 Kaley M. Major1*
Kaley M. Major1* Bethany M. DeCourten1,2
Bethany M. DeCourten1,2 Jie Li3
Jie Li3 Monica Britton3Matthew L. Settles3Alvine C. Mehinto4Richard E. Connon5
Monica Britton3Matthew L. Settles3Alvine C. Mehinto4Richard E. Connon5 Susanne M. Brander1
Susanne M. Brander1- 1Department of Environmental and Molecular Toxicology, Oregon State University, Corvallis, OR, United States
- 2Department of Biology and Marine Biology, University of North Carolina Wilmington, Wilmington, NC, United States
- 3Bioinformatics Core, Genome Center, University of California, Davis, Davis, CA, United States
- 4Southern California Coastal Water Research Project, Costa Mesa, CA, United States
- 5Department of Anatomy, Physiology & Cell Biology, School of Veterinary Medicine, University of California, Davis, Davis, CA, United States
The inland silverside, Menidia beryllina, is a euryhaline fish and a model organism in ecotoxicology. We previously showed that exposure to picomolar (ng/L) levels of endocrine disrupting chemicals (EDCs) can cause a variety of effects in M. beryllina, from changes in gene expression to phenotypic alterations. Here we explore the potential for early life exposure to EDCs to modify the epigenome in silversides, with a focus on multi- and transgenerational effects. EDCs included contaminants of emerging concern (the pyrethroid insecticide bifenthrin and the synthetic progestin levonorgestrel), as well as a commonly detected synthetic estrogen (ethinylestradiol), and a synthetic androgen (trenbolone) at exposure levels ranging from 3 to 10 ng/L. In a multigenerational experiment, we exposed parental silversides to EDCs from fertilization until 21 days post hatch (dph). Then we assessed DNA methylation patterns for three generations (F0, F1, and F2) in whole body larval fish using reduced representation bisulfite sequencing (RRBS). We found significant (α = 0.05) differences in promoter and/or gene body methylation in treatment fish relative to controls for all EDCs and all generations indicating that both multigenerational (F1) and transgenerational (F2) effects that were caused by strict inheritance of DNA methylation alterations and the dysregulation of epigenetic control mechanisms. Using gene ontology and pathway analyses, we found enrichment in biological processes and pathways representative of growth and development, immune function, reproduction, pigmentation, epigenetic regulation, stress response and repair (including pathways important in carcinogenesis). Further, we found that a subset of potentially EDC responsive genes (EDCRGs) were differentially methylated across all treatments and generations and included hormone receptors, genes involved in steroidogenesis, prostaglandin synthesis, sexual development, DNA methylation, protein metabolism and synthesis, cell signaling, and neurodevelopment. The analysis of EDCRGs provided additional evidence that differential methylation is inherited by the offspring of EDC-treated animals, sometimes in the F2 generation that was never exposed. These findings show that low, environmentally relevant levels of EDCs can cause altered methylation in genes that are functionally relevant to impaired phenotypes documented in EDC-exposed animals and that EDC exposure has the potential to affect epigenetic regulation in future generations of fish that have never been exposed.
Introduction
Endocrine disrupting chemicals (EDCs) include a variety of compound classes such as pesticides, pharmaceuticals, industrial chemicals, and metals, that are grouped together on the basis of their tendency to alter hormone signaling. Many EDCs enter the aquatic environment through runoff and wastewater, allowing them to move into estuarine and marine systems (Ribeiro et al., 2009; Bayen et al., 2013; Brander, 2013; Cole et al., 2016; DeCourten et al., 2019b; Zhou et al., 2019). Even at low levels (ng/L), EDC exposure during early development has been implicated in causing a variety of effects in fish across biological scales, including changes in growth and development, reproduction, immune function, sex ratio, gene expression, and DNA methylation (Hinck et al., 2008; Schug et al., 2016). For example, a growing body of literature has linked molecular endpoints to physiological and behavioral endpoints of EDC exposure in the ecologically and toxicologically relevant inland silverside, Menidia beryllina, an estuarine species common in North America (Brander et al., 2016; Cole et al., 2016; DeCourten and Brander, 2017; DeCourten et al., 2019b; Frank et al., 2019). Multi- and transgenerational effects of EDC exposure have been documented in M. beryllina, sometimes with latent effects occurring in the F1 generation (indirectly exposed as primordial germ cells), reinforcing concern for longterm population-level impacts from these ubiquitous environmental chemicals (DeCourten and Brander, 2017; DeCourten et al., 2019a; DeCourten et al., unpublished).
Epigenetic modifications can modulate gene expression stably (i.e., heritably) without alteration to the primary DNA sequence and are now considered to be a common mechanism for transgenerational inheritance of physiological phenotypes (Jablonka and Raz, 2009). Aside from meiotically stable (i.e., germline) modifications, mitotically stable (i.e., somatic) epigenetic modifications are essential to cellular differentiation and development, as well as important in maintaining epigenetically controlled disease phenotypes within an individual (Best et al., 2018). In fact, a variety of disease phenotypes (metabolic syndromes, neurological disorders, infertility, and cancer) originating during development have also been linked to epigenetic mechanisms (Head, 2014; Bhandari, 2016). Some of the most well-studied structural mechanisms of epigenetic control include DNA methylation [the addition of a methyl group to the cytosine in a cytosine-guanine dinucleotide (CpG)] and histone modifications (i.e., acetylation, methylation, ubiquination). Both of these epigenetic mechanisms physically alter the ability of transcriptional machinery to access DNA, thus impacting gene expression (Brander et al., 2017; Alavian-Ghavanini and Ruegg, 2018). The effect of DNA methylation varies with its relative location to genes. DNA methylation near transcription start sites (TSS) often suppresses vertebrate gene expression by blocking transcription machinery, while methylation within the gene body may actually stimulate transcription or alter splice variants (Jones, 2012). The area of environmental epigenomics has arisen to specifically study the effects of environmental exposures, including EDCs, on the perturbation of epigenetic mechanisms. Although EDCs have been well established to alter DNA methylation, particularly in fish (Aniagu et al., 2008; Mirbahai et al., 2011; Olsvik et al., 2014, 2019; Aluru et al., 2018) the mechanism(s) by which that disruption occurs remains under investigation (Xin et al., 2015; Alavian-Ghavanini and Ruegg, 2018).
Altered methylation states may be caused by general epigenetic dysregulation (i.e., changes in the functioning of methylation machinery) or transgenerational epigenetic inheritance. In mammals, there are generally two waves of global genome demethylation associated with development: after fertilization, and again before primordial germ cell (PGC) differentiation. However, fishes differ in their patterns of developmental DNA methylation erasure, with medaka (Oryzias latipes), for example, having a pattern that is the same as that of mammals (Wang and Bhandari, 2019), but zebrafish (Danio rerio) lacking both reprogramming stages, with embryos eventually harboring the paternal methylome (Jiang et al., 2013; Potok et al., 2013; Ortega-Recalde et al., 2019). In M. beryllina, the developmental pattern of DNA methylation erasure has not been established, although its pattern could undoubtedly affect the way that environmental exposures are passed down through methylation. Understanding the linkages between molecular changes caused by environmental EDC exposure and altered phenotypes is essential to strengthening current and informing new adverse outcome pathways (AOPs) for EDCs, which will help define their risk to wild populations (Ankley et al., 2009; Perkins et al., 2019).
Exploring the epigenetic effects of EDC exposure in estuarine fish is essential, not only from a risk assessment perspective, but also from the standpoint of understanding the mechanisms underlying the effects of EDC exposure across a range of different species. To explore how DNA methylation may be influenced by EDC exposure in estuarine fish, we exposed a parental generation (F0) of M. beryllina to a suite of EDCs at environmentally relevant concentrations during early life [8 hours post fertilization (hpf) to 21 days post hatch (dph)]. EDCs included contaminants of emerging concern: the pyrethroid insecticide bifenthrin (Bif) and the synthetic progestin levonorgestrel (Levo), as well as the commonly detected synthetic estrogen ethinylestradiol (EE2), and the synthetic androgen trenbolone (Tren) at exposure levels between 3 and 10 ng/L. These chemicals have been linked to a variety of effects in fish, such as decreased egg production, reproductive impairment, skewed sex ratios, developmental deformities, alterations in behavior, changes in protein and/or gene expression, and DNA methylation (Jobling et al., 1998; Kidd et al., 2007; Jin et al., 2009; Brander et al., 2012; Forsgren et al., 2013; Svensson et al., 2013, 2014; Ellestad et al., 2014; Orlando and Ellestad, 2014; Schwindt et al., 2014; Bertram et al., 2015; Hua et al., 2015; Runnalls et al., 2015; Orn et al., 2016; DeCourten and Brander, 2017; Robinson et al., 2017). In our companion paper, DeCourten et al. (unpublished) demonstrated that EDC exposure caused physiological effects and sometimes changes in gene expression and DNA methylation in a limited analysis of 20 genes. These effects could be measured in the offspring (F1 generation), which were indirectly exposed to EDCs as primordial germ cells, as well as in second-generation offspring (F2), which were never exposed to EDCs. Here we explore the changes in DNA methylation for a larger collection of potentially EDC responsive genes and determine the scope and functional implications of changes in DNA methylation that may underlie physiological changes. We use reduced representation bisulfite sequencing (RRBS) to examine differential methylation in a subset of larval fish across three generations, from the larger study by DeCourten et al. (unpublished). In doing so we capitalize on a unique opportunity to relate apical endpoints, gene expression, and epigenetic modification data to get a fuller picture of the effects of EDC exposure across biological scales. The goals of the present study were to (1) quantify differential methylation caused by exposure to four EDCs across generations of fish that have been directly exposed (F0), indirectly exposed (F1), or unexposed (F2); (2) use focused analyses to determine which biological processes, pathways, and/or select genes are most impacted by differential methylation while relating methylation effects to physiological endpoints measured in larval fish; and (3) determine the extent that differential methylation is present in a multigenerational (F1) or transgenerational (F2) context.
Materials and Methods
EDC Exposures, Chemical Analyses, and Multigenerational Rear-Out
A complete account of EDC exposures in the fish analyzed herein will be published elsewhere (DeCourten et al., unpublished). Briefly, embryos were obtained from an adult brood stock and processed according to methods in Porazinski et al. (2010), but without dechorionation. Fish [aged 8 hours post fertilization (hpf) – 21 days post hatch (dph)] in the parental (F0) generation were exposed to environmentally relevant concentrations of each of four chemicals separately: bifenthrin (Bif, 3.02 ng/L; Chem Services, West Chester, PA, United States; 99.5% pure mix of isomers), EE2 (6.79 ng/L; Sigma-Aldrich, St. Louis, MO, United States; CAS 57-63-6, >98% purity), levonorgestrel (Levo, 9.27 ng/L; USP, Rockville, MD, United States; 100% purity) and trenbolone (Tren, 9.60 ng/L; Spectrum Laboratory Products, Gardena, CA, United States; 100% purity). Concentrations for all chemicals are reported as average measured concentrations of exposure water, verified analytically using liquid chromatography-spectrometry described elsewhere (DeCourten et al., unpublished). Control groups were exposed to 10 μL/L of methanol to control for any effects of the solvent used for lipophilic chemicals and five experimental replicates were used for each treatment. New treatment water was mixed just prior to the daily 75% water changes. During periods in the 25 mL beakers (8 hpf-hatch at day 7–10) new water was mixed everyday by adding 10 μL (for BF or EE2) or 20 μL (for levonorgestrel or trenbolone) of EDC stock solutions in MeOH (0.1 mcg/mL). During exposure periods in the 1.4 L jars (hatch-21dph) water was mixed in 2–3 L batches by adding of 5 μL (bifenthrin and EE2) and 10 μL (levonorgestrel and trenbolone) for each liter made of EDC stock solutions in MeOH (1 mcg/L). Each replicate (n = 4–5) was maintained independently as described above throughout the course of the 3-generation study, with EDC exposures continuing until the 21-dph sampling time point (for the F0 generation only; Figure 1. After the initial 21-dph sampling time point of the F0 animals, a subset of animals was sampled for molecular endpoints, while the rest of the animals were transferred to clean water and no further EDC exposure occurred. At 21 dph remaining fish were transferred to 6 L glass containers where they were fed a diet of Hikari tropical fish food and live Artemia nauplii twice per day, with a 60% water change daily and reared to ∼120 dph. After 120 dph, fish were transferred to 20-gallon recirculating tanks where they were spawned in groups of ∼50 individuals (50:50 sex ratio) by placing strands of dye-free acrylic yarn into tanks overnight, allowing for spawning for roughly 3 h. After spawning, embryos were transferred to the laboratory where they were treated the same as F0 animals, but without chemical exposure, through the F2 generation. Thus, F1 animals were exposed to EDCs indirectly as primordial germ cells within F0 parents, while F2 animals were not exposed to EDCs at all. Any effects noted in the F1 animals would be considered “multigenerational” effects given that both F0 and F1 were either directly or indirectly exposed to the chemicals. Any effects noted in F2 animals, which were never exposed to the chemicals, are considered “transgenerational” effects.
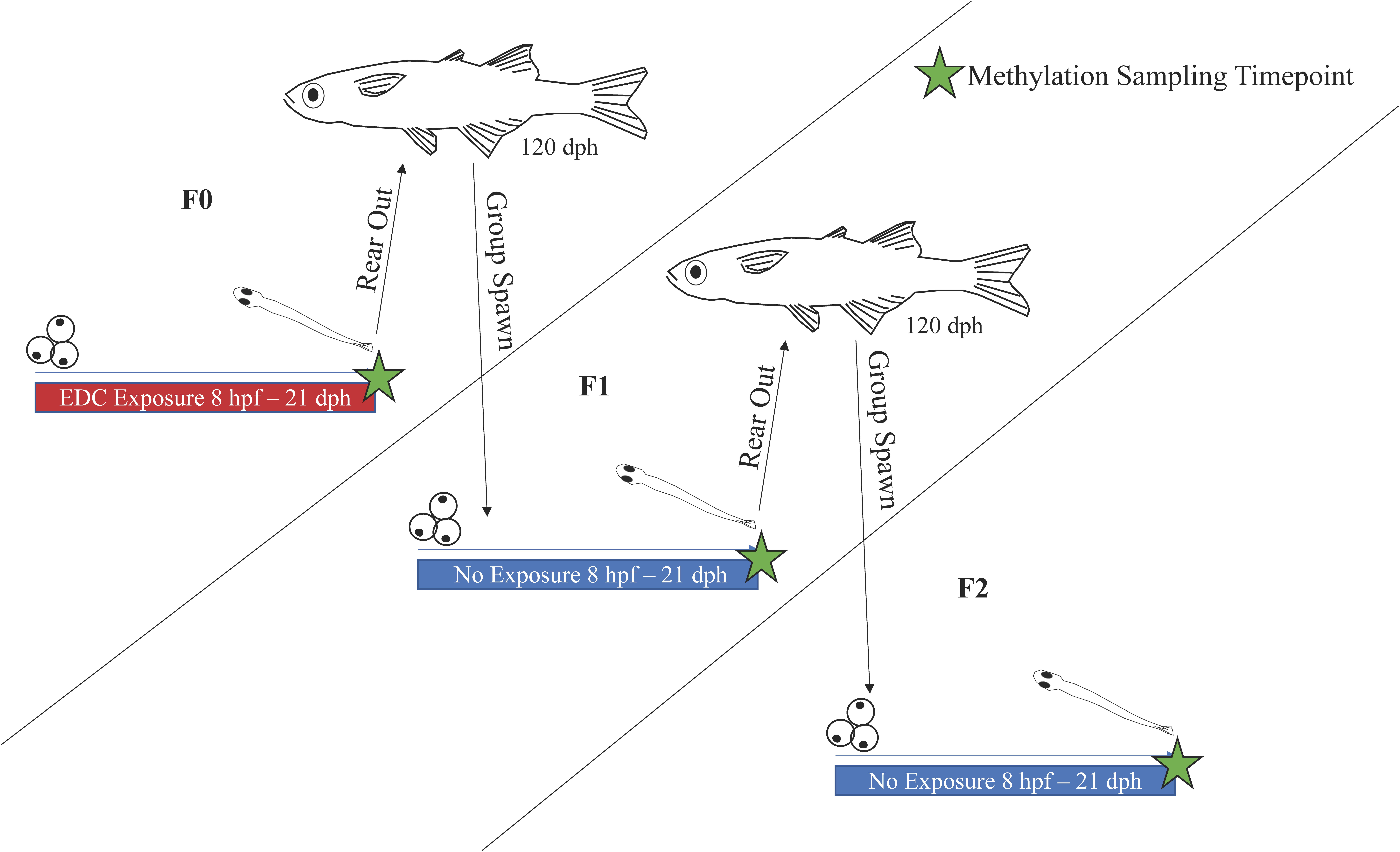
Figure 1. Multigenerational experimental design for M. beryllina exposed to one of four different EDCs (Bif, EE2, Levo, or Tren) in the parental generation (F0) from 8 hpf through 21 dph. At 21 dph, a subset of larval fish were sampled for molecular endpoints, while the rest were reared in clean water to approximately 120 dph, after which time they were group spawned (∼50 individuals per tank). Conditions and sampling timepoints remained the same for F1 and F2 generation fish but without chemical exposure during early life.
DNA Extraction, Genome Sequencing, RRBS
DNA from fish in the EDC exposure experiment was extracted from two whole 21-dph larvae from each replicate tank (n = 4–5) using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Samples were quantified using a Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) and purity was assessed via electrophoresis on a 1% agarose gel and visualized on a Gel DocTM XR + Gel Documentation system (Bio-Rad, Hercules, CA, United States). After DNA extraction, approximately 500 ng of genomic DNA was treated with 1 μL of RNAseIF (NEB, Ipswich, MA, United States) for 30 min at 37°C followed by a 0.8X AmPure XP (Beckman Coulter, Carlsbad, CA, United States) clean up.
Genomic DNA was adjusted to a concentration of 1.0 ng/μl, and 1.25 ng of template gDNA was loaded on a Chromium Genome Chip. Whole genome sequencing libraries were prepared using Chromium Genome Library & Gel Bead Kit v.2 (10X Genomics, cat. 120258), Chromium Genome Chip Kit v.2 (10X Genomics, cat. 120257), Chromium i7 Multiplex Kit (10X Genomics, cat. 120262) and Chromium controller according to manufacturer’s instructions with one modification. Briefly, gDNA was combined with Master Mix, a library of Genome Gel Beads, and partitioning oil to create Gel Bead-in-Emulsions (GEMs) on a Chromium Genome Chip. The GEMs were isothermally amplified with primers containing an Illumina Read 1 sequencing primer, a unique 16-bp 10x bar-code and a 6-bp random primer sequence, and bar-coded DNA fragments were recovered for Illumina library construction. The amount and fragment size of post-GEM DNA was quantified prior using a Bioanalyzer 2100 with an Agilent High sensitivity DNA kit (Agilent, cat. 5067-4626). Prior to Illumina library construction, the GEM amplification product was sheared on an E220 Focused Ultrasonicator (Covaris, Woburn, MA, United States) to approximately 350 bp (55 s at peak power = 175, duty factor = 10, and cycle/burst = 200). Then, the sheared GEMs were converted to a sequencing library following the 10X standard operating procedure. The library was quantified by qPCR with a Kapa Library Quant kit (Kapa Biosystems-Roche) and sequenced on one lane of HiSeq4000 sequencer (Illumina, San Diego, CA, United States) with paired-end 150 bp reads. Using 420M sequencing reads from the genomic library, the Menidia genome was assembled using supernova (version 2.0.0) with default parameters, and annotated using GMAP with default parameters1 and with the M. beryllina transcriptome (14,393 genes: Supplementary File “transcript_gene_list.txt”) established by Jeffries et al. (2015).
Reduced representation bisulfite sequencing (RRBS) libraries were generated using the Premium RRBS Kit from Diagenode (Liege, Belgium) following the manufacturer’s instructions. Fragment size distribution of resulting library pools was assessed via micro-capillary gel electrophoresis on a Bioanalyzer 2100 (Agilent, Santa Clara, CA, United States). The library pools were quantified by qPCR with a Kapa Library Quant kit (Kapa Biosystems/Roche, Basel Switzerland) and each pool was randomized and sequenced on six lanes of an Illumina HiSeq 4000 (Illumina, San Diego, CA, United States) run with single-end 100 bp reads.
Differential Gene Methylation and Functional Enrichment Analyses
Illumina reads were subjected to quality control using trim_galore2 (version 0.4.5) under RRBS mode. Bases with quality higher than 30 were trimmed from the 3′ end of the reads first, followed by the removal of any adapter sequences from the reads. Reads less than 30 bp in length after trimming were discarded. Bismark (Krueger and Andrews, 2011) (version 0.19.0) with default parameters was used to map all reads that passed the quality control to the M. beryllina reference genome that was assembled using 10X linked read technology (not published). Methylated regions were extracted from the alignment using DMRfinder (version 0.3) (Gaspar and Hart, 2017) using default parameters (maximum 500 bp in length with no less than 3 CpG sites within). Differential methylation in each methylated region was analyzed using a binomial test. Multiple test correction was carried out using Benjamini–Hochberg procedure (Benjamini and Hochberg, 1995) which has been applied as an appropriate method for minimizing false positives in methylation datasets (Asomaning and Archer, 2012). We analyzed differential methylation by treatment (Bif, EE2, Levo, Tren) and generation (F0, F1, F2) relative to the control for that generation, and differential methylation was considered significant relative to the control if the Benjamini–Hochberg adjusted p-value was less than 0.05 (Supplementary Data Analysis). The genomic context of methylated regions with respect to genes of interest was generated by annotating the assembled genome using previously published transcriptomic data (Jeffries et al., 2015; Brander et al., 2016) using gmap (Wu and Watanabe, 2005). The relationship between a methylated region and a gene was generated by overlapping the methylated region with any defined genomic regions. We will focus our discussion on two different regions with respect to genes: (1) Upstream 1000 bp, which we considered generally representative of the gene promoter region and will be referred to as “promoter” for brevity, and (2) within gene body, which comprised the exonic gene region. While the true location of the promoter region varies by individual gene, others have also designated the promoter as 1000 bp upstream of the TSS (Brenet et al., 2011; Wan et al., 2016). Further, in a survey of eukaryotic genomes, vertebrate promoters were frequently identified in less than 1000 bp upstream of the corresponding gene (Yella et al., 2018).
To elucidate the effects of EDCs on differential methylation across generations, we performed three different types of analyses. First, we tracked differential methylation by treatment and generation at the level of the gene, to determine a snapshot of differentially methylated genes overall (promoter and gene body combined) and by region (promoter and gene body analyzed separately).
Next, we performed functional enrichment analyses to determine whether gene ontology (GO) terms or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were enriched in the treatment groups based on differential methylation. Genes of interest for each functional enrichment analysis were chosen based on a significant directional change of methylation (hyper, hypo) as well as gene region (promoter versus gene body) relative to controls. GO Term enrichment analyses for biological processes was performed using the R package topGO (version 2.37) (Alexa and Rahnenfuhrer, 2019). GO Term enrichment analyses for cellular component and molecular function were not included in the present work because we chose to focus on the biological processes that were affected by changes in methylation. KEGG Pathways Enrichment analyses were obtained using the R package KEGGREST (version 1.26.1) (Tenenbaum, 2020), based on the D. rerio pathways database from the R package org.Dr.eg.db (Carlson, 2019). D. rerio Ensembl ID’s were converted to KEGG IDs using the bioDBnet database3 (Mudunuri et al., 2009). Enriched GO terms or KEGG pathways were considered significant at the level of α = 0.01 (Fisher’s exact test) (Supplementary Data Analysis).
Finally, we focused on the differential methylation by gene region for a suite of 109 genes that have either been responsive to EDC exposure or those that have the potential to be responsive based on previous work and phenotypes associated with EDC disruption in M. beryllina (Jeffries et al., 2015; Frank et al., 2018; DeCourten et al., 2019a). These genes included hormone receptors, genes essential to osmoregulation and immune function, steroid metabolism, DNA methylation, oxidative stress (Supplementary Table S1).
Results and Discussion
Through RRBS analysis, we found evidence of differential methylation for all four EDC treatments and all generations, indicating direct (F0), multigenerational (F1) and transgenerational (F2) effects of Bif, EE2, Levo, and Tren. Given the abundance of differential methylation and functional enrichment we identified, we focus on the most prominent trends in those data overall, including evidence of both transgenerational epigenetic dysregulation as well as strict epigenetic inheritance.
Genome and RRBS Data Quality
The genome assembly included 52,786 scaffolds that sums up to 568,309,548 base pairs (bp) (Supplementary Figure S2). The N50 of the assembly was 4,365,106 bp, in 27 scaffolds. The completeness of the assembly in gene space was assessed using BUSCO (version 3.0.2) with the vertebrate database. Out of 2,586 core BUSCO genes, 82.9% were found to be complete, with 2,023 genes in single copy and 121 genes in duplicated copies. There were 224 fragmented BUSCO genes, and 218 BUSCO genes were missing from the assembly.
Analysis of RRBS data yielded the number of CpG sites between 1.7 million and 2.3 million in all samples. These CpG sites belonged to 315,410 methylated regions. The coverage information for each sample is summarized as a box plot (Supplementary Figure S3). The methylated regions that did not have more than 10 reads covered from at least 146 samples were removed before the differential methylation analysis. This filtering process reduced the number of methylated regions to 66,983.
Overall Trends in Differential Methylation
All treatments (Bif, EE2, Levo, Tren) and generations (F0, F1, F2) showed evidence of differential methylation relative to the control in the analysis of all genes (14,393 genes informed by the M. beryllina transcriptome). The percentage of genes that were differentially methylated in their promoter and/or gene body ranged from 6% (Levo F0) to 11% (Bif F2) of all annotated genes (Figure 2). Differential methylation at the gene level was nominally higher in the F1and F2 generation animals (8–11% of all annotated genes) of each treatment compared with F0 animals (6–8% of all annotated genes) (Figure 2), suggesting a possible increase in multigenerational methylation effects (F1) and transgenerational methylation effects (F2) compared to direct parental effects (F0) caused by all four EDCs. A similar trend has been noted for other EDCs in transgenerational experiments. For an exposure occurring in the parental generation, oviparous fishes, including M. beryllina and zebrafish, the F2 generation would be the first generation to display transgenerational effect, compared the F3 generation in viviparous animals (Brander et al., 2017). In one study examining transgenerational epigenetic inheritance in gestating female rats exposed to the agricultural fungicide vinclozin, differential DNA methylation was compared between F3 and F1 generation sperm. Beck et al. (2017) found significantly more DMRs in F3 compared to F1 sperm, with no shared overlap between those DMRs, indicating distinct transgenerational epigenetic methylation patterns. The authors attributed the distinct methylation patterns between generations to the sensitive developmental period of direct exposure in F1s given that these animals were exposed during a period of PGC deprogramming and subsequent reprogramming. Another study on methylmercury-exposed zebrafish also showed more epimutations in F2 (transgenerational effect) compared with embryonically exposed F0 sperm, and most DMRs were unique between the two generations (Carvan et al., 2017). Thus, it seems that transgenerational epigenetic effects of EDCs can be highly variable, and dependent on epigenetic disruption during critical developmental stages. If epigenetic machinery (methylation, histones, ncRNAs, etc.) are disrupted during critical developmental windows in early generations, these perturbations appear to display an exacerbation of epigenetic changes in subsequent generations. Although the pattern of epigenetic erasure and reprogramming post fertilization and PGC differentiation that occurs in medaka (same phylogenetic group – Atherinomorpha – as Menidia) but not zebrafish (Jiang et al., 2013; Potok et al., 2013; Ortega-Recalde et al., 2019; Wang and Bhandari, 2019) has not been established in M. beryllina, evidence of transgenerational epigenetic effects as a result of dysregulation of epigenetic programming appears to be evident in both models (medaka, which shares the pattern with mammals and zebrafish, which does not) as well as in M. beryllina.
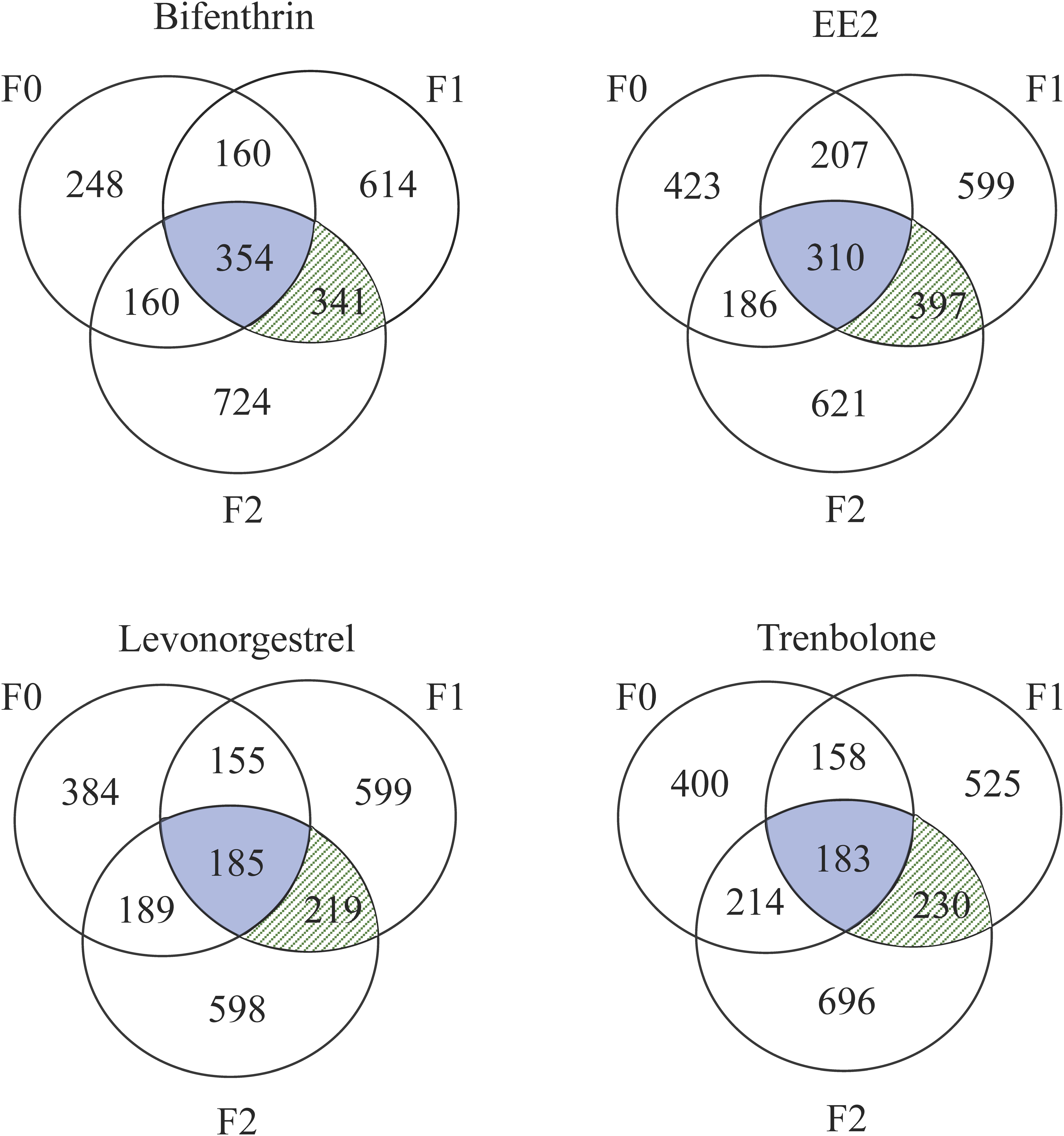
Figure 2. Venn diagrams showing the number of genes (promoter and gene body regions combined) displaying differential methylation relative to control by treatment and generation as informed by the M. beryllina transcriptome. F0 animals were treated with an EDC (Bif, EE2, Levo, or Tren) during early development. F1 animals were exposed to these treatments indirectly as primordial germ cells within the F0 animals, and F2 animals were not exposed at all. The differential methylation in F1 animals is indicative of a multigenerational effect of EDCs, while the differential methylation in the F2 generation indicates a transgenerational effect. Differentially methylated genes shared by F1 and F2 (shaded green stripes), sometimes originating in F0 generations (shaded blue), is indicative of potential transgenerational epigenetic inheritance.
Aside from the evidence of epigenetic dysregulation driving differential methylation among larval M. beryllina, a subset of differential methylation effects (3–5% of all annotated genes, depending on treatment) were shared between F1 and F2 generations, sometimes originating in the F0 generation (Figure 2), and can be considered as candidates for evidence of transgenerational epigenetic inheritance in the traditional, strict sense. For example, Rissman and Adli (2014) discuss strict transgenerational epigenetic inheritance as comparable to imprinting, when a specific epimutation is established in the germ line and maintained in subsequent generations. The directional and positional methylation changes at the gene level among EDC treated M beryllina would need to be individually verified to confirm this phenomenon. Some genes (∼1% for each treatment) were differentially methylated in the F0 and F2 generations, but not in the F1, thus, the potential for those methylation changes to be directly inherited is unclear. Specific evidence for transgenerational epigenetic inheritance in the strict sense is discussed further below (see Differential Methylation in EDC Responsive Genes).
When distinguishing between gene regions (promoter, gene body), differential methylation was more frequent within the gene body compared with the promoter region for all treatments and generations (Figure 3). Despite our use of RRBS to detect differential methylation, which operationally favors promoter regions (Yong et al., 2016), we were still able to detect more gene body differential methylation than promoter methylation. CpG methylation in the promoter region has been correlated with decreased gene expression in fish (Wan et al., 2016). Methylation in the gene body, however, is not correlated with gene repression and has the potential to affect splicing (Laurent et al., 2010) and sometimes lead to increased gene expression (Jones, 2012). It is possible that our definition of the promoter region as only 1 kb upstream was too narrow to capture many of the CpG islands in functional promoters. Gene promoters vary in their proximity relative to TSS by individual gene, so any nominal cutoff for promoters will not accurately represent the functional promoter for any given gene. Further, gene body sizes are typically larger than promoters, which would favor gene body differential methylation detection in our study. Still, we captured a great amount of gene body methylation given our use of RRBS methods. Most differentially methylated genes showed clear evidence of either hypo- or hypermethylation relative to the control, but a minority (<6%) showed evidence of both hypo and hyper methylation in different loci within the same gene region (Figure 3), indicating that directional methylation changes were locally specific.
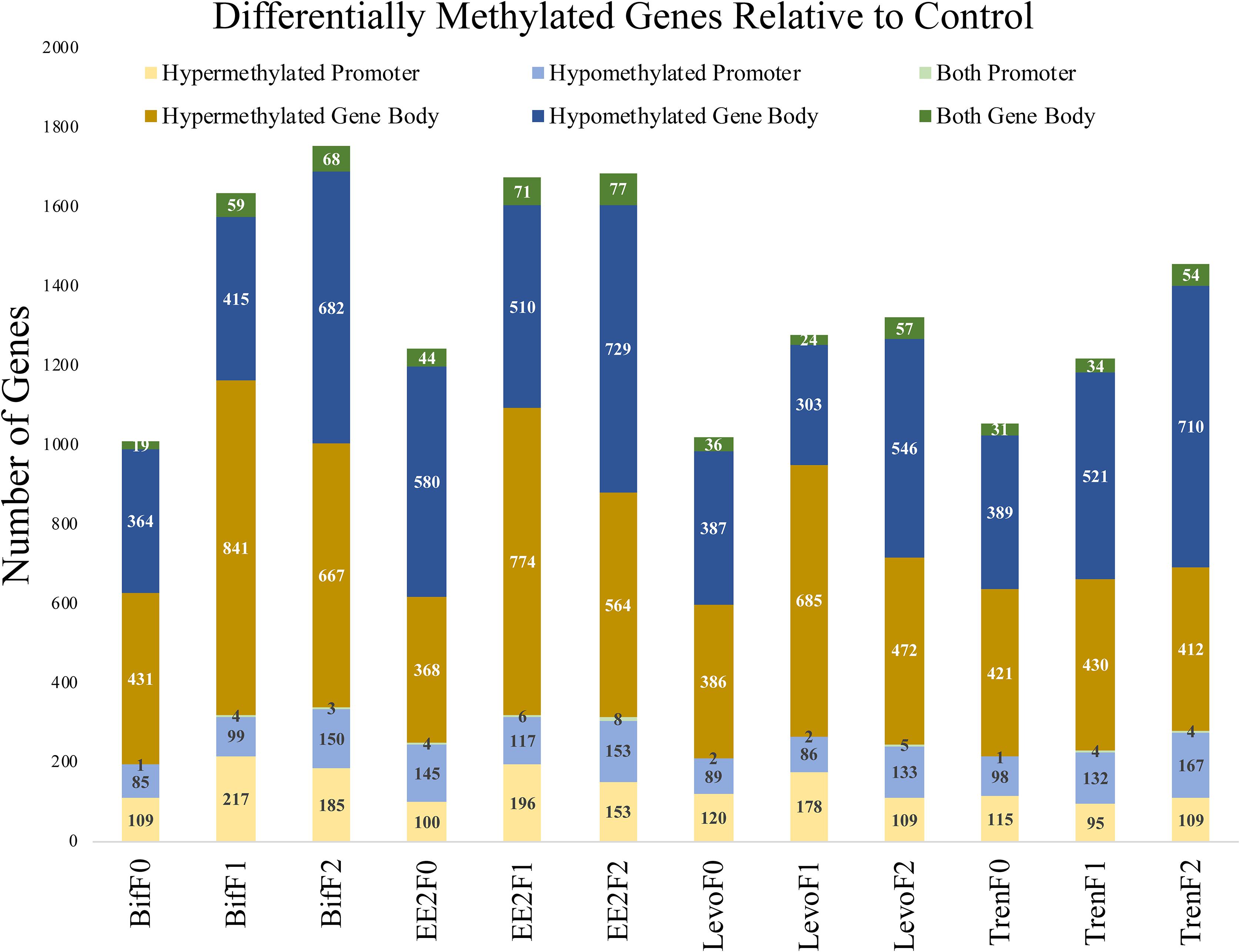
Figure 3. Bar graph depicting the differential methylation relative to control by treatment (Bif, EE2, Levo, Tren), generation (F0, F1, F2), directional methylation change (hyper, hypo) and region (promoter, gene body) for all genes as informed by the M. beryllina transcriptome. Differential methylation was more prominent in the gene body than the promoter regions for all treatments and generations. While most genes only showed a singular directional change in methylation by region relative to the control, a minority showed both hypo and hyper methylation at different loci in the same gene.
GO Term and KEGG Pathway Enrichment Analyses
GO Term and KEGG pathway enrichment analyses use a standardized classification system and vocabulary to place genes into groups, making it easier to understand the overall functional implications of genes that, in this case, are differentially methylated more than would be expected from a random selection of genes. Our GO Term analysis focused on biological processes (and not molecular function or cellular component) while KEGG pathway analysis groups genes by their inclusion in a given pathway (Nguyen et al., 2019; The Gene Ontology Consortium, 2019). Functional analyses of GO term (biological process) enrichment based on differentially methylated gene regions yielded significant enrichment (α = 0.01) for a total of 144 GO terms when considering all treatments, generations, directional methylation, gene regions, while KEGG pathway analysis yielded 66 significantly enriched pathways (Supplementary Tables S4, S5). Differential gene body methylation produced a greater number of significantly enriched GO terms and KEGG pathways than did differential promoter methylation. As with overall differential methylation analyses, this skew may be related to the limited promoter window screened in the present study and the greater length in the gene body compared to the promoter (see above). Enrichment of the same term or pathway sometimes occurred across multiple treatments and generations (Table 1 and Figures 4, 5). It is important to note, however, that our use of a non-model organism with limited annotation for GO and KEGG enrichment analyses requires cautious interpretation, as enrichment results may be inflated by limited annotation information. Thus, we focus our discussion on enrichment of generalized categories that contain many enriched terms or those in which the number of differentially methylated annotated genes was high compared to the number of background genes expected in those gene ontology groups or pathways.

Table 1. Most frequently detected functional analysis enrichments considering gene region (promoter and gene body) directional differential methylation relative to the control (hyper, hypo), treatment (Bif, EE2, Levo, Tren), and generation (F0, F1, F2) for Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway codes.
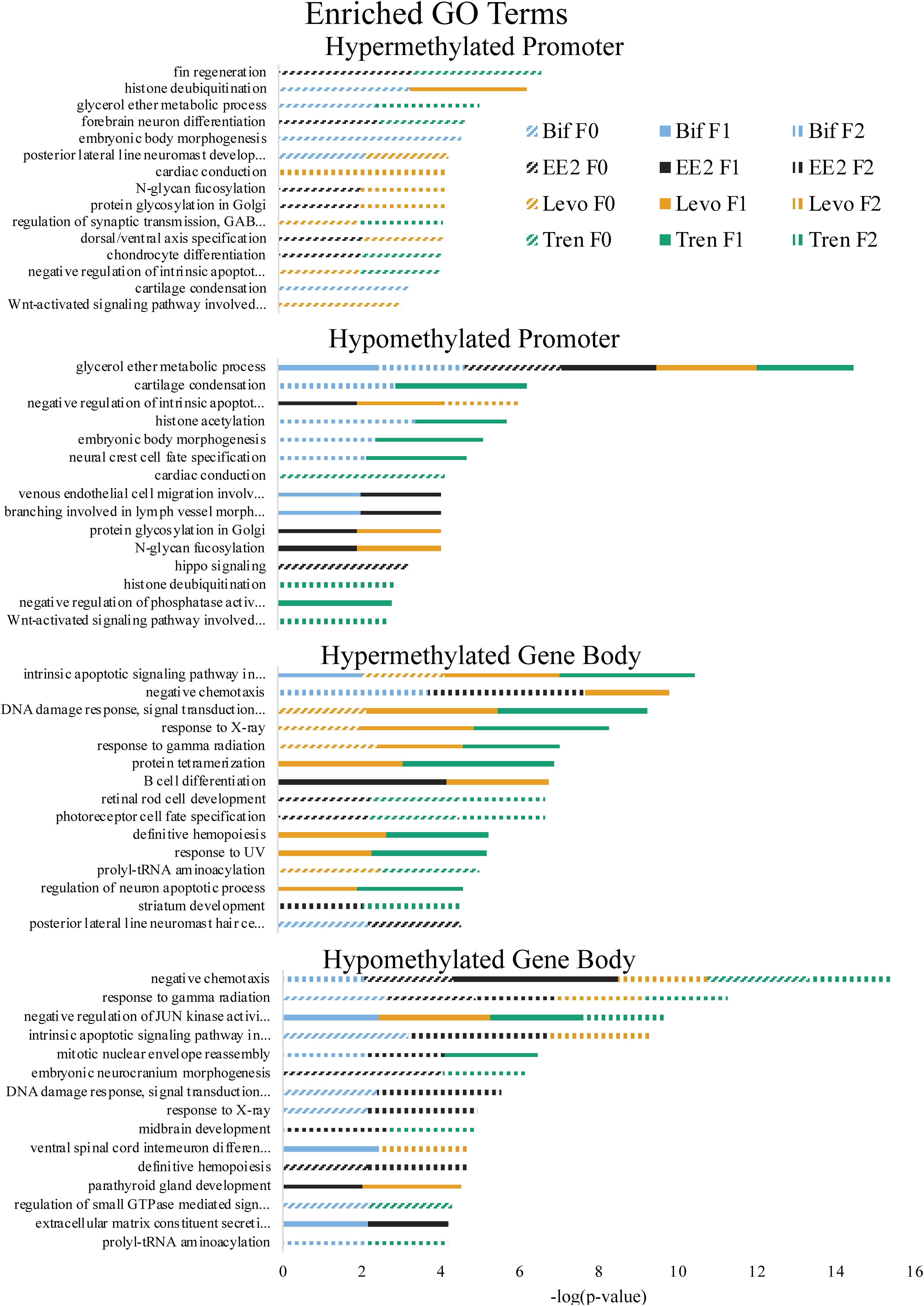
Figure 4. Enriched Gene Ontology (GO) Terms by directional methylation change relative to the control (hyper, hypo), gene region (promoter, gene body), treatment (Bif, EE2, Levo, Tren), and generation (F0, F1, F2) showing level of significance based on Fisher’s exact test p-values.
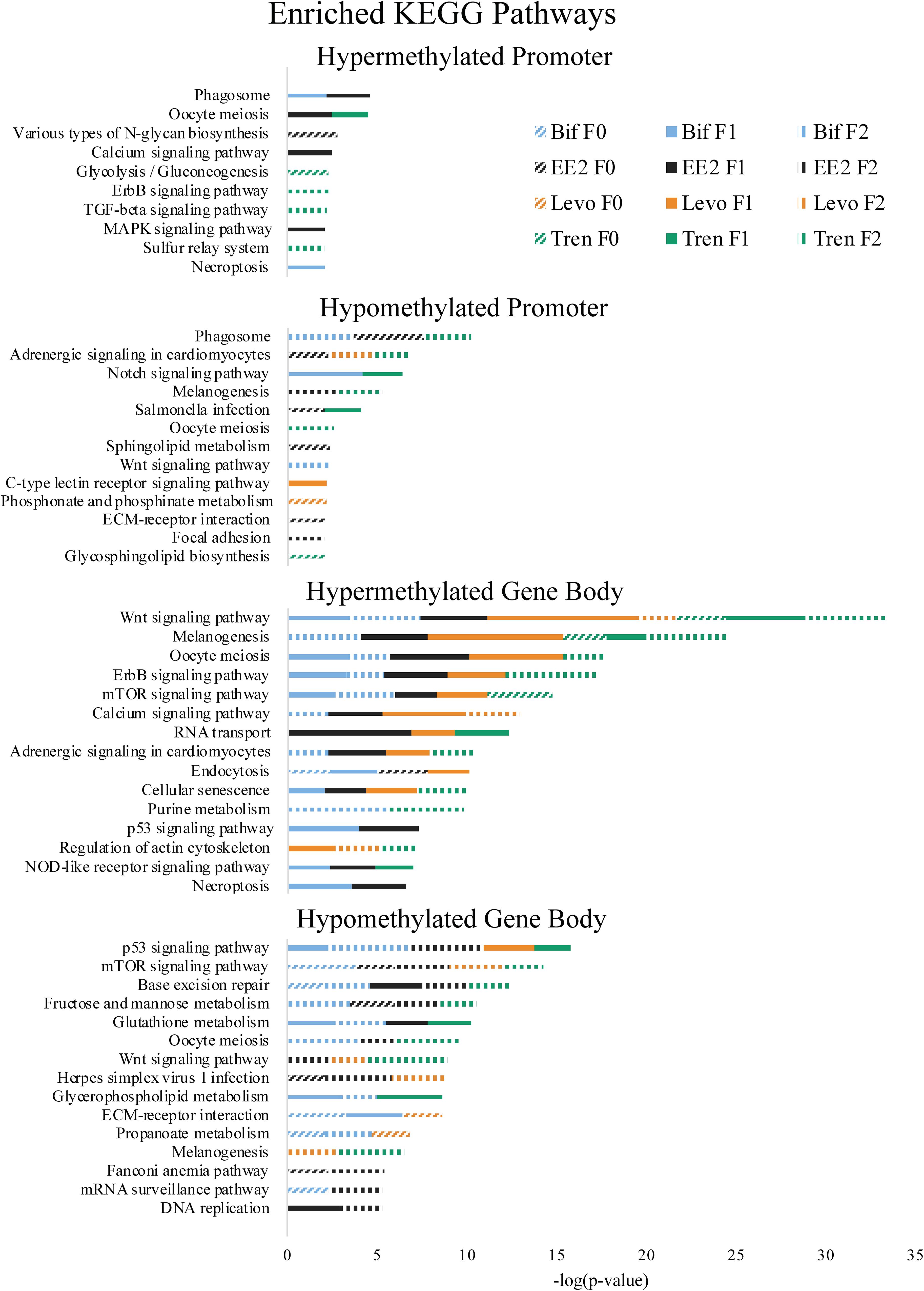
Figure 5. Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways by directional methylation change relative to the control (hyper, hypo), gene region (promoter, gene body), treatment (Bif, EE2, Levo, Tren), and generation (F0, F1, F2) showing level of significance based on Fishers exact test p-values.
As part of our larger study, DeCourten et al. (unpublished) found craniofacial and/or skeletal deformities in at least one generation of all four EDC treatments. Our analysis of DNA methylation of enrichment of GO term and KEGG pathways produced changes in methylation for chondrocyte differentiation, cartilage condensation, embryonic morphogenesis, Wnt signaling (which is essential to embryonic development; Steinhart and Angers, 2018), and regulation of actin cytoskeleton (Figures 4, 5), overlapping with all treatments and generations for which craniofacial and/or skeletal deformity phenotypes were measured (Bif F0, EE2 F1, Levo F2, Tren F0 and F1) (DeCourten et al., unpublished). Wnt signaling is essential for embryonic development. Other studies have documented phenotypic growth and development effects caused by EDC exposure linked to changes in DNA methylation. For example, prenatal exposure of the EDC cadmium led to an overrepresentation of DNA methylation in genes that were essential for organ and morphological development and bone mineralization, although only in females (Kippler et al., 2013). Further, DNA methylation has been associated with craniofacial abnormalities (Alvizi et al., 2017). We also found altered cardiovascular deformities in Bif F0, EE2 F1, and Tren F0 animals, some of the same treatments for which we have identified corresponding DNA methylation enrichment in GO terms and KEGG pathways including cardiac conduction (TrenF0) and adrenergic signaling of cardiomyocytes (Bif F2, EE2 F1, Levo F1, and Tren F2 (Figures 4, 5), suggesting that altered methylation of cardiac genes and pathways may be linked to the physical deformities caused by EDC exposure. While we did not find perfect correspondence between the treatments/generations with cardiac deformities and the altered DNA methylation GO terms and KEGG pathways, the overlap suggests that DNA methylation may play a role in altered cardiac developmental phenotypes. Although information on the relationship between EDC exposure, cardiac phenotypes, and DNA methylation is lacking, early life exposure to bifenthrin and EE2 has been linked to altered cardiac phenotypes (Jin et al., 2009; Salla et al., 2016). The overlap between the physiological whole organism endpoints resulting from EDC exposure and the functional enrichment of related GO terms and KEGG pathways in our differential methylation analysis suggest that some of the multi- and transgenerational effects observed in DeCourten et al. (unpublished) may be the result of EDC altered epigenetic control, although further work would be needed to mechanistically confirm the connection.
We found evidence of a variety of methylation enriched GO terms and KEGG pathways that are critical to neurodevelopment (e.g., wnt-activated signaling pathway involved in forebrain neuron fate commitment, neural crest cell fate specification, embryonic neurocranium morphogenesis, mTOR signaling pathway, calcium signaling pathway; Figures 4, 5). One study (Frank et al., 2019) found that early life exposure to low (picomolar) concentrations of bifenthrin altered gene expression in calcium signaling pathways, and led to a latent olfactory predator avoidance cue behavioral phenotype in M. beryllina. The authors concluded that early-life effects of bifenthrin on neurodevelopment had effects later in life. Negative chemotaxis, or the movement away from chemicals, was the most frequently enriched GO term with differential methylation within the gene body across all treatments, affecting multiple generations within each treatment (Supplementary Table S1). It is possible that the alteration of methylation of genes involved in negative chemotaxis is yet another way in which EDCs have impacted neurodevelopmental processes. Others have documented other non-reproductive behavioral effects of estrogens and androgens in fish. Lagesson et al. (2019) exposed eastern mosquitofish (Gambusia holbrooki) to environmentally relevant levels (3 ng/L) of trenbolone at one of two temperature regimes for 21 days and found that trenbolone increased fish boldness behavior and altered predator escape behavior (but only at the higher temperature). Three-spined sticklebacks (Gasterosteus aculeatus) exposed to EE2 during early development and assayed for behavioral endpoints 8 months after exposures showed a reduction in anxiety behavior related to scototaxis (light/dark preference) (Porseryd et al., 2019). Zebrafish exposed to BPA (0.1 nM to 30 μM) as embryos showed evidence of hyperactivity in mid-range concentrations and altered transcription of genes involved in methylation (dnmt1 and cbs). The authors also found differential DNA methylation in many regions of the genome, primarily within gene bodies, and particularly in genes involved in neurodevelopment, suggesting the potential for differential DNA methylation of neurodevelopmental genes to affect observed differences in swimming behavior (Olsvik et al., 2019).
A number of GO terms and KEGG pathways involved in the regulation of the p53 tumor suppressor gene, generalized response to DNA damage, and regulation of the Wnt signaling pathway were enriched for at least one generation in each treatment. Indeed, Brander et al. (2016) showed that pathways involved in carcinogenesis were differentially expressed within M. beryllina exposed to bifenthrin. Further, trenbolone (Boettcher et al., 2011) and binary mixtures of tributyltin and EE2 (Micael et al., 2007) are known genotoxicants in fish. EDCs also play a role in dysregulating DNA methylation for genes important in carcinogenesis (Serman et al., 2014), a finding also supported elsewhere (Mirbahai et al., 2011). One study in mammals showed that most Wnt pathway gene bodies (and not promoters) were differentially methylated in cancer cells (Galamb et al., 2016). We found that Wnt pathway differential methylation was prominent in the gene bodies for all treatments, further strengthening the idea that EDCs may alter methylation in genes and pathways relevant to carcinogenesis. Gene body methylation of TP53 (which encodes p53) in somatic cells contributes to many types of cancer (Rideout et al., 1990; Jones, 2012). We found differential methylation in the gene bodies p53 pathways for all four EDCs. Exposure to EDCs has been widely linked with cancer phenotypes (Soto and Sonnenschein, 2010; Rachon, 2015; Karoutsou et al., 2017), and those cancer phenotypes linked to epigenetic changes (Bhandari, 2016), providing support that the differential methylation caused by the four EDCs in the present study may play a functional role in carcinogenesis.
Altered DNA methylation in some biological functions may explain the transgenerational transfer of altered methylation patterns. The enriched GO terms related to epigenetic control mechanisms (histone deubiquination, histone acetylation) occurred in F1 or F2 animals for three out of four EDC treatments, suggesting that genes important in epigenetic programming have been dysregulated in these treatments due to early life exposure during sensitive windows, thus potentially perpetuating EDC-induced epigenetic dysregulation in subsequent generations as in Beck et al. (2017) and Carvan et al. (2017).
Another mechanism potentially responsible for the differential DNA methylation that we observed in larval M. beryllina may be an EDC-induced change in metabolism, particularly glutathione metabolism. Glutathione conjugation is a common detoxification mechanism for estrogens (Zhu and Conney, 1998) and has been established as a primary pathway involved in bifenthrin (Brander et al., 2016), trenbolone (Evrard and Maghuin-Rogister, 1987), and levonorgestrel metabolism (Stanczyk and Roy, 1990). Adult M. beryllina exposed to 0.5, 5, and 50 ng L–1 of bifenthrin showed a great deal of differential gene expression for metabolic processes (Brander et al., 2016), in line with our observed GO term and KEGG enrichment for differentially methylated metabolic processes and pathways. We found hypomethylated glutathione metabolism to be enriched in gene bodies for three of four EDC treatments (Bif, EE2, and Tren), which potentially affected glutathione metabolism. Evidence that metabolic alterations can contribute to epigenetic dysregulation is building. For example, more resources diverted to glutathione metabolism may compete with the resources needed by epigenetic machinery to methylate DNA and histones, thereby leading to a generalized hypomethylation (Lee et al., 2009; Oppold and Müller, 2017; Sharma and Rando, 2017). While we found both hyper- and hypomethylation as a result of EDC exposure, it is possible that some of the hypomethylation we observed was produced as a result of the demands of glutathione metabolism. Thus, the general glutathione pathway utilized to detoxify EDCs could be contributing to the differential methylation observed throughout the genome, and/or the differential methylation specific to the glutathione metabolism pathway may also be affecting the balance of methylation/demethylation in other parts of the genome.
Differential Methylation in Potentially EDC-Responsive Genes
The analysis of differential methylation in 109 potentially EDC-responsive genes (EDCRG) yielded 29 genes that were differentially methylated in at least one treatment/generation within their promoter and/or gene body regions. All treatments, generations, and gene regions (promoter, gene body) showed some differentially methylated EDCRGs, with the exception of the Bif F2 promoter region, for which no EDCRG differential methylation was noted (Supplementary Figures S6, S7). While a total of 18 EDCRG’s were differentially methylated in the promoter and/or gene body across all F0 treatments, 20 EDCRG showed a multigenerational (F1) differential methylation effect while 22 showed a transgenerational (F2) effect depending on treatment and generation (Supplementary Figures S6, S7).
Differentially methylated EDCRGs included those involved in calcium signaling (mtor, ryr1, calr, tgfb), osmoregulation (atnb233, atp1a1, atp1a1b, atp1a2), steroidogenesis (17ß-hsd, 3ß -hsd), hormone receptors (ar, esr2, esr3, gpr30), immune function and/or inflammation (cbr1, inha, stat1, tgfb), structure (krt13, pdlm1), DNA methylation (dnmt3a), metabolism (alas2, cyp1a1, smpd3), sex determination (dmrt1), protein translation (noa1), proteolysis and degradation (htra3, usp42), zinc transport (slc39a14), and more general signaling pathway regulation (ywhab) (Supplementary Figures S6, S7). By frequency, the genes with the most alterations to their methylation patterns across all treatments and generations were dmrt1, ar, 17ß-hsd, atp1a1, atnb233, atp1a1b, ywhab, gpr30, cbr1, mtor, atp1a2, htra3 (Supplementary Table S8). The differential methylation we observed in these genes showed concordance with the generalized patterns of altered methylation enrichment in GO term and KEGG pathway analyses. Many of those genes showed differential methylation across generations within the same treatment, primarily within the gene body. Differential methylation occurring across generations was sometimes consistent (e.g., hypomethylation in the promoter of dmrt1 for Tren F0 through F2 generations), although directional methylation often alternated by generation (e.g., hypomethylation of atp1a1 within the gene body in EE2 F0 and F2, but hypermethylation of the same gene in F1), with hypomethylation in the gene body prominent in generations F0 and F2, but hypermethylation often prominent in the F1 generation (Figure 6). As with the overall analysis of all genes, a subset of EDCRGs showed patterns of both hyper- and hypomethylation, within the same greater region (promoter, gene body), although a singular directional differential methylation was the most common.
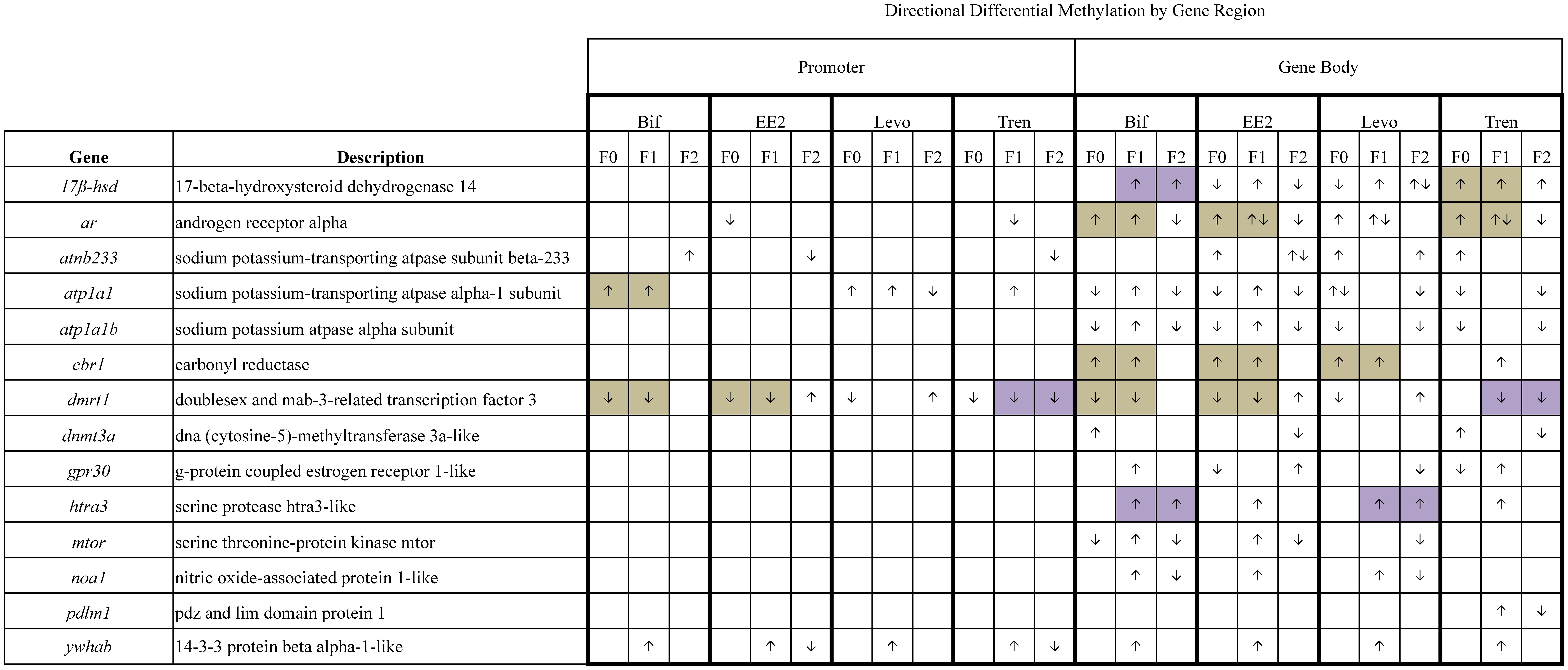
Figure 6. Potential endocrine disrupting compound-responsive genes (EDCRGs) with differential methylation across more than one generation within the same treatment (Bif, EE2, Levo, Tren) relative to the control in Supplementary Table S1. Arrows indicate the change in methylation relative to the control (↑ is hypermethylation; ↓ is hypomethylation) in the gene region indicated (promoter or gene body). In instances for which both hyper- and hypo-methylation were noted for different loci in the same gene region, both arrows are indicated (↑↓). A change in methylation in F1 animals indicates a multigenerational effect, while a change in methylation in F2 animals indicates a transgenerational effect of EDC exposure. Methylation changes are shaded in tan the same locus that was differentially methylated across the F0 and F1 generations, indicating identical effects occurring from exposure in both generations or potentially a multigenerational inheritance pattern. Purple shading indicates the maintenance of specific methylation direction and loci from an exposed generation (F1) to an unexposed generation (F2) in support of transgenerational epigenetic inheritance.
The majority of differential gene methylation documented for EDCRGs did not occur at the same sites within a given gene across the generations. Or, if they did, the direction of methylation change relative to the control often alternated (Supplementary Table S8), potentially indicating the interplay of epigenetic feedback loops (e.g., Bonasio et al., 2010). However, the same directional methylation changes (hypo, hyper) at the same loci were sometimes noted in F0 and F1 generations of the same treatment (17ß-hsd in Tren; ar in Bif, EE2, and Levo; atp1a1 in Bif; cbr1 in Bif, EE2, and Levo; dmrt1 in Bif and EE2) indicating identical effects occurring from exposure in both generations or potentially a multigenerational inheritance pattern. Although this phenomenon was less prevalent in F1 and F2 generations of the same treatment (17ß-hsd in Bif; dmrt1 in Tren; htra3 in Bif and Levo), the maintenance of specific methylation from an exposed generation (F1) to an unexposed generation (F2) indicates a strict form of transgenerational epigenetic inheritance (Figure 6). These examples of transgenerational epigenetic inheritance provide evidence of germline-established differential methylation, in contrast to the evidence of epigenetic programming dysregulation that we also found.
Doublesex And Mab-3 Related Transcription Factor 1 (dmrt1) was one of the genes that showed evidence of transgenerational epigenetic inheritance and was the most frequently differentially methylated gene across all treatments and generations. Dmrt1 is an important gene in sexual determination across many species, including those that exhibit temperature dependent sex determination, as Menidia species do (DeCourten et al., 2017; Huang et al., 2017) and in the closely related Japanese medaka (Otake et al., 2008). In the marine half-smooth tongue sole (Cynoglossus semilaevis), the level of dmrt1 demethylation was responsible for male gonad development (Shao et al., 2014). It is possible that the skewed sex ratios that are common in EDC exposed populations (Jobling et al., 1998; Kidd et al., 2007; Hua et al., 2015; Orn et al., 2016; DeCourten et al., 2017) may be the result of epigenetic reprogramming at the dmrt1 locus, as evidenced from the differential methylation caused by both estrogenic and androgenic EDCs in the present study.
We found differential methylation in genes involved in calcium signaling (calr, ryr1, mtor, tgfb) depending on treatment and generation, although all four EDCs yielded at least one calcium signaling gene with differential methylation (Supplementary Figures S6, S7). Alterations in calcium signaling gene expression as a result of bifenthrin exposure in M. beryllina and zebrafish have been established (Frank et al., 2018, 2019), and in response to other EDCs as well (Brander, 2013). Early life exposures of EE2 in coho salmon (Oncorhynchus kisutch) led to changes in transcription for both tgfb and calcium signaling pathways (Harding et al., 2013). The functional significance of the altered methylation in calcium signaling genes requires further study, but our preliminary results suggest that DNA methylation may play a regulatory role in altered calcium signaling pathway function in EDC-exposed fish.
We found differential methylation in the gene body of dnmt3a, the DNA methyl transferase responsible for de novo methylation (Okano et al., 1999; Hermann et al., 2004), in three different treatments, thus providing a potential mechanism to help explain DNA methylation dysregulation in exposed animals, although more work would need to be performed to confirm the functional relevance of this differential methylation. Other EDCs (TCDD, DES, PCB153) have been shown to alter transcription of Dnmts, which may in turn have direct effects on methylation (Wu et al., 2006).
Hormone receptors and regulators of steroidogenesis play an important role in development and reproduction. A recent multigenerational study with M. beryllina showed that exposure to 1 ng/L EE2 produced gene expression (gpr30, 17ß-hsd) changes in directly exposed generations of fish. Exposure to 1 ng/L bifenthrin instead produced latent effects in indirectly exposed F1 generation fish (GPR30) (DeCourten et al., 2019a). We found evidence of differential methylation for gpr30 in EE2 F0 and F2 and Bif F1 animals, while 17ß-hsd was differentially methylated in nearly all treatments and generations, indicating that a change in methylation state for these genes may be related to gene expression differences observed previously (DeCourten et al., 2019a). We also documented frequent differential methylation of the ar, another hormone receptor, across most treatments and generations. Casati et al. (2013) found that ar expression was modulated by other EDCs (PCBs), and that the altered expression had a basis in epigenetic mechanisms. Changes in 17ß-hsd, gpr30, and ar methylation have been correlated with cancer (Bhavani et al., 2008; Tian et al., 2012; Manjegowda et al., 2017), thus there is the potential for differential methylation of these genes to have significant phenotypic effects during both development and later in life.
A limited analysis of differential gene methylation in larval M. beryllina from the same treatments as the present study showed little correlation between changes in gene expression and DNA methylation in a suite of twenty genes (measured at 21 dph). In fact, only one gene, 17ß-hsd, showed hypomethylation within the gene body and significantly decreased expression in EE2 F0 animals (DeCourten et al., unpublished). The lack of concordance between gene expression changes and methylation could indicate that the differential methylation we observed is not functionally relevant and does not, therefore, translate to altered gene expression. Or, the lack of correlation could be a result of the single time point at which gene expression was measured, with epigenetic modifications being more stable than gene expression responses to EDCs. It is also possible that our approach of tracking methylation in the whole body larval fish, rather than specific tissues or cell-types created a dilution effect, making it difficult to track and correlate gene expression and DNA methylation. Future studies involving specific cell types (i.e., gonad) may allow for more prominent differential methylation trends to be revealed on a more mechanistically relevant basis. Other studies have also documented a lack of correspondence between DNA methylation and gene expression, even with tracking tissue-specific methylation (Aluru et al., 2018; Ryu et al., 2018). Further work is needed to determine the functional relationship between EDC-altered gene expression and differential methylation, ideally with multiple timepoint measurements and tissue and/or cell-specific DNA methylation profiling. Still, our detection of differential DNA methylation in all treatments and generations relative to the control provides evidence that EDCs drive changes in methylation on a multi- and transgenerational scale – a phenomenon that has already been established in mouse and human models (Susiarjo et al., 2007).
Conclusion
Here, we show that early life exposure to environmentally relevant (low parts per trillion) concentrations of EDCs (Bif, EE2, Tren, or Levo) caused differential methylation of mechanistically relevant genes (in promoter and/or gene body regions) in directly exposed (F0), indirectly exposed (F1), and unexposed (F2) generations of fish. We show evidence of strict epigenetic transgenerational inheritance as well as more generalized epigenetic transgenerational effects that may be driven by dysregulation in epigenetic programming during sensitive windows of development. Our functional and gene level analyses are in accordance with one another as well as with the physiological endpoints measured in DeCourten et al. (unpublished) and elsewhere as a result of EDC exposure. We show that growth and developmental, epigenetic regulation, and carcinogenic pathways are affected by differential methylation, primarily in the gene body compared with the promoter region of genes. All of these data provide evidence that DNA methylation may be altered by EDC exposure in early life, and that more work should be performed to better understand the mechanisms behind epigenetic alteration from exposure to environmentally relevant levels of EDCs.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioSample Accession – SAMN14992539. All data are stored online at: https://datadryad.org/stash/dataset/doi:10.5061/dryad.4f4qrfj8h.
Ethics Statement
The animal study was reviewed and approved by the UNCW Institutional Animal Care and Use Committee under protocol number A1415-010.
Author Contributions
KM was the lead author and performed the RRBS data analysis downstream of DMR identification and statistical analysis. SB and BD designed the original study. BD performed the multi and transgenerational experiments with M. beryllina. SB secured funding, advised KM and BD, and facilitated the overall study at UNCW and OSU. AM performed the analytical chemistry to quantify EDC levels in exposure solutions. MB, JL, and MS performed the genome sequencing, assembly and annotation and/or RRBS processing, and primary data analysis. RC assisted with funding the study, advised on research, and served as a liaison between UNCW and the UC Davis genome center.
Funding
Funding for this work was provided by the U.S. Environmental Protection Agency, grant no. 835799 (to SB, associate investigators RC and AM) and no. 83950301 (to SB), the California Department of Fish and Wildlife, grant no. P1796002 (to SB and RC), and the Delta Stewardship Council contract no. 18206 (to SB and RC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank colleagues and staff at the University of North Carolina Wilmington and the associated Center for Marine Science for supporting the empirical work that made these analyses possible. We thank the UC Davis Genome Center for sequencing and analysis support and the Oregon State Center for Genome Research and Biocomputing for valuable advice on analyses. We also thank Josh Forbes, Hunter Roark, and many undergraduate researchers at UNCW, student researchers Jordan Laundry, Yvonne Rericha, and Lindsay Wilson at OSU, as well as Keith Maruya, Ellie Wenger, Wayne Lao (SCCWRP), and Shane Snyder (University of Arizona) for their contributions to the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00471/full#supplementary-material
Footnotes
- ^ https://github.com/juliangehring/GMAP-GSNAP/
- ^ http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- ^ https://biodbnet-abcc.ncifcrf.gov/db/db2db.php
References
Alavian-Ghavanini, A., and Ruegg, J. (2018). Understanding epigenetic effects of endocrine disrupting chemicals: from mechanisms to novel test methods. Basic Clin. Pharmacol. Toxicol. 122, 38–45. doi: 10.1111/bcpt.12878
Alexa, A., and Rahnenfuhrer, J. (2019). topGO: Enrichment Analysis for Gene Ontology. Available online at: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed September 11, 2019).
Aluru, N., Karchner, S. I., Krick, K. S., Zhu, W., and Liu, J. (2018). Role of DNA methylation in altered gene expression patterns in adult zebrafish (Danio rerio) exposed to 3, 3’, 4, 4’, 5-pentachlorobiphenyl (PCB 126). Environ. Epigenet. 4:dvy005.
Alvizi, L., Ke, X., Brito, L. A., Seselgyte, R., Moore, G. E., Stanier, P., et al. (2017). Differential methylation is associated with non-syndromic cleft lip and palate and contributes to penetrance effects. Sci. Rep. 7:2441.
Aniagu, S. O., Williams, T. D., Allen, Y., Katsiadaki, I., and Chipman, J. K. (2008). Global genomic methylation levels in the liver and gonads of the three-spine stickleback (Gasterosteus aculeatus) after exposure to hexabromocyclododecane and 17-beta oestradiol. Environ. Int. 34, 310–317. doi: 10.1016/j.envint.2007.03.009
Ankley, G. T., Bencic, D. C., Breen, M. S., Collette, T. W., Conolly, R. B., Denslow, N. D., et al. (2009). Endocrine disrupting chemicals in fish: developing exposure indicators and predictive models of effects based on mechanism of action. Aquat. Toxicol. 92, 168–178. doi: 10.1016/j.aquatox.2009.01.013
Asomaning, N., and Archer, K. J. (2012). High-throughput DNA methylation datasets for evaluating false discovery rate methodologies. Comput. Stat. Data Anal. 56, 1748–1756. doi: 10.1016/j.csda.2011.10.020
Bayen, S., Zhang, H., Desai, M. M., Ooi, S. K., and Kelly, B. C. (2013). Occurrence and distribution of pharmaceutically active and endocrine disrupting compounds in Singapore’s marine environment: influence of hydrodynamics and physical-chemical properties. Environ. Pollut. 182, 1–8. doi: 10.1016/j.envpol.2013.06.028
Beck, D., Sadler-Riggleman, I., and Skinner, M. K. (2017). Generational comparisons (F1 versus F3) of vinclozolin induced epigenetic transgenerational inheritance of sperm differential DNA methylation regions (epimutations) using MeDIP-Seq. Environ. Epigenet. 3:dvx016.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the flase discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bertram, M. G., Saaristo, M., Baumgartner, J. B., Johnstone, C. P., Allinson, M., Allinson, G., et al. (2015). Sex in troubled waters: widespread agricultural contaminant disrupts reproductive behaviour in fish. Horm. Behav. 70, 85–91. doi: 10.1016/j.yhbeh.2015.03.002
Best, C., Ikert, H., Kostyniuk, D. J., Craig, P. M., Navarro-Martin, L., Marandel, L., et al. (2018). Epigenetics in teleost fish: from molecular mechanisms to physiological phenotypes. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 224, 210–244. doi: 10.1016/j.cbpb.2018.01.006
Bhandari, R. K. (2016). Medaka as a model for studying environmentally induced epigenetic transgenerational inheritance of phenotypes. Environ. Epigenet. 2:dvv010. doi: 10.1093/eep/dvv010
Bhavani, V., Srinivasulu, M., Ahuja, Y. R., and Hasan, Q. (2008). Role of BRCA1, HSD17B1, and HSD17B2 methylation in breast cancer tissue. Cancer Biomark. 5, 207–213. doi: 10.3233/cbm-2009-0105
Boettcher, M., Kosmehl, T., and Braunbeck, T. (2011). Low-dose effects and biphasic effects profiles: Is trenbolone a genotoxicant? Mutat. Res. 732, 152–157. doi: 10.1016/j.mrgentox.2011.04.012
Bonasio, R., Tu, S., and Reinberg, D. (2010). Molecular signals of epigenetic states. Science 330, 612–616. doi: 10.1126/science.1191078
Brander, S. M. (2013). “Thinking outside the box: assessing endocrine disruption in aquatic life A2,” in Monitoring Water Quality: Pollution Assessment, Analysis, and Remediation, ed. S. Ahuja (Amsterdam: Elsevier), 103–147.
Brander, S. M., Biales, A. D., and Connon, R. E. (2017). The role of epigenomics in aquatic toxicology. Environ. Toxicol. Chem. 36, 2565–2573. doi: 10.1002/etc.3930
Brander, S. M., Cole, B. J., and Cherr, G. N. (2012). An approach to detecting estrogenic endocrine disruption via choriogenin expression in an estuarine model fish species. Ecotoxicology 21, 1272–1280. doi: 10.1007/s10646-012-0879-2
Brander, S. M., Jeffries, K. M., Cole, B. J., DeCourten, B. M., White, J. W., Hasenbein, S., et al. (2016). Transcriptomic changes underlie altered egg protein production and reduced fecundity in an estuarine model fish exposed to bifenthrin. Aquat. Toxicol. 174, 247–260. doi: 10.1016/j.aquatox.2016.02.014
Brenet, F., Moh, M., Funk, P., Feierstein, E., Viale, A. J., Socci, N. D., et al. (2011). DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One 6:e14524. 10.1371/journal.pone.0014524 doi: 10.1371/journal.pone.0014524
Carlson, M. (2019). org.Dr.eg.db: Genome Wide Annotation for Zebrafish. R package version 3.8.2. Available online at: https://bioconductor.org/packages/org.Dr.eg.db/ (accessed September 11, 2019).
Carvan, M. J. III, Kalluvila, T. A., Klingler, R. H., Larson, J. K., Pickens, M., Mora-Zamorano, F. X., et al. (2017). Mercury-induced epigenetic transgenerational inheritance of abnormal neurobehavior is correlated with sperm epimutations in zebrafish. PLoS One 12:e0176155. doi: 10.1371/journal.pone.0176155
Casati, L., Sendra, R., Poletti, A., Negri-Cesi, P., and Celotti, F. (2013). Androgen receptor activation by polychlorinated biphenyls: epigenetic effects mediated by the histone demethylase Jarid1b. Epigenetics 8, 1061–1068. doi: 10.4161/epi.25811
Cole, B. J., Brander, S. M., Jeffries, K. M., Hasenbein, S., He, G., Denison, M. S., et al. (2016). Changes in Menidia beryllina gene expression and in vitro hormone-receptor activation after exposure to estuarine waters near treated wastewater outfalls. Arch. Environ. Contam. Toxicol. 71, 210–223. doi: 10.1007/s00244-016-0282-8
DeCourten, B. M., and Brander, S. M. (2017). Combined effects of increased temperature and endocrine disrupting pollutants on sex determination, survival, and development across generations. Sci. Rep. 7:9310. doi: 10.1038/s41598-017-09631-1
DeCourten, B. M., Connon, R. E., and Brander, S. M. (2019a). Direct and indirect parental exposure to endocrine disruptors and elevated temperature influences gene expression across generations in a euryhaline model fish. PeerJ 7:e6156. doi: 10.7717/peerj.6156
DeCourten, B. M., Romney, A., and Brander, S. (2019b). “The heat is on: complexities of aquatic endocrine disruption in a changing global climate,” in Evaluating Water Quality to Prevent Future Disasters, ed. S. Ahuja (Amsterdam: Elsevier), 13–49. doi: 10.1016/b978-0-12-815730-5.00002-8
Ellestad, L. E., Cardon, M., Chambers, I. G., Farmer, J. L., Hartig, P., Stevens, K., et al. (2014). Environmental gestagens activate fathead minnow (Pimephales promelas) nuclear progesterone and androgen receptors in vitro. Environ. Sci. Technol. 48, 8179–8187. doi: 10.1021/es501428u
Evrard, P., and Maghuin-Rogister, G. (1987). In vitro metabolism of trenbolone: study of the formation of covalently bound residues. Food Addit. Contam. 5, 59–65. doi: 10.1080/02652038809373663
Forsgren, K. L., Riar, N., and Schlenk, D. (2013). The effects of the pyrethroid insecticide, bifenthrin, on steroid hormone levels and gonadal development of steelhead (Oncorhynchus mykiss) under hypersaline conditions. Gen. Comp. Endocrinol. 186, 101–107. doi: 10.1016/j.ygcen.2013.02.047
Frank, D. F., Brander, S. M., Hasenbein, S., Harvey, D. J., Lein, P. J., Geist, J., et al. (2019). Developmental exposure to environmentally relevant concentrations of bifenthrin alters transcription of mTOR and ryanodine receptor-dependent signaling molecules and impairs predator avoidance behavior across early life stages in inland silversides (Menidia beryllina). Aquat. Toxicol. 206, 1–13. doi: 10.1016/j.aquatox.2018.10.014
Frank, D. F., Miller, G. W., Harvey, D. J., Brander, S. M., Geist, J., Connon, R. E., et al. (2018). Bifenthrin causes transcriptomic alterations in mTOR and ryanodine receptor-dependent signaling and delayed hyperactivity in developing zebrafish (Danio rerio). Aquat. Toxicol. 200, 50–61. doi: 10.1016/j.aquatox.2018.04.003
Galamb, O., Kalmar, A., Peterfia, B., Csabai, I., Bodor, A., Ribli, D., et al. (2016). Aberrant DNA methylation of WNT pathway genes in the development and progression of CIMP-negative colorectal cancer. Epigenetics 11, 588–602. doi: 10.1080/15592294.2016.1190894
Gaspar, J. M., and Hart, R. P. (2017). DMRfinder: efficiently identifying differentially methylated regions from MethylC-seq data. BMC Bioinformatics 18:528. doi: 10.1186/s12859-017-1909-0
Harding, L. B., Schultz, I. R., Goetz, G. W., Luckenbach, J. A., Young, G., Goetz, F. W., et al. (2013). High-throughput sequencing and pathway analysis reveal alteration of the pituitary transcriptome by 17alpha-ethynylestradiol (EE2) in female coho salmon, Oncorhynchus kisutch. Aquat. Toxicol. 142-143, 146–163. doi: 10.1016/j.aquatox.2013.07.020
Head, J. A. (2014). Patterns of DNA methylation in animals: an ecotoxicological perspective. Integr. Comp. Biol. 54, 77–86. doi: 10.1093/icb/icu025
Hermann, A., Gowher, H., and Jeltsch, A. (2004). Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 61, 2571–2587. doi: 10.1007/s00018-004-4201-1
Hinck, J. E., Blazer, V. S., Denslow, N. D., Echols, K. R., Gale, R. W., Wieser, C., et al. (2008). Chemical contaminants, health indicators, and reproductive biomarker responses in fish from rivers in the Southeastern United States. Sci. Total Environ. 390, 538–557. doi: 10.1016/j.scitotenv.2007.10.026
Hua, J., Han, J., Guo, Y., and Zhou, B. (2015). The progestin levonorgestrel affects sex differentiation in zebrafish at environmentally relevant concentrations. Aquat. Toxicol. 166, 1–9. doi: 10.1016/j.aquatox.2015.06.013
Huang, S., Ye, L., and Chen, H. (2017). Sex determination and maintenance: the role of DMRT1 and FOXL2. Asian J. Androl. 19, 619–624.
Jablonka, E., and Raz, G. (2009). Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 84, 131–176. doi: 10.1086/598822
Jeffries, K. M., Brander, S. M., Britton, M. T., Fangue, N. A., and Connon, R. E. (2015). Chronic exposures to low and high concentrations of ibuprofen elicit different gene response patterns in a euryhaline fish. Environ Sci. Pollut. Res. 22, 17397–17413. doi: 10.1007/s11356-015-4227-y
Jiang, L., Zhang, J., Wang, J. J., Wang, L., Zhang, L., Li, G., et al. (2013). Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell 153, 773–784. doi: 10.1016/j.cell.2013.04.041
Jin, M., Zhang, X., Wang, L., Huang, C., Zhang, Y., Zhao, M., et al. (2009). Developmental toxicity of bifenthrin in embryo-larval stages of zebrafish. Aquat. Toxicol. 95, 347–354. doi: 10.1016/j.aquatox.2009.10.003
Jobling, S., Nolan, M., Tyler, C. R., Brighty, G., and Sumpter, J. P. (1998). Widespread sexual disruption in wild fish. Environ. Sci. Technol. 32, 2498–2506. doi: 10.1021/es9710870
Jones, P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature 13, 484–492. doi: 10.1038/nrg3230
Karoutsou, E., Karoutsos, P., and Karoutsos, D. (2017). Endocrine disruptors and carcinogenesis. Arch. Cancer Res. 5:1. doi: 10.1002/9781118891094.ch1
Kidd, K. A., Blanchfield, P. J., Mills, K. H., Palace, V. P., Evans, R. E., Lazorchak, J. M., et al. (2007). Collapse of a fish population after exposure to a synthetic estrogen. Proc. Natl. Acad. Sci. U.S.A. 104, 8897–8901. doi: 10.1073/pnas.0609568104
Kippler, M., Engstrom, K., Mlakar, S. J., Bottai, M., Ahmed, S., Hossain, M. B., et al. (2013). Sex-specific effects of early life cadmium exposure on DNA methylation and implications for birth weight. Epigenetics 8, 494–503. doi: 10.4161/epi.24401
Krueger, F., and Andrews, S. R. (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. doi: 10.1093/bioinformatics/btr167
Lagesson, A., Saaristo, M., Brodin, T., Fick, J., Klaminder, J., Martin, J. M., et al. (2019). Fish on steroids: temperature-dependent effects of 17beta-trenbolone on predator escape, boldness, and exploratory behaviors. Environ. Pollut. 245, 243–252. doi: 10.1016/j.envpol.2018.10.116
Laurent, L., Wong, E., Li, G., Huynh, T., Tsirigos, A., Ong, C. T., et al. (2010). Dynamic changes in the human methylome during differentiation. Genome Res. 20, 320–331. doi: 10.1101/gr.101907.109
Lee, D. H., Jacobs, D. R. Jr., and Porta, M. (2009). Hypothesis: a unifying mechanism for nutrition and chemicals as lifelong modulators of DNA hypomethylation. Environ. Health Perspect. 117, 1799–1802. doi: 10.1289/ehp.0900741
Manjegowda, M. C., Gupta, P. S., and Limaye, A. M. (2017). Hyper-methylation of the upstream CpG island shore is a likely mechanism of GPER1 silencing in breast cancer cells. Gene 614, 65–73. doi: 10.1016/j.gene.2017.03.006
Micael, J., Reis-Henriques, M. A., Carvalho, A. P., and Santos, M. M. (2007). Genotoxic effects of binary mixtures of xenoandrogens (tributyltin, triphenyltin) and a xenoestrogen (ethinylestradiol) in a partial life-cycle test with Zebrafish (Danio rerio). Environ. Int. 33, 1035–1039. doi: 10.1016/j.envint.2007.06.004
Mirbahai, L., Yin, G., Bignell, J. P., Li, N., Williams, T. D., Chipman, J. K., et al. (2011). DNA methylation in liver tumorigenesis in fish from the environment. Epigenetics 6, 1319–1333. doi: 10.4161/epi.6.11.17890
Mudunuri, U., Che, A., Yi, M., and Stephens, R. M. (2009). bioDBnet: the biological database network. Bioinformatics 25, 555–556. doi: 10.1093/bioinformatics/btn654
Nguyen, T. M., Shafi, A., Nguyen, T., and Draghici, S. (2019). Identifying significantly impacted pathways: a comprehensive review and assessment. Genome Biol. 20:203.
Okano, M., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferass Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. doi: 10.1016/s0092-8674(00)81656-6
Olsvik, P. A., Whatmore, P., Penglase, S. J., Skjaerven, K. H., Angles d’Auriac, M., Ellingsen, S., et al. (2019). Associations between behavioral effects of Bisphenol A and DNA Methylation in Zebrafish Embryos. Front. Genet. 10:184. doi: 10.3389/fgene.2019.00184
Olsvik, P. A., Williams, T. D., Tung, H. S., Mirbahai, L., Sanden, M., Skjaerven, K. H., et al. (2014). Impacts of TCDD and MeHg on DNA methylation in zebrafish (Danio rerio) across two generations. Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 165, 17–27. doi: 10.1016/j.cbpc.2014.05.004
Oppold, A.-M., and Müller, R. (2017). Epigenetics: a hidden target of insecticides. Adv. Insect Physiol. 53, 313–324. doi: 10.1016/bs.aiip.2017.04.002
Orlando, E. F., and Ellestad, L. E. (2014). Sources, concentrations, and exposure effects of environmental gestagens on fish and other aquatic wildlife, with an emphasis on reproduction. Gen. Comp. Endocrinol. 203, 241–249. doi: 10.1016/j.ygcen.2014.03.038
Orn, S., Holbech, H., and Norrgren, L. (2016). Sexual disruption in zebrafish (Danio rerio) exposed to mixtures of 17alpha-ethinylestradiol and 17beta-trenbolone. Environ. Toxicol. Pharmacol. 41, 225–231. doi: 10.1016/j.etap.2015.12.010
Ortega-Recalde, O., Day, R. C., Gemmell, N. J., and Hore, T. A. (2019). Zebrafish preserve global germline DNA methylation while sex-linked rDNA is amplified and demethylated during feminisation. Nat. Commun. 10:3053.
Otake, H., Hayashi, Y., Hamaguchi, S., and Sakaizumi, M. (2008). The Y chromosome that lost the male-determining function behaves as an X chromosome in the medaka fish, Oryzias latipes. Genetics 179, 2157–2162. doi: 10.1534/genetics.108.090167
Perkins, E. J., Gayen, K., Shoemaker, J. E., Antczak, P., Burgoon, L., Falciani, F., et al. (2019). Chemical hazard prediction and hypothesis testing using quantitative adverse outcome pathways. ALTEX 36, 91–102. doi: 10.14573/altex.1808241
Porazinski, S. R., Wang, H., and Furutani-Seiki, M. (2010). Dechorionation of medaka embryos and cell transplantation for the generation of chimeras. J. Vis. Exp. 46:2055.
Porseryd, T., Larsson, J., Kellner, M., Bollner, T., Dinnetz, P., Hällström, I. P., et al. (2019). Altered non-reproductive behavior and feminization caused by developmental exposure to 17alpha-ethinylestradiol persist to adulthood in three-spined stickleback (Gasterosteus aculeatus). Aquat. Toxicol. 207, 142–152. doi: 10.1016/j.aquatox.2018.11.024
Potok, M. E., Nix, D. A., Parnell, T. J., and Cairns, B. R. (2013). Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell 153, 759–772. doi: 10.1016/j.cell.2013.04.030
Rachon, D. (2015). Endocrine disrupting chemicals (EDCs) and female cancer: informing the patients. Rev. Endocr. Metab. Disord. 16, 359–364. doi: 10.1007/s11154-016-9332-9
Ribeiro, C., Pardal, M. A., Tiritan, M. E., Rocha, E., Margalho, R. M., Rocha, M. J., et al. (2009). Spatial distribution and quantification of endocrine-disrupting chemicals in Sado River estuary, Portugal. Environ. Monit. Assess. 159, 415–427. doi: 10.1007/s10661-008-0639-1
Rideout, W. M. I., Coetzee, G. A., Olumi, A. F., and Jones, P. A. (1990). 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 249, 1288–1290. doi: 10.1126/science.1697983
Rissman, E. F., and Adli, M. (2014). Minireview: transgenerational epigenetic inheritance: focus on endocrine disrupting compounds. Endocrinology 155, 2770–2780. doi: 10.1210/en.2014-1123
Robinson, J. A., Staveley, J. P., and Constantine, L. (2017). Reproductive effects on freshwater fish exposed to 17alpha-trenbolone and 17alpha-estradiol. Environ. Toxicol. Chem. 36, 636–644. doi: 10.1002/etc.3526
Runnalls, T. J., Beresford, N., Kugathas, S., Margiotta-Casaluci, L., Scholze, M., Scott, A. P., et al. (2015). From single chemicals to mixtures–reproductive effects of levonorgestrel and ethinylestradiol on the fathead minnow. Aquat. Toxicol. 169, 152–167. doi: 10.1016/j.aquatox.2015.10.009
Ryu, T., Veilleux, H. D., Donelson, J. M., Munday, P. L., and Ravasi, T. (2018). The epigenetic landscape of transgenerational acclimation to ocean warming. Nat. Clim. Change 8, 504–509. doi: 10.1038/s41558-018-0159-0
Salla, R. F., Gamero, F. U., Rissoli, R. Z., Dal-Medico, S. E., Castanho, L. M., dos Santos Carvalh, C., et al. (2016). Impact of an environmental relevant concentration of 17alpha-ethinylestradiol on the cardiac function of bullfrog tadpoles. Chemosphere 144, 1862–1868. doi: 10.1016/j.chemosphere.2015.10.042
Schug, T. T., Johnson, A. F., Birnbaum, L. S., Colborn, T., Guillette, L. J. Jr., Crews, D. P., et al. (2016). Minireview: endocrine disruptors: past lessons and future directions. Mol. Endocrinol. 30, 833–847. doi: 10.1210/me.2016-1096
Schwindt, A. R., Winkelman, D. L., Keteles, K., Murphy, M., Vajda, A. M., Frid, C., et al. (2014). An environmental oestrogen disrupts fish population dynamics through direct and transgenerational effects on survival and fecundity. J. Appl. Ecol. 51, 582–591. doi: 10.1111/1365-2664.12237
Serman, L., Nikuseva, T. M., Serman, A., and Vranic, S. (2014). Epigenetic alternations of the Wnt signaling pathway in cancer: a mini review. Bosn. J. Basic Med. Sci. 14, 191–194.
Shao, C., Li, Q., Chen, S., Zhang, P., Lian, J., Hu, Q., et al. (2014). Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 24, 604–615. doi: 10.1101/gr.162172.113
Sharma, U., and Rando, O. J. (2017). Metabolic inputs into the epigenome. Cell Metab. 25, 544–558. doi: 10.1016/j.cmet.2017.02.003
Soto, A. M., and Sonnenschein, C. (2010). Environmental causes of cancer: endocrine disruptors as carcinogens. Nat. Rev. Endocrinol. 6, 363–370. doi: 10.1038/nrendo.2010.87
Stanczyk, F. Z., and Roy, S. (1990). Metabolism of levonorgestrel, norethindrone, and structurally related contraceptive steroids. Contraception 42, 67–96. doi: 10.1016/0010-7824(90)90093-b
Steinhart, Z., and Angers, S. (2018). Wnt signaling in development and tissue homeostasis. Development 145:dev146589. doi: 10.1242/dev.146589
Susiarjo, M., Hassold, T. J., Freeman, E., and Hunt, P. A. (2007). Bisphenol A exposure in utero disrupts early oogenesis in the mouse. PLoS Genet. 3:e5. doi: 10.1371/journal.pgen.0030005
Svensson, J., Fick, J., Brandt, I., and Brunstrom, B. (2013). The synthetic progestin levonorgestrel is a potent androgen in the three-spined stickleback (Gasterosteus aculeatus). Environ. Sci. Technol. 47, 2043–2051. doi: 10.1021/es304305k
Svensson, J., Fick, J., Brandt, I., and Brunstrom, B. (2014). Environmental concentrations of an androgenic progestin disrupts the seasonal breeding cycle in male three-spined stickleback (Gasterosteus aculeatus). Aquat. Toxicol. 147, 84–91. doi: 10.1016/j.aquatox.2013.12.013
Tenenbaum, D. (2020). KEGGREST: Client-Side REST Access to KEGG. R package version 1.28.0. Available online at: https://bioconductor.org/packages/release/bioc/html/KEGGREST.html
The Gene Ontology Consortium (2019). The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338.
Tian, J., Lee, S. O., Liang, L., Luo, J., Huang, C. K., Li, L., et al. (2012). Targeting the unique methylation pattern of androgen receptor (AR) promoter in prostate stem/progenitor cells with 5-aza-2’-deoxycytidine (5-AZA) leads to suppressed prostate tumorigenesis. J. Biol. Chem. 287, 39954–39966. doi: 10.1074/jbc.m112.395574
Wan, Z. Y., Xia, J. H., Lin, G., Wang, L., Lin, V. C., Yue, G. H., et al. (2016). Genome-wide methylation analysis identified sexually dimorphic methylated regions in hybrid tilapia. Sci. Rep. 6:35903.
Wang, X., and Bhandari, R. K. (2019). DNA methylation dynamics during epigenetic reprogramming of medaka embryo. Epigenetics 14, 611–622. doi: 10.1080/15592294.2019.1605816
Wu, Q., Zhou, Z. J., and Ohsako, S. (2006). Effect of environmental contaminants on DNA methyltransferase activity of mouse preimplantation embryos. Wei Sheng Yan Jiu 35, 30–32.
Wu, T. D., and Watanabe, C. K. (2005). GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875. doi: 10.1093/bioinformatics/bti310
Xin, F., Susiarjo, M., and Bartolomei, M. S. (2015). Multigenerational and transgenerational effects of endocrine disrupting chemicals: a role for altered epigenetic regulation? Semin. Cell Dev. Biol. 43, 66–75. doi: 10.1016/j.semcdb.2015.05.008
Yella, V. R., Kumar, A., and Bansal, M. (2018). Identification of putative promoters in 48 eukaryotic genomes on the basis of DNA free energy. Sci. Rep. 8:4520.
Yong, W. S., Hsu, F. M., and Chen, P. Y. (2016). Profiling genome-wide DNA methylation. Epigenet. Chromatin 9:26.
Zhou, G. J., Li, X. Y., and Leung, K. M. Y. (2019). Retinoids and oestrogenic endocrine disrupting chemicals in saline sewage treatment plants: removal efficiencies and ecological risks to marine organisms. Environ. Int. 127, 103–113. doi: 10.1016/j.envint.2019.03.030
Keywords: epigenetics, Menidia beryllina, endocrine disruptors, transgenerational epigenetic inheritance, multigenerational exposure, DNA methylation, RRBS
Citation: Major KM, DeCourten BM, Li J, Britton M, Settles ML, Mehinto AC, Connon RE and Brander SM (2020) Early Life Exposure to Environmentally Relevant Levels of Endocrine Disruptors Drive Multigenerational and Transgenerational Epigenetic Changes in a Fish Model. Front. Mar. Sci. 7:471. doi: 10.3389/fmars.2020.00471
Received: 28 January 2020; Accepted: 27 May 2020;
Published: 24 June 2020.
Edited by:
Hollie Putnam, University of Rhode Island, United StatesReviewed by:
Taewoo Ryu, Okinawa Institute of Science and Technology Graduate University, JapanMoises A. Bernal, Auburn University, United States
Heather Diana Veilleux, University of Alberta, Canada
Copyright © 2020 Major, DeCourten, Li, Britton, Settles, Mehinto, Connon and Brander. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaley M. Major, a2FsZXkubWFqb3JAZ21haWwuY29t