 Adriana Messyasz1,2*
Adriana Messyasz1,2* Stephanie M. Rosales1,3,4
Stephanie M. Rosales1,3,4 Ryan S. Mueller1
Ryan S. Mueller1 Teresa Sawyer5
Teresa Sawyer5 Adrienne M. S. Correa6Andrew R. Thurber1,7
Adrienne M. S. Correa6Andrew R. Thurber1,7 Rebecca Vega Thurber1
Rebecca Vega Thurber1- 1Department of Microbiology, Oregon State University, Corvallis, OR, United States
- 2Environmental Science Graduate Program, Oregon State University, Corvallis, OR, United States
- 3Cooperative Institute for Marine and Atmospheric Studies, University of Miami, Miami, FL, United States
- 4Atlantic Oceanographic and Meteorological Laboratory, National Oceanographic and Atmospheric Administration, Miami, FL, United States
- 5Electron Microscopy Facility, Oregon State University, Corvallis, OR, United States
- 6BioSciences Department, Rice University, Houston, TX, United States
- 7College of Earth, Ocean, and Atmospheric Sciences, Oregon State University, Corvallis, OR, United States
Eukaryotic viruses and bacteriophage have been implicated in disease and bleaching in corals, but the compositional and functional diversity of these viruses in healthy and compromised hosts remains underexplored. To investigate whether viral assemblages differ in concert with coral bleaching, we collected bleached and non-bleached conspecific pairs of corals during a minor bleaching event in 2016 from reefs on the island of Mo’orea, French Polynesia. Using electron microscopy (EM), we identified several viral particle types, all reminiscent of medium to large-sized nucleocytoplasmic large DNA viruses (NCLDV). We also found that viral metagenomes from bleached corals have significantly more eukaryotic virus sequences, whereas bacteriophage sequences are significantly more abundant in viral metagenomes from non-bleached colonies. In this study, we also initiated the assembly of the first eukaryotic dsDNA coral virus genome. Based on our EM imagery and our taxonomic annotations of viral metagenome sequences, we hypothesize that this genome represents a novel, phylogenetically distinct member of the NCLDVs, with its closest sequenced relative being a distant marine flagellate-associated virus. We also showed that this NCLDV is abundant in bleached corals, but it is also present in apparently healthy corals, suggesting it plays a role in the onset and/or severity of coral bleaching.
Introduction
Viruses, both bacteriophage (phage) and eukaryotic, are vital components of ocean ecosystems, including coral reefs (Sweet and Bythell, 2017; Thurber et al., 2017). Coral reefs are experiencing severe ecosystem declines that result from climate change-mediated and local stressors (Hoegh-Guldberg, 1999; Pendleton et al., 2016; Hughes et al., 2017). The role of viruses in these biologically and culturally valuable habitats (Costanza et al., 2014) may provide insight into mechanisms for their loss. However, viral communities are just beginning to be characterized in corals, and their functional roles in reef health and disease are far from understood.
Viruses are hypothesized to drive several aspects of disease processes in marine ecosystems. For example, phage are thought to limit bacterial virulence in abalone (Friedman et al., 2014) and pathogen adherence in some coral hosts (Efrony et al., 2007; Marhaver et al., 2008). Increased viral activity and abundance has been linked with white diseases in corals (Soffer et al., 2013; Pollock et al., 2014), providing at least one clear example of viral activities being linked to coral disease. However, the diversity of potential outcomes resulting from viral infection of a coral colony is poorly understood, as are the factors that contribute to specific types (e.g., mutualistic, antagonistic) of virus-host interactions (van Oppen et al., 2009).
Viruses are also hypothesized to play a role in coral bleaching, which occurs when dinoflagellate symbionts (family Symbiodiniaceae) that live inside coral tissues are expelled from the coral host or if the symbionts lose their photosynthetic pigments. This causes the coral tissue to lose color and ultimately limits the energy available to the coral colony (Hayes and Bush, 1990; Hughes et al., 2003). Massive coral bleaching events, driven by rising ocean temperatures, are becoming more frequent and often lead to widespread coral death (Carpenter et al., 2008; Pendleton et al., 2016; Hughes et al., 2017). Interestingly, extensive heterogeneity in coral bleaching responses has been recorded (Edmunds, 1994; Ware et al., 1996; Guest et al., 2012; Wooldridge, 2014) and is often attributed to the community structure and function of the algal symbiont within corals.
Some evidence implicates a role for eukaryotic viruses in a natural coral bleaching event (Marhaver et al., 2008; Correa et al., 2016) and during thermal stress (Thurber et al., 2008; Thurber et al., 2009; Correa et al., 2012; Levin et al., 2017). For example, coupling transmission electron microscopy (TEM) imagery with viral genomic information Correa et al. (2016) were able to describe a viral outbreak characterized by an abundance of atypical herpes-like viruses and nucleocytoplasmic large DNA viruses (NCLDV) that was concomitant with a local bleaching event (Correa et al., 2016). Lawrence et al. (2017) also found NCLDV-like genes upregulated within Symbiodiniaceae cultures under UV stress, providing further evidence of coral-associated viruses that may play a role in coral bleaching. The NCLDVs are members of a newly proposed viral order of giant viruses, because of similarities in several of their physical features (jelly-roll capsid, viral factories, capsid diameters >100 nm), their relatively large genome size (100 kB to an excess of 1 Mb), and their encoding of transcription-related genes (Colson et al., 2012, 2013). Correa et al. (2016), Lawrence et al. (2017), and other works (Levin et al., 2017; Weynberg et al., 2017), provide initial evidence that one or multiple NCLDVs can infect members of the coral holobiont (host, dinoflagellates, microbiome, and virome), however, a full or partial genome of a coral giant virus has yet to be recovered.
Understanding the roles of viruses in corals is in part limited by the difficulty in establishing methods for viral identification. For example, a fundamental challenge of viral phylogenetics is that no single gene sequence is shared among all taxa (Rohwer and Edwards, 2002), preventing the application of common phylogenetic approaches used for bacteria, archaea, and eukaryotes (e.g., analyses of universal ribosomal genes; Schmidt et al., 1991). This has led to the development of different approaches to characterize the presence and abundance of viral populations in natural environments and host-associated niches. PCR-based methods to amplify conserved genes do exist for viruses, but these are restricted to the analysis of known genes, which have similar protein structures and are only encoded by sets of closely-related viruses (Mokili et al., 2012). Alternatively, shot-gun sequencing of viral metagenomes does not exclusively focus on known viral genes, and instead can generate sequences from all the viral genomes within a sample (Streit and Schmitz, 2004; Edwards and Rohwer, 2005; Delwart, 2007; Tang and Chiu, 2010; Mokili et al., 2012). A limitation of applying shotgun viral metagenomic approaches to the analysis of coral samples, though, is that viral nucleic acids make up less than 1% of the total reads from a given non-cultured and host-based metagenome (Yamashita et al., 2016). Improved bioinformatics algorithms are needed to identify and parse viral reads among the more numerous eukaryotic genomic and bacterial sequences in order to identify evidence of disease- or bleaching-associated viruses on coral reefs.
Despite the difficultly of analyzing viral communities it is essential to try to identify the roles of viruses on coral health. Whether viruses are involved in exacerbating or directly causing some forms of coral bleaching remains unknown. Thus, the main objective of this study is to investigate the role of viruses in coral bleaching, while optimizing existing viral bioinformatic workflows for viral metagenomics, and complementing these sequence-based approaches with electron microscopy (EM). Other studies have compared viral communities between bleached and healthy coral (Marhaver et al., 2008) or between stressed and healthy corals (Vega Thurber et al., 2009 used metagenomics after microbial enrichment), but have relied on targeted metagenomic approaches. Here, using non-targeted metagenomics, we evaluate a natural stress event, in which some colonies bleached and others did not, to characterize and compare the viruses associated with bleached and apparently healthy coral colonies in situ on a reef. We used immediately adjacent, but phenotypically distinct, coral pairs to test the hypothesis that the abundance of NCLDVs is linked to coral bleaching. We posit that viral infection may help explain the sometimes-paradoxical bleaching patterns on coral reefs, especially in the case of two adjacent corals that differ drastically in the extent of their bleaching.
Materials and Methods
Coral Collections and Preservation
Bleached (B) Pocillopora spp. coral colonies, with no other signs of disease, were photographed (along with a Coral Health Chart as a color standard) and sampled from the north shore forereef 17°28′ S 149°49′W off Mo’orea, French Polynesia, between March 9 and 16, 2016. After each bleached coral was sampled, an immediately adjacent non-bleached (NB) pocilloporid colony of the same morphotype, with no signs of disease, was similarly photographed and sampled, generating three B-NB paired samples (Figure 1). Pocillopora spp., rather than a specific species, was targeted because on Mo’orea, pocilloporids appear to represent a species complex, rather than morphologically identifiable species (Gélin et al., 2017). A small fragment of each colony was collected on SCUBA using bone-cutters and placed in separate sterile whirlpaks. At the surface, samples were placed on ice and transported back to shore, and then further fragmented using flame sterilized bone cutters. For each coral sample, a small piece from one fragment was preserved in 0.5% Electron Microscopy (EM) grade glutaraldehyde and 10X PBS for EM. The remaining fragments for each sample were airbrushed with 1.5 mL of Qiagen Dneasy Kit lysis buffer and the resulting coral tissue slurry was used for whole community DNA extraction with a Dneasy Kit (Qiagen, Germantown, MD). Equivalent volumes of lysis buffer for the larger amounts of tissue extraction were amended appropriately. The elutions were suspended in Tris-EDTA and frozen for transport to Oregon State University (OSU, Corvallis, OR, United States).

Figure 1. The three coral colony pairs used for viral metagenomic analysis. Corals were found on the northern forereef off the island of Mo’orea, French Polynesia. Coral Health Chart colors indicate extent of bleaching for each coral colony (#1 bleached to #6 healthy). All corals were identified as Pocillopora spp.
Fixation, Decalcification, and Staining for Electron Microscopy
Six fixed coral fragments (N = 3 B-NB colony pairs) were decalcified with 500 μL of 10% EDTA pH 7. Once decalcified, the tissues were embedded in agarose for post-fixation staining. The samples were rinsed with 0.1 M sodium cacodylate buffer and post-fixation processing was conducted in 1.5% potassium ferrocyanide and 2% osmium tetroxide in water. Then T-O-T-O staining was followed by uranyl acetate and lead nitrate staining (Pfeifer et al., 2015). Samples were then dehydrated in serial acetone baths (10, 30, 50, 70, 90, 95, 100%) for 10–15 min each. Dehydrated samples were infiltrated with araldite resin and sectioned using a RMC ultra-microtome. Images were taken with a FEI Helios Nanolab 650 Microscope in scanning transmission electron microscopy (STEM) mode.
Metagenome Preparation and Sequencing
For metagenome generation, the extracted DNA from the six coral colonies (3 coral pairs) from Mo’orea (Gump Research Station) were cleaned with Zymo DNA Clean and Concentrator-5 Kits (Zymo, Irvine, CA) and eluted into Ultra-pure 18 Ω H2O for sequencing. DNA from the three colony pairs were prepared for sequencing with the PrepX DNA Library Kit (WaferGen, Biosystems, Fremont, CA), then sequenced on the Illumina HiSeq 3000 platform (2 × 150 paired-end), at the Center for Genome Research and Biocomputing (CGRB) at OSU.
Bioinformatics Quality Filtering, Host Removal, and Contig Assembly
An average of 70 million reads were produced for each metagenome library. Changes in read and contig numbers and other parameters were recorded after each step of our contig assembly bioinformatic workflow (Supplementary Table 1). Initial quality control (QC) measures included Illumina adapter/index trimming and quality filtering using the program fqtrim (parameters: -R – pid5 – pid3 -D -A -q 20 -w -m -l 50 -P33) (Geo Pertea, 2018). At the time of the analysis, there were no congeneric reference genomes available. Thus, we used the genomes of two other coral species Orbicella faveolata and Acropora digitifera, for data filtration. To remove host and symbiont contaminating reads, Bowtie2 (Langmead and Salzberg, 2012) was used to globally align metagenome library reads to a database of Orbicella faveolata (GenBank: MPSW00000000.1), Acropora digitifera (GenBank: BACK00000000.1), and Symbiodiniaceae sp. clade A MAC-Cass KB8 (Tax ID: 671378, UniProt) sequences (parameters: max penalty for mismatch: -mp 4, max fragment length: -X 1000). All aligned reads putatively identified as host and symbiont derived were discarded. The remaining reads were then used to assemble contigs with MegaHit using the default parameters (Li et al., 2015). Raw read sequences have been submitted to the NCBI SRA (BioProject ID: PRJNA647466).
Viral Taxonomy Classification and Estimates of Relative Viral Similarities
To annotate genes within the individual coral viral metagenomes, all contigs were analyzed with the Open Reading Frame (ORF) finder program Prodigal (Hyatt et al., 2010) (default parameters), and resulting ORFs were compared to the Viral RefSeq Protein database (from May 2018) using DIAMOND BLASTx (Buchfink et al., 2015). As viruses contain sequence homology to both their hosts and other organisms, we took a conservative sequence alignment approach to decrease the number of false positives and used an e-value of 1.7e–13 after analyzing a simulated bacterial negative control metagenome sample (Angly et al., 2012) and allowing for a 5% false positive rate (for details see Supplementary Methods). Viral taxonomic identifications [i.e., group, family, species] for contigs were generated using the taxonomy assignments from the Viral RefSeq, Viral Zone, and Uniprot databases. Relative percent abundance of viral similarities (i.e., BLAST hits) was generated to compare the B and NB samples with R (Version 1.1.456) using ggplot2 (Wickham, 2016). Statistical analysis of similarity counts to each virus family was conducted using DeSeq2 (RStudio, Wald test and parametric fit type) (Love et al., 2014).
NCLDV Genome Recruitment Methods and Visualization
Putative NCDLV viral contigs from all 6 libraries were further identified using tBLASTx against known NCLDVs with the greatest number of similarities generated (from previous section): Acanthamoeba polyphaga mimivirus (NC_014649.1), Cafeteria roenbergensis virus BV-PW1 (NC_014637.1), Lymphocystis disease virus (NC_005902.1), Paramecium bursaria Chlorella virus (NC_000852.5), and Megavirus chiliensis (NC_016072.1). Best matches to individual genomes were summarized in metagenomic recruitment plots, which were viewed using the default settings of MGAViewer (Zhu et al., 2013).
Reference-Based de novo Assembly of Coral dsDNA NCLDV
To target putative coral-associated NCLDVs, merge redundant contigs, and reduce the time of downstream computational analyses, we conducted a meta-assembly of all 6 metagenomes generated above. A two-part workflow was used to generate a combined reference and de novo-based assembly of a novel coral dsDNA NCLDV (Supplementary Figure 1). In part one, an initial reference-based assembly against Cafeteria roenbergensis virus BV-PW1 (CroV; NC_014637.1) was conducted because CroV had the highest number of MegaHit contig recruits of all NCLDVs analyzed (Table 1). For this analysis, post QC reads of B and NB metagenomes were used and aligned against the full CroV BV-PW1 genome using Bowtie2 (Langmead and Salzberg, 2012) with the parameters: -p 10 -X 1000. Reads aligning to CroV BV-PW1 were then selected for further analysis.
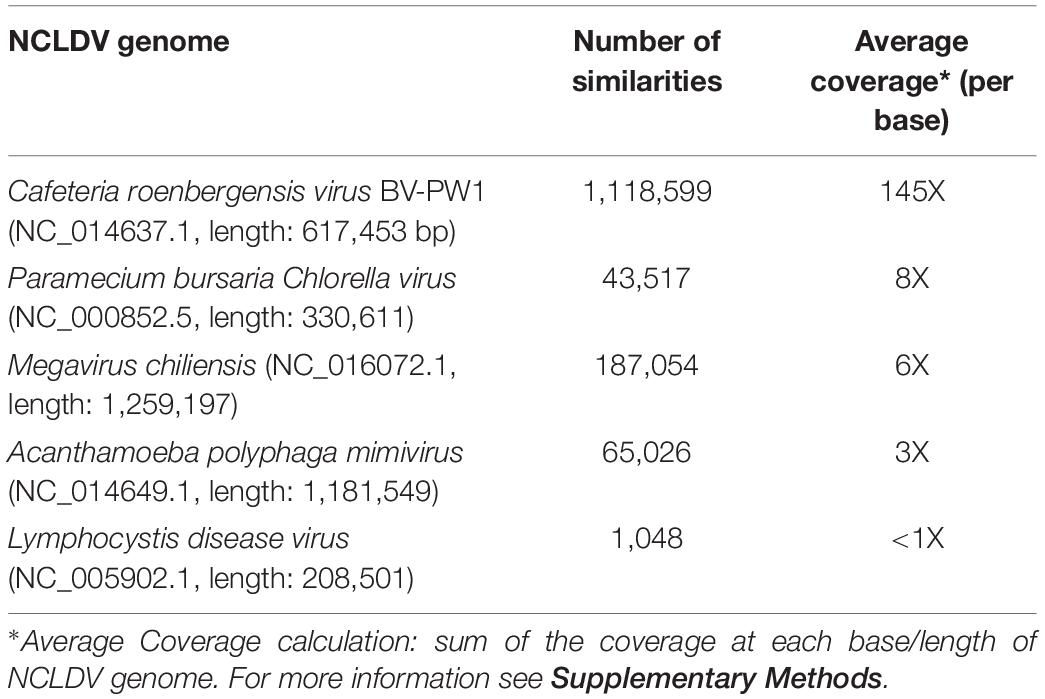
Table 1. Results of MegaHit contig recruitments to NCLDV.
As part of this workflow, we also created a “meta-assembly” via GAM-NGS tools (Vicedomini et al., 2013). Briefly, all B and NB quality controlled (host-removed) reads were combined into one file and then aligned to MegaHit contigs from each sample (via BWA; Li and Durbin, 2009). The process was run iteratively for each B sample contig, for each NB samples contig, and finalized with a B/NB meta-assembly via GAM-NGS to remove redundancy between contigs from each library. Once the contigs from the meta-assembly were created, they were then aligned using BLASTx to the NCVOG (NCLDV clusters of orthologous genes) database (Yutin et al., 2009) to identify those with significant similarity to known NCLDV genes (e-value: <1e–5). These putative NCVOG contigs were further assembled via SPAdes (Bankevich et al., 2012) (version 3.11.1; careful and only assembler was used, kmer: 21,33,55,77), using the reads similar to CroV BV-PW1. These contigs were then further elongated via PriceTI (Ruby et al., 2013) (parameters: contig.fasta 3 10 5 -nc 60 -dbmax 75 -mol 30 -tol 20 -mpi 80 -target 90 2 1 1).
In part two of our workflow, reads from a single sample library (5B) were used to extend the CroV/NCVOG assembly using Price TI (from cycle 1, with the same parameters as above). This informative library was selected as it recruited the most reads to identified NCLDV contigs of the meta-assembly (Supplementary Figure 2). Resulting contigs >500 bp and 5B aligned reads were used for a final SSPACE (Boetzer et al., 2011) elongation (parameters: -x 1 -m 50 -r 0.7 -g 1 -o 20 -p 1 -z 120). Raw reads were aligned back to the genome to assess assembly quality using QUAST (Gurevich et al., 2013) and chimeras were checked using VSEARCH (v2.7.1; parameters: – uchime_de novo, – chimeras) (Rognes et al., 2016).
Final Assembly Search Against Environmental and Viral Databases
The final 192 contigs were searched against the NCBI metagenomic protein or environmental (env_nr) database (BLASTx, e-value: 1e-5) and compared (tBLASTx) to the genomes of known NCLDVs. Comparisons to CroV, Megavirus chiliensis, and Acanthamoeba polyphaga mimivirus were visualized using the CGView GUI interface (BLAST alignment) (Stothard and Wishart, 2005). Final assembly ORFs were found (Prodigal) and searched against the NCBI non-redundant (nr) database (BLASTp, e-value: 1e-10, -max_target_seqs 1). These ORFs were also aligned to genes of the NCVOG database (BLASTp) to identify contigs that included viral NCLDV genes of interest (e.g., transcription factors, ubiquitin, topoisomerase, etc.). These ORFs were also searched against the JGI IMG VR all proteins database (DIAMOND BLASTp, parameters: -k1, -e 1e-10) (Paez-Espino et al., 2017) and the KEGG Genes (database: genus_prokaryotes + family_eukaryotes + viruses) using the KOALA (KEGG Orthology and Links Annotation) GHOSTX search (GhostKOALA) (Kanehisa et al., 2016) to identify functional annotations.
Phylogenetic Analysis of Conserved NCLDV Genes
Based on homology results, three identified coral-associated NCLDV orthologs were used for phylogenetic analysis: ribonucleoside-diphosphate reductase, DnaJ/Hsp40, and DNA topoisomerase 2. Only three previously analyzed NCLDV genomes were found to contain all three genes: Acanthamoeba polyphaga mimivirus (NC_014649.1), Cafeteria roenbergensis virus BV-PW1 (NC_014637.1), and Megavirus chiliensis (JN258408.1; Supplementary Table 2 provides NCBI accession numbers of genes used for each NCLDV). The three genes from each NCDLV were end-to-end concatenated in the same order, resulting in ∼2,000 amino acid long sequences. To find homologs of these genes within our assembly, we first created a BLAST database for our assembly and then aligned it against a query consisting of the three genes from the three NCLDVs (tBLASTn, -e-value: 1), resulting in 8 assembly contigs with similarities. ORFs were found in these contigs (via Prodigal), however, no single ORF was greater than 1kbp long. To identify which ORFs had best similarities to one of the three genes, they were aligned against the Viral RefSeq database (BLASTp). ORFs with the highest bitscore against one of the three genes were chosen for phylogenetic analysis of the genes from the three NCLDVs. These ORFs were concatenated together forming a 1,190 amino acid long sequence, which was considerably shorter than the concatenation of the genes from the known NCLDVs.
After conducting a multiple sequence alignment or MSA (via MAFFT (Katoh and Standley, 2013) protein alignment default parameters: BLOSUM62 matrix, 1.53 gap open penalty, 0.123 gap extension penalty), we input our MSA into the GBlocks (Castresana, 2000) GUI interface. This program eliminated poorly aligned portions and divergent regions of our protein MSA, reducing all gene comparisons to more similar amino acid lengths and making our MSA more suitable for phylogenetic analysis. We ran GBlocks allowing for gap positions in the final blocks. These final blocks were then used to build a phylogenetic tree (PhyML; Guindon et al., 2010) using the default LG substitution model and 100 boot-strap replicates (parameters: -d aa -b 100 – run_id GB -n 1 -f m -v e -c 4 -a e -o tlr -s BEST). The results of this phylogenetic tree (Supplementary Figure 3A) were corroborated by running the same steps without GBlocks (Supplementary Figure 3B), and running a phylogenetic tree without our assembly contigs (Supplementary Figure 3C).
Results
Bleached Corals Contain a Unique Set of Virus-Like Particles in Their Gastroderm and Associated With Symbiodiniaceae Cells
Virus-like particles (VLPs) were present in both the bleached (B) and non-bleached (NB) coral samples based on EM images. VLPs were identified as densely stained or dark nucleo-rich spheres or polyhedrons often with an icosahedral shape. VLPs were found in tissues undergoing extensive cellular dissociation (Supplementary Figure 4). Numerous large VLPs were identified in coral cells and cellular debris throughout the coral gastroderm. These VLPs found in the B corals, had a size range of 200–360 nm and appeared either within necrotic coral cells or in proximity to viral factories (Figure 2).
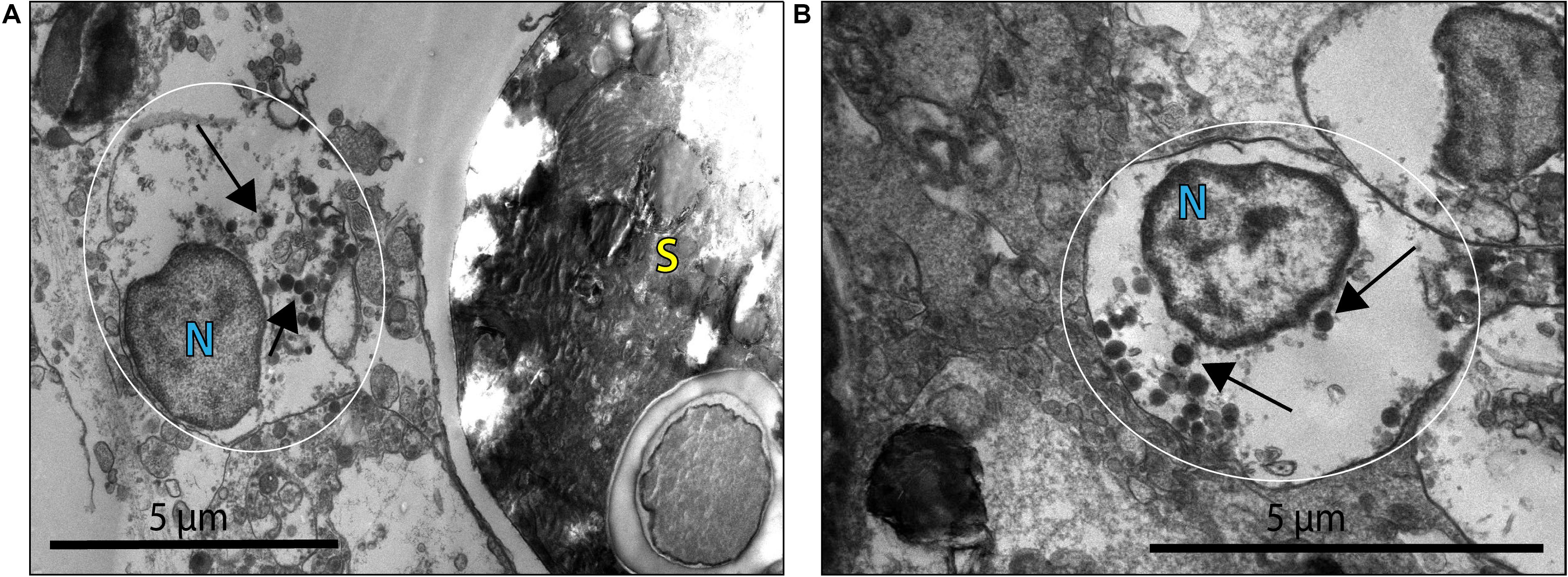
Figure 2. Virus-like particles (VLP) indicated by black arrows within what may be a coral cell or viral factory [general placement of this structure is highlighted by the white circle; N denotes possible coral nuclei or a viral factory (VF)] associated with B coral cells. (A) VLPs ∼200–300 nm in diameter within an apparently necrotic coral cell or in proximity to a VF near a Symbiodiniaceae cell (S). (B) VLPs ∼260 nm in diameter within an apparently necrotic coral host cell or VF.
This morphology and the large capsid diameter suggests the bleached coral-associated VLPs are within the NCLDV (Colson et al., 2013). We also found VLPs with an even larger diameter, ∼400–500 nm (Figure 3), and concentric circular structures within the capsid that were often, but not always, enveloped (Figure 3A). Additionally, one of these larger VLPs had hair-like projections or fibrils and an atypical, spherical, rather than icosahedral, shape (Figure 3B). Along with host-associated VLPs, some VLPs within the gastroderm were closely associated with Symbiodiniaceae (Figure 4). These VLPs were large and icosahedral with capsids ∼170–200 nm in diameter and seen alongside Symbiodiniaceae (Figures 4A,B). Other VLPs were small with spherical capsids that were ∼40 nm and appeared within the symbiosome (Figure 4C).
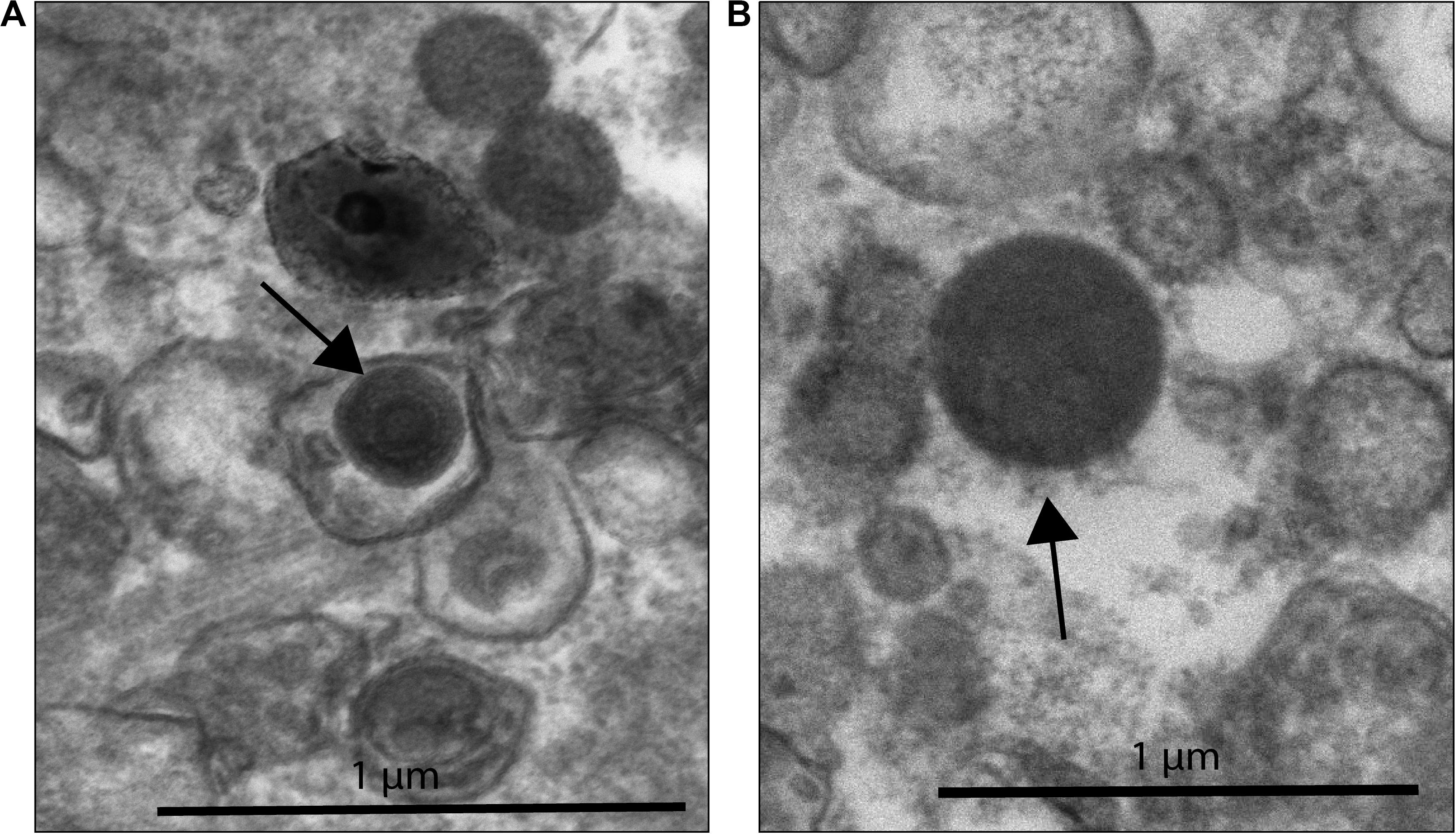
Figure 3. NCLDV-like VLP within B coral samples. (A) VLP surrounded by a vacuole-like structure with a total diameter of ∼410 nm (black arrow) found outside of coral cells. This VLP has concentric circles within the capsid structure, a characteristic observed in known NCLDV-like Mimiviridae. (B) VLP ∼480 nm in diameter, with possible fibrils (black arrow). Zoomed out image of 3B can be found in Supplementary Figure 4.
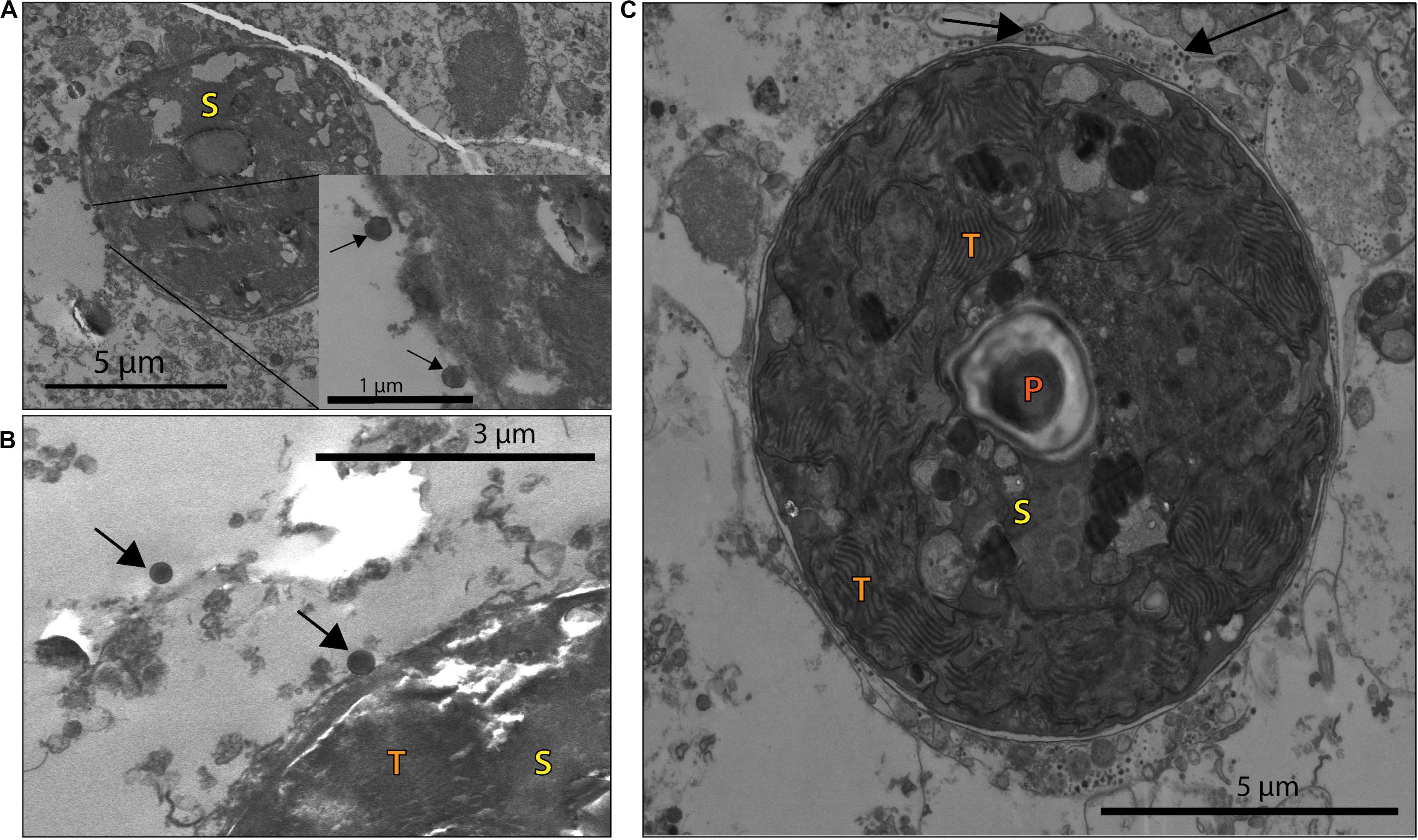
Figure 4. VLP associated with coral B and NB Symbiodiniaceae cells (S), with visible pyrenoid (P) and thylakoid membrane (T) structures. (A) VLPs ∼170 nm in diameter (black arrows) closely associated with a Symbiodiniaceae cell in a NB sample. (B) Larger VLPs (∼200 nm in diameter, arrows) close to a B sample Symbiodiniaceae cell. (C) Whole Symbiodiniaceae cell, from NB sample, with VLPs (∼40 nm in diameter, arrows) around and within the symbiosome.
Bleached Corals Contain More Eukaryotic Virus Sequence Similarities Than Non-bleached Corals
Metagenomic analyses were used to infer the identity and function of VLPs in the B and NB corals using a custom workflow (Supplementary Figure 5) and the viral RefSeq database. Statistical differential abundance analysis (Love et al., 2014) of raw viral contig counts showed significant differences between viral sequences that define host (eukaryotic and bacterial), genome types (dsDNA or retro-viral), and between viral families found within B and NB samples (Supplementary Table 3). Overall there were more similarities to dsDNA eukaryotic virus sequences in the B viral metagenomes (white bars), whereas NB viral metagenomes (green bars) had more similarities to bacteriophage sequences (phage; Figure 5).
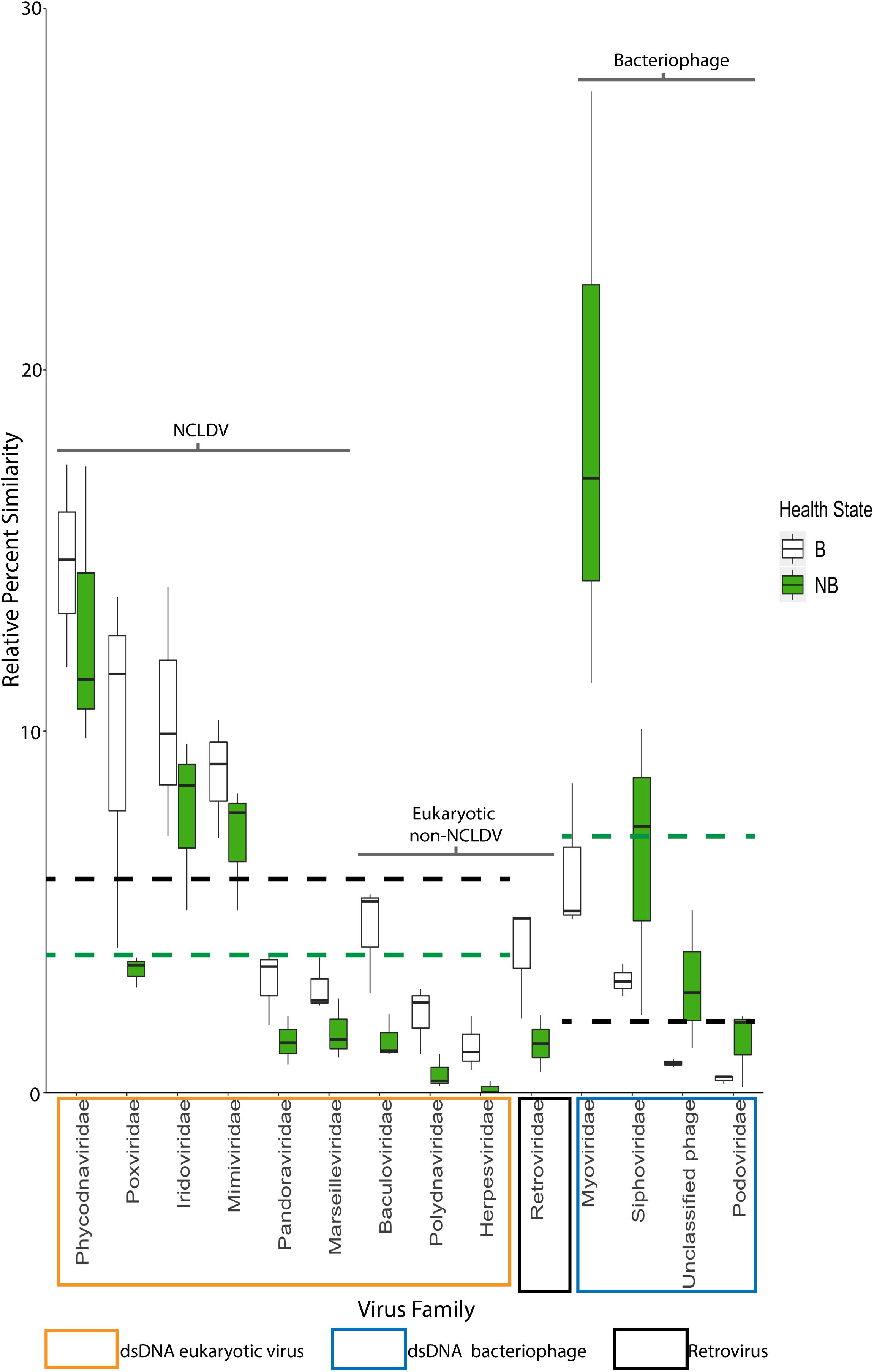
Figure 5. Relative percent of similarities (y-axis) to virus families (x-axis) identified after viral homology analysis of each individual metagenome (N = 3). Of the 26 viral families annotated, these 14 families represent those with >5% similarities and/or families with significant differences between B and NB viral metagenomes (Baculoviridae, Herpesviridae, Polydnaviridae, Retroviridae, and Myoviridae, Podoviridae, and Siphoviridae). Viral families are boxed by viral group: eukaryotic viruses (orange), retroviruses (black) and bacteriophage (blue). NCLDV families are indicated by the black bracket. Dashed lines indicate average mean across eukaryotic and bacteriophage families for B (black) and NB (green) samples. A general trend of more similarities to dsDNA eukaryotic viruses for B than NB viral metagenomes, and more similarities to dsDNA phage for NB than B viral metagenomes is visible. Additionally, there is a significant increase (p < 0.05) in the relative percent of similarities to Retroviridae in the B (3.9%) compared to the NB (1.4%).
Analysis of the relative percent of all viral contig similarities, regardless of their library origin, resulted in annotation to 26 described and accepted viral families (Supplementary Table 3). The relative percent of similarities to seven individual viral families were significantly different between the two coral health categories: Baculoviridae, Herpesviridae, Polydnaviridae, Retroviridae, Myoviridae, Podoviridae, and Siphoviridae. The NCLDV similarities (including sequence annotations to the families Ascoviridae, Iridoviridae, Mimiviridae, Phycodnaviridae, and Pandoraviridae) represented the largest percent of dsDNA eukaryotic virus contig similarities (42.4 ± 4.38% SE NCLDV; 6.1 ± 0.70% SE non-NCLDV eukaryotic dsDNA viruses) (Table 2). Of the NCLDV annotations, the highest percent of similarities were to Phycodnaviridae. When comparing B vs NB viral metagenomes, there were significantly more (p = 0.02; t-test, t-stat = 1, DF = 1) NCLDV similarities in B corals (50.4 ± 3.5% SEM) than NB corals (34.4 ± 4.4%). Two viral families, Lavidaviridae (virophage to Mimiviridae) and Nimaviridae, were only found in B contigs, but these contributed to less than 0.08% of the total relative percent similarities of viral contigs.
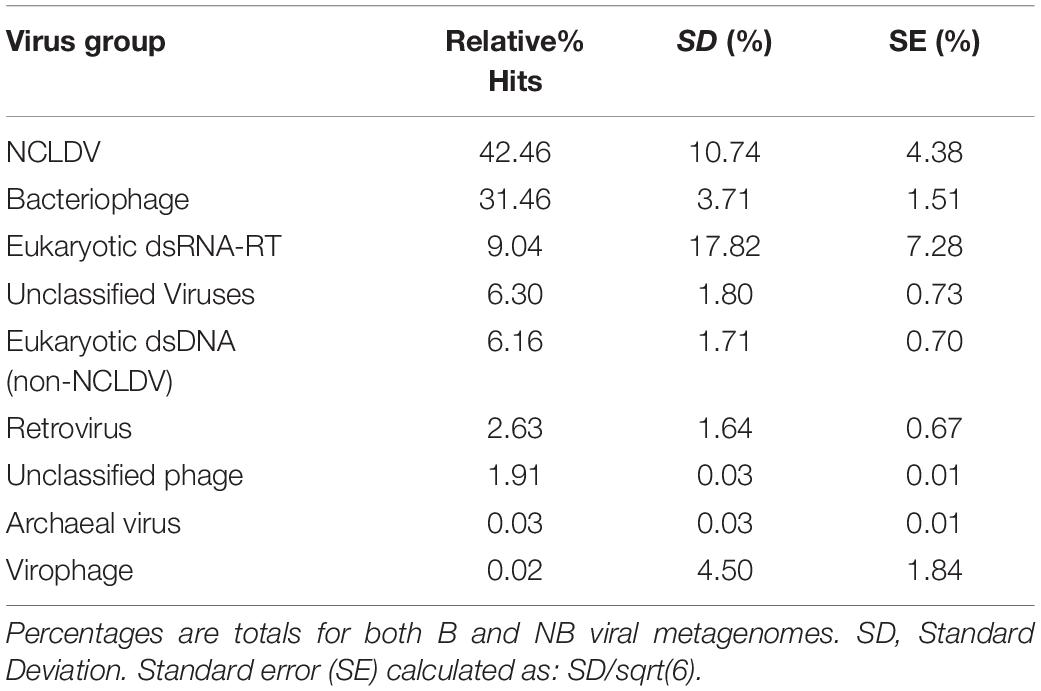
Table 2. Viral contig similarities (noted as “hits”) to viral groups.
Recruitment-Based Analysis of Coral Contigs Identifies Deep and Broad Coverage to NCLDV
Since NCLDVs were the dominant eukaryotic virus annotations in all viral metagenomes, we recruited MegaHit contigs (from all B and NB sample viral metagenomes) to the genomes of known NCLDV strains (Colson et al., 2013): Acanthamoeba polyphaga mimivirus (NC_014649.1), Cafeteria roenbergensis virus BV-PW1 (NC_014637.1), Lymphocystis disease virus (NC_005902.1), Paramecium bursaria Chlorella virus (NC_000852.5), and Megavirus chiliensis (NC_016072.1). Although some genomes had extensive recruitment across the whole length, most alignments were generally below the 70% identity level (Supplementary Figure 6). However, the Cafeteria roenbergensis virus (CroV) BV-PW1 genome recruited more than 1 million B and NB contigs, resulting in an average of 145X coverage per base of the CroV genome (Table 1). Regions of the CroV genome that had over 10,000X coverage include: ∼15,000x coverage at a putative deoxyuridine 5’-triphosphate (DUT) nucleotidohydrolase (between 65 kb and 70 kb), ∼15,000x coverage at a hypothetical protein near a putative CPD class I photolyase (at ∼113 kb), ∼14,000x coverage at a putative phosphoglycerate mutase (at ∼125 kb), and ∼15,000x coverage at a putative DnaK/Hsp70 (at ∼ 451 kb) (Supplementary Figure 6). We compared recruitment plots against various dsDNA viruses and found that the number of recruits was not dependent on genome size. For example, there was less contig recruitment to Megavirus chiliensis (∼1.2M bp) than to CroV (∼700K bp) (Table 1).
Corals Contain a Unique Giant Virus
Given that the CroV BV-PW1 genome recruited the largest number of contigs overall, we used the recruited contigs to assemble and annotate an uncultivated virus genome (UViG) using Minimum Information (MI) UViG standards (Roux et al., 2019). A meta-assembly produced a total of 554,561 non-redundant contigs from the >3 million contigs from all initial MegaHit assemblies (Supplementary Figure 5). From these, a total of 192 contigs were assigned to a draft genome of approximately 900,000 bp in length with no chimeras detected. From all host-filtered reads, 8% mapped to this draft genome, of which 54% were properly mapped, 31% were singletons, and 10% were misjoint mates or mapped as incorrect pairs. These results show that this genome is incomplete and/or these contigs represent several coral giant virus genomes.
A search of these contigs against the NCBI env_nr database (BLASTx) produced no sequence similarities. There were 652 ORFs found in the 192 contigs in the final assembly. These ORFs aligned to known genes and genetic elements of the CroV and other NCLDV genomes, and included the following: inteins (ribonucleoside-triphosphate reductase, ribonucleotide reductase small subunit), transcription genes (putative ATP-dependent RNA helicase, putative superfamily II helicase/eIF-4AIII, helicase-2, helicase 45, ATP-dependent DNA helicase PIF1, HrpA-like helicase, DNA topoisomerase 2, DNA gyrase/DNA topoisomerase IV subunit A), and ubiquitin components (putative ubiquitin-conjugating enzyme E2, ubiquitin family protein). Yet out of 652 ORFs, only 171 (26%) had similarities to known NCVOGs. When compared to the NCBI nr database, most of these ORFs (70%) were similar to hypothetical/uncharacterized proteins from Symbiodiniaceae, Pocillopora spp., and bacteria and the only viral hits were to phage. Additionally, 72% of hits (n = 291 ORF similarities) were to Pocillopora damicornis proteins. Of these Pocillopora damicornis hits, 55% (n = 161 ORF similarities) were to hypothetical or uncharacterized proteins, and the remaining hits (n = 130) were to functional genes. From the 171 functional annotations of our assembly to the NCVOG database, 39% of annotated genes (n = 66 ORF similarities) appear to share homology with the Pocillopora damicornis functional genes from the NCBI nr annotations.
For the IMG VR search, 124 of the 652 ORFs, or 19%, had similarities to viral proteins. Based on the IMG VR annotations we categorized the similarities based on viral cluster: 43% were environmental (n = 53 ORF similarities), 27% were iVG (isolate viral genomes) and prophage (n = 33 ORF similarities), 17% were host-associated (n = 21 ORF similarities), and 13% (n = 16 ORF similarities) were engineered. Of the 33 iVG annotations, 30 were to dsDNA (2 RNA-RT and 1 undefined), of which 10 annotations were to Iridoviridae, 10 to Poxviridae, 4 to Mimiviridae, and 4 to Pandoraviridae. The second category was host: 52% NA (n = 64 ORF similarities), 29% Bacteria (n = 36 ORF similarities), and 19% Eukaryote (n = 24 ORF similarities). The third category was ecosystem: the highest percent 27% were to iVG (n = 33 ORF similarities), followed by 23% aquatic marine (n = 28 ORF similarities), 15% aquatic freshwater (n = 18 ORF similarities), 8% were Green algae (n = 10 ORF similarities) (for full results see Supplementary Table 4).
For functional annotation, 161 ORFs (25%) annotated to KEGG genes (Supplementary Figure 7). Of these annotations, almost half (49%) annotated to genetic information and processing (transcription: RNA polymerase, messenger RNA biogenesis; translation: Aminoacyl-tRNA biosynthesis, RNA tranport; folding, sorting and degradation: protein processing in endoplasmic reticulum, ubiquitin mediated proteolysis; and replication and repair: DNA replication, base exicision repair). Another 14% annotated to environmental information processing (membrane transport, signal transduction, and signaling molecules and interaction). A total of 21% of annotation were to metabolic functions (metabolism protein families, metabolism of cofactors, vitamins, nucleotides, lipids, amino acid, glycans, and carbohydrates). Approximately 9% of annotations were to cellular processes and signaling (transport and catabolism, endocystosis, cell cycle, cell motility). The final 7% annotated to human diseases including viral infectious diseases (measles, Herpes simplex virus 1 infection, Epstein-Barr virus infection). The taxonomic breakdown of these annotations resulted in 58% undefined annotations, 38% cnidarian annotations, and 1% annotations each to Alveolates, Bacteroidetes, Arthropods, and Vertebrates. From the 161 functional annotations of our assembly to the KEGG gene database, 45% of annotated genes (n = 72 ORF similarities) appear to share homology with the Pocillopora damicornis functional genes from the NCBI nr annotations.
A phylogenetic tree of conserved genes within known NCLDV genomes (i.e., Ribonucleoside-diphosphate reductase, DNAJ/Hsp40, and DNA topoisomerase II), failed to resolve the placement of the putative coral NCLDV (Supplementary Figure 3), limiting the interpretation of the evolutionary history of the coral UViG sequences. Thus, our draft genome contigs were compared to three other NCLVDs: CroV, Acanthamoeba polyphaga mimivirus and Megavirus chiliensis (Figure 6). Acanthamoeba polyphaga mimivirus and Megavirus chiliensis exhibited higher coverage to CroV than our draft coral virus genome. Based on our genomic and phylogenetic analyses, our coral virus is most phylogenetically similar to CroV (Supplementary Figure 3), yet has low coverage and low percent identity to CroV and other NCLDVs (Table 3). This indicates that our draft coral virus is a unique eukaryotic dsDNA giant virus with no sequenced relatives or a completely novel and uncharacterized DNA virus. We predict that the genome is roughly 900,000 bp long.
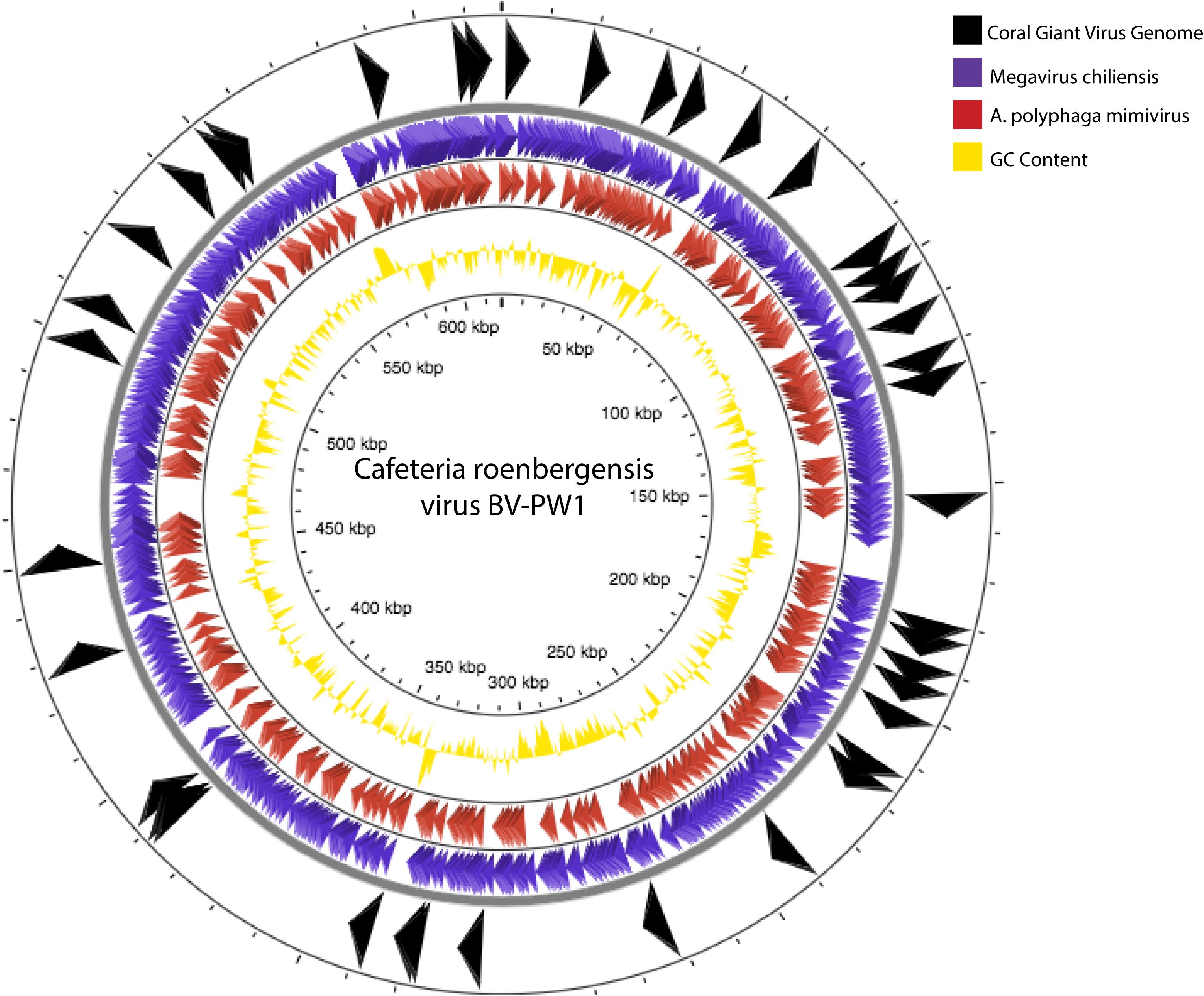
Figure 6. Genome comparisons of our assembly to Cafeteria roenbergensis virus (CroV) BV-PW1 (length: 617,453 bp). Arrows indicate BLAST similarities between CroV and our assembled coral giant virus genome (black arrows, outermost track), A. polyphaga mimivirus (purple arrows, second outermost track) and Megavirus chiliensis (red arrows, third outermost track). GC content of CroV is indicated by track with yellow peaks (fourth track).
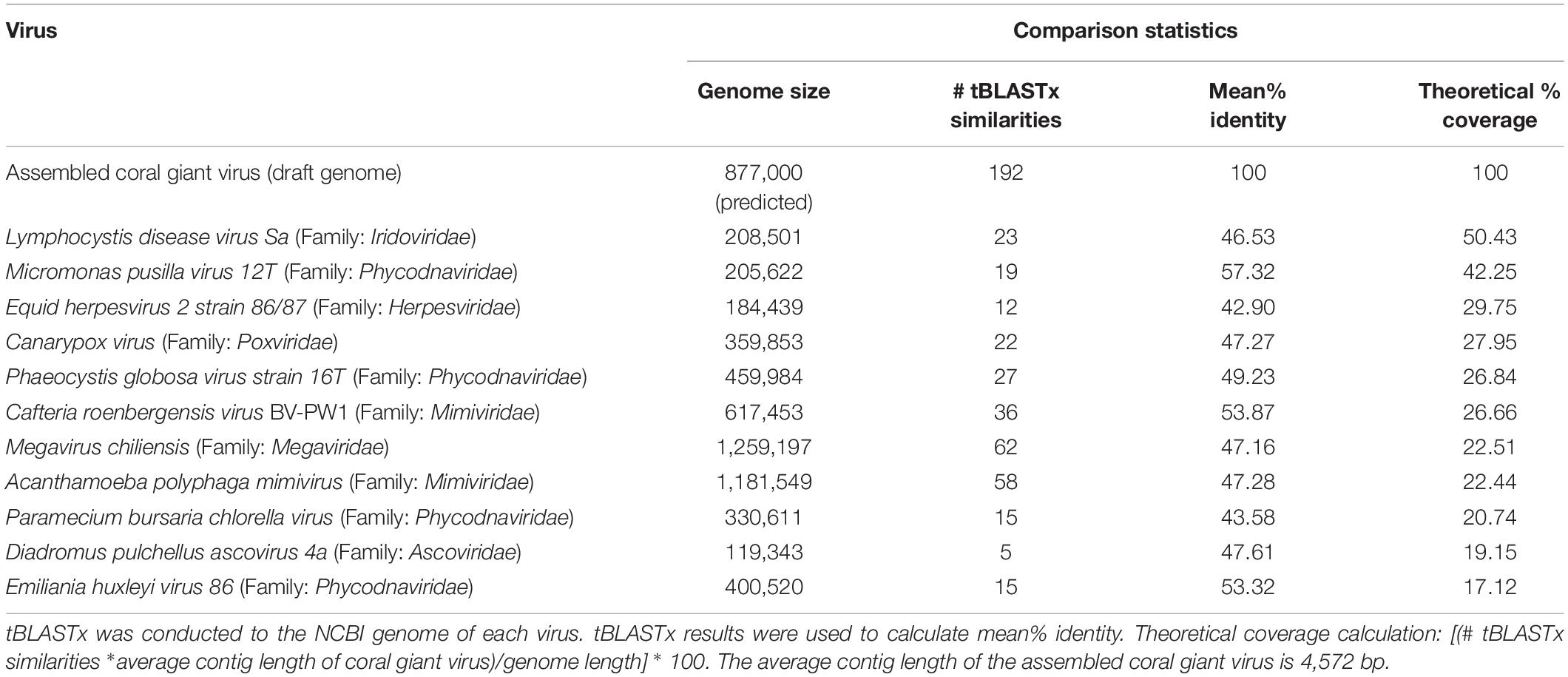
Table 3. Comparison of assembled coral virus draft genome with other known dsDNA viruses.
Discussion
Coral reefs are increasingly suffering from thermal anomalies and disease (Smith et al., 2006; Bruno et al., 2007; Lawrence et al., 2015; Soffer et al., 2015; Hughes et al., 2017). However, corals also demonstrate heterogeneity in response to various disturbances (Vega Thurber et al., 2014; Daniels et al., 2015; Zaneveld et al., 2016). This heterogeneity could result from genetic variation of the corals themselves and/or variation in the microbiome or virome of different hosts. Using conspecific Pocilloporidae coral samples from a natural bleaching event, we used metagenomics to show that corals exhibiting more extreme bleaching phenotypes have viromes with increased percentages of viral genes associated with NCLDVs or giant viruses. We then constructed a novel coral giant virus draft genome from these libraries. We additionally showed visual evidence of giant virus-like particles associated with host and algal symbiont cells. These combined data suggest that eukaryotic viral infection may be exacerbated by and/or contribute directly to bleaching phenotypes. This led us to formulate the following hypothesis to test with future experiments: Induction of viral infection during moderate temperature stress may result in earlier and potentially more severe loss of symbiont and host cells. Therefore, corals with these viral infections become visually bleached earlier in a thermal stress event despite being in the same environment as their non-bleached conspecific neighbors.
Microscopy Indicates Corals Are Infected With Multiple Eukaryotic Viral Types
Based on qualitative assessments, our EM imagery supports the presence of nucleocytoplasmic large DNA virus particles (Figures 2, 3) residing within corals cells. Images of what we interpret as either necrotic coral cells (i.e., ruptured cell membranes with intact and non-condensed nuclei) filled with ∼15–20 large (200–300 nm in diameter) VLPs, or viral factories producing these VLPs (Sicko-Goad and Walker, 1979; Raoult and Forterre, 2008; Lawrence et al., 2014; Akashi and Takemura, 2019; Rolland et al., 2019), indicate viral infection of bleaching corals (Figure 2). Similarly-shaped particles, which are larger in size (410–480 nm in diameter), are also found extracellularly (Figure 3), suggesting that these could be different VLPs or the same VLPs at different stages of their replication cycle. The imperfect icosahedral and concentric circle capsid morphology, as well as the size (200 to over 400 nm in diameter) and number (∼20 cell) of these particles, are diagnostic of NCLDVs (Colson et al., 2012, 2013). However, while one observed VLP was found with capsid fibrils, a feature sometimes seen in Mimiviruses (Rodrigues et al., 2015), most observed VLPs were not (Figure 3B). Further, these projections are not morphologically similar to the numerous and long (125–140 nm) protein filaments seen in Mimiviruses as they are relatively short (∼50 nm), less dense, and do not encircle the entire capsid (Rodrigues et al., 2015; Andrade et al., 2017). Additionally, the VLP in 3B does not have the typical icosahedral shape of Mimiviridae (Rodrigues et al., 2015). It has a more spherical shape reminiscent of Molliviridae, giant viruses infecting Acanthamoeba (Legendre et al., 2015).
EM also identified two types of VLPs alongside Symbiodiniaceae cells and within the symbiosome (Figure 4). Smaller than the giant VLPs discussed above, one of these algal-symbiont-associated VLPs are also structurally similar to NCLDV (e.g., icosahedral non-enveloped capsid structures in Figure 4A inset). Their 170–200 nm size range suggests these particles are more closely related to Phycodnaviruses, such as Chloroviruses, Coccolithoviruses, and Prasinoviruses. The second type of VLP was strikingly small (∼40 nm in diameter) and numerically more abundant on the exterior of the symbiont cell (Figure 4C). A Symbiodiniaceae + ssRNA virus that infects and kills algal symbionts in culture has been described based on genomic analysis (Correa et al., 2012; Levin et al., 2017), but to date no images of these viruses have been published. Members of the + ssRNA viruses that infect free-living dinoflagellates such as Heterocapsa circularisquama (HcRNAv) are similarly shaped and have capsids in the 40 nm size range of those observed within the bleaching corals here (Tomaru et al., 2004; Lawrence et al., 2014; Montalvo-Proaño et al., 2017; Weynberg et al., 2017). Thus, we speculate that these viruses may be small RNA viruses associated with the bleaching event. Since we did not sequence RNA from these samples, we cannot test this hypothesis from a genomic standpoint in this study.
Bleaching Corals Contain Relatively More Eukaryotic Viruses Than Neighboring Non-bleached Congenerics
Bleaching events are becoming increasingly common around the world. However, across a reef, observed patterns in bleaching may be heterogeneous, especially when bleaching intensity is moderate or mild (Edmunds, 1994; Ware et al., 1996; Guest et al., 2012; Wooldridge, 2014). In many cases localized stressors, including water flow, nutrient stress, or light exposure expedite bleaching (Hoegh-Guldberg, 1999; Houk et al., 2014). However, in this study we focused on corals that shared the local environment and appeared to experience near identical environmental conditions. We found that bleached and non-bleached coral metagenomes have significantly different viral communities (Figure 5). A shift in the relative percent similarities of certain viruses depending on health state may help explain, with further quantification, the role that these various viral groups play in reef health.
It is hypothesized that viruses have both beneficial and detrimental associations with corals. Coral mucosal phage are thought to protect the animal from bacterial pathogens (Leruste et al., 2012; Barr et al., 2013; Nguyen-Kim et al., 2015), while active viral infections within the coral may lead to coral disease (Soffer et al., 2013; Weynberg et al., 2015). However, there has been little empirical evidence that specific viral types (phage or eukaryotic) are associated with declines in coral health. Marhaver et al. (2008) first described the composition of eukaryotic viruses and phage in both bleached and healthy coral, but the authors could not specify whether viral assemblages significantly differed between these health states. Further in 2016, we showed that NCLDV-like particles and gene sequences were associated with a natural bleaching event in Australia, but we were unable to link these to variable phenotypes (Correa et al., 2016). In this experiment, bleached (B) viral metagenomes exhibited a significant relative increase in similarities to eukaryotic dsDNA viruses compared to those of non-bleached (NB) viral metagenomes (Figure 5 black vs. green dashed lines). In contrast, B viral metagenomes exhibited a significant relative decrease in similarities to dsDNA phage compared to NB viral metagenomes (Figure 5 green vs. black dashed lines). This provides the first quantitative evidence of a shift in viral assemblages between coral bleaching states.
As previously hypothesized these data suggest that phage production either protects the coral from bacterial pathogens and/or provides limiting nutrients to the system via the viral shunt (Rohwer and Thurber, 2009; Weitz et al., 2015; Thurber et al., 2017). Additionally, only B coral viral metagenomes had virophage annotations, supporting the inference that there is a bleaching-related shift in viral communities, whereby the coral microbiome is dominated by phage in the non-bleached state and eukaryotic viruses in the bleached state. During stress, eukaryotic viruses may take advantage of the weakened coral and Symbiodiniaceae states, potentially negatively impacting this symbiotic relationship through viral production and/or infection.
Due to the compositional nature of relative abundance data, we cannot confirm whether the reduction in phage genes is due to a decrease in host bacterial abundance during stress or an increase in gene sequences from eukaryotic viruses during bleaching. The mechanisms behind an increase of eukaryotic viruses during bleaching also remain unclear. Latent eukaryotic viruses may exist in non-bleached corals, and sublethal thermal stress could trigger lytic viral production that ultimately contribute to bleaching. Conversely, an increase in eukaryotic viruses could result from external viral opportunism of the weakened holobiont during thermal stress.
Metagenomics and UViG Analysis Suggests Corals Contain a Novel Giant Virus
Based on the presence of giant VLPs in the electron microscopy data (Figure 3) and the high percentage of similarities to NCLDV in the metagenomic data, we worked to assemble the genome of the first coral giant virus. While eukaryotic non-NCLDV virus families were significantly different between the bleached and non-bleached metagenomes, the low percent similarity (=5%) of these families may have predisposed them to the detection of significant differences between the bleaching phenotypes and thus the biological relevance of these differences is difficult to interpret. We therefore chose to focus on the NCLDV group for assembly because our viral metagenomes had the highest percent of similarities to this viral group (Table 2).
As measured by MI UViG standards (Roux et al., 2019) the final 192 contigs represent a low-quality draft genome of the first coral dsDNA virus. We compared our novel draft genome to known NCLDVs, like CroV and other marine eukaryotic viruses (Figure 6). If these contigs represent one coral giant virus, we have yet to resolve gaps within the genome and its sequence order. Therefore, there is the possibility that these contigs represent several coral metagenomic giant virus lineages (Schulz et al., 2020b). Based on our high percent similarities to Phycodnaviridae and our high contig recruitment to Mimiviridae, these lineages are likely from one or more unique viruses within the NCLDV. Community level classification of our final assembly (Table 3 and Supplementary Table 4) also suggests that this assembly represents a hybrid signal of distantly related eukaryotic viruses.
Additionally, while no chimeras were found in the final assembly, it does appear to be incomplete and many genes and sequences remain undefined. Comparison of our assembly to the NCBI environmental nr database resulted in no significant similarities, even when using a less conservative e-value cutoff. Only ∼20–25% of final assembly ORFs annotated to known NCVOG’s, IMG VR proteins, or KEGG genes, with most annotations being to cellular and viral genetic information and processing and no annotations to hallmark viral genes like the major capsid protein. Furthermore, taxonomic characterization of KEGG annotations resulted in 58% of annotations to an undefined organism. The next highest percentage, 38%, were to cnidarians. Similarly, the search to IMG VR proteins resulted in 52% of annotations being categorized into an undefined host. The search against the comprehensive NCBI nr database had a majority of hits to coral proteins (72%), however, overall, 70% of hits were to uncharacterized or hypothetical proteins. The highly undefined nature of this assembly makes it difficult to fully characterize. When cross-referencing our assembly functional annotations to assembly ORFs with similarities to Pocillopora damicornis genes, we found that 40–45% of these annotations shared homology with P. damicornis genes. This indicates that ∼40–45% of functional genes within this viral genome may be derived from a coral host.
Standardized bioinformatic pipelines for uncovering viruses via metagenomics are being developed, however assembling eukaryotic viral genomes from uncultured, multicellular, host-based systems is still an uncommon practice (Castro et al., 2019; Ponsero and Hurwitz, 2019; Schulz et al., 2020a). For our dataset, some techniques, such as metagenomic binning using nucleotide composition, contig abundance, and reference genomes (Schulz et al., 2020b), did not work for identifying viral contigs. This is likely due to the lack of known reference coral, or other closely related, virus genomes to help identify viral bins within the metagenomic sequencing depth used. Further, some eukaryotic viruses rely on and often share genomic elements with their host, limiting our ability to distinguish viral sequences from host (Pride et al., 2006). Additionally, a review of eukaryotic algae viruses by Coy et al. (2018) notes that omics approaches are better suited for novel viral characterization and assembly, but that such approaches remain challenging because of processes like amplification bias and chimeras during assembly (Coy et al., 2018). Such challenges have been resolved for phage (Roux et al., 2016, 2017), yet few solutions have been created for eukaryotic viruses.
Despite these challenges, we used a custom bioinformatic workflow and both reference-based and de novo techniques to generate this coral giant virus draft genome. Based on our genome comparison, this coral giant virus genome has no known close relatives. The phylogenetic placement of a coral giant virus is challenging due to the highly uncharacterized state of viral diversity, particularly from corals (R. V. Thurber et al., 2017). NCLDVs, like CroV, Acanthamoeba polyphaga mimivirus, and Megavirus chiliensis (Figure 6), are all very distantly related, originating from hosts that diverged from one another millions of years ago (Colson et al., 2012, 2013; Yutin et al., 2013; Gallot-Lavallée and Blanc, 2017; Schulz et al., 2017; Wilson et al., 2017). For example, if this coral virus does infect Symbiodiniaceae (an alveolate), the closest described and sequenced virus genome is known to infect a Stramenopile (i.e., Cafeteria roenbergensis), which is in a group of protists highly divergent from alveolates. Further, to our knowledge, there are no viral genomes published that infect the phylogenetically closer Rhizaria (Needham et al., 2019). Thus, the paucity of discovered protist viruses in the database (Needham et al., 2019) makes it difficult to conduct phylogenetic analyses of these viral genomes and genes. Regardless of the challenges of host-based viral metagenomics, here we began the genomic characterization a new coral giant virus, providing a pathway to better understand the role of the coral virome beyond what can be obtained through observational approaches.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA647466.
Author Contributions
The permits protocol OCE-1619697. RV, AT, AC, and AM designed the research. AT and AC conducted the field research. AM, SR, and RM performed the genomic and phylogenetic analysis. TS performed the electron microscopy. AM and RV wrote the manuscript. All authors contributed to revisions.
Funding
This work was funded by the NSF Ocean Sciences grant (#1635913) to RV, AC, AT, used for sequencing data generation, a Dimensions of Biodiversity NSF grant (#1442306) to RV used for sample collection, and NSF NRT grant (#1545188) to AM, used for a research assistantship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.555474/full#supplementary-material
References
Akashi, M., and Takemura, M. (2019). Co-isolation and characterization of two pandoraviruses and a Mimivirus from a riverbank in Japan. Viruses 11:1123. doi: 10.3390/v11121123
Andrade, A. C. D. S. P., Rodrigues, R. A. L., Oliveira, G. P., Andrade, K. R., Bonjardim, C. A., Scola, B. L., et al. (2017). Filling knowledge gaps for Mimivirus entry, uncoating, and morphogenesis. J. Virol. 91:e01335-17. doi: 10.1128/JVI.01335-17
Angly, F., Willner, D., Rohwer, F., Philip, H., and Tyson, G. (2012). Grinder: a versatile amplicon and shotgun sequence simulator. Nucleic Acids Res. 40:e94. doi: 10.1093/nar/gks251
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barr, J., Auro, R., Furlan, M., Whiteson, K., Erb, M., Pogliano, J., et al. (2013). Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. U.S.A. 110, 10771–10776. doi: 10.1073/pnas.1305923110
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Bruno, J. F., Selig, E. R., Casey, K. S., Page, C. A., Willis, B. L., Harvell, C. D., et al. (2007). Thermal stress and coral cover as drivers of coral disease outbreaks. PLoS Biol. 5:e124. doi: 10.1371/journal.pbio.0050124
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Carpenter, K. E., Abrar, M., Aeby, G., Aronson, R. B., Banks, S., Bruckner, A., et al. (2008). One-third of reef-building corals face elevated extinction risk from climate change and local impacts. Science 321, 560–563. doi: 10.1126/science.1159196
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Castro, C. J., Marine, R. L., Ramos, E., and Ng, T. F. F. (2019). The effect of variant interference on de novo assembly for viral deep sequencing. bioRxiv [Preprint]. doi: 10.1101/815480
Colson, P., de Lamballerie, X., Fournous, G., and Raoult, D. (2012). Reclassification of giant viruses composing a fourth domain of life in the new order megavirales. INT 55, 321–332. doi: 10.1159/000336562
Colson, P., De Lamballerie, X., Yutin, N., Asgari, S., Bigot, Y., Bideshi, D. K., et al. (2013). “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 158, 2517–2521. doi: 10.1007/s00705-013-1768-6
Correa, A. M. S., Ainsworth, T. D., Rosales, S. M., Thurber, A. R., Butler, C. R., and Vega Thurber, R. L. (2016). Viral outbreak in corals associated with an in situ bleaching event: atypical herpes-like viruses and a new megavirus infecting symbiodinium. Front. Microbiol. 7:127. doi: 10.3389/fmicb.2016.00127
Correa, A. M. S., Welsh, R. M., and Thurber, R. L. V. (2012). Unique nucleocytoplasmic dsDNA and +ssRNA viruses are associated with the dinoflagellate endosymbionts of corals. ISME J. 7:ismej201275. doi: 10.1038/ismej.2012.75
Costanza, R., de Groot, R., Sutton, P., van der Ploeg, S., Anderson, S. J., Kubiszewski, I., et al. (2014). Changes in the global value of ecosystem services. Glob. Environ. Change 26, 152–158. doi: 10.1016/j.gloenvcha.2014.04.002
Coy, S. R., Gann, E. R., Pound, H. L., Short, S. M., and Wilhelm, S. W. (2018). Viruses of eukaryotic algae: diversity, methods for detection, and future directions. Viruses 10:487. doi: 10.3390/v10090487
Daniels, C., Baumgarten, S., Yum, L. K., MIchell, C. T., Bayer, T., Arif, C., et al. (2015). Metatranscriptome analysis of the reef-building coral Orbicella faveolata indicates holobiont response to coral disease. Front. Mar. Sci. 2:62. doi: 10.3389/fmars.2015.00062
Edmunds, P. J. (1994). Evidence that reef-wide patterns of coral bleaching may be the result of the distribution of bleaching-susceptible clones. Mar. Biol. 121, 137–142. doi: 10.1007/BF00349482
Edwards, R. A., and Rohwer, F. (2005). Viral metagenomics. Nat. Rev. Microbiol. 3:nrmicro1163. doi: 10.1038/nrmicro1163
Efrony, R., Loya, Y., Bacharach, E., and Rosenberg, E. (2007). Phage therapy of coral disease. Coral Reefs 26, 7–13. doi: 10.1007/s00338-006-0170-1
Friedman, C. S., Wight, N., Crosson, L. M., VanBlaricom, G. R., and Lafferty, K. D. (2014). Reduced disease in black abalone following mass mortality: phage therapy and natural selection. Front. Microbiol. 5:78. doi: 10.3389/fmicb.2014.00078
Gallot-Lavallée, L., and Blanc, G. (2017). A glimpse of nucleo-cytoplasmic large DNA virus biodiversity through the eukaryotic genomics window. Viruses 9:17. doi: 10.3390/v9010017
Gélin, P., Postaire, B., Fauvelot, C., and Magalon, H. (2017). Reevaluating species number, distribution and endemism of the coral genus Pocillopora Lamarck, 1816 using species delimitation methods and microsatellites. Mol. Phylogenet. Evol. 109, 430–446. doi: 10.1016/j.ympev.2017.01.018
Geo Pertea (2018). gpertea/fqtrim: Fqtrim Release v0.9.7. Geneva: Zenodo, doi: 10.5281/zenodo.1185412
Guest, J. R., Baird, A. H., Maynard, J. A., Muttaqin, E., Edwards, A. J., Campbell, S. J., et al. (2012). Contrasting patterns of coral bleaching susceptibility in 2010 suggest an adaptive response to thermal stress. PLoS One 7:e33353. doi: 10.1371/journal.pone.0033353
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hayes, R. L., and Bush, P. G. (1990). Microscopic observations of recovery in the reef-building scleractinian coral, Montastrea annularis, after bleaching on a Cayman reef. Coral Reefs 8, 203–209. doi: 10.1007/BF00265012
Hoegh-Guldberg, O. (1999). Climate change, coral bleaching and the future of the world’s coral reefs. Mar. Freshwater Res. 50, 839–866. doi: 10.1071/mf99078
Houk, P., Benavente, D., Iguel, J., Johnson, S., and Okano, R. (2014). Coral reef disturbance and recovery dynamics differ across gradients of localized stressors in the Mariana Islands. PLoS One 9:e105731. doi: 10.1371/journal.pone.0105731
Hughes, T. P., Baird, A. H., Bellwood, D. R., Card, M., Connolly, S. R., Folke, C., et al. (2003). Climate Change, human impacts, and the resilience of coral reefs. Science 301, 929–933. doi: 10.1126/science.1085046
Hughes, T. P., Kerry, J. T., Álvarez-Noriega, M., Álvarez-Romero, J. G., Anderson, K. D., Baird, A. H., et al. (2017). Global warming and recurrent mass bleaching of corals. Nature 543:nature21707. doi: 10.1038/nature21707
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment Software Version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:nmeth.1923. doi: 10.1038/nmeth.1923
Lawrence, S. A., Davy, J. E., Wilson, W. H., Hoegh-Guldberg, O., and Davy, S. K. (2015). Porites white patch syndrome: associated viruses and disease physiology. Coral Reefs 34, 249–257. doi: 10.1007/s00338-014-1218-2
Lawrence, S. A., Floge, S. A., Davy, J. E., Davy, S. K., and Wilson, W. H. (2017). Exploratory analysis of Symbiodinium transcriptomes reveals potential latent infection by large dsDNA viruses. Environ. Microbiol. 19, 3909–3919. doi: 10.1111/1462-2920.13782
Lawrence, S. A., Wilson, W. H., Davy, J. E., and Davy, S. K. (2014). Latent virus-like infections are present in a diverse range of Symbiodinium spp. (Dinophyta). J. Phycol. 50, 984–997. doi: 10.1111/jpy.12242
Legendre, M., Lartigue, A., Bertaux, L., Jeudy, S., Bartoli, J., Lescot, M., et al. (2015). In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting acanthamoeba. PNAS 112, E5327–E5335. doi: 10.1073/pnas.1510795112
Leruste, A., Bouvier, T., and Bettarel, Y. (2012). Enumerating viruses in coral mucus. Appl. Environ. Microbiol. 78, 6377–6379. doi: 10.1128/AEM.01141-12
Levin, R. A., Voolstra, C. R., Weynberg, K. D., and van Oppen, M. J. H. (2017). Evidence for a role of viruses in the thermal sensitivity of coral photosymbionts. ISME J. 11, 808–812. doi: 10.1038/ismej.2016.154
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 106–110. doi: 10.1186/s13059-014-0550-8
Marhaver, K. L., Edwards, R. A., and Rohwer, F. (2008). Viral communities associated with healthy and bleaching corals. Environ. Microbiol. 10, 2277–2286. doi: 10.1111/j.1462-2920.2008.01652.x
Mokili, J. L., Rohwer, F., and Dutilh, B. E. (2012). Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2, 63–77. doi: 10.1016/j.coviro.2011.12.004
Montalvo-Proaño, J., Buerger, P., Weynberg, K. D., and van Oppen, M. J. H. (2017). A PCR-based assay targeting the major capsid protein gene of a dinorna-like ssRNA virus that infects coral photosymbionts. Front. Microbiol. 8:1665. doi: 10.3389/fmicb.2017.01665
Needham, D. M., Yoshizawa, S., Hosaka, T., Poirier, C., Choi, C. J., Hehenberger, E., et al. (2019). A distinct lineage of giant viruses brings a rhodopsin photosystem to unicellular marine predators. PNAS 116, 20574–20583. doi: 10.1073/pnas.1907517116
Nguyen-Kim, H., Bettarel, Y., Bouvier, T., Bouvier, C., Doan-Nhu, H., Nguyen-Ngoc, L., et al. (2015). Coral mucus is a hot spot for viral infections. Appl. Environ. Microbiol. 81, 5773–5783. doi: 10.1128/AEM.00542-15
Paez-Espino, D., Chen, I.-M. A., Palaniappan, K., Ratner, A., Chu, K., Szeto, E., et al. (2017). IMG/VR: a database of cultured and uncultured DNA Viruses and retroviruses. Nucleic Acids Res. 45, D457–D465. doi: 10.1093/nar/gkw1030
Pendleton, L., Comte, A., Langdon, C., Ekstrom, J. A., Cooley, S. R., Suatoni, L., et al. (2016). Coral reefs and people in a high-CO2 world: where can science make a difference to people? PLoS One 11:e0164699. doi: 10.1371/journal.pone.0164699
Pfeifer, C. R., Shomorony, A., Aronova, M. A., Zhang, G., Cai, T., Xu, H., et al. (2015). Quantitative analysis of mouse pancreatic islet architecture by serial block-face SEM. J. Struct. Biol. 189, 44–52. doi: 10.1016/j.jsb.2014.10.013
Pollock, F. J., Wood-Charlson, E. M., van Oppen, M. J. H., Bourne, D. G., Willis, B. L., and Weynberg, K. D. (2014). Abundance and morphology of virus-like particles associated with the coral Acropora hyacinthus differ between healthy and white syndrome-infected states. Mar. Ecol. Prog. Ser. 510, 39–43. doi: 10.3354/meps10927
Ponsero, A. J., and Hurwitz, B. L. (2019). The promises and pitfalls of machine learning for detecting viruses in aquatic metagenomes. Front. Microbiol. 10:806. doi: 10.3389/fmicb.2019.00806
Pride, D. T., Wassenaar, T. M., Ghose, C., and Blaser, M. J. (2006). Evidence of host-virus co-evolution in tetranucleotide usage patterns of bacteriophages and eukaryotic viruses. BMC Genomics 7:8. doi: 10.1186/1471-2164-7-8
Raoult, D., and Forterre, P. (2008). Redefining viruses: lessons from Mimivirus. Nat. Rev. Microbiol. 6, 315–319. doi: 10.1038/nrmicro1858
Rodrigues, R. A. L., dos Santos Silva, K., Dornas, F. P., de Oliveira, D. B., Magalhães, T. F. F., Santos, D. A., et al. (2015). Mimivirus fibrils are important for viral attachment to the microbial world by a diverse glycoside interaction repertoire. J. Virol. 89, 11812–11819. doi: 10.1128/JVI.01976-15
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Rohwer, F., and Edwards, R. (2002). The phage proteomic tree: a genome-based taxonomy for Phage. J. Bacteriol. 184, 4529–4535. doi: 10.1128/JB.184.16.4529-4535.2002
Rohwer, F., and Thurber, R. V. (2009). Viruses manipulate the marine environment. Nature 459, 207–212. doi: 10.1038/nature08060
Rolland, C., Andreani, J., Cherif Louazani, A., Aherfi, S., Francis, R., Rodrigues, R., et al. (2019). Discovery and further studies on giant viruses at the IHU mediterranee infection that modified the perception of the virosphere. Viruses 11:312. doi: 10.3390/v11040312
Roux, S., Adriaenssens, E. M., Dutilh, B. E., Koonin, E. V., Kropinski, A. M., Krupovic, M., et al. (2019). minimum information about an uncultivated virus genome (MIUViG). Nat. Biotechnol. 37, 29–37. doi: 10.1038/nbt.4306
Roux, S., Emerson, J. B., Eloe-Fadrosh, E. A., and Sullivan, M. B. (2017). Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 5:e3817. doi: 10.7717/peerj.3817
Roux, S., Solonenko, N. E., Dang, V. T., Poulos, B. T., Schwenck, S. M., Goldsmith, D. B., et al. (2016). Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 4:e2777. doi: 10.7717/peerj.2777
Ruby, J. G., Bellare, P., and Derisi, J. L. (2013). PRICE: software for the targeted assembly of components of (Meta) genomic sequence data. G3 3, 865–880. doi: 10.1534/g3.113.005967
Schmidt, T. M., DeLong, E. F., and Pace, N. R. (1991). Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J. Bacteriol. 173, 4371–4378. doi: 10.1128/jb.173.14.4371-4378.1991
Schulz, F., Andreani, J., Francis, R., Khalil, J. Y. B., Lee, J., Scola, B. L., et al. (2020a). Advantages and limits of metagenomic assembly and binning of a giant virus. bioRxiv [Preprint]. doi: 10.1101/2020.01.10.902254
Schulz, F., Roux, S., Paez-Espino, D., Jungbluth, S., Walsh, D. A., Denef, V. J., et al. (2020b). Giant virus diversity and host interactions through global metagenomics. Nature 578, 432–436. doi: 10.1038/s41586-020-1957-x
Schulz, F., Yutin, N., Ivanova, N. N., Ortega, D. R., Lee, T. K., Vierheilig, J., et al. (2017). Giant viruses with an expanded complement of translation system components. Science 356, 82–85. doi: 10.1126/science.aal4657
Sicko-Goad, L., and Walker, G. (1979). Viroplasm and large virus-like particles in the dinoflagellateGymnodinium uberrimum. Protoplasma 99, 203–210. doi: 10.1007/BF01275735
Smith, J. E., Shaw, M., Edwards, R. A., Obura, D., Pantos, O., Sala, E., et al. (2006). Indirect effects of algae on coral: algae-mediated, microbe-induced coral mortality. Ecol. Lett. 9, 835–845. doi: 10.1111/j.1461-0248.2006.00937.x
Soffer, N., Brandt, M. E., Correa, A. M., Smith, T. B., and Thurber, R. V. (2013). Potential role of viruses in white plague coral disease. ISME J. 8:ismej2013137. doi: 10.1038/ismej.2013.137
Soffer, N., Zaneveld, J., and Thurber, R. V. (2015). Phage–bacteria network analysis and its implication for the understanding of coral disease. Environ. Microbiol. 17, 1203–1218. doi: 10.1111/1462-2920.12553
Stothard, P., and Wishart, D. S. (2005). Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539. doi: 10.1093/bioinformatics/bti054
Streit, W. R., and Schmitz, R. A. (2004). Metagenomics – the key to the uncultured microbes. Curr. Opin. Microbiol. 7, 492–498. doi: 10.1016/j.mib.2004.08.002
Sweet, M., and Bythell, J. (2017). The role of viruses in coral health and disease. J. Inverteb. Pathol. 147, 136–144. doi: 10.1016/j.jip.2016.12.005
Tang, P., and Chiu, C. (2010). Metagenomics for the discovery of novel human viruses. Future Microbiol. 5, 177–189. doi: 10.2217/fmb.09.120
Thurber, R. L. V., Barott, K. L., Hall, D., Liu, H., Rodriguez-Mueller, B., Desnues, C., et al. (2008). Metagenomic analysis indicates that stressors induce production of herpes-like viruses in the coral Porites compressa. PNAS 105, 18413–18418. doi: 10.1073/pnas.0808985105
Thurber, R. V., Payet, J. P., Thurber, A. R., and Correa, A. M. S. (2017). Virus–host interactions and their roles in coral reef health and disease. Nat. Rev. Microbiol. 15, 205–216. doi: 10.1038/nrmicro.2016.176
Thurber, R. V., Willner-Hall, D., Rodriguez-Mueller, B., Desnues, C., Edwards, R. A., Angly, F., et al. (2009). Metagenomic analysis of stressed coral holobionts. Environ. Microbiol. 11, 2148–2163. doi: 10.1111/j.1462-2920.2009.01935.x
Tomaru, Y., Katanozaka, N., Nishida, K., Shirai, Y., Tarutani, K., Yamaguchi, M., et al. (2004). Isolation and characterization of two distinct types of HcRNAV, a single-stranded RNA virus infecting the bivalve-killing microalga Heterocapsa circularisquama. Aquat. Microb. Ecol. 34, 207–218. doi: 10.3354/ame034207
van Oppen, M. J. H., Leong, J.-A., and Gates, R. D. (2009). Coral-virus interactions: a double-edged sword? Symbiosis 47, 1–8. doi: 10.1007/BF03179964
Vega Thurber, R. L., Burkepile, D. E., Fuchs, C., Shantz, A. A., McMinds, R., and Zaneveld, J. R. (2014). Chronic nutrient enrichment increases prevalence and severity of coral disease and bleaching. Glob. Chang Biol. 20, 544–554. doi: 10.1111/gcb.12450
Vicedomini, R., Vezzi, F., Scalabrin, S., Arvestad, L., and Policriti, A. (2013). GAM-NGS: genomic assemblies merger for next generation sequencing. BMC Bioinformatics 14(Suppl. 7):S6. doi: 10.1186/1471-2105-14-S7-S6
Ware, J. R., Fautin, D. G., and Buddemeier, R. W. (1996). Patterns of coral bleaching: modeling the adaptive bleaching hypothesis. Ecol. Model. 84, 199–214. doi: 10.1016/0304-3800(94)00132-4
Weitz, J. S., Stock, C. A., Wilhelm, S. W., Bourouiba, L., Coleman, M. L., Buchan, A., et al. (2015). A multitrophic model to quantify the effects of marine viruses on microbial food webs and ecosystem processes. ISME J. 9, 1352–1364. doi: 10.1038/ismej.2014.220
Weynberg, K. D., Neave, M., Clode, P. L., Voolstra, C. R., Brownlee, C., Laffy, P., et al. (2017). Prevalent and persistent viral infection in cultures of the coral algal endosymbiont Symbiodinium. Coral Reefs 36, 773–784. doi: 10.1007/s00338-017-1568-7
Weynberg, K. D., Voolstra, C. R., Neave, M. J., Buerger, P., and van Oppen, M. J. H. (2015). From cholera to corals: viruses as drivers of virulence in a major coral bacterial pathogen. Sci. Rep. 5, 1–9. doi: 10.1038/srep17889
Wilson, W. H., Gilg, I. C., Moniruzzaman, M., Field, E. K., Koren, S., LeCleir, G. R., et al. (2017). Genomic exploration of individual giant ocean viruses. ISME J. 11, 1736–1745. doi: 10.1038/ismej.2017.61
Wooldridge, S. A. (2014). Assessing coral health and resilience in a warming ocean: why looks can be deceptive. BioEssays 36, 1041–1049. doi: 10.1002/bies.201400074
Yamashita, A., Sekizuka, T., and Kuroda, M. (2016). VirusTAP: viral genome-targeted assembly pipeline. Front. Microbiol. 7:32. doi: 10.3389/fmicb.2016.00032
Yutin, N., Colson, P., Raoult, D., and Koonin, E. V. (2013). Mimiviridae: clusters of orthologous genes, reconstruction of gene repertoire evolution and proposed expansion of the giant virus family. Virol. J. 10:106. doi: 10.1186/1743-422X-10-106
Yutin, N., Wolf, Y. I., Raoult, D., and Koonin, E. V. (2009). Eukaryotic large nucleo-cytoplasmic DNA viruses: clusters of orthologous genes and reconstruction of viral genome evolution. Virol. J. 6:223. doi: 10.1186/1743-422X-6-223
Zaneveld, J. R., Burkepile, D. E., Shantz, A. A., Pritchard, C. E., McMinds, R., Payet, J. P., et al. (2016). Overfishing and nutrient pollution interact with temperature to disrupt coral reefs down to microbial scales. Nat. Commun. 7, 1–12. doi: 10.1038/ncomms11833
Keywords: marine metagenomics, nucleocytoplasmic large DNA virus, giant virus, virus like particle, virome, Symbiodiniaceae
Citation: Messyasz A, Rosales SM, Mueller RS, Sawyer T, Correa AMS, Thurber AR and Vega Thurber R (2020) Coral Bleaching Phenotypes Associated With Differential Abundances of Nucleocytoplasmic Large DNA Viruses. Front. Mar. Sci. 7:555474. doi: 10.3389/fmars.2020.555474
Received: 24 April 2020; Accepted: 28 August 2020;
Published: 06 October 2020.
Edited by:
Michael Sweet, University of Derby, United KingdomReviewed by:
Karen Dawn Weynberg, The University of Queensland, AustraliaPatrick William Laffy, Australian Institute of Marine Science (AIMS), Australia
Copyright © 2020 Messyasz, Rosales, Mueller, Sawyer, Correa, Thurber and Vega Thurber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adriana Messyasz, bWVzc3lhc3phQGdtYWlsLmNvbQ==