 Gretchen E. Hofmann
Gretchen E. Hofmann- Department of Ecology, Evolution and Marine Biology, University of California, Santa Barbara, Santa Barbara, CA, USA
In this horizon scan article, I review the emerging area of ecological epigenetics in marine animals with studies of DNA methylation as the primary focus. Epigenetic mechanisms such as DNA methylation have the capacity to create rapid changes in phenotypic plasticity. Epigenetic modifications of DNA are mechanisms that modify gene expression, and may do so in marine animals in an environmentally-induced manner. Thus, changes in the transcriptome that are driven by DNA methylation in response to environmental change may be one process by which marine animals can respond to anthropogenic, environmental change. From a technical standpoint, assays to quantify levels of DNA methylation, and next-generation sequencing approaches are opening up new avenues of exploration, such as simultaneously profiling the transcriptome and the methylome. A horizon scan for this topic suggests that the use of epigenomics and other methods to quantify DNA methylation will likely reveal important mechanisms of response to rapid environmental change in marine metazoans.
Introduction
Within ocean global change biology, a critical research challenge is to understand the processes that might support organismal resistance to environmental change. Although the level of organismal response can vary (Reusch, 2014; Sunday et al., 2014), phenotypic plasticity has emerged as an important area of study as it occurs on ecological rather than evolutionary time scales (i.e., more rapidly than evolutionary adaptation). Recently, there has been an emergence of studies exploring how epigenetic and epigenomic mechanisms might contribute to phenotypic plasticity in marine metazoans (Roberts and Gavery, 2012; Ledón-Rettig, 2013; Schrey et al., 2013; Dixon et al., 2014; Metzger and Schulte, 2016). Here, epigenetic modifications of DNA, such as DNA methylation, regulate and/or change gene expression, and thus change the transcriptome without changing DNA sequence (Szulwach and Jin, 2014). Since the transcriptome itself is a trait, it is a molecular-level mechanism that influences phenotypic plasticity within cells and organisms, and is one process that can mediate environmentally-induced plasticity. Overall, epigenetic processes alter gene expression in a manner that can potentially influence plastic and compensatory responses to environmental change in marine ecosystems such as ocean warming and ocean acidification.
With this mechanistic link in mind, I review studies that are leading the way in the area of ecological epigenetics and epigenomics in marine metazoans (see Table 1). For this horizon scan, I will use the definition of epigenetics (sensu Deans and Maggert, 2015), that is “the study of phenomena and mechanisms that cause chromosome-bound, heritable changes to gene expression that are not dependent on changes to DNA sequence” (Deans and Maggert, 2015). In order to focus the discussion, this horizon scan will highlight studies that examined changes in DNA methylation, a covalent modification of genomic DNA. This choice was made because it seems at the moment to be one of the most studied epigenetic mechanisms in marine metazoans, and numerous technical approaches are becoming available to support investigations in non-model marine organisms. Finally, I highlight future directions in this emerging area of studying organism-environment interactions in a global change context in marine metazoans.
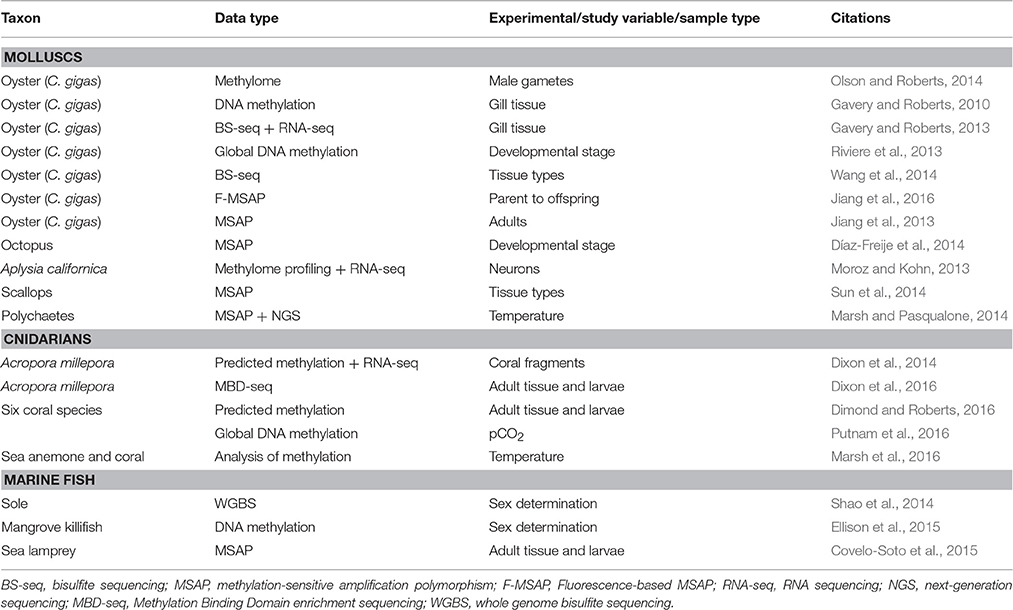
Table 1. Recent studies that assess DNA methylation in marine metazoans.
Epigenetics—A Brief Overview
Epigenetic mechanisms have been of interest in evolutionary biology (Richards et al., 2010; Zhang et al., 2013; Deans and Maggert, 2015), in the field of development (Beldade et al., 2011), and in such specialized fields as ecotoxicology (Vandegehuchte and Janssen, 2014). Historically, the research community has debated how to exactly define epigenetics (Deans and Maggert, 2015). Depending on the perspective of the investigator, a range of potential mechanisms are recognized, including covalent modifications of histones (Bannister and Kouzarides, 2011), DNA methylation (Bird, 2002), and gene regulation by a variety of non-coding RNAs (Jiao and Slack, 2014). Some biomedical research venues exemplify this broader definition of epigenetic mechanisms, and include both heritable and with-in generation processes (e.g., the U.S. National Institute of Health's Roadmap Epigenetics project - http://www.roadmapepigenomics.org/overview).
Epigenetic studies using non-model animals have increased in the last few years (Perfus-Barbeoch et al., 2014; Bonasio, 2015; Metzger and Schulte, 2016; Verhoeven et al., 2016). This emerging data has allowed investigators to assess the role of epigenetic mechanisms in an environmental and ecological context (Ledón-Rettig, 2013). Overall, the emergence for the appreciation of the role of epigenetics in an ecological context has developed in both aquatic and terrestrial systems (Bonduriansky et al., 2012). Studies on plants have dominated the literature on ecological epigenetics (Schrey et al., 2013; Baulcombe and Dean, 2014; Verhoeven et al., 2016), especially in response to stress (Wang et al., 2011; Grativol et al., 2012; Al-Lawati et al., 2016). On the marine side, epigenetic and epigenomic approaches are being used to explore areas such as (1) sources of phenotypic plasticity, as created by differential gene expression (Dixon et al., 2014; Dimond and Roberts, 2016; Marsh et al., 2016), (2) mechanisms involved in development (Riviere et al., 2013), and (3) a possible rapid response to anthropogenic environmental change (Putnam et al., 2016). These studies are mechanistic, but importantly also bridge to understanding adaptive capacity of marine organisms in response to environmental change (Calosi et al., 2016).
DNA Methylation
DNA methylation was one of the first epigenetic modifications of DNA to be discovered (Bird, 2002; Deans and Maggert, 2015) and has been the subject of extensive studies in numerous systems. This epigenetic mark (sensu Feng et al., 2010) has increasingly been the focus of studies in ecological epigenetics in non-model systems and plants (Schrey et al., 2013; Bonasio, 2015; Verhoeven et al., 2016; Metzger and Schulte, 2016) and increasingly in marine metazoans. Biochemically, DNA methylation involves the addition of a methyl group (CH3) to the pyrimidine ring of cytosines within CG dinucleotides (CpGs) resulting in 5-methylcytosine (5mC). This epigenetic modification alters transcription, and has implications for rates of DNA sequence changes, thereby also having evolutionary consequences in marine animals (Dixon et al., 2016). Patterns of DNA methylation vary significantly amongst taxa, with plants and animals showing divergent patterns (Feng et al., 2010). Within metazoans, variation exists across taxa, with vertebrate genomes being highly methylated and invertebrate genomes possessing variable levels of DNA methylation (Feng et al., 2010; Flores et al., 2013).
Measuring DNA Methylation
There are several techniques that can be used to quantify changes in DNA methylation, and excellent reviews covering various approaches have recently appeared (Plongthongkum et al., 2014; Metzger and Schulte, 2016). Each method has advantages and disadvantages and vary in terms of resolution, cost, and computational needs. In many ways, advances in biomedical science, e.g., cancer epigenetics, have driven the availability of resources to support studies of DNA methylation in non-model organisms. For the purposes of this horizon scan, I will focus on 3 basic approaches used to assess the methylation state of DNA. These are: (1) the methylation-sensitive amplification polymorphism (MSAP) method, (2) kit-based quantitative tools such as ELISAs that capture global DNA methylation patterns, and (3) full genome sequencing via bisulfite sequencing that profiles the genomic-scale methylation patterns, or the methylome.
The MSAP approach, a modified AFLP (amplified fragment length polymorphism) that employs restriction enzymes and separates fragments on a gel, has been available for decades and, while useful to investigators (Schrey et al., 2013), the results can be challenging to interpret. Recommendations for scoring bands and best practices are available (Pérez-Figueroa, 2013; Schulz et al., 2013), and this method will likely be of use in marine systems. On the positive side, MSAP results provide information on pattern and changes of methylation state (e.g., changes in the environment—see Schulz et al., 2014) and the method is well-suited for studies with many samples, e.g., in a population-scale study, when other methods may be too costly. The MSAP assay is somewhat limited in its diagnostic power as it does not provide any information regarding what functional gene might be methylated. However, some studies have used next-generation sequencing to examine the fragments that are obtained after the use of restriction enzymes in the MSAP process (Marsh and Pasqualone, 2014), thereby increasing the resolution of the MSAP approach.
Second in the list of methods are various ELISA-like, spectrophotometric assays that can detect the levels of methylated cytosines in a sample of genomic DNA. These methods are exemplar of the tools that have become commercially available as the field of epigenetics expands in biomedical science and have already been used in studies of marine metazoans (Riviere et al., 2013; Putnam et al., 2016). The limitation here is that the data are only global levels with no information about changes in specific regions of the genome; however again, this is an affordable and approachable method to gain insight into changes in global patterns (e.g., tissue-wide, or as a function of developmental stage) of DNA methylation between discrete samples.
Lastly, bisulfite sequencing (BS or Bs), a process where treatment of DNA with sodium bisulfite chemically converts unmethylated cytosines to uracil, has recently been coupled with next generation sequencing (NGS) approaches, e.g., whole genome sequencing, (Laird, 2010; Li et al., 2010) and also with RAD sequencing (BsRADseq) (Trucchi et al., 2016). NGS-based approaches have greatly increased the level of resolution of quantifying DNA methylation, and linking the patterns to the genome and gene regulation (Marsh et al., 2016). However, these epigenomic approaches come with the commensurate need for bioinformatics. Fortunately, these bioinformatics tools are increasingly available (Kishore et al., 2015). Within marine systems, investigations combining NGS techniques with DNA methylation screening have recently occurred in studies of the Pacific oyster (Olson and Roberts, 2014) and scleractinian coral (Dixon et al., 2016).
Pioneering Studies
To anchor this horizon scan, it is worth calling out some pioneering studies within marine ecological epigenetics and epigenomics that have set the stage for future investigations in marine metazoans. To begin, the most studied marine animal with regard to epigenetics seems to be the Pacific oyster, Crassostrea gigas (Table 1). Gavery and Roberts were the first to report DNA methylation in C. gigas (Gavery and Roberts, 2010), and continued in a second study with BS-seq analysis. In the latter study, Gavery and Roberts correlated methylated genes in the C. gigas methylome with high transcript abundance in the transcriptome (Gavery and Roberts, 2013). The abundance of publications with C. gigas as the study organism is likely due to its economic importance in combination with the early availability of genomic resources (Zhang et al., 2012). Oysters also featured in some of the first studies to examine the role of DNA methylation during development of a marine metazoan, and in early stage marine invertebrates. Studies have been conducted on oyster early development (Riviere et al., 2013) and gametes (Olson and Roberts, 2014), and on early stage Octopus vulgaris (Díaz-Freije et al., 2014).
A second pioneering advance in marine ecological epigenetics can be found in investigations that assessed epigenetic mechanisms in an ocean global change context. Here, two studies stand out. Working on an Antarctic marine polychaete, DNA methylation was linked to regulation of energy metabolism in response to temperature acclimation (Marsh and Pasqualone, 2014). Using NGS in combination with computational tools, Marsh and Pasqualone found that differential temperature acclimation (here, +4 °C vs. −1.5 °C temperature treatments) of adult polychaetes led to a change in the methylation state of the worm genome. Of the epigenetic marks they identified in the genome, 85% of the changes between the treatment groups reflected increased DNA methylation. Overall, the technical approach in the Marsh and Pasqualone study—the ability to map DNA methylation patterns and assess changes on a genomic scale—is a major step in understanding how and whether DNA methylation changes in response to abiotic conditions. In a second notable study in a global change context, Putnam and colleagues measured changes in global levels of DNA methylation in the scleractinian coral, Pocillopora damicornis, in response to high pCO2 levels in laboratory experiments (Putnam et al., 2016). Using a commercially available kit (the MethylFlash kit from Epigentek), these investigators demonstrated that global levels of methylation increased in coral genomic DNA in response to acclimation to low pH conditions that mimic ocean acidification. The results are suggestive that epigenetic mechanisms could create plasticity in corals to resist low pH in situ.
Lastly, one of the more informative advances in understanding how DNA methylation modulates gene expression, and thus phenotypic plasticity in marine animals, combines comparative transcriptomics with field studies in the natural habitat of the study organism. A study on the coral, Acropora millepora is exemplar of this approach (Dixon et al., 2014). Using samples generated in a reciprocal transplant experiment conducted in the field on the Great Barrier Reef, these investigators identified correlations of differential gene expression and DNA methylation in corals across populations, and between the outplant environments that varied in abiotic conditions. In addition to assessing the relationship of gene expression to gene methylation, the study also revealed a pattern where weak methylation increased the flexibility, or plasticity of gene expression, in response to environmental cues, something that had been proposed earlier by other investigators (Roberts and Gavery, 2012). Studies such as that conducted by Dixon and colleagues will begin to reveal how and whether epigenetic mechanisms operate in natural populations to support phenotypic plasticity.
Future Studies
The use of epigenomic tools will generate new insights into the response of marine organisms to changing environments. Further, these advancing applications of epigenetics tools will shed light on a critical mechanism, the role of phenotypic plasticity in response to global change, with influences extending into management (Marshall et al., 2016). At the moment, there are emerging lines of investigation that indicate epigenetic mechanisms are either at work, or merit investigation. For example, in marine metazoans, there is abundant evidence that physiological plasticity created by transgenerational plasticity (TGP) are “first response” options for organisms to adjust to rapid environmental change. In marine ecosystems, evidence is accruing that environmental stress can be buffered by TGP, a situation where the environmental experience of the adults can influence the phenotype of the progeny (Salinas and Munch, 2012; Munday, 2014), with maternal effects being a prime example (Marshall, 2008). Notably, TGP has been documented in marine organisms in response to variable pH and/or elevated pCO2 conditions in an ocean acidification context, for example (see Table 2). Studies such as these all point to the possibility that epigenetic mechanisms may modulate the environment-organism interaction.
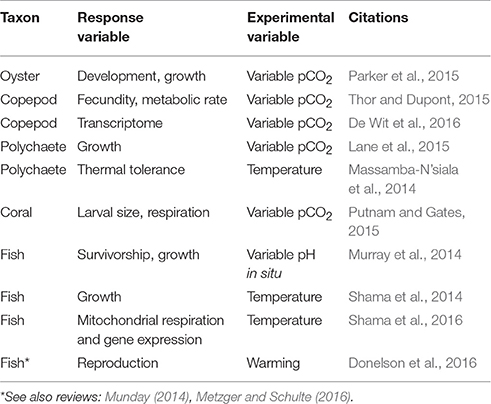
Table 2. Recent studies reporting transgenerational plasticity in a global change context.
Ecological epigenetics field studies stand to advance our knowledge of the role of the epigenome in modulating the G × E (Gene × Environment) interaction and thereby altering gene expression patterns. To what extent does the methylome and variable DNA methylation mediate rapid response to variable abiotic conditions? Are these mechanisms a source of “soft inheritance” in an environmental change context? Such work has been highlighted in plant molecular ecology (Bossdorf and Zhang, 2011; Herrera and Bazaga, 2011) and will likely begin to appear in marine ecology as more epigenomic tools are utilized in different systems. Notably, epigenomic mechanisms could be operating in organisms that experience routine variation in abiotic factors (e.g., intertidal invertebrates or vertically migrating zooplankton), or in animals that are sessile and do not have behavioral mechanisms to escape changes in their physical environment (e.g., corals or intertidal mussels). Interesting field studies here would be to examine patterns across environmental temperature gradients, e.g., comparing high intertidal to low intertidal mussels, or comparing corals found across similar local-scale thermal gradients (Palumbi et al., 2014).
One of the most promising areas for advancement in marine ecological epigenetics is the use of NGS technology and bioinformatics approaches. With the advent of combined RNA-seq and RAD-seq datasets with bisulfite sequencing data, new insights into environmentally relevant gene expression will likely emerge (Olson and Roberts, 2014; Dixon et al., 2016) along with population-level differences in DNA methylation in natural populations (Trucchi et al., 2016).
Summary
Within the “genome to phenome” pathway, epigenomic processes alter the manner in which DNA is transcribed into mRNA without altering DNA sequence, and thus contributes to physiological plasticity of the organism in an intergenerational or intragenerational context (Burggren, 2014, 2015). In general, ecological epigenomics is under-studied in marine systems. However, examining function at this level will likely reveal important mechanisms of response to rapid environmental change, something this is important as epigenomic-driven changes in organism tolerance occur on ecological, and not evolutionary, time scales. These targeted mechanisms are highly relevant in a global change context as they allow rapid adjustment of physiological tolerances and performance. Epigenomic mechanisms, in addition to other changes such as changes in the transcriptome, may together act as powerful mechanisms in species that experience variable conditions in nature. Although some of the techniques are nascent and some of the early results may be correlational, it seems these approaches are worthwhile and serve as a “first-cut” analysis of mechanisms that drive phenotypic plasticity in a global change context (Calosi et al., 2016; Verhoeven et al., 2016).
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Funding
During the writing of this mini-review, GH was supported by U.S. National Science Foundation award PLR-1246202, and by funds from a UC Climate Champion award from the University of California.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Al-Lawati, A., Al-Bahry, S., Victor, R., Al-Lawati, A. H., and Yaish, M. W. (2016). Salt stress alters DNA methylation levels in alfalfa (Medicago spp.). Genet. Mol. Res. 15:gmr.15018299. doi: 10.4238/gmr.15018299
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Baulcombe, D. C., and Dean, C. (2014). Epigenetic regulation in plant responses to the environment. Cold Spring Harb. Perspect. Biol. 6:a019471. doi: 10.1101/cshperspect.a019471
Beldade, P., Mateus, A. R., and Keller, R. A. (2011). Evolution and molecular mechanisms of adaptive developmental plasticity. Mol. Ecol. 20, 1347–1363. doi: 10.1111/j.1365-294X.2011.05016.x
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. doi: 10.1101/gad.947102
Bonasio, R. (2015). The expanding epigenetic landscape of non-model organisms. J. Exp. Biol. 218, 114–122. doi: 10.1242/jeb.110809
Bonduriansky, R., Crean, A. J., and Day, T. (2012). The implications of nongenetic inheritance for evolution in changing environments. Evol. Appl. 5, 192–201. doi: 10.1111/j.1752-4571.2011.00213.x
Bossdorf, O., and Zhang, Y. (2011). A truly ecological epigenetics study. Mol. Ecol. 20, 1572–1574. doi: 10.1111/j.1365-294X.2011.05044.x
Burggren, W. W. (2014). Epigenetics as a source of variation in comparative animal physiology – or – Lamarck is lookin' pretty good these days. J. Exp. Biol. 217, 682–689. doi: 10.1242/jeb.086132
Burggren, W. W. (2015). Dynamics of epigenetic phenomena: intergenerational and intragenerational phenotype ‘washout’. J. Exp. Biol. 218, 80–87. doi: 10.1242/jeb.107318
Calosi, P., De Wit, P., Thor, P., and Dupont, S. (2016). Will life find a way? Evolution of marine species under global change. Evol. Appl. 9, 1035–1042. doi: 10.1111/eva.12418
Covelo-Soto, L., Saura, M., and Morán, P. (2015). Does DNA methylation regulate metamorphosis? The case of the sea lamprey (Petromyzon marinus) as an example. Comp. Biochem. Physiol. B Biochemi. Mol. Biol. 185, 42–46. doi: 10.1016/j.cbpb.2015.03.007
Deans, C., and Maggert, K. A. (2015). What do you mean, “Epigenetic”? Genetics 199, 887–896. doi: 10.1534/genetics.114.173492
De Wit, P., Dupont, S., and Thor, P. (2016). Selection on oxidative phosphorylation and ribosomal structure as a multigenerational response to ocean acidification in the common copepod Pseudocalanus acuspes. Evol. Appl. 9, 1112–1123. doi: 10.1111/eva.12335
Díaz-Freije, E., Gestal, C., Castellanos-Martínez, S., and Morán, P. (2014). The role of DNA methylation on Octopus vulgaris development and their perspectives. Fronti. Physiol. 5:62. doi: 10.3389/fphys.2014.00062
Dimond, J. L., and Roberts, S. B. (2016). Germline DNA methylation in reef corals: patterns and potential roles in response to environmental change. Mol. Ecol. 25, 1895–1904. doi: 10.1111/mec.13414
Dixon, G. B., Bay, L. K., and Matz, M. V. (2014). Bimodal signatures of germline methylation are linked with gene expression plasticity in the coral Acropora millepora. BMC Genomics 15:1109. doi: 10.1186/1471-2164-15-1109
Dixon, G. B., Bay, L. K., and Matz, M. V. (2016). Evolutionary consequences of DNA methylation in a basal metazoan. Mol. Biol. Evol. 33, 2285–2293. doi: 10.1093/molbev/msw100
Donelson, J. M., Wong, M., Booth, D. J., and Munday, P. L. (2016). Transgenerational plasticity of reproduction depends on rate of warming across generations. Evol. Appl. 9, 1072–1081. doi: 10.1111/eva.12386
Ellison, A., Rodríguez López, C. M., Moran, P., Breen, J., Swain, M., Megias, M., et al. (2015). Epigenetic regulation of sex ratios may explain natural variation in self-fertilization rates. Proc. R. Soc. B Biol. Sci. 282:20151900. doi: 10.1098/rspb.2015.1900
Feng, S., Cokus, S. J., Zhang, X., Chen, P.-Y., Bostick, M., Goll, M. G., et al. (2010). Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. U.S.A. 107, 8689–8694. doi: 10.1073/pnas.1002720107
Flores, K. B., Wolschin, F., and Amdam, G. V. (2013). The role of methylation of DNA in environmental adaptation. Integr. Comp. Biol. 53, 359–372. doi: 10.1093/icb/ict019
Gavery, M. R., and Roberts, S. B. (2010). DNA methylation patterns provide insight into epigenetic regulation in the Pacific oyster (Crassostrea gigas). BMC Genomics 11:483. doi: 10.1186/1471-2164-11-483
Gavery, M. R., and Roberts, S. B. (2013). Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 1:e215. doi: 10.7717/peerj.215
Grativol, C., Hemerly, A. S., and Ferreira, P. C. (2012). Genetic and epigenetic regulation of stress responses in natural plant populations. Biochim. Biophys. Acta 1819, 176–185. doi: 10.1016/j.bbagrm.2011.08.010
Herrera, C. M., and Bazaga, P. (2011). Untangling individual variation in natural populations: ecological, genetic and epigenetic correlates of long-term inequality in herbivory. Mol. Ecol. 20, 1675–1688. doi: 10.1111/j.1365-294X.2011.05026.x
Jiang, Q., Li, Q., Yu, H., and Kong, L. (2016). Inheritance and variation of genomic DNA methylation in diploid and triploid Pacific oyster (Crassostrea gigas). Mar. Biotechnol. 18, 124–132. doi: 10.1007/s10126-015-9674-4
Jiang, Q., Li, Q., Yu, H., and Kong, L.-F. (2013). Genetic and epigenetic variation in mass selection populations of Pacific oyster Crassostrea gigas. Genes Genomics 35, 641–647. doi: 10.1007/s13258-013-0114-4
Jiao, A. L., and Slack, F. J. (2014). RNA-mediated gene activation. Epigenetics 9, 27–36. doi: 10.4161/epi.26942
Kishore, K., de Pretis, S., Lister, R., Morelli, M. J., Bianchi, V., Amati, B., et al. (2015). Methylpipe and compEpiTools: a suite of R packages for the integrative analysis of epigenomics data. BMC Bioinformatics 16:313. doi: 10.1186/s12859-015-0742-6
Laird, P. W. (2010). Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 11, 191–203. doi: 10.1038/nrg2732
Lane, A., Campanati, C., Dupont, S., and Thiyagarajan, V. (2015). Trans-generational responses to low pH depend on parental gender in a calcifying tubeworm. Sci. Rep. 5:10847. doi: 10.1038/srep10847
Ledón-Rettig, C. C. (2013). Ecological epigenetics: an introduction to the symposium. Integr. Comp. Biol. 53, 307–318. doi: 10.1093/icb/ict053
Li, N., Ye, M., Li, Y., Yan, Z., Butcher, L. M., Sun, J., et al. (2010). Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods 52, 203–212. doi: 10.1016/j.ymeth.2010.04.009
Marsh, A. G., Hoadley, K. D., and Warner, M. E. (2016). Distribution of CpG motifs in upstream gene domains in a reef coral and sea anemone: implications for epigenetics in cnidarians. PLoS ONE 11:e0150840. doi: 10.1371/journal.pone.0150840
Marsh, A. G., and Pasqualone, A. A. (2014). DNA methylation and temperature stress in an Antarctic polychaete, Spiophanes tcherniai. Front. Physiol. 5:173. doi: 10.3389/fphys.2014.00173
Marshall, D. J. (2008). Transgenerational plasticity in the sea: context-dependent maternal effects across the life history. Ecology 89, 418–427. doi: 10.1890/07-0449.1
Marshall, D. J., Burgess, S. C., and Connallon, T. (2016). Global change, life-history complexity and the potential for evolutionary rescue. Evol. Appl. 9, 1189–1201. doi: 10.1111/eva.12396
Massamba-N'siala, G., Prevedelli, D., and Simonini, R. (2014). Trans-generational plasticity in physiological thermal tolerance is modulated by maternal pre-reproductive environment in the polychaete Ophryotrocha labronica. J. Exp. Biol. 217, 2004–2012. doi: 10.1242/jeb.094474
Metzger, D. C. H., and Schulte, P. M. (2016). Epigenomics in marine fishes. Mar. Genomics 30, 43–54. doi: 10.1016/j.margen.2016.01.004
Moroz, L. L., and Kohn, A. B. (2013). “Single-neuron transcriptome and methylome sequencing for epigenomic analysis of aging,” in Biological Aging: Methods and Protocols, ed T. O. Tollefsbol (Totowa, NJ: Humana Press), 323–352.
Munday, P. L. (2014). Transgenerational acclimation of fishes to climate change and ocean acidification. F1000Prime Rep. 6:99. doi: 10.12703/P6-99
Murray, C., Malvezzi, A., Gobler, C., and Baumann, H. (2014). Offspring sensitivity to ocean acidification changes seasonally in a coastal marine fish. Mar. Ecol. Prog. Ser. 504, 1–11. doi: 10.3354/meps10791
Olson, C. E., and Roberts, S. B. (2014). Genome-wide profiling of DNA methylation and gene expression in Crassostrea gigas male gametes. Front. Physiol. 5:224. doi: 10.3389/fphys.2014.00224
Palumbi, S. R., Barshis, D. J., Traylor-Knowles, N., and Bay, R. A. (2014). Mechanisms of reef coral resistance to future climate change. Science 344, 895–898. doi: 10.1126/science.1251336
Parker, L. M., O'Connor, W. A., Raftos, D. A., Pörtner, H.-O., and Ross, P. M. (2015). Persistence of positive carryover effects in the oyster, Saccostrea glomerata, following transgenerational exposure to ocean acidification. PLoS ONE 10:e0132276. doi: 10.1371/journal.pone.0132276
Pérez-Figueroa, A. (2013). msap: a tool for the statistical analysis of methylation-sensitive amplified polymorphism data. Mol. Ecol. Resour. 13, 522–527. doi: 10.1111/1755-0998.12064
Perfus-Barbeoch, L., Castagnone-Sereno, P., Reichelt, M., Fneich, S., Roquis, D., Pratx, L., et al. (2014). Elucidating the molecular bases of epigenetic inheritance in non-model invertebrates: the case of the root-knot nematode Meloidogyne incognita. Front. Physiol. 5:211. doi: 10.3389/fphys.2014.00211
Plongthongkum, N., Diep, D. H., and Zhang, K. (2014). Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat. Rev. Genet. 15, 647–661. doi: 10.1038/nrg3772
Putnam, H. M., Davidson, J. M., and Gates, R. D. (2016). Ocean acidification influences host DNA methylation and phenotypic plasticity in environmentally susceptible corals. Evol. Appl. 9, 1165–1178. doi: 10.1111/eva.12408
Putnam, H. M., and Gates, R. D. (2015). Preconditioning in the reef-building coral Pocillopora damicornis and the potential for trans-generational acclimatization in coral larvae under future climate change conditions. J. Exp. Biol. 218, 2365–2372. doi: 10.1242/jeb.123018
Reusch, T. B. (2014). Climate change in the oceans: evolutionary versus phenotypically plastic responses of marine animals and plants. Evol. Appl. 7, 104–122. doi: 10.1111/eva.12109
Richards, C. L., Bossdorf, O., and Pigliucci, M. (2010). What role does heritable epigenetic variation play in phenotypic evolution? Bioscience 60, 232–237. doi: 10.1525/bio.2010.60.3.9
Riviere, G., Wu, G.-C., Fellous, A., Goux, D., Sourdaine, P., and Favrel, P. (2013). DNA methylation is crucial for the early development in the oyster C. gigas. Mar. Biotechnol. 15, 739–753. doi: 10.1007/s10126-013-9523-2
Roberts, S. B., and Gavery, M. R. (2012). Is there a relationship between DNA methylation and phenotypic plasticity in invertebrates? Front. Physiol. 2:116. doi: 10.3389/fphys.2011.00116
Salinas, S., and Munch, S. B. (2012). Thermal legacies: transgenerational effects of temperature on growth in a vertebrate. Ecol. Lett. 15, 159–163. doi: 10.1111/j.1461-0248.2011.01721.x
Schrey, A. W., Alvarez, M., Foust, C. M., Kilvitis, H. J., Lee, J. D., Liebl, A. L., et al. (2013). Ecological epigenetics: beyond MS-AFLP. Integr. Comp. Biol. 53, 340–350. doi: 10.1093/icb/ict012
Schulz, B., Eckstein, R. L., and Durka, W. (2013). Scoring and analysis of methylation-sensitive amplification polymorphisms for epigenetic population studies. Mol. Ecol. Resour. 13, 642–653. doi: 10.1111/1755-0998.12100
Schulz, B., Eckstein, R. L., and Durka, W. (2014). Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Mol. Ecol. 23, 3523–3537. doi: 10.1111/mec.12835
Shama, L. N., Mark, F. C., Strobel, A., Lokmer, A., John, U., and Mathias Wegner, K. (2016). Transgenerational effects persist down the maternal line in marine sticklebacks: gene expression matches physiology in a warming ocean. Evol. Appl. 9, 1096–1111. doi: 10.1111/eva.12370
Shama, L. N. S., Strobel, A., Mark, F. C., and Mathias Wegner, K. (2014). Transgenerational plasticity in marine sticklebacks: maternal effects mediate impacts of a warming ocean. Funct. Ecol. 28, 1482–1493. doi: 10.1111/1365-2435.12280
Shao, C., Li, Q., Chen, S., Zhang, P., Lian, J., Hu, Q., et al. (2014). Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 24, 604–615. doi: 10.1101/gr.162172.113
Sun, Y., Hou, R., Fu, X., Sun, C., Wang, S., Wang, C., et al. (2014). Genome-wide analysis of DNA methylation in five tissues of zhikong scallop, Chlamys farreri. PLoS ONE 9:e86232. doi: 10.1371/journal.pone.0086232
Sunday, J. M., Calosi, P., Dupont, S., Munday, P. L., Stillman, J. H., and Reusch, T. B. (2014). Evolution in an acidifying ocean. Trends Ecol. Evol. (Amst). 29, 117–125. doi: 10.1016/j.tree.2013.11.001
Szulwach, K. E., and Jin, P. (2014). Integrating DNA methylation dynamics into a framework for understanding epigenetic codes. Bioessays 36, 107–117. doi: 10.1002/bies.201300090
Thor, P., and Dupont, S. (2015). Transgenerational effects alleviate severe fecundity loss during ocean acidification in a ubiquitous planktonic copepod. Glob. Chang. Biol. 21, 2261–2271. doi: 10.1111/gcb.12815
Trucchi, E., Mazzarella, A. B., Gilfillan, G. D., Lorenzo, M. T., Schönswetter, P., and Paun, O. (2016). BsRADseq: screening DNA methylation in natural populations of non-model species. Mol. Ecol. 25, 1697–1713. doi: 10.1111/mec.13550
Vandegehuchte, M. B., and Janssen, C. R. (2014). Epigenetics in an ecotoxicological context. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 764–765, 36–45. doi: 10.1016/j.mrgentox.2013.08.008
Verhoeven, K. J. F., Vonholdt, B. M., and Sork, V. L. (2016). Epigenetics in ecology and evolution: what we know and what we need to know. Mol. Ecol. 28, 1631–1638. doi: 10.1111/mec.13617
Wang, W.-S., Pan, Y.-J., Zhao, X.-Q., Dwivedi, D., Zhu, L.-H., Ali, J., et al. (2011). Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot. 62, 1951–1960. doi: 10.1093/jxb/erq391
Wang, X., Li, Q., Lian, J., Li, L., Jin, L., Cai, H., et al. (2014). Genome-wide and single-base resolution DNA methylomes of the Pacific oyster Crassostrea gigas provide insight into the evolution of invertebrate CpG methylation. BMC Genomics 15:1119. doi: 10.1186/1471-2164-15-1119
Zhang, G., Fang, X., Guo, X., Li, L., Luo, R., Xu, F., et al. (2012). The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54. doi: 10.1038/nature11413
Keywords: epigenetics, mechanisms of plasticity, epigenomics and epigenetics, DNA methylation, global change biology, global change and organismal response
Citation: Hofmann GE (2017) Ecological Epigenetics in Marine Metazoans. Front. Mar. Sci. 4:4. doi: 10.3389/fmars.2017.00004
Received: 15 October 2016; Accepted: 05 January 2017;
Published: 18 January 2017.
Edited by:
Carlos M. Duarte, King Abdullah University of Science and Technology, Saudi ArabiaReviewed by:
Jennifer L. Bowen, University of Massachusetts Boston, USAMichael Yu Roleda, Norwegian Institute of Bioeconomy Research, Norway
Copyright © 2017 Hofmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gretchen E. Hofmann, Z3JldGNoZW4uaG9mbWFubkBsaWZlc2NpLnVjc2IuZWR1