led to a revision of the original morphological taxonomy (Darling et al., 2006; Aurahs et al., 2011; Weiner et al., 2015).
These single-cell molecular studies have further revealed a detailed picture of the distribution patterns of the cryptic species of planktonic foraminifera, enabling comparison of the largely globally distributed morphospecies with the more restricted and ecologically specialized biogeography of their cryptic counterparts (e.g., de Vargas et al., 1999; Darling et al., 2007; Aurahs et al., 2009a; Morard et al., 2011; Seears et al., 2012; Weiner et al., 2012, 2014). This approach provides a high resolution perspective for the construction of hypotheses on species dispersal, gene flow between populations and potential speciation mechanisms in the open ocean (de Vargas et al., 1999; Darling et al., 2000; Weiner et al., 2012, 2014).
In recent years, screening of planktonic foraminifera diversity and distribution has become feasible on a much larger scale by using high throughput sequencing (HTS) of environmental samples, without the time-consuming isolation of single individuals (de Vargas et al., 2015). However, in order to avoid over-interpretation of sequence differences caused by sequencing errors or intra-individual variability, interpretation of HTS studies relies on the existence of a comprehensive sequence database that allows identification of HTS sequences with a high taxonomic resolution (Guillou et al., 2013; Yilmaz et al., 2014). Such a combined sequence and taxon database can only be established when based on sequences obtained by using the single-cell approach, where an accurate taxonomic description of molecular sequences can be obtained (Morard et al., 2015).
Since the advent of molecular studies on planktonic foraminifera, the methods used to recover genetic information from individual specimens have progressively improved. From the start, planktonic foraminifera have proved to be difficult target organisms with regard to both DNA yield and amplification success. In addition, one of the greatest challenges was in avoiding contamination by exogenous biological material. Planktonic foraminifera are associated with algal symbionts, parasites, and food particles, which occur on the surface as well as within the shell (Hemleben et al., 1989). Since planktonic foraminifera cannot be cultured in axenic conditions, their DNA extracts are often contaminated by undesired alien DNA, which masks the foraminiferal signal in gene amplification. This impeded early attempts to isolate foraminifera DNA, and continues to affect the success rates of foraminiferal genetic studies. Consequently, new methods have been developed and optimized to allow a more efficient collection of specimens and the preservation and extraction of DNA without dissolution of the shell. Unfortunately, the descriptions of the different methods are scattered throughout the literature, are inconsistent in detail and their efficiency has not been formally evaluated.
Following a large multi-laboratory effort to merge single-cell sequence data into a comprehensive database (Morard et al., 2015), we realized a need to review the different methodologies developed throughout the era of single-cell molecular analysis of planktonic foraminifera and to provide guidelines for method standardization for future research. To this end, we review all such methods and evaluate them using a dataset on success rates obtained in multiple laboratories over two decades, reflecting a range of methodological parameters. This dataset allows a first-order quantitative evaluation of factors affecting DNA amplification success rates of extractions from single planktonic foraminiferal specimens. The methods we present and evaluate could potentially be utilized for other protists and serve as an example of best practice. Specifically, we have focused on the issue of compatibility of data obtained from different methods and evaluated specimen degradation during recovery, transport and storage of the samples. In this way, we provide a benchmark for future studies on planktonic foraminifera, as well as a useful resource for molecular studies of other protist groups.
Compilation of Methods
In the following sections we present all methods that have been developed and applied for (1) sampling, isolation and preservation of planktonic foraminifera, (2) extraction of DNA from single specimens and (3) their genetic analysis (Figures 1, 2). The methods are shortly introduced in the main text and additionally, in the Supplementary Material we provide standardized protocols with a detail well above that typically shown in the Materials and Methods sections of research papers.
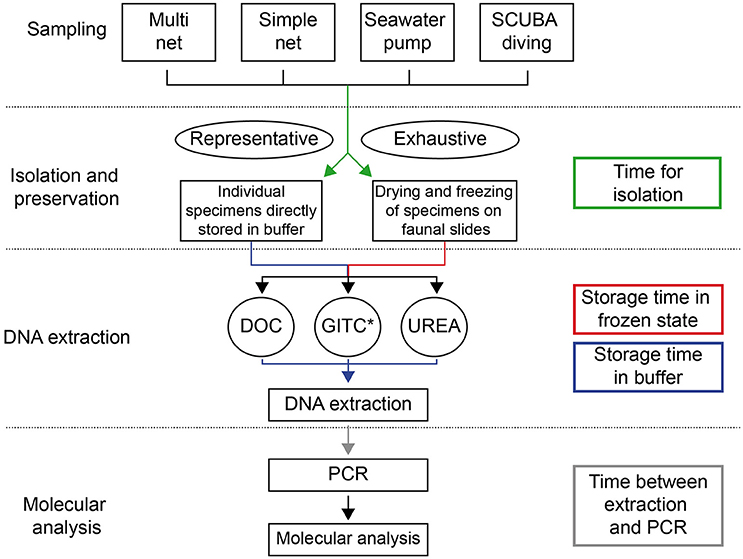
Figure 1. Methodological pathway for the genetic analysis of planktonic foraminifera, representing the different existing methods and their linkage. The processing/storage times between different steps, which potentially influence success rates of DNA amplification, are represented by colored arrows.

Figure 2. Photographs of the methodological pathway for sampling, isolation, preservation and culture maintenance of planktonic foraminifera. The linkage between the different methods is represented by connecting arrows. Section A shows the four different ways of collecting planktonic foraminifera: multiple-closing net (A1), simple plankton net (A2), seawater pump (A3) and scuba diving (A4). Section B depicts the isolation of specimens from the bulk plankton sample (B1–3) by showing them in various stages of cleaning with and without attached contaminants (B4–6). Photographs of section C show the culture maintenance of planktonic foraminifera: culture vials in water baths with flow-through system (C1), living individual of Hastigerina pelagica (C2), individual of Trilobatus sacculifer feeding on an Artemia salina nauplius (C3). Section D represents the preservation methods “drying and freezing” (D1) and “direct storage in buffer” (D2). Scale bars measure 500 μm.
In order to conduct molecular analysis on planktonic foraminifera, a standard microbiology laboratory with usual equipment and containment level one is sufficient. Due to the danger of cross-contamination, it is recommended to spatially separate the steps of DNA extraction, Polymerase Chain Reaction (PCR) set-up and post-PCR analysis. It would be desirable to use different laboratories for these steps, but when space is limited, a partitioned bench or sterile hood combined with rigorous laboratory practice is sufficient. A strict laboratory routine, which enhances reproducibility, is important for the successful molecular analysis of planktonic foraminifera.
Sampling, Isolation, and Preservation of Foraminifera
Planktonic foraminifera occur in the open ocean off the continental shelf and therefore sampling generally has to be conducted from research vessels or small boats in regions where a narrow shelf allows rapid access to the open ocean.
Living planktonic foraminifera can be sampled in multiple ways (Figures 1, 2). Multiple-closing plankton nets, which allow depth stratified sampling, can only be deployed from larger research vessels, because they require winches with coaxial cable. The maximum sampling depth usually does not exceed 1000 m, since very few planktonic foraminifera specimens are found below that depth (Arnold and Parker, 2002). Simple plankton nets, which sometimes feature a closing mechanism, can be deployed from research vessels or small boats and typically only reach the top 100 meters of the water column. All plankton nets can be towed vertically, horizontally or obliquely. The mesh size of plankton nets should be no larger than 100 μm in order to avoid a bias toward larger specimens and/or larger species. For small high latitude species, plankton net mesh may be reduced to 83 μm mesh (Darling et al., 2007). In addition, planktonic foraminifera can be collected by filtering seawater supplied by a shipboard pump. Finally, they can be sampled individually by scuba diving in surface waters by capturing single specimens in jars directly out of their natural surroundings without damaging them (e.g., Huber et al., 1997).
Isolation of Individual Specimens
After collection, the bulk plankton sample is diluted with freshly filtered sea water in order to prevent deoxygenation, which would lead to the deterioration of foraminiferal specimens. Nevertheless, foraminifera should be isolated from the bulk sample as quickly as possible in order to avoid degradation of cellular material. At the same time, care has to be taken to minimize contamination by other organisms. Individual specimens are separated from the bulk sample by screening subsamples under a stereomicroscope and isolating the foraminifera using brushes and needles. Detailed steps for this process are outlined in Supplementary Data Sheet 1.
The isolation process can be conducted according to two different approaches:
Representative picking
In this approach, only selected specimens are taken from the bulk plankton sample. Usually large specimens full of cytoplasm are considered alive and chosen for processing. Small individuals and those with reduced cytoplasm are not selected, unless rare. A representative number of individuals of every morphospecies present are picked, while the remaining bulk sample is either discarded or preserved in formalin for future reference. This method allows for quick processing of samples and focuses on specimens which are most likely to yield positive PCR results.
Exhaustive picking
The aim of this approach is to isolate all foraminiferal specimens present in the plankton sample, including empty shells. The screening process of the bulk sample is thus repeated until no further planktonic foraminifera are found in the entire sample and the residue is then discarded. This approach yields larger sample sizes for genetic and biogeographic studies and allows immediate quantitative analysis of the species composition and abundance in the sample. On the other hand, it is labor intensive and time-consuming, which can be detrimental for DNA preservation during isolation.
Preservation of Specimens
Following isolation, two different approaches are used to preserve specimens for transport and long-term storage. Specimens can be stored either individually, directly in DNA extraction buffer or dried and frozen on cardboard faunal slides (Figure 2, details in Supplementary Data Sheet 1). Preservation in ethanol, a hypothetical third option commonly used for metazoans, is not recommended for foraminifera as it inhibits the amplification of DNA by PCR (Holzmann and Pawlowski, 1996; Lecroq, 2014). Drying and freezing has been used preferably in the last years, since it allows faster handling of foraminiferal specimens (André et al., 2013; Weiner et al., 2014, 2015). A considerably larger number of specimens can be preserved using this method, as it does not involve individual selection and placement in microtubes immediately after sampling.
Culture Maintenance
Planktonic foraminifera have never been successfully reproduced under laboratory conditions, yet they can be maintained in culture to increase DNA yield for molecular analysis by producing gametogenic individuals. Planktonic foraminifera reproduce sexually by the formation of a large number of flagellated gametes and just prior to gamete release, individual shells comprise multiple genomes with minimal contamination by symbionts and food particles (Darling et al., 1996b). Prior to gametogenesis, they seem to consume most symbionts as an energy source for DNA replication and repel the remaining symbionts and food particle debris (Bé and Anderson, 1976; Darling et al., 1996b). Working with gametogenic individuals thus increases the amount of DNA, while the risk of contamination is kept as low as possible. A detailed description of the culturing procedure can be found in Supplementary Data Sheet 1.
DNA Extraction
DNA is extracted from each isolated individual separately for single-cell genetic analysis. This is carried out using one of three methods: (1) DOC DNA extraction buffer containing sodium-deoxycholate (Holzmann and Pawlowski, 1996; Pawlowski, 2000), which does not allow preservation of the calcite shells, (2) GITC* buffer containing guanidinium isothiocyanate (e.g., Morard, 2010) or (3) urea buffer with high concentrations of urea (Seears and Wade, 2014; Weiner et al., 2014). Both of the latter methods are non-destructive for the calcite shells (Figure 3), which can thus be used for morphometric and geochemical analyses. It is therefore highly preferable for future studies to use methods that allow the preservation of intact shells. Detailed description of the buffer compositions, preparation and extraction procedures are given in Supplementary Data Sheet 1. Due to the danger of contamination it is essential to include negative controls in every step of the molecular analysis.
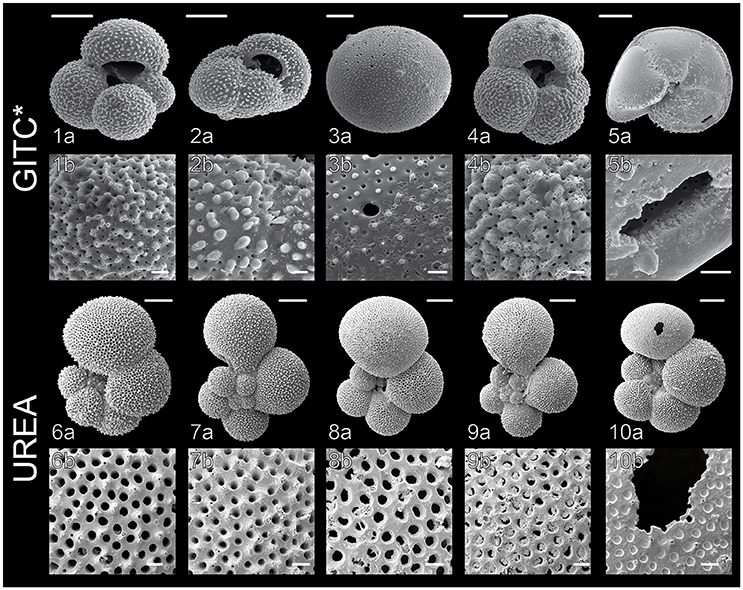
Figure 3. Scanning Electron Microscope (SEM) images of planktonic foraminiferal shells after DNA extraction, carried out according to the GITC* and urea protocols. (1) G. bulloides Type IIb, South Indian Ocean (from Morard et al., 2013), (2) G. inflata Type I, South Indian Ocean, (3) O. universa Type III, West Pacific, (4) G. bulloides Type IIg, South Indian Ocean, (5) G. truncatulinoides Type I, South Pacific, (6) G. siphonifera Type IIa5, Mozambique Channel (from Weiner et al., 2015), (7) G. calida Type IIIb, Mozambique Channel, (8) G. siphonifera Type Ib, Mozambique Channel, (9) G. radians Type Ia, Mozambique Channel (from Weiner et al., 2015), (10) G. siphonifera Type Ib, Mozambique Channel. Images 1a,b, 2a,b, 6a,b and 7a,b show completely intact, clean shells, whereas the shells in images 3a,b and 8a,b are slightly covered by buffer precipitates and the shells in images 4a,b and 9a,b show signs of slight dissolution. Parts of the shells in images 5a,b and 10a,b were mechanically damaged during the DNA extraction. Scale bars measure 100 μm in the upper panels and 10 μm in the lower panels.
Analysis of Single-Cell DNA Extracts
Genetic studies on planktonic foraminifera focus on the ribosomal RNA gene complex, which occurs in all domains of life and as multiple copies in the genome, making it a useful marker for phylogenetic studies and barcoding of protists (Pawlowski et al., 2012). Although the general structure of rDNA in foraminifera is the same as in all other eukaryotes, comprising the large subunit (LSU) and the small subunit (SSU) separated by internal transcribed spacer (ITS) regions (e.g., Pawlowski, 2000), it is marked by its exceptionally long length, compared to other organisms. The complete SSU fragment of some planktonic foraminiferal species measures more than 4 kb in sequence length, which is about twice the length found in other eukaryotes (de Vargas et al., 1997). This can be attributed to highly variable expansion segments integrated as loops in the helices of the SSU rDNA. The foraminifera SSU rDNA contains three unique variable regions (37/f, 41/f, and 47/f; Pawlowski and Lecroq, 2010) that differ widely between the different groups, complicating automated sequence alignments (de Vargas et al., 1997; Aurahs et al., 2009b). Furthermore, rates of SSU rDNA evolution in planktonic foraminifera vary significantly between the different species and are one of the highest known (de Vargas et al., 1997). Due to these high rates of evolution, the SSU rDNA has proved to be an excellent marker gene to study the genetic diversity of planktonic foraminifera, providing sufficient resolution well below the level of morphospecies (e.g., Darling et al., 1999; de Vargas et al., 1999; Darling and Wade, 2008; Morard et al., 2009; André et al., 2014; Weiner et al., 2014).
DNA Amplification
In most genetic studies on planktonic foraminifera, either a ~1000 bp fragment of the 3′ end of the SSU rDNA or a fragment of the ITS is amplified by PCR (Figure 4). A detailed description of the amplification procedure is given in Supplementary Data Sheet 1.
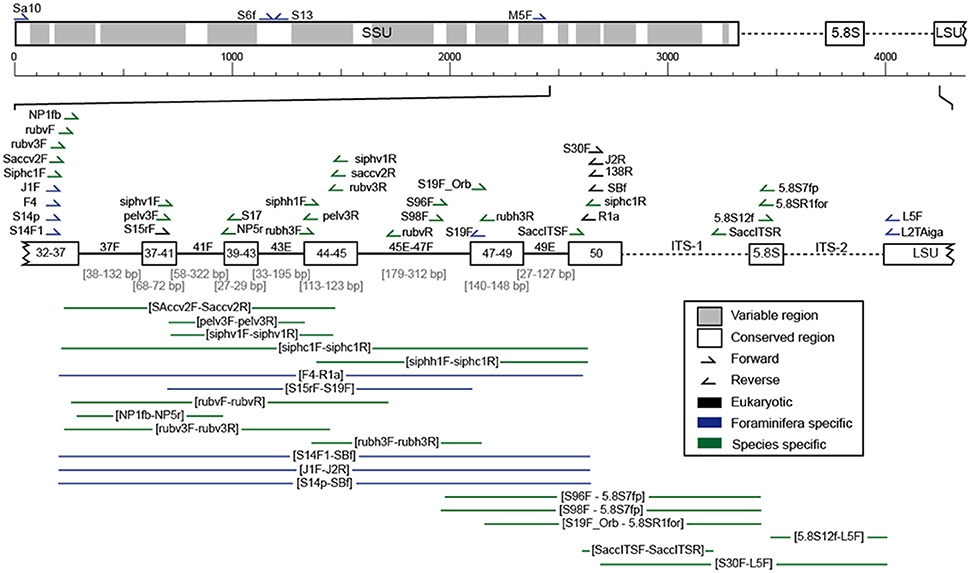
Figure 4. Scheme of the ribosomal DNA with its variable (gray) and conserved (white) regions. The fragment mostly analyzed in planktonic foraminifera, located at the 3′ end of the SSU up to the ITS regions, is shown in more detail (variable regions as lines and conserved regions as boxes) and includes the position of all primer pairs used for the amplification of planktonic foraminiferal rDNA in the here evaluated studies (Supplementary Table 1).
Over the years, a large number of different primer sets has been developed for the amplification of planktonic foraminiferal rDNA (Figure 4, Supplementary Table 1). In early investigations, universal primers were used that would amplify the rDNA of most protists (Medlin et al., 1988; White et al., 1990), since the genes of planktonic foraminifera were still unknown. Following the successful amplification of foraminiferal DNA (e.g., Pawlowski et al., 1994; Darling et al., 1996a,b) more specific primers were designed to selectively amplify foraminiferal DNA. Throughout later years, highly specific primers have been designed that only amplify specific groups, species or genotypes of planktonic foraminifera. Such primers are designed to amplify these rDNA regions, which are most characteristic for the target species.
Nested and Semi-Nested PCR
In many cases, PCR products are not visible on a gel after the first (primary) PCR run, since the quantity of rDNA template within a single foraminiferal cell is very low. Thus, nested (two new internal primers) or semi-nested (one new internal primer) PCR is conducted as an additional step to amplify the template further (see Supplementary Data Sheet 1 for details).
Dilution
A further optimization strategy to enhance amplification success is the dilution of the original DNA extract by a factor of 2–10 with H2Obidest prior to the PCR reaction. This is especially recommended for the DOC extraction protocol (see Supplementary Data Sheet 1), which does not include a purification step prior to the PCR analysis. Dilution of the DNA extract leads to a simultaneous reduction in PCR inhibitors such as EDTA, which increases target DNA amplification success. Equally, instead of diluting the original DNA extract, the primary PCR product can be diluted before using it for the secondary PCR reaction. This generally leads to higher amplification success in the secondary PCR.
Cloning
A further peculiarity of foraminiferal rDNA, in addition to its long length, is the particularly high number of gene copies in the genome. Since large amounts of rRNA are required in each cell, rDNA genes occur in the genome in tandem repeats, which in eukaryotic cells usually add up to several hundred copies (Long and Dawid, 1980). A study conducted on three benthic foraminiferal species showed there are as many as 10,000–30,000 copies of the genes (Weber and Pawlowski, 2014), which are marked by intra-individual sequence divergence (Pawlowski, 2000; Weber and Pawlowski, 2014). In planktonic foraminifera, the phenomenon of intra-individual variability in the SSU rDNA appears less common, being mostly restricted to some groups of non-spinose and microperforate species. However, it is essential to clone the rDNA of at least two individuals of each planktonic foraminiferal genotype, to evaluate the extent of intra-individual variability (André et al., 2014). A standardized protocol of this process is provided in Supplementary Data Sheet 1.
Restriction Fragment Length Polymorphism
The Restriction Fragment Length Polymorphism (RFLP) approach is an alternative to sequencing, for screening the genetic diversity among a large number of individuals. After PCR amplification, restriction enzymes are used to cut the amplified DNA fragment at specific sequence motifs into several smaller nucleotide fragments. The digested DNA is then run on a gel, where its fragments are separated by size and migrate in a specific pattern that can be used to discriminate the different genotypes present within the sample. An RFLP protocol is established once the extent of genetic diversity within a foraminiferal morphospecies has been identified through genotyping of specimens collected across a wide biogeographic and ecological range (de Vargas et al., 1999, 2001, 2002; Darling et al., 2007; Morard et al., 2009, 2011, 2013; Quillévéré et al., 2013). Although the RFLP approach is cost effective and rapid, it may overlook some rare genotypes present within the sampled population (Morard et al., 2011) and should be regarded as complementary to genotyping studies when particularly large numbers of specimens are required. All enzymes that have been used for RFLP analysis on planktonic foraminifera are listed in Supplementary Table 2.
Evaluation of Methods
In order to evaluate the methods applied for single-cell genetic analysis of planktonic foraminifera, we compared the DNA amplification success rates in studies carried out by the authors of the present study throughout the last 20 years. These studies applied different combinations of the methods and parameters described above, and although they have not been designed specifically for the purpose of comparison, their evaluation allows a first-order quantitative assessment of factors affecting success rate. We note that the data do not allow quantifying or eliminating interaction terms between different parameters, but nevertheless we consider a comparison and evaluation of the methods of high value for future methods-standardization. Being aware of this shortcoming of the data, we thoroughly discuss problems related to potential interactions of parameters and try to deduce their impact as detailed as possible. Only studies for which all parameters described above were known and which analyzed a sufficient number of foraminiferal specimens were included in the analysis (Supplementary Table 3). In total, the influence of eight parameters on the success rates of these genetic studies has been investigated. Success rate has been defined as the proportion of specimens from a single sample that yielded a positive PCR product. A sample is hereby defined as a set of specimens from a single sampling station belonging to the same morphospecies.
Influence of Methodological Parameters on Amplification Success Rates
The influence of five nominal analytical factors (i.e., such factors that can only have discrete values, hereafter called levels) was determined on the success rates of the genetic studies: target species, isolation method, preservation method, DNA extraction method, and sampling region. Thereto, each sample was treated as one observation. This analysis is dependent on sample size (i.e., number of specimens per station), since smaller sample sizes lead to a much greater uncertainty in the estimate of the success rates. In our dataset, the mean sample sizes (specimens per station) and also the total sample sizes (total number of specimens in analysis) are highly variable among morphospecies (Supplementary Data Sheet 2). Therefore, the dataset was limited to those morphospecies, which show an average sample size of at least 10 specimens per station (Globigerinoides ruber, Orbulina universa, Globigerina bulloides, Neogloboquadrina pachyderma, Globoconella inflata, Globorotalia truncatulinoides; Supplementary Data Sheet 2). It was then possible to calculate an average success rate, which is the average of the individual success rates per sample and factor level (Supplementary Data Sheet 2). We then performed a non-parametric multivariate analysis of variances (NPMANOVA, Anderson, 2001) as implemented in the R-package “vegan” v. 2.2-1, using the binomial distance metric (Anderson and Millar, 2004). In case of a significant overall NPMANOVA, we applied a pairwise NPMANOVA as post-hoc test (corrected for the false discovery rate after Benjamini and Hochberg, 1995).
The identity of the target morphospecies was tested as the first influencing factor on the success rates, using a hypothesis derived from our observations, that some species are more likely to yield a positive PCR product. The analysis revealed that success rates are indeed not equal among species (p < 0.001). From the species tested, G. ruber shows consistently lower and O. universa consistently higher success rates than the other morphospecies (Supplementary Data Sheet 2, Figure 5). However, despite the differences in the average success rates, the success rate can fluctuate between 0 and 100% in nearly all morphospecies.
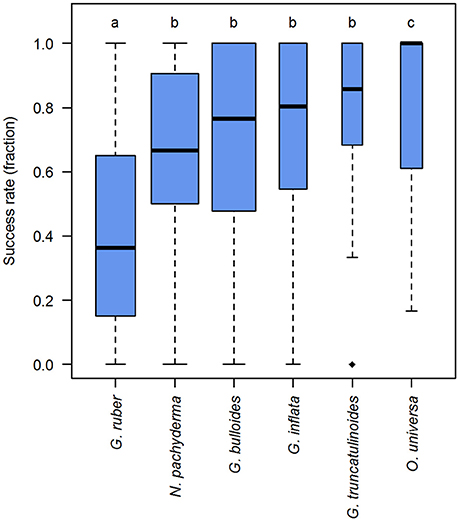
Figure 5. DNA amplification success rates in planktonic foraminifera in dependence of morphospecies identity. Boxplot showing the median (thick black lines), interquartile range (blue boxes), 1.5 × interquartile range (whiskers), and outliers (black diamonds) of the success rates in molecular analysis of planktonic foraminifera. Box widths are scaled to the number of observations within morphospecies. Lower case letters above boxes indicate groups between which success rates differ significantly (compare Supplementary Data Sheet 2).
The specimen isolation method was found to influence the amplification success rates significantly (p < 0.001). Representative sampling yields average success rates that are 20–30% higher than in samples obtained by exhaustive picking (Figure 6).
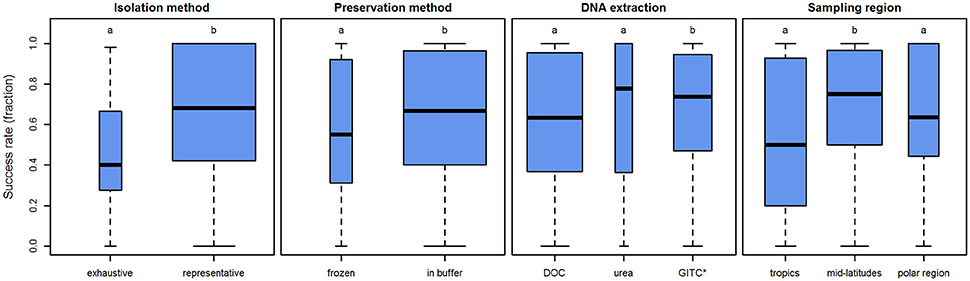
Figure 6. Influence of several parameters on the amplification success rates in planktonic foraminifera. Boxplots showing the median (thick black lines), interquartile range (blue boxes), and 1.5 × interquartile range (whiskers) of the success rates in molecular analysis of planktonic foraminifera. Box width is scaled to the number of observations within the respective level. Lower case letters above boxes indicate groups between which success rates differ significantly.
Additionally, the influence of the preservation method was tested. Direct storage in buffer yields significantly higher average success rates than drying and freezing the sample (p = 0.026). It has to be noted, however, that the average success rate only differs by c.10% (Figure 6).
Application of different DNA extraction buffers also influences the DNA amplification success rates (p < 0.001). Our analysis implies that the DOC and urea buffers show similar success rates (p = 0.350), but GITC* performs better than both DOC (p = 0.003) and urea (p = 0.005). Yet again, the effect is small (Figure 6). The average success rate when using GITC* as opposed to DOC is higher by less than 10%. Interestingly, the average success rate of urea is even slightly higher (5%) than that of GITC*, but both DOC and urea have a higher amount of samples with lower success rates (the interquartile range reaches further down). This means that with GITC*, the chances of getting higher success rates in all samples are better, compared to the other buffers.
As a further potential factor, the sampling region was tested. Here, we speculate that higher temperatures lead to faster degradation of DNA, so that samples from regions with cooler surface waters may be assumed to preserve better and thus show higher success rates. Thus, we categorized the samples into three regions according to their latitude: tropics 0–30°, mid-latitudes 30–60°, and polar regions 60–90°. Using this categorical approach we see that success rates are not the same in all regions (p = 0.002). In contrast to what would have been expected, success rates are highest in the mid-latitude samples (ppolar = 0.038, ptropics = 0.003) and show no significant difference between the tropics and polar regions (p = 0.449; Figure 6). This result could theoretically be influenced by the fact that the two species, which work significantly better or worse than others (O. universa and G. ruber, respectively), are not homogeneously distributed across all regions. However, when disregarding these two species and using only species with similar, medium success rates, the results remain virtually the same (Supplementary Data Sheet 3). The effect of this parameter is 25% difference between the mid-latitudes and tropics and approximately 10% between the mid-latitudes and polar regions.
Influence of Processing/Storage Times on Amplification Success Rates
The genetic analysis of planktonic foraminifera includes several time-consuming processing steps and periods of sample storage (Figure 1). It was therefore of interest whether the duration of these steps would influence the success rates of DNA amplification. To examine this, we applied a generalized linear model (GLM, Nelder and Wedderburn, 1972), using a binomial fit with the logit as link-function. In contrast to the analysis of the nominal analytical factors, this method treats every specimen instead of every sample as one observation. We were therefore able to use the complete data set instead of being limited to only a few morphospecies. We also tested all GLMs for their inference against the null-model that the points vary randomly (Faraway, 2006) based on the deviance. This allows us to evaluate whether the predictors in our models explain the data better than the assumption of random variation, which would indicate that the predictor is truly meaningful. This method is more liberal than determining inference based on the distribution of the model-deviance alone, but it is more suitable in this case because we know that success rates are influenced by several factors and no single predictor alone could explain the data well enough under this assumption.
The first step of the analysis is the isolation of specimens from a plankton sample. Its duration usually depends on the abundance of foraminifera in the sample and the number of scientists available for this work. It is reasonable to assume that longer processing times lead to degradation of DNA, and thus negatively impact success rates. We had information on the processing time for only a small subset of the data, containing the morphospecies G. ruber (white), O. universa, G. bulloides, G. inflata, and G. truncatulinoides. A GLM of these data indicates that longer processing times have a negative effect on success rates (one-tailed pneg < 0.001; pinference < 0.001; Figure 7). It seems that 100% success rates cannot be achieved when processing is not completed on the day of sampling, but this observation could also result from the fact that relatively few samples are available for which processing took longer than one day.
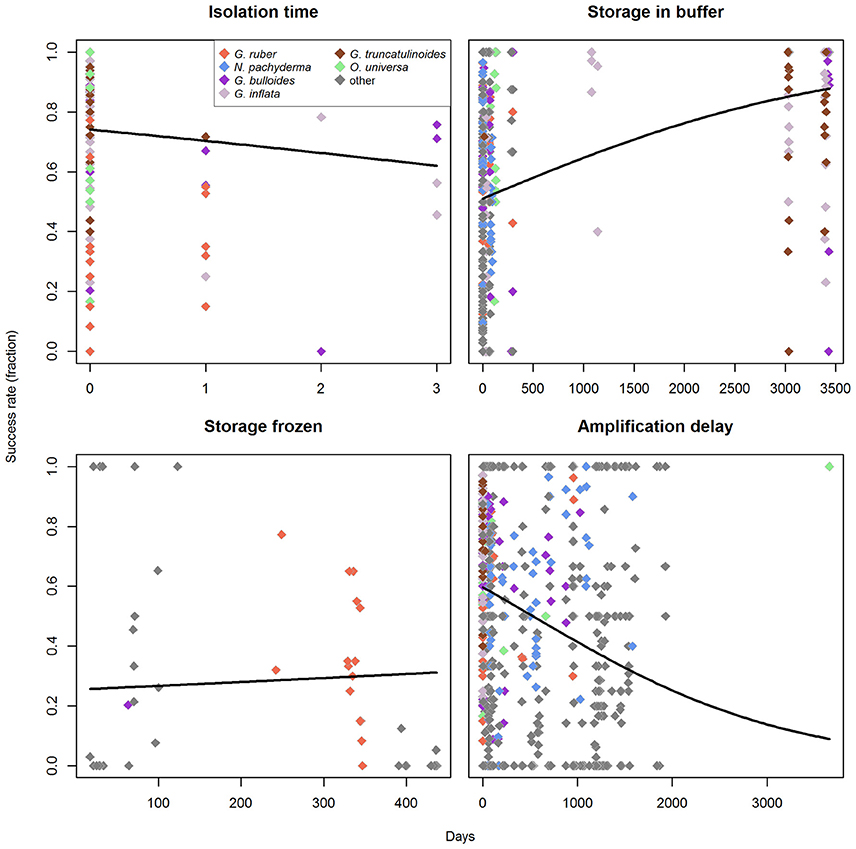
Figure 7. Influence of the processing/storage times on amplification success rates in the molecular analysis of planktonic foraminifera. Regression lines represent a binomial generalized linear model with logit as link-function. The morphospecies used for the analysis of nominal factors are indicated in different colors. Zero days corresponds to the respective process being completed within 1 day.
Specimens are then stored using one of two inherently different preservation methods (storage in buffer vs. drying and freezing). Since these two storage methods presumably affect the success rates very differently over time, the analysis for the effect of storage-time was done for each one individually. For buffer storage, we found that storage time has no negative effect (one-tailed pneg = 1; Figure 7), but rather seems to increase success rates (one-tailed ppos < 0.001), with a significant influence of the predictor (pinference < 0.001). This apparent increase in success rates is likely an artifact and could be influenced by the fact that we have nearly no observations available for storage times between 150 and 3000 days and only very few (in comparison) for storage times > 3000 days. However, when the data are limited to the rather continuous spectrum of storage times between 0 and 150 days, the same pattern prevails (one-tailed pneg = 1; Supplementary Data Sheet 3). The observed positive trend with storage time in buffer may further be a relic of the fact that the samples with larger storage times are biased toward the species that have higher success rates. G. ruber (marked by low success rates) has generally short storage times in buffer in our dataset (on average 23 days), while the species with medium to high success rates all have longer average storage times (O. universa: 80 days, G. bulloides: 483 days, G. inflata: 1850 days, G. truncatulinoides: 3028 days). However, the only species with significantly higher success rates (O. universa) also has very short mean storage times, and thus counteracts the effect of G. ruber on the data. For storage by drying we find no correlation of the storage time with amplification success (two-tailed p = 0.254; Figure 7).
After DNA extraction, in some cases samples are stored again before DNA amplification and the elapsed time might also influence the success rates. Indeed, an analysis of our data implies that there is a significant relationship (p < 0.001, pinference < 0.001) and that longer timespans between extraction and amplification likely have a negative effect on the expected success rates (Figure 7). Here it should be noted that 100% success rates are still possible even for long timespans between extraction and amplification, but the proportion of successful amplification is higher when this timespan is kept shorter.
Binomial linear models can be heavily influenced by overdispersion of the measurement values in regard to the predictor, which may lead to wrong results. We therefore tested all GLMs for overdispersion on the basis of the Pearson residuals (Faraway, 2006). For storage time in buffer and delay between extraction and amplification of the DNA we find no problem (p < 0.001). For storage in frozen state we detect a significant overdispersion (p = 0.595), but here this is expected since the predictor itself is already insignificant. Only for the isolation time we find that overdispersion might be a problem (p = 0.072), but we argue that it can be expected that DNA deteriorates over time when the sample is not stored in a frozen state. This observed trend is therefore intuitively most reasonable, and since the overdispersion-test is only marginally insignificant, we conclude that the trend most likely really exists.
Discussion and Future Guidelines
While comparing the results of single-cell genetic analyses on planktonic foraminifera from the last two decades, it became obvious that the success rates of these studies depend to a certain degree on the methodological parameters chosen (Table 1).

Table 1. Factors influencing DNA amplification success rates and recommendations for successful single-cell genetic analysis of planktonic foraminifera.
Success Rates Depend on Target Species
First, we note that different morphospecies show different amplification success, regardless of the parameters applied for the molecular analysis. While we have no data for exhaustive picking in O. universa, all possible levels of all other factors contain G. ruber (marked by low success rates), O. universa (high success rates), and at least one of the species with medium success rates. Therefore, it is unlikely that the observed signal is an artifact of the dataset, but some species in fact are more likely to yield positive PCR products than others. This finding corresponds to the personal experiences of the authors that some species are notoriously resistant to DNA amplification (e.g., Turborotalita humilis) and thus for some species, no DNA sequences exist to date. The most likely explanation for these differences is the existence of highly divergent substitution rates among planktonic foraminiferal species (de Vargas et al., 1997), resulting in an unusually strong primer bias. The varying success rates could also reflect differences in buffer digestion efficiency between species featuring shells of different architecture. This result is certainly affecting further analyses concerning the influence of other factors on amplification success, but due to the nature of the dataset we have no possibility to quantify the degree of interaction between the factors. We note, however, that the different species are well distributed across the methods and parameters tested, so that this interaction is limited to a minimum.
Representative Picking Yields Higher Success Rates at the Cost of Quantitative Bias
The isolation method influences amplification success rates severely, with representative picking yielding much higher success rates than exhaustive picking. Exhaustive picking represents the whole spectrum of specimens present in the water column, including small and deteriorating specimens, so that the relative proportion of viable templates in the sample is greatly reduced. The difference between the two methods seems to have the largest influence on amplification success rates (up to 30%) of all factors tested, thus arguing for the preferential application of representative picking. Yet, this isolation method bears the risk of misrepresenting the diversity in the water column by only selecting certain specimens. In addition, such datasets cannot be used for a quantitative analysis of the genotype/species composition and abundance in the sample. The decision to choose one method or the other is a trade-off between the requirement to obtain better qualitative or quantitative data and the work load to achieve it. As a compromise, exhaustive picking may be carried out only on a subsample of the total sample.
Preservation in Buffer Yields Higher Success Rates at the Cost of Longer Processing Times
Our data imply that direct storage of specimens in DNA extraction buffer leads to higher amplification success rates than drying and freezing of specimens. It may thus be concluded that buffer storage is the better choice for specimen preservation. Yet, this method is highly time-consuming while on the ship and especially disadvantageous when combined with exhaustive sampling, while its positive effect on success rates is very small (increase in amplification success: 11% for the average, 4–9% for the interquartile range). In addition, no reduction of amplification success over time is detectable when samples are dried and frozen (Figure 7), which might thus be the better choice due to its time-efficiency in preserving a large number of individuals. This especially holds when exhaustive sampling is carried out in regions with high population densities, where the processing time for storage in buffer would be prohibitively long due to the large number of specimens.
GITC* DNA Extraction Buffer Yields Highest Success Rates at the Cost of Complex Handling
We find evidence for a slightly different performance between the three DNA extraction buffers, i.e., DOC, urea, and GITC*, although the urea sample size is small (123 observations for GITC*, 256 for DOC and 25 for urea). Our analysis implies that the GITC* buffer exhibits the best general performance with the additional benefit of allowing the retention of the shell. Although this would support the general use of GITC* buffer in the future, the differences between buffers are relatively small regarding their DNA amplification success rates. Therefore, it is important to consider their other qualities when choosing which buffer to use. In this context we highlight the logistical advantage of the urea buffer, which can be kept at room temperature during transportation. Similarly, the quick and uncomplicated laboratory protocols for the DOC buffer have to be considered a great advantage in comparison with both the GITC* and urea buffers. The decision on which buffer to choose for DNA extraction is therefore complex and should be dictated by considerations on the logistics of sample transport, the time available to conduct the extraction protocol and the necessity to retain the calcite shells.
Importantly, the small difference in performance among the three buffers implies that they are largely compatible and that there is no reason to assume that prior studies using different buffers are biased concerning their reported genetic diversity.
Generally, it is preferable to use freshly prepared buffers, since their extraction efficiency seems to reduce with time as observed by the authors. In addition, it is advisable to store frozen samples at a constant temperature once the DNA is extracted, since repeated freezing and thawing leads to progressive denaturation of the DNA. We therefore recommend dividing the sample into working and stock aliquots (half of the total available sample volume each), so that at least one portion of the sample remains at constant temperature for long-term storage. It is recommended to keep the working aliquot at −20°C, while the long-term storage material should be kept at −80°C.
Samples from Temperate Regions Yield Higher Success Rates
Concerning the ambient water temperature, we observe lower success rates in the tropics compared to the mid-latitudes. This was expected, since the higher temperatures in the tropics lead to faster deoxygenation of the sample and faster DNA decay. Yet interestingly, in the polar regions we observe success rates similar to the tropics, which is counter-intuitive when the ambient sea surface temperature is being held responsible for this effect. This trend could be influenced by the fact that samples from the tropics and mid-latitudes both contain O. universa specimens, while the polar samples only contain G. bulloides and N. pachyderma, two species with medium amplification success rates. However, we rigorously tested this trend for the exclusion of species and found it to be stable, making this explanation very unlikely (Supplementary Data Sheet 3). A further influencing factor could be the difference between water temperature and ambient temperature during the picking process, which may negatively affect success rates. In colder regions this difference is especially pronounced, leading to increased degradation of DNA in the case of extended picking time without cooling the sample material. It is therefore recommended to cool plankton samples from low temperature waters on ice or cooling blocks during the entire picking process. Overall, these results confirm that samples from tropical and polar regions will probably always suffer from lower amplification success, a fact which can partly be countered by increasing the sampling intensity and decreasing sample processing times.
Rapid Isolation of Specimens Enhances Success Rates
Tests for the influence of sample processing and storage times on amplification success rate expectedly reveal the necessity for fast isolation of specimens from the bulk plankton sample. This necessitates the prudent adjustment of workforce to match the number of specimens required in order to finish their isolation within 1 day.
Storage Times Do Not Influence Success Rates
Our dataset reveals that prolonged storage times of samples before DNA extraction do not negatively impact amplification success. Long-time preservation of specimens in a dried and frozen state does not influence success rates at all, while preservation in buffer even seems to have a beneficial effect on success rates over time. While surprising, this positive trend unlikely is an artifact of our dataset, as it is stable over different time frames and does not seem to be influenced by morphospecies identity. In this regard, it has to be noted that a positive correlation between storage time in buffer and amplification success has also been observed by Seears and Wade (2014), who obtained better PCR results after 6 weeks of storage compared to immediate DNA extraction for several different buffers. While we still cannot be absolutely certain that the observed trend is not an artifact, this prior finding implies that it may be real and may reflect the positive effect of a more intensive incubation of the cellular material in the extraction buffer. It can therefore be concluded that there seems to be no apparent limit for sample storage times before completing the entire DNA extraction process, and even old samples are likely to provide an unbiased assessment of the genetic diversity of planktonic foraminifera if they have been stored under the recommended conditions.
Prompt Amplification after DNA Extraction Enhances Success Rates
In contrast to the storage time of samples before DNA extraction, the amount of time between DNA extraction and amplification shows a significant inverse effect on the success rates. While the full range from 0 to 100% is possible for any timespan, at least up to 2000 days, we observe significantly greater chances for higher success rates when this timespan is kept shorter. Based on our data, we can therefore recommend performing DNA amplification at the latest between 50 and 100 days after its extraction, but we reiterate that even samples left after extraction for several years can show high success rates.
Large Specimens, Suitable Primers and Minimum Contamination Enhance Success Rates
In addition to the statistically evaluated parameters mentioned above, the extensive fund of knowledge gathered by the authors throughout the years when carrying out single-cell genetic analysis of planktonic foraminifera is also of considerable value.
Firstly, the size of the specimens used for DNA extraction influences success rates. Very small specimens have often proved difficult to amplify, which may be due to loss during transfer into the DNA extraction buffer. A further risk concerning small specimens lies in the possibility that they attach to larger specimens and contaminate the samples.
A further crucial factor influencing amplification success is the choice of primers. In early studies, universal eukaryotic primers had to be used, since the foraminiferal rRNA gene sequences were unknown and could not be directly targeted. Now that sequences are known for most of the morphospecies, it is possible (and often necessary) to specifically target them by designing morphospecies or even genotype specific primers. Such a high level of primer specificity is possible due to a combination of unusually high evolutionary rates of the planktonic foraminiferal rDNA (de Vargas et al., 1997) and also due to the presence of highly variable regions specifically associated with foraminiferal SSU rDNA. However, using highly specific primers also bears the risk of failing to amplify DNA from unknown genotypes with derived sequences. Primer design thus represents a delicate balance that requires continual fine-tuning in order to achieve the best possible outcome for DNA amplification. In the future this issue might be overcome by applying single cell whole genome sequencing, thus circumventing the PCR-based amplification of specific genes (e.g., Gawad et al., 2016). Yet, the application of this method on planktonic foraminifera so far is still hampered by the low amounts of foraminiferal DNA present in a single cell compared to the large amount of contaminant DNA.
The problem of contamination is generally one of the main issues in single-cell genetic analysis of protists. Samples of planktonic foraminifera can be contaminated by prey particles, symbionts or even small foraminiferal specimens which may impede the successful amplification of the foraminiferal rDNA. Counteracting this problem by working with gametogenic individuals was important during the first attempts to amplify foraminiferal DNA (Darling et al., 1996b). This time-consuming approach is currently unnecessary for screening of genetic diversity, yet it is useful if large quantities of clonal DNA are required, e.g., for the discovery of new genes or whole genome approaches.
Remarkably, while comparing the methods and protocols applied for genetics on planktonic foraminifera it became clear that under all circumstances, no matter which particular species and protocol is used, success rates are always highly variable. This observation may be attributed to the fact that for unicellular organisms only limited amounts of DNA are available, whereas in multicellular organisms much larger amounts of DNA can be obtained from multiple cells, thus making them more reliable for providing high amplification success rates.
Conclusion
In this manuscript we compile and review all protocols that have been applied for single-cell genetic analysis of planktonic foraminifera. We present observations by the authors and objectively analyze a dataset of success rates of molecular studies from the last two decades, in which different combinations of methods have been applied, against eight potential predictors. We find that all factors tested (i.e., choice of target species, methods for specimen isolation and preservation, choice of DNA extraction buffer, sampling region as well as storage and handling times of samples) influence DNA amplification success rates. Yet, the effects between different methods are small and in all categories, cases with low and high success rates occur at both ends of the factor gradient. Whilst several recommendations for future studies can be made on the basis of these analyses, we conclude that the bias incurred by these parameters is likely to be small, allowing comparisons of studies that are based on different methods. In addition, we show that genetic analysis on planktonic foraminifera can be carried out on samples stored for several years. Both of these aspects facilitate the assembly of large datasets, even when originating from different working groups. By presenting the methods in great detail, the present manuscript serves both as a comprehensive resource for future molecular studies on planktonic foraminifera, but also allows transfer of methods to the genetic analysis of other protists groups.
Author Contributions
All authors contributed their methods, experiences, and success rates of genetic analyses to this review. AW combined these contributions, MW conducted the statistical analysis, and RM contributed to the preparation of the figures. AW, MW, KD, and MK drafted the manuscript and all authors critically revised it. The final version was approved by all authors.
Funding
This work was supported by the DFG-Research Centre/Cluster of Excellence “The Ocean in the Earth System.” Further support came from the Natural Environment Research Council of the United Kingdom (NER/J/S2000/00860 and NE/D009707/1), the Leverhulme Trust and the Carnegie Trust for the Universities of Scotland, the Agence National pour la Recherche ANR-JCJC06-0142-PALEO- CTD, the INSU INTERRVIE program, and the French Government “Investissements d'Avenir” programme OCEANOMICS (ANR-11-BTBR-0008).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SL and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
This study is a contribution to the efforts of the SCOR/IGBP Working Group 138 “Modern planktonic foraminifera and ocean changes.” We thank the reviewers Thomas Stach and Sébastien Lavoué for providing constructive comments that helped us to improve the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2016.00255/full#supplementary-material
References
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46. doi: 10.1046/j.1442-9993.2001.01070.x
Anderson, M. J., and Millar, R. B. (2004). Spatial variation and effects of habitat on temperate reef fish assemblages in northeastern New Zealand. J. Exp. Mar. Biol. Ecol. 305, 191–221. doi: 10.1016/j.jembe.2003.12.011
André, A., Quillévéré, F., Morard, R., Ujiié, Y., Escarguel, G., de Vargas, C., et al. (2014). SSU rDNA divergence in planktonic Foraminifera: molecular taxonomy and biogeographic implications. PLoS ONE 9:e104641. doi: 10.1371/journal.pone.0104641
André, A., Weiner, A., Quillévéré, F., Aurahs, R., Morard, R., Douady, C. J., et al. (2013). The cryptic and the apparent reversed: lack of genetic differentiation within the morphologically diverse plexus of the planktonic foraminifer Globigerinoides sacculifer. Paleobiology 39, 21–39. doi: 10.1666/0094-8373-39.1.21
Arnold, A. J., and Parker, W. C. (2002). “Biogeography of planktonic Foraminifera,” in Modern Foraminifera ed B. K. Sen Gupta (Dordrecht: Kluwer Academic Publishers), 103–122.
Aurahs, R., Göker, M., Grimm, G. W., Hemleben, V., Hemleben, C., Schiebel, R., et al. (2009b). Using the multiple analysis approach to reconstruct phylogenetic relationships among planktonic Foraminifera from highly divergent and lenght-polymorphic SSU rDNA sequences. Bioinform. Biol. Insights 3, 155–177.
Aurahs, R., Grimm, G. W., Hemleben, V., Hemleben, C., and Kucera, M. (2009a). Geographical distribution of cryptic genetic types in the planktonic foraminifer Globigerinoides ruber. Mol. Ecol. 18, 1692–1706. doi: 10.1111/j.1365-294X.2009.04136.x
Aurahs, R., Treis, Y., Darling, K., and Kucera, M. (2011). A revised taxonomic and phylogenetic concept for the planktonic foraminifer species Globigerinoides ruber based on molecular and morphometric evidence. Mar. Micropal. 79, 1–14. doi: 10.1016/j.marmicro.2010.12.001
Aze, T., Ezard, T. H. G., Purvis, A., Coxall, H. K., Stewart, D. R. M., Wade, B. S., et al. (2011). A phylogeny of Cenozoic macroperforate planktonic Foraminifera from fossil data. Biol. Rev. Camb. Philos. Soc. 86, 900–927. doi: 10.1111/j.1469-185X.2011.00178.x
Bé, A. W., and Anderson, O. R. (1976). Gametogenesis in planktonic Foraminifera. Science 192, 890–892. doi: 10.1126/science.946914
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Methodol. 57, 289–300.
Darling, K. F., Kroon, D., Wade, C. M., and Leigh Brown, A. J. (1996a). Molecular phylogeny of the planktic Foraminifera. J. Foram. Res. 26, 324–330. doi: 10.2113/gsjfr.26.4.324
Darling, K. F., Kroon, D., Wade, C. M., and Leigh Brown, A. J. (1996b). “The isolation and amplification of the 18S ribosomal RNA gene from planktonic Foraminifers using gametogenic specimens,” in Microfossils and Oceanic Environments, eds R. C. Whatley and A. Moguilevsky (Aberystwyth: University of Wales; Aberystwyth Press), 249–259.
Darling, K. F., Kucera, M., Kroon, D., and Wade, C. M. (2006). A resolution for the coiling direction paradox in Neogloboquadrina pachyderma. Paleoceanography 21:PA2011. doi: 10.1029/2005PA001189
Darling, K. F., Kucera, M., and Wade, C. M. (2007). Global molecular phylogeography reveals persistent Arctic circumpolar isolation in a marine planktonic protist. Proc. Natl. Acad. Sci. U.S.A. 104, 5002–5007. doi: 10.1073/pnas.0700520104
Darling, K. F., and Wade, C. M. (2008). The genetic diversity of planktic foraminifera and the global distribution of ribosomal RNA genotypes. Mar. Micropal. 67, 216–238. doi: 10.1016/j.marmicro.2008.01.009
Darling, K. F., Wade, C. M., Kroon, D., Leigh Brown, A. J., and Bijma, J. (1999). The diversity and distribution of modern planktic foraminiferal small subunit RNA genotypes and their potential as tracers of present and past ocean circulations. Paleoceanography 14, 3–12. doi: 10.1029/1998PA900002
Darling, K. F., Wade, C. M., Steward, I. A., Kroon, D., Dingle, R., and Leigh Brown, A. J. (2000). Molecular evidence for genetic mixing of Arctic and Antarctic subpolar populations of planktonic Foraminifers. Nature 405, 43–47. doi: 10.1038/35011002
de Vargas, C., Audic, S., Henry, N., Decelle, J., Mahé, F., Logares, R., et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348, 1261605. doi: 10.1126/science.1261605
de Vargas, C., Bonzon, M., Rees, N. W., Pawlowski, J., and Zaninetti, L. (2002). A molecular approach to biodiversity and biogeography in the planktonic Foraminifer Globigerinella siphonifera (d'Orbigny). Mar. Micropal. 45, 101–116. doi: 10.1016/S0377-8398(02)00037-3
de Vargas, C., Norris, R., Zaninetti, L., Gibb, S. W., and Pawlowski, J. (1999). Molecular evidence of cryptic speciation in planktonic foraminifers and their relation to oceanic provinces. Proc. Natl. Acad. Sci. U.S.A. 96, 2864–2868. doi: 10.1073/pnas.96.6.2864
de Vargas, C., Renaud, S., Hilbrecht, H., and Pawlowski, J. (2001). Pleistocene adaptive radiation in Globorotalia truncatulinoides: genetic, morphologic, and environmental evidence. Paleobiology 27, 104–125. doi: 10.1666/0094-8373(2001)027<0104:PARIGT>2.0.CO;2
de Vargas, C., Sáez, A., Medlin, L., and Thierstein, H. (2004). “Super-species in the calcareous plankton,” in Coccolithophores: From Molecular Processes to Global Impact, eds H. Thierstein and J. Young (Berlin: Springer Verlag), 271–298.
de Vargas, C., Zaninetti, L., Hilbrecht, H., and Pawlowski, J. (1997). Phylogeny and rates of molecular evolution of planktonic Foraminifera: SSU rDNA sequences compared to the fossil record. J. Mol. Evol. 45, 285–294. doi: 10.1007/PL00006232
Faraway, J. J. (2006). Extending the Linear Model with R: Generalized Linear, Mixed Effects and Nonparametric Regression Models. Boca Raton, FL: Chapman & Hall/CRC.
Gawad, C., Koh, W., and Quake, S. R. (2016). Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17, 175–188. doi: 10.1038/nrg.2015.16
Guillou, L., Bachar, D., Audic, S., Bass, D., Berney, C., Bittner, L., et al. (2013). The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41, D597–D604. doi: 10.1093/nar/gks1160
Harper, J. T., Gile, G. H., James, E. R., Carpenter, K. J., and Keeling, P. J. (2009). The Inadequacy of morphology for species and genus delineation in microbial eukaryotes: an example from the parabasalian termite symbiont coronympha. PLOS ONE 4:e6577. doi: 10.1371/journal.pone.0006577
Hemleben, C., Spindler, M., and Anderson, O. R. (1989). Modern Planktonic Foraminifera. Heidelberg: Springer.
Heywood, J. L., Sieracki, M. E., Bellows, W., Poulton, N. J., and Stepanauskas, R. (2011). Capturing diversity of marine heterotrophic protists: one cell at a time. ISME J. 5, 674–684. doi: 10.1038/ismej.2010.155
Holzmann, M., and Pawlowski, J. (1996). Preservation of foraminifera for DNA extraction and PCR amplification. J. Foram. Res. 26, 264–267. doi: 10.2113/gsjfr.26.3.264
Huber, B. T., Bijma, J., and Darling, K. F. (1997). Cryptic speciation in the living planktonic Foraminifer Globigerinella siphonifera (d' Orbigny). Paleobiology 23, 33–62. doi: 10.1017/S0094837300016638
Lecroq, B. (2014). “Molecular assessment of benthic foraminiferal diversity,” in Approaches to Study Living Foraminifera: Collection, Maintenance and Experimentation, eds H. Kitazato and J. M. Bernhard (Japan: Springer), 91–102.
Long, E. O., and Dawid, I. B. (1980). Repeated genes in eukaryotes. Annu. Rev. Biochem. 49, 727–764. doi: 10.1146/annurev.bi.49.070180.003455
Medlin, L., Elwood, H. J., Stickel, S., and Sogin, M. L. (1988). The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71, 491–499. doi: 10.1016/0378-1119(88)90066-2
Morard, R. (2010). For A Better Characterization of the Fossil Pelagic Record: Molecular, Biogeographical and Ecological Diversity of Planctonic Foraminifers Cryptic Species. Dissertation, Lyon, France: Université Claude Bernard Lyon, I. Available online at: http://www.theses.fr/2010LYO10213
Morard, R., Darling, K. F., Mahé, F., Audic, S., Ujiié, Y., Weiner, A. K. M., et al. (2015). PFR2: a curated database of planktonic Foraminifera18S ribosomal DNA as a resource for studies of plankton ecology, biogeography, and evolution. Mol. Ecol. Res. 49, 1–14. doi: 10.1111/1755-0998.12410
Morard, R., Quillévéré, F., Douady, C. J., de Vargas, C., de Garidel-Thoron, T., and Escarguel, G. (2011). Worldwide genotyping in the planktonic foraminifer Globoconella inflata: implications for life history and paleoceanography. PLoS ONE 6:e26665. doi: 10.1371/journal.pone.0026665
Morard, R., Quillévéré, F., Escarguel, G., de Garidel-Thoron, T., de Vargas, C., and Kucera, M. (2013). Ecological modeling of the temperature dependence of cryptic species of planktonic Foraminifera in the Southern Ocean. Palaeogeogr. Palaeoclimatol. Palaeoecol. 391, 13–33. doi: 10.1016/j.palaeo.2013.05.011
Morard, R., Quillévéré, F., Escarguel, G., Ujiie, Y., de Garidel-Thoron, T., Norris, R. D., et al. (2009). Morphological recognition of cryptic species in the planktonic foraminifer Orbulina universa. Mar. Micropal. 71, 148–165. doi: 10.1016/j.marmicro.2009.03.001
Nelder, J. A., and Wedderburn, R. W. M. (1972). Generalized linear models. J. R. Stat. Soc. Ser. A 135, 370–384. doi: 10.2307/2344614
Pawlowski, J. (2000). Introduction to the molecular systematics of Foraminifera. Micropaleontology 46 Suppl. 1, 1–12.
Pawlowski, J., Audic, S., Adl, S., Bass, D., Belbahri, L., Berney, C., et al. (2012). CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biol. 10:e1001419. doi: 10.1371/journal.pbio.1001419
Pawlowski, J., Bolivar, I., Guiard-Maffia, J., and Gouy, M. (1994). Phylogenetic position of Foraminifera inferred from LSU rRNA gene sequences. Mol. Biol. Evol. 11, 929–938.
Pawlowski, J., and Lecroq, B. (2010). Short rDNA Barcodes for species identification in Foraminifera. J. Eukar. Microbiol. 57, 197–205. doi: 10.1111/j.1550-7408.2009.00468.x
Quillévéré, F., Morard, R., Escarguel, G., Douady, C. J., Ujiié, Y., de Garidel-Thoron, T., et al. (2013). Global scale same-specimen morpho-genetic analysis of Truncorotalia truncatulinoides: a perspective on the morphological species concept in planktonic Foraminifera. Palaeogeogr. Palaeoclimatol. Palaeoecol. 391, 2–12. doi: 10.1016/j.palaeo.2011.03.013
Seears, H. A., Darling, K. F., and Wade, C. M. (2012). Ecological partitioning and diversity in tropical planktonic Foraminifera. BMC Evol. Biol. 12:54. doi: 10.1186/1471-2148-12-54
Seears, H. A., and Wade, C. M. (2014). Extracting DNA from within intact Foraminiferal shells. Mar. Micropal. 109, 46–53. doi: 10.1016/j.marmicro.2014.04.001
Weber, A. A.-T., and Pawlowski, J. (2014). Wide occurrence of SSU rDNA intragenomic polymorphism in Foraminifera and its implications for molecular species identification. Protist 165, 645–661. doi: 10.1016/j.protis.2014.07.006
Weiner, A., Aurahs, R., Kurasawa, A., Kitazato, H., and Kucera, M. (2012). Vertical niche partitioning between cryptic sibling species of a cosmopolitan marine planktonic protist. Mol. Ecol. 21, 4063–4073. doi: 10.1111/j.1365-294X.2012.05686.x
Weiner, A. K. M., Weinkauf, M. F. G., Kurasawa, A., Darling, K. F., and Kucera, M. (2015). Genetic and morphometric evidence for parallel evolution of the Globigerinella calida morphotype. Mar. Micropal. 114, 19–35. doi: 10.1016/j.marmicro.2014.10.003
Weiner, A. K. M., Weinkauf, M. F. G., Kurasawa, A., Darling, K. F., Kucera, M., and Grimm, G. W. (2014). Phylogeography of the tropical planktonic Foraminifera lineage Globigerinella reveals isolation inconsistent with passive dispersal by ocean currents. PLoS ONE 9:e92148. doi: 10.1371/journal.pone.0092148
White, T. J., Bruns, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in Pcr Protocols: A Guide to Methods and Applications, eds M. Innis, D. Gelfland, J. Sninsky and T. J. White (San Diego: Harcourt Brace Jovanovich), 315–322.
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Keywords: foraminifera, protists, single-cell genetics, cryptic diversity, comparability, methods, laboratory protocols, standardization
Citation: Weiner AKM, Morard R, Weinkauf MFG, Darling KF, André A, Quillévéré F, Ujiie Y, Douady CJ, de Vargas C and Kucera M (2016) Methodology for Single-Cell Genetic Analysis of Planktonic Foraminifera for Studies of Protist Diversity and Evolution. Front. Mar. Sci. 3:255. doi: 10.3389/fmars.2016.00255
Received: 28 September 2016; Accepted: 23 November 2016;
Published: 15 December 2016.
Edited by:
Wei-Jen Chen, National Taiwan University, TaiwanReviewed by:
Thomas Stach, Humboldt University of Berlin, GermanySébastien Lavoué, National Taiwan University, Taiwan
Copyright © 2016 Weiner, Morard, Weinkauf, Darling, André, Quillévéré, Ujiie, Douady, de Vargas and Kucera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnes K. M. Weiner, YXdlaW5lckBqYW1zdGVjLmdvLmpw