 Cory C. Padilla1
Cory C. Padilla1 Anthony D. Bertagnolli1
Anthony D. Bertagnolli1 Laura A. Bristow2
Laura A. Bristow2 Neha Sarode3
Neha Sarode3 Jennifer B. Glass4
Jennifer B. Glass4 Bo Thamdrup5
Bo Thamdrup5 Frank J. Stewart1*
Frank J. Stewart1*- 1School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, USA
- 2Department of Biogeochemistry, Max Planck Institute for Marine Microbiology, Bremen, Germany
- 3Biological Laboratories, Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA, USA
- 4School of Earth and Atmospheric Sciences, Georgia Institute of Technology, Atlanta, GA, USA
- 5Department of Biology and Nordic Center for Earth Evolution, University of Southern Denmark, Odense, Denmark
Diverse planktonic microorganisms play a crucial role in mediating methane flux from the ocean to the atmosphere. The distribution and composition of the marine methanotroph community is determined partly by oxygen availability. The low oxygen conditions of oxygen minimum zones (OMZs) may select for methanotrophs that oxidize methane using inorganic nitrogen compounds (e.g., nitrate, nitrite) in place of oxygen. However, environmental evidence for methane-nitrogen linkages in OMZs remains sparse, as does our knowledge of the genomic content and metabolic capacity of organisms catalyzing OMZ methane oxidation. Here, binning of metagenome sequences from a coastal anoxic OMZ recovered the first near complete (95%) draft genome representing the methanotroph clade OPU3. Phylogenetic reconstruction of concatenated single copy marker genes confirmed the OPU3-like bacterium as a divergent member of the type Ia methanotrophs, with an estimated genome size half that of other sequenced taxa in this group. The proportional abundance of this bacterium peaked at 4% of the total microbial community at the top of the anoxic zone in areas of nitrite and nitrate availability but low methane concentrations. Genes mediating dissimilatory nitrate and nitrite reduction were identified in the OPU3 genome, and transcribed in conjunction with key enzymes catalyzing methane oxidation to formaldehyde and the ribulose monophosphate (RuMP) pathway for formaldehyde assimilation, suggesting partial denitrification linked to methane oxidation. Together, these data provide the first field-based evidence for methanotrophic partial denitrification by the OPU3 cluster under anoxic conditions, supporting a role for OMZs as key sites in pelagic methane turnover.
Introduction
Methane (CH4) is a potent greenhouse gas with 25 times the warming potential per-mol compared to CO2 (IPCC, 2013). The oceans contribute up to 4% of annual global methane emissions (Kirschke et al., 2013), with the sea-to-air flux of methane determined largely by the balance of methane production and consumption by marine microbes. Methane production has been observed under both oxic and anoxic conditions, although the mechanisms of production differ, via either archaeal methanogenesis in sediments (Reeburgh, 2007; Valentine, 2011) or aerobic catabolism of methylated phosphorous-containing compounds (Karl et al., 2008; Damm et al., 2010; Carini et al., 2014). Likewise, the metabolic pathways used to consume methane include both aerobic methanotrophy and anaerobic oxidation of methane (AOM). The latter process is mediated by prokaryotes using alternative oxidants such as sulfate (; Knittel and Boetius, 2009) or through the putative generation of intracellular oxygen from nitrate () and nitrite (; Raghoebarsing et al., 2006; Ettwig et al., 2010). While marine methanotrophy has been studied extensively over the past decades (Reeburgh, 2007; Valentine, 2011), the variables controlling the diversity, distribution, and activity of the dominant pelagic methanotrophs are still not well-understood, and genomic data for the diverse members of the marine methanotroph community remain sparse.
Oceanic oxygen minimum zones (OMZs) may be important sites for pelagic methane cycling. OMZs form at mid-water depths where heterotrophic respiration rates exceed the introduction of oxygen (Wyrtki, 1962; Helly and Levin, 2004; Wright et al., 2012). In the major OMZs of the Eastern Pacific, oxygen falls below the detection of modern sensors (Thamdrup et al., 2012; Tiano et al., 2014), creating anoxic conditions dominated by anaerobic microbial metabolisms (Naqvi et al., 2010; Stewart et al., 2012; Ulloa et al., 2012). Notably, OMZs contribute up to half of oceanic nitrogen loss, primarily through the anaerobic processes of denitrification, or anaerobic ammonia oxidation (anammox; Codispoti et al., 2001; Thamdrup, 2012). OMZs are also the largest pool of pelagic methane in the global ocean (Sansone et al., 2001), and represent potentially important sources of methane to the atmosphere (Naqvi et al., 2010). Methane maxima in OMZs may be due either to advection from nearby sediments (Sansone et al., 2001; Pack et al., 2015) or potentially from internal production by methanogenesis (Padilla et al., 2016), although the latter remains to be verified experimentally. Overlapping zones of elevated methane and oxidized nitrogen concentrations in OMZs suggest a pelagic niche for microbes conducting AOM coupled to reductive nitrogen transformations. With OMZs predicted to expand with increasing seawater temperatures (Stramma et al., 2008; Long et al., 2016), characterizing methane-consuming microbial populations in OMZs is critical for understanding greenhouse gas and nutrient budgets during global warming.
Diverse bacteria may be responsible for linking methane oxidation to pathways of nitrogen loss under anoxia. A recent study confirmed that OMZs harbor transcriptionally active bacteria of the candidate division NC10 (Padilla et al., 2016), a group hypothesized to dismutate nitric oxide (NO) into N2 and O2 gas, with the latter used as the terminal oxidant in an intra-aerobic methanotrophy pathway (Ettwig et al., 2010). Bacterial groups canonically associated with aerobic methanotrophy may also play a role in nitrogen loss by directly using nitrate or nitrite as terminal oxidants in AOM. Evidence for these groups, predominantly of the gammaproteobacterial order Methylococcales, has been found in both culture-dependent and -independent studies of diverse low oxygen environments (Kalyuzhnaya et al., 2013; Chistoserdova, 2015; Kits et al., 2015a,b; Danilova et al., 2016), including meromictic lakes (Biderre-Petit et al., 2011; Blees et al., 2014) and marine water columns (Hayashi et al., 2007; Tavormina et al., 2013). Studies with isolates of two methanotrophic Methylococcales genera (Methylomonas and Methylomicrobium) show that hypoxic conditions stimulate denitrification to produce N2O accompanied by increased cellular ATP yields (Kits et al., 2015a,b). Thus, such low oxygen-adapted methanotrophs may act as both a source of nitrous oxide and a sink for methane, depending on oxygen availability.
It remains unclear whether similar physiological mechanisms are used by planktonic methanotrophs in natural OMZ communities. While NC10 bacteria occur in OMZs, our prior work showing low NC10 abundance in a zone of comparatively high methane oxidation rates in a coastal OMZ suggests that other microbial players contribute to OMZ methanotrophy (Padilla et al., 2016). Indeed, PCR-based surveys of the methanotroph marker gene particulate methane monooxygenase (pmo) have identified diverse marine clades of the Methylococcales, designated as operational pmo units (OPUs), as being widely distributed through pelagic and sediment low oxygen environments (Tavormina et al., 2008, 2010). The OPU3 clade has been detected in a wide range of marine habitats, including the deep sea (Jensen et al., 2008; Lesniewski et al., 2012), methane seeps and oil spills (Wasmund et al., 2009; Tavormina et al., 2010; Kessler et al., 2011; Rivers et al., 2013), and OMZs (Hayashi et al., 2007; Tavormina et al., 2013). These studies suggest a low-to-no oxygen niche for OPU3, with members of this group being particularly prevalent in Eastern Pacific OMZs (Tavormina et al., 2008, 2010, 2013; Knief, 2015). These OPU clades cluster apart from other denitrifying methanotrophs (Figure 1; Tavormina et al., 2008, 2010). However, no information regarding the genomic capacities and in situ activity of these groups has been reported.
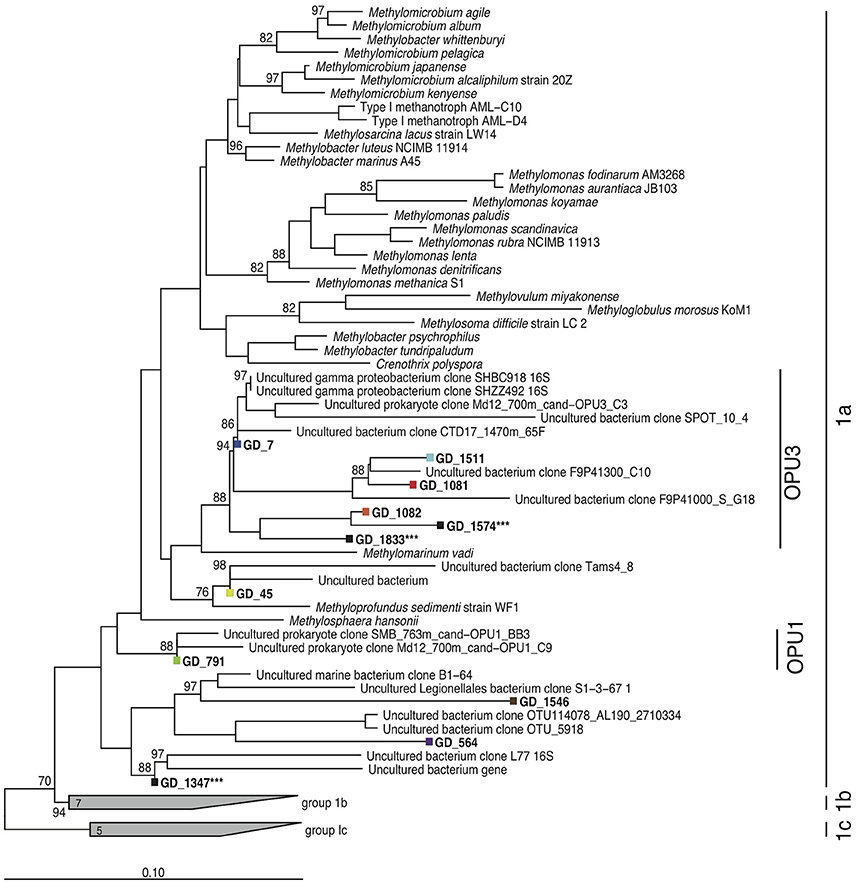
Figure 1. Methanotroph 16S rRNA gene phylogeny. Neighbor-joining phylogeny based on partial full-length 16S rRNA genes, with phylogenetic approximation of OTUs (colors) recovered from pooled gDNA and cDNA amplicon fragments assessed by parsimonious placement. Scale bar denotes nucleotide substitutions. Asterisks denote OTUs that were present at low abundance in non-rarefied datasets but lost when the datasets were subsampled to an even sequence depth. Bootstrap values ≥70 are displayed.
Here, we report a near-complete genome from the OPU3 clade. The genome was recovered by binning of metagenome sequences from the anoxic coastal OMZ in Golfo Dulce (GD), Costa Rica. Like other anoxic OMZs, the GD anoxic, non-sulfidic zone is enriched in nitrite (~0.75 μM at the time of sampling) and supports an active anaerobic microbial community mediating nitrogen loss, notably through both the anammox process and denitrification (Dalsgaard et al., 2003). As reported in Padilla et al. (2016), methane concentrations at the time of sampling for the current study increased with depth into the GD anoxic zone, from <5 nM at the base of the oxycline (~80 m) to >70 nM near the sediment-water interface (~190 m), suggesting methane efflux from sediments (Figure 2). In contrast, methane-oxidation rates were highest (2.6 ± 0.7 nM d−1) at the top of the anoxic zone below the oxycline (90 m), but below detection deeper in the water column. This pattern is consistent with the periphery of OMZs as sites of active methane consumption (Ward et al., 1989; Sansone et al., 2001; Naqvi et al., 2010; Pack et al., 2015), potentially with methanotrophs at these transition depths adapted for both aerobic and anaerobic methane oxidation to accommodate fluctuation in oxycline depth or oxygen intrusions. Here, metagenomic data, interpreted alongside 16S rRNA amplicon sequences and phylogenetic analyses, identify an OPU3 bacterium as a dominant member of the anoxic GD community. Mapping of coupled mRNA transcripts to the OPU3 genome provides environmental transcriptional evidence of methanotrophic denitrification outside of the NC10 phylum in an OMZ, further confirming OMZs as key sites of oceanic methane-nitrogen linkages.
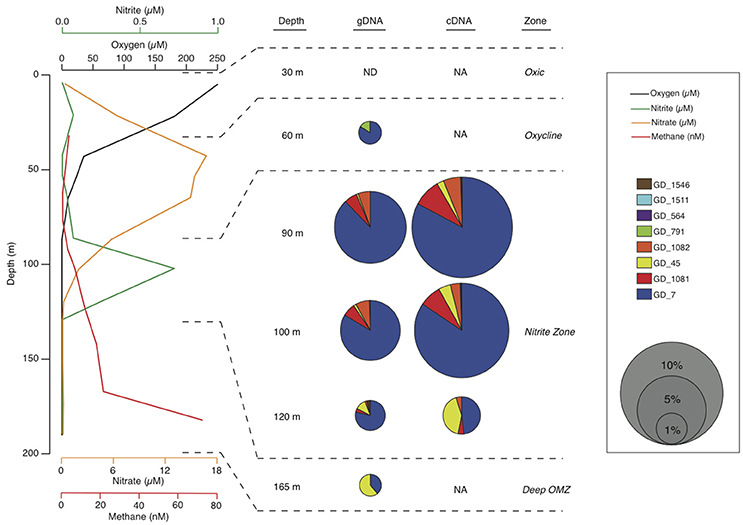
Figure 2. Water column chemistry and Methylococcales OTU abundances in the GD water column. Dissolved oxygen (measured by Clark type electrodes; black line), nitrite (green), nitrate (yellow), and methane (red) concentrations through the oxic zone (~0–30 m), oxycline (~30–80 m), anoxic nitrite-enriched zone (~80–140 m), and deeper anoxic zone (~140 m to sediment-water interface at ~190 m). Chemical data are from Padilla et al. (2016). Bubble plots show abundances of Methylococcales OTUs as a percentage of total community 16S rRNA gene (gDNA) and transcript (cDNA) amplicons. ND, Not detected; NA, No available data for that sample.
Methods and Materials
Sample Collection
The collection of samples used in this study was described in Padilla et al. (2016). Briefly, discrete water samples from depths of 30, 60, 90, 100, 120, and 165 m were collected using a hand-deployed Niskin bottle in late January from a site at the northern head of the coastal basin of GD, Costa Rica (Supplementary Figure 1). Microbial biomass was collected for molecular analysis by filtration of seawater (~1–3 L) through a glass fiber disc pre-filter (GF/A, 47 mm, 1.6 μm pore-size, Whatman) and a primary collection filter (Sterivex™, 0.22 μm pore-size, Millipore) using a peristaltic pump immediately after recovery. Filters were preserved in RNA stabilizing buffer (25 mM Sodium Citrate, 10 mM EDTA, 5.3M Ammonium sulfate, pH 5.2), flash-frozen in liquid nitrogen, and stored at −80°C. Replicate filters for DNA analysis were collected from similar water volumes following RNA collection, preserved with lysis buffer (50mM Tris-HCl, 40 mM EDTA, 0.73 M Sucrose), and stored at −80°C. Dissolved oxygen concentrations were measured with a Clark-type O2 electrode (Revsbech, 1989) mounted on a hand-deployed CTD (Sea & Sun Technology).
Chemical Analysis
Nitrite and dissolved methane concentrations at the time of biomass collection were first reported in Padilla et al. (2016) and are presented again here for context (Figure 2). Samples for nitrate were filtered (0.45 μm cellulose acetate) and frozen until analysis. Concentrations of nitrate + nitrite were determined using chemiluminescence after reduction to nitric oxide with acidic vanadium (III; Braman and Hendrix, 1989).
DNA Extraction
DNA was extracted from Sterivex filters (>0.2 μm biomass size fraction) using a phenol:chloroform protocol. Cells were lysed by adding lysozyme (2 mg in 40 μl of lysis buffer per filter) directly to the Sterivex cartridge, sealing the ends, and incubating for 45 min at 37°C. Proteinase K (1 mg in 100 μl lysis buffer, with 100 μl 20% SDS) was added, and cartridges were resealed and incubated for 2 h at 55°C. The lysate was removed, and the DNA was extracted once with phenol:chloroform:isoamyl alcohol (25:24:1) and once with chloroform:isoamyl alcohol (24:1) and then concentrated by spin dialysis using Ultra-4 (100 kDA, Amicon) centrifugal filters.
RNA Extraction
RNA was extracted from Sterivex filters using a modification of the mirVana™ miRNA Isolation kit (Ambion). Filter cartridges were thawed on ice, RNA stabilizing buffer was then expelled and discarded, and cells were lysed by adding Lysis buffer and miRNA Homogenate Additive (Ambion) directly to the cartridges. Following vortexing and incubation on ice, lysates were transferred to RNAase-free tubes and processed via acid-phenol:chloroform extraction according to the kit protocol. The TURBO DNA-free™ kit (Ambion) was used to remove DNA, and the extract was purified using the RNeasy MinElute Cleanup Kit (Qiagen).
16s rRNA Gene and Transcript Analysis
Fragments of the 16S rRNA molecule were analyzed to assess the taxonomic composition in both the community DNA (gDNA) and RNA (cDNA) pools. gDNA amplicon data were generated previously (Padilla et al., 2016) and re-analyzed here to focus on the methanotrophic community. cDNA amplicon data were generated in this study, as follows. Approximately 50 ng of RNA from each sample was reverse transcribed into cDNA using the Superscript First-Strand synthesis system for RT-PCR (Invitrogen), following protocols of Campbell et al. (2009). Amplicon generation and sequencing was done using an established pipeline in our lab (e.g., Padilla et al., 2015, 2016). Briefly, amplicons were synthesized using Platinum® PCR SuperMix (Life Technologies) with primers F515 and R806, encompassing the V4 region of the 16S rRNA gene (Caporaso et al., 2011). Both forward and reverse primers were barcoded and appended with Illumina-specific adapters according to Kozich et al. (2013). Equal amounts of cDNA were used for each PCR reaction to avoid biases due to variable template concentrations (Kennedy et al., 2014). Thermal cycling involved: denaturation at 94°C (3 min), followed by 30 cycles of denaturation at 94°C (45 s), primer annealing at 55°C (45 s), and primer extension at 72°C (90 s), followed by extension at 72°C for 10 min. Amplicons were analyzed by gel electrophoresis to verify size (~400 bp, including barcodes and adaptor sequences) and purified using the Diffinity RapidTip2 PCR purification tips (Diffinity Genomics, NY). Amplicons from different samples were pooled at equimolar concentrations and sequenced on an Illumina MiSeq using a 500-cycle kit.
For both gDNA datasets (from Padilla et al., 2016) and cDNA datasets (this study), barcoded sequences were de-multiplexed, trimmed (length cutoff 100 bp), and filtered to remove low quality reads (average Phred score <25) using Trim Galore! (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Paired-end reads were then merged using FLASH (Magoč and Salzberg, 2011), with program parameters of a minimum average length of 250 bp for each read, minimum average length of 300 bp for paired read fragments, and a maximum allowable fragment standard deviation of 30 bp. The number of trimmed and merged reads per sample ranged from 3844 to 95,351 for both gDNA and cDNA amplicon pools. Chimeric sequences were detected by reference-based searches using USEARCH (Edgar, 2010) against the SILVA rRNA database 119. Amplicon sequences were then processed using “UPARSE” as implemented in USEARCH. Briefly, sequences were dereplicated, sorted by size, and clustered into operational taxonomic units (OTUs) at 97% sequence similarity. Sequences from all samples were then mapped back to the OTU file using “usearch_global,” and sample/OTU matrices were produced from the resulting output with the python script “uc2otutable.py.” Methanotrophic sequences were identified in the amplicon OTUs by BLASTN against SILVA 119, followed by keyword parsing to identify all sequences matching a known methanotroph-containing genus. Top database matches (hits) to these sequences were also identified via BLASTN against the nt database. The SINA aligner with the ARB output designation was used to align sequences representing all identified putative methanotroph OTUs in our data (n = 11; both gDNA and cDNA amplicons pooled), the sequences most closely related to these OTUs via BLASTN, and the methanotrophs isolated in culture in Knief (2015). The resulting “.arb” file was imported into ARB (Ludwig et al., 2004). A reference phylogenetic tree was compiled using near full-length sequences (1300 bp or greater) with the Feldstein correction, a custom filter based on the associated nucleotide-alignment, and bootstrap support of 1000 re-samplings. Short sequences (1300 or less), including the amplicon sequences, were then inserted into the near full-length reference tree using the parsimony tool in the ARB environment. Proportional abundances of identified methanotroph OTUs (Figure 2) were calculated after rarefaction based on the sample with the lowest number of reads (3844 reads) for both gDNA and cDNA. In rarefying the data some OTUs that were either in low abundance or unique to specific samples were removed; their contribution to the total (all samples) community is expected to be negligible.
Metagenome (DNA) Sequencing
A single depth, 90 m at the peak of measured OMZ methane oxidation rates (Padilla et al., 2016) and methanotroph abundance based on amplicon sequence data, was selected for metagenome sequencing. Community DNA was processed using the Nextera XT DNA Sample Prep kit and sequenced using a paired-end Illumina MiSeq 600 kit.
Metatranscriptome (cDNA) Sequencing
Metatranscriptomes from three depths within the anoxic zone (90, 100, 120 m) were analyzed to assess methanotroph transcriptional activity. Three datasets representing these depths were generated previously (Padilla et al., 2016) and were reanalyzed in this study. To increase the number of sequences representing the methanotroph-enriched 90 m depth, a fourth dataset was generated using an aliquot of the total RNA used in Padilla et al. (2016). For this aliquot, the Ribo-Zero™ rRNA Removal Kit for bacteria (Epicenter) was used to deplete ribosomal RNA (rRNA) sequences prior to sequencing. rRNA was not depleted from the RNA aliquots used to generate the Padilla et al. (2016) datasets. For all samples, cDNA was prepared for sequencing using the ScriptSeq™ v2 RNA-Seq Library preparation kit (Epicenter) and sequenced on an Illumina MiSeq using a 500 cycle kit. Sequencing statistics are listed in Supplementary Table 1.
Metagenomic Analysis
Sequences were trimmed using the same methods as described above for the amplicon analysis. Quality trimmed forward and reverse sequences were merged and assembled into contigs using SPAdes 3.7.0 (Nurk et al., 2013) with the “-meta” option. The number of contigs, contig length, GC content, N50, and L50 assembly statistics were calculated with metaQUAST (Mikheenko et al., 2016; Supplementary Table 2). Contigs ≥ 500 bp were organized into genome bins based on tetranucleotide frequency and sequence coverage using MaxBin 2.0 (Wu et al., 2016). Bin completeness and contamination levels were estimated based on the representation of lineage-specific marker gene sets using CheckM (Parks et al., 2015). Assembly and genome bin statistics are in Supplementary Table 3. For each bin with ≥50% completeness and ≤10% contamination, the program Prodigal (Hyatt et al., 2010) was used to predict open reading frames (ORFs). Predicted ORFs were queried via BLASTP against the NCBI-nr database (April 2016; criteria for a significant match: amino acid ID ≥40% over ≥70% of the alignment, and bit score ≥50) and assigned to a prokaryotic KEGG Orthology (KO) identifier (Kanehisa and Goto, 2000) using the MetaGenome Analyzer 5 (MEGAN5; Hudson et al., 2011), with taxonomic classification in MEGAN5 via the Lowest Common Ancestor (LCA) algorithm based on the NCBI taxonomy using the default settings. Annotations are reported in Supplementary Tables 4, 5.
Based on high completeness and low contamination (see below), a single bin (bin 010) taxonomically affiliated with OPU3-like Methylococcales bacteria was selected for comparative analysis. The identity of this bin was further confirmed based on phylogenetic analysis of 107 concatenated single copy marker genes present in this bin and in the genomes (n = 36) of known methanotrophic taxa from the Gammaproteobacteria, Alphaproteobacteria, Verrucomicrobia, and the NC10 candidate phylum. Marker genes were identified using Hidden Markov Models (HMMs) via HMMER3 (http://hmmer.org/; Finn et al., 2011) with default settings. Identified genes were aligned using clustalW (Sievers et al., 2011), then concatenated using the alignment tool “Aln.cat.rb” in the enve-omics package (Rodriguez-R and Konstantinidis, 2016). The resulting alignment was inspected manually for errors and then used to generate a maximum likelihood phylogeny inferred with the Dayhoff substitution model with 100 bootstrap iterations using MEGA6 (Tamura et al., 2013). For each genome in the phylogeny, the presence/absence of key genes of dissimilatory and assimilatory nitrogen metabolism was evaluated via BLASTX using the KEGG Automated Annotation Server (KAAS). Each genome was individually assessed with the bi-directional best-hit option on default settings and the results were manually searched for subunits of nitrogen metabolism genes, with the results (presence/absence) mapped onto the concatenated marker gene phylogeny. Gene content in bin 010 relative to the 36 other methanotroph genomes included in this analysis was evaluated via reciprocal BLASTP using the two-way amino-acid identity script in the enve-omics packages (Konstantinidis and Tiedje, 2005) with a bit score threshold of 50, and match criteria of >35% amino acid identity over >65% of each gene (Supplementary Table 4).
Phylogenetic analysis was used to further examine the taxonomic affiliation of key genes of the denitrification process recovered in bin 010: narG and nirK. The amino acid sequence from each gene was aligned against a representative sequence set, identified based on top BLASTP results, using MUSCLE with default settings (Edgar, 2004). The alignments were manually inspected and used to generate maximum likelihood phylogenies using MEGA6.0 with the Dayhoff substitution model and 1000 bootstrap iterations. To further characterize taxonomic affiliations, the taxonomic annotation of all genes occurring on the same contig as key marker genes of denitrification and methanotrophy (nar, nir, pmo) was determined via BLASTX against NCBI-nr (Supplementary Figure 2).
Metatranscriptome Analysis
Reads were filtered by quality and merged as described above. The program ribopicker (Schmieder et al., 2012) was used to remove rRNA sequences from metatranscriptome datasets. To characterize the transcript pool affiliated with the OPU3 genomic bin described above, transcripts were mapped to bin 010 ORFs using BLASTX with match criteria of >90% amino acid identity over >60% of the transcript length and bit score >50. To compare transcript levels among bin 010 ORFS, the number of transcripts with significant BLASTX matches per ORF was normalized to variation in ORF length, and then expressed as a proportion of total transcripts mapping to bin 010. To also characterize the transcript pool at the community level, merged non-rRNA datasets were queried via DIAMOND (Buchfink et al., 2015) against the NCBI-nr database (April 2016) using the sensitive search setting. DIAMOND-identified protein-coding transcripts were assigned to functional categories as above based on Kegg Ortholog (KO) identifiers and taxonomically classified according to the NCBI taxonomy using the LCA algorithm in MEGAN5. Transcript counts per KO were normalized to the total number of mRNA transcripts assigned to a prokaryotic KO. To show variations in contributions of bin 010 to community mRNA transcription with depth, the number of transcripts with significant BLASTX matches to bin 010 ORFs was expressed as a proportion of total prokaryotic mRNA transcripts assignable to a KO.
All sequence data, including the bin 010 assembly, have been submitted to the Sequence Read Archive at NCBI under the following BioProject ID's: PRJNA328797 and PRJNA277357.
Results and Discussion
Methanotroph Community Composition and Transcription
Deep-coverage sequencing of 16S rRNA gene (gDNA) and transcript (cDNA) amplicons revealed an abundant, diverse, and active methanotroph community in the Golfo Dulce OMZ. All sequences that matched putative aerobic methanotrophs were classified as type Ia Gammaproteobacteria methanotrophs. Across all depths, 11 OTUs from the gDNA dataset were identified as Methylococcales by BLASTN. Eight of these OTUs remained after rarefaction (3844 reads) of the total gDNA and cDNA amplicon pools. Phylogenetic analysis classified these eight OTUs as most closely related to methanotrophs of the OPU3 and OPU1 clusters and the genus Methyloprofundus (Figure 1), with six of the eight belonging to the OPU3 clade. A single OPU3 OTU (GD_7) dominated the methanotroph sequence dataset, constituting ~35–75% of the Methylococcales pool in both the gDNA and cDNA datasets at all depths (Figure 2). Only one OTU from the OPU1 clade (GD_791) was detected and was confined primarily to the 60 m sample where O2 concentration was ~25 μM. Together, these eight OTUs, which collectively represented 1.8% of the total gDNA combined over all depths, were undetected or at low abundance (0.4%) above or within the oxycline (30 and 60 m, respectively) but increased to 4–5% of the gDNA pool in the upper anoxic zone (90 and 100 m) where O2 concentrations fell below detection, <50 nM, and nitrite accumulated (Figure 2). Notably, OTU GD_7, the dominant OPU3 bacterium, constituted 4% of the total microbial community (gDNA pool) at 90 m. These OTUs then decreased to <1% of the community deeper in the anoxic zone at 120 m, and at 165 m where nitrate and nitrite were depleted and methane accumulated. The proportional representation of these eight OTUs was ~2-fold higher in the cDNA compared to the gDNA datasets at all depths, indicating a transcriptionally active Methylococcales community.
Recovery of the Genome of an OPU3-Like Methanotrophic Denitrifier
Metagenomic analysis confirmed the potential for coupled methanotrophy and denitrification in the Golfo Dulce OMZ. Genomic DNA from within the secondary nitrite maximum at 90 m was deeply sequenced, assembled into contigs, and binned into OTUs resulting in 52 draft genomes (bins) ranging from 12.4 to 100% completeness and 0–86% contamination (Supplementary Table 3). Of these, bin 010 contained a single near-full length (1405 bp) 16S rRNA gene fragment identical to that of OTU GD_7 identified in the amplicon analysis (above). The contig containing this fragment consisted solely of the 16S rRNA gene and exhibited an average per-base coverage of 6.6, nearly identical to the bin-wide average (all contigs) of 6.7. The bin contamination level was 6% based on duplicated marker genes, a level consistent with that reported in other studies drawing conclusions of genome content based on binning [see Sekiguchi et al., 2015 (6–7%); Campanaro et al., 2016 (3–5%); Güllert et al., 2016 (5–10%)]. On average, duplicate marker genes in bin 010 shared 81% amino acid identity, within the genome-wide range observed for bacterial strains categorized in the same species (Konstantinidis and Tiedje, 2005), suggesting that contaminant sequences in this bin are from closely related taxa.
Given the high abundance of OTU GD_7 at >4% in both the gDNA and cDNA amplicon pools (Figure 2), suggesting a substantial contribution to OMZ community processes, bin 010 was selected for in-depth characterization. The genome represented by this bin, hereafter designated GD_7, was estimated to be 95.3% complete, with 2011 coding sequences distributed over 305 contigs and a total of 2,106,486 bp (Supplementary Table 2). The estimated GD_7 genome size, at 2.2 Mbp, is consistent with that of pelagic free-living bacteria (Raes et al., 2007; Shi et al., 2011), but substantially smaller than that of most other gammaproteobacterial methanotrophs (~4–5 Mbp; Figure 3). LCA-based taxonomic classification of GD_7 coding sequences indicated an affiliation with the order Methylococcales. Phylogenetic analysis of 107 housekeeping genes (concatenated) from available methanotroph genomes confirmed GD_7 as basal but most closely related to the type-Ia methanotrophs of the Methylococcales, a diverse group of bacteria with some members shown experimentally to couple methane oxidation to denitrification (Figure 3; Kits et al., 2015a,b).
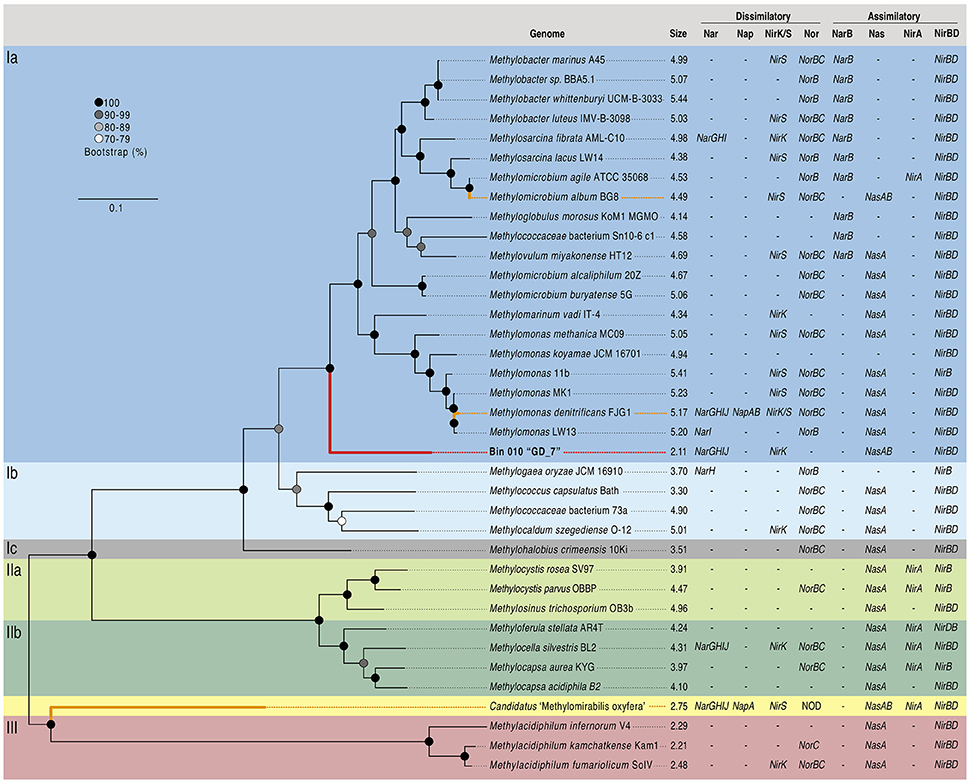
Figure 3. Concatenated gene phylogeny of methanotrophic genomes and description of N utilizing genes. Phylogeny based on the concatenated alignment of 107 single copy housekeeping genes from OTU GD_7 (bin 010) and known methanotroph genomes (n = 36). Maximum likelihood phylogeny was inferred based on the Dayhoff substitution model, bootstrapped 100 times; bootstrap support values ≥70 are displayed. Phylogenetic classification (Ia, Ib, Ic, IIa, IIb, III) is based on a recent review (Knief, 2015). Genome size in megabases and presence/absence of key genes of dissimilatory and assimilatory nitrogen metabolism are displayed next to taxon names. Experimentally confirmed denitrifying taxa are highlighted with bold orange branches, with the assembled bin 010 denoted by the bold red branch. The scale bar represents amino acid substitutions per site. Abbreviations are as follows: Nar, membrane bound nitrate/nitrite oxidoreductase; Nap, periplasmic nitrate/nitrite oxidoreductase; NirK, copper containing nitrite reductase; NirS, cytochrome cd1 nitrite reductase; Nor, nitric oxide reductase; NOD, nitric oxide dismutase (putative); NarB, ferredoxin nitrate reductase; Nas, assimilatory nitrate reductase; NirA, assimilatory nitrite reductase; NirBD, assimilatory nitrite reductase.
Below, we discuss a subset of key functional genes recovered in the GD_7 bin. As the inferred genome is fragmentary and exhibits 6% contamination (above), confirmation that the recovered genes are co-localized on the same chromosome in OPU3 requires further analysis of a pure culture or a closed genome. Similarly, as the recovered GD_7 genome is not 100% complete, we cannot definitively conclude that specific genes are absent from this genome. However, the gene content described below, when contextualized relative to that of other methanotrophs, prior results, and the environmental conditions of Golfo Dulce, is strongly suggestive of key metabolic functions in GD_7.
The genome content of GD_7 indicates the potential for methanotrophic denitrification. GD_7 contains genes required for energy generation by methane oxidation, including those encoding the three subunits of particulate methane monooxygenase (pmoCAB) for methane oxidation to methanol, which together co-occur on a contig with diverse other genes having close homologs in other gammproteobacterial methanotrophs (inferred from top BLASTX matches; Supplementary Figure 2, Supplementary Table 4). Also present are genes for the lanthanide-dependent (xoxF) methanol dehydrogenase involved in methanol oxidation to formaldehyde, NAD(P)-dependent methylene-H4MPT dehydrogenase (mtd) involved in formaldehyde oxidation to formate, and formate dehydrogenase (fdh) for formate oxidation to CO2. The presence of XoxF type methanol dehydrogenases, but absence of the calcium-dependent MxaF type, has also been reported for pelagic non-methanotrophic methylotrophs with relatively small genomes (Giovannoni et al., 2008) and is consistent with recent studies suggesting the prevalence of XoxF compared to MxaF in natural systems (Keltjens et al., 2014; Ramachandran and Walsh, 2015).
GD_7 also contains two operons encoding a dissimilatory nitrate reductase (nar), as well as genes for synthesis of the essential nar molybdenum cofactor. Of the downstream genes of the denitrification pathway, only the nirK gene encoding nitric oxide (NO)-producing nitrite reductase was detected in GD_7. The two nar operons, as well as nir, were recovered on contigs alongside other functional genes with homology to those of gammaproteobacterial methanotrophs (Supplementary Figure 2). Phylogenetic analysis of marker genes nirK and narG, with the latter encoding the alpha subunit of Nar, also supports a methanotroph affiliation. One of the narG copies (ORF 1321) clusters with high support in a clade containing denitrifying gammaproteobacterial methanotrophs, including Methylomonas denitrificans (Figure 4A). Likewise, GD_7 nirK clusters with a sequence from the gammaproteobacterial Methylothermaceae and that of the nitrite-oxidizing bacterium Nitrospina gracilis, although the function of NirK in Nitrospina remains unclear (Lüker et al., 2013; Figure 4B). In contrast, the second nar operon may have an origin outside of the gammaproteobacteria, as analysis of the corresponding narG copy (ORF 1413) reveals a closer affiliation to narG from a genus of Chlorobi bacteria (Ignavibacterium) containing facultatively anaerobic chemoheterotrophic members (Figure 4A).
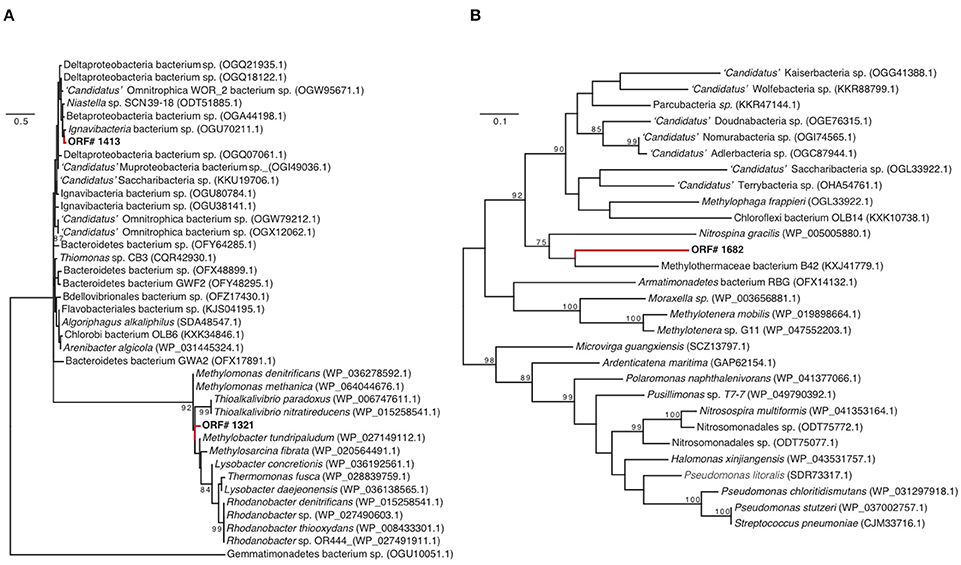
Figure 4. Nitrate reductase (A) (narG) and nitrite reductase (B) (nirK) phylogenies. The trees include closely related sequences identified as top matches to GD_7 narG and nirK amino sequences through BLASTP queries against NCBI-nr. Unrooted phylogenies were inferred based on maximum likelihood analysis using the Dayhoff substitution model with 1000 bootstrap iterations. Bootstrap values >70 are displayed, along with NCBI accession numbers. Scale bars for narG and nirK represent 50 and 10 changes per 100 amino acids (respectively).
These results suggest that certain genes for partial denitrification in GD_7 share ancestry with those of gammaproteobacterial methanotrophs, while others, notably ORF 1413, may have been acquired horizontally (Figure 4A). Horizontal acquisition of denitrification genes in methanotrophs may be common, as the presence of nar and nir genes is independent of methanotrophic phylogenetic histories (Figure 3). Indeed, the co-occurrence of divergent denitrification genes in the same genome is not unusual. Many bacteria, for example, encode divergent nar copies (Philippot, 2002; Tsementzi et al., 2016), potentially as an adaptation to variable oxygen conditions (Iobbi-Nivol et al., 1990). Although multiple nar operons have yet to be reported in other methanotrophs, a marine methylotroph was shown to encode two nar enzymes, with one copy playing a role in regulating the other depending on oxygen availability (Mauffrey et al., 2015). Some denitrifying methanotrophs also encode the perisplasmic dissimilatory nitrate reductase (Nap), hypothesized to function primarily under nitrate-limiting conditions (Figure 3; Ferguson and Richardson, 2004); however, nap genes were not detected in GD_7. In contrast to Methylomonas denitrificans FJG1, Methylomicrobium album BG8, and Candidatus “Methylomirablis oxyfera,” GD-7 does not appear to harbor an NO-reductase (nor), and is therefore likely incapable of producing N2O as an intermediate or end-product (Figure 3; Kits et al., 2015a,b). In M. denitrificans FJG1, M. album BG8, and potentially GD-7, nitrate is likely utilized for respiratory purposes. This is in contrast to NC10 members, which likely utilize nitric oxide (derived from nitrate and nitrite reduction) for dismutation with concomitant production of intracellular oxygen. Rather, our data identify a partial methanotrophic denitrification pathway in GD_7, with NO as the likely respiratory end product.
GD_7 also contains genes encoding key enzymes of C1 carbon assimilation from methane, including those of the ribulose monophosphate (RuMP) pathway for incorporation of methanol-derived formaldehyde and regeneration of ribulose-5-phosphate (Supplementary Figure 3). Key enzymes of the serine pathway for formaldehyde assimilation were also detected (Table 1, Supplementary Tables 4, 5), with 10 of the serine cycle genes recovered on the same contig. Diagnostic enzymes of the ethylmalonyl-CoA (EMC) pathway (ethylmalonyl-CoA mutase, crotonyl-CoA reductase/carboxylase), used by some serine cycle-utilizing methanotrophs for glyoxylate regeneration (Chistoserdova, 2011), were not detected in GD_7. Neither were isocitrate lyase and malate synthase, suggesting glyoxylate generation does not occur via the glyoxylate cycle in GD_7. This pattern, the presence of serine cycle genes and absence of an EMC pathway and glyoxylate shunt, has been reported in other type 1 methanotrophs (Poehlein et al., 2013; de la Torre et al., 2015). In GD_7, all enzymes of the Citric Acid (TCA) cycle are present, as are genes of glycolysis and for pyruvate dehydrogenase for acetyl-CoA generation (Supplementary Table 4). However, GD_7 appears to lack the genes encoding the Entner-Doudoroff Pathway, which would be atypical of Type I methanotrophs that have been hypothesized to depend primarily on Entner-Doudoroff cleavage reactions over glycolysis (Kalyuzhnaya et al., 2013). However, we cannot rule out that these or other genes were not detected due to incomplete coverage (~95%) of this genome bin. Together, these patterns suggest C1 incorporation from methane via the RuMP pathway, with acetyl-CoA synthesis via pyruvate for entry into the TCA cycle for biosynthesis.
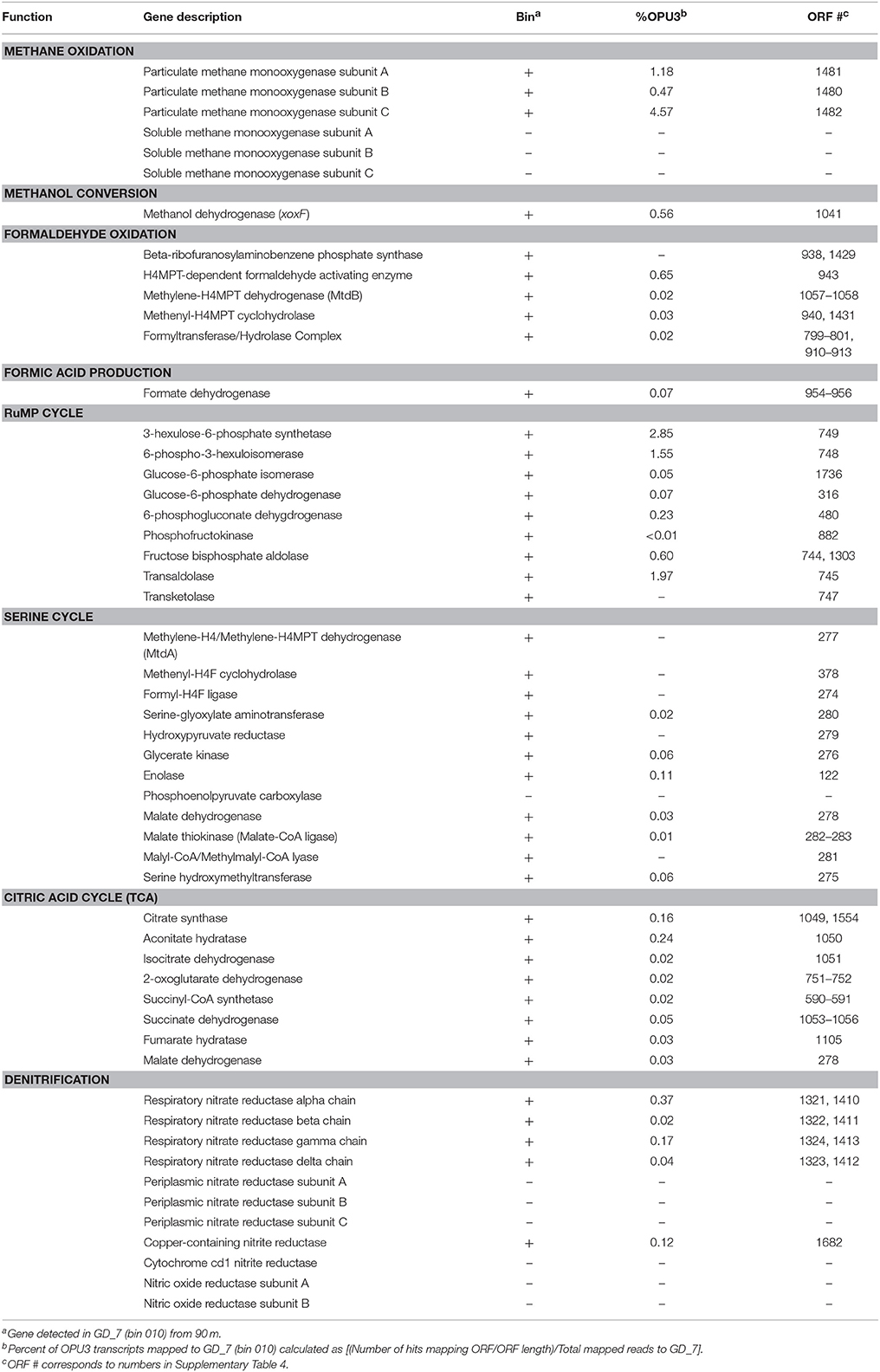
Table 1. Representation of methane oxidation and denitrification genes present (±) in GD_7 (bin 010) and abundance in the coupled 90 m metatranscriptome.
Reciprocal BLASTP against all methanotrophic bacteria in Figure 3 identified 102 protein-coding genes in GD_7 that lack close homologs in other methanotroph genomes (based on the imposed BLAST criteria: bit score >50, with >35% amino acid identity across >65% of the gene). Of this divergent gene set, 47 encode hypothetical proteins (Supplementary Table 4). The remaining annotated genes are associated with diverse functions, including flagellum synthesis or regulation, phage tail synthesis, and diverse functions of amino acid or peptide metabolism, as well as an arginase potentially involved in urea generation. Genes in this divergent set may be candidates for follow-up studies to better understand niche-specific adaptations in GD_7.
OPU3 Transcription in the OMZ
To characterize the Methylococcales-like transcript pool, we examined transcripts from 90, 100, and 120 m that mapped with high identity to GD_7. To increase coverage across the bin, we analyzed an additional 90 m dataset from which rRNA was removed prior to sequencing. This step increased data yield but altered the abundance of mapped transcripts (expressed as a % of total prokaryotic mRNA; Figure 5), suggesting that rRNA subtraction may also have removed certain mRNA transcripts. This procedure does not affect our conclusions, however, as our goal was to characterize the range of genes actively transcribed by GD_7; variation in transcription among genes is presented in only a general sense (e.g., Figure 5). Nonetheless, the rRNA-subtracted dataset is not considered when evaluating changes across depths (below).
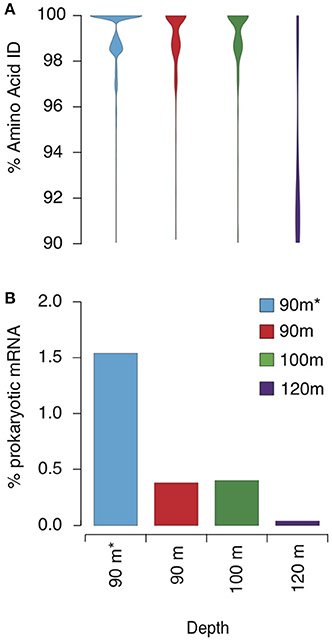
Figure 5. Transcripts mapping to GD_7. (A) Violin plots indicating the proportion of mRNA reads at each amino acid %ID in each sample. (B) Percentage of mRNA recruited to the bin from each sample as a proportion of prokaryotic mRNA assigned by MEGAN5. Asterisk (*) 90 m sample denotes rRNA removal prior to sequencing using Ribo-Zero™ rRNA Removal Kit for bacteria (Epicentre). Transcript mapping to GD_7 is based on BLASTX with the following criteria: bit score ≥ 50, amino acid ID ≥ 90%, and alignment length ≥ 60% of the gene.
Metatranscriptome analysis confirmed that Methylococcales bacteria were transcriptionally active in the Golfo Dulce OMZ. Across all datasets, a total of 24,989 reads representing 764 genes were recruited via BLASTX to GD_7 contigs with high identity (>90% AAI; bit scores >50) across 60% of the transcript fragment (Supplementary Table 5). The proportional representation of these mapped transcripts peaked at 90 and 100 m at ~0.5% of total mRNA reads (non-rRNA-subtracted dataset) before decreasing to <0.1% deeper in the anoxic zone (120 m sample; Figure 5B). Indeed, at 90 and 100 m, the vast majority (~90%) of recruited transcripts share >98% AAI with genes of GD_7 (Figure 5A; Supplementary Figure 4). In contrast, at 120 m, the majority of mapped reads share <94% AAI. These patterns suggest discrete OPU3-like populations based on depth, with GD_7 confined primarily to the upper OMZ where both nitrite and nitrate are enriched compared to greater depths in the anoxic zone (Figure 2).
Transcripts involved in methanotrophy and denitrification were among the most abundant of those recruiting to GD_7, based on the mRNA-enriched 90 m dataset (Supplementary Figures 4, 5; Supplementary Table 5). Mapped transcripts included all genes of the aerobic methane oxidation pathway (Table 1), with pmoA and pmoC being particularly abundant, as well as genes of the RuMP pathway. Denitrification transcripts included narG and nirK, both of which were detected at 90 and 100 m, but absent from the 120 m dataset. Interestingly, only transcripts mapping to narG ORF 1413 were detected (Supplementary Table 5), suggesting differential regulation of Nar expression and therefore the potential for functional variation between the two enzyme variants (ORF 1321, 1413). Terminal oxidase genes, though present in GD_7, were not detected in the mapped transcript pool, suggesting that any oxygen being utilized is for methane oxidation rather than respiration. Genes for fatty acid metabolism were also among the most abundant transcripts recruiting to GD_7, as were those mediating flagellar biosynthesis and regulation. The high transcription of flagellar genes is consistent with prior reports of the importance of motility in type 1 methanotrophs from low oxygen environments (Danilova et al., 2016), suggesting a need to traverse redox gradients to access zones of optimal substrate (e.g., methane) or oxygen conditions, potentially including those at the micron scale on suspended or sinking particles. Enzymes involved in amino acid metabolism were also abundant in the GD_7 transcript pool (Supplementary Table 5, Supplementary Figure 5), with proline dehydrogenase being among the most highly transcribed GD_7-specific protein at 90 m. This enzyme is involved in proline catabolism, producing reductant in the form of reduced co-enzyme Q10 (i.e., ubiquinol). Although use of proline for reductant has been reported for diverse bacteria under both oxic and anoxic conditions (Deutch and Soffer, 1975; Barker, 1981), the potential contribution of amino acid catabolism to reductant pools in methanotrophs, including OPU3 members, remains uncharacterized. Genes encoding enzymes of the methylcitrate pathway (MCA) involved in propionate catabolism were also highly transcribed (Supplementary Table 5, Supplementary Figure 5). These included methylcitrate synthase, methylcitrate dehydrogenase and aconitate hydratase, although the MCA gene encoding methylisocitrate lyase was not detected. MCA genes and transcripts have been reported in aquatic betaproteobacterial methylotrophs, raising the hypothesis that propionate, which can be produced via demethylation of compounds such as dimethylsulfoniopropionate, may be an important carbon substrate in these bacteria (Kalyuzhnaya et al., 2008, 2010). The potential for propionate catabolism in OPU3 should be further evaluated.
Taxonomic classification of the bulk metatranscriptome based on lowest common ancestry (LCA) corroborated the transcriptional activity of a Methylococcales community in the OMZ. The majority (~80%) of LCA-identified Methylococcales transcripts could not be assigned to a genus (Supplementary Figure 6). Of those transcripts that could be classified, most are affiliated with Methylomonas, Methylobacter, or Methylomicrobium. The proportional abundance of total Methylococcales transcripts across depths was nearly identical to that of the transcript pool mapping to GD_7 (Figure 5B, Supplementary Figure 6), indicating that the vast majority of Methylococcales transcripts were affiliated with this OPU3 taxon and that its contribution to community transcription peaked at 90 and 100 m just below the oxycline. As in the GD_7 transcript pool, biochemical functions highly represented in the bulk Methylococcales transcripts were predominantly associated with methane oxidation and C1 assimilation, with transcripts encoding pmoCAB among the most abundant (data not shown).
Conclusion
These results provide the first environmental meta-omic evidence for coupled methanotrophy and partial denitrification in the OPU3 bacterial clade. The recovery of a near complete genome for GD_7, interpreted alongside community composition data and linked metatranscriptome analysis identifying transcripts of methane-oxidation, C1 assimilation via the RuMP pathway, and partial denitrification, implicate this abundant OTU as a potentially significant player in methane-driven nitrogen transformations in the Golfo Dulce. The localized distribution at 90 m of transcripts with high identity to GD_7 (Figure 5), along with the recovery of diverse other OPU3 OTUs, suggests the potential for temporal or spatial variation in the contribution of different OPU3 ecotypes to community metabolism, likely driven by local concentrations of methane, oxidized nitrogen, or dissolved oxygen.
Our results suggest that GD_7 is adapted to conditions at the upper OMZ periphery where oxygen is absent but nitrate, nitrite, and methane are all available. Prior studies have suggested a low-to-no oxygen niche for the OPU3 group as a whole, notably as pelagic methane concentrations typically increase with decreasing oxygen content (Sansone et al., 2001; Pack et al., 2015). Indeed, the abundance of OPU3-like OTUs has been shown to increase with decreasing oxygen further off the Costa Rican coast (Tavormina et al., 2013). However, the activity of methanotrophic denitrifiers in the OMZ water column may also be driven strongly by nitrite conditions; for example, in Methylomicrobium album, transcription of methanotrophy genes and nitrous oxide production via denitrification was dependent on both nitrite availability and low oxygen conditions, whereas denitrification gene transcription was stimulated only by nitrite availability (Kits et al., 2015a). In the GD, the distribution of OPU3 amplicons and transcriptional activity, notably those of GD_7, mirror that of the nitrite profile (Figure 2). The high abundance of OPU3 genes and transcripts in the upper OMZ corresponds with the previously reported maximum in methane oxidation rates at 90 m, whereas rates lower in the OMZ were undetectable at the time of collection (Padilla et al., 2016). Despite the anoxia at 90 m, it remains possible that the rates reported in Padilla et al. (2016) represent a combination of both aerobic and denitrification-dependent methane oxidation, potentially due to inter-individual variation in an OPU population or variation among OPU ecotypes. Genomes and gene expression studies targeting diverse OPU phylotypes from the GD and other low oxygen marine habitats would be valuable for resolving these possibilities. Characterizing the metabolic diversity of pelagic methane utilizing bacteria is critical for predicting linkages between greenhouse gas, carbon, and nutrient fluxes. This is particularly important for microbial communities along the steep redox gradients of OMZs, as these habitats are predicted to expand in response to climate change.
Author Contributions
CP, BT, JG, and FS conceived and designed the study. NS, FS, LB, and BT collected field samples. FS, CP, and AB designed the sequencing and analysis approach. CP, AB, and NS extracted nucleic acids and performed sequencing reactions and sequence processing and conducted bioinformatics analyses. LB and BT conducted chemical measurements. CP, AB, JG, BT, LB, and FS analyzed the data. CP, AB, and FS wrote the paper with contributions from all authors.
Funding
This research was supported by the National Science Foundation (1151698, 1558916, 1564559 to FS), the Sloan Foundation (RC944 to FS), the Danish National Research Foundation DNRF53, the Danish Council of Independent Research, and the European Research Council “Oxygen” grant (267233; supporting LB and BT).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Alvaro Morales for coordinating work at the GD field site and Pete Girguis for analysis of methane concentrations.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2017.00023/full#supplementary-material
References
Barker, H. A. (1981). Amino acid degradation by anaerobic bacteria. Ann. Rev. Biochem. 50, 23–40. doi: 10.1146/annurev.bi.50.070181.000323
Biderre-Petit, C., Jézéquel, D., Dugat-Bony, E., Lopes, F., Kuever, J., Borrel, G., et al. (2011). Identification of microbial communities involved in the methane cycle of a freshwater meromictic lake. FEMS Microbiol. Ecol. 77, 533–545. doi: 10.1111/j.1574-6941.2011.01134.x
Blees, J., Niemann, H., Wenk, C. B., Zopfi, J., Schubert, C. J., Kirf, M. K., et al. (2014). Micro-aerobic bacterial methane oxidation in the chemocline and anoxic water column of deep south-Alpine Lake Lugano (Switzerland). Limnol. Oceanogr. 59, 311–324. doi: 10.4319/lo.2014.59.2.0311
Braman, R. S., and Hendrix, S. A. (1989). Nanogram nitrite and nitrate determination in environmental and biological materials by vanadium (III) reduction with chemiluminescence detection. Anal. Chem. 61, 2715–2718. doi: 10.1021/ac00199a007
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Campanaro, S., Treu, L., Kougias, P. G., De Francisci, D., Valle, G., and Angelidaki, I. (2016). Metagenomic analysis of functional characterization of the biogas microbiome using high throughput shotgun sequencing and a novel binning strategy. Biotech. Biofuel. 9:1. doi: 10.1186/s13068-016-0441-1
Campbell, B. J., Yu, L., Straza, T. R. A., and Kirchman, D. L. (2009). Temporal changes in bacterial rRNA and rRNA genies in Delaware (USA) coastal waters. Aquat. Microb. Ecol. 57, 123–135. doi: 10.3354/ame01335
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Carini, P., White, A. E., Campbell, E. O., and Giovannoni, S. J. (2014). Methane production by phosphate-starved SAR11 chemoheterotrophic marine bacteria. Nat. Comm. 5, 4346. doi: 10.1038/ncomms5346
Chistoserdova, L. (2011). Modularity of methylotrophy, revisited. Environ. Microb. 13, 2603–2622. doi: 10.1111/j.1462-2920.2011.02464.x
Chistoserdova, L. (2015). Methylotrophs in natural habitats: current insights through metagenomics. Appl. Microbiol. Biot. 99, 5763–5779. doi: 10.1007/s00253-015-6713-z
Codispoti, L. A., Brandes, J. A., Christensen, J. P., Devol, A. H., Naqvi, S. W. A., Paerl, H. W., et al. (2001). The oceanic fixed nitrogen and nitrous oxide budgets: moving targets as we enter the anthropocene? Sci. Mar. 65, 85–105. doi: 10.3989/scimar.2001.65s285
Dalsgaard, T., Canfield, D. E., Petersen, J., Thamdrup, B., and Acuña-González, J. (2003). N2 production by the anammox reaction in the anoxic water column of Golfo Dulce, Costa Rica. Nature 422, 606–608. doi: 10.1038/nature01526
Damm, E., Helmke, E., Thoms, S., Schauer, U., Nothig, E., Bakker, K., et al. (2010). Methane production in aerobic oligotrophic surface water in the central Arctic Ocean. Biogeosciences 7, 1099–1108. doi: 10.5194/bg-7-1099-2010
Danilova, O. V. Suzina, N. E., Van De Kamp, J., Svenning, M. M., Bodrossy, L., and Dedysh, S. N. (2016). A new cell morphotype among methane oxidizers: a spiral-shaped obligately microaerophilic methanotroph from northern low oxygen environments. ISME J. 10, 2734–2743. doi: 10.1038/ismej.2016.48
de la Torre, A., Metivier, A., Chu, F., Laurens, L. M. L., Beck, D. A. C., Pienkos, P. T., et al. (2015). Genome-scale metabolic reconstructions and theoretical investigation of methane conversion in Methylomicrobium buryatense strain 5G(B1). Microb. Cell Fact. 14, 188. doi: 10.1186/s12934-015-0377-3
Deutch, C. E., and Soffer, R. L. (1975). Regulation of proline catabolism by leucyl, phenylalanyl-tRNA-protein transferase. Proc. Natl. Acad. Sci. U.S.A. 72, 405–408. doi: 10.1073/pnas.72.1.405
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548. doi: 10.1038/nature08883
Ferguson, S. J., and Richardson, D. J. (2004). “The enzymes and bioenergetics of bacterial nitrate, nitrite, nitric oxide and nitrous oxide respiration,” in Respiration in Archaea and Bacteria, ed D. Zannoni (Springer), 169–206.
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Giovannoni, S. J., Hayakawa, D. H., Tripp, H. J., Stingl, U., Givan, S. A., Cho, J.-C., et al. (2008). The small genome of an abundant coastal ocean methylotroph. Environ. Microbiol. 10, 1771–1782. doi: 10.1111/j.1462-2920.2008.01598.x
Güllert, S., Fischer, M. A., Turaev, D., Noebauer, B., Ilmberger, N., Wemheuer, B., et al. (2016). Deep metagenome and transcriptome analyses of microbial communities affiliated with an industrial biogas fermenter, a cow rumen, and elephant feces reveal major differences in carbohydrate hydrolysis strategies. Biotech. Biofuels 9:121. doi: 10.1186/s13068-016-0534-x
Hayashi, T., Obata, H., Gamo, T., Sano, Y., and Naganuma, T. (2007). Distribution and phylogenetic characteristics of the genes encoding enzymes relevant to methane oxidation in oxygen minimum zones of the Eastern Pacific Ocean. Res. J. Environ. Sci. 1, 275–284. doi: 10.3923/rjes.2007.275.284
Helly, J. J., and Levin, L. A. (2004). Global distribution of naturally occurring marine hypoxia on continental margins. Deep Sea Res. Pt. I. 51, 1159–1168. doi: 10.1016/j.dsr.2004.03.009
Hudson, D. H., Mitra, S., Ruscheweyh, H. J., Weber, N., and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. doi: 10.1101/gr.120618.111
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Iobbi-Nivol, C., Santini, C. L., Blasco, F., and Giordano, G. (1990). Purification and further characterization of the second nitrate reductase of Escherichia coli K12. Eur. J. Biochem. 188, 679–687. doi: 10.1111/j.1432-1033.1990.tb15450.x
IPCC (2013). Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge; New York, NY: Cambridge University Press.
Jensen, S., Neufeld, J. D., Birkeland, N. K., Hovland, M., and Murrell, J. C. (2008). Insight into the microbial community structure of a Norwegian deep-water coral reef environment. Deep Sea Res. Pt. I. 55, 1554–1563. doi: 10.1016/j.dsr.2008.06.008
Kalyuzhnaya, M. G., Beck, D. A., Suciu, D., Pozhitkov, A., Lidstrom, M. E., and Chistoserdova, L. (2010). Functioning in situ: gene expression in Methylotenera mobilis in its native environment as assessed through transcriptomics. ISME J. 4, 388–398. doi: 10.1038/ismej.2009.117
Kalyuzhnaya, M. G., Lapidus, A., Ivanova, N., Copeland, A. C., McHardy, A. C., Szeto, E., et al. (2008). High-resolution metagenomics targets specific functional types in complex microbial communities. Nat. Biotechnol. 26, 1029–1034. doi: 10.1038/nbt.1488
Kalyuzhnaya, M. G., Yang, S., Rozova, O. N., Smalley, N. E., Clubb, J., Lamb, A., et al. (2013). Highly efficient methane biocatalysis revealed in a methanotrophic bacterium. Nat. Commun. 4, 2785. doi: 10.1038/ncomms3785
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Karl, D. M., Beversdorf, L., Bjorkman, K. M., Church, M. J., Martinez, A., and DeLong, E. F. (2008). Aerobic production of methane in the sea. Nat. Geosci. 1, 473–478. doi: 10.1038/ngeo234
Keltjens, J. T., Pol, A., Reimann, J., and Op den Camp, H. J. (2014). PQQ-dependent methanol dehydrogenases: rare-earth elements make a difference. Appl. Microb. Biotech. 98, 6163–6183. doi: 10.1007/s00253-014-5766-8
Kennedy, K., Hall, M. W., Lynch, M. D. J., Moreno-Hagelsieb, G., and Neufeld, J. D. (2014). Evaluating bias of Illumina-based bacterial 16S rRNA gene profiles. Appl. Environ. Microb. 80, 5717–5722. doi: 10.1128/AEM.01451-14
Kessler, J. D., Valentine, D. L., Redmond, M. C., Du, M. R., Chan, E. W., Mendes, S. D., et al. (2011). A persistent oxygen anomaly reveals the fate of spilled methane in the deep Gulf of Mexico. Science 331, 312–315. doi: 10.1126/science.1199697
Kirschke, S., Bousquet, P., Ciais, P., Saunois, M., Canadell, J. G., Dlugokencky, E. J., et al. (2013). Three decades of global methane sources and sinks. Nat. Geosci. 6, 813–823. doi: 10.1038/ngeo1955
Kits, K. D., Campbell, D. J., Rosana, A. R., and Stein, L. Y. (2015a). Diverse electron sources support denitrification under hypoxia in the obligate methanotroph Methylomicrobium album strain BG8. Front. Microb. 6:1072. doi: 10.3389/fmicb.2015.01072
Kits, K. D., Klotz, M. G., and Stein, L. Y. (2015b). Methane oxidation coupled to nitrate reduction under hypoxia by the Gammaproteobacterium Methylomonas denitrificans, sp nov type strain FJG1. Environ. Microb. 17, 3219–3232. doi: 10.1111/1462-2920.12772
Knief, C. (2015). Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front. Microb. 6:1346. doi: 10.3389/fmicb.2015.01346
Knittel, K., and Boetius, A. (2009). Anaerobic oxidation of methane: Progress with an unknown process. Ann. Rev. Microb. 63, 311–344. doi: 10.1146/annurev.micro.61.080706.093130
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microb. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Lesniewski, R. A., Jain, S., Anantharaman, K., Schloss, P. D., and Dick, G. J. (2012). The metatranscriptome of a deep-sea hydrothermal plume is dominated by water column methanotrophs and lithotrophs. ISME J. 6, 2257–2268. doi: 10.1038/ismej.2012.63
Long, M. C., Deutsch, C., and Ito, T. (2016). Finding forced trends in oceanic oxygen. Global Biogeochem. Cyc. 30, 381–397. doi: 10.1002/2015GB005310
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Lüker, S., Nowka, B., Rattei, T., Spieck, E., and Daims, H. (2013). The genome of Nitrospina gracilis illuminates the metabolism and evolution of the major marine nitrite oxidizer. Front. Microb. 4:27. doi: 10.3389/fmicb.2013.00027
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mauffrey, F., Martineau, C., and Villemur, R. (2015). Importance of the two dissimilatory (Nar) nitrate reductases in the growth and nitrate reduction of the methylotrophic marine bacterium Methylophaga nitratireducenticrescens JAM1. Front. Microb. 6:1475. doi: 10.3389/fmicb.2015.01475
Mikheenko, A., Saveliev, V., and Gurevich, A. (2016). MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 32, 1088–1090. doi: 10.1093/bioinformatics/btv697
Naqvi, S. W. A., Bange, H. W., Farias, L., Monteiro, P. M. S., Scranton, M. I., and Zhang, J. (2010). Marine hypoxia/anoxia as a source of CH4 and N2O. Biogeosciences 7, 2159–2190. doi: 10.5194/bg-7-2159-2010
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 20, 714–737. doi: 10.1089/cmb.2013.0084
Pack, M. A., Heintz, M. B., Reeburgh, W. S., Trumbore, S. E., Valentine, D. L., Xu, X. M., et al. (2015). Methane oxidation in the eastern tropical North Pacific Ocean water column. J. Geophys. Res. Biogeo. 120, 1078–1092. doi: 10.1002/2014JG002900
Padilla, C. C., Bristow, L. A., Sarode, N., Garcia-Robledo, E., Ramírez, E. G., Benson, C. R., et al. (2016). NC10 bacteria in marine oxygen minimum zones. ISME J. 10, 2067–2071. doi: 10.1038/ismej.2015.262
Padilla, C. C., Ganesh, S., Gantt, S., Huhman, A., Parris, D. J., Sarode, N., et al. (2015). Standard filtration practices may significantly distort planktonic microbial diversity estimates. Front. Microb. 6:547. doi: 10.3389/fmicb.2015.00547
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Philippot, L. (2002). Denitrifying genes in Bacterial and Archaeal genomes. Biochim. Biophys. Acta 1577, 355–376. doi: 10.1016/S0167-4781(02)00420-7
Poehlein, A., Deutzmann, J. S., Daniel, R., and Simeonova, D. D. (2013). Draft genome sequence of the methanotrophic gammaproteobacterium Methyloglobulus morosus DSM 22980 strain KoM1. Genome Ann. 1, e01078–e01013. doi: 10.1128/genomeA.01078-13
Raes, J., Korbel, J. O., Lercher, M. J., von Mering, C., and Bork, P. (2007). Prediction of effective genome size in metagenomic samples. Genome Biol. 8:R10. doi: 10.1186/gb-2007-8-1-r10
Raghoebarsing, A. A., Pol, A., van de Pas-Schoonen, K. T., Smolders, A. J. P., Ettwig, K. F., Rijpstra, W. I. C., et al. (2006). A microbial consortium couples anaerobic methane oxidation to denitrification. Nature 440, 918–921. doi: 10.1038/nature04617
Ramachandran, A., and Walsh, D. A. (2015). Investigation of XoxF methanol dehydrogenases reveals new methylotrophic bacteria in pelagic marine and freshwater ecosystems. FEMS Microbiol. Ecol. 97, 10. doi: 10.1093/femsec/fiv105
Reeburgh, W. S. (2007). Oceanic methane biogeochemistry. Chem. Rev. 107, 486–513. doi: 10.1021/cr050362v
Revsbech, N. P. (1989). An oxygen microsensor with a guard cathode. Limnol. Oceanogr. 34, 474–478. doi: 10.4319/lo.1989.34.2.0474
Rivers, A. R., Sharma, S., Tringe, S. G., Martin, J., Joye, S. B., and Moran, M. A. (2013). Transcriptional response of bathypelagic marine bacterioplankton to the Deepwater Horizon oil spill. ISME J. 7, 2315–2329. doi: 10.1038/ismej.2013.129
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Preprints e1900v1. doi: 10.7287/peerj.preprints.1900v1. Available online at: https://peerj.com/preprints/1900/
Sansone, F. J., Popp, B. N., Gasc, A., Graham, A. W., and Rust, T. M. (2001). Highly elevated methane in the eastern tropical North Pacific and associated isotopically enriched fluxes to the atmosphere. Geophys. Res. Lett. 28, 4567–4570. doi: 10.1029/2001GL013460
Schmieder, R., Lim, Y. W., and Edwards, R. (2012). Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 28, 433–435. doi: 10.1093/bioinformatics/btr669
Sekiguchi, Y., Osashi, A., Parks, D. H., Yamauchi, T., Tyson, G. W., and Hugenholtz, P. (2015). First genomics insights into members of a candidate bacterial phylum responsible for wastewater bulking. PeerJ. 3:e740. doi: 10.7717/peerj.740
Shi, Y. M., Tyson, G. W., Eppley, J. M., and DeLong, E. F. (2011). Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J. 5, 999–1013. doi: 10.1038/ismej.2010.189
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W. Z., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi: 10.1038/msb.2011.75
Stewart, F. J., Ulloa, O., and DeLong, E. F. (2012). Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ. Microbiol. 14, 23–40. doi: 10.1111/j.1462-2920.2010.02400.x
Stramma, L., Johnson, G. C., Sprintall, J., and Mohrholz, V. (2008). Expanding oxygen-minimum zones in the tropical oceans. Science 320, 655–658. doi: 10.1126/science.1153847
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tavormina, P. L., Ussler, W., Joye, S. B., Harrison, B. K., and Orphan, V. J. (2010). Distributions of putative aerobic methanotrophs in diverse pelagic marine environments. ISME J. 4, 700–710. doi: 10.1038/ismej.2009.155
Tavormina, P. L., Ussler, W., and Orphan, V. J. (2008). Planktonic and sediment-associated aerobic methanotrophs in two seep systems along the North American margin. Appl. Environ. Microbiol. 74, 3985–3995. doi: 10.1128/AEM.00069-08
Tavormina, P. L., Ussler, W., Steele, J. A., Connon, S. A., Klotz, M. G., and Orphan, V. J. (2013). Abundance and distribution of diverse membrane-bound monooxygenase (Cu-MMO) genes within the Costa Rica oxygen minimum zone. Environ. Microbiol. Rep. 5, 414–423. doi: 10.1111/1758-2229.12025
Thamdrup, B. (2012). New pathways and processes in the global nitrogen cycle. Ann. Rev. Ecol. Evol. Syst. 43, 407–428. doi: 10.1146/annurev-ecolsys-102710-145048
Thamdrup, B., Dalsgaard, T., and Revsbech, N. P. (2012). Widespread functional anoxia in the oxygen minimum zone of the Eastern South Pacific. Deep Sea Res. Pt. I. 65, 36–45. doi: 10.1016/j.dsr.2012.03.001
Tiano, L., Garcia-Robledo, E., Dalsgaard, T., Devol, A. H., Ward, B. B., and Ulloa, O. (2014). Oxygen distribution and aerobic respiration in the north and south eastern tropical Pacific oxygen minimum zones. Deep Sea Res. Pt. I. 94, 173–183. doi: 10.1016/j.dsr.2014.10.001
Tsementzi, D., Wu, J., Deutsch, S., Nath, S., Rodriguez-R, L. M., Burns, A. S., et al. (2016). SAR11 bacteria linked to ocean anoxia and nitrogen loss. Nature 536, 179–183. doi: 10.1038/nature19068
Ulloa, O., Canfield, D. E., DeLong, E. F., Letelier, R. M., and Stewart, F. J. (2012). Microbial oceanography of anoxic oxygen minimum zones. Proc. Natl. Acad. Sci. U.S.A. 109, 15996–16003. doi: 10.1073/pnas.1205009109
Valentine, D. L. (2011). Emerging topics in marine methane biogeochemistry. Annu. Rev. Mar. Sci. 3, 147–171. doi: 10.1146/annurev-marine-120709-142734
Ward, B. B., Kilpatrick, K. A., Wopat, A. E., Minnich, E. C., and Lidstrom, M. E. (1989). Methane oxidation in Saanich Inlet during summer stratification. Cont. Shelf Res. 9, 65–75. doi: 10.1016/0278-4343(89)90083-6
Wasmund, K., Kurtböke, D. I., Burns, K. A., and Bourne, D. G. (2009). Microbial diversity in sediments associated with a shallow methane seep in the tropical Timor Sea of Australia reveals a novel aerobic methanotroph diversity. FEMS Microbiol. Ecol. 68, 142–151. doi: 10.1111/j.1574-6941.2009.00667.x
Wright, J. J., Konwar, K. M., and Hallam, S. J. (2012). Microbial ecology of expanding oxygen minimum zones. Nat. Rev. Microbiol. 10, 381–394. doi: 10.1038/nrmicro2778
Wu, Y. W., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Keywords: methane, bacteria, oxygen minimum zone, metagenomics, nitrogen cycle
Citation: Padilla CC, Bertagnolli AD, Bristow LA, Sarode N, Glass JB, Thamdrup B and Stewart FJ (2017) Metagenomic Binning Recovers a Transcriptionally Active Gammaproteobacterium Linking Methanotrophy to Partial Denitrification in an Anoxic Oxygen Minimum Zone. Front. Mar. Sci. 4:23. doi: 10.3389/fmars.2017.00023
Received: 29 August 2016; Accepted: 18 January 2017;
Published: 07 February 2017.
Edited by:
Gordon T. Taylor, Stony Brook University, USAReviewed by:
Ludmila Chistoserdova, University of Washington, USAMaria Pachiadaki, Bigelow Laboratory for Ocean Sciences, USA
Copyright © 2017 Padilla, Bertagnolli, Bristow, Sarode, Glass, Thamdrup and Stewart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank J. Stewart, ZnJhbmsuc3Rld2FydEBiaW9sb2d5LmdhdGVjaC5lZHU=