 Haydn Rubelmann1
Haydn Rubelmann1 David J. Karlen
David J. Karlen James R. Garey
James R. Garey- 1Department of Cell Biology, Microbiology and Molecular Biology, University of South Florida, Tampa, FL, United States
- 2Environmental Protection Commission of Hillsborough County, Tampa, FL, United States
The biocomplexity of sediment communities along a 120 m transect near an arsenic-rich, shallow marine hydrothermal vent at Tutum Bay, Papua New Guinea was thoroughly examined. A count of macro- and meiofaunal organisms was combined with bacterial and eukaryotic SSU rRNA gene surveys to assess biodiversity. Each site along the transect had distinct microbial communities. Near-vent sites were more similar to each other than sites further from the vent. Some species, such as Ignavibacterium, Caldilinea, and Capitella were only found near-vent. Biodiversity generally increased with distance from the vent. The community composition responded to the presence of hydrothermal fluids with a clear correlation between temperature and thermophilic organisms. Primary production appeared to be a mix of chemo- and phototrophy. Association analyses suggest many potential interactions between organisms occur at certain sites, and that species distributions and interactions occur in the context of complex spatial relationships related to the geochemistry of the hydrothermal vent fluids. While Tutum Bay is heavily influenced by arsenic, no specific correlation between bacteria that metabolize arsenic and the concentration of different oxidation states of arsenic ions was observed, perhaps because very little of the arsenic present was bioavailable. The observed homogeneous distribution of arsenic reducers along the transect could be due to background arsenic metabolism. This study represents a holistic study of biocomplexity on a broad phylogenetic range across a 120 m transect associated with a marine shallow-water hydrothermal vent.
Subject Category: Microbial ecology and functional diversity of natural habitats.
Introduction
Deep-sea hydrothermal systems are light restricted and rely on chemosynthesis for energy input, using both inorganic forms of carbon and organic detritus as the carbon source in the synthesis of biomolecules. Oxidation reactions of reduced chemical species (H2S, H2, CH4, etc.) provide electron donors necessary for chemotrophic growth. In seawater, where sulfur species are readily available, the oxidation of reduced sulfur can provide energy to synthesize organic carbon, highlighting the importance of the sulfur cycle in sustaining these communities (Tsutsumi et al., 2001).
In contrast, shallow-sea hydrothermal systems are not as light limited and are not characterized by an abundance of vent-specific taxa. Here, chemosynthesis runs parallel to photosynthesis as a means of primary production. Many shallow-sea hydrothermal vent systems are characterized by bacterial mats, similar to deep-sea vents, and chemosynthesis occurs near the vent itself and in the superficial layer of water above the vents (Tarasov et al., 2005). Diatoms, cyanobacterial mats, and other algae aid in this primary production.
The shallow-sea hydrothermal system of Tutum Bay, Ambitle Island, Papua New Guinea serves as a natural laboratory to study the biocomplexity of prokaryotic and eukaryotic species in an arsenic-rich environment. The metabolic repertoire and subsequent biodiversity of species found at the site differs from other commonly studied shallow hydrothermal-sea vent systems. As an arsenic-rich system, the potentially toxic qualities of arsenic represent a hurdle for sustaining communities at Tutum Bay's hydrothermal systems (Pichler and Dix, 1996; Price and Pichler, 2005; Pichler et al., 2006). Not only is arsenate a structural analog of phosphate that can inhibit oxidative phosphorylation, but arsenite binds to sulfhydryl groups and vicinal thiols of many proteins, disrupting the function of numerous proteins. Arsenic has 4 oxidation states: As (−III), As (0), As (III), and As (V)—the latter two being most prevalent in nature (Oremland and Stolz, 2003). Arsenate readily adsorbs onto aluminum and iron hydroxides, forming inorganic compounds that reduce their bioavailability. Arsenite does this less readily, leaving it mobile, with more potential for toxicity in hydrological systems. Further complicating arsenic mobility, Fe (III) reducing bacteria are able to reduce ferric oxyhydroxides, and subsequently release arsenate back into the hydrological system (Konhauser, 2007).
Both prokaryotes and eukaryotes have developed methods to deal with the toxic nature of arsenic. The discovery that microorganisms synthesized and excreted trimethylarsine suggested that other methylated forms of arsenic exist (Cullen and Reimer, 1989; Michalke et al., 2000). Further studies showed that some animals have the enzymes (Lin et al., 2002) to convert inorganic forms of arsenic into both methyl arsenic and dimethyl arsenic (Doak and Freedman, 1970) which are excreted in the urine (Thomas et al., 2007). In addition to methylation, prokaryotes have evolved enzymes that allow both the oxidation and reduction of arsenic. Dissimilatory arsenate-respiring prokaryotes reduce arsenic (V) to arsenic (III) when other more favorable terminal electron acceptors are not present and couple the reaction to the oxidation of organic compounds. Heterotrophic arsenite oxidizers convert As (III) found on the cell membrane to less toxic As (V). This process is seen as a defense mechanism against the more toxic arsenite, which easily enters the cell. Lastly, chemoautotrophic arsenite oxidizers reduce oxygen or nitrate during the oxidation of arsenite and use the energy to fix CO2 (Oremland and Stolz, 2003).
Extensive chemical analysis has been performed along transects at Tutum Bay, specifically at hydrothermal Vent 4 (Price and Pichler, 2005; Akerman et al., 2011). Mechanisms have been proposed for the biogeochemical cycling of arsenic and other inorganic metabolites based on Gibbs free energy calculations at this vent (Akerman et al., 2011), and the distribution and speciation of arsenic has been well-characterized with particular attention to the element's bioavailability (Price and Pichler, 2005). Our study focuses on the biocomplexity along a specific transect at hydrothermal Vent 4, V4-B, (Figure 1) with attention to the phylogenetic biodiversity and functional repertoire present in pore samples at particular sites along the transect. Bacterial 16S rRNA genes and eukaryote 18S rRNA gene were sequenced from clone libraries and distributed into functional guilds and trophic groups. This data was combined with morphological data scored from the same sites. These ecological patches were linked to the physical parameters previously described and particular attention was given to the evidence of arsenic and iron metabolizing organisms throughout the transect. It was thought that in areas of the transect with high concentrations of arsenic, the overall biodiversity would be lower while the richness and diversity of organisms capable of metabolizing arsenic would be higher.
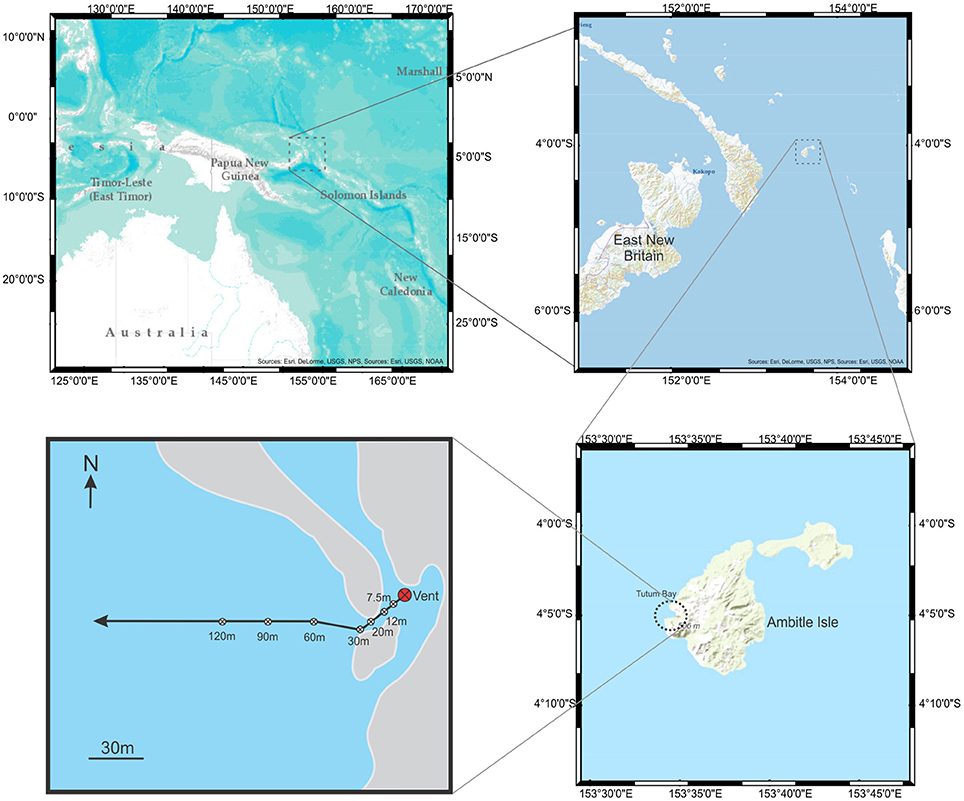
Figure 1. Maps showing Ambitle Island and Tutum Bay with transect and sample sites. From the vent, the transect extended southwest to avoid reef outcroppings before extending 300 m west.
Materials and Methods
Sampling Site and Collection
The Tutum Bay site has a number of focused vents surrounded by areas of diffuse venting. Transect 4B extends 30 m southwest from Vent 4 along soft-bottom and then directly westward to 120 m (Pichler and Dix, 1996; Price and Pichler, 2005; Pichler et al., 2006; Akerman et al., 2011). The transect and location for this study is shown in Figure 1. Sediment samples for macrofaunal and meiofaunal analyses were collected in replicates of 5 as described by Karlen et al. (2010). Samples were sieved through a 500 μm mesh screen to collect macrofauna and then onto a 50 μm screen to collect meiofauna. Core samples for bacterial DNA were collected from 5 to 10 cm as described by Meyer-Dombard et al. (2012). Five samples for eukaryotic DNA analyses were collected at each site along the transect with a 60 ml syringe corer to a depth of 5 cm and the replicates pooled. Sediment was passed through a 4 mm sieve to eliminate large macrofauna. Samples for sequencing eukaryote DNA were collected at sites 0, 30, 60, 90, 120 m from the vent and frozen until used. Samples for sequencing bacterial DNA were collected at sites 7.5, 12, 20, 30, 60, 90, and 120 m, from the vent and frozen until used.
Porewater and Sediment Analysis
Porewater samples were collected 5 cm beneath the sediment surface using a 60 ml plastic syringe connected to a plastic probe. Temperature was measured in situ using a digital thermometer. Oxidation-reduction potential and pH were measured using a Myron-L pH meter on site. Fe2+, NH4+, and SO42− were measured on site using a spectrophotometer (HACH, Colorado). Arsenic species were measured using analytical instrumentation and methods off site described in detail in Price et al. (2007) and again by Akerman et al. (2011). The geochemical data used in the analyses here is from Table 1 of Akerman et al. (2011). The organic carbon content of the sediment was determined off-site by loss on ignition from 2 g of sample at 550°C for 4 h (Heiri et al., 2001).
Macrofaunal and Meiofaunal Identification
Macrofaunal samples were rinsed onto a 500 μm mesh sieve and identified to the lowest practical taxonomic level (Karlen et al., 2010). Each specimen was archived and voucher specimens were photographed.
Meiofaunal samples were decanted from sieved sediment onto a 50 μm mesh plankton net placed in a funnel. The remaining sediment was resuspended, and meiofauna again decanted onto the plankton net. This process was repeated 5 times and the meiofaunal specimens sorted to the lowest practical taxonomic level. The specimens were archived and photographed.
Eukaryote and Bacterial Sequencing and Identification
Sediment DNA was extracted from 0.5 g sediment using a MoBio UltraClean Soil DNA extraction kit (Mo Bio Laboratories, Carlsbad, CA, USA). PCR was performed using ID Labs IDProof™ Polymerase Enzyme (ID Labs, London, ON, Canada) optimized for 50 μL reactions. Primers used for the 18s rRNA gene PCR were 18S4 (5′-CCGGAATTCAAGCTTGCTTGCTTGTCTCAAAGATTAAGCC-3′) and 18S5 (5′-CCGGAATTCAAGCTTACCATACTCCCCCCGGAACC-3′) (Mackey et al., 1996). Primers used for the 16s rRNA gene PCR were 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTCTTACGACTT-3′) (DeLong, 1992). The 18s PCR consisted of an initial 2 min 95°C denaturation, 45 cycles of 15 s at 95°C, 1 min at 50°C, 1 min at 72°C, followed by a 72°C extension for 7 min. The protocol for 16s PCR differed in that 40 cycles with a 45°C annealing temperature and a 2 min extension step was used.
Cloning was done with a PCR4-TOPO-TA Cloning Kit (Invitrogen, Carlsbad, CA, USA). Clones were isolated into 96-well-culture plates, cultured and DNA extracted using the Eppendorf Perfectprep Plasmid 96 Vac miniprep kit (Eppendorf, Hamburg, Germany). The resulting plasmid DNAs were sequenced by Polymorphic DNA Technologies, Inc. (Alameda, CA, USA). Sequences were filtered for quality (Wu et al., 2009) and submitted to Genbank (bacterial accession numbers JN837712–JN839729, eukaryotic accession numbers JQ241793–JQ244768).
Sequences were clustered into Operational Taxonomic Units (OTUs) using SEQUENCHER (Gene Codes, Ann Arbor, MI, USA). Eukaryote OTUs were established according to a 99% similarity (Wu et al., 2009). Prokaryotic OTUs were established using 97% similarity. OTUs and singletons were identified to the nearest identifiable match using NCBI's database and BLAST algorithm (uncultured organism matches were not considered). This identification was used to provisionally identify the genus of each OTU (Supplemental Materials 1, 2) in further analyses (Wu et al., 2011).
Classification of Prokaryotic Functional Guilds
Potential metabolic capabilities of each provisional bacterial genus were assessed from the literature (Supplemental Material 3). In some cases, a genus would fall into more than one functional guild (e.g., sulfur reducing, iron reducing, and arsenic oxidizing), and was scored more than once. Facultative aerobes or anaerobes were similarly considered in both categories. Further categorization of arsenic metabolism was performed by searching genomes of genera found for arsenic reducing or oxidizing genes within the Joint Genome Institute's Integrated Microbial Genome database (Supplemental Material 4).
Biodiversity Calculations, Association Analyses, and Networks
Primer-E (Clarke and Gorley, 2006) was used to calculate Shannon diversity at each site. Chao 1 indexes was calculated and rarefaction analysis was accomplished using Estimate-S (Colwell and Elsensohn, 2014). Cluster analysis was performed in Primer-E for abiotic factors, macro- and meiofauna, and at the phylum level, genus level, and sequence level for bacteria and eukaryotes across all sites. The abundance data were square root transformed and clustered by Bray-Curtis similarity. Abiotic data were normalized and clustered according to Euclidean distance. Association analyses were performed on species found at more than one site together and dendograms were made using the Primer-E software.
Phylogenetic networks were created using the clustered OTUs and singletons for both prokaryotes and eukaryotes with the Cytoscape software package (Shannon et al., 2003). A functional guild network of prokaryotes and a trophic food web were also created using the Cytoscape software.
The Surfer software package (Golden Software Inc., Golden, CO, USA) was used to interpolate a gradient of abiotic factors using an x,y coordinate system corresponding to the transect V4-B according to chemical analyses done at each site.
Results and Discussion
Abiotic Factors
Abiotic factors across the transect (Figure 2) show a distribution that reflects the hydrothermal venting found at the site. Sites from 0 to 12 m are heavily influenced by hydrothermal fluids, while sites further out are more closely related to that of normal sea water. Although diffuse venting is present and may be seen in the pH and oxidation-reduction potential differences found from site to site and a small temperature rise at 60 m, the abiotic factors generally formed a gradient along the transect toward that of normal seawater away from the vent. Arsenic concentrations remained higher than normal seawater at all sites.
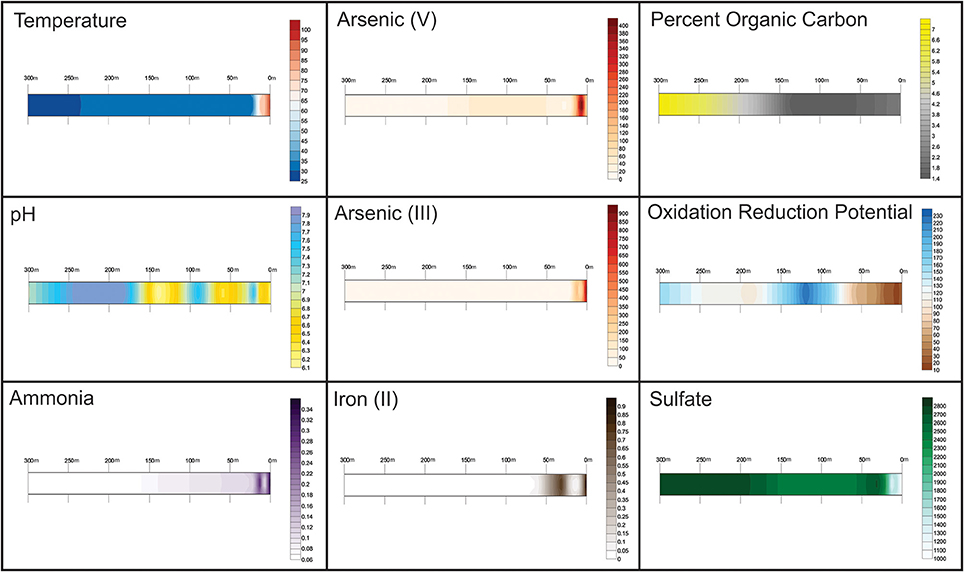
Figure 2. Abiotic factors across the transect at Tutum Bay. Sampling sites are labeled in meters away from vent. Values between sample sites were interpolated within the Surfer program. Contour maps from left to right: Top, Temperature (pH); Arsenic (V) in μg L−1, Percent organic carbon; Middle: pH, Arsenic (III) in μg L−1, Oxidation Reduction Potential (mV); Bottom: Ammonia (aq) in mg L−1, Fe (II) in μg L−1, Sulfate in mg L−1.
The temperature ranged from 99°C at the vent to 29°C 300 m from the vent. Arsenic (V) and Iron (II) (Figure 2) show an inverse correlation at near-vent sites, with Arsenic (V) concentrations peaking at 7.5 m and Iron (II) concentrations below detection levels. Arsenic (III) has the highest concentration at the vent (944 μg L−1) which gradually diminishes away from the vent. Lastly, the concentration of sulfate increases with increased distance from the vent.
Similarity clustering of biotic and abiotic factors (Figure 3) shows the general trend visualized in Figure 2. Near-vent sites cluster together more closely than sites distant from the vent. While not all data was available for each factor at every site, 90 and 120 m exclusively cluster together, showing the same dendogram topology for all biotic data. With the exception of eukaryotic sequence data, contiguous sites appear more similar in biota than noncontiguous sites. The abiotic data shows the incongruity of the 7.5 m site with the rest of the near-vent sites, which can also be seen in Figure 2 primarily due to Arsenic (V) and Iron (II) concentrations. However, because there are biotic similarities among them, the 0, 7.5, and 12 m sites will be referred to as near-vent while sites 20 m and greater from the vent will be considered far-vent.
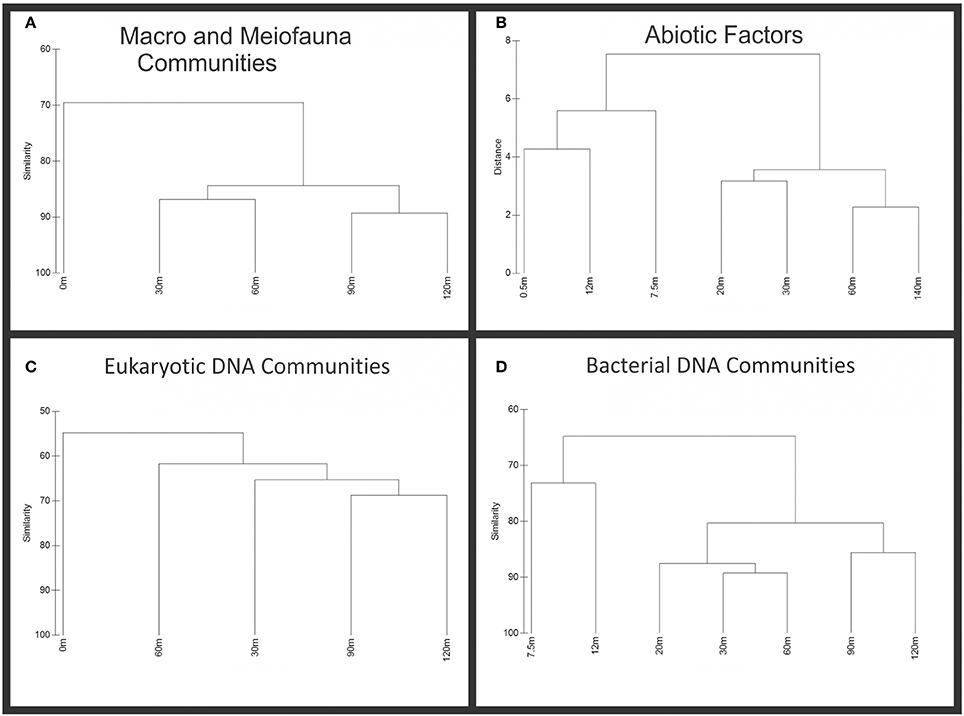
Figure 3. Cluster analysis of abiotic and biotic data. Biotic samples were standardized by total, transformed by square root and had resemblance calculated using Bray-Curtis similarity. Abiotic factors were normalized and clustered based on Euclidean distance. (A) Macrofauna and Meiofauna dendogram showing similarity among transect sites. (B) Dendogram of abiotic factors showing similarity along transect sites. (C) Eukaryotic sequence dendogram, showing similarity among transect sites. (D) Bacterial sequence dendogram, showing similarity among transect sites.
Biodiversity
A total of 1,692 bacterial sequences (720 nucleotides each) were obtained averaging 240 sequences per site while 1,481 eukaryotic sequences (576 nucleotides each) were obtained with an average of 296 sequences per site along the transect. The BLAST results for Eukaryotic and Bacterial sequences with match percentages and accession numbers are provided as Supplemental Materials 1, 2. Given the limited number of sequences from each site, rarefaction curves were carried out for the combined near-vent and combined far-vent communities using the Chao1 index (Figure 4). A lower number of OTUs was estimated at near-vent communities with upper-bound estimates reaching just above 200 for eukaryotes and around 1,000 for bacteria. At the far vent sites the number of OTUs are estimated with upper bounds of approximately 1,000 eukaryote and 4,000 bacterial OTUs. In comparison, studies of other shallow hydrothermal vent systems using conventional sequencing (Giovannelli et al., 2013) show much lower Chao1 estimates of bacterial diversity while next gen sequencing methods show comparable estimates (Wang et al., 2015).
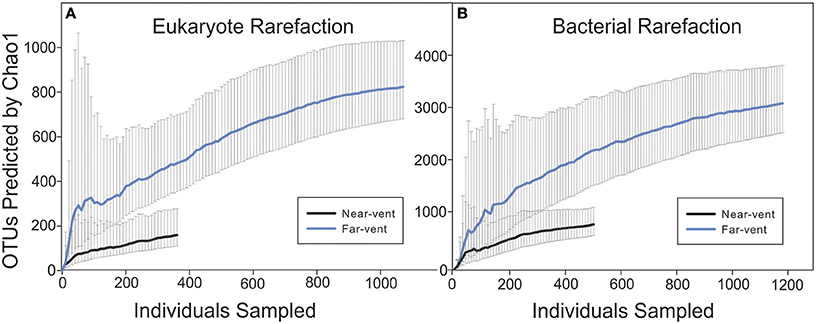
Figure 4. Eukaryotic and bacterial rarefaction curves. Chao 1 indexes were calculated for both eukaryotic and bacterial sequence data. (A) The eukaryotic near-vent community consisted only of OTUs obtained from the 0 m site. OTUs from 30, 60, 90, and 120 m were used in the eukaryotic far-vent community analysis. (B) The near-vent bacterial community consisted of OTUs from 7.5 to 12 m while the far-vent community consisted of OTUs from 20 to 120 m.
Bacterial species found along the transect are represented in Figure 5. The patch assemblages found at 7.5 m and 12 m are easily distinguishable from the sites found further away from the vent. The phylum Chlorobi is found only at the two near-vent sites and Proteobacteria diversity is greatly reduced compared to the 20–120 m sites. Actinobacteria was the most abundant phylum found at 7.5 m from the vent (68 individuals representing 37 species) with Proteobacteria following with 64 individuals representing 49 species. At 12 m from the vent, Chlorobi, predominantly found as the genus Ignavibacterium, and Chloroflexi are more abundant than anywhere else along the transect. The diversity and relative abundance of Proteobacteria dominates the 20–120 m sites, with Gammaproteobacteria having the highest abundance (348 individuals) followed by Deltaproteobacteria (287 individuals). Distribution lines show species similarity between sites, showing the greatest sharing of species at sites away from the vent (20–120 m). Only one OTU was found at all sites (black distribution line) and matched most closely to Acidomicrobium ferrooxidans (87% similarity). Figure 6 shows the distribution of the most common genera along the transect. Ignavibacterium and Caldilinea are exclusively found at the near-vent sites, while Steroidobacter, Thiohalomonas, Pelagibius, and Thiohalospira are exclusive to the 20–120 m sites. Many genera are represented throughout the whole transect but tend to be in low abundance at the 12 m site.
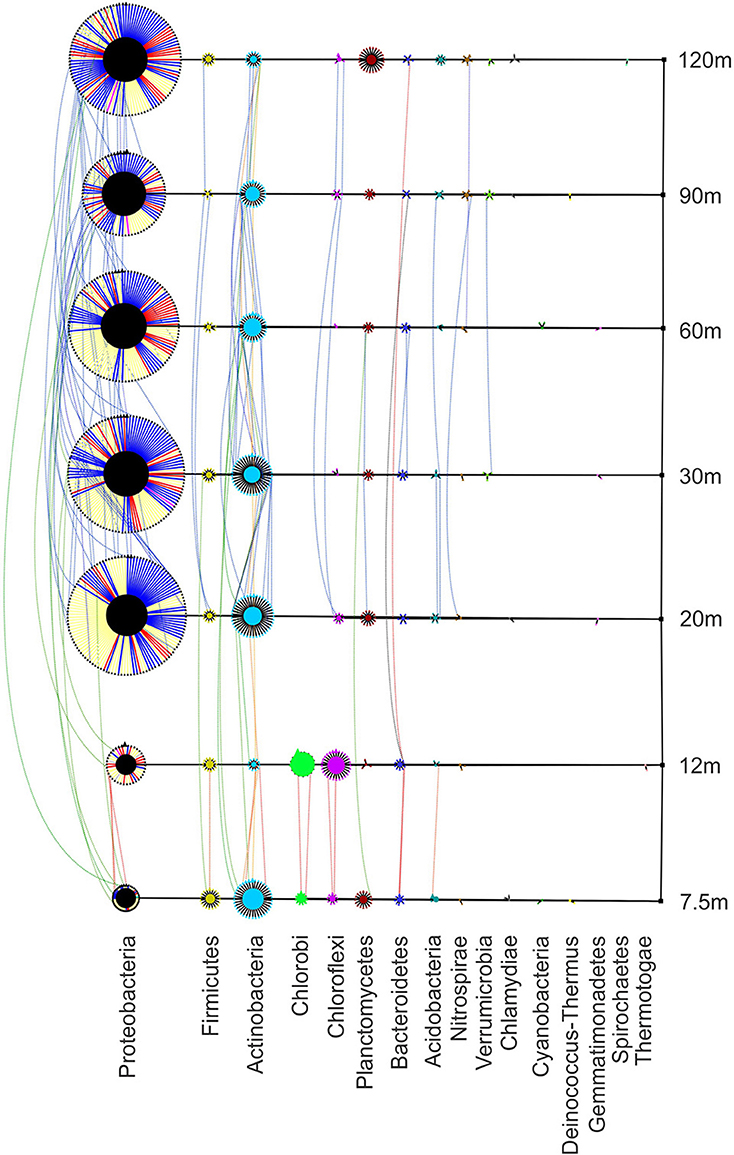
Figure 5. Bacterial phylogenetic network showing richness, abundance and distribution of phyla at each site. Transect nodes (square) designate sample sites (right). Edges connect transect nodes to phyla nodes (circles). Phyla nodes are connected to individual species nodes (triangles). This connection is colored in Proteobacteria to designate class (α–red, β–gray, δ–yellow, ε–pink,γ–blue, ζ–teal). Phyla and species node sizes are proportional to relative abundance. Species nodes are connected by dotted edges if they are found at more than one site. These distribution edges are orange (near vent), blue (away from vent), green (near and away from vent), or yellow (all sites) to show any homogeneity across the transect. Phyla nodes are color coded to facilitate differentiation.
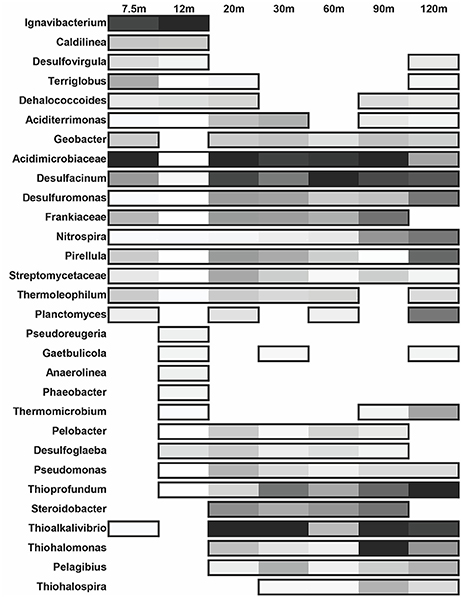
Figure 6. Distribution of the most abundant bacterial genera across each site. The shading is determined by relative abundance where lighter colors represent a lower abundance in respect to the given site. Non-boxed regions represent an absence of the genus at that site.
Bacterial diversity (Shannon's diversity index = 4.76) at 7.5 m is dominated by thermophilic Actinobacteria and Proteobacteria (Figure 6). Members of Acidimicrobiacea (including the genus Aciditerrimonas) within Actinobacteria have the highest relative abundance within the community at 7.5 m. These bacteria are commonly found in areas of extreme acidity and high sulfur concentration. They are either mesophilic or moderately thermophilic and capable of iron oxidation (Itoh et al., 2011). The 7.5 m site is slightly acidic (Figure 2) with sulfide concentrations roughly 3 ppm. The 12 m site has the lowest bacterial diversity of all sites (Shannon's diversity index = 4.07), where members of Actinobacteria decrease significantly. Chlorobi and Chloroflexi, particularly the genus Ignavibacterium, become dominant. Members of Ignavibacterium, while belonging to the genus Chlorobi, are obligate chemoheterotrophs, grow anaerobically, and are moderate thermophiles (Iino et al., 2010). At both near-vent sites, members of the genus Caldilinea are relatively abundant, similar to sediments of the nearby transect 4-A (Meyer-Dombard et al., 2012). Type species for Caldilinea were isolated from a deep-hot aquifer in France and from a hot-spring in Japan. While Caldilinea belongs to Chloroflexi (green non-sulfur bacteria), neither type species can grow under phototrophic conditions (Sekiguchi et al., 2003; Gregoire et al., 2011). The biodiversity of bacteria 20–120 m from the vent is greater than near-vent (Shannon's diversity index = 4.78–5.23). Proteobacteria dominate while members of Chlorobi are absent. The Actinobacteria family Acidimicrobiacea, and Proteobacteria genera Desulfacinum and Thioalkalivibrio are found in greatest relative abundance. A type species of the genus Desulfacinum was isolated from a shallow-water hydrothermal vent in Greece (Sievert and Kuever, 2000). It is thermophilic, anaerobic, mixotrophic, and can reduce sulfate (Rees et al., 1995). Thioalkalivibrio species are tolerant to hypersaline and alkaline environments. Their metabolic capabilities include sulfur oxidation and reduction, denitrification, and thiocyanate metabolism (Muyzer et al., 2011). They tend to be mesophilic, explaining why they are found in greatest relative abundance away from the vent.
Eukaryotic organisms, both sequenced from environmental DNA and scored by morphological identification, show an abundance of nematodes and arthropods throughout the transect (Figure 7). The vent site has the highest abundance of annelids, primarily due to the abundance of one species of Capitella. Playthelminthes, fungi, and heterokonts are found throughout, with fungi and heterokonts showing extreme heterogeneity from site to site.
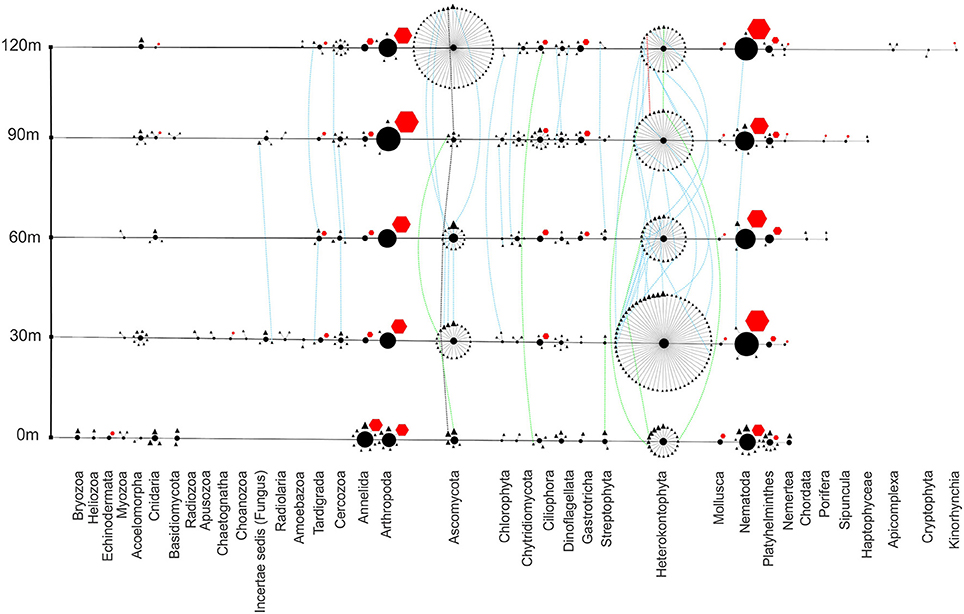
Figure 7. Eukaryote phylogenetic network showing richness, abundance and distribution of phyla at each site. Transect nodes (square) designate sample sites (left). Edges connect transect nodes to phyla nodes (circles). Phyla nodes are connected to individual species nodes. Species nodes are either triangles or octagons to describe the method of identification as either sequencing or scoring, respectively. Phyla and species node sizes are proportional to relative abundance. Species nodes are connected by dotted distribution edges if they are found at more than one site. These distribution edges are blue if found far from vent (30–120 m), green if found both near-vent (0 m) and any other site or black (all sites) to show any homogeneity across the transect.
The dominant eukaryotic genera from sequence data shows many vent specific (0 m) genera: Capitella, Linuche, Schlerochilius, Cladophora, Neochromadora, Oblongichytrium, and Thoracostomopsidae (Figure 8). Other genera such as Pauliella, Penicillium, and Stauroneis are found at all sites along the transect.
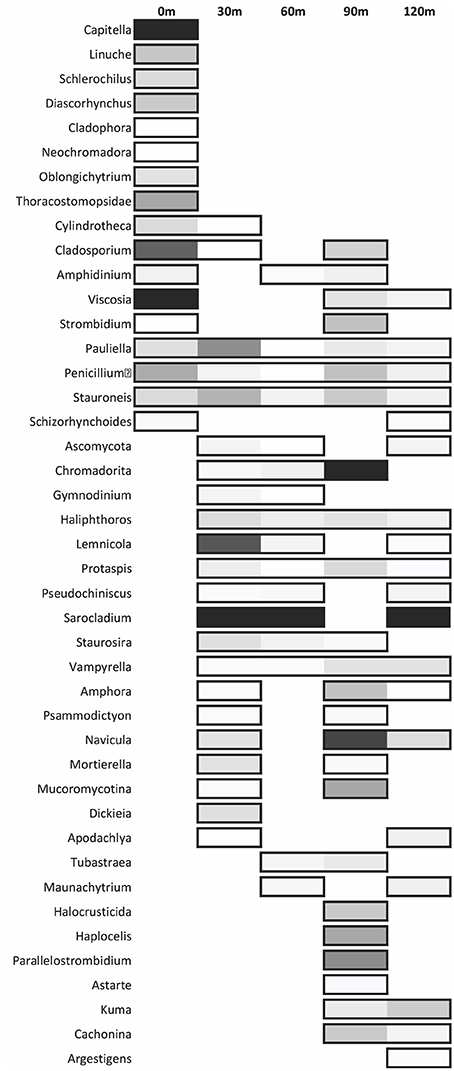
Figure 8. Distribution of the most abundant eukaryote genera across each site. The shading is determined by relative abundance where lighter colors represent a lower abundance in respect to the given site. Non-boxed regions represent an absence of the genus at that site.
Eukaryotic diversity along the transect varies greatly (Shannon's diversity index = 2.7–4.47) with the highest diversity furthest from the vent (120 m site) (Figure 7). At the 0 m site (Shannon's diversity index = 3.47), members of the genera Capitella and Viscosia have the highest relative abundance (Figure 8). Capitella species are opportunistic polychaete annelids present in unstable habitats. Certain capitellids inhabit shallow-hydrothermal areas in Greece and have adapted to survive sulfidic, low organic environments (Gamenick et al., 1998). Viscosia, an r-strategist nematode, exploits low diversity niches (Semprucci et al., 2013) and are often found in disturbed environments. The genus Cladosporium, a cosmopolitan fungus, has the third highest relative abundance at the 0 m site. Members of the genus are typically plant pathogens, but also decompose decaying organic material (Bensch et al., 2012). The molecular data showed that 30 m from the vent (Shannon's diversity index = 4.22), the fungus Sarocladium had the highest relative abundance followed by the diatom, Lemnicola (Figure 8). Sarocladium dominates the sequence data at 30, 60, and 120 m. Sarocladium is a fungus found in soda lakes and closely related to the genus Acremonium which is found in marine environments (Grum-Grzhimaylo et al., 2013). Our most prevalent Sarocladium 18S gene sequence is a 100% match to Sarocladium kiliense (HQ232198) and a 96% match to Acremonium brevis (HQ232183). Eukaryote diversity was lowest 60 m from the vent (Shannon's diversity index = 2.7). Sequencing data at 90 m shows that nematodes of the genus Chromadorita were in highest relative abundance, followed by the diatom Navicula. Data from the hand-sorted meiofauna and macrofauna did not always agree with the molecular data (Wu et al., 2009). For example, only a few nematode sequences were found at the 30 m site while the bulk of hand-sorted specimens from the 30 m site were nematodes. This is most likely due to the fact that hand-sorting of macrofauna and meiofauna excludes organisms such as filamentous fungi and heterokonts, abundant in the molecular analysis but absent from the hand-sorted data set.
Overall, the geochemistry of the shallow hydrothermal system at Vent 4-B in Tutum Bay appears to strongly influence the biodiversity found throughout the transect. BIOENV analysis suggests temperature and alkalinity best explain the patterns of bacterial communities while arsenic (V) is a strong influence on eukaryotic communities. These factors establish a distinct near-vent community consisting of sites <20 m from the vent (Figures 3, 5, 7) and a far-vent community consisting of sites ≥20 m. While these communities are unique at every site, some similarity is evident. Network distribution lines linking species found at more than one site show more similarity among either the near-vent or far-vent community while having little or no similarly between the two. The bacterial communities near the vent (7.5 and 12 m) reveal increased heterogeneity compared to sites further from the vent. This is also indicated by abiotic factors suggesting that there are more microhabitats within the near-vent community at Vent 4-B than revealed within this study.
Functional Analyses
The functions of bacteria along the transect show several trends (Figure 9). Thermophilic bacteria are found at near-vent sites in high abundance and decrease at 20 m and outward. The abundance and diversity of mesophiles peaks at 20 and 30 m and generally are higher away from the vent. Nitrogen, sulfur, and arsenic metabolizers are abundant at near-vent sites, although not as diverse as at sites 20 m outwards. Iron metabolizers, while present throughout the transect, show the lowest abundance and diversity at the 12 m site.
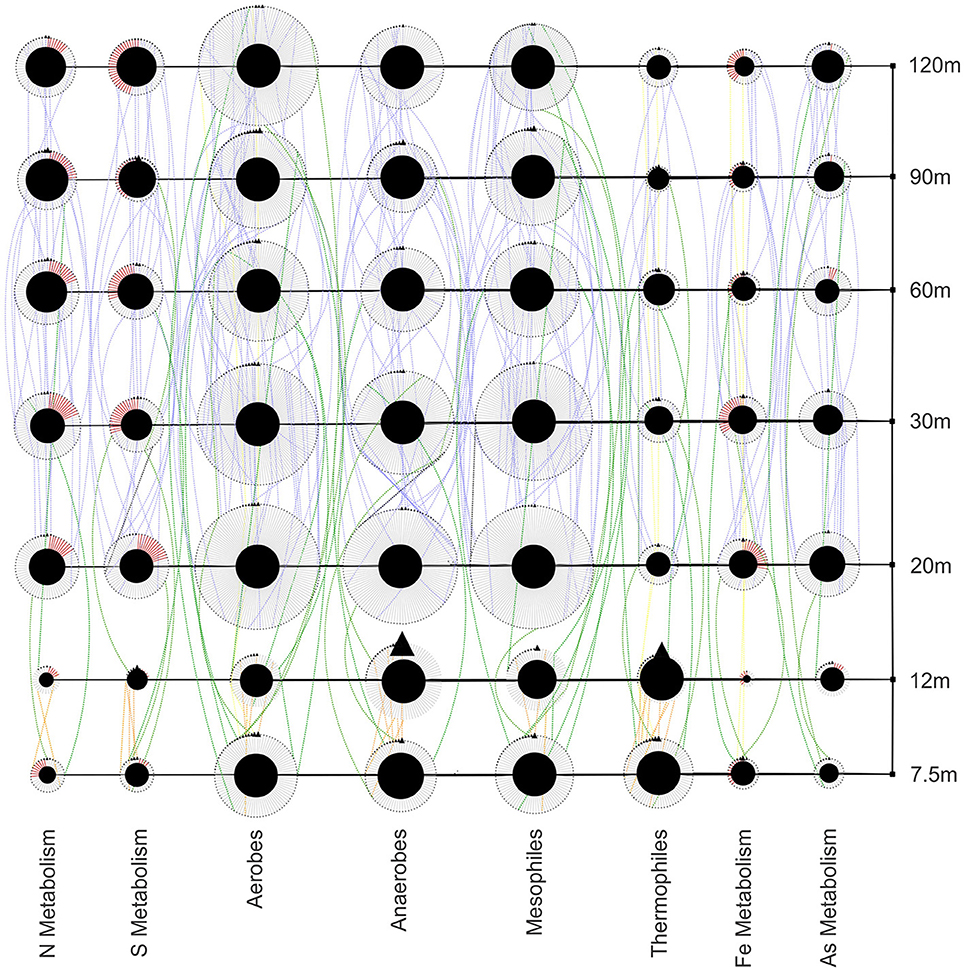
Figure 9. Bacterial functional guild network showing richness, abundance and distribution at each site. Red edges represent oxidizers within metabolic guilds. Transect nodes (square) designate sample sites (right). Edges connect transect nodes to guild nodes (circles). Guild nodes are connected to individual species nodes (triangles). Species nodes are connected by dotted edges if they are found at more than one site. These distribution edges are orange (near vent), blue (away from vent), green (near and away from vent) or yellow (all sites) to show any homogeneity across the transect.
Analyzing the functional guilds along with the abiotic factors at each site reveals additional trends (Figure 10). The temperature of the pore water remains high until after 12 m where it sharply decreases. A small rise in temperature around 60 m can be attributed to observed diffuse venting. The relative abundance of thermophiles also follows this trend with a minor peak at 60 m that matches a local rise in temperature. The relative abundance of mesophiles follows an inverse trend, increasing as the temperature decreases. Oxidation-reduction potential increases with distance from the vent and corresponds to an increase in relative abundance of aerobically-respiring organisms. An exception at 7.5 m was observed, where the relative abundance of aerobic organisms was higher than at 12 m, perhaps because facultative organisms were scored into both guilds. A low relative abundance of nitrogen oxidizers was found, while nitrogen reducers increased with distance from the vent (Figure 10). Nitrite concentrations were low (<1ppm) at all sites and nitrate concentrations generally increased with distance from the vent following a trend similar to nitrogen-reducing bacteria. The relative abundance of sulfur-reducing bacteria varied greatly while sulfur-oxidizing bacteria increased further from the vent (Figure 10). While sulfur-reducing bacteria did not correspond to concentrations of either sulfide or sulfate, sulfur-oxidizers appeared to increase as the concentration of sulfate increased at least for the first 60 m of the transect. Arsenic (III) and arsenic (V) species, primarily found as arsenite and arsenate, respectively, did not follow a clear trend along the transect. Arsenic (V) was found at higher concentration than arsenic (III) at the 7.5 m site while arsenic (III) was highest at the 12 m site (Figure 10). Arsenic-reducing bacteria were more abundant than arsenic-oxidizing bacteria, which were nearly absent along the transect. There did not appear to be a relationship with arsenic (III) or arsenic (V) to arsenic-oxidizing, arsenic-reducing or arsenic-methylating bacteria, although this could be due to the limitations of our method of inferring function. Previous studies have measured the amount of bioavailable arsenic along the 4B transect (Price and Pichler, 2005). The amount of bioavailable arsenic represents only a small amount of the total arsenic present in the samples and appears to correspond to changes in the relative abundance of arsenic reducers along the first 60 m of the transect (Figure 10). Iron reduction, which introduces arsenate back into the system through the reduction of iron (III) oxyhydroxides, was similarly examined. The relative abundance of iron reducers and oxidizers increases and decreases together along the transect, generally corresponding to the amount of Iron (II) in the system (Figure 10). No data was available for Iron (III).
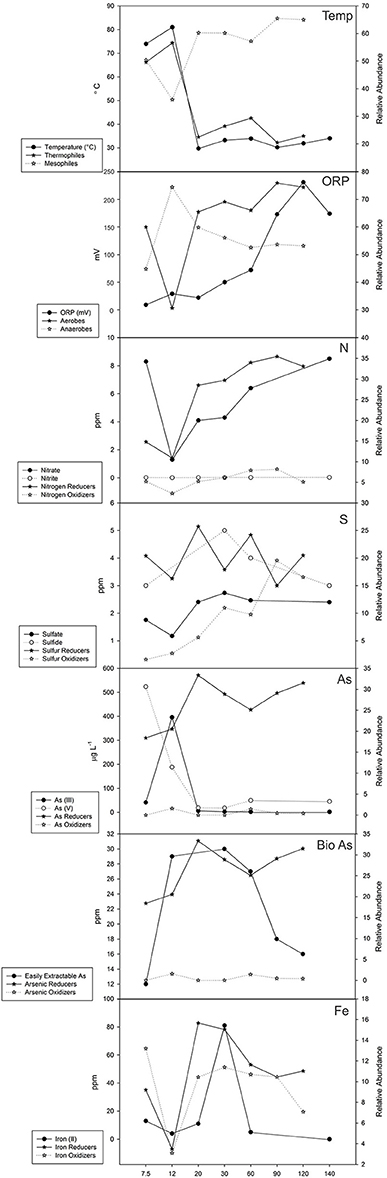
Figure 10. Geochemical measurements and functional guild relative abundance values. Temperature (°C), oxidation-reduction potential (mV), nitrogen (ppm), sulfur (ppm), arsenic (μgL−1), easily extracted arsenic (ppm), and iron concentrations (ppm) are plotted against related functional guilds.
Arsenic Cycling at Tutum Bay
A schematic of arsenic cycling at Tutum Bay may be seen in Figure 11. It reflects the finding that most arsenic at Tutum Bay is adsorbed onto iron oxyhydroxides (Price and Pichler, 2005). Biological data from this study are depicted as blue arrows and suggest a mechanism by which biota may interact with arsenic compounds at Tutum Bay. The largest arrow (black) points to the abiotic process of As (V) adsorbing onto iron (III) oxyhydroxides. The reverse process can happen both abiotically (smaller black arrows) and biotically (blue arrows). This is facilitated by iron-reducing bacteria, which reduce ferric oxyhydroxides, remobilizing arsenate. Orpiment (As2S3) formation can occur during the mineralization of sulfide in the presence of arsenite. This is usually indirectly related to the presence of sulfur-reducing bacteria. Members of the genus Desulfotomaculum (found at Tutum Bay) have been shown to reduce both As (V) and S (VI) to produce orpiment (Newman et al., 1997). The methylation of arsenic species by a range of organisms is shown as blue arrows. Although we found biological support for this model, neither aluminum oxyhydroxides nor orpiment were assayed at Tutum Bay. Microbes known to produce DMA and MMA were detected, but these compounds were not found in Tutum Bay porewater samples (Price and Pichler, 2005). It is possible that volatile forms of arsenic or other forms of organic arsenic were produced or that organisms we identified capable of forming DMA and MMA were inactive.
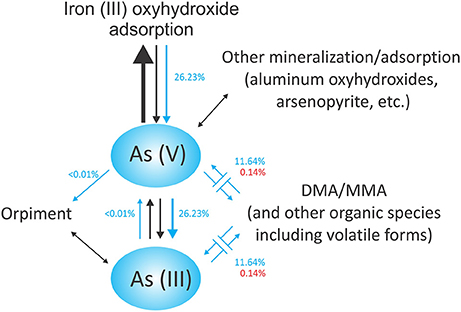
Figure 11. Proposed arsenic cycle of Tutum Bay. Abiotic processes are shown as black arrows. Biotic processes are shown with blue arrows. The sizes of the arrows propose a relative rate at which the processes occur. Percentages represent the number of individuals scored out of the total number of organisms used in the functional classification analysis. Blue percentages are bacterial while red are eukaryotic.
Association Analysis
An association analysis was carried out with combined bacterial and eukaryotic species found at more than one site together (Figure 12). This analysis was limited to the 30, 60, 90, and 120 m sites where both bacterial and eukaryotic data were available. There were a total of 130 OTUs that were found at more than one site together. These OTUs can be seen clustered into a similarity dendogram representing their relative distribution along the transect. Six clades formed with correlation values >0.5. Node 1 contains species found mostly at both the 30 and 90 m sites. Node 2 contains the least number of taxa which are found mainly at the 30 and 120 m site. Node 3 represents taxa found both at 30 and 60 m while node 4 represents taxa found both at 60 and 120 m. Node 5 contains taxa primarily found at sites 60 and 90 m. Lastly, node 6 is the largest and contains taxa found mainly at the 90 and 120 m sites. The association analysis shows that relatively few taxa were found associated together at discontiguous sites. Most associations are either contiguous at 30 and 60 m (node 3) or contiguous away from vent at 90 and 120 m (node 6). These associations demonstrate that there are likely many interactions among phylogenetically diverse organisms at these sites, possibly including predator-prey relationships, commensalism, parasitism, and symbiotic relationships. For example, Capitella sp. are found in a similar distribution pattern to many chemolithoautotrophs, such as Thioalkivibrio sp. Capatellids have been shown to have a symbiotic association with chemoautotrophic bacteria, even suggesting the burrows created by capatellids increase bacterial activity around the burrows (Tsutsumi et al., 2001).
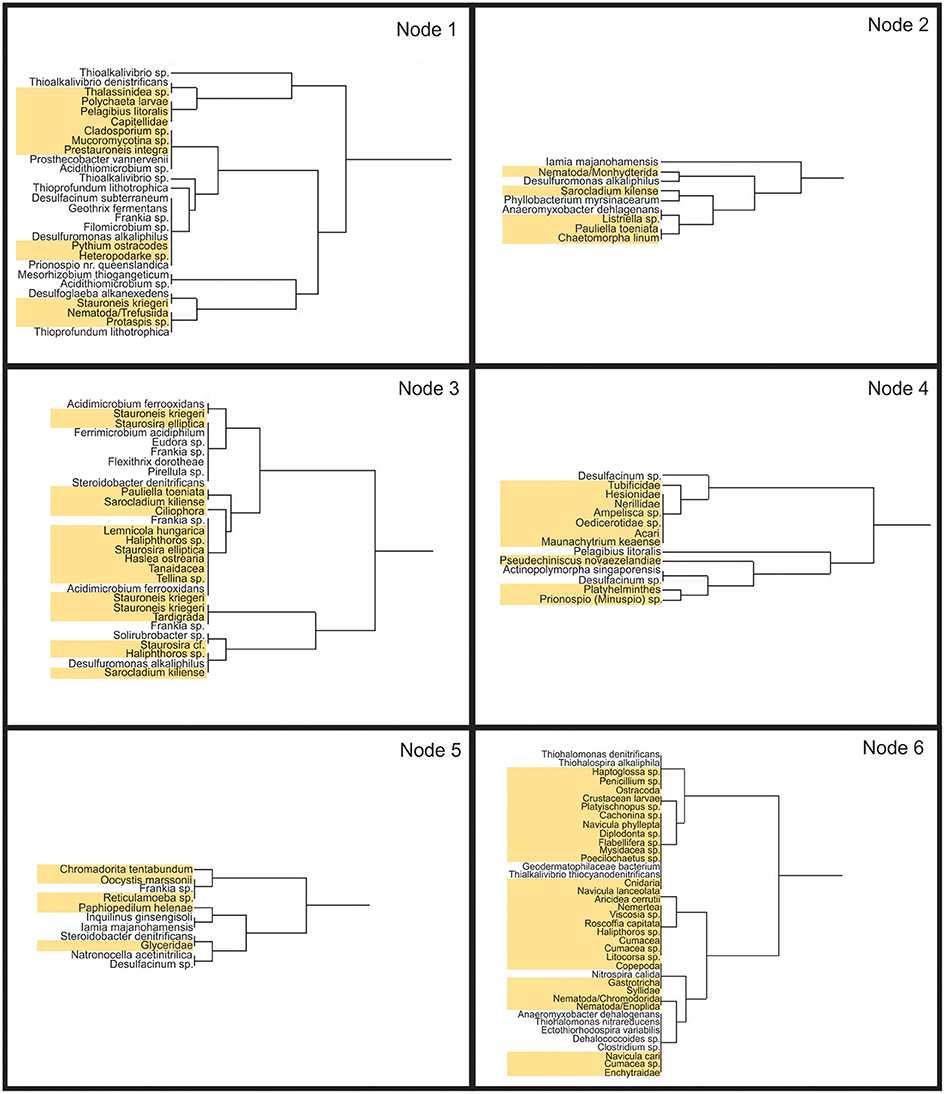
Figure 12. Association Meta Analysis between bacteria and eukaryotes. Six nodes (correlation values > 0.5) represent species with similar distribution patterns. Taxa highlighted in yellow are eukaryotic. Only species found together at more than one site were used in this analysis. Node 1 represents organisms found together primarily at sites 30 and 90: nodes 2 primarily 30 and 120 m; node 3, primarily 30 and 60 m; node 4, 60 and 120 m; node 5, 60 and 90 m; node 6, 90 and 120 m.
Comparison to Other Studies
Similarities exist between the Tutum Bay communities and those of vent areas from other studies, but most studies do not sample as far from the vent as ours. One example of similar findings involves the Capitella genus described above. In addition, Sievert et al. (1999) found a high relative abundance of dissimilatory iron-reducing bacteria in the transition zone of a shallow-water hydrothermal vent near Milos Island, Greece. At the Tutum Bay transect, an increased relative abundance of dissimilatory iron-reducing bacteria is also seen in the transition zone from near-vent to far-vent (Figure 9). DNA analysis at the transect of Milos Island revealed the first report of Acidobacterium at a hydrothermal site (Sievert et al., 2000). At Tutum Bay, DNA analyses found Acidobacterium at near-vent sites but also throughout the transect. In a shallow-water hydrothermal system in Japan, sulfur-oxidizing Chlorobi appeared to dominate the near-vent microbial mats (Hirayama et al., 2007). While the Chlorobi species found in Japan are photoautotrophic, our study finds all but 1 of 106 sequences at Tutum Bay belong to the genus Ignavibacterium which grows heterotrophically. A recent shallow-water pyrosquencing study in the Okinawa Trough found bacterial communities dominated by sulfur-metabolizing organisms. They found members of the genus Nautilia and Thiomicrospira as the major component of the two hydrothermal vents sampled (Zhang et al., 2012). Neither genus was found in our study. Members of Thiohalomonas and Thioprofundum were the most abundant representatives of sulfur oxidizers at our Tutum Bay transect. Our study yielded 1930 bacterial 16s rRNA gene sequences across the Tutum Bay transect, but only 158 represented sulfur oxidizers with the majority (n = 85) found at the 90 and 120 m sites, suggesting that sulfur oxidation is not a major metabolic process in Tutum Bay. The samples in the Taiwan study were taken from the water column of a vent heavily colored with sulfur precipitants, while our samples were from sediment pore water.
Conclusions
This study is one of the first to assess biocomplexity across a broad phylogenetic range including bacteria, microbial eukaryotes, meiofauna and macrofauna from a shallow-water hydrothermal vent using both molecular and morphological data. Biodiversity generally increased with distance from the vent, with an exception where mid-transect diffuse venting was found. Hydrothermal influences were found across the entire 120 m transect, suggesting that hydrothermal vents can influence a larger region than previously thought. While abiotic data show higher similarity among the near-vent sites, the larger changes in biodiversity in the near vent area compared to the far vent area suggests that there are smaller scale changes that we have not detected. Overall, the biodiversity of the communities responded to the presence of hydrothermal fluids with a clear correlation between temperature and thermophilic organisms. Functional guilds display heterogeneity between the 7.5 and 12 m sites, again suggesting more community diversity than we observed with our limited number of near-vent sample sites. However, compared to far-vent sites, near-vent sites have less functional redundancy. No specific correlation between bacteria that metabolize arsenic and the concentration of different oxidation states of arsenic ions was observed, perhaps because very little of the arsenic present was bioavailable. The homogeneous distribution of arsenic reducers may represent background arsenic metabolism. Eukaryote biodiversity appeared to be more affected by arsenic and temperature than bacteria. Association analysis of organisms at the site coupled with the varied geochemical environment across the transect suggest many potential interactions between phylogenetically diverse organisms occur at certain sites, and that species distributions and interactions occur in the context of complex spatial relationships. Most biodiversity studies at hydrothermal sites focus on effluent or communities growing in the immediate vicinity of the vents. This study shows that hydrothermal vents may influence greater distances, with distinct micro communities in this case ranging up to 20 m from the vent with influences as far as 120 m.
Author Contributions
JG participated in the planning, field work, analysis, and writing of the manuscript. DK was a graduate student who participated in the field work, analyses and writing of the manuscript. He carried out all the morphological identification of benthic invertebrates. HR was a graduate student who carried out the bulk of the molecular and statistical analysis as well as the writing presented in this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Pamela Hallock-Muller, Thomas Pichler, Brian McCloskey, Jan Amend, Roy Price and Darcy Meyer-Dombard for their field assistance. We also thank the crew of the M/V Star Dancer and the government of Papua New Guinea. This work was supported by the U.S. National Science Foundation under BE/CBC grant CHE-0221834 to T. Pichler and by internal grants from the University of South Florida.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2017.00177/full#supplementary-material
References
Akerman, N. H., Price, R. E., Pichler, T., and Amend, J. P. (2011). Energy sources for chemolithotrophs in an arsenic- and iron-rich shallow-sea hydrothermal system. Geobiology 9, 436–445. doi: 10.1111/j.1472-4669.2011.00291.x
Bensch, K., Braun, U., Groenewald, J. Z., and Crous, P. W. (2012). The genus Cladosporium. Stud. Mycol. 72, 1–401. doi: 10.3114/sim0003
Colwell, R. K., and Elsensohn, J. E. (2014). EstimateS turns 20: statistical estimation of species richness and shared species from samples, with non-parametric extrapolation. Ecography 37, 609–613. doi: 10.1111/ecog.00814
Cullen, W. R., and Reimer, K. J. (1989). Arsenic speciation in the environment. Chem. Rev. 89, 713–764. doi: 10.1021/Cr00094a002
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 89, 5685–5689.
Doak, G. O., and Freedman, L. D. (1970). Organometallic Compounds of Arsenic, Antimony, and Bismuth. New York, NY: Wiley-Interscience.
Gamenick, I., Abbiati, M., and Giere, O. (1998). Field distribution and sulphide tolerance of Capitella capitata (Annelida: Polychaeta) around shallow water hydrothermal vents off Milos (Aegean Sea). A new sibling species? Mar. Biol. 130, 447–453. doi: 10.1007/s002270050265
Giovannelli, D., d'Errico, G., Manini, E., Yakimov, M., and Vetriani, C. (2013). Diversity and phylogenetic analyses of bacteria from a shallow-water hydrothermal vent in Milos island (Greece). Front. Microbiol. 4:184. doi: 10.3389/fmicb.2013.00184
Gregoire, P., Bohli, M., Cayol, J. L., Joseph, M., Guasco, S., Dubourg, K., et al. (2011). Caldilinea tarbellica sp. nov., a filamentous, thermophilic, anaerobic bacterium isolated from a deep hot aquifer in the Aquitaine Basin. Int. J. Syst. Evol. Microbiol. 61(Pt 6), 1436–1441. doi: 10.1099/ijs.0.025676-0
Grum-Grzhimaylo, A. A., Georgieva, M. L., Debets, A. J., and Bilanenko, E. N. (2013). Are alkalitolerant fungi of the Emericellopsis lineage (Bionectriaceae) of marine origin? IMA Fungus 4, 213–228. doi: 10.5598/imafungus.2013.04.02.07
Heiri, O., Lotter, A. F., and Lemcke, G. (2001). Loss on ignition as a method for estimating organic and carbonate content in sediments: reproducibility and comparability of results. J. Paleolimnol. 25, 101–110. doi: 10.1023/A:1008119611481
Hirayama, H., Sunamura, M., Takai, K., Nunoura, T., Noguchi, T., Oida, H., et al. (2007). Culture-dependent and -independent characterization of microbial communities associated with a shallow submarine hydrothermal system occurring within a coral reef off Taketomi Island, Japan. Appl. Environ. Microbiol. 73, 7642–7656. doi: 10.1128/AEM.01258-07
Iino, T., Mori, K., Uchino, Y., Nakagawa, T., Harayama, S., and Suzuki, K. (2010). Ignavibacterium album gen. nov., sp. nov., a moderately thermophilic anaerobic bacterium isolated from microbial mats at a terrestrial hot spring and proposal of Ignavibacteria classis nov., for a novel lineage at the periphery of green sulfur bacteria. Int. J. Syst. Evol. Microbiol. 60(Pt 6), 1376–1382. doi: 10.1099/ijs.0.012484-0
Itoh, T., Yamanoi, K., Kudo, T., Ohkuma, M., and Takashina, T. (2011). Aciditerrimonas ferrireducens gen. nov., sp. nov., an iron-reducing thermoacidophilic actinobacterium isolated from a solfataric field. Int. J. Syst. Evol. Microbiol. 61(Pt 6), 1281–1285. doi: 10.1099/ijs.0.023044-0
Karlen, D. J., Price, R. E., Pichler, T., and Garey, J. R. (2010). Changes in benthic macrofauna associated with a shallow-water hydrothermal vent gradient in Papua New Guinea. Pac. Sci. 64, 391–404. doi: 10.2984/64.3.391
Lin, S., Shi, Q., Nix, F. B., Styblo, M., Beck, M. A., Herbin-Davis, K. M., et al. (2002). A novel S-adenosyl-L-methionine: arsenic(III) methyltransferase from rat liver cytosol. J. Biol. Chem. 277, 10795–10803. doi: 10.1074/jbc.M110246200
Mackey, L. Y., Winnepenninckx, B., De Wachter, R., Backeljau, T., Emschermann, P., and Garey, J. R. (1996). 18S rRNA suggests that Entoprocta are protostomes, unrelated to Ectoprocta. J. Mol. Evol. 42, 552–559.
Meyer-Dombard, D. R., Price, R. E., Pichler, T., and Amend, J. P. (2012). Prokaryotic populations in arsenic-rich shallow-sea hydrothermal sediments of ambitle Island, Papua New Guinea. Geomicrobiol. J. 29, 1–17. doi: 10.1080/01490451.2010.520073
Michalke, K., Wickenheiser, E. B., Mehring, M., Hirner, A. V., and Hensel, R. (2000). Production of volatile derivatives of metal(loid)s by microflora involved in anaerobic digestion of sewage sludge. Appl. Envrion. Microbiol. 66, 2791–2796. doi: 10.1128/AEM.66.7.2791-2796.2000
Muyzer, G., Sorokin, D. Y., Mavromatis, K., Lapidus, A., Foster, B., Sun, H., et al. (2011). Complete genome sequence of Thioalkalivibrio sp. K90mix. Stand. Genomic Sci. 5, 341–355. doi: 10.4056/sigs.2315092
Newman, D. K., Beveridge, T. J., and Morel, F. (1997). Precipitation of Arsenic Trisulfide by Desulfotomaculum auripigmentum. Appl. Environ. Microbiol. 63, 2022–2028.
Oremland, R. S., and Stolz, J. F. (2003). The ecology of arsenic. Science 300, 939–944. doi: 10.1126/science.1081903
Pichler, T., Amend, J. P., Garey, J. R., Hallock, P., Hsia, N. P., Karlen, D., et al. (2006). A Natural Laboratory to Study Arsenic Geobiocomplexity. Eos Trans. Am. Geophys. Union 87, 221–225. doi: 10.1029/2006EO230002
Pichler, T., and Dix, G. R. (1996). Hydrothermal venting within a coral reef ecosystem, Ambitle island, Papua New Guinea. Geology 24, 435–438. doi: 10.1130/0091-7613(1996)024<0435:Hvwacr>2.3.Co;2
Price, R. E., Amend, J. P., and Pichler, T. (2007). Enhanced geochemical gradients in a marine shallow-water hydrothermal system: unusual arsenic speciation in horizontal and vertical pore water profiles. Appl. Geochem. 22, 2595–2605. doi: 10.1016/j.apgeochem.2007.06.010
Price, R. E., and Pichler, T. (2005). Distribution, speciation and bioavailability of arsenic in a shallow-water submarine hydrothermal system, Tutum Bay, Ambitle Island, PNG. Chem. Geol. 224, 122–135. doi: 10.1016/j.chemgeo.2005.07.017
Rees, G. N., Grassia, G. S., Sheehy, A. J., Dwivedi, P. P., and Patel, B. K. C. (1995). Desulfacinum infernurn gem nov., sp. nov., a thermophilic sulfate-reducing bacterium from a petroleum reservoir. Int. J. Syst. Bacteriol. 45, 85–89.
Sekiguchi, Y., Yamada, T., Hanada, S., Ohashi, A., Harada, H., and Kamagata, Y. (2003). Anaerolinea thermophila gen. nov., sp. nov. and Caldilinea aerophila gen. nov., sp. nov., novel filamentous thermophiles that represent a previously uncultured lineage of the domain Bacteria at the subphylum level. Int. J. Syst. Evol. Microbiol. 53(Pt 6), 1843–1851. doi: 10.1099/ijs.0.02699-0
Semprucci, F., Moreno, M., Sbrocca, S., Rocchi, M., Albertelli, G., and Balsamo, M. (2013). The nematode assemblage as a tool for the assessment of marine ecological quality status: a case-study in the Central Adriatic Sea. Mediterr. Mar. Sci. 14, 48–57. doi: 10.12681/mms.366
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sievert, S. M., Brinkhoff, T., Muyzer, G., Ziebis, W., and Kuever, J. (1999). Spatial heterogeneity of bacterial populations along an environmental gradient at a shallow submarine hydrothermal vent near Milos Island (Greece). Appl. Environ. Microbiol. 65, 3834–3842.
Sievert, S. M., and Kuever, J. (2000). Desulfacinum hydrothermale sp. nov., a thermophilic, sulfate-reducing bacterium from geothermally heated sediments near Milos Island (Greece). Int. J. Syst. Evol. Microbiol. 50(Pt 3), 1239–1246. doi: 10.1099/00207713-50-3-1239
Sievert, S. M., Kuever, J., and Muyzer, G. (2000). Identification of 16S ribosomal DNA-defined bacterial populations at a shallow submarine hydrothermal vent near Milos Island (Greece). Appl. Environ. Microbiol. 66, 3102–3109. doi: 10.1128/AEM.66.7.3102-3109.2000
Tarasov, V. G., Gebruk, A. V., Mironov, A. N., and Moskalev, L. I. (2005). Deep-sea and shallow-water hydrothermal vent communities: two different phenomena? Chem. Geol. 224, 5–39. doi: 10.1016/j.chemgeo.2005.07.021
Thomas, D. J., Li, J., Waters, S. B., Xing, W., Adair, B. M., Drobna, Z., et al. (2007). Arsenic (+3 Oxidation State) Methand the Methylation of Arsenicals. Exp. Biol. Med. 232, 3–13.
Tsutsumi, H., Wainwright, S., Montani, S., Sagal, M., Ichihara, S., and Kogure, K. (2001). Exploitation of a chemosynthetic food resource by the polychaete Capitella sp. I. Mar. Ecol. Prog. Ser., 216, 119–127. doi: 10.3354/meps216119
Wang, L., Cheung, M. K., Kwan, H. S., Hwang, J. S., and Wong, C. K. (2015). Microbial diversity in shallow-water hydrothermal sediments of Kueishan Island, Taiwan as revealed by pyrosequencing. J. Basic Microbiol. 55, 1308–1318. doi: 10.1002/jobm.201400811
Wu, T., Ayres, E., Bardgett, R. D., Wall, D. H., and Garey, J. R. (2011). Molecular study of worldwide distribution and diversity of soil animals. Proc. Nat. Acad. Sci. U.S.A. 108, 17720–17725. doi: 10.1073/pnas.1103824108
Wu, T., Ayres, E., Li, G., Bardgett, R. D., Wall, D. H., and Garey, J. R. (2009). Molecular profiling of soil animal diversity in natural ecosystems: incongruence of molecular and morphological results. Soil Biol. Biochem. 41, 849–857. doi: 10.1016/j.soilbio.2009.02.003
Keywords: microbial community structure and function, benthic communities, invertebrates, hydrothermal vents, arsenic
Citation: Rubelmann H, Karlen DJ and Garey JR (2017) Changes in Eukaryotic and Bacterial Communities along a 120 m Transect Associated with a Shallow Marine Hydrothermal Vent. Front. Mar. Sci. 4:177. doi: 10.3389/fmars.2017.00177
Received: 11 February 2017; Accepted: 22 May 2017;
Published: 06 June 2017.
Edited by:
Iliana B. Baums, Pennsylvania State University, United StatesReviewed by:
Emre Keskin, Ankara University, TurkeyAdam Michael Reitzel, University of North Carolina at Charlotte, United States
Copyright © 2017 Rubelmann, Karlen and Garey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James R. Garey, Z2FyZXlAdXNmLmVkdQ==