 Juliane Germer
Juliane Germer Nicolas Cerveau
Nicolas Cerveau Daniel J. Jackson
Daniel J. Jackson- Department of Geobiology, Georg-August University of Göttingen, Göttingen, Germany
Symbiotic interactions are widespread throughout the animal kingdom and are increasingly recognized as an important trait that can shape the evolution of a species. Sponges are widely understood to be the earliest branching clade of metazoans and often contain dense, diverse yet specific microbial communities which can constitute up to 50% of their biomass. These bacterial communities fulfill diverse functions influencing the sponge's physiology and ecology, and may have greatly contributed to the evolutionary success of the Porifera. Here we have analyzed and characterized the holo-transcriptome of the hypercalcifying demosponge Vaceletia sp. and compare it to other sponge transcriptomic and genomic data. Vaceletia sp. harbors a diverse and abundant microbial community; by identifying the underlying molecular mechanism of a variety of lipid pathway components we show that the sponge seems to rely on the supply of short chain fatty acids by its bacterial community. Comparisons to other sponges reveal that this dependency may be more pronounced in sponges with an abundant microbial community. Furthermore, the presence of bacterial polyketide synthase genes suggests bacteria are the producers of Vaceletia's abundant mid-chain branched fatty acids, whereas demospongic acids may be produced by the sponge host via elongation and desaturation of short-chain precursors. We show that the sponge and its microbial community have the molecular tools to interact through different mechanisms including the sponge's immune system, and the presence of eukaryotic-like proteins in bacteria. These results expand our knowledge of the complex gene repertoire of sponges and show the importance of metabolic interactions between sponges and their endobiotic microbial communities.
Introduction
Sponges (Porifera) are widely thought to be the earliest branching clade of metazoans (Pick et al., 2010; Pisani et al., 2015), with a simple body plan that has remained essentially unchanged for hundreds of millions of years (Yin et al., 2015). They have a widespread distribution and are important members of benthic communities. Due to their position on the metazoan tree of life sponges can be used to test hypotheses regarding the evolution of a variety of important traits such as biologically controlled biomineralisation (Jackson et al., 2007, 2010, 2011; Germer et al., 2015), signaling pathways and gene regulation (Fernandez-Valverde and Degnan, 2016; Levin et al., 2016), gastrulation and mesoderm formation (Nakanishi et al., 2014), and genome evolution.
Sponges are known for hosting dense and diverse microbial communities in their tissues (Taylor et al., 2007) and although it is unclear when this association was established (Taylor et al., 2007, and references therein) it could be reasonably assumed that sponges and microorganisms have been interacting since the emergence of sponges (Hentschel et al., 2012). Sponge-associated microorganisms have been reported to constitute up to 50% of the sponge's total biomass at densities several orders of magnitude higher than the microbial density in the surrounding seawater (Reitner et al., 1997; Taylor et al., 2007). For these reasons sponges are no longer considered to be individual organisms, but as “holobionts” comprising the sponge host and the sum total of its endobiotic microbial community (Webster and Thomas, 2016). Research over the last decade has characterized the phylogenetic composition and diversity of the microbial communities of many shallow water sponges from many different habitats (Taylor et al., 2007). Despite the fact that sponges are surrounded by, and ingest, a plethora of microorganisms due to their filter-feeding lifestyle, many sponge–associated microorganisms are endemic to sponges (Hentschel et al., 2002; Simister et al., 2012). Knowledge regarding the functional roles of sponge-associated microorganisms is also growing. Bacterial symbionts can, for example, provide supplementary nutrition (Thomas et al., 2010; Fiore et al., 2015), and can remove metabolic waste products such as ammonia, nitrite and nitrate by producing bioactive secondary metabolites (Piel et al., 2004; Hochmuth et al., 2010; Wilson et al., 2014) which can in turn be used by the sponge as chemical defenses (Pawlik, 2011). How these sponge-microbe relationships are established and maintained, and to what extant these relationships are a genuine symbiosis, remains unclear.
Most sponge-associated microorganisms reside in the sponge mesohyl, an extracellular matrix comprising most of the sponge's body, which is also the site of digestion (Taylor et al., 2007). This implies that sponge-associated microorganisms must either have a mechanism to avoid digestion by the sponge, for example by producing slime capsules (Wilkinson et al., 1984), and/or sponges are able to recognize their symbionts. The surprisingly well-developed innate immune system of sponges (Müller et al., 2003) is likely to play an important role in distinguishing pathogenic from symbiotic microbe.
Vaceletia is a monospecific, hypercalcified sponge genus with a single described species, Vaceletia crypta (Vacelet, 1977), belonging to the Dictyoceratida within the Demospongiae (Wörheide, 2008; Erpenbeck et al., 2009; Morrow and Cárdenas, 2015). Vaceletia first appears in the Middle Triassic (Reitner et al., 1997) and has been referred to as a “living fossil”. Today, Vaceletia inhabits cryptic niches, such as caves or deeper fore reef areas that are light reduced and have oligotrophic conditions (Reitner, 1992; Wörheide, 1998). Vaceletia harbors a dense and diverse microbial community that can make up to 50% of the sponges' biomass (Reitner et al., 1997). The bacterial community composition of V. crypta has been previously studied using 16S rRNA clone libraries and DGGE (Karlińska-Batres and Wörheide, 2013). This microbial community was highly diverse and shared features with other sponge derived microbial communities. Vaceletia occurs in different growth forms (solitary vs. colonial), which are likely to represent different species, however their taxonomy is not yet fully resolved (Wörheide, 2008). DGGE cluster analyses indicate distinct microbial communities exist in the solitary and colonial species (Karlińska-Batres and Wörheide, 2013). In this study we characterize the transcriptome of the yet to be described colonial-branching Vaceletia sp. species. We previously generated this sequence resource to assist a proteomic survey of Vaceletia's calcified skeleton (Germer et al., 2015). Due to the high abundance and diversity of microbes in this sponge, we reason that interactions between the sponge and its microbial community may also play some role in fabricating its calcified skeleton. This reasoning also follows from our previous work that demonstrated Astrosclera willeyana (another hyperclacified demosponge) degrades a proportion of its microbial community via the autophagy pathway (Jackson and Wörheide, 2014), and then initiates calcification on the organic remains of these microbes (Jackson et al., 2010). Here we characterize the transcriptome of an individual adult Vaceletia sp. and search for evidence of a variety of sponge-microbe interactions including mechanisms relating to innate immunity, eukaryotic-like proteins in bacteria, and metabolic interactions between the sponge host and its associated bacteria. We also identify the presence of a variety of signaling pathway components and transcription factors that further support the notion that sponges possess a complex gene regulatory repertoire more than adequate to both specify sponge the cell types that must interact with and manage the microbiome, and to coordinate the deposition of an intricate and elaborate calcified skeleton.
Materials and Methods
Sample Collection and RNA Extraction
Specimens of Vaceletia sp. were collected in 2009 during the Deep DownUnder Expedition (http://www.deepdownunder.de) at Osprey- and Bougainville Reefs (Coral Sea, Australia) in depths from 5 to 24 m by SCUBA diving. Sponges do not fall under the German animal protection act §8. This work was therefore exempt from the University of Göttingen Ethics Committee. Samples for RNA extraction were preserved in RNAlater and stored at −20°C. Before processing, samples were carefully inspected under a stereomicroscope for contaminating organisms which were carefully removed. Total RNA derived from the head of one individual was extracted using the miRNeasy Kit (Qiagen) according to the manufacturer's instructions. RNA was checked by gel electrophoresis on a 1.5% TAE agarose gel and by spectrometry using a Nanodrop spectrometer.
Library Preparation, RNA Sequencing and Sequence Assembly
Library preparation and sequencing was carried out at the Transcriptome and Genome Analysis Laboratory, University Medical Center Göttingen, Germany. A TrueSeq RNA Sample Kit (Illumina, Cat. N°RS-122-2002) was used to generate an mRNA library using 500 ng of total RNA. During TrueSeq Illumina library construction rRNA was depleted (oligo dT beads capture poly adenylated mRNAs). Accurate quantitation of cDNA libraries was performed using the QuantiFluor™ dsDNA System (Promega) and the size range of the final cDNA libraries was determined using a Bioanalyzer 2,100 from Agilent (280 bp). Sequencing was conducted on the Illumina HiSeq 2000 platform to generate 100 bp paired-end reads. Read qualities were assessed using FastQC (Andrews, 2014), and Trimmomatic (Bolger et al., 2014) was used to remove low quality reads and adapter sequences. Reads were assembled de novo using our recently developed assembly pipeline (Cerveau and Jackson, 2016). Briefly, we employed three assembly packages with unique assembly strategies: Trinity V2.0.3 (Haas et al., 2013), CLC Genomics Workbench (www.qiagenbioinformatics.com) and IDBA-tran V1.1.1 (Peng et al., 2013). The CLC assembler was run using a word size of 20 base pairs (bp), a bubble size of 50 bp, with reads mapped back to the transcriptome using default parameters. IDBA_tran was run with kmer values ranging from 20 to 100 bp with a step size of 10 bp. Trinity assemblies were run with default parameters and a k-mer value of 25. The coding transcriptomes of these three assemblies were merged into a concatenated assembly, and redundancy was removed. The raw reads are deposited to NCBI under SRA accession number SRR4423080.
Transcriptome Annotation and Characterization
The assembled Vaceletia sp. transcriptome was annotated using BLASTx searches (with an e-value cut-off of 1e-5) against the NCBI non-redundant database, using Trinotate (Haas et al., 2013), BLASTx and BLASTp searches (with an e-value cut-off of 1e-5) against the Swissprot/Uniprot database, and against Pfam (Finn et al., 2014) using Hmmer (Finn et al., 2011). SignalP 4.0 (Petersen et al., 2011) and TMHMM (Krogh et al., 2001) were used to predict signal peptides and transmembrane regions, respectively.
Transcripts were assigned a putative taxonomic origin using the lowest common ancestor algorithm implemented in MEGAN5 (Huson et al., 2011) with the following parameters: MinScore = 50, MaxExpected = 0.01, TopPercent 10, and MinSupport = 20. In order to separate and compare contigs with bacteria and metazoan origin, contigs identified as such were extracted from MEGAN and used in individual BLASTx searches. The results were visualized in MEGAN, and a comparison between the bacterial and metazoan contigs was constructed using the “compare” option with normalized counts. Functional annotation was performed using the KEGG database implemented in MEGAN. Sequences that were assigned by MEGAN to a bacterial origin were extracted and further characterized using BLAST searches as described above. Potential eukaryotic-like proteins were identified via keyword search of the BLASTx output file using a customized Perl script. Matching contigs were further evaluated using hmmer against the Pfam 28.0 database (with a cut-off value of 1e-5) for the presence of eukaryotic-like protein domains (ankyrin repeat (ANK): PF00023, tetratricopeptide repeat (TPR): PF00515.24, PF13374.2, PF13414.2, PF13424.2, PF13428.2, PF13429.2, PF13432.2, PF13431.2, PF13512.2, PF14559.2, PF07719.13, PF09976.5, PF07720.8, PF07721.10, PF13174.2, PF13176.2, PF13181.2, PF13371.2, SEL1: PF08238, leucine rich repeat (LRR): PF00560.29, PF07723.9, PF07725.8, PF12799.3, PF13306.2, PF13516.2, PF13855.2, PF14580.2, fibronectin: PF00041, cadherin: PF00028, NHL (named after NCL-1, HT2A, and Lin-41): PF01436).
Pattern Recognition Receptors (PRRs) were identified from the Vaceletia sp. BLASTx output file using a customized Perl script. The script scanned the description lines of the BLASTx output file for key words and collected all positive contigs. These contigs were further evaluated for the presence of the PRR domains using hmmer against the Pfam 28.0 database with a cut-off value of 1e-5.
Genes involved in short-chain fatty acid biosynthesis were identified via the blast based KEGG pathway map coverage implemented in MEGAN. To get an overview of the type I fatty acid synthase (FAS), type II FAS and polyketide synthase (PKS) diversity the transcriptomes of Vaceletia sp., Amphimedon queenslandica, Stylissa cateri, Petrosia ficiformis, Spongilla lacustris, Pseudospongosorites suberitoides, Xestospongia testudinaris, Sycon coactum, and Corticum candelabrum were screened for contigs containing a ketosynthase (KS) domain using hmmer against the KS HMM profiles PF00109 and PF02801 with a cut-off value of 1e-5. Positive matches were extracted from the respective transcriptomes. For phylogenetic analyses protein reference sequences were downloaded from NCBI genebank and the NaPDoS database (Ziemert et al., 2012). Searching animal derived type I FAS sequences against porifera sequences (taxid: 6040) stored in the NCBI nr database did not yield any significant hits. Since only KS domains were used in the construction of a phylogenetic tree, the NaPDoS server (Ziemert et al., 2012) was used to extract the KS domain from all sequences. Many Vaceletia-derived sequences only span a small region of the KS domain and were discarded. Full and partial KS amino acid sequences (at least 160 amino acids) were aligned using clustalO (Sievers et al., 2011). Gblocks (Castresana, 2000) was used to identify conserved sites. Phylogenetic analysis were conducted using MrBayes v 3.2.6 (Ronquist et al., 2012) with the following parameters: lset rates = gamma, preset aamodelpr = mixed, mcmcp, nruns = 4, ngen = 10,000,000, relburn = yes, burninfrac = 0.25, printfreq = 1,000, samplefreq = 100, nchains = 4, saveprlens = yes and RAxML on the RAxML blackbox webserver (Stamatakis et al., 2008) using the combined bootstrapping and maximum likelihood search algorithm with the WGA model and 100 bootstraps.
Bacterial PKS genes were identified by searching a list of sponge-specific PKS sequences retrieved from Genbank against the bacterial derived contigs of the Vaceletia sp. transcriptome. Each predicted Vaceletia sp. sequence was then searched against the nr database using BLASTx. Domain structure was evaluated with hmmer against the Pfam 28.0 database with a cut-off value of 1e-5.
Genes involved in fatty acid elongation and desaturation were identified from the Vaceletia sp. BLASTx output file using a customized Perl script. The script scanned the description lines of the BLASTx output file for key words and collected all positive contigs. Additionally, genes were identified by searching a list of fatty acid elongation and desaturation sequences retrieved from Genebank against Vaceletia sp. transcriptome. Positive and matching contigs were further evaluated using hmmer against the Pfam 28.0 database with a cut-off value of 1e-5.
Sterol-24/28-methyltransferase (SMT) genes were identified by searching a list of sponge-specific SMT sequences retrieved from Genbank against Vaceletia sp. transcriptome. Each predicted Vaceletia sp. sequence was then searched against the NCBI-nr database using BLASTx. Positive matches were aligned to the SMT protein alignment provided by Gold et al. (2016) using clustalO. Phylogenetic analyses were conducted using MrBayes v 3.2.6 (Ronquist et al., 2012) with the following parameters: lset rates = gamma, preset aamodelpr = mixed, mcmcp, nruns = 4, ngen = 10,000,000, relburn = yes, burninfrac = 0.25, printfreq = 1,000, samplefreq = 100, nchains = 4, saveprlens = yes, and RAxML on the RAxML blackbox webserver (Stamatakis et al., 2008) using the combined bootstrapping and maximum likelihood search algorithm with the GTA model and 100 bootstraps.
Protein sequences from a collection of recently reported sponge transcriptomes (Riesgo et al., 2014) were used in local BLAST searches against the Vaceletia sp. transcriptome to identify conserved signaling proteins. Matching contigs were further evaluated using hmmer against the Pfam 28.0 database with a cut-off value of 1e-5.
Homeobox containing genes were identified by searching the homeobox domain (PF00046) and homeobox KN domain (PF05920) against the Vaceletia sp. transcriptome using hmmer with a cut-off value of 1e-5. Contigs containing these domains were extracted and further characterized using the Homeobox database (Zhong et al., 2008; Zhong and Holland, 2011).
Comparison of the Vaceletia sp. Transcriptome to Other Sponge Transcriptomes and Transcriptome Completeness
In order to place our results within a broader poriferan context, we included a range of other datasets. Riesgo reported eight transcriptomes derived from representatives of all poriferan classes (Riesgo et al., 2014). These were BLAST searched against the nr database, loaded into MEGAN and annotated against KEGG as described in section Transcriptome Annotation and Characterization. Raw Illumina reads for Amphimedon queenslandica (SRR1511621), Xestospongia testudinaris (SRR1738066/68) and Stylissa cateri (SR1738063) were assembled as described for the Vaceletia data, and also BLAST searched against the nr database, loaded into MEGAN and annotated against KEGG as described above. In addition, to evaluate the completeness of all the transcriptomes we investigated, we performed a BUSCO analysis (Simão et al., 2015) (Table S1).
Lipid Extraction and GC/MS Analysis
Lipids were extracted from crushed, freeze-dried heads of Vaceletia sp. by ultrasonication with DCM/methanol (1:3, v/v), DCM and n-hexane, respectively. The total organic extract was derivatized using trimethylchlorsilane (TMCS)/methanol (1:9; v:v; 1.5 h at 80°C), yielding fatty acid methyl esters (FAME). Separation was achieved by column chromatography. The extract was dried on silica gel 60, loaded onto the column and eluted successively with n-hexane/DCM (5:1, v/v), DCM, DCM/acetone (9:1, v/v) and DCM/methanol (1:1, v/v) to yield hydrocarbons, fatty acid, alcohols and polar residue, respectively. Gas chromatography-mass spectrometry (GC/MS) analyses were carried out using a Varian CP-3800 gas chromatograph coupled to a Varian 1200L mass spectrometer. The system was equipped with a silica column (Phenomenex Zebron ZB-5MS, 30 m, 0.25 μm film thickness, inner diameter 0.32 mm). Samples were injected on column. The carrier gas was helium. The temperature programme was 80°C (3 min) to 310°C at 4°C min per minute (held 25 min). Mass spectra were produced using an electron energy of 70 eV, a mass range of m/z 50–650 and a scan time of 0.25 s in full scan mode.
Results and Discussion
Transcriptome Characterization
Quality filtering of raw reads resulted in 34,592,975 paired end reads that were assembled as described by Cerveau and Jackson (2016), see Methods section. The assembly generated 91,443 contigs with an average length of 933 bp and an N50 value of 1,307 (Table 1). Assembled contigs were compared to the NCBI non-redundant database and to the Swissprot/Uniprot database using BLASTx (with an e-value of 1e-5). In total 76,207 contigs (83.3%) and 70,734 (65.7%) were annotated, respectively. Using TransDecoder 107,655 coding sequences (cds) were identified and used in BLASTp searches against the Swissprot/Uniprot database which resulted in 65,722 (61.0%) hits. Further, conserved Pfam domains were assigned to 69,083 (64.2%) of the translated queries. In total 24,893 coding sequences (23.1%) could not be annotated by any of these sequence similarity searches (Table 2). This is a relatively high proportion compared to other sponge transcriptomes (Riesgo et al., 2014).

Table 1. Vaceletia. sp. transcriptome assembly metrics.
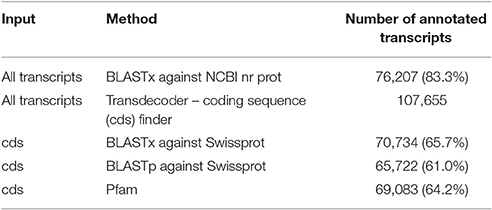
Table 2. Results of sequence similarity searches.
Taxonomic Distribution of Vaceletia sp. Contigs
To obtain information regarding the taxonomic origin of the assembled contigs, BLASTx output files were used as input for MEGAN (Huson et al., 2011). MEGAN uses a Lowest Common Ancestor algorithm to assign protein-coding (mRNA) genes to a taxon, which can then be visualized in a phylogenetic tree. A MEGAN output of Vaceletia sp. contigs is shown in Figure 1. 367,018 Vaceletia sp. contigs were assigned to Bacteria (40%), 29,947 contigs to Metazoa (33%) and 18,221 contigs (20%) could not be taxonomically assigned.
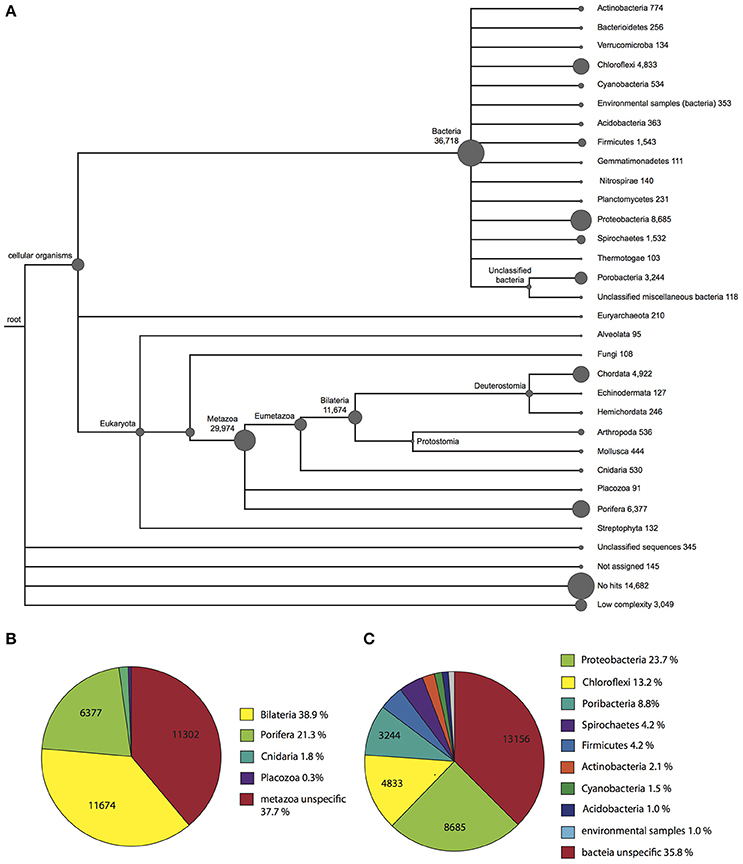
Figure 1. Taxonomic assignment of Vaceletia sp. contigs using MEGAN. (A) A phylogenetic tree reflecting the relationships and abundance of the taxonomic assignments. (B) Further phylogenetic breakdown of contigs assigned to a metazoan origin. (C) Further phylogenetic breakdown of contigs assigned to a bacterial origin.
Among the Vaceletia sp. sequences that shared significant similarity with a metazoan sequence, a high proportion (6,377 contigs, 21%) were assigned to sponges (mostly Amphimedon). However, the majority shared the highest similarity either with a specific bilaterian sequence (11,674 contigs, 39%), or with an unspecific sequence and were therefore assigned to the metazoan node (11,302 contigs, 38%) (Figure 1B). A recent characterization of eight sponge transcriptomes gave similar results (Riesgo et al., 2014). Although, the sequence similarity hit rate for our Vaceletia transcriptome is higher, the significant proportion of contigs assigned to the “unspecific” category highlights the lack of curated database entries for sponges.
A striking number of contigs in our dataset (40%) were taxonomically assigned to Bacteria. Some sponges are known to harbor a remarkable diversity of microorganisms. In Vaceletia crypta the total biomass derived from bacteria has been reported to be as high as 50% (Reitner et al., 1997). Recent molecular studies using 16S rRNA characterized the microbial communities of sponges from many marine habitats. This work paints a picture of a handful of dominant sponge-associated bacterial phyla, including Proteobacteria (especially the classes Alpha-, Gamma- and Deltaproteobacteria), Chloroflexi, Actinobacteria, Acidobacteria, Nitrospirae, and the candidate phylum Poribacteria (Hentschel et al., 2012). All of these sponge-associated Bacteria phyla are present in our Vaceletia sp. transcriptome dataset (Figure 1C). As much as 24% of the bacterial contigs were assigned to Proteobacteria, followed by 13% to Chloroflexi and 9% to the candidate phylum Poribacteria. Traditionally, 16S rRNA is used for both taxonomic characterization of microbial communities, as was previously performed for V. crypta, the solitary sister species to Vaceletia sp. (Karlińska-Batres and Wörheide, 2013). Taxonomic assignment as performed by MEGAN employs protein-coding genes. Interestingly, in comparison to previously reported sponge transcriptomes (Riesgo et al., 2014; Ryu et al., 2016) the Vaceletia sp. transcriptome contains by far the highest proportion of bacterially derived contigs (40%) in comparison to high microbial abundance (HMA) sponges such as X. testudinaris (1.4%), C. nucula (7.5%), and C. candelabrum (13.7%). It must be kept in mind that comparisons between studies such as this are likely to suffer from some systematic error associated with library preparation and downstream data handling. However broadly similar library preparation (ribosomally depleted mRNA), sequencing (Illumina platform) and sequence data handling methods were employed by all of these studies.
Metabolic Interactions between Vaceletia sp. and Its Microbial Community
It has been suggested, that the evolutionary history and the ecological success of sponges is closely intertwined with their microbial community (Taylor et al., 2007). Sponge-microbe symbioses are likely evolutionary ancient, and have probably co-evolved to yield benefits to both sponge and microbe. Microbes living within sponges are provided with a nutrient rich environment through the products of the host's metabolism (Fan et al., 2012). In return, microbes can provide the sponge host with access to novel metabolic pathways and chemical defenses (Kennedy et al., 2007; Fan et al., 2012; Liu et al., 2012; Radax et al., 2012; Li et al., 2014; Fiore et al., 2015). Taking a closer look at the metabolic pathways present in the Vaceletia sp. transcriptome, a significant proportion of all metabolic pathways are derived from contigs originating from bacteria (Figure 2). Comparing bacterial and metazoan features in these metabolic pathways reveals similar results to those reported by Fiore et al for the sponge Xetospongia muta and its symbiotic community (Fiore et al., 2015). Because the metabolism of fatty acids has received some attention in sponges we focused on this aspect of the sponge-microbe metabolic relationship.
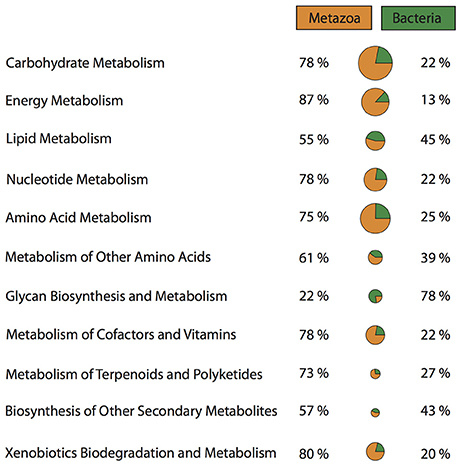
Figure 2. Proportions of metazoan derived and bacterial derived contigs involved in a variety of metabolic pathways.
Lipid Metabolism: Biosynthesis of Short Chain Fatty Acids
Sponges are a rich source of unusual lipids that play a functional and structural role in their membranes. It has been proposed that a significant amount of these lipids are of bacterial origin (Gillan et al., 1988). The fatty acid (FA) compounds of Vaceletia sp. show that this sponge is rich in short chain FAs, mid-chain branched fatty acids (MBFAs) and the sponge specific demospongic acids (Figure S1). Cell fraction experiments demonstrated the occurrence of short chain FAs in both sponge cells and bacteria, with increased levels in the bacterially derived matter (Zimmerman et al., 1989, 1990). In animals, fatty acid biosynthesis is catalyzed by the monomodular type I fatty acid synthase (FAS), whereas in bacteria it is usually catalyzed by individual enzymes, the type II FAS (Lu et al., 2004). Mapping Vaceletia sp.'s transcriptome against the KEGG lipid metabolism pathways shows that most components of the short chain FA biosynthesis pathway are present (Figure 3). All compounds are represented by bacterial derived contigs and are mapped to the type II FAS, suggesting a purely bacterial origin for short-chain FAs in Vaceletia. To determine whether this is generally the case for sponges, the transcriptomes of I. fasciculate, C. nucula, P. ficiformis, S. lacustris, P. suberitoides, X. testudinaris, S. cateri, S. coacatum, and C. candelabrum were also mapped against the KEGG lipid metabolism pathway (Figure 4). Only C. candelabrum gave similar results to Vaceletia sp. with three components represented by both bacterial and metazoan derived contigs. Both sponges are HMA sponges with the highest percentage of bacterial derived contigs of all transcriptomes. In X. testudinaris, also a HMA sponge, the majority of components are present but in contrary to Vaceletia they map to the type I FAS pathway, implying a metazoan origin of short chain FAs. X. testudinaris is the only sponge to possess an animal type FAS. The lack of bacterial derived components might also be the result of the extremely low percentage of bacterially derived contigs (1.4%) in that transcriptome. Although, the short chain FA biosynthesis pathway is incomplete or missing in the other sponge transcriptomes, a general trend is observable. HMA sponges tend to have more bacterial derived pathway components then LMA sponges (Figure 4). No literature regarding the classification of P. suberitoides, A. vastus, and S. coactum as HMA or LMA sponges is available, but the low percentage of bacterial derived sequences (0.8, 1.1, and 1.7%, respectively) suggests that they are LMA sponges. Genomic data from A. queenslandica indicates an incomplete short chain biosynthesis pathway, also suggesting that this sponge is not itself capable of producing short chain fatty acids (Srivastava et al., 2010). Additionally, we identified contigs containing a KS domain from Vaceletia sp., Amphimedon queenslandica, Stylissa cateri, Petrosia ficiformis, Spongilla lacustris, Pseudospongosorites suberitoides, Xestospongia testudinaris, Sycon coactum, and Corticum candelabrum. This domain is present in type I FASs, in the component FabF of the type II FASs and in polyketide synthases (PKSs), a protein class that is evolutionary linked to type I and type II FASs and resembles the architecture of these (Jenke-Kodama, 2005). Bayesian and maximum likelihood phylogenetic analyses were conducted on the conserved KS domains. Both phylogenetic reconstructions show, that the type II sequences identified via the KEGG pathway-mapping cluster together with type II FAS sequences from bacteria within the Type II FAS clade (Figure S2). No sequence recovered by our analysis falls within the animal type I FAS clade. However, our results suggest that a variety of sponges rely on the supply of short chain FAs from their endobiotic microbial communities. Whether these communities present a final FA product to the host, or whether the sponge farms and then harvests these communities for their FA resources is as yet unknown.
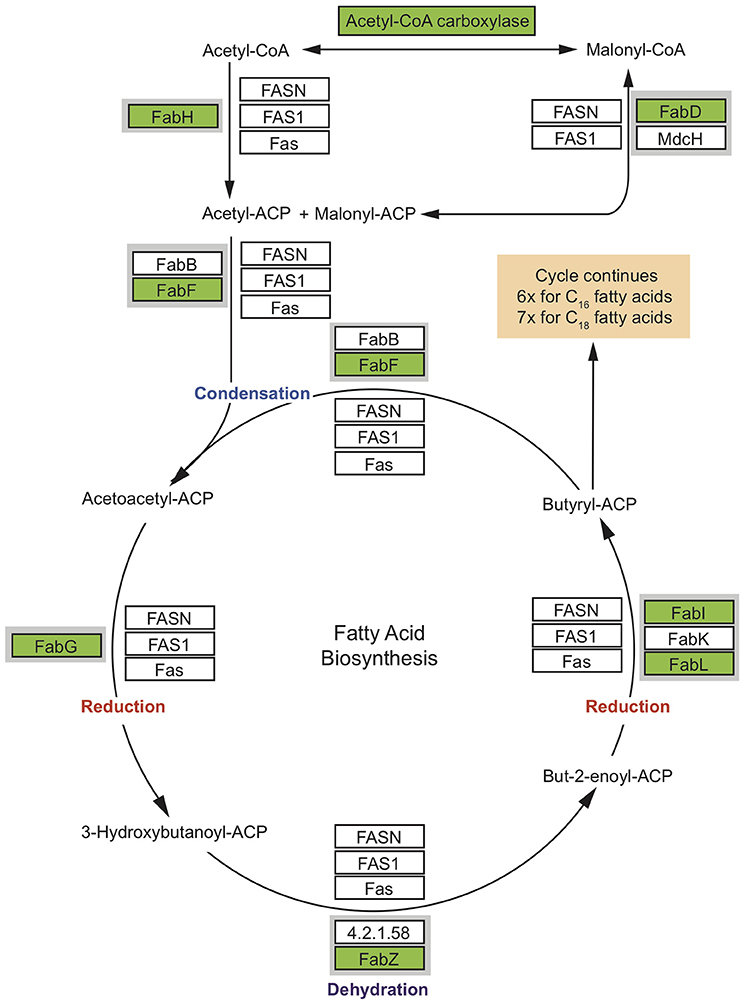
Figure 3. Condensed short chain fatty acid biosynthesis pathway. Coverage of the short chain fatty acid biosynthesis pathway. MEGAN was used to visualize contigs on the KEGG map. Rectangles represent proteins of the pathway while arrows represent signaling routes. Pathway components showing significant similarity to contigs in the Vaceletia sp. transcriptome are highlighted in green. Type II FAS is highlighted in gray. Condensation of malonyl –ACP with acetyl-ACP initiates fatty acid synthesis. Successive steps of reduction, dehydration and reduction lead to chain elongation via the type I FAS used by animals or the type II FAS used by bacteria. Butyryl-ACP, with the addition of malonyl-ACP, then undergoes subsequent cycles of elongation. Each round elongates the FA to have two more carbon units.
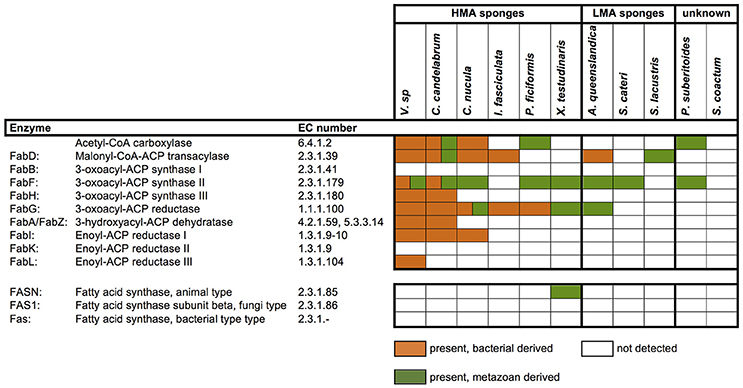
Figure 4. Presence and absence of metazoan and/or bacterial derived short-chain fatty acid biosynthesis components. Components were identified by mapping the transcriptomes to the corresponding KEGG map.
Lipid Metabolism: Biosynthesis of Methyl-Branched Fatty Acids
Another prominent compound class of Vaceletia's biomarker content are the mid-chain branched fatty acids (Figure S1). MBFAs are often abundant compounds in HMA sponges and are believed to have a bacterial origin (Thiel et al., 1999). A recent study positively correlated the presences of a sponge specific PKS in bacteria-rich sponges with the presence of MBFAs and proposed Poribateria as the potential producers of these compounds (Hochmuth et al., 2010). As Vaceletia sp. contains large amounts of MBFAs (Figure S1) and hosts Poribacteria (Figure 1), we examined the bacteria derived subset of Vaceltia's transcriptome for the occurrence of a bacterial-type PKS named “sponge symbiont ubiquitous PKS” (Sup-type PKS) (Fieseler et al., 2007). This group of genes is commonly found in sponges, but is also highly specific to sponges (Fieseler et al., 2007), and has also been found in the genome of a representative Poribacterium (Siegl et al., 2010). Various Vaceletia sp. contigs share significant sequence similarity with the SupA gene isolated from a bacterial symbiont of the sponge Theonella swinhoei. Two of these contigs (Table S2) contain all of the previously identified domains (Fieseler et al., 2007), including the methyltransferase domain, which most probably is responsible for the mid-chain methyl branch. In Mycobacterium tuberculosis complex lipids are part of the cell wall and seem to be potent virulence factors by modulating the host cell function (Cox et al., 1999). The question has been raised if Sup genes might play a similar role in sponge symbionts and thus be important for establishing and maintaining symbioses in sponges (Fieseler et al., 2007), but so far the function of MBFAs in sponge symbionts is still unknown.
Lipid Metabolism: Biosynthesis of Demospongic Acids
The most abundant FA compounds of Vaceletia's biomarker content are demospongic acids (Figure S1). Demospongic acids are non-methylene interrupted fatty acids with an atypical 5,9-diunsaturation pattern within a carbon number range from C20 to C34 (Kornprobst and Barnathan, 2010). They are named after their first proven presence in different demosponges and although not exclusive to sponges (Kornprobst and Barnathan, 2010), they are a characteristic feature of demosponges and hexactinellids (Litchfield et al., 1976). Early studies with 14-C-labbled FAs on the sponge Microciona prolifera demonstrated that demospongic acids are biosynthesized via elongation of short chain precursors mainly derived from exogenous sources, followed by desaturation (Figure 5A; Morales and Litchfield, 1977; Hahn et al., 1988; Djerassi and Lam, 1991). Probably the most remarkable feature of the Δ5, 9-FA biosynthesis is the desaturation process as it was found that the double bond can be introduced at either the Δ5 or Δ9 loci, meaning that the second double bond may be inserted on either site of the existing one, which is contrary to the formation of polyunsaturated FAs in animals (Hahn et al., 1988; Djerassi and Lam, 1991). These studies demonstrate that sponges must possess an active elongation and desaturation enzyme system, however the underlying molecular processes remain largely unstudied (Monroig et al., 2013). The end product of the short chain fatty acid synthesis via FAS is palmitic acid (C16) and to a lesser extent myristic (C14) and stearic (C18) acid. To produce FAs with chain length > C18 animals use a system involving four membrane-bound enzymes (Leonard et al., 2004). In silico searches of the Vaceletia sp. transcriptome for these enzymes, namely elongation of very long chain fatty acids (Elovl) proteins, ketoacyl-CoA reductase (KAR), β-hydroxyacyl-CoA dehydratase (HADC) and trans-2,3-enoyl-CoA reductase (TER), reveal a variety of contigs (Figure 5B, Table S3). As elongation is followed by desaturation in sponges we additionally searched for Δ5,9 desaturases in Vaceletias's transcriptome and found contigs showing high similarities to Δ5 desaturase and stearoyl-CoA desaturase (Table S3). Stearoyl-CoA desaturase is an enzyme that is ubiquitous present in living organism (Castro et al., 2011) showing Δ9 desaturase activity. Almost all compounds are represented by metazoan derived contigs confirming that sponges possess an active elongation and desaturation enzyme system analogous to what is known from animals to produce endogenous demospongic acids and other long chain saturated and unsaturated FAs.
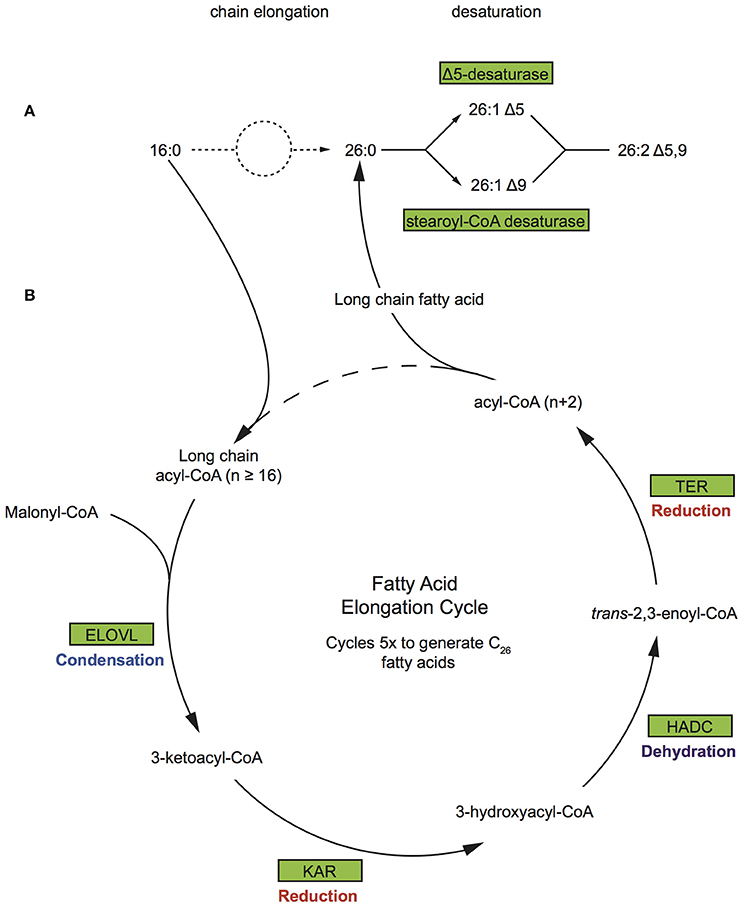
Figure 5. Biosynthetic pathway of demospongic acids and fatty acid elongation pathway (A) As demonstrated in the sponge Microciona prolifera (Morales and Litchfield, 1977; Hahn et al., 1988; Djerassi and Lam, 1991). (B) The FA elongation cycle consists of four steps and each round elongates the FA to have two more carbon units. Rectangles represent proteins of the pathway while arrows represent signaling routes. Pathway components showing significant similarity to contigs in the Vaceletia sp. transcriptome are highlighted in green. ELOVL, elongation of very long chain fatty acids; KAR, ketoacyl-CoA reductase; HADC, β-hydroxyacyl-CoA dehydratase; TER, trans-2,3-enoyl-CoA reductase.
Lipid Metabolism: Biosynthesis of Sterols
Sterols occur ubiquitously among eukaryotes and fulfill important biologically functions; they are important for membrane structure and fluidity and function as precursors to signaling molecules or hormones. Sponges are a rich source of a great variety of sterol compounds with unusual structural features (Djerassi and Silva, 1991) that sometimes are so unique that they might be used to provide taxonomical information of these organisms (Bergquist et al., 1991). Sponges can acquire sterols in different ways; by de novo biosynthesis, by dietary uptake, by modifications of dietary sterols or by biosynthesis with associated microorganisms (Goad, 1981; Djerassi and Silva, 1991).
Mapping Vaceletia's transcriptome against the KEGG steroid biosynthesis pathway shows that the majority of the components to synthesize cholesterol are present and are mostly represented by metazoan derived contigs (Table S4). However, components in the KEGG pathway to produce lanosterol are mapped to bacterial derived contigs (Table S4). Lanosterol is an intermediate product of the steroid biosynthesis pathway and is generally regarded as the precursor to all animal sterols (Djerassi and Silva, 1991). Sterol biosynthesis is considered a eukaryotic key feature, nevertheless sterol production has been observed recently in a few bacterial species (Bird et al., 1971; Bode et al., 2003; Desmond and Gribaldo, 2009). Interrogation of Vaceletia's transcriptome indicate that the sterol precursor is not synthesized by the sponge de novo but rather acquired by dietary uptake or produced by associated microorganism and then further modified by the sponge. Searching transcriptomes of I. fasciculate, C. nucula, P. ficiformis, S. lacustris, P. suberitoides, X. testudinaris, S. cateri, S. coacatum, and C. candelabrum against the KEGG steroid biosynthesis pathway confirms the results of a previous study (Gold et al., 2016) that the sterol precursor is either synthesized by the sponge de novo or acquired by dietary uptake. These transcriptomes show no evidence that associated microorganisms might somehow be involved in the biosynthesis of sterols in these sponges (Table S4).
Characterizing sterols and their biosynthetic origin is interesting and important as their diagenetically derived counterparts, steranes, can be used by geochemists as “molecular fossils”. One unique C30 sponge sterol that is of particular interest is 24-isopropylcholesterol and its geological counterpart 24-isopropylcholestane (McCaffrey et al., 1994). 24-isopropylcholestane is widely accepted as a sponge-specific biomarker. It is found abundantly in certain Neoproterozoic to Early Cambrian rocks (~650–540 mya) representing the oldest evidence for animal life in the geological record (Love et al., 2008; Gold et al., 2016). 24-isopropylcholesterol is produced by certain demosponges (Love et al., 2008) and in trace amounts by pelagophyte algae (Antcliffe, 2013) by using the enzyme sterol-24/28-methyltransferase (SMT). Molecular clock analysis suggest that sponge SMT evolved before the Phanerozoic whereas SMTs of the algaea evolved approximately 100 mya after that, indicating that 24-isopropylcholestane indeed is a sponge specific biomarker (Gold et al., 2016). Across eukaryotes a strong correlation between C-24 side chain alkylation and the number of copies of the gene SMT exists in the genome, implicating that each SMT copy is responsible for one alkylation step (Gold et al., 2016). Sponges seem to be the exception as fewer SMT copies as expected were consistently recovered to produce C29 and C30 sterols (Gold et al., 2016). Hence, to produce 24-isopropylcholesterol sponges need two SMT copies (named SMT1 and SMT2) whereas other eukaryotes would need at least three SMT copies (Gold et al., 2016). BLAST searches for SMT genes recovered two potential SMT1 contigs from the Vaceletia transcriptome. These contigs show significant similarities to a predicted cycloartenol-C-24-metyltransferase from Populus euphratica (eudicot) and to a hypothetical protein of Sphaeroforma artica (unicellular organism), respectively. Phylogenetic analyses cluster contig CLC_28234 with SMTs from trypanosomes (parasitic protozoans) and contig idb_20522 with Ichthyosporea (parasitic Holozoa) (Figure S3). These results suggest that the detected SMT genes are not sponge derived but rather a contamination from an endo- or epibiont.
Characterization of the Expressed Immune System of Vaceletia sp.
Due to the high proportion of bacterially derived contigs from the Vaceletia sp. transcriptome, genes that regulate interactions between the sponge host and these microbes must also be expressed and present in this transcriptome. Our interest in the evolution of mechanisms that control metazoan biomineralization previously uncovered deep relationships between sponges and their microbial communities (Jackson et al., 2010), and also revealed connections with biomineralization that have deep evolutionary origins (Jackson et al., 2011). In order to better understand how Vaceletia sp. manages its microbial community, with the prospect of understanding how this relationship may influence the construction of its highly calcified skeleton (Germer et al., 2015), we also surveyed and characterized some components of Vaceletia's innate immune system.
The first contact point between entering bacteria and the sponge host are so-called germ line encoded pattern recognition receptor (PRR) (Gordon, 2002). PRRs detect and bind microbial ligands known as microbial- or pathogen-associated molecular pattern (MAMPs and PAMPs, respectively), which typically trigger a signal transduction cascade that leads to the transcription of immune response effector genes (Janeway and Medzhitov, 2002). Ligands that are recognized by PRR include microbial glycolipids and glycoproteins, peptidoglycans of gram-positive and gram-negative bacteria, and lipopolysaccharides of gram-negative bacteria (Gordon, 2002) as well as recognition mechanisms for fungi and viral DNA (Akira et al., 2006).
Toll-Like Receptors
Toll-like receptors (TLR) are among the most prominent and best characterized members of PRRs in animals. They are membrane bound receptors canonically composed of an intracellular Toll/interleukin-1 receptor (TIR) domain and extracellular leucine-rich-repeat (LRR) domains, which are responsible for the extracellular pattern recognition. So far, no conventional TLRs have been found in sponges (Gauthier et al., 2010; Hentschel et al., 2012), and we find no conventional TLRs in the transcriptome of Vaceletia s p. We detected several sequences with similarity to TLRs, and domain searches revealed that the majority of these sequences contain a single TIR domain but are devoid of the characteristic LRR domains and therefore lack the MAMP-binding site. Instead of the LRR domain, three contigs possess an extracellular Immunoglobulin (IG) domain (Table S5). In Amphimedon queenslandica the presence of TLR related receptors with an intracellular TIR domain and an extracellular Immunoglobulin (Ig) domain instead of LRR domains have been reported (Gauthier et al., 2010; Srivastava et al., 2010). In general Toll-like receptor proteins are reduced in number in sponges. Surveys of eight sponge transcriptomes revealed the presence of toll-like receptor 2 in only three sponges, Ircinia fasciculata, Petrosia ficiformis and Corticum candelabrum (Riesgo et al., 2014; Figure 6). Although, it has been suggested that the Ig domains may interact with microorganisms, the lack of diversity of these receptors suggest that they play little or no role as PRRs in sponges (Gauthier et al., 2010). The myeloid differentiation primary response gene 88 (MyD88) is another TIR domain containing protein with a characteristic DEATH domain and could be identified in the Vaceletia sp. transcriptome (Table S5). MyD88 functions downstream of TLRs and has previously been identified in other sponges (Wiens et al., 2005; Srivastava et al., 2010; Riesgo et al., 2014; Figure 6).
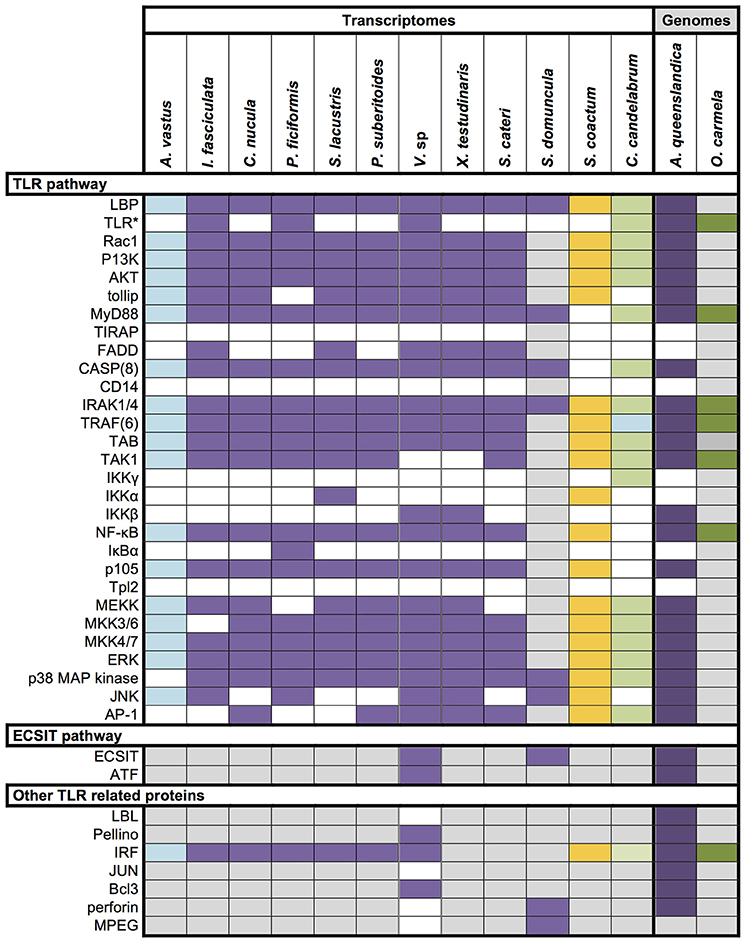
Figure 6. Overview of innate immunity components in Porifera. Data derives from recent papers (Wiens et al., 2005, 2007; Gauthier et al., 2010; Riesgo et al., 2014) and our own analysis. Different colors represent different sponge class; blue, Hexactinellid; purple, Demosponge; yellow, Calcarea; green, Homoscleromorpha; Colored square, component present; white square, component absent; gray square, unknown; *, includes unconventional Toll-like receptor with IG domains instead of LRR domains.
Nucleotide-Binding Domain and Leucine-Rich Repeat Proteins (NLR)
TLRs are responsible for detecting extracellular MAMPs and PAMPs. Most cytosolic counterparts of TLRs are NLRs (Nucleotide-binding domain and Leucine-rich repeat protein also known as Nucleotide Oligorimerisation Domain (NOD)-like receptors). NLRs react to bacteria that invade the cell and to bacterial products remaining after phagocytosis (Franchi et al., 2009), or to endogenous damage-associated molecular patterns (DAMPS) that indicate cellular stress or injury (Stuart et al., 2013). Surprisingly, intracellular NLRs are not only important for the detection of intracellular bacteria, but can also react to extracellular bacteria or their components (Ferrand and Ferrero, 2013). How extracellular bacteria are detected is not yet fully understood, but based on this ability it has been hypothesized that NLRs play an important role in distinguishing pathogenic from commensal bacteria (Ferrand and Ferrero, 2013). The A. queenslandica genome encodes a surprisingly large and diverse repertoire of NLRs with 135 genes possessing the NLR's typical NACHT domain and C-terminal LRRs (Yuen et al., 2014). Of these, approximately one third have the typical tripartite architecture with an N-terminal DEATH or CARD domain (Yuen et al., 2014). This unexpected abundance and diversity might indicate an NLR-based immune system response that is sophisticated and highly specialized (Degnan, 2015). In contrary to A. queenslandica, Vaceletia sp. seems to have a much reduced (if any) NLR-based immune system response. We could find no contig that contained both NACHT and LRR domains, and only three contigs possessed a NACHT domain and a C-terminal DEATH domain but lacked detectable LRRs (Table S5).
Toll-Like Receptor Signaling Pathway
The recognition of PAMPs through TLRs results in the initialization of signaling cascades such as the Toll-like receptor signaling pathway. Depending on which adapter molecule [e.g., MyD88 or TRIF-related adapter molecule (TRAM)] is recruited, the activation of the pathway leads to the production of inflammatory cytokines or the induction of type I interferon (Akira et al., 2006). In addition to the pathway leading to nuclear localization of NF-κB, the TLR signaling cascade can also activate the Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) resulting in the activation of AP1 (activating protein 1). NF-κB and AP1 are both transcription factors, which when activated result in the transcription of genes involved in immune responses. The link between these pathways seems to require the participation of the adaptor protein ECSIT (evolutionary conserved signaling intermediate in Toll pathway) (Kopp et al., 1999), which have also been shown to link the TLR signaling pathway to the TGF-β (transforming growth factor-beta)/BMP (bone morphogenetic protein) pathway (Xiao et al., 2003).
To obtain a more comprehensive picture of what is already known for sponge innate immunity components we surveyed the literature and summarized the present state of knowledge in Figure 6. All components listed were either obtained by transcriptomic, genomic or experimentally focused studies. Additionally, we mapped the transcriptomes of Aphrocallistes vastus, Ircinia fasciculate, Chondrilla nucula, Petrosia ficiformis, Spongilla lacustris, Pseudospongosorites suberitoides, Xestospongia testudinaris, Stylissa cateri, Sycon coactum, and Corticum candelabrum against the KEGG TLR signaling pathway. A recent study of the genome and expressed sequence tags of A. queenslandica showed that the majority of the molecules involved in TLR signaling are present (Gauthier et al., 2010). Mapping the Vaceletia sp. transcriptome against the KEGG TLR signaling pathway shows that most of the components in the pathway from MyD88 to NF-κB and AP-1 are present, with all mapped contigs being of metazoan origin (Figure 7). TLR, FADD and MEKK3/6 were not detected by KEGG, however manual BLASTing of these components against the Vaceletia sp. transcriptome indicated the presence of contigs with significant similarities to these components (Table S6). The other sponge transcriptomes that were mapped to the KEGG TLR signaling pathway show a similar pattern of presences and absence of pathway components (Figure 6).
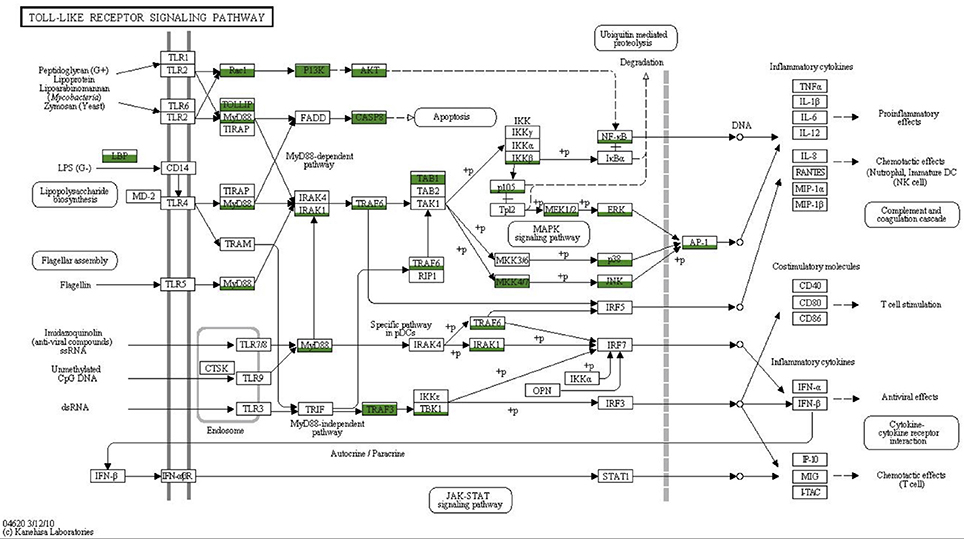
Figure 7. Coverage of the TLR signaling pathway. MEGAN was used to visualize contigs on the KEGG map. Rectangles represent proteins of the pathway while arrows represent signaling routes. Pathway components showing significant similarities to contigs in the Vaceletia sp. transcriptome are highlighted in green. Different fill levels indicate the number of contigs assigned to the corresponding pathway component.
As discussed above, canonical TLRs are missing from the Vaceletia sp. transcriptome. Wiens et al. (2005) proposed an alternative pattern recognition system for antimicrobial defense, a cell surface protein named SLIP (sponge lipopolysaccharide (LPS)-interacting protein), which was suggested to be a substitute for the missing TLRs as it interacts in vivo with MyD88. The Vaceletia sp. transcriptome contains contigs that share similarity with SLIP (CAI68017.1), however without experimental validation it is impossible to say if this protein substitutes for the missing TLRs.
Although, it is not entirely clear how sponges recognize PAMPs, the presence of a mostly complete TLR signaling pathway in all investigated sponges to date suggests this pathway plays an important role in the sponge immune response. To gain deeper insight into how sponges manage their relationships with bacteria, functional analyses are now needed to dissect the roles that these TLR components play.
Eukaryotic-Like Domains
Recent metagenomic studies on sponges have revealed an abundance of eukaryotic-like proteins in sponge-associated bacteria (Siegl et al., 2010; Thomas et al., 2010; Fan et al., 2012; Liu et al., 2012; Kamke et al., 2014; Li et al., 2014). Notably, genes encoding proteins containing ankyrin repeats (ANKs) and tetratricopeptide repeats (TPRs) are abundant. These classes of proteins have been found in various facultative and obligate symbionts, as well as in intracellular pathogens and are thought to play a role in host microbe interactions by interfering with eukaryotic protein-protein interactions. For example, recent work provides evidence that ANK proteins of an uncultured gamma-proteobacterial sponge symbiont can interfere with phagocytosis which could provide a mechanism for bacteria to avoid digestion by their host (Nguyen et al., 2013). Vaceletia sp. contigs assigned by MEGAN to a bacterial origin were searched for the occurrence of eukaryotic-like proteins. This subset was rich in proteins containing ANK and TPR domains (Table S7). Other eukaryotic-like proteins with potential host-bacteria communication functions are also present (Table S7). NHL domain (named after NCL-1, HT2A, and Lin-41) containing proteins were abundant and are thought to play a role in protein-protein interactions. Fibronectin and Cadherin domains, likely to be involved in cell adhesion (Hoffmann et al., 2011), were also found in the bacterially derived subset. A recent study demonstrated that sponge symbionts generally possess more eukaryotic-like proteins than bacteria from the surrounding seawater (Fan et al., 2012). The presence of a variety of eukaryotic-like proteins within Vaceletia's bacterially derived transcriptome supports that observation. Although, evidence is accumulating that eukaryotic-like domains are enriched in sponge endobiotic bacteria, the role of these proteins remains unknown and awaits the development of functional assays.
Conserved Developmental Signaling Pathways and Homeobox Genes
It is now well recognized that the genetic repertoire of sponges is more complex than their morphological simple bodyplan implies. To check the completeness of the Vaceletia sp. transcriptome we surveyed it for the occurrence of important signaling pathway components belonging to the Hedgehog, TGF-β, Wnt, and Notch/Delta pathways that have recently been reported from other sponge transcriptomes (Riesgo et al., 2014). Components were identified using KEGG mapping and/or BLASTx searches. Our results show that almost all signaling components known from other sponges are present in Vaceletia sp. (Figure 8). Some components of the Wnt pathway appear to be missing. Sequences showing similarity to TCF, Gro and SFRP are present but could not be unambiguously identified.
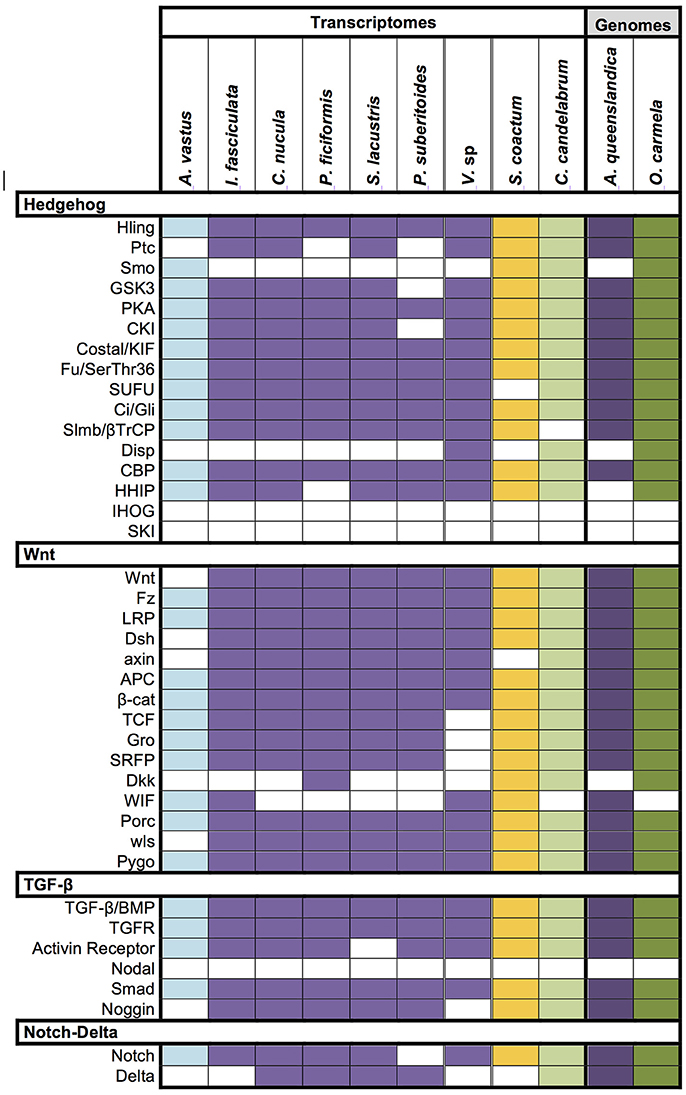
Figure 8. Conserved signaling pathway components identified in Vaceletia sp. Presence/absence of genes belonging to the Hedgehog, TGF-β, Wnt, and Notch/Delta pathways in Vaceletia sp. compared to the presence of these genes in other sponges recently reported from Riesgo et al. (2014). Different colors represent different sponge class; blue, Hexactinellid; purple, Demosponge; yellow, Calcarea; green, Homoscleromorpha; Colored square, component present; white square, component absent.
Homeobox genes play crucial roles in developmental processes in all animals. The genome of the demosponge Amphimedon queenslandica contains several NK genes linked in a cluster but no Hox or ParaHox genes (Larroux et al., 2007). Due to the lack of Hox and ParaHox genes in both Amphimedon and the ctenophore Mnemiopsis leidyi the origin of Hox and ParaHox genes have been proposed to have occurred after the divergences of sponges and ctenophores from all other animals (Larroux et al., 2007; Ryan et al., 2010). On the other hand, the lack of Hox and ParaHox genes has been interpreted as gene loss as the genome of Amphimedon possesses distinct Hox and ParaHox neighborhoods (so called ghost loci) (Ramos et al., 2012). The presence of the ParaHox gene Cdx in the calcisponges Sycon ciliatum and Leucosolenia complicata supports the ghost locus hypothesis and pushes the origin of Hox and ParaHox genes prior to the divergence of sponges from the rest of the animals (Fortunato et al., 2014).
We have identified 40 homeobox containing genes in the Vaceletia transcriptome (Table S8). Of these, 11 show significant similarity to ANPT-, nine to PRD-, six to LIM-, three to POU-, two to SINE- and nine to TALE-class genes. However, this classification of Vaceletia's homeobox genes relies solely on similarity searches and should only be considered an initial classification. Further lines of evidence (e.g., careful phylogenetic analyses) need to be performed before these classifications could be considered complete. The current knowledge of homeobox containing genes in sponges is summarized in Figure 9. As already described by Fortunato et al. (2014), the ANTP-class gene repertoire of the two calcisponges is very similar but differs notable from the repertoire of the two demosponges. Neither Hox nor ParaHox genes are present in the transcriptome of Vaceletia. Both Amphimedon and Vaceletia contain genes of all homeobox gene classes but differ in their composition (Figure 9). No further data (except for the ANTP-class) is currently available for the Calcispongiae. The number of homeobox genes identified in Vaceletia also confirms the significantly reduced demosponge homeobox gene content in comparison to eumatazoan lineages (Larroux et al., 2007).
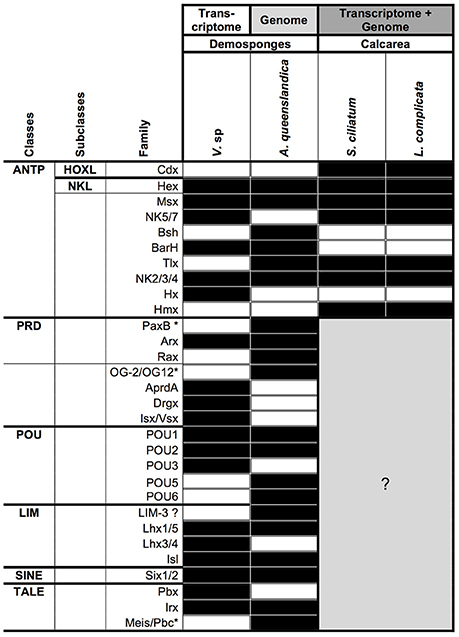
Figure 9. Presence/absence of homeobox genes in sponges. Black square, component present; white square, component absent; gray square, unknown. Data for A. queenslandica and the two Calcisponges was taken from recent publications (Larroux et al., 2007; Fortunato et al., 2014). ANTP, Antennapedia; HOXL, HOX-like; NKL, NK-like; PRD, name derives from the Paired gene of Drosophila; POU, name derives from the mammalian Pou1F1, Pou2F1, Pou2F2, and nematode unc-86 genes; LIM, derives from the nematode lin-11, mammalian Isl1 and nematode mec-3 genes; SINE, named after the Drosophila gene sine oculis; TALE, Three Amino Acid Loop Extension. *Gene name not present in the Homeobox Database (Zhong et al., 2008; Zhong and Holland, 2011).
Conclusions
In characterizing and comparing the transcriptome of Vaceletia sp. to other sponge transcriptomes and genomes we demonstrate that this genus is likely to be interacting with its extensive microbial community in a variety of ways. Our data further highlights the important role the sponge innate immune system is likely to play in managing its microbial community, and the possible role that bacterial eukaryotic-like proteins may also play in this interaction. Metabolic interactions between sponge-associated bacteria and the sponge host are also likely to be more complex than currently appreciated. Our data also highlights the discrepancy between our understanding of how complex these interactions are, and how these interactions are functionally fulfilled. Many other interesting questions remain unanswered such as how is bacterial growth within a host regulated, to what extent are these relationships mutualistic, commensal or parasitic, or how do sponges determine the composition of their bacterial community? As sponge-microbe symbioses are likely to be one of the most ancient within the animal kingdom addressing these questions will assist our understanding of how these relationships can evolve.
Ethics Statement
All experiments described here comply with the current laws and regulations of the country they were conducted in.
Author Contributions
JG carried out bioinformatic analyses and wrote the manuscript. NC conducted the de novo transcriptome assemblies and the BUSCO analysis. DJ collected the samples for transcriptome sequencing, conceived, and supervised the study, and planned and drafted the manuscript. All authors read and approved the final manuscript.
Funding
This research was funded by a grant from the Deutsche Forschungsgemeinschaft to DJ (JA2108/2-1), and from the Open Access Publication Fund provided by the University of Göttingen.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Transcriptome and Genome Analysis Laboratory (University of Göttingen) provided Illumina sequencing services. Samples for transcriptome sequencing were collected during the “Deep Down Under” expedition (DFG project Wo896/7-1), and we acknowledge Professor Gert Wörheide's (LMU, Munich) role in leading that expedition. Professor Volker Thiel and Dr. Martin Blumenberg are gratefully acknowledged for assistance with the GC/MS work. Samples for GC/MS analysis were collected through work funded by DFG projects Mi 157/10; Re 665/4,8; Th 713/1.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2017.00081/full#supplementary-material
Abbreviations
ANK, ankyrin repeats; ANTP, Antennapedia; AP1, activating protein 1; BLAST, Basic Local Alignment Search Tool; BMP, bone morphogenetic protein; Cds, coding sequences; DAMPS, damage-associated molecular patterns; DGGE, Denaturing gradient gel electrophoresis; ECSIT, evolutionary conserved signaling intermediate in Toll pathway; Elovl, elongation of very long chain fatty acids; FA, fatty acid; FAS, fatty acid synthase; GC/MS, gas chromatography/mass spectrometry; HADC, β-hydroxyacyl-CoA dehydratase; HMA, high microbial abundance; HOXL, HOX-like; IG, Immunoglobulin; JNK, Jun N-terminal kinase; KAR, ketoacyl-CoA reductase; KEEG, Kyoto Encyclopedia of Genes and Genomes; LIM, derives from the nematode lin-11; mammalian Isl1 and nematode mec-3 genes; LMA, low microbial abundance; LPS, lipopolysaccharide; LRR, leucine-rich-repeat; MAMP, microbial-associated molecular pattern; MAPK, p38 mitogen-activated protein kinase; MBFA, mid-chain branched fatty acid; My D88, myeloid differentiation primary response gene 88; NKL, NK-like; NLR, Nucleotide-binding domain and Leucine-rich repeat protein; NOD, Nucleotide Oligorimerisation Domain; PAMP, pathogen-associated molecular pattern; PKS, polyketide synthase; POU, name derives from the mammalian Pou1F1, Pou2F1, Pou2F2, and nematode unc-86 genes; PRD, name derives from the Paired gene of Drosophila; PRR, Pattern Recognition Receptor; SINE, named after the Drosophila gene sine oculis; SLIP, sponge lipopolysaccharide-interacting protein; SMT, sterol-24/28-methyltransferase; Sup-type PKS, sponge symbiont ubiquitous PKS; TALE, Three Amino Acid Loop Extension; TCF, T-cell specific transcription factor; TER, trans-2,3-enoyl-CoA reductase; TGF-β, transforming growth factor- β; TIR, Toll/interleukin-1 receptor; TLR, toll-like receptor; TPR, tetratricopeptide repeats; TRAM, TRIF-related adapter molecule; TRIF, TIR-domain-containing adapter-inducing interferon-β.
References
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Andrews, S. (2014). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Antcliffe, J. B. (2013). Questioning the evidence of organic compounds called sponge biomarkers. Palaeontology 56, 917–925. doi: 10.1111/pala.12030
Bergquist, P. R., Karuso, P., Cambie, R. C., and Smith, D. J. (1991). Sterol composition and classification of the Porifera. Biochem. Syst. Ecol. 19, 17–24. doi: 10.1016/0305-1978(91)90109-D
Bird, C. W., Lynch, J. M., Pirt, F. J., Reid, W. W., Brooks, C. J., and Middleditch, B. S. (1971). Steroids and squalene in Methylococcus capsulatus grown on methane. Nature 230, 473–474. doi: 10.1038/230473a0
Bode, H. B., Zeggel, B., Silakowski, B., Wenzel, S. C., Reichenbach, H., and Möller, R. (2003). Steroid biosynthesis in prokaryotes: identification of myxobacterial steroids and cloning of the first bacterial 2, 3 (S)-oxidosqualene cyclase from the myxobacterium Stigmatella aurantiaca. Mol. Microbiol. 47, 471–481. doi: 10.1046/j.1365-2958.2003.03309.x
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Castro, L. F., Wilson, J. M., Gonçalves, O., Galante-Oliveira, S., Rocha, E., and Cunha, I. (2011). The evolutionary history of the stearoyl-CoA desaturase gene family in vertebrates. BMC Evol. Biol. 11:132. doi: 10.1186/1471-2148-11-132
Cerveau, N., and Jackson, D. J. (2016). Combining independent de novo assemblies optimizes the coding transcriptome for nonconventional model eukaryotic organisms. BMC Bioinformatics 17:525. doi: 10.1186/s12859-016-1406-x
Cox, J. S., Chen, B., McNeil, M., and Jacobs, W. R. (1999). Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402, 79–83. doi: 10.1038/47042
Degnan, S. M. (2015). The surprisingly complex immune gene repertoire of a simple sponge, exemplified by the NLR genes: a capacity for specificity? Dev. Comp. Immunol. 506, 58–62. doi: 10.1016/j.dci.2014.07.012
Desmond, E., and Gribaldo, S. (2009). phylogenomics of sterol synthesis: insights into the origin, evolution, and diversity of a key eukaryotic feature. Genome Biol. Evol. 1, 364–381. doi: 10.1093/gbe/evp036
Djerassi, C., and Lam, W. K. (1991). Phospholipid studies of marine organisms. Part 25. Sponge phospholipids. Accounts Chem. Res. 24, 69–75. doi: 10.1021/ar00003a002
Djerassi, C., and Silva, C. J. (1991). Biosynthetic studies of marine lipids. 41. Sponge sterols: origin and biosynthesis. Accounts Chem. Res. 24, 371–378. doi: 10.1021/ar00012a003
Erpenbeck, D., Voigt, O., Wörheide, G., and Lavrov, D. V. (2009). The mitochondrial genomes of sponges provide evidence for multiple invasions by Repetitive Hairpin-forming Elements (RHE). BMC Genomics 10:591. doi: 10.1186/1471-2164-10-591
Fan, L., Reynolds, D., Liu, M., Stark, M., Kjelleberg, S., Webster, N. S., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. U.S.A. 109, E1878–E1887. doi: 10.1073/pnas.1203287109
Fernandez-Valverde, S. L., and Degnan, B. M. (2016). Bilaterian-like promoters in the highly compact Amphimedon queenslandica genome. Sci. Rep. 6, 22496–22496. doi: 10.1038/srep22496
Ferrand, J., and Ferrero, R. L. (2013). Recognition of extracellular bacteria by NLRs and its role in the development of adaptive immunity. Front. Immunol. 4:344. doi: 10.3389/fimmu.2013.00344
Fieseler, L., Hentschel, U., Grozdanov, L., Schirmer, A., Wen, G., Platzer, M., et al. (2007). widespread occurrence and genomic context of unusually small polyketide synthase genes in microbial consortia associated with marine sponges. Appl. Environ. Microbiol. 73, 2144–2155. doi: 10.1128/AEM.02260-06
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Fiore, C. L., Labrie, M., Jarett, J. K., and Lesser, M. P. (2015). Transcriptional activity of the giant barrel sponge, Xestospongia muta Holobiont: molecular evidence for metabolic interchange. Front. Microbiol. 6:364. doi: 10.3389/fmicb.2015.00364
Fortunato, S. A., Adamski, M., Ramos, O. M., Leininger, S., Liu, J., Ferrier, D. E., et al. (2014). Calcisponges have a ParaHox gene and dynamic expression of dispersed NK homeobox genes. Nature 514, 620–623. doi: 10.1038/nature13881
Franchi, L., Eigenbrod, T., Muñoz-Planillo, R., and Nunez, G. (2009). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247. doi: 10.1038/ni.1703
Gauthier, M. E. A., Du Pasquier, L., and Degnan, B. M. (2010). The genome of the sponge Amphimedon queenslandica provides new perspectives into the origin of Toll-like and interleukin 1 receptor pathways. Evol. Dev. 12, 519–533. doi: 10.1111/j.1525-142X.2010.00436.x
Germer, J., Mann, K., Wörheide, G., and Jackson, D. J. (2015). The skeleton forming proteome of an early branching metazoan: a molecular survey of the biomineralization components employed by the coralline sponge Vaceletia sp. PLoS ONE 10:e0140100. doi: 10.1371/journal.pone.0140100
Gillan, F. T., Stoilov, I. L., Thompson, J. E., Hogg, R. W., Wilkinson, C. R., and Djerassi, C. (1988). Fatty acids as biological markers for bacterial symbionts in sponges. Lipids 23, 1139–1145. doi: 10.1007/BF02535280
Goad, L. J. (1981). Sterol biosynthesis and metabolism in marine invertebrates. Pure Appl. Chem. 53, 837–852. doi: 10.1351/pac198153040837
Gold, D. A., Grabenstatter, J., de Mendoza, A., Riesgo, A., Ruiz-Trillo, I., and Summons, R. E. (2016). Sterol and genomic analyses validate the sponge biomarker hypothesis. Proc. Natl. Acad. Sci. U.S.A. 113, 2684–2689. doi: 10.1073/pnas.1512614113
Gordon, S. (2002). Pattern recognition receptors. Cell 111, 927–930. doi: 10.1016/S0092-8674(02)01201-1
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hahn, S., Stoilov, I. L., Ha, T. B. T., Raederstorff, D., Doss, G. A., Li, H. T., et al. (1988). Biosynthetic studies of marine lipids. 17. The course of chain elongation and desaturation in long-chain fatty acids of marine sponges. J. Am. Chem. Soc. 110, 8117–8124. doi: 10.1021/ja00232a025
Hentschel, U., Hopke, J., Horn, M., Friedrich, A. B., Wagner, M., Hacker, J., et al. (2002). Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 68, 4431–4440. doi: 10.1128/AEM.68.9.4431-4440.2002
Hentschel, U., Piel, J., Degnan, S. M., and Taylor, M. W. (2012). Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 10, 641–654. doi: 10.1038/nrmicro2839
Hochmuth, T., Niederkrüger, H., Gernert, C., Siegl, A., Taudien, S., Platzer, M., et al. (2010). Linking chemical and microbial diversity in marine sponges: possible role for Poribacteria as producers of methyl-branched fatty acids. ChemBioChem. 11, 2572–2578. doi: 10.1002/cbic.201000510
Hoffmann, C., Ohlsen, K., and Hauck, C. R. (2011). Integrin-mediated uptake of fibronectin-binding bacteria. Eur. J. Cell Biol. 90, 891–896. doi: 10.1016/j.ejcb.2011.03.001
Huson, D. H., Mitra, S., Ruscheweyh, H. J., Weber, N., and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. doi: 10.1101/gr.120618.111
Jackson, D. J., Macis, L., Reitner, J., Degnan, B. M., and Wörheide, G. (2007). Sponge paleogenomics reveals an ancient role for carbonic anhydrase in skeletogenesis. Science 316, 1893–1895. doi: 10.1126/science.1141560
Jackson, D. J., Macis, L., Reitner, J., and Wörheide, G. (2011). A horizontal gene transfer supported the evolution of an early metazoan biomineralization strategy. BMC Evol. Biol. 11:238. doi: 10.1186/1471-2148-11-238
Jackson, D. J., Thiel, V., and Wörheide, G. (2010). An evolutionary fast-track to biocalcification. Geobiology 8, 191–196. doi: 10.1111/j.1472-4669.2010.00236.x
Jackson, D. J., and Wörheide, G. (2014). Symbiophagy and biomineralization in the “living fossil” Astrosclera willeyana. Autophagy 10, 408–415. doi: 10.4161/auto.27319
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216. doi: 10.1146/annurev.immunol.20.083001.084359
Jenke-Kodama, H. (2005). Evolutionary Implications of Bacterial Polyketide Synthases. Mol. Biol. Evol. 22, 2027–2039. doi: 10.1093/molbev/msi193
Kamke, J., Rinke, C., Schwientek, P., Mavromatis, K., Ivanova, N., Sczyrba, A., et al. (2014). The candidate phylum Poribacteria by single-cell genomics: new insights into phylogeny, cell-compartmentation, eukaryote-like repeat proteins, and other genomic features. PLoS ONE 9:e87353. doi: 10.1371/journal.pone.0087353
Karlińska-Batres, K., and Wörheide, G. (2013). Microbial diversity in the coralline sponge Vaceletia crypta. Antonie Van Leeuwenhoek 103, 1041–1056. doi: 10.1007/s10482-013-9884-6
Kennedy, J., Marchesi, J. R., and Dobson, A. D. W. (2007). Metagenomic approaches to exploit the biotechnological potential of the microbial consortia of marine sponges. Appl. Microbiol. Biotechnol. 75, 11–20. doi: 10.1007/s00253-007-0875-2
Kopp, E., Medzhitov, R., Carothers, J., Xiao, C., Douglas, I., Janeway, C. A., et al. (1999). ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev. 13, 2059–2071. doi: 10.1101/gad.13.16.2059
Kornprobst, J.-M., and Barnathan, G. (2010). Demospongic acids revisited. Mar. Drugs 8, 2569–2577. doi: 10.3390/md8102569
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Larroux, C., Fahey, B., Degnan, S. M., Adamski, M., Rokhsar, D. S., and Degnan, B. M. (2007). The NK homeobox gene cluster predates the origin of Hox genes. Curr. Biol. 17, 706–710. doi: 10.1016/j.cub.2007.03.008
Leonard, A. E., Pereira, S. L., Sprecher, H., and Huang, Y.-S. (2004). Elongation of long-chain fatty acids. Prog. Lipid Res. 43, 36–54. doi: 10.1016/S0163-7827(03)00040-7
Levin, M., Anavy, L., Cole, A. G., Winter, E., Mostov, N., Khair, S., et al. (2016). The mid-developmental transition and the evolution of animal body plans. Nature 531, 637–641. doi: 10.1038/nature16994
Li, Z.-Y., Wang, Y.-Z., He, L.-M., and Zheng, H.-J. (2014). Metabolic profiles of prokaryotic and eukaryotic communities in deep-sea sponge Neamphius huxleyi indicated by metagenomics. Sci. Rep. 4, 3895–3895. doi: 10.1038/srep03895
Litchfield, C., Greenberg, A. J., Noto, G., and Morales, R. W. (1976). Unusually high levels of C24 - C30 fatty acids in sponges of the class demospongiae. Lipids 11, 567–570. doi: 10.1007/BF02532903
Liu, M., Fan, L., Zhong, L., Kjelleberg, S., and Thomas, T. (2012). Metaproteogenomic analysis of a community of sponge symbionts. ISME J. 6, 1515–1525. doi: 10.1038/ismej.2012.1
Love, G. D., Grosjean, E., Stalvies, C., Fike, D. A., Grotzinger, J. P., Bradley, A. S., et al. (2008). Fossil steroids record the appearance of Demospongiae during the Cryogenian period. Nature 457, 718–721. doi: 10.1038/nature07673
Lu, Y.-J., Zhang, Y.-M., and Rock, C. O. (2004). Product diversity and regulation of type II fatty acid synthases. Biochem. Cell Biol. 82, 145–155. doi: 10.1139/o03-076
McCaffrey, M. A., Moldowan, J. M., Lipton, P. A., Summons, R. E., Peters, K. E., Jeganathan, A., et al. (1994). Paleoenvironmental implications of novel C 30 steranes in Precambrian to Cenozoic age petroleum and bitumen. Geochim. Cosmochim. Ac. 58, 529–532. doi: 10.1016/0016-7037(94)90481-2
Monroig, Ó., Tocher, D., and Navarro, J. (2013). Biosynthesis of polyunsaturated fatty acids in marine invertebrates: recent advances in molecular mechanisms. Mar. Drugs 11, 3998–4018. doi: 10.3390/md11103998
Morales, R. W., and Litchfield, C. (1977). Incorporation of 1-14C-Acetate into C26 fatty acids of the marine sponge Microciona prolifera. Lipids 12, 570–576. doi: 10.1007/BF02533383
Morrow, C., and Cárdenas, P. (2015). Proposal for a revised classification of the Demospongiae (Porifera). Front. Zool. 12:7. doi: 10.1186/s12983-015-0099-8
Müller, W. E. G., and Müller, I. M. (2003). Origin of the metazoan immune system: identification of the molecules and their functions in sponges. Integr. Comp. Biol. 43, 292–281. doi: 10.1093/icb/43.2.281
Nakanishi, N., Sogabe, S., and Degnan, B. M. (2014). Evolutionary origin of gastrulation: insights from sponge development. BMC Biol. 12:26. doi: 10.1186/1741-7007-12-26
Nguyen, M. T., Liu, M., and Thomas, T. (2013). Ankyrin-repeat proteins from sponge symbionts modulate amoebal phagocytosis. Mol. Ecol. 23, 1635–1645. doi: 10.1111/mec.12384
Pawlik, J. R. (2011). The chemical ecology of sponges on caribbean reefs: natural products shape natural systems. BioScience 61, 888–898. doi: 10.1525/bio.2011.61.11.8
Peng, Y., Leung, H. C. M., Yiu, S.-M., Lv, M.-J., Zhu, X.-G., and Chin, F. Y. (2013). IDBA-tran: a more robust de novo de Bruijn graph assembler for transcriptomes with uneven expression levels. Bioinformatics 29, i326–i334. doi: 10.1093/bioinformatics/btt219
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pick, K. S., Philippe, H., Schreiber, F., Erpenbeck, D., Jackson, D. J., Wrede, P., et al. (2010). Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Mol. Biol. Evol. 27, 1983–1987. doi: 10.1093/molbev/msq089
Piel, J., Hui, D., Wen, G., Butzke, D., Platzer, M., Fusetani, N., et al. (2004). Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc. Natl. Acad. Sci. U.S.A. 101, 16222–16227. doi: 10.1073/pnas.0405976101
Pisani, D., Pett, W., Dohrmann, M., Feuda, R., Rota-Stabelli, O., Philippe, H., et al. (2015). Genomic data do not support comb jellies as the sister group to all other animals. Proc. Natl. Acad. Sci. U.S.A. 112, 15402–15407. doi: 10.1073/pnas.1518127112
Radax, R., Rattei, T., Lanzen, A., Bayer, C., Rapp, H. T., Urich, T., et al. (2012). Metatranscriptomics of the marine sponge Geodia barretti: tackling phylogeny and function of its microbial community. Eviron. Microbiol. 14, 1308–1324. doi: 10.1111/j.1462-2920.2012.02714.x
Ramos, O. M., Barker, D., and Ferrier, D. E. K. (2012). Ghost loci imply Hox and ParaHox existence in the last common ancestor of animals. Curr. Biol. 22, 1951–1956. doi: 10.1016/j.cub.2012.08.023
Reitner, J. (1992). Coralline spongien: der versuch einer phylogenetisch-taxonomischen analyse. Berl. Geowisschenschaftliche Abh. E 1, 1–352.
Reitner, J., Wörheide, G., Lange, R., and Thiel, V. (1997). Biomineralization of calcified skeletons in three Pacific coralline demosponges - an approach to the evolution of basal skeletons. Cour. Forsch-Inst. Senckenberg 201, 371–383
Riesgo, A., Farrar, N., Windsor, P. J., Giribet, G., and Leys, S. P. (2014). The analysis of eight transcriptomes from all poriferan classes reveals surprising genetic complexity in sponges. Mol. Biol. Evol. 31, 1102–1120. doi: 10.1093/molbev/msu057
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Ryan, J. F., Pang, K., Mullikin, J. C., Martindale, M. Q., and Baxevanis, A. D. (2010). The homeodomain complement of the ctenophore Mnemiopsis leidyi suggests that Ctenophora and Porifera diverged prior to the ParaHoxozoa. Evol. Dev. 1:9. doi: 10.1186/2041-9139-1-9
Ryu, T., Seridi, L., Moitinho-Silva, L., Oates, M., Liew, Y. J., Mavromatis, C., et al. (2016). Hologenome analysis of two marine sponges with different microbiomes. BMC Genomics 17:158. doi: 10.1186/s12864-016-2501-0
Siegl, A., Kamke, J., Hochmuth, T., Piel, J. O., Richter, M., Liang, C., et al. (2010). Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5, 61–70. doi: 10.1038/ismej.2010.95
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Simister, R. L., Deines, P., Botté, E. S., Webster, N. S., and Taylor, M. W. (2012). Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Eviron. Microbiol. 14, 517–524. doi: 10.1111/j.1462-2920.2011.02664.x
Srivastava, M., Simakov, O., Chapman, J., Fahey, B., Gauthier, M. E. A., Mitros, T., et al. (2010). The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466, 720–726. doi: 10.1038/nature09201
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Stuart, L. M., Paquette, N., and Boyer, L. (2013). Effector-triggered versus pattern-triggered immunity: how animals sense pathogens. Nat. Rev. Immunol. 13, 199–206. doi: 10.1038/nri3398
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-Associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/MMBR.00040-06
Thiel, V., Jenisch, A., Wörheide, G., Löwenberg, A., Reitner, J., and Michaelis, W. (1999). Mid-chain branched alkanoic acids from “living fossil” demosponges: a link to ancient sedimentary lipids? Org. Geochem. 30, 1–14. doi: 10.1016/S0146-6380(98)00200-9
Thomas, T., Rusch, D., DeMaere, M. Z., Yung, P. Y., Lewis, M., Halpern, A., et al. (2010). Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 4, 1557–1567. doi: 10.1038/ismej.2010.74
Vacelet, J. (1977). Une nouvelle relique du secondaire: un représentant actuel des eponges fossiles sphinctozoaires. C. R. L'Academie Des Sci. Paris D 285, 509–511.
Webster, N. S., and Thomas, T. (2016). The sponge hologenome. MBio 7, e00135–e00116. doi: 10.1128/mBio.00135-16
Wiens, M., Korzhev, M., Krasko, A., Thakur, N. L., Perovi -Ottstadt, S., Breter, H. J., et al. (2005). Innate immune defense of the sponge Suberites domuncula against bacteria involves a MyD88-dependent signaling pathway induction of a perforin-like molecule. J. Biol. Chem. 280, 27949–27959. doi: 10.1074/jbc.M504049200
Wiens, M., Korzhev, M., Perovic-Ottstadt, S., Luthringer, B., Brandt, D., Klein, S., et al. (2007). Toll-like receptors are part of the innate immune defense system of sponges (Demospongiae: Porifera). Mol. Biol. Evol. 24, 792–804. doi: 10.1093/molbev/msl208
Wilkinson, C. R., Garrone, R., and Vacelet, J. (1984). Marine sponges discriminate between food bacteria and bacterial symbionts: electron microscope radioautography and in situ evidence. Proc. R. Soc. Lond. B. Biol. Sci. 220, 528–519. doi: 10.1098/rspb.1984.0018
Wilson, M. C., Mori, T., Rückert, C., Uria, A. R., Helf, M. J., Takada, K., et al. (2014). An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506, 58–62. doi: 10.1038/nature12959
Wörheide, G. (1998). The reef cave dwelling ultraconservative coralline demosponge Astrosclera willeyana from the Indo-Pacific. Facies 38, 1–88. doi: 10.1007/BF02537358
Wörheide, G. (2008). A hypercalcified sponge with soft relatives: Vaceletia is a keratose demosponge. Mol. Phylogenet. Evol. 47, 433–438. doi: 10.1016/j.ympev.2008.01.021
Xiao, C., Shim, J. H., Klüppel, M., Zhang, S. S. M., Dong, C., Flavell, R. A., et al. (2003). Ecsit is required for Bmp signaling and mesoderm formation during mouse embryogenesis. Genes Dev. 17, 2933–2949. doi: 10.1101/gad.1145603
Yin, Z., Zhu, M., Davidson, E. H., Bottjer, D. J., Zhao, F., and Tafforeau, P. (2015). Sponge grade body fossil with cellular resolution dating 60 Myr before the Cambrian. Proc. Natl. Acad. Sci. U.S.A. 112, E1453–E1460. doi: 10.1073/pnas.1414577112
Yuen, B., Bayes, J. M., and Degnan, S. M. (2014). The characterization of sponge NLRs provides insight into the origin and evolution of this innate immune gene family in animals. Mol. Biol. Evol. 31, 106–120. doi: 10.1093/molbev/mst174
Zhong, Y.-F., Butts, T., and Holland, P. W. (2008). HomeoDB: a database of homeobox gene diversity. Evol. Dev. 10, 516–518. doi: 10.1111/j.1525-142X.2008.00266.x
Zhong, Y.-F., and Holland, P. W. H. (2011). HomeoDB2: functional expansion of a comparative homeobox gene database for evolutionary developmental biology. Evol. Dev. 13, 567–568. doi: 10.1111/j.1525-142X.2011.00513.x
Ziemert, N., Podell, S., Penn, K., Badger, J. H., Allen, E., and Jensen, P. R. (2012). The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 7:e34064. doi: 10.1371/journal.pone.0034064.s007
Zimmerman, M. P., Hoberg, M., Ayanoglu, E., and Djerassi, C. (1990). Cell separation of Tethya aurantia, an analytical study of embryonic and differentiated sponge cells. Lipids 25, 383–390. doi: 10.1007/BF02537981
Keywords: sponge, microbes, symbiosis, transcriptome, immunity, metabolism, fatty acid, signaling pathway
Citation: Germer J, Cerveau N and Jackson DJ (2017) The Holo-Transcriptome of a Calcified Early Branching Metazoan. Front. Mar. Sci. 4:81. doi: 10.3389/fmars.2017.00081
Received: 31 October 2016; Accepted: 10 March 2017;
Published: 28 March 2017.
Edited by:
Dennis A. Bazylinski, University of Nevada, Las Vegas, USAReviewed by:
Torsten Thomas, University of New South Wales, AustraliaEric E. Allen, Scripps Institution of Oceanography, USA
Copyright © 2017 Germer, Cerveau and Jackson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel J. Jackson, ZGphY2tzb0B1bmktZ29ldHRpbmdlbi5kZQ==