 Marcela Herrera
Marcela Herrera Maren Ziegler
Maren Ziegler Christian R. Voolstra
Christian R. Voolstra Manuel Aranda
Manuel Aranda- Division of Biological and Environmental Science and Engineering, Red Sea Research Center, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Exaiptasia is a laboratory sea anemone model system for stony corals. Two clonal strains are commonly used, referred to as H2 and CC7, that originate from two genetically distinct lineages and that differ in their Symbiodinium specificity. However, little is known about their other microbial associations. Here, we examined and compared the taxonomic composition of the bacterial assemblages of these two symbiotic Exaiptasia strains, both of which have been cultured in the laboratory long-term under identical conditions. We found distinct bacterial microbiota for each strain, indicating the presence of host-specific microbial consortia. Putative differences in the bacterial functional profiles (i.e., enrichment and depletion of various metabolic processes) based on taxonomic inference were also detected, further suggesting functional differences of the microbiomes associated with these lineages. Our study contributes to the current knowledge of the Exaiptasia holobiont by comparing the bacterial diversity of two commonly used strains as models for coral research.
Introduction
Microbes are an essential part of life; they interact with their hosts in such a way that it not only affects their fitness and survival but also defines them as a unique biological entity (McFall-Ngai et al., 2013). Increasing research has focused on understanding these interactions, looking at organisms that range from basal metazoans like Hydra sp. (Fraune and Bosch, 2007) to humans (Ursell et al., 2012). Contribution of the microbiota to the evolution, development, and molecular and physiological functioning of animals has been studied for different groups (McFall-Ngai et al., 2013; Rosenberg and Zilber-Rosenberg, 2013); but particularly for corals (Thompson et al., 2015; Bourne et al., 2016), as they form the basis of one of the most productive and diverse ecosystems on earth.
Much work has been done on identifying coral-associated bacteria that can cause or prevent bleaching (Rosenberg et al., 2009) and diseases (Rosenberg et al., 2007; Bourne et al., 2009), highlighting how the coral microbiome can disrupt the holobiont equilibrium. Ultimately, the health and persistence of coral reef ecosystems depends on the symbiotic partnership between the coral animal, its photosynthetic dinoflagellates and a specific consortium of microbes (Bourne et al., 2016). A more comprehensive approach that takes into account the whole coral holobiont is therefore critical to understand the adaptation and resilience of these organisms (Voolstra et al., 2015). However, working with corals is challenging; they are difficult to culture in aquariums and in situ studies are not always feasible (Voolstra, 2013).
Advancements in molecular, cell, and developmental biology have been achieved through the establishment of different model systems (Davis, 2004), from which the sea anemone Exaiptasia (Grajales and Rodríguez, 2014) has been used as a laboratory model organism for corals (Weis et al., 2008). Exaiptasia lives in a symbiotic relationship with dinoflagellates of the genus Symbiodinium, but unlike scleractinian corals, it can also live in a symbiont-free (i.e., aposymbiotic) state. Further, it grows fast and propagates easily; its undemanding culturing conditions have facilitated coral research for more than 30 years now (Muller-Parker, 1984; Weis et al., 2008; Voolstra, 2013).
Recently, two genetically distinct populations of Exaiptasia have been characterized, a globally distributed lineage (found in Japan, Mexico, Hawaii, the Mediterranean, and Australia) and a local lineage (found in the United States South Atlantic coast as defined in Thornhill et al., 2013), that harbor different Symbiodinium species (Thornhill et al., 2013; Grawunder et al., 2015). This specificity and stability of the Exaiptasia-Symbiodinium association has been suggested to be the result of coevolutionary processes (Thornhill et al., 2013; Voolstra, 2013), raising the question of how specific the relationship with their microbes is. Microbial community relationships can be highly conserved as observed in the cnidarian Hydra sp., where species-specific bacterial phylotypes were maintained over long periods of time and across different environments (Fraune and Bosch, 2007). Bacterial associations of Exaiptasia have, however, not been examined until recently (Röthig et al., 2016a), showing that despite its simple body organization, this sea anemone can be associated with diverse bacterial communities. Here, we characterized the bacterial assemblage of symbiotic Exaiptasia H2 (originally isolated from Hawaii, USA) and compared it to the previously published microbiome of Exaiptasia CC7 (originally obtained from North Carolina, USA; Röthig et al., 2016a), which belong to the “global” and “local” lineages, respectively (Grawunder et al., 2015). Knowledge on the bacterial composition of Exaiptasia will be vital for its establishment as a model system for coral research.
Materials and Methods
Exaiptasia Culture Conditions
Symbiotic Exaiptasia of the clonal strain H2 (Xiang et al., 2013) were cultured in clear polycarbonate containers (2 L capacity; Cambro Camwear, USA) filled with autoclaved natural seawater collected from the Red Sea (~39 PSU salinity) as described in Röthig et al. (2016a). Tanks were kept in Intellus Control System Incubators (Model I-22LLVL, Percival Scientific, USA) at 25°C on a diurnal 12 h light: 12 h dark cycle under white-light (20–40 μmol photons m−2 s−1 of photosynthetically active radiation). Anemones were fed 3 times per week with freshly hatched Artemia brine shrimps, and seawater in the tanks was stagnant and exchanged 1–2 times per week.
DNA Isolation and 16S rRNA Gene Sequencing
Five anemones and one water sample were processed as described in Röthig et al. (2016a). Briefly, polyps of ~1.0 cm size were collected and transferred into 1.5 mL microtubes, and remaining water was carefully removed. DNA was extracted using a spin column DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's protocol. Animals were not fed 1 week prior to the DNA extraction in order to avoid potential Artemia contamination. For the water sample, 500 mL water was collected from the tank in which the animals were reared and filtered through a 0.22 μm Durapore PVDF filter (Millipore, Billerica, MA, USA). The filter was then cut in strips using a sterile razorblade, transferred into a 1.5 mL microtube with 400 μL AP1 buffer (DNease Plant Mini Kit, Qiagen, Hilden, Germany) and incubated on a rotating wheel for 20 min before subsequent DNA extraction procedures. DNA concentrations of all samples were quantified on a NanoDrop 2,000C spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Primers 784F and 1061R (Andersson et al., 2008) were used to amplify the V5–V6 hypervariable regions of the 16S rRNA gene. PCRs were performed in triplicate per sample (using 15–30 ng of DNA from each anemone sample and ~5 ng DNA of the water sample) with a Qiagen Multiplex PCR Kit (Qiagen, Hilden, Germany) and a final primer concentration of 0.5 μM for a final volume of 15 μL. Thermal profiles consisted of an initial activation step of 15 min at 95°C, followed by 27 cycles each of 30 s at 95°C, 90 s at 55°C, and 30 s at 72°C, and a final extension step of 10 min at 72°C. Amplified PCR products were pooled for each sample and subsequently cleaned using the Agencourt AMPure XP magnetic bead system (BeckmanCoulter, Indianapolis IN, USA), indexed with Nextera XT sequencing adapters (Illumina, San Diego, CA, USA), and quality checked on a BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA). The final library was sequenced at 8 pM with 10% phiX on the Illumina MiSeq, 2 × 300 bp paired-end v3 chemistry according to the manufacturer's instructions.
Bacterial Community Analysis
Sequencing data was analyzed using the software mothur (v1.36.1; Schloss et al., 2009). To allow direct comparisons between the microbiomes of Exaiptasia H2 (this study) and Exaiptasia CC7 (Röthig et al., 2016a), data obtained in this study was processed following the same procedure described by Röthig et al. (2016a) for the previously published CC7 data. Sequences were demultiplexed, quality trimmed, preclustered (2 bp difference; Huse et al., 2010), and aligned against the SILVA database (release 119; Pruesse et al., 2007). Chimeric sequences were removed using UCHIME (Edgar et al., 2011), and sequences assigned to chloroplasts, mitochondria, archaea, eukaryotes, and other unwanted sequences, were excluded. Phylogenetically classified sequences (Greengenes database release gg_13_8_99; bootstrap = 60; McDonald et al., 2012) were then used to evaluate the bacterial taxonomic composition of samples and plotted with Krona (Ondov et al., 2011). For further analyses, we subsampled to 20,816 sequences per sample, as this was the total number of sequences obtained for the water sample. Sequences were then clustered into Operational Taxonomic Units (OTUs) using a 97% similarity cutoff. Alpha diversity indices and principal coordinate analysis (PCoA) were performed as implemented in mothur. A list of all determined OTUs and their distribution over samples was created using the commands make.shared, classify.OTU, and get.OTUrep. Based on this we obtained a putative core microbiome (i.e., OTUs that were present in 100% of the Exaiptasia samples). For each of these OTUs, the representative sequence was BLASTed against GenBank nr database to identify previous occurrences of identical or highly similar bacterial sequences. Furthermore, shared OTUs between symbiotic H2 and symbiotic CC7 core microbiomes (Röthig et al., 2016a) were identified. Differences in the average relative abundance of these OTUs were assessed using t-tests.
Taxonomy-based functional profiling of the bacterial communities of H2 and CC7 was done using METAGENassist (Arndt et al., 2012), an automated taxonomic-to-phenotypic mapping package. Input files were created using the relative abundances of all OTUs present in both core microbiomes combined. These were further filtered based on interquartile range (Hackstadt and Hess, 2009) and normalized over samples by sum and over taxa by Pareto scaling. Data was then analyzed for “metabolism by phenotype” using Spearman distance measure and average clustering algorithm to visualize the results in a heatmap.
Results
The Microbiome of Exaiptasia H2
We sequenced 16S rRNA gene amplicon libraries from five Exaiptasia H2 anemones and one water sample, yielding a total of 788,277 sequences. Quality filtering steps removed 423,408 sequences, while subsequent chimera detection removed further 23,675 sequences. Lastly, removal of undesirable sequences matching non-bacterial targets (18,311) resulted in a final set of 322,883 sequences with an average length of 292 bp that annotated to bacteria, and were used for further analyses. Rarefaction analysis showed that a subsample of 20,816 sequences from each sample was sufficient to account for the majority of bacterial diversity (Figure S1). Clustering at the 97% similarity level resulted in 211 OTUs (Table S1), from which 176 were associated with Exaiptasia and 106 with water. From these, 105 and 35 OTUs were exclusively found in the anemone and water samples, respectively. A specific microbiome was observed for Exaiptasia H2 (Figure S2), showing a clear separation from the water sample (Figure S3).
At least 10 bacterial phyla, 18 classes, and 41 orders were recovered from the symbiotic H2 microbiome (Figure 1). Of these phyla, Proteobacteria (52.6%) and Bacteroidetes (36.6%) were the most dominant. Actinobacteria comprised only 10.2%, whereas the other phyla were present at <1% of the bacterial community. Moreover, within Proteobacteria, Gammaproteobacteria (57.3%) was the most represented class followed by Alphaproteobacteria (28.3%) and Deltaproteobacteria (13.5%). At a finer scale, Flavobacteriaceae (35.0%) and Alteromonadaceae (15.2%) were the most abundant families, contributing to half of the total bacterial diversity. In contrast, families Rhodobacteraceae (order Rhodobacterales, class Alphaproteobacteria) and Vibrionaceae (order Vibrionales, class Gammaproteobacteria) dominated the water sample with 34.6 and 32.1%, respectively. We further identified a core microbiome that consisted of 49 OTUs (Table 1, Table S1), and that included the 10 most abundant bacterial taxa, accounting for >80% of all OTU sequence counts. These bacteria represented abundant and rare members of the Exaiptasia H2 microbiome, with mean abundances that range from 2 to 7,309 counts. Kordia sp. (Flavobacteriaceae) was the most dominant OTU (34%) across samples.
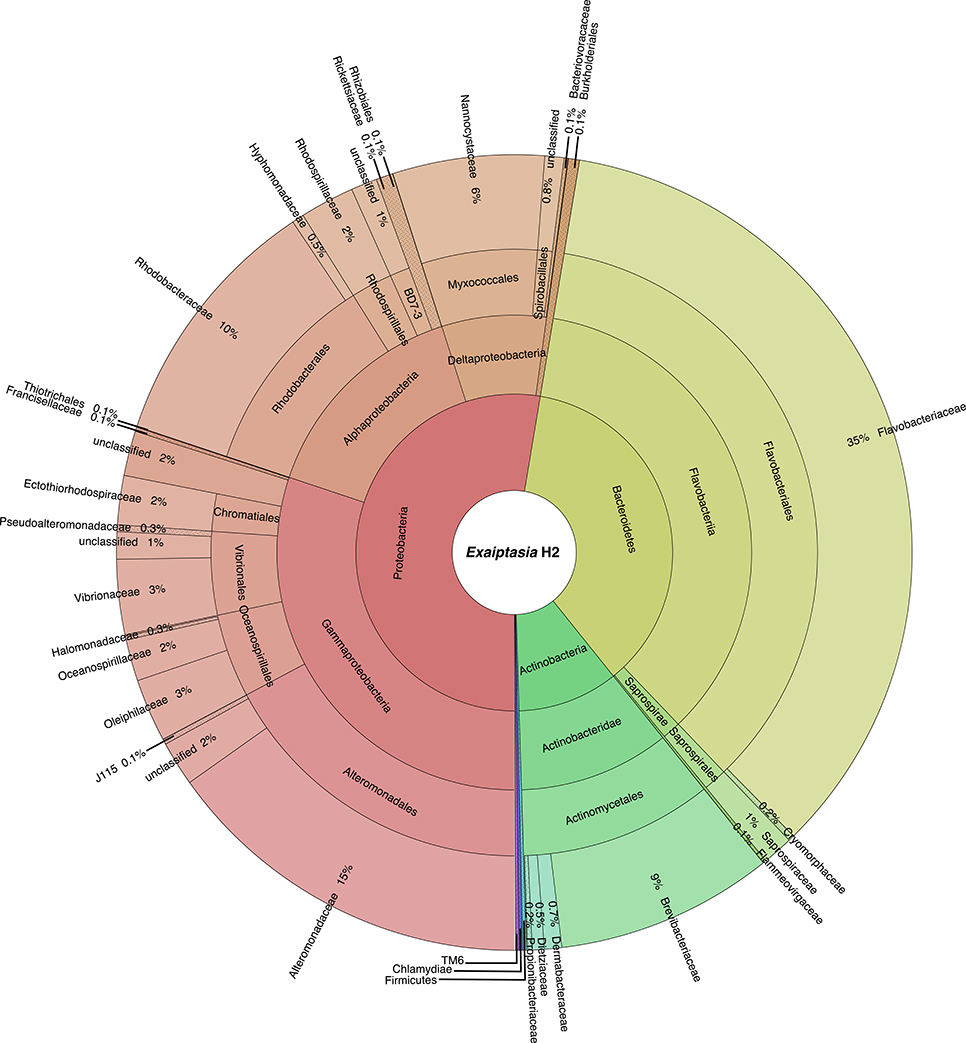
Figure 1. Taxonomic community composition of bacteria associated with Exaiptasia H2. The inner ring represents different phyla, and as the plot progresses outwards, there is an increasing taxonomic resolution for each ring (i.e., class, order, family, respectively). Only OTUs with relative abundance >0.1% are reported.
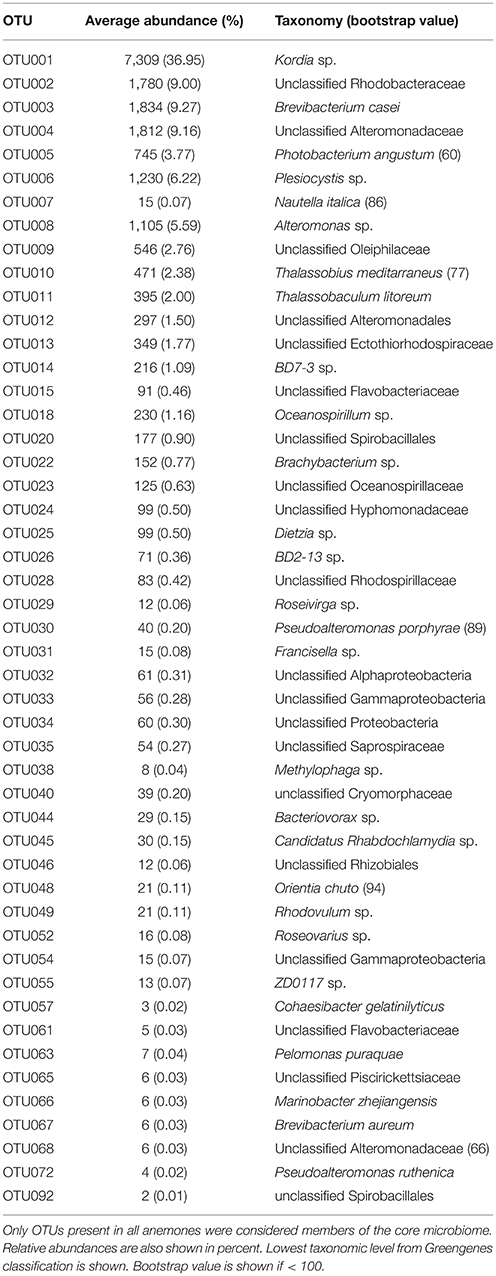
Table 1. Exaiptasia H2 49 core microbial OTUs.
Distinct Bacterial Communities in Exaiptasia H2 and CC7
We compared the taxonomic composition of the microbial assemblages of symbiotic Exaiptasia H2 and CC7 strains infected with the Symbiodinium strain SSB01 that was previously isolated from H2, thus allowing for a comparison of the two Exaiptasia strains without potential symbiont effects (Xiang et al., 2013; Röthig et al., 2016a). We examined the diversity and relative abundance of bacterial OTUs and found significant differences (t-test, p < 0.05 post Benjamini–Hochberg correction). More than 17 phyla were represented in CC7 (Röthig et al., 2016a), depicting a more taxonomically diverse microbiome than the 10 phyla detected in H2. Average abundances of Proteobacteria (69.7%), Actinobacteria (25.9%), and Firmicutes (1.5%) were notably higher in CC7 than H2, with 52.6, 10.2, and 0.1%, respectively. In contrast, Bacteroidetes (1.63%) showed more than a 20-fold decrease in CC7 compared to H2. A detailed analysis of the Proteobacteria assembly in CC7 showed higher and lower abundances in Betaproteobacteria (8.9%) and Alphaproteobacteria (7.2%), respectively. Furthermore, Pseudomonadaceae (19.7%) was the most abundant family in CC7, yet present at only very low numbers in H2 (<0.1%). On the OTU level, the bacterial diversity of CC7 and H2 was also different (Table 2). The average number of OTUs (118), and the inverse Simpson (11.78) and Simpson's evenness (0.10) indices were higher in CC7 than H2 (96.00, 5.87, and 0.06, respectively). Similarly, the Chao1 estimator of species richness was lower in H2 (114.76) than in CC7 (124.20). Comparisons amongst core microbiomes revealed 13 OTUs in common (Table 3), of which only four were observed to also be significantly more abundant in CC7 than H2 (t-test, p < 0.05 post Benjamini–Hochberg correction). The occurrence of Propionibacterium acnes (Actinobacteria) and Staphylococcus epidermis (Firmicutes) bacteria in both H2 and CC7 core microbiomes called for precaution when interpreting results, as these taxa are known for being common contaminants (e.g., human skin-associated organisms, contamination of DNA extraction kits, and other laboratory reagents) in sequenced-based microbiome studies (Salter et al., 2014). For this reason, and due to their low abundance, we removed these bacterial taxa from further taxonomy-based functional analysis.
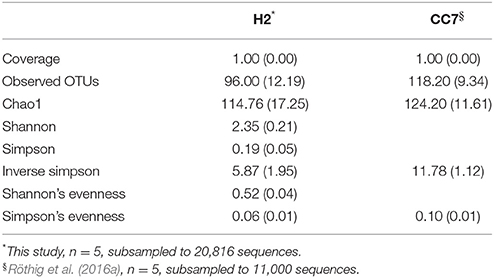
Table 2. Summary of alpha diversity indices (mean ± SD) of the microbial communities associated with Exaiptasia H2 and CC7 strains.
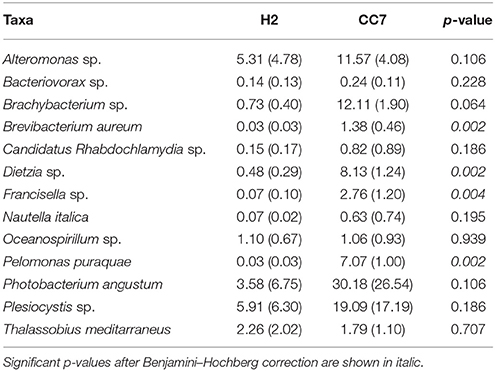
Table 3. Differences in the average relative abundance in percent (± SD) of shared bacterial taxa (OTUs) between Exaiptasia H2 and CC7 core microbiomes.
Taxonomy-Based Functional Profiling of Bacterial Communities in Exaiptasia
We also characterized the functional potential of the bacterial communities of Exaiptasia by examining the enrichment and depletion of different metabolic processes based on phylogenetic inference using METAGENassist (Arndt et al., 2012). Putative functions were inferred from the taxonomic composition underlying the microbiome of H2 and CC7. A total of 84 OTUs were assigned, mapped and condensed into 73 distinct functional taxa. This analysis showed that the bacterial community associated with Exaiptasia H2 covered a range of nutrient cycling pathways, in particular sulfur (“sulfate reduction,” “sulfide oxidation”) and nitrogen (“nitrogen fixation,” “nitrite reduction,” “ammonia oxidation”) cycling. Further, other metabolic processes involving the breakdown of complex organic molecules were observed (“xylan,” “chitin and chlorophenol degraders,” and “sugar fermenters”).
Overall, the putative functional profile of H2 samples appeared to be more variable than CC7 (Figure 2). In particular two samples (H2_1 and H2_4) were different from the remaining samples, yet they still clustered together (Figure S4). Processes such as “sulfate reducer,” “sulfide oxidizer,” and “nitrite reducer” were consistently enriched in H2, whereas up regulation of processes like “chitin degradation,” “atrazine metabolism,” “sulfur oxidizer,” “xylan degrader,” “sugars fermentor,” and “propionate metabolism” was observed for CC7. Other processes, “degrades aromatic hydrocarbons,” “ammonia oxidizer,” and “dehalogenation” were inconsistently enriched in H2, reflecting a greater variation across individuals (Figure 2).
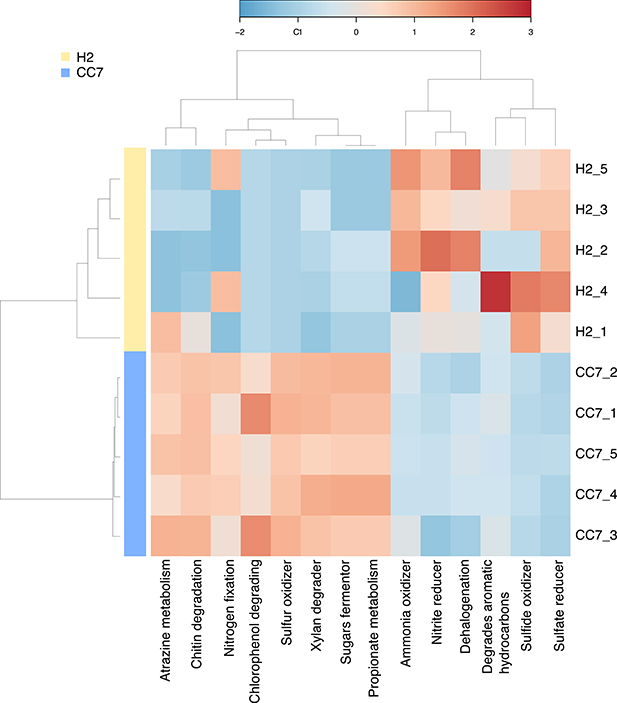
Figure 2. Taxonomy-based functional profiling of Exaiptasia H2 and CC7 bacterial communities. Changes in putative functional differences between both populations are displayed on a relative scale with enrichment in red and depletion in blue. H2 is a female clonal strain originally isolated from Hawaii, USA. CC7 is a male clonal strain originally from North Carolina, USA.
Discussion
Exaiptasia H2 and CC7 Strains Only Share a Small Common Microbiome
Interestingly, we found significant differences in the bacterial diversity of the two Exaiptasia strains that have been cultured in the laboratory at the same conditions for long-term. Several studies (e.g., Littman et al., 2009; Pantos et al., 2015; Roder et al., 2015) have suggested that geographic location and environmental settings can determine the holobiont microbiome, whereas others (e.g., Hentschel et al., 2002; Fraune and Bosch, 2007; Montalvo and Hill, 2011; Lawler et al., 2016) have shown that bacterial communities are species-specific associations conserved over time and space. Moreover, coevolution of bacteria and coral has been proposed (Thompson et al., 2015), as it was demonstrated for Endozoicomonas symbionts in corals with contrasting life-history traits and across global scales (Bayer et al., 2013; Neave et al., 2016, 2017). Detailed analysis of these studies revealed fine scale host-Endozoicomonas specific patterns suggesting phylosymbiosis between corals and this bacterial genus. On the other hand, other bacteria have been universally found in different coral lineages from widely separated geographical habitats (Ainsworth et al., 2015), supporting the idea that consistent, specific host-bacterial interactions occur.
The presence of distinct microbial communities in Exaiptasia H2 and CC7 strains aligns with differences in algal symbiont specificities observed for the populations these lab strains were originally obtained from, i.e., Hawaii and North Carolina, respectively (Thornhill et al., 2013; Grawunder et al., 2015) and lends further support to the distinction of these Exaiptasia lineages. It should be noted that the CC7 anemones (Röthig et al., 2016a) used for these comparisons were also symbiotic with Symbiodinium minutum (SSB01), which has been previously isolated from H2 anemones (Xiang et al., 2013).
Our results showed a comparably lower richness at the OTU level for Exaiptasia H2, with a larger core microbiome (50 OTUs) than CC7 (37 OTUs). Only a small fraction (~15%) of the microbiomes was shared between both groups, and it did not include the dominant bacterial symbionts Kordia sp. or Pseudomonas veronii, present in H2 and CC7, respectively. Kordia is an algicidal bacterium (Sohn et al., 2004) capable of inhibiting the growth of diatoms, and even killing them by active lysis (Paul and Pohnert, 2011). Its ubiquity and high abundance (37%) in the H2 core microbiome indicates that these bacteria might be important for the host; however, this has to be studied in more depth. P. veronii, on the other hand, was the most abundant taxon in CC7. This has been repeatedly found in corals from the Red Sea (Röthig et al., 2016b; Ziegler et al., 2016), and yet it was absent in H2. It is also possible that differences in dominant taxa in Exaiptasia are due to sex-specific factors, since H2 are females and CC7 males (Grawunder et al., 2015). For example, studies on mice (Yurkovetskiy et al., 2013) have shown that sex hormones drive differences in the microbiota of males and females during growth; and that sex-specific dietary requirements of crickets determine their microbial symbioses (Schmid et al., 2014). Changes in the bacterial communities before and after spawning have also been documented for different coral species (Ceh et al., 2012), suggesting that certain taxa might be correlated with reproduction.
Noteworthy is that 2 out of the 13 OTUs (Table 3) that were defined as shared core microbes between both Exaiptasia strains are involved in sulfur cycling processes. Both Alteromonas sp. and Oceanospirillum sp. bacteria can degrade dimethylsufoniopropionate (DMSP), a compound that has been found within animals that harbor symbiotic algae, such as corals, marine bivalves, and polychaetes (Raina et al., 2009; Morrow et al., 2012). It has also been shown that these bacteria can be vertically transmitted in corals (Sharp et al., 2012), implying a strong role in symbioses. Further, bacteria of the genus Photobacterium are remarkably resistant to ultraviolet radiation (e.g., UVB; Matallana-Surget et al., 2009) and have been associated with coral bleaching (Thompson et al., 2005; Moreira et al., 2014). Also linked to bleaching is Nautella italica, a bacterial pathogen that can invade, colonize, and induce bleaching in marine macroalgae (Fernandes et al., 2011). Knowledge of the ecology and functional importance of these bacteria is still lacking, and is crucial to better understand their role in shaping the core microbiome of Exaiptasia.
Functional Differences Associated with the Bacterial Community of Different Hosts
A clear separation between H2 and CC7 was observed, depicting differences in the putative functional profiles of their bacterial communities. Consistent enrichment of carbon cycling related functions (e.g., “chitin degradation,” “xylan degrader,” and “sugars fermentor”) in CC7 indicate potential differences in carbon utilization pathways. It should be noted that CC7 harbored the heterologous Symbiodinium SSB01 strain, which has previously been isolated from Exaiptasia H2. It can therefore not be excluded that nutrient interactions within the holobiont might have changed to allow the microbiome to take advantage of different carbon pathways, as has been observed in corals under stress (Bourne et al., 2016).
Changes in sulfur (DMSP degradation) and nitrogen cycling processes are also dependent on Symbiodinium (Raina et al., 2009; Bourne et al., 2016). It assimilates and converts most of the sulfur pool in the holobiont to DMSP, a more stable and soluble molecule that is later used as a source of reduced sulfur and carbon for marine microorganisms (Raina et al., 2009), and likely determines their abundance and diversity in the host (Bourne et al., 2016). Nitrogen availability, as well as other nutrients, in the holobiont is also controlled by the algal symbionts (Rädecker et al., 2015; Bourne et al., 2016), and thus, increased nitrogen availability may promote the activity of nitrogen-fixating bacteria (i.e., diazotrophs). Interestingly, a higher diversity of diazotrophs was observed in CC7; at least 18 OTUs within the group Rhizobiales were part of the bacterial community whereas only 8 were present in H2.
Although, we see functional differences in the bacterial community between Exaiptasia H2 and CC7, caution should be applied. It has to be stressed that the functional classification of OTUs is based on comparison to well-characterized bacterial groups and the assumption that these functions are conserved in the related groups present in the microbiome of Exaiptasia. Further, functional groups might not be retained purely due to their metabolic pathways but because of host-specific preferences, even under the same environmental conditions. Different bacteria can provide similar functions; for example, nitrogen-fixating bacteria in the H2 core microbiome (e.g., unclassified Rhizobiales and Cohaesibacter gelatinilyticus) are different from the ones found in CC7 (e.g., unclassified Phyllobacteriaceae and Methylobacterium hispanicum).
Conclusions
Exaiptasia is a cnidarian-dinoflagellate symbiosis model system on the rise, yet until recently, little was known about its bacterial associations. This study provides a baseline on the bacterial diversity of the widely used laboratory strains Exaiptasia H2 and compared it to the bacterial microbiome of Exaiptasia CC7. Our data suggest the presence of a host-specific microbiome. We believe that knowledge on the specific microbiota of these two laboratory-cultured strains, which are commonly used for diverse experiments worldwide, is a valuable contribution to further our understanding of the Exaiptasia holobiont system. We also provided a basic level of functional understanding of the microbiome by assigning functional profiles based on taxonomic data. Our taxonomically inferred functional data showed a clear difference in the metabolic processes of H2 and CC7, which implies that there is still more research needed to better understand how hosts actively select their microbial symbionts and how the different bacterial taxa contribute to the overall functioning of the holobiont.
Data Accessibility
Sequence data have been deposited on NCBI under BioProject Accession no. PRJNA3060672 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA360672).
Ethics Statement
Study was exempt due to the use of invertebrate animals.
Author Contributions
MA and CRV designed and conceived the experiments. MA and CRV contributed with reagents/materials/analysis tools. MH conducted the laboratory work. MH and MZ analyzed the data. MH and MA wrote the manuscript with input from all authors. All authors gave approval for publication.
Funding
This study was supported by KAUST baseline research funds to MA and CRV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Craig Michell for MiSeq sequencing library generation, Till Röthig, Yi Jin Liew, and Shobhit Agrawal for helpful discussions that greatly improved the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2017.00115/full#supplementary-material
References
Ainsworth, T. D., Krause, L., Bridge, T., Torda, G., Raina, J. B., Zakrzewski, M., et al. (2015). The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2274. doi: 10.1038/ismej.2015.39
Andersson, A. F., Lindberg, M., Jakobsson, H., Bäckhed, F., Nyrén, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3:e2836. doi: 10.1371/journal.pone.0002836
Arndt, D., Xia, J., Liu, Y., Zhou, Y., Guo, A. C., Cruz, J. A., et al. (2012). METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res. 40, 1–9. doi: 10.1093/nar/gks497
Bayer, T., Neave, M. J., Alsheikh-Hussain, A., Aranda, M., Yum, L. K., Mincer, T., et al. (2013). The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl. Environ. Microbiol. 79, 4759–4762. doi: 10.1128/AEM.00695-13
Bourne, D. G., Garren, M., Work, T. M., Rosenberg, E., Smith, G. W., and Harvell, C. D. (2009). Microbial disease and the coral holobiont. Trends Microbiol. 17, 554–562. doi: 10.1016/j.tim.2009.09.004
Bourne, D. G., Morrow, K. M., and Webster, N. S. (2016). Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 15:18. doi: 10.1146/annurev-micro-102215-095440
Ceh, J., Raina, J. B., Soo, R. M., van Keulen, M., and Bourne, D. G. (2012). Coral-bacterial communities before and after a coral mass spawning event on Ningaloo Reef. PLoS ONE 7:e36920. doi: 10.1371/journal.pone.0036920
Davis, R. H. (2004). Timeline: the age of model organisms. Nat. Rev. Genet. 5, 69–76. doi: 10.1038/nrg1250
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fernandes, N., Case, R. J., Longford, S. R., Seyedsayamdost, M. R., Steinberg, P. D., Kjelleberg, S., et al. (2011). Genomes and virulence factors of novel bacterial pathogens causing bleaching disease in the marine red alga Delisea pulchra. PLoS ONE 6:e27387. doi: 10.1371/journal.pone.0027387
Fraune, S., and Bosch, T. C. (2007). Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc. Natl. Acad. Sci. U.S.A. 104, 13146–13151. doi: 10.1073/pnas.0703375104
Grajales, A., and Rodríguez, E. (2014). Morphological revision of the genus Exaiptasia and the family Aiptasiidae (Cnidaria, Actiniatia, Metridioidea). Zootaxa 3826, 55–100. doi: 10.11646/zootaxa.3826.1.2
Grawunder, D., Hambleton, E. A., Bucher, M., Wolfowicz, I., Bechtoldt, N., and Guse, A. (2015). Induction of Gametogenesis in the Cnidarian endosymbiosis model Exaiptasia sp. Sci. Rep. 5:15677. doi: 10.1038/srep15677
Hackstadt, A. J., and Hess, A. M. (2009). Filtering for increased power for microarray data analysis. BMC Bioinformatics 10:11. doi: 10.1186/1471-2105-10-11
Hentschel, U., Hopke, J., Horn, M., Friedrich, A. B., Wagner, M., Hacker, J., et al. (2002). Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 68, 4431–4440. doi: 10.1128/AEM.68.9.4431-4440.2002
Huse, S. M., Welch, D. M., Morrison, H. G., and Sogin, M. L. (2010). Ironing out the wrinkles in the rare biosphere through improved OUT clustering. Environ. Microbiol. 12, 1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x
Lawler, S. N., Kellogg, C. A., France, S. C., Clostio, R. W., Brooke, S. D., and Ross, S. W. (2016). Coral-associated bacterial diversity is conserved across two deep-sea Anthothela species. Front. Microbiol. 7:458. doi: 10.3389/fmicb.2016.00458
Littman, R. A., Willis, B. L., Pfeffer, C., and Bourne, D. G. (2009). Diversities of coral-associated bacteria differ with location, but not species, for three acroporid corals on the Great Barrier Reef. FEMS Microbiol. Ecol. 68, 152–163. doi: 10.1111/j.1574-6941.2009.00666.x
Matallana-Surget, S., Douki, T., Cavicchioli, R., and Joux, F. (2009). Remarkable resistance to UVB of the marine bacterium Photobacterium angustum explained by an unexpected role of photolyase. Photochem. Photobiol. Sci. 8, 1313–1320. doi: 10.1039/b902715g
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., Desantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U.S.A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
Montalvo, N. F., and Hill, R. T. (2011). Sponge-associated bacteria are strictly maintained in two closely related but geographically distant sponge hosts. Appl. Environ. Microbiol. 77, 7207–7216. doi: 10.1128/AEM.05285-11
Moreira, A. P., Chimetto Tonon, L. A., Pereira Cdo, V., Alves, N. Jr., Amado-Filho, G. M., Francini-Filho, R. B., et al. (2014). Culturable heterotrophic bacteria associated with healthy and bleached scleractinian Madracis decactis and the fireworm Hermodice carunculata from the remote St. Peter and St. Paul Archipelago, Brazil. Curr. Microbiol. 68, 38–46. doi: 10.1007/s00284-013-0435-1
Morrow, K. M., Moss, A. G., Chadwick, N. E., and Liles, M. R. (2012). Bacterial associated of two Caribbean coral species reveal species-specific distribution and geographic variability. Appl. Environ. Microbiol. 78, 6438–6449. doi: 10.1128/AEM.01162-12
Muller-Parker, G. (1984). Photosynthesis-irradiance responses and photosynthetic periodicity in the sea anemone Exaiptasia pulchella and its zooxanthellae. Mar. Biol. 82:225. doi: 10.1007/BF00392403
Neave, M. J., Michell, C., Apprill, A., and Voolstra, C. R. (2017). Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci. Rep. 7:40579. doi: 10.1038/srep40579
Neave, M. J., Rachmawati, R., Xun, L., Michell, C. T., Bourne, D. G., Apprill, A., et al. (2016). Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 11, 186–200. doi: 10.1038/ismej.2016.95
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385
Pantos, O., Bongaerts, P., Dennis, P. G., Tyson, G. W., and Hoegh-Guldberg, O. (2015). Habitat-specific environmental conditions primarily control the microbiomes of the coral Seriatopora hystrix. ISME J. 9, 1916–1919. doi: 10.1038/ismej.2015.3
Paul, C., and Pohnert, G. (2011). Interactions of the algicidal bacterium Kordia algicidal with diatoms: regulated protease excretion for specific algal lysis. PLoS ONE 6:e21032. doi: 10.1371/journal.pone.0021032
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Rädecker, N., Pogoreutz, C., Voolstra, C. R., Wiedenmann, J., and Wild, C. (2015). Nitrogen cycling in corals: the key to understanding holobiont functioning? Trends Microbiol. 23, 490–497. doi: 10.1016/j.tim.2015.03.008
Raina, J. B., Tapiolas, D., Willis, B. L., and Bourne, D. G. (2009). Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl. Environ. Microbiol. 75, 3492–3501. doi: 10.1128/AEM.02567-08
Roder, C., Bayer, T., Aranda, M., Kruse, M., and Voolstra, C. R. (2015). Microbiome structure of the fungid coral Ctenactis echinata aligns with environmental differences. Mol. Ecol. 24, 3501–3511. doi: 10.1111/mec.13251
Rosenberg, E., Koren, O., Reshed, L., Efrony, R., and Zilberg-Rosenberg, I. (2007). The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microb. 5, 355–362. doi: 10.1038/nrmicro1635
Rosenberg, E., Kushmaro, A., Kramarsky-Winter, E., Banin, E., and Loya, Y. (2009). The role of microorganisms in coral bleaching. ISME J. 3, 139–146. doi: 10.1038/ismej.2008.104
Rosenberg, E., and Zilber-Rosenberg, I. (2013). “Role of microorganisms in adaptation, development, and evolution of animals and plants: the hologenome concept,” in The Prokaryotes, eds E. Rosenberg, E. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 347–405.
Röthig, T., Costa, R. M., Simona, F., Baumgarten, S., Torres, A. F., Radhakrishnan, A., et al. (2016a). Distinct bacterial communities associated with the coral model Aiptasia in aposymbiotic and symbiotic states with Symbiodinium. Front. Mar. Sci. 3:234. doi: 10.3389/fmars.2016.00234
Röthig, T., Ochsenkühn, M. A., Roik, A., van der Merwe, R., and Voolstra, C. R. (2016b). Long-term salinity tolerance is accompanied by major restructuring of the coral bacterial microbiome. Mol. Ecol. 25, 1308–1323. doi: 10.1111/mec.13567
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffat, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequenced-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schmid, R. B., Lehman, R. M., and Lundgren, J. G. (2014). Sex-specific interactions of microbial symbioses on cricket dietary selection. Environ. Entomol. 43, 896–902. doi: 10.1603/EN13311
Sharp, K. H., Distel, D., and Paul, V. J. (2012). Diversity and dynamics of bacterial communities in early life stages of the Caribbean coral Porites astreoides. ISME J. 6, 790–801. doi: 10.1038/ismej.2011.144
Sohn, J. H., Lee, J. H., Yi, H., Chun, J., Bae, K. S., Ahn, T. Y., et al. (2004). Kordia algicida gen. nov., sp. an algicidal bacterium isolated from red tide. Int. J. Syst. Evol. Microbiol. 54, 675–680. doi: 10.1099/ijs.0.02689-0
Thompson, F. L., Thompson, C. C., Naser, S., Hoste, B., Vandemeulebroecke, K., Munn, C., et al. (2005). Photobacterium rosenbergii sp. nov. and Enterovibrio coralii sp. nov., vibrios associated with coral bleaching. Int. J. Syst. Evol. Microbiol. 55, 913–917. doi: 10.1099/ijs.0.63370-0
Thompson, I. R., Rivera, H. E., Closek, C. J., and Medina, M. (2015). Microbes in the coral holobiont: partners through evolution, development, and ecological interactions. Front. Cell. Infect. Microbiol. 4:176. doi: 10.3389/fcimb.2014.00176
Thornhill, D. J., Xiang, Y., Pettay, D. T., Zhong, M., and Santos, S. R. (2013). Population genetic data of a model symbiotic cnidarian system reveal remarkable symbiotic specificity and vectored introductions across ocean basins. Mol. Ecol. 22, 4499–4515. doi: 10.1111/mec.12416
Ursell, L. K., Metcalf, J. L., Parfrey, L. W., and Knight, R. (2012). Defining the human microbiome. Nutr. Rev. 70, S38–S44. doi: 10.1111/j.1753-4887.2012.00493.x
Voolstra, C. R. (2013). A journey into the wild of the cnidarian model system Exaiptasia and its symbionts. Mol. Ecol. 22, 4366–4368. doi: 10.1111/mec.12464
Voolstra, C. R., Miller, D. J., Ragan, M. A., Hoffmann, A., Hoegh-Guldberg, D., Bourne, E., et al. (2015). The ReFuGe 2020 consortium—using ‘omics’ approaches to explore the adaptability and resilience of coral holobionts to environmental change. Front. Mar. Sci. 2:68. doi: 10.3389/fmars.2015.00068
Weis, M. V., Davy, S. K., Hoegh-Guldberg, O., Rodriguez-Lanetty, M., and Pringle, J. R. (2008). Cell biology in model systems as the key to understanding corals. Trends Ecol. Evol. 23, 369–376. doi: 10.1016/j.tree.2008.03.004
Xiang, T., Hambleton, E. A., DeNofrio, J. C., Pringle, J. R., and Grossman, A. R. (2013). Isolation of clonal axenic strains of the symbiotic dinoflagellate Symbiodinium and their growth and host specificity1. J. Phycol. 29, 447–458. doi: 10.1111/jpy.12055
Yurkovetskiy, L., Burrows, M., Khan, A. A., Graham, L., Volchkov, P., Becker, L., et al. (2013). Gender bias in autoimmunity is influenced by microbiota. Immunity 39, 400–412. doi: 10.1016/j.immuni.2013.08.013
Keywords: 16S rRNA gene, coral, core microbiome, host-specific, model organism
Citation: Herrera M, Ziegler M, Voolstra CR and Aranda M (2017) Laboratory-Cultured Strains of the Sea Anemone Exaiptasia Reveal Distinct Bacterial Communities. Front. Mar. Sci. 4:115. doi: 10.3389/fmars.2017.00115
Received: 13 November 2016; Accepted: 10 April 2017;
Published: 02 May 2017.
Edited by:
Sebastian Fraune, University of Kiel, GermanyReviewed by:
Linda Louise Blackall, University of Melbourne, AustraliaJulie L. Meyer, University of Florida, USA
Copyright © 2017 Herrera, Ziegler, Voolstra and Aranda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel Aranda, bWFudWVsLmFyYW5kYUBrYXVzdC5lZHUuc2E=