 Ying Chen
Ying Chen Xiaoyuan Feng1,2
Xiaoyuan Feng1,2 Ying He
Ying He Fengping Wang
Fengping Wang- 1State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
- 2State Key Laboratory of Ocean Engineering, Shanghai Jiao Tong University, Shanghai, China
- 3Department of Biochemistry and Molecular Biology, Guilin Medical University, Guilin, China
Species of Limnobacter genus are widespread in a variety of environments, yet knowledges upon their metabolic potentials and mechanisms of environmental adaptation are limited. In this study, a cell aggregate containing Limnobacter and anaerobic methanotrophic archaea (ANME) was captured from an enriched anaerobic methane oxidizing (AOM) microbial community. A genomic bin of Limnobacter was obtained and analyzed, which provides the first metabolic insights into Limnobacter from an AOM environment. This Limnobacter was found to contain genes involved in the Embden-Meyerhof pathway, the citrate cycle, citronellol degradation, and transporters of various organic substances, indicating a potentially heterotrophic lifestyle. A number of genes involved in sulfur oxidization, oxidative phosphorylation and ethanol fermentation that serve both aerobic and anaerobic purposes have been found in Limnobacter. This work suggests that in the AOM environment, Limnobacter strains may live on the organic substances produced through AOM activity and subsequently may contribute to the AOM community by providing sulfate from sulfur oxidation.
Introduction
Members of Limnobacter genus have often been detected in various environments, such as surface sea water, the deep ocean, the human intestine, and volcanic deposits (Lu et al., 2008; Eloe et al., 2010; Rigsbee et al., 2010; Vedler et al., 2013). Currently very few species of this genus have been isolated and characterized, and only two Limnobacter species, L. thiooxidans and L. litoralis, have been described (Spring et al., 2001; Lu et al., 2011). Both species are heterotrophic and capable of aerobically utilizing thiosulfate as an energy source. Till present, only one genome from Limnobacter genus is available but without insight analysis (Limnobacter sp. MED105, genome analysis has not yet been reported). Therefore, the metabolic potentials and environmental relevance of this genus are not well-understood.
Anaerobic oxidation of methane (AOM) is critical for controlling the emission of methane (Reeburgh, 2007), the second most important greenhouse gas, from anoxic environments. Anaerobic methanotrophic archaea (ANME-1, -2, and -3) are able to mediate the AOM mostly in association with sulfate-reducing bacteria (SRB) of the class Deltaproteobacteria, or by directly coupling AOM and incomplete sulfate reduction with the passage of zero-valent sulfur to sulfur-disproportionating Deltaproteobacteria (Knittel and Boetius, 2009; Milucka et al., 2012). In addition to SRB of Deltaproteobacteria, Alphaproteobacteria, and Betaproteobacteria are also thought to be potential bacterial partners of ANME (Pernthaler et al., 2008).
To understand the molecular mechanism of AOM, we have utilized a method of micromanipulation and single-aggregate metagenome sequencing to analyze AOM microbial communities in an AOM enrichment culture (Wang et al., 2014). The micromanipulation allows the isolation of a single aggregate composed of ANME and their partners. De novo assembly of genomic reads of a cell aggregate and binning by tetranucleotide signatures (Dick et al., 2009) separate the bacterial bins from the archaeal ones. In the present study, a cell aggregate containing ANME-2a/SRB and Betaproteobacteria assigned to Limnobacter spp. was isolated from an AOM enrichment by micromanipulation. After Illumina sequencing of the multiple displacement amplification (MDA) product, a partial genome assembly of a Limnobacter sp. was obtained. This assembly provides an unprecedented chance to understand the metabolic capabilities of a Limnobacter spp. from the AOM environment. It also implicates a potential interaction between ANME-2a archaea and Limnobacter spp. bacteria.
Materials and Methods
Sample Description
The enrichment sample was obtained from a continuous bioreactor supplemented with methane and sulfate (Zhang et al., 2010, 2011). The original sedimentary sample was taken in 2006 from Captain Arutyunov Mud Volcano (N35°39.700″ W07°20.012″) in the Gulf of Cadiz, Atlantic Ocean (Zhang et al., 2010). The bioreactor was supplied with methane-saturated artificial seawater medium at a pressure of approximately 8 MPa. As described in our previous work (Zhang et al., 2010), every liter of artificial seawater medium contains NaCl 26 g, MgCl2 6H2O 5 g, CaCl2 2H2O 1.4 g, Na2SO4 1.3 g, NH4Cl 0.3 g, KH2PO4 0.1 g, and KCl 0.5 g supplemented with 30 ml bicarbonate solution, 1 ml trace element solution, 1 ml vitamin mixture solution, 1 ml thiamine solution, and 1 ml vitamin B12 solution. The anaerobic condition was maintained via the addition of a reducing agent, sodium sulfide, in the medium at a final concentration of ~150 μM. During the incubation, the production of endogenous sulfide by AOM-SR occurred at a rate of approximately 9.22 μmol sulfide production/gdw/day, and the resulting concentration of sulfide in the medium ranged from 0.5 to 2.0 mM (Zhang et al., 2010). The enrichment culture maintained high AOM-SR activity when it was retrieved for use (in November 2011) in the present analysis.
Cell-Aggregate Isolation and Metagenome Sequencing
The culturing of microbial aggregates, genome amplification, and sequencing followed methods described previously (Wang et al., 2014). Multiple displacement amplification (MDA) was performed in each tube to obtain a single aggregate using REPLI-g Mini Kit reagents (Qiagen, Hilden, Germany) following the manufacturer's protocol. The cell lysis procedure followed the manual supplied with the REPLI-g Mini Kit; briefly, the cell aggregate was incubated with Buffer D2 on ice for 10 min, and the reaction was terminated by the addition of Stop Solution. MDA was carried out at 30°C for 12 h and inactivated by heating at 65°C for 3 min. The single aggregate genomes were then analyzed primarily by 16S rRNA gene sequencing using primers Arch21F and Arch958R for archaea (DeLong, 1992), as well as Bac27F and Bac1492R for bacteria (Lane, 1991). Finally, the samples of interest were sent to Macrogen, Inc. (South Korea) for genome sequencing.
Metagenome Assembly and Binning
Illumina 2*100 bp paired-end sequencing was performed on a single aggregate of M12. For Illumina sequencing, a 500-bp insert size library was constructed. Initially, 21,868,024 reads, totaling 2,208,670,424 bp, were generated for the M12 aggregate. The raw shotgun sequencing reads were dereplicated (100% identity over 100% lengths) and trimmed using Sickle (https://github.com/najoshi/sickle). Dereplicated, trimmed and paired-end Illumina reads were assembled using SPAdes version 3.5.0 with k-mer sizes of 21, 33, 55, and 77 (Bankevich et al., 2012). Binning of the assembled metagenome sequences was initially performed using tetranucleotide frequencies in emergent self-organizing maps (ESOMs) (Figure S1) with 4 to 8K as the fragment cutoff. Raw sequence data from the cell aggregate metagenome were submitted to the Sequence Read Archive at the National Center for Biotechnology Information (NCBI) under accession number SAMN04004108. The genome assembly of M12 has been incorporated into the Integrated Microbial Genomes (IMG) system under submission ID 68701. The completeness of the genomic bin was estimated based on CheckM (Parks et al., 2015).
Annotation of the SCA Genome
Gene prediction was carried out using MetaGene and fraggene_scan (Noguchi et al., 2006; Rho et al., 2010). Accordingly, 3800 and 4052 ORFs were predicted for the M12 assembly, and, for each predicted ORF, functional information was collected from similarity searches against the NCBI non-redundant protein database using BLASTP with an expectation cut-off value of <10−5. Sequences that had reliable hits with the non-redundant database were compared against the KEGG and COG sequence databases using an expectation cut-off value of <10−5.
Results
Cell Aggregate Isolation and Characterization
Cell aggregates were isolated from an AOM enrichment incubated in a high-pressure continuous flow bioreactor (Zhang et al., 2010). Within the culture, cell aggregates were mostly formed between ANME and SRB, with diameters ranging from 3 to 50 μm, and each cell aggregate contained several to hundreds of cells (Chen et al., 2014). Two hundred cell aggregates were captured by micromanipulations as previously described (Wang et al., 2014) (see also Materials and Methods). All were assayed by 16S rRNA gene fragments, and 11 ANME-positive cell aggregate metagenomes were obtained. The associated bacteria of these metagenomes varied, with five aggregates containing SRB of Deltaproteobacteria, two containing unclassified obsidian pool 1 (OP1) group bacteria, one being bacteria-negative, and the rest containing Acinetobacter of Gammaproteobacteria, Acidobacteria, and Limnobacter of Betaproteobacteria.
The cell aggregate metagenome named M12 containing ANME2 and Limnobacter of Betaprobacteria was analyzed in this study. The ANME-2a identified from M12 shares 100% 16S rRNA gene sequence identity with that of M25, a cell aggregate composed solely of ANME-2a (Wang et al., 2014). The Limnobacter from M12 (Limnobacter sp. M12) showed 99.6% 16S rRNA gene sequence identity to that of Limnobacter sp. MED105 (NZ_ABCT00000000.1), which was isolated from surface waters of the East Mediterranean Sea (Pinhassi and Berman, 2003).
General Features of the Genome Assembly
The cell aggregate M12 was subjected to Illumina sequencing, and in total, 21,868,024 reads with 2,208,670,424 bp were generated. Two well-resolved bins were recovered after binning M12 (for details, see Materials and Methods and Table 1). Bin24 was assigned to Limnobacter, Bin20 to ANME-2a and no Bins that assigned to SRB was characterized. Based on the CheckM analysis (Parks et al., 2015), an estimated 96.65% of the complete genome draft of Limnobacter was recovered from Bin24 (Table 1). Because genomic and metabolic analyses of ANME-2a from a single aggregate metagenome (M25) have been conducted previously (Wang et al., 2014), and the average nucleotide identity (ANI) between Bin20 and M25 is 99.2%, Bin20 was not further analyzed in this study.

Table 1. Overview of genome features.
Genomic Analysis and Metabolic Potential of Limnobacter sp. M12
General Genomic Features of Limnobacter sp. M12
The obtained genome of Limnobacter sp. M12 has an estimated genome completeness of ~96.65%. It was compared with the other available genome from Limnobacter species, Limnobacter sp. MED 105. Genomic comparison between Limnobacter sp. M12 and Limnobacter sp. MED105 showed that the ANI between Limnobacter sp. M12 and Limnobacter MED105 is 87.2%, and the alignment fraction value (AF) is 0.47 (Table 1). Based on the current standard for species description (Konstantinidis and Tiedje, 2005; Varghese et al., 2015), Limnobacter sp. M12 and MED105 could be considered as two species, although their 16S rRNA gene sequence identity reached 99.6%.
The genomes of Limnobacter sp. M12 and Limnobacter sp. MED105 shared ~90% of common predicted ORFs (with >30% identity and >50% coverage), and Limnobacter sp. M12 hold 52 unique ORFs (Table S1). Majority of these M12-unique genes are related to amino acids, benzoate, and starch and sucrose metabolism; defense against viral infection, gene transposition, insertion and horizontal gene transfer. Details of these gene information are listed in Table S1.
Metabolic Potentials of Limnobacter sp. M12
Carbon Metabolism
The assembled genome of Limnobacter sp. M12 contains almost all genes that encode enzymes involved in the Betaproteobacteria Embden-Meyerhof pathway and ethanol fermentation in which glucose is oxidized via a step-wise process anaerobically to produce pyruvate, which can be converted to ethanol with acetaldehyde as an intermediate (Figure 1 and Table S2).
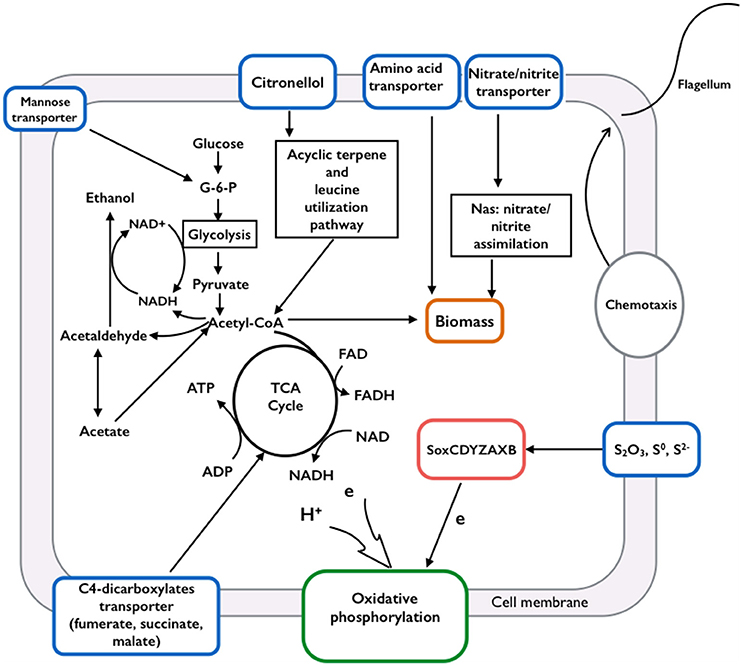
Figure 1. Schematic of the metabolic pathways identified from Limnobacter sp. M12. Genome analysis suggested that Limnobacter sp. M12 is able to utilize organic carbon and nitrogen sources through glycolysis, the citrate cycle, acyclic terpene, leucine utilization pathways, the nitrate/nitrite assimilation system, and their associated transport systems. Through carbon metabolism, the generated electrons and NADH could support energy conservation via ethanol fermentation or oxidative phosphorylation. The electron could also be derived from a sulfur-oxidizing system (soxCDYZAXB) to the oxygen via oxidative phosphorylation. Limnobacter sp. M12 is motile because it contains genes encoding proteins involved in chemotaxis and flagella movement.
The citrate cycle and oxidative respiratory chains (Tables S3, S4) were also identified. Through these pathways, pyruvate derived from the Embden-Meyerhof pathway can be oxidized completely, generating carbon dioxide and water when oxygen is available. Genes encoding tripartite ATP-independent periplasmic transporters (TRAPs) for dicarboxylate, C4-dicarboxylate (e.g., fumarate, malate and succinate) and mannitol/chloroaromatic compounds were also identified (see Table S5 in the Supplemental material). These findings indicate a heterotrophic lifestyle for Limnobacter sp. M12.
Citronellol is a naturally occurring aromatic component with antibacterial effects (Bakkali et al., 2008). It is produced by plants as well as certain marine organisms, such as sponges, microalgae, and coral (Bakkali et al., 2008). Citronellol contains a 3-methyl substitution, making degradation difficult. Only a few bacteria can use citronellol as the sole carbon source (Tozoni et al., 2010). Citronellol can be metabolized to acetyl coenzyme A and acetoacetate via the acyclic terpene utilization and leucine utilization pathways (Förster-Fromme et al., 2006). The majority of genes involved in citronellol utilization were identified in Limnobacter sp. M12 (as shown in Figure 2 and in Table S6 of the Supplemental Material), suggesting the ability of Limnobacter sp. M12 to metabolize citronellol.
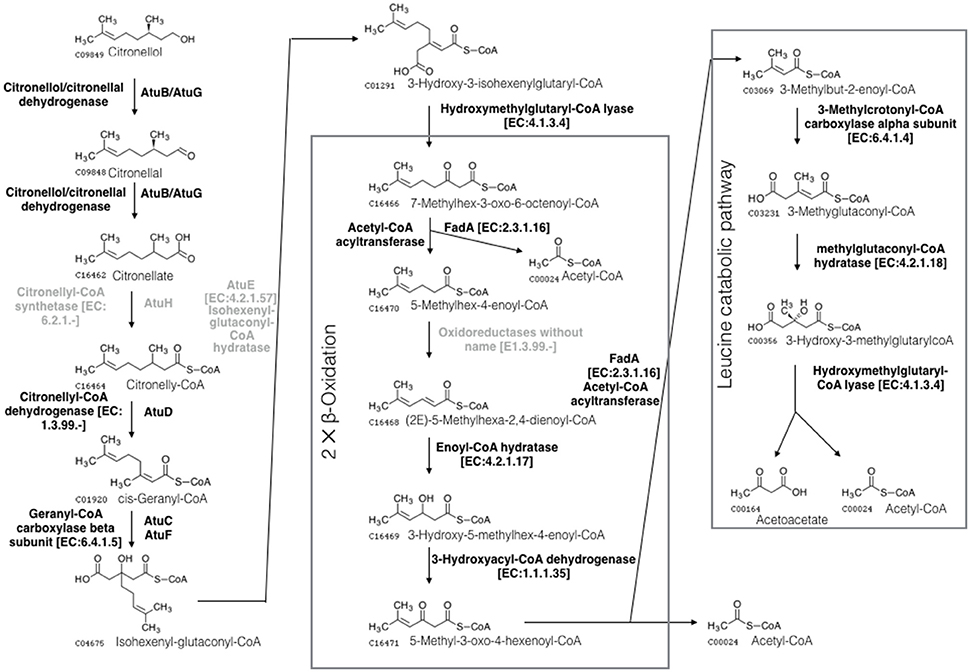
Figure 2. Schematic of the microbial citronellol degradation pathways according to Förster-Fromme et al. (2006). Citronellol is oxidized to its corresponding acid (citronellate) by the citronellol/citronellal dehydrogenase AtuB or AtuG. Citronellate is subjected to carboxylation, hydration and the removal of acetyl-CoA, producing 7-methyloctanoyl-3-oxo-6-octenol-CoA. This component is subjected to two rounds of β-oxidation that lead to the production of 3-methylbut-2-enoyl-CoA. This metabolite then flows into the leucine catabolic pathway, generating acetyl-CoA and acetoacetate. Three molecules of acetyl-CoA are generated during the pathways, which can be utilized as carbon and energy sources by the microorganism. The enzymes missing in the M12 SAG are shown in gray.
Sulfur Oxidation
Sulfur oxidase is an enzyme containing a molybdopterin cofactor and a heme group enzyme. It allows microorganisms to utilize reduced inorganic sulfur components (for example, S2− and S0) as an electron donor for the energy-generating system (Friedrich et al., 2005). A complete gene cluster (soxCDYZAXB) encoding sulfur oxidase (Sox) was identified in a 151-kb fragment of M12 (see Figure 3 and Table S7 in the Supplemental Material). All encoded Sox protein sequences displayed the highest sequence identity (approximately 95%) with those from Limnobacter sp. MED105 (ABCT00000000), with a similar gene order. Upstream of the M12 soxCDYZAXB cluster, genes encoding cytochrome o ubiquinol oxidase (CyoABCD) were identified (Figure 3). The CyoABCD complex belongs to the oxidative phosphorylation system, which transfers electrons from ubiquinol to oxygen (Riley et al., 2006). The presence of the sox and cyo genes suggests the capability of aerobic sulfur oxidization by Limnobacter sp. M12.
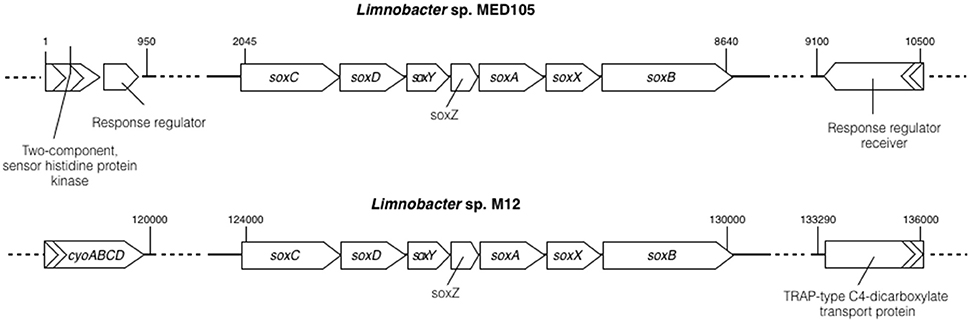
Figure 3. Comparison of the sulfur-oxidizing system locus (sox) in Limnobacter sp. MED105 and Limnobacter sp. M12. The organization of sox genes, including soxCDYZAXB, is similar in both MED105 and M12, but the flanking genes are different. CyoABCD represents genes encoding cytochrome o ubiquinol oxidase, which is involved in oxidative phosphorylation. Numbers on the top of the gene clusters represent the base numbers on the assembly.
Nitrogen Metabolism
Genes encoding transporters of general L-amino acids and branched-chain amino acids were found in Limnobacter sp. M12 (see Table S8 in the Supplemental Material). The general L-amino acid transporter system, which belongs to the ABC transporter, is responsible for transporting a wide range of L-amino acids including those with acid, base, amide, and aliphatic side chains. This transporter is composed of four genes (aapJMPQ), namely aapJ, which encodes a substrate-binding protein, aapQ and aapM, which encode two permeases serving in the cross-membrane transport of L-amino acids, and aapP, which encodes an ATP-binding protein. The branched-chain amino acid transporters require five genes, livFGHKM, which are responsible for the transport of extracellular branched-chain amino acids, such as leucine, isoleucine, and valine (Winters et al., 1991; Ribardo and Hendrixson, 2011). All aapJMQ genes were identified from the partial genome of Limnobacter sp. M12; however, aapP was not recovered, and all livFHGKM genes were contained in the partial genome of L. sp. M12.
In addition, genes involved in nitrate/nitrite assimilation (nasAB) were identified from the partial genome of Limnobacter sp. M12, suggesting that this strain is able to take up nitrogen from nitrate/nitrite.
Cell Motility
A nearly complete gene set of the polar flagella in Limnobacter sp. M12 was identified (see Table S9 in the Supplemental Material), which contains genes encoding flagella biosynthesis regulators (FlhCD) and flagella structural proteins (FlhAB, FlgA-L, FliC-K, and FliM-T). Two of the genes involved in the flagella system, the negative regulator of flagellin synthesis and the flagella synthesis protein flgN, were missing from the partial genome sequence of Limnobacter sp. M12. In addition, an analysis of the partial genome of Limnobacter sp. M12 suggests that switching of the flagella is likely to be regulated by a chemotaxis system. A typical chemotaxis system includes a methyl-accepting chemotaxis protein (MCP), an aerotaxis receptor (Aer) and chemotaxis proteins (CheA-D, CheRWVXYZ, and MotAB). The partial genome of Limnobacter sp. M12 encodes all of these components except for CheC and CheX (see Table S10 in the Supplemental Material). In addition, the BqsS and bqsR genes are a two-component system that enables microbial cells to switch between biofilm and planktonic lifestyles in Pseudomonas aeruginosa (Dong et al., 2008). The homologs of the bqsS and bqsR genes were identified from the partial genome of Limnobacter sp. M12.
Discussion
Genomic analysis revealed a heterotrophic lifestyle of Limnobacter sp. M12, with the capacity to utilize dicarboxylate, mannitol and citronellol, as well as performing amino acid and nitrate/nitrite assimilation and sulfur oxidation (as shown in Figure 1). It is noted that Limnobacter sp. M12 has the potential for oxygen respiration, as it possesses genes involved in oxidative phosphorylation. However, aerobic respiration is unlikely considering the anoxic condition of the bioreactor. During AOM enrichment, anaerobic artificial seawater was continuously supplied to the bioreactor, and a considerable sulfide-producing rate (9.22 μmol sulfide production/gdw/day) was constantly observed (Zhang et al., 2010). Hence, free oxygen may not be present in the culture. However, we cannot exclude the possibility that oxygen synthesis may occur within the anaerobic culture; some microorganisms, such as Candidatus Methylomirabilis oxyfera have been reported to generate oxygen as an intermediate when using nitrite to metabolize methane (Ettwig et al., 2010). On the other hand, fermentation could be a reasonable strategy for Limnobacter sp. M12 to survive in this anaerobic condition. Limnobacter sp. M12 contains genes that encode essential enzymes for ethanol fermentation (aldehyde dehydrogenase and alcohol dehydrogenase).
Compared to the Limnobacter sp. MED105 genome, Limnobacter sp. M12 contains many specific proteins that may benefit the cell against hazards from the environment. These include MqsR (motility quorum-sensing regulator), a part of the toxin/antitoxin system that influences quorum sensing, biofilm formation and the general stress response (Kim and Wood, 2010; Wang and Wood, 2011); HipA protein, which mediates multi-drug tolerance (Schumacher et al., 2009); Phasins, a granule-associated protein participating in the formation of intracellular granules, which enhance the fitness and stress resistance of bacteria (Pötter et al., 2004; de Almeida et al., 2007; Neumann et al., 2008); and abortive infection protein and virulence-associated protein, which serve in phage defense (Gerdes et al., 2005; Dy et al., 2014).
According to the AOM enrichment where the M12 consortia were isolated, methane and carbon dioxide were supplied as the only carbon sources, and sulfate was the only electron acceptor. The organic carbon could be produced by the ANME and SRB cells from methane and carbon dioxide. Limnobacter sp. M12 is heterotrophic, as proposed in this study; it might utilize the organic carbon produced from the AOM community as its carbon and energy source. In particular, the presence of the soxCDYZAXB genes and the reduced sulfur components (for example, S0 and S2−) generated from AOM-SR (Milucka et al., 2012) could be used as an additional energy source for Limnobacter sp. M12. Under strictly anaerobic conditions, Limnobacter sp. M12 may thrive on the fermentation of organic carbon compounds produced by the ANME archaea. However, in the presence of trace oxygen concentrations that might appear in the reactor or in shallow AOM active sediments, these Betaproteobacteria may thrive on the oxidation of sulfur compounds. Thereby, Limnobacter may protect oxygen-sensitive ANME archaea in the environment.
Author Contributions
FW and YC designed the project, YC performed the molecular biology experiment, XF, YH, and YC analyzed the genome, and all the authors involved in the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been financially supported by the Natural Science Foundation of China (grant 91228201, 91428308, 41576129), China Ocean Mineral Resources R&D Association (grant DY125-22-04).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2016.00257/full#supplementary-material
References
Bakkali, F., Averbeck, S., Averbeck, D., and Idaomar, M. (2008). Biological effects of essential oils–a review. Food Chem. Toxicol. 46, 446–475. doi: 10.1016/j.fct.2007.09.106
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Chen, Y., Li, Y.-L., Zhou, G.-T., Li, H., Lin, Y.-T., Xiao, X., et al. (2014). Biomineralization mediated by anaerobic methane-consuming cell consortia. Sci. Rep. 4:5696. doi: 10.1038/srep05696
de Almeida, A., Nikel, P. I., Giordano, A. M., and Pettinari, M. J. (2007). Effects of granule-associated protein PhaP on glycerol-dependent growth and polymer production in poly(3-hydroxybutyrate)-producing Escherichia coli. Appl. Environ. Microbiol. 73, 7912–7916. doi: 10.1128/AEM.01900-07
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 89, 5685–5689. doi: 10.1073/pnas.89.12.5685
Dick, G. J., Andersson, A. F., Baker, B. J., Simmons, S. L., Thomas, B. C., Yelton, A. P., et al. (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10:R85. doi: 10.1186/gb-2009-10-8-r85
Dong, Y. H., Zhang, X. F., An, S. W., Xu, J. L., and Zhang, L. H. (2008). A novel two-component system BqsS-BqsR modulates quorum sensing-dependent biofilm decay in Pseudomonas aeruginosa. Commun. Integr. Biol. 1, 88–96. doi: 10.4161/cib.1.1.6717
Dy, R. L., Przybilski, R., Semeijn, K., Salmond, G. P., and Fineran, P. C. (2014). A widespread bacteriophage abortive infection system functions through a Type IV toxin-antitoxin mechanism. Nucleic Acids Res. 42, 4590–4605. doi: 10.1093/nar/gkt1419
Eloe, E. A., Shulse, C. N., Fadrosh, D. W., Williamson, S. J., Allen, E. E., and Bartlett, D. H. (2010). Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ. Microbiol. Rep. 3, 449–458. doi: 10.1111/j.1758-2229.2010.00223.x
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548. doi: 10.1038/nature08883
Förster-Fromme, K., Höschle, B., Mack, C., Bott, M., Armbruster, W., and Jendrossek, D. (2006). Identification of genes and proteins necessary for catabolism of acyclic terpenes and leucine/isovalerate in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 72, 4819–4828. doi: 10.1128/AEM.00853-06
Friedrich, C. G., Bardischewsky, F., Rother, D., Quentmeier, A., and Fischer, J. (2005). Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8, 253–259. doi: 10.1016/j.mib.2005.04.005
Gerdes, K., Christensen, S. K., and Løbner-Olesen, A. (2005). Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 3, 371–382. doi: 10.1038/nrmicro1147
Kim, Y., and Wood, T. K. (2010). Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli. Biochem. Biophys. Res. Commun. 391, 209–213. doi: 10.1016/j.bbrc.2009.11.033
Knittel, K., and Boetius, A. (2009). Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63, 311–334. doi: 10.1146/annurev.micro.61.080706.093130
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY: John Wiley & Sons), 115–175.
Lu, H., Fujimura, R., Sato, Y., Nanba, K., Kamijo, T., and Ohta, H. (2008). Characterization of herbaspirillum- and limnobacter-related strains isolated from young volcanic deposits in miyake-jima island, japan. Microbes Environ. 23, 66–72. doi: 10.1264/jsme2.23.66
Lu, H., Sato, Y., Fujimura, R., Nishizawa, T., Kamijo, T., and Ohta, H. (2011). Limnobacter litoralis sp. nov., a thiosulfate-oxidizing, heterotrophic bacterium isolated from a volcanic deposit, and emended description of the genus Limnobacter. Int. J. Syst. Evol. Microbiol. 61(Pt 2), 404–407. doi: 10.1099/ijs.0.020206-0
Milucka, J., Ferdelman, T. G., Polerecky, L., Franzke, D., Wegener, G., Schmid, M., et al. (2012). Zero-valent sulphur is a key intermediate in marine methane oxidation. Nature 1–10. doi: 10.1038/nature11656
Neumann, L., Spinozzi, F., Sinibaldi, R., Rustichelli, F., Pötter, M., and Steinbüchel, A. (2008). Binding of the major phasin, PhaP1, from Ralstonia eutropha H16 to poly(3-hydroxybutyrate) granules. J. Bacteriol. 190, 2911–2919. doi: 10.1128/JB.01486-07
Noguchi, H., Park, J., and Takagi, T. (2006). MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 34, 5623–5630. doi: 10.1093/nar/gkl723
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pernthaler, A., Dekas, A. E., Brown, C. T., Goffredi, S. K., Embaye, T., and Orphan, V. J. (2008). Diverse syntrophic partnerships from deep-sea methane vents revealed by direct cell capture and metagenomics. Proc. Natl. Acad. Sci. U.S.A. 105, 7052–7057. doi: 10.1073/pnas.0711303105
Pinhassi, J., and Berman, T. (2003). Differential growth response of colony-forming α-and γ-proteobacteria in dilution culture and nutrient addition experiments from Lake Kinneret (Israel), the eastern Mediterranean Sea, and the Gulf of Eilat. Appl. Environ. Microbiol. 69, 199–211. doi: 10.1128/AEM.69.1.199-211.2003
Pötter, M., Müller, H., Reinecke, F., Wieczorek, R., Fricke, F., Bowien, B., et al. (2004). The complex structure of polyhydroxybutyrate (PHB) granules: four orthologous and paralogous phasins occur in Ralstonia eutropha. Microbiology 150(Pt 7), 2301–2311. doi: 10.1099/mic.0.26970-0
Reeburgh, W. S. (2007). Oceanic methane biogeochemistry. Chem. Rev. 107, 486–513. doi: 10.1021/cr050362v
Rho, M., Tang, H., and Ye, Y. (2010). FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38, e191. doi: 10.1093/nar/gkq747
Ribardo, D. A., and Hendrixson, D. R. (2011). Analysis of the LIV system of campylobacter jejuni reveals alternative roles for LivJ and LivK in commensalism beyond branched-chain amino acid transport. J. Bacteriol. 193, 6233–6243. doi: 10.1128/JB.05473-11
Rigsbee, L., Agans, R., Foy, B. D., and Paliy, O. (2010). Optimizing the analysis of human intestinal microbiota with phylogenetic microarray. FEMS Microbiol. Ecol. 75, 332–342. doi: 10.1111/j.1574-6941.2010.01009.x
Riley, M., Abe, T., Arnaud, M. B., Berlyn, M. K. B., Blattner, F. R., Chaudhuri, R. R., et al. (2006). Escherichia coli K-12: a cooperatively developed annotation snapshot—2005. Nucleic Acids Res. 34, 1–9. doi: 10.1093/nar/gkj405
Schumacher, M. A., Piro, K. M., Xu, W., Hansen, S., Lewis, K., and Brennan, R. G. (2009). Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323, 396–401. doi: 10.1126/science.1163806
Spring, S., Kämpfer, P., and Schleifer, K. H. (2001). Limnobacter thiooxidans gen. nov., sp. nov., a novel thiosulfate-oxidizing bacterium isolated from freshwater lake sediment. Int. J. Syst. Evol. Microbiol. 51, 1463–1470. doi: 10.1099/00207713-51-4-1463
Tozoni, D., Zacaria, J., Vanderlinde, R., Delamare, A. P. L., and Echeverrigaray, S. (2010). Degradation of citronellol, citronellal and citronellyl acetate by Pseudomonas mendocina IBPse 105. Electron. J. Biotechnol. 13, 2–3. doi: 10.2225/vol13-issue2-fulltext-8
Varghese, N. J., Mukherjee, S., Ivanova, N., Konstantinidis, K. T., Mavrommatis, K., Kyrpides, N. C., et al. (2015). Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771. doi: 10.1093/nar/gkv657
Vedler, E., Heinaru, E., Jutkina, J., Viggor, S., Koressaar, T., Remm, M., et al. (2013). Limnobacter spp. as newly detected phenol-degraders among Baltic Sea surface water bacteria characterised by comparative analysis of catabolic genes. System. Appl. Microbiol. 36, 525–532. doi: 10.1016/j.syapm.2013.07.004
Wang, F.-P., Zhang, Y., Chen, Y., He, Y., Qi, J., Hinrichs, K.-U., et al. (2014). Methanotrophic archaea possessing diverging methane-oxidizing and electron-transporting pathways. ISME J. 8, 1069–1078. doi: 10.1038/ismej.2013.212
Wang, X., and Wood, T. K. (2011). Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 77, 5577–5583. doi: 10.1128/AEM.05068-11
Winters, D. A., Poolman, B., Hemme, D., and Konings, W. N. (1991). Branched-Chain amino acid transport in cytoplasmic membranes of Leuconostoc mesenteroides subsp. dextranicum CNRZ 1273. Appl. Environ. Microbiol. 57, 3350–3354.
Zhang, Y., Henriet, J.-P., Bursens, J., and Boon, N. (2010). Stimulation of in vitro anaerobic oxidation of methane rate in a continuous high-pressure bioreactor. Bioresour. Technol. 101, 3132–3138. doi: 10.1016/j.biortech.2009.11.103
Keywords: limnobacter, anaerobic methane oxidation, high-pressure, multiple displacement amplification, mud volcano
Citation: Chen Y, Feng X, He Y and Wang F (2016) Genome Analysis of a Limnobacter sp. Identified in an Anaerobic Methane-Consuming Cell Consortium. Front. Mar. Sci. 3:257. doi: 10.3389/fmars.2016.00257
Received: 07 June 2016; Accepted: 24 November 2016;
Published: 08 December 2016.
Edited by:
Sandie M. Degnan, University of Queensland, AustraliaReviewed by:
Haiwei Luo, The Chinese University of Hong Kong, Hong KongRui Zhang, Xiamen University, China
Roland Hatzenpichler, Montana State University, USA
Copyright © 2016 Chen, Feng, He and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengping Wang, ZmVuZ3Bpbmd3QHNqdHUuZWR1LmNu