 Jun Li
Jun Li Yuehuan Zhang†
Yuehuan Zhang†- Key Laboratory of Tropical Marine Bio-Resources and Ecology, Guangdong Provincial Key Laboratory of Applied Marine Biology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China
Thermal exposure of sessile marine animals inhabiting estuarine intertidal regions is a serious concern with respect to their physiological processes. Organisms living in this region can be exposed to high temperature (>40°C) during the low tide, which may affect the survival of these organisms. The Hong Kong oyster, Crassostrea hongkongensis, distributed along the coast waters of the South China Sea, is one of the dominant sessile inhabitants of marine intertidal region which undergoes large seasonal temperature fluctuations (up to 10–20°C during diurnal cycles) every year. To cope with acute thermal stress, it has developed several adaptation mechanisms, e.g., the firmly shut of the shells. However, the genetic basis of these mechanisms remain largely unclear. To better understand how acute thermal exposure affects the biology of the oyster, two cDNA libraries obtained from the gill of oysters exposed to 37°C thermal stress and ambient temperature were sequenced using the Digital Gene Expression (DGE) tag profiling strategy. In total, 5.9 and 6.2 million reads were obtained from thermal stress and control libraries, respectively, with approximately 74.25 and 75.02% of the reads mapping to the Crassostrea hongkongensis reference sequence. A total of 605 differentially expressed transcripts could be detected in the thermal stress group as compared to the control group, of which 378 are up-regulated and 227 are down-regulated. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis indicated that these Differentially Expressed Genes (DEGs) were enriched with a broad spectrum of biological processes and pathways, including those associated with chaperones, antioxidants, immunity, apoptosis and cytoskeletal reorganization. Among these significantly enriched pathways, protein processing in the endoplasmic reticulum was the most affected metabolic pathway, which plays an important role in the unfolded protein response (UPR) and ER-associated degradation (ERAD) processes. Our results demonstrate the complex multi-modal cellular response to thermal stress in C. hongkongensis.
Introduction
Oysters are marine bivalve belonging to the phylum mollusca, which contains the largest number of described animal species (Jones and Blaxter, 2005). They can inhabit estuarine or intertidal zones characterized by dramatic changes in salinity, oxygen concentration and temperature. Among these, temperature is regarded as the most important factor that potentially regulates the physiological processes in these marine organisms (Prokofiev, 2001). With the intertidal zone's high exposure to the sun, the temperature can range from very high during the summer to near freezing in the winters. Such drastic changes in temperature can affect the animal's health leading to decreased growth rate and increased mortality. However, these organisms have developed several adaptations to resist such extreme temperature fluctuations. Oysters close their shells which can isolate themself from exogenous heat shock.
The oyster, Crassostrea hongkongensis is endemic to the coastal waters of the South China Sea (Lam and Morton, 2003), which often experience rapid and dramatic temperature fluctuations during diurnal/tidal cycles (up to 10–20°C within a few hours) and seasonal changes (from 10 to 35–40°C). C. hongkongensis have evolved to acquire powerful defense mechanisms to cope with thermal stress enabling it to survive dynamic changes in climatic conditions of the intertidal region. Thus, C. hongkongensis can serve as a model organism for studying thermal stress response (Zhang G. et al., 2015). Additionally, study of thermal stress tolerance in these organisms holds great significance in recent years. Global warming is a major concern, which could adversely affect oyster aquaculture. Various studies have employed quantitative polymerase chain reaction (qRT-PCR) to identify some important molecular mechanisms and key processes during stress adaptation or acclimatization in oyster. And a series of key thermal tolerance related genes including heat shock proteins (HSPs), HSP-related and the genes regulating energy metabolism have been identified in oysters. (Meistertzheim et al., 2007; Kawabe and Yokoyama, 2012; Zhang and Zhang, 2012; Li et al., 2015; Wang et al., 2015). Some other studies have utilized high-throughput techniques like microarray assay to estimate the expression profiles of hundreds of thousands of genes at the genome wide scale (Farcy et al., 2009; Lang et al., 2009). After heat shock, pacific oyster could immediately increase transcription of genes which encode heat shock proteins, and genes whose products synthesize lipids, participate in cellular immunity, and influence reproductive activity. Although our understanding of the general mechanism of thermal tolerance is enriched from these previous studies in pacific oyster, specific information on the thermal stress adaptations of Hong Kong oyster C. hongkongensis remains scant.
Recently, high-throughput transcriptomic studies in C. hongkongensis have focused on reproduction related genes, thus providing valuable resources to the oyster research community (Tong et al., 2015). However, none of the transcriptomic studies investigated the gene expression changes associated with thermal stress in C. hongkongensis. Digital Gene Expression Profiling (DGE) is a high throughput low cost sequencing technique used for quick estimation of tissue specific gene expression levels under specific conditions. It is more advantageous than previous microarrays technology which has several limitations, including the accuracy of low abundance expression measurements, the differences of hybridization properties, and limitation to known genes (Wheelan et al., 2008). This technique has been widely used in transcriptomic studies in various species, including Caenorhabditis elegans (Nesbitt et al., 2010), Physcomitrella patens (Nishiyama et al., 2012), Cucumis sativus (Qi et al., 2012), Microsporum canis (Xiao et al., 2015), Bombyx mori (Zhang et al., 2014), and Mus musculus (Zhang et al., 2013). Aiming to elucidate the molecular mechanisms of thermal stress tolerance in C. hongkongensis, a DGE based gene expression analysis was performed with Hong Kong oysters. Our results yielded sets of up-regulated and down-regulated genes in response to thermal stress. Differentially expressed genes were analyzed by the follow-up gene ontology (GO) and KEGG pathway analysis, shedding light into the pathways involved in thermal stress adaptation in C. hongkongensis.
Materials and Methods
Experimental Oysters and Thermal Stress
Crassostrea hongkongensis specimens were obtained from an oyster farm in Zhanjiang, Guangdong Province, China. The healthy adult oysters which will quickly close and spray seawater upon stimulation with stirring the seawater were then aerated in sand-filtered natural seawater in a 500-L tank at 23° ± 2°C for 2 weeks prior to the experiment. The oysters were fed twice daily with microalgae, Tetraselmis suecica and Isochrysis galbana. For thermal stress treatment, 100 oysters were subjected to heat shock for 1 h in a 100-L tank with pre-warmed (37°C) seawater followed by transfer to ambient seawater (23° ± 2°C). The seawater is static during the treatment with salinity at 20 ppt and pH at 7.2. Previous studies indicate that 1 h thermal exposure is stressful to this species (Lang et al., 2009). The control group was maintained in ambient seawater (23° ± 2°C) throughout the study. After 24 h, 10 oysters were randomly sampled from thermal-stressed and control groups.
Samples Collection and RNA Extraction
Gill tissue of 10 oysters from each of the two groups was sampled in this study. We chose to study gill tissue as it is a large organ with high surface area exposed to the environment and has been shown to be perturbed by thermal stress (Lang et al., 2009). Total RNA was extracted from gill suspensions using 1 mL TRIzol Reagent (Invitrogen, Carlsbad, CA) per 100 mg of tissue following the manufacturer's instructions. RNA concentrations were measured using a Nanodrop 2000c Spectrophotometer (Thermo Scientific, USA) and the quality of the RNA was analyzed by Bioanalyzer 2100.
DGE Library Preparation and Sequencing
DGE Libraries were prepared using the Illumina Digital Gene Expression Tag Profiling Kit, according to the manufacturer's protocol. Briefly, the total RNA samples were treated with deoxyribonuclease I (DNase I) to remove any possible DNA contamination. Then 8 μg of total RNA was treated with Oligo (dT) magnetic beads to purify mRNA. Purified mRNA was mixed with fragmentation buffer, to yield short fragments. The first strand cDNA was synthesized from the cleaved RNA fragments using random primers. RNase H and DNA polymerase I were subsequently added to remove the RNA fragments and synthesize the second strand of cDNA, respectively. The cDNA fragments were then end repaired, followed by 3′ single nucleotide A (adenine) addition and ligation of adapters. Finally, products were purified and amplified by PCR to create the sequencing libraries. The libraries were quantified using Agilent 2100 Bioanalyzer and ABI Step One Plus Real-Time PCR System and then sequenced by Illumina HiSeq 2000 sequencer at the Beijing Genomics Institute (BGI, Shenzhen, China). The sequencing data obtained were deposited in NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE85707, accession number: GSE85707).
Analysis and Mapping of DGE Tags
The raw image outputs from the sequencer were transformed into base calls using the Illunina pipeline. Prior to mapping the DGE tags, empty tags (no tag sequence between the adaptors), adaptors, low quality tags (tags with unknown sequences “N”), tags with a copy number of 1 (likely sequencing error), and tags that were too long or too short were excluded from the analysis, leaving behind clean tags of 21 nt in length. The clean tags were mapped to the transcriptome of C. hongkongensis sequence (Tong et al., 2015) using Bowtie (Langmead et al., 2009). Only tags with a perfect match or 1 nt mismatch were further considered and annotated. To monitor mapping events on both strands, tags mapping to both sense and antisense sequences were included in the data analysis. All tags mapped to the reference sequences were filtered, and the remaining clean tags were designated as unambiguous clean tags. Gene expression was normalized using the Reads Per kb per Million (RPKM) method (Mortazavi et al., 2008).
Characterization and Functional Annotation of DEGs
To identify DEGs, the number of raw clean tags in each library was normalized to RPKM. To examine expression differences between the thermal treatment group and the control group, the RPKM of the two groups were compared. Identification of the DEGs were performed following a rigorous algorithm described previously (Benjamini and Yekutieli, 2001). False discovery rate (FDR) ≤ 0.001 and log2 fold change ≥ 1 were used as cut-off to determine significant differentially expressed genes (DEGs). All DEGs were further subjected to Gene Ontology (GO) and KEGG Orthology (KO) enrichment analysis by mapping the DEGs to the GO (http://www.geneontology.org/) and KEGG (http://www.genome.jp/kegg/) databases followed by hypergeometric tests. A 5% significance level following the Bonferroni correction was used as the threshold for considering enriched terms for DEGs.
Validation of the DEGs by qRT-PCR
The oyster samples from another independent treatment groups were used for quantitative real-time PCR (qRT-PCR) assays. 5 oysters were randomly sampled for each group. The qRT-PCR analysis was performed on selected DEGs using Lightcycler480 (Roche, Basel, Switzerland). All reactions were performed using a SYBR Green MasterMix (SYBR Premix EX Taq™, TaKaRa) according to the manufacturer's instructions. EF1α gene was used as the internal control. Primers used for qRT-PCR are listed in Table 1. PCR conditions were as follows: initial denaturation at 95°C for 3 min; followed by 45 amplification cycles of 95°C for 15 s, 55°C for 15 s, 72°C for 10 s; 85°C for 20 s for signal collection in each cycle. Melting curve analysis was performed at the end of each PCR reaction to confirm specificity of the amplicons. Reactions were performed in triplicate, and the relative expression levels were calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001). Significance of the difference in the expression levels of selected genes between the control and treatment were estimated using Student's t-test.

Table 1. Sequences of primers used in this study.
Results and Discussion
Analysis of DGE Libraries and Tag Mapping
DGE analysis is a powerful tool to identify and quantify gene expression at the whole genome level and could provide comprehensive transcript data for studying differentially expressed genes (Mardis, 2008). Recently, DGE techniques have been successfully utilized to study the molecular mechanism of thermal adaptation in Chlorostoma funebralis, Cryptolaemus montrouzieri, and Delia antiqua (Gleason and Burton, 2015; Zhang Y. H. et al., 2015; Hao et al., 2016). To identify genes involved in thermal stress tolerance in the Hong Kong oyster, a cDNA library was constructed from the gill tissues of 10 pooled oysters subjected to a 1 h heat shock in pre-warmed (37°C) seawater. The control library was also prepared using gills of 10 pooled oysters from a group maintained in ambient seawater (23°C). A total of 5,908,582 and 6,178,057 reads were generated from the thermal stress and the control libraries, respectively (Table 2). Following adapter trimming and clean-up, 4,387,274 (74.25% of total reads) and 4,635,084 (75.02% of total reads) reads could be successfully mapped to the reference transcriptome for the treatment and control, respectively (Figure S1). Among these, 3,400,605 (thermal stress) and 3,691,555 (control) reads matched perfectly to the reference.
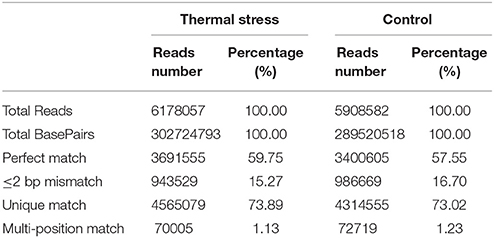
Table 2. Summary of alignment statistics of DGE in two libraries mapped to reference transcriptome.
Sequencing saturation data can reflect the sequencing quality to some extent. Saturation analysis of the clean reads showed that the genes that the clean tags were mapped to were saturated when the tag count approached 3 million (Figure S2). Therefore, the sequencing depth used in our study is sufficient to obtain appropriate transcriptome coverage. Additionally, the unigene coverage, defined by the number of clean reads aligned to a particular reference transcript, was analyzed for the transcriptome reference for each sample. As showed in Figure S3, the distributions of the unigene coverage obtained in the two libraries are more or less uniform, with a >50% coverage obtained for 72 and 76% of the total number of genes for thermal stress and control groups, respectively. This implies that the DGE technology is sensitive enough to detect transcriptomic changes in our model species and can be exploited for gene expression studies at the genome wide scale.
Analysis of Differentially Expressed Genes (DEGs) after Thermal Stress
To analyze the gene expression changes associated with thermal stress in oysters, a rigorous algorithm was utilized to identify DEGs from the normalized DGE datasets using pairwise comparison between the two groups (thermal stress and control). To obtain the authentic DEGs, 10 pooled oysters were used to reduce the variation among individuals. DGE analysis of the two libraries detected transcripts of 20,824 and 20,751 genes under thermal stress and control conditions, respectively (Table S1). The transcripts showing at least 2-fold change (FDR≤0.001 and |log2-Ratio (thermal stress/control)|≥1) in the thermal stress library compared to the control library were considered to be differentially regulated. Using this cut-off criterion, a total of 605 genes were found to be differentially regulated (DEGs) in oysters exposed to thermal stress in comparison to the controls (Table S2). Among these DEGs, 378 genes were up-regulated and 227 were down-regulated in response to thermal stress (Figure 1, Table 3).
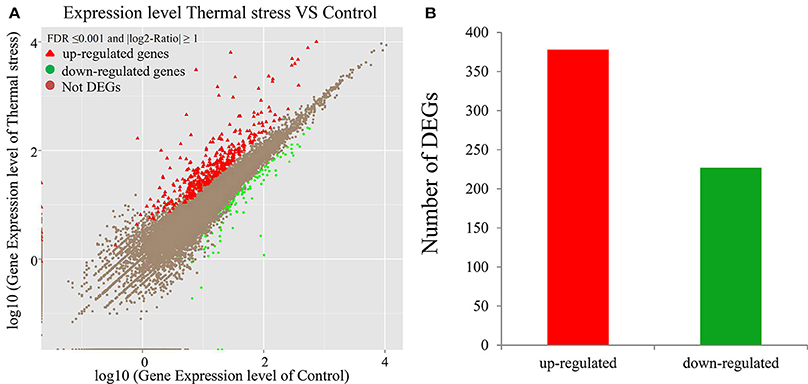
Figure 1. Differentially-expressed genes (DEGs) in thermal stress and control group (P ≤ 0.05). (A) Scatter plot showing the relationship between the gene expression level in oysters in presence and absence of thermal stress. The red triangles and the green dots represent the up-regulated and down-regulated genes, respectively. The brown dots represent the genes that are not differentially expressed. (B) Number of differentially expressed transcripts based on pairwise comparison: 378 (red) among the 605 differentially expressed genes were up-regulated whereas the rest (green) were down-regulated (P ≤ 0.05).
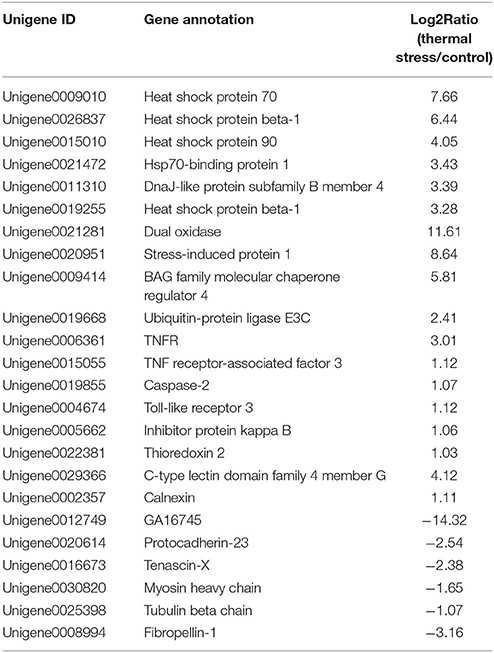
Table 3. Selected DEGs between the thermal tress and control libraries.
Go and Signaling Pathway Enrichment Analysis of DEGs
To gain a better insight into the possible mechanisms of thermal stress response all the 605 differentially expressed genes were mapped to the terms in GO database. The main GO terms included “cellular component,” “molecular function,” and “biological process.” In this study, 110 differentially expressed genes were mapped to a GO ID and 9 functional groups were found to be significantly enriched (Bonferroni-corrected P ≤ 0.05; Table S4) (Figure 2). The genes in the enriched functional groups participated in various metabolic and cellular functions including endoplasmic reticulum membrane (GO:0005789), nuclear outer membrane-endoplasmic reticulum membrane network (GO:0042175), endoplasmic reticulum (GO:0005783), endoplasmic reticulum part (GO:0044432), purine nucleoside binding (GO:0001883), ribonucleoside binding (GO:0032549), purine ribonucleoside binding (GO:0032550), nucleoside binding (GO:0001882) and small molecule binding (GO:0036094). These functional groups were thus inferred to participate in thermal stress response of the oyster.
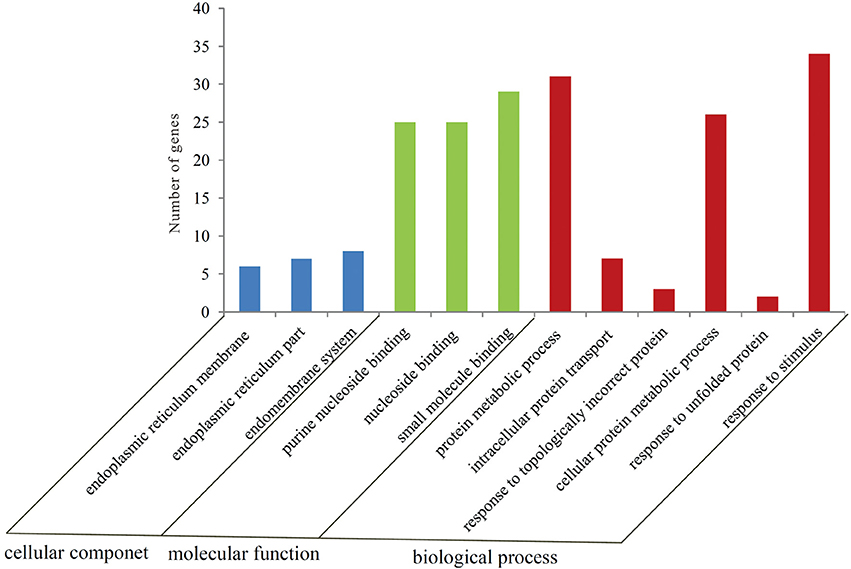
Figure 2. Number of genes in each GO category as found from the Gene Ontology classification of DEGs in the thermally stressed oysters. The results are summarized in three major categories: biological process, cellular component and molecular function.
To further investigate the function of differentially expressed genes during thermal stress, KEGG based pathway analysis of the DEGs were performed. The DEGs were mapped to annotated genes in the KEGG database to identify significantly enriched genes involved in metabolic and/or signal transduction pathways. A BLAST search against the KEGG database indicated that 605 DEGs were assigned to 150 KEGG pathways, and 18 of these pathways were significantly enriched with a cut-off Q ≤ 0.05 (Table S4). A variety of signaling pathways including protein processing in endoplasmic reticulum, apoptosis, ubiquitin mediated proteolysis, endocytosis, spliceosome, RIG-I-like receptor signaling pathway and MAPK signaling pathway were found to be significantly affected (Figure 3). Out of the 18 significantly enriched pathways, protein processing in endoplasmic reticulum was the most affected metabolic pathway, with 40 unigenes mapping to the genes of this pathway (Figure 4). Amongst these 40 unigenes, 25 (mapped to 16 genes) are the main players of unfolded protein response (UPR) and ER-associated degradation (ERAD) processes. After thermal stress, several major components of the UPR and ERAD process were up-regulated, including BiP, PERK, Derlin, HSP40, HSP70, NEF, sHSF, and Prg1. Misfolded proteins resulting from thermal stress are retained within the ER lumen in complex with the above mentioned molecular chaperones, and then directed for degradation through the proteasome in the ERAD process (Fewell and Brodsky, 2009). Results from our pathway analysis are consistent with this mechanism of misfolded protein removal under thermal stress. Corresponding with our results, previous reports in Oncorhynchus tshawytscha and Stylophora pistillata suggest that the genes involved in protein processing in endoplasmic reticulum are significantly up-regulated under heat stress (Maor-Landaw et al., 2014; Tomalty et al., 2015). Thus, our transcriptomic study indicated that protein processing in endoplasmic reticulum may be involved in thermal stress response in the Hong Kong oysters which are vital for survival of the organism in such unfavorable conditions.
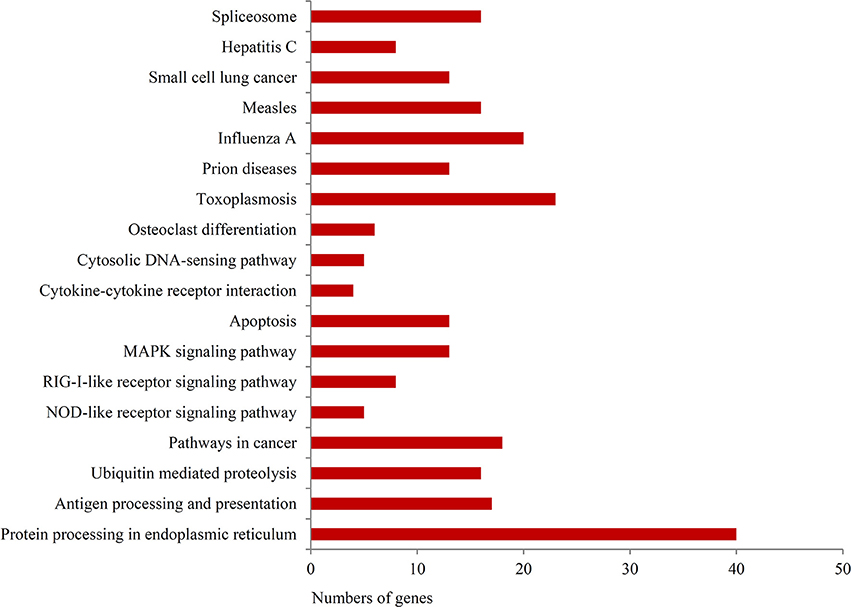
Figure 3. KEGG pathway classification of identified genes.
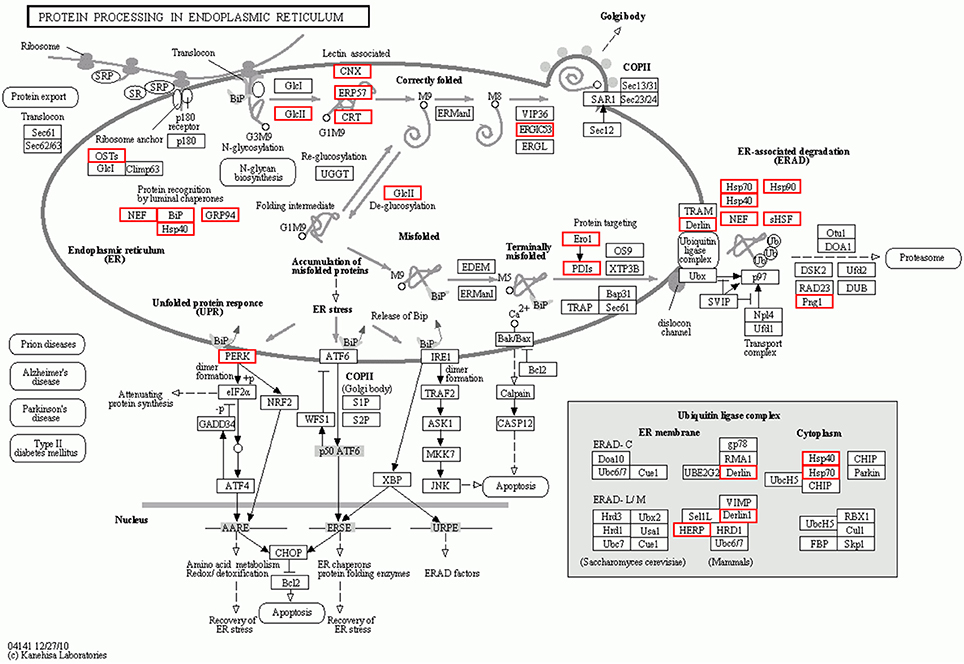
Figure 4. Interaction network of differentially expressed genes involved in protein processing in the endoplasmic reticulum during thermal stress. Genes up-regulated during thermal stress are marked with the red boxes.
Validation of DEGs by qRT-PCR
To validate the digital gene expression (DGE) results, 8 up-regulated (Heat shock protein 70, HSPbeta-1, DNA polymerase, C-type lectin, apoptosis inhibitor protein, cAMP-responsive element-binding protein-like 2, Rad17, metallothionein) and 4 down-regulated (PATS1, Alkaline phosphatase, Hemicentin-1, C1q1) genes representing different biological processes were selected for qRT-PCR analysis (Figure 5). The results indicated a strong correlation (R2 = 0.908) between the expression fold changes detected by DGE and qRT-PCR (Figure 6). This finding is comparable with other studies analyzing correlations between RNA-seq and qRT-PCR results (Camarena et al., 2010; Zhang et al., 2013; Zhai et al., 2016). The qRT-PCR validation results confirmed the accuracy and reliability of the expression changes detected by DGE analysis. Thus, the DEGs functional enrichment analysis could be used to make reasonable deductions. We hypothesize that these DGEs could play an important role in the response to thermal stress in the Hong Kong oyster.
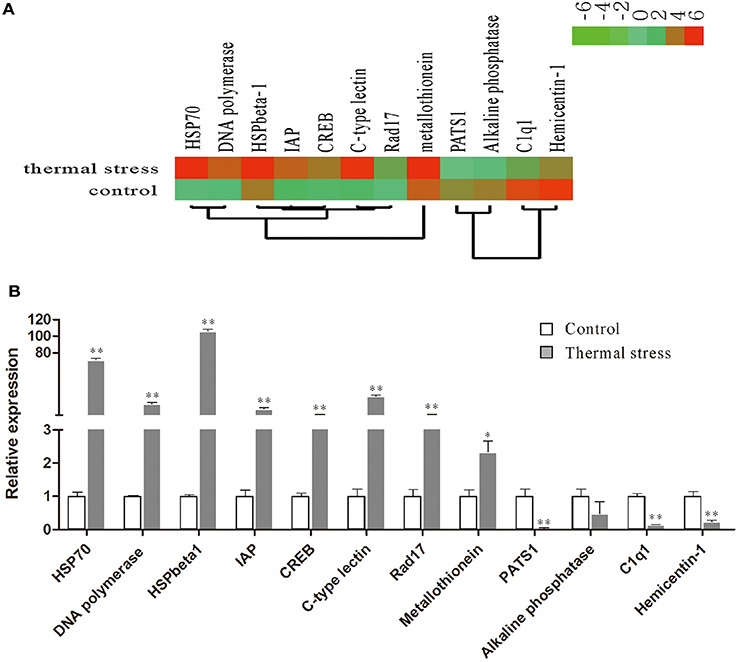
Figure 5. Relative expression levels of selected DEGs in the thermal stress and the control group. (A) Hierarchical clustering of DEGs in the thermal stress and the control group. Red indicates up-regulation and green indicates down-regulation; (B) relative expression of DEGs verified by qRT-PCR. Each qRT-PCR reaction was run in five replicates with EF1α as an internal control. Significant differences are indicated: *p < 0.05 and **p < 0.01.
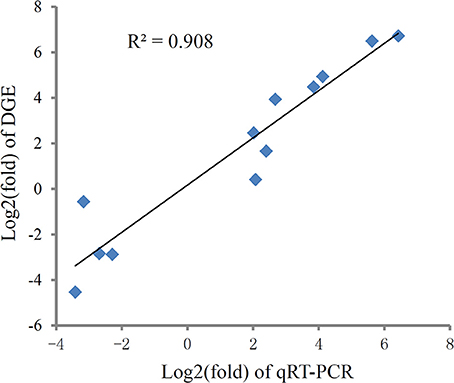
Figure 6. Correlation of expression level analyzed by log2 RNA-Seq (y axis) with data obtained using log2 qRT-PCR, (x axis) from thermal and control group. The selected genes cover different expression levels of both up- and down-regulated genes.
Genes Associated with Thermal Stress Tolerance
Thermal stress is one of the most prominent forms of abiotic stress which increases the susceptibility of oysters to diseases, thereby affecting oyster production in the aquaculture industry (Hofmann and Todgham, 2010; Shiel et al., 2015). Therefore, functions related to thermal stress response form the major focus of this study. Gene expression analysis is a useful method to discover genes involved in the regulation of thermal stress. Techniques for gene expression analysis include northern blots, qRT-PCR, DNA microarray and RNA-seq DGE). Northern blots and qRT-PCR are good for detecting whether a single gene is being expressed. With DNA microarrays, it could only use the available network knowledge for investigation of possible pathways (Wheelan et al., 2008). And the advantages of DGE technique in profiling lower expressed genes and measuring greater dynamic range have made it an attractive alternative to other next-generation sequencing techniques. To generate an information platform for thermal stress response, representative transcripts showing differential expression of 2 fold or more were analyzed under control and thermal stress condition in C. hongkongensis. Functional analysis for these DEGs showed that genes related to protein folding (molecular chaperones or HSP family), antioxidants, immune response, and cytoskeleton was differentially expressed between the thermal treatment and control group respectively.
Involvement of Chaperones, Heat Shock and Ubiquitin Proteins
Among the thermal stress-induced transcripts, genes associated with protein folding and chaperone binding were the most significantly enriched. Exposure to high temperature can trigger the synthesis of heat shock proteins (HSPs) and other molecular chaperones. Many HSPs function as molecular chaperones to protect thermally damaged proteins from aggregation, unfold aggregated proteins, and refold damaged proteins or target them for efficient degradation (Verghese et al., 2012). This explains the significant enrichment of the GO associated with protein folding and chaperone binding in the present study. We found that 33 genes belonging to HSP family, including HSP beta-1, SIP1, AHSA1, alpha-crystallin B, HSPB1, HSP40, HSP68, HSP70, and HSP90, were dramatically induced after thermal stress. Upon heat shock, HSPs are activated by a change in their oligomerization state and through phosphorylation; subsequently, they bind to denaturing proteins and prevent their aggregation (Haslbeck et al., 2005; Zeng et al., 2015). Previous reports on various aquaculture species of oysters suggested a rapid induction in the expression level of HSPs when the animals were subjected to thermal stress (Dong et al., 2006; Ueda and Boettcher, 2009; Huang et al., 2011; Cho and Jeong, 2012; Wan et al., 2012; Wu et al., 2012; Li et al., 2016). In the present study, it was found that 10 paralogs of both HSP70 and HSP40 were simultaneously up-regulated under thermal stress. It is well known that HSP70 plays several essential roles in cellular protein metabolism which include folding and assembly of newly synthesized proteins, refolding of misfolded and aggregated proteins, membrane translocation of organelle and secretory proteins, and controlling the activity of regulatory proteins (Ryan and Pfanner, 2001; Pratt and Toft, 2003). HSP40 on the other hand facilitates cellular recovery from proteotoxic stress primarily by regulating the ATPase activity of HSP70 proteins through a well conserved J domain (Kelley, 1998). The concerted activity of HSP70 and HSP40 in response to thermal stress is also evidenced by Li et al. (2016) who observed that HSP40 and HSP70 are co-expressed in response to thermal stress in the mollusk Pinctada martensii. Thermal stress adaptation requires massive induction in the expression and synthesis of HSPs. However, excessive energy expenditure in synthesis of HSPs could have deleterious effect on oysters by compromising their physiological and immune status. Thus, although the oyster needs HSP induction, it would also need a parallel mechanism to control HSP overproduction. In the present study, the HSP suppressor, BAG family molecular chaperone regulator 4 (BAG4), was up-regulated by 31 fold during thermal stress treatment, strongly suggesting in vivo concerted regulation of HSPs in maintaining cellular homeostasis in the oyster.
Although HSPs could help rescue the misfolded proteins, some damages caused by thermal stress are irreversible and such misfolded proteins need to be eliminated through proteolysis to avoid forming cytotoxic aggregates (Parsell and Lindquist, 1993; Kültz, 2005). Unfolded or damaged proteins that are not rescued through chaperone stabilization or refolding are directed for degradation in the ubiquitin/proteasome pathway. Such proteins are marked for removal by the covalent attachment of multiple ubiquitin molecules and are subsequently degraded by the 26S proteasome or the lysosome (Ciechanover, 1994, 1998).
Several genes involved in the attachment of ubiquitin to target proteins were upregulated in C. hongkongensis under thermal stress. These included the Ubiquitin-protein ligase E3C, HERC2, MIB2, and Ubiquitin conjugation factor E4. Ubiquitin-conjugating enzyme receives ubiquitin and forms a thioester-linked intermediate with ubiquitin. This is followed by binding of E2 and the substrate to ubiquitin ligase which catalyzes the transfer of ubiquitin to the substrate (Richly et al., 2005). Ubiquitin conjugation factor E4 binds to the ubiquitin moieties of these conjugates and, in conjunction with the E1, E2, and E3 enzymes, drives multiubiquitin chain assembly, yielding long chains (Koegl et al., 1999). The increased expression of genes involved in ubiquitin/proteosome pathway in the present study indicates that elimination of denatured proteins in the oyster begins very early during thermal exposure.
Increased Expression of Antioxidants
Rise in environmental temperature can often induce production of reactive oxygen species (ROS) in aquatic organisms (Madeira et al., 2013; Li et al., 2015). The accumulation of ROS can damage important biomolecules, such as DNA, proteins and lipids, and thereby impair normal cellular function (Meng et al., 2014). In response to ROS production, these organisms activate the cellular scavenging system that involves enzymes, such as thioredoxins. Thioredoxins can act as antioxidants by reducing ROS-damaged proteins via cysteine thiol-disulfide exchange (Nordberg and Arner, 2001). In the present study, up-regulation of Thioredoxin-1 and Thioredoxin-2 were observed under thermal stress, indicating their critical role in cellular redox homeostasis and detoxification in the oyster.
Induction of Innate Immunity and Apoptosis
Acute thermal stress has been shown to induce the innate immune system and inflammatory response in mollusks, such as Pomacea canaliculata (Mu et al., 2015) and Crassostrea gigas (Zhang Y. et al., 2015). In the present study, expression of several immune-related genes was upregulated, such as the pattern recognition receptors, retinoic acid inducible I (RIG-I), C-type lectin, leucine-rich repeat-containing protein, and toll-like receptor 3 (TLR3), cellular transducer signaling gene inhibitor protein kappa B (IκB) and p38. Previous reports suggest that high temperature could impair the immune system, reducing the phagocytic ability of the immune cells, which in turn increases the susceptibility of the oysters to opportunistic infections (Hegaret et al., 2004). In such oysters with impaired immunity, bacterial pathogens secrete proteases which facilitate the infection process (Romestand et al., 2002). Elevated expression of RIG-I, C-type lectin, TLR3, p38, and IκB in oysters exposed to thermal stress may help prevent pathogen attack and increases the resistance to pathogens. Apoptosis-related gene, tumor necrosis factors receptor (TNFR), TNF receptor-associated factor 3 (TRAF3), caspase-2, and inhibitor of apoptosis protein (IAP) were also upregulated in the present study, suggesting that heat shock might increase the rate of apoptosis. Apoptosis is an important immune response in oysters that allow the immune system to remove dead cells produced by thermal stress. Our results suggest that up-regulation of immune regulatory proteins could compensate for the impairment of innate immunity in oysters facing thermal stress.
Cytoskeletal Reorganization under Thermal Stress
Cytoskeletal organization also undergoes pronounced transformation under thermal stress (Richter et al., 2010). Cytoskeleton proteins have dynamic structures, which help maintain cell shape, and aid in cellular division, cell movement and support. Changes in the expression of cytoskeletal gene transcripts in response to abiotic stress have been reported in many marine organisms (Lockwood and Somero, 2011; Evans and Hofmann, 2012). In the present study, exposure to thermal stress reduced the expression of genes involved in the formation of major cytoskeletal structures, such as tubulin (0.47-fold compared to the control group), and myosin (0.31-fold compared to the control group). Also, there is a decline in the expression of the matrix metalloproteinase genes, zinc metalloproteinase nas-7, disintegrin and metalloproteinase domain-containing protein 10, which are involved in extracellular matrix synthesis and remodeling. Additional evidence reflective of changes in cytoskeletal organization in the present study comes from the decreased expression of protocadherins Tenascin X, and an extracellular matrix protein, plays a crucial architectural function by modulating cell adhesion (Bristow et al., 2005). Protocadherins constitute the largest subfamily of the cadherin superfamily of cell-adhesion molecules (Chen and Maniatis, 2013). Our findings strongly suggest extensive cytoskeletal reorganization in the oyster in response to thermal stress.
Conclusions
In the present study, we have used Digital Gene Expression (DGE) tag profiling strategy to identify the differentially expressed genes during thermal stress in the Hong Kong oyster, C. hongkongensis. We found that among the 605 differentially expressed transcripts, 378 were up-regulated and 227 were down-regulated in the thermal stress group as compared to the control group. Additionally, gene ontology and KEGG pathway analysis indicated that these differentially expressed genes (DEGs) were enriched with a broad spectrum of biological processes and pathways among which, protein processing in the endoplasmic reticulum was the most affected metabolic pathway. Moreover, some differentially expressed genes including HSP family, ubiquitin proteins, and antioxidants are involved in the thermal stress. This study thus provides insights into the molecular mechanisms underlying thermal stress tolerance in the Hong Kong oyster.
Author Contributions
Conceived and designed the experiments: JL and YZ. Performed the experiments: YHZ, YL, and FM. Analyzed the data: YT, YL, and YZ. Contributed reagents/materials/analysis tools: YL, FM, and YT. Wrote the paper: JL and ZY.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Joint Funds of NSFC-Guangdong of China (U1201215), National Science Foundation of China (No. 31502193 and 31572640), Guangdong Natural Science Foundation (No. 2014A030310402) and Science and Technology Planning Project of Guangdong Province, China (No. 2014B030301064), Guangdong Natural Science Funds for Distinguished Young Scholar's (No. 2015A030306003), and the Guangdong Special Support Program of Youth Scientific and Technological Innovation (No. 510143757060).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2017.00112/full#supplementary-material
Figure S1. Quality assessment of the raw reads from the thermal stress and control libraries. As shown here, 74% and 75% of the total reads from (A) Thermal stress and (B) Control mapped to the reference sequences.
Figure S2. (A) Saturation analysis of the clean reads obtained from the thermal stress library. The number of genes increases rapidly with the number of clean reads until 1.5 million reads are sampled, with the total number of newly found genes plateauing at approximately 6 million reads. (B) Same relationship between number of genes and reads sampled as shown in (A) are observed in case of the control library.
Figure S3. Percentage of reads mapping to the genes for the (A) thermal stress library and (B) the control library. As evidenced here, more than 50% of the genes are covered by high read coverage.
Table S1. The list of unigenes mapped to the reference transcriptome.
Table S2. The up- and down-regulated transcirpts bewteen thermal stress and control libraries.
Table S3. Gene classification based on gene ontology (GO) for DEGs in thermal stress and control libraries.
Table S4. KEGG analysis for DEGs in thermal stress and control libraries.
References
Benjamini, Y., and Yekutieli, D. (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–1188. doi: 10.1214/aos/1013699998
Bristow, J., Carey, W., Egging, D., and Schalkwijk, J. (2005). Tenascin-X, collagen, elastin, and the Ehlers-Danlos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 139C, 24–30. doi: 10.1002/ajmg.c.30071
Camarena, L., Bruno, V., Euskirchen, G., Poggio, S., and Snyder, M. (2010). Molecular mechanisms of ethanol-induced pathogenesis revealed by RNA-sequencing. PLoS Pathog. 6:e1000834. doi: 10.1371/journal.ppat.1000834
Chen, W. V., and Maniatis, T. (2013). Clustered protocadherins. Development 140, 3297–3302. doi: 10.1242/dev.090621
Cho, E. S., and Jeong, H. D. (2012). Effect of environmental impact to molecular expression of heat-shock protein (HSP70) in oyster Crassostrea gigas from Gamak bay, Korea. J. Environ. Biol. 33, 609–615.
Ciechanover, A. (1994). The ubiquitin-proteasome proteolytic pathway. Cell 79, 13–21. doi: 10.1016/0092-8674(94)90396-4
Ciechanover, A. (1998). The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 17, 7151–7160. doi: 10.1093/emboj/17.24.7151
Dong, C. W., Zhang, Y. B., Zhang, Q. Y., and Gui, J. F. (2006). Differential expression of three Paralichthys olivaceus Hsp40 genes in responses to virus infection and heat shock. Fish Shellfish Immunol. 21, 146–158. doi: 10.1016/j.fsi.2005.11.002
Evans, T. G., and Hofmann, G. E. (2012). Defining the limits of physiological plasticity: how gene expression can assess and predict the consequences of ocean change. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1733–1745. doi: 10.1098/rstb.2012.0019
Farcy, E., Voiseux, C., Lebel, J. M., and Fievet, B. (2009). Transcriptional expression levels of cell stress marker genes in the Pacific oyster Crassostrea gigas exposed to acute thermal stress. Cell Stress Chaperones 14, 371–380. doi: 10.1007/s12192-008-0091-8
Fewell, S. W., and Brodsky, J. L. (2009). “Entry into the endoplasmic reticulum: protein translocation, folding and quality control,” in Trafficking Inside Cells, ed N. Segev (Pittsburgh, PA: Springer), 119–142.
Gleason, L. U., and Burton, R. S. (2015). RNA-seq reveals regional differences in transcriptome response to heat stress in the marine snail Chlorostoma funebralis. Mol. Ecol. 24, 610–627. doi: 10.1111/mec.13047
Hao, Y. J., Zhang, Y. J., Si, F. L., Fu, D. Y., He, Z. B., and Chen, B. (2016). Insight into the possible mechanism of the summer diapause of Delia antiqua (Diptera: Anthomyiidae) through digital gene expression analysis. Insect Sci. 23, 438–451. doi: 10.1111/1744-7917.12323
Haslbeck, M., Franzmann, T., Weinfurtner, D., and Buchner, J. (2005). Some like it hot: the structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 12, 842–846. doi: 10.1038/nsmb993
Hegaret, H., Wikfors, G. H., Soudant, P., Delaporte, M., Alix, J. H., Smith, B. C., et al. (2004). Immunological competence of eastern oysters, Crassostrea virginica, fed different microalgal diets and challenged with a temperature elevation. Aquaculture 234, 541–560. doi: 10.1016/j.aquaculture.2004.01.010
Hofmann, G. E., and Todgham, A. E. (2010). Living in the now: physiological mechanisms to tolerate a rapidly changing environment. Annu. Rev. Physiol. 72, 127–145. doi: 10.1146/annurev-physiol-021909-135900
Huang, W. J., Leu, J. H., Tsau, M. T., Chen, J. C., and Chen, L. L. (2011). Differential expression of LvHSP60 in shrimp in response to environmental stress. Fish Shellfish Immunol. 30, 576–582. doi: 10.1016/j.fsi.2010.12.001
Jones, M., and Blaxter, M. (2005). Evolutionary biology: animal roots and shoots. Nature 434, 1076–1077. doi: 10.1038/4341076a
Kawabe, S., and Yokoyama, Y. (2012). Role of hypoxia-inducible factor alpha in response to hypoxia and heat shock in the Pacific oyster Crassostrea gigas. Mar. Biotechnol. 14, 106–119. doi: 10.1007/s10126-011-9394-3
Kelley, W. L. (1998). The J-domain family and the recruitment of chaperone power. Trends Biochem. Sci. 23, 222–227. doi: 10.1016/S0968-0004(98)01215-8
Koegl, M., Hoppe, T., Schlenker, S., Ulrich, H. D., Mayer, T. U., and Jentsch, S. (1999). A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96, 635–644. doi: 10.1016/S0092-8674(00)80574-7
Kültz, D. (2005). Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 67, 225–257. doi: 10.1146/annurev.physiol.67.040403.103635
Lam, K., and Morton, B. (2003). Mitochondrial DNA and morphological identification of a new species of Crassostrea (Bivalvia: Ostreidae) cultured for centuries in the Pearl River Delta, Hong Kong, China. Aquaculture 228, 1–13. doi: 10.1016/S0044-8486(03)00215-1
Lang, R. P., Bayne, C. J., Camara, M. D., Cunningham, C., Jenny, M. J., and Langdon, C. J. (2009). Transcriptome profiling of selectively bred pacific oyster Crassostrea gigas families that differ in tolerance of heat shock. Mar. Biotechnol. 11, 650–668. doi: 10.1007/s10126-009-9181-6
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Li, A. J., Leung, P. T., Bao, V. W., Lui, G. C., and Leung, K. M. (2015). Temperature-dependent physiological and biochemical responses of the marine medaka Oryzias melastigma with consideration of both low and high thermal extremes. J. Therm. Biol. 54, 98–105. doi: 10.1016/j.jtherbio.2014.09.011
Li, J., Zhang, Y., Liu, Y., Xiao, S., and Yu, Z. (2016). Co-expression of heat shock protein (HSP) 40 and HSP70 in Pinctada martensii response to thermal, low salinity and bacterial challenges. Fish Shellfish Immunol. 48, 239–243. doi: 10.1016/j.fsi.2015.11.038
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lockwood, B. L., and Somero, G. N. (2011). Transcriptomic responses to salinity stress in invasive and native blue mussels (genus Mytilus). Mol. Ecol. 20, 517–529. doi: 10.1111/j.1365-294X.2010.04973.x
Madeira, D., Narciso, L., Cabral, H. N., Vinagre, C., and Diniz, M. S. (2013). Influence of temperature in thermal and oxidative stress responses in estuarine fish. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 166, 237–243. doi: 10.1016/j.cbpa.2013.06.008
Maor-Landaw, K., Karako-Lampert, S., Waldman Ben-Asher, H., Goffredo, S., Falini, G., Dubinsky, Z., et al. (2014). Gene expression profiles during short-term heat stress in the red sea coral Stylophora pistillata. Glob. Chang. Biol. 20, 3026–3035. doi: 10.1111/gcb.12592
Mardis, E. R. (2008). The impact of next-generation sequencing technology on genetics. Trends Genet. 24, 133–141. doi: 10.1016/j.tig.2007.12.007
Meistertzheim, A. L., Tanguy, A., Moraga, D., and Thebault, M. T. (2007). Identification of differentially expressed genes of the Pacific oyster Crassostrea gigas exposed to prolonged thermal stress. FEBS J. 274, 6392–6402. doi: 10.1111/j.1742-4658.2007.06156.x
Meng, X. L., Liu, P., Li, J., Gao, B. Q., and Chen, P. (2014). Physiological responses of swimming crab Portunus trituberculatus under cold acclimation: antioxidant defense and heat shock proteins. Aquaculture 434, 11–17. doi: 10.1016/j.aquaculture.2014.07.021
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Mu, H. W., Sun, J., Fang, L., Luan, T. G., Williams, G. A., Cheung, S. G., et al. (2015). Genetic basis of differential heat resistance between two species of congeneric freshwater snails: insights from quantitative proteomics and base substitution rate analysis. J. Proteome Res. 14, 4296–4308. doi: 10.1021/acs.jproteome.5b00462
Nesbitt, M. J., Moerman, D. G., and Chen, N. S. (2010). Identifying novel genes in C. elegans using SAGE tags. BMC Mol. Biol. 11:96. doi: 10.1186/1471-2199-11-96
Nishiyama, T., Miyawaki, K., Ohshima, M., Thompson, K., Nagashima, A., Hasebe, M., et al. (2012). Digital gene expression profiling by 5‗-end sequencing of cDNAs during reprogramming in the moss Physcomitrella patens. PLoS ONE 7:e36471. doi: 10.1371/journal.pone.0036471
Nordberg, J., and Arner, E. S. (2001). Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 31, 1287–1312. doi: 10.1016/S0891-5849(01)00724-9
Parsell, D. A., and Lindquist, S. (1993). The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu. Rev. Genet. 27, 437–496. doi: 10.1146/annurev.ge.27.120193.002253
Pratt, W. B., and Toft, D. O. (2003). Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. (Maywood). 228, 111–133. doi: 10.1177/153537020322800201
Prokofiev, V. V. (2001). [The effect of water temperature and salinity on the longevity of cercaria of marine littoral trematodes Podocotyle atomon (Opecoelidae) and Renicola thaidus (renicolidae)]. Parazitologia. 35, 69–76.
Qi, X. H., Xu, X. W., Lin, X. J., Zhang, W. J., and Chen, X. H. (2012). Identification of differentially expressed genes in cucumber (Cucumis sativus L.) root under waterlogging stress by digital gene expression profile. Genomics 99, 160–168. doi: 10.1016/j.ygeno.2011.12.008
Richly, H., Rape, M., Braun, S., Rumpf, S., Hoege, C., and Jentsch, S. (2005). A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120, 73–84. doi: 10.1016/j.cell.2004.11.013
Richter, K., Haslbeck, M., and Buchner, J. (2010). The heat shock response: life on the verge of death. Mol Cell 40, 253–266. doi: 10.1016/j.molcel.2010.10.006
Romestand, B., Corbier, F., and Roch, P. (2002). Protease inhibitors and haemagglutinins associated with resistance to the protozoan parasite, Perkinsus marinus, in the Pacific oyster, Crassostrea gigas. Parasitology 125, 323–329. doi: 10.1017/S0031182002002135
Ryan, M. T., and Pfanner, N. (2001). Hsp70 proteins in protein translocation. Adv. Protein Chem. 59, 223–242. doi: 10.1016/S0065-3233(01)59007-5
Shiel, B. P., Hall, N. E., Cooke, I. R., Robinson, N. A., and Strugnell, J. M. (2015). De novo characterisation of the greenlip abalone transcriptome (Haliotis laevigata) with a focus on the heat shock protein 70 (HSP70) family. Mar. Biotechnol. 17, 23–32. doi: 10.1007/s10126-014-9591-y
Tomalty, K. M., Meek, M. H., Stephens, M. R., Rincon, G., Fangue, N. A., May, B. P., et al. (2015). Transcriptional response to acute thermal exposure in juvenile chinook salmon determined by RNAseq. G3 (Bethesda). 5, 1335–1349. doi: 10.1534/g3.115.017699
Tong, Y., Zhang, Y., Huang, J., Xiao, S., Zhang, Y., Li, J., et al. (2015). Transcriptomics analysis of Crassostrea hongkongensis for the Discovery of reproduction-related genes. PLoS ONE 10:e0134280. doi: 10.1371/journal.pone.0134280
Ueda, N., and Boettcher, A. (2009). Differences in heat shock protein 70 expression during larval and early spat development in the Eastern oyster, Crassostrea virginica (Gmelin, 1791). Cell Stress Chaperones 14, 439–443. doi: 10.1007/s12192-008-0096-3
Verghese, J., Abrams, J., Wang, Y., and Morano, K. A. (2012). Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol. Mol. Biol. Rev. 76, 115–158. doi: 10.1128/MMBR.05018-11
Wan, Q., Whang, I., and Lee, J. (2012). Molecular and functional characterization of HdHSP20: a biomarker of environmental stresses in disk abalone Haliotis discus discus. Fish Shellfish Immunol. 33, 48–59. doi: 10.1016/j.fsi.2012.03.034
Wang, F., Xiao, S., Zhang, Y., Liu, Y., Yan, Y., Xiang, Z., et al. (2015). ChAkt1 involvement in orchestrating the immune and heat shock responses in Crassostrea hongkongensis: molecular cloning and functional characterization. Fish Shellfish Immunol. 47, 1015–1023. doi: 10.1016/j.fsi.2015.11.009
Wheelan, S. J., Martinez Murillo, F., and Boeke, J. D. (2008). The incredible shrinking world of DNA microarrays. Mol. Biosyst. 4, 726–732. doi: 10.1039/b706237k
Wu, C. X., Zhao, F. Y., Zhang, Y., Zhu, Y. J., Ma, M. S., Mao, H. L., et al. (2012). Overexpression of Hsp90 from grass carp (Ctenopharyngodon idella) increases thermal protection against heat stress. Fish Shellfish Immunol. 33, 42–47. doi: 10.1016/j.fsi.2012.03.033
Xiao, C. W., Ji, Q. A., Wei, Q., Liu, Y., Pan, L. J., and Bao, G. L. (2015). Digital gene expression analysis of microsporum canis exposed to berberine chloride. PLoS ONE 10:e124265. doi: 10.1371/journal.pone.0124265
Zeng, T., Zhang, L., Li, J., Wang, D., Tian, Y., and Lu, L. (2015). De novo assembly and characterization of Muscovy duck liver transcriptome and analysis of differentially regulated genes in response to heat stress. Cell Stress Chaperones 20, 483–493. doi: 10.1007/s12192-015-0573-4
Zhai, L., Xu, L., Wang, Y., Zhu, X., Feng, H., Li, C., et al. (2016). Transcriptional identification and characterization of differentially expressed genes associated with embryogenesis in radish (Raphanus sativus L.). Sci. Rep. 6:21652. doi: 10.1038/srep21652
Zhang, G., Li, L., Meng, J., Qi, H., Qu, T., Xu, F., et al. (2015). Molecular basis for adaptation of oysters to stressful marine intertidal environments. Annu. Rev. Anim. Biosci. 4, 357–381. doi: 10.1146/annurev-animal-022114-110903
Zhang, S. D., Liu, X. M., Zhu, B., Yin, X. M., Du, M. F., Song, Q. S., et al. (2014). Identification of differentially expressed genes in the pheromone glands of mated and virgin bombyx mori by digital gene expression profiling. PLoS ONE 9:e111003. doi: 10.1371/journal.pone.0111003
Zhang, X., Hao, L., Meng, L., Liu, M., Zhao, L., Hu, F., et al. (2013). Digital gene expression tag profiling analysis of the gene expression patterns regulating the early stage of mouse spermatogenesis. PLoS ONE 8:e58680. doi: 10.1371/journal.pone.0058680
Zhang, Y. H., Wu, H. S., Xie, J. Q., Jiang, R. X., Deng, C. S., and Pang, H. (2015). Transcriptome responses to heat-and cold-stress in ladybirds (Cryptolaemus montrouzieri Mulasnt) analyzed by deep-sequencing. Biol. Res. 48, 66. doi: 10.1186/s40659-015-0054-3
Zhang, Y., Sun, J., Mu, H., Li, J., Xu, F., Xiang, Z., et al. (2015). Proteomic basis of stress responses in the gills of the pacific oyster Crassostrea gigas. J. Proteome Res. 14, 304–317. doi: 10.1021/pr500940s
Keywords: digital gene expression, Crassostrea hongkongensis, thermal stress, GO, KEGG
Citation: Li J, Zhang Y, Mao F, Tong Y, Liu Y, Zhang Y and Yu Z (2017) Characterization and Identification of Differentially Expressed Genes Involved in Thermal Adaptation of the Hong Kong Oyster Crassostrea hongkongensis by Digital Gene Expression Profiling. Front. Mar. Sci. 4:112. doi: 10.3389/fmars.2017.00112
Received: 28 February 2017; Accepted: 10 April 2017;
Published: 27 April 2017.
Edited by:
Sandie M. Degnan, The University of Queensland, AustraliaReviewed by:
Bin Tang, Hangzhou Normal University, ChinaGary H. Dickinson, The College of New Jersey, USA
Copyright © 2017 Li, Zhang, Mao, Tong, Liu, Zhang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ziniu Yu, Y2FybHp5dUBzY3Npby5hYy5jbg==
Yang Zhang, eXpoYW5nQHNjc2lvLmFjLmNu
†These authors have contributed equally to this work.