 Tianxiang Gao1
Tianxiang Gao1 Yongshuang Xiao
Yongshuang Xiao- 1Fisheries College, Zhejiang Ocean University, Zhoushan, China
- 2Key Laboratory of Sustainable Development of Polar Fishery of Ministry of Agriculture, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, China
- 3Center for Ocean Mega-Science, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 4The Key Laboratory of Mariculture (Ocean University of China), Ministry of Education, Qingdao, China
The Pleistocene climate oscillations have been proven to have profound influences in the evolutionary processes of marine species and left significantly tractable footprints in genomes of marine fauna. However, such history remains unknown for the Japanese grenadier anchovy Coilia nasus, a relatively large fish found in marine, freshwater and brackish water. In order to provide insight into the phylogeographic pattern and historical demography of C. nasus, we employed the mtDNA D-loop and Cyt b as molecular markers to estimate the genetic variation among populations of C. nasus. We took samples from 18 populations throughout the distribution of C. nasus. Partial sequences of mtDNA D-loop (n = 424) and Cyt b (n = 129) were obtained. Four phylogeographic lineages were revealed by the mtDNA D-loop analysis. Frequency changes of the four lineages throughout the species’ distribution indicated limited genetic exchange among three major geographic regions. Patterns of isolation-by-distance were detected among the populations. Neutrality test, mismatch distribution, and network analyses consistently showed that all the four lineages had populations with relatively stable over time and then subjected to sudden expansion during the late-Pleistocene era (c. 20,000–75,000 years ago). The phylogeographic and demographic history of C. nasus was highly influenced by climatic fluctuations in the Pleistocene. Geographic isolation resulted from Pleistocene climatic fluctuations might have profound impacts on the distribution pattern of genetic variation of C. nasus. Our result also indicated that the population expansion event for C. nasus occured along with habitat continuous expansion during the inter-glacial period.
Introduction
In the Northern Hemisphere, the Pleistocene climate oscillations have been proved to have profound influences in the evolutionary processes of marine species and left significantly tractable footprints in genomes of marine fauna (Hewitt, 1996, 1999, 2004; Bernatchez and Wilson, 1998; Kitamura et al., 2000; Schmitt, 2007). Representative researches of the origins and evolutions corresponding to glacial refugia and recolonization route have been well studied in many terrestrial and aquatic species (Teacher et al., 2011).
The marine species were always thought to be less sensitive to the effects of Pleistocene climatic oscillations (Hewitt, 2000), however, recent studies have indicated that the Pleistocene glaciations had profound influences on the evolutionary process of many coastal marine species (Avise, 2000; Liu et al., 2007, Wilson and Veraguth, 2010; Liu et al., 2011a). The northwestern (NW) Pacific is characterized by marginal seas and shallow continental shelves, which were deeply impacted by Pleistocene glacial cycles. As one of the largest marginal seas in the northwest Pacific Ocean, most areas of the East China Sea Shelf were exposed during the period of the Pleistocene glaciation maximum (Xu and Oda, 1999). During the last interglacial period, the transgression has happened in the East China Sea Shelf, which caused the coastline to migrate from the Okinawa Trough to the coast of Bohai bay with about 1200 km (Wang, 1999; Xu and Oda, 1999). The sea level rose together to create a large area of newly formed marine habitats, allowing coastal marine species to expand their habitat ranges (Dodson et al., 2007; Liu et al., 2007; Xu et al., 2009; Wilson and Veraguth, 2010). Suitable DNA markers can be used to examine the genetic effects originated from the climatic oscillations. Compared with marine species, anadromous species were more vulnerable to the periodic climatic oscillations in the Northern Hemisphere (Consuegra et al., 2002; Mäkinen and Merilä, 2008; Aldenhoven et al., 2010). For example, recent study shows that the divergence between the resident freshwater and anadromous pairs of the threespine stickleback (Gasterosteus aculeatus) dates to the Late Pleistocene or Early Holocene periods (Bell and Foster, 1994). Phylogeographic studies of the anadromous Atlantic salmon showed that the Baltic Sea basin populations were possibly originated from south-eastern glacial-refugia existed during the Last Glacial Maximum (LGM) period (Tonteri et al., 2005). Historically, the Yangtze River was greatly influenced by the Late Pleistocene climatic oscillations and was likely to be an inland river due to the low sea level, headward erosion and dry climates during the LGM (Yang, 1986; Xiao et al., 2003). The Huai River is much younger than the Yangtze River. The existence of special sediment proves that the Huai River was formed later than 13 ka (Yang et al., 2010a, 2012). However, a series of geological activities have resulted in the decline of the water level of the Huai River by 13 meters since 2 ka BP (Yang et al., 2010a, 2012). For these reasons, studies on the phylogeographic and demographic histories of the anadromous species that inhabit Yangtze River, Huai River and the NW Pacific are helpful to illuminate the effects of Pleistocene climatic fluctuations and geographical environment.
The Japanese grenadier anchovy, Coilia nasus, is one of the most commercially important fishery resources along the NW Pacific Ocean coasts. C. nasus is widely distributed along the coastal waters of China, the western coast of the Korea, and the Ariake Sea of Japan. The scientific name of this species is always written as C. ectenes. However, as a priority, C. ectenes should be synonymized with C. nasus (Jordan and Seale, 1905). The resources for C. nasus have dramatically declined in the past few years, due to the overexploitation and deterioration of the habitat environment (Cai et al., 1980; Duan et al., 2012).
Coilia nasus has two ecotypes with anadromous form and freshwater form (Cheng, 2011). The anadromous grenadier anchovy spends most of its life in the inshore sea and ascends estuaries and rivers for breeding during its extensive migration, whereas freshwater grenadier anchovy always inhabits freshwater (Yuan et al., 1980; Cheng, 2011). In Japan, C. nasus was only found in the Ariake Sea, which spawned in rivers flowing into the Ariake Bay (Takita, 1967; Whitehead et al., 1988). In China, the anadromous form of C. nasus even penetrated to the Yangtze River for more than 1000 km during the spawning season, whereas the freshwater form inhabited the streams and lakes in the middle reaches of the Yangtze River and the Huai River (Yuan et al., 1976, 1980; Cheng, 2011). Compared to anadromous form, freshwater form was characterized by earlier age at maturity, shorter maxilla, smaller egg size, lower level of growth rate and smaller body size (Yuan et al., 1976; Ni et al., 1990). Although the anadromous and freshwater forms used to be regarded as different species according to their morphological and ecological differences (Yuan et al., 1976; Cheng, 2011), recent studies demonstrated that the genetic differentiation between the two forms was below the species level (Tang et al., 2007; Yang et al., 2010b; Xu et al., 2020). Until now, enough information about the phylogeographic pattern and historical demography among the freshwater and anadromous populations is missing in the Yangtze River. Previous study on C. nasus has shown that the genetic differentiation between Japanese and Chinese populations may result from life-history traits, ecology factors, and genetic isolation following the glacial retreat (Yang et al., 2011). A deep genetic break was observed among populations of C. nasus based on mtDNA Cyt b information (Ma et al., 2010). The existence of some highly diverged sequences (29 fixed substitutions) in the Fuzhou and Wuhan populations was the main reason for the deep genetic break (Ma et al., 2010).
Population demography and distribution of C. nasus might have been greatly impacted by the Pleistocene climatic oscillations. The anadromous populations of C. nasus prefer estuarine habitats. Most areas of this type are located in the East China Shelf, which was exposed by the lower sea level during the LGM. The Yangtze River, which was also profoundly affected by the lowed sea level in the LGM, is the species’ prime spawning area. Thus, we speculate that the phylogeographic and demographic history of C. nasus might be greatly influenced by the Pleistocene climatic fluctuations. Furthermore, the freshwater forms inhabiting in the middle reaches of the Yangtze River might also have originated from the freshwater refugium in the Yangtze River during the last glaciation. We also presumed that the habitat range expansion following the last interglacial period might result in population expansion for C. nasus.
Intraspecific phylogeographical studies have been proven to be important in testing biogeographical hypotheses and making insight into historical events (Bermingham and Martin, 1998). In addition, few studies have elaborated the genetic structure, demography, and evolutionary process for the freshwater and anadromous populations of C. nasus. The mtDNA is useful for phylogeography and evolutionary studies of animal populations. The mtDNA was characterized with non-recombining rapidly evolving rate, which can be used to detect the past isolations (e.g., in glacial refugia) and secondary contacts from isolated populations (Avise, 2000; Hewitt, 2000). Furthermore, variation in nucleotide level was effective on the inferences of historical demography analysis (Drummond et al., 2005; Ho and Shapiro, 2011). In this study, we analyzed the partial mtDNA D-loop and Cyt b sequences to reveal the phylogeographic pattern and historical demography of C. nasus across its range. The objective of this study was to obtain a comprehensive description of the phylogeographic pattern and historical demography, and then clarify the influence of Pleistocene climate oscillations on the evolutionary history of this species. Also, we aim to determine the glacial refugia for the freshwater populations during the Pleistocene period.
Materials and Methods
Ethics Statement
All methods were carried out in line with the relevant principle and directions in China.
Sampling and Sequencing
A total of 424 individuals were collected from 18 localities throughout the distribution of C. nasus from 2006 to 2013 (Table 1 and Figure 1). Five specimens of C. grayii were also collected from the Pearl River as the outgroup. All individuals were identified based on morphological features, and a piece of muscle tissue was obtained from each individual and preserved in 95% ethanol or frozen for DNA extraction. Genomic DNA was isolated from the muscle tissue by proteinase K digestion and purified by a standard phenol-chloroform method. The first hypervariable segment of the mtDNA D-loop was amplified with fish primers CR-F (5′-CCACCACTTAGCTCCCAA-3′) and CR-R (5′-ACCAGATGCCAGGAATAG-3′). The 5′ end of the mtDNA Cyt b gene was amplified for 129 individuals with the primers L14734 (5′-AAC CAC CGT TGT TAT TCA ACT-3′) and H15149 (5′-CTC AGA ATG ACA TTT GTC CTC A-3′; Zhang et al., 2007).
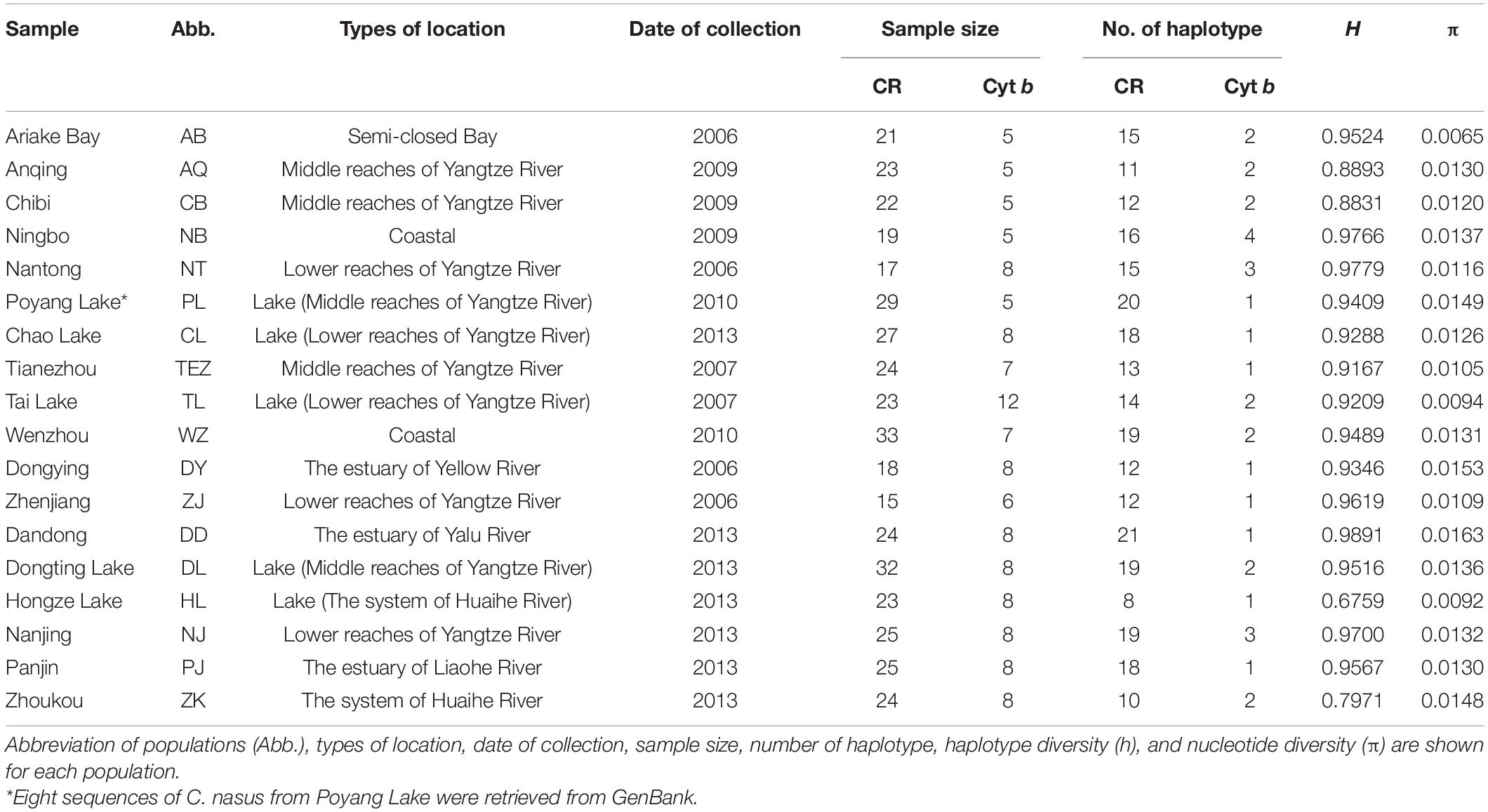
Table 1. Sampling information and molecular diversity indices for the mtDNA D-loop sequences of Coilia nasus.
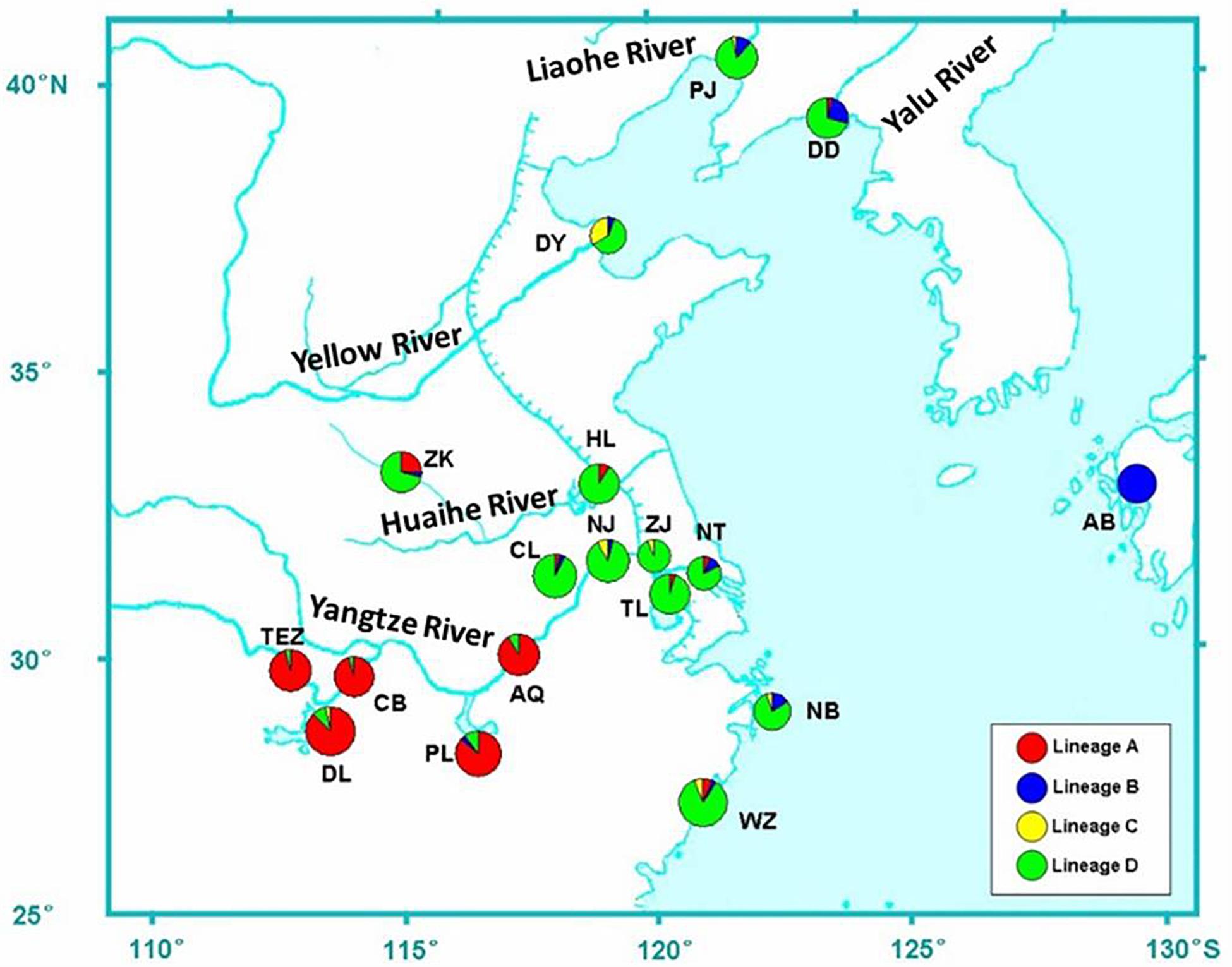
Figure 1. Sample locations of C. nasus. Populations are marked by abbreviations that correspond to Table 1. Pie charts show the frequencies of lineages A, B, C, and D (see results for determining the four lineages).
The PCR system with 1.25 U Taq DNA polymerase (Takara Co.), 10–100 ng template DNA, 200 nM forward and reverse primers, 200 μM of each dNTP, 10 mM Tris–HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, was used in the present study. The PCR profile included denaturation at 95°C for 3 min; 40 cycles of denaturation at 94°C for 45 s, annealing at 50°C for45 s, and extension at 72°C for 45 s; and extension at 72°C for 5 min in a Biometra thermal cycler. PCR products were purified using the AxyPrep PCR Clean-up Kit [Axygen Biotechnology (Hangzhou) Limited] and were further sequenced on an ABI 3730XL automated sequencer (Sunny Life Technologies Corp., Shanghai, China and Life Technologies, Shanghai, China). Additionally, mtDNA Cyt b sequences for 12 individuals of C. nasus and two individuals of C. grayii were also retrieved from GenBank (Table 2).

Table 2. The origin of the homologous cytochrome b sequences of Coilia nasus and Coilia grayii.
Data Analysis
We used the DNASTAR software (DNASTAR, Inc.) to edit and align all sequences in the study. In order to prevent bias in the estimation of genetic parameters for mtDNA Cyt b, we deleted the tRNAGlu sequence from the 5′end of mtDNA Cyt b fragment. We used the program ARLEQUIN to calculate the population genetic indices such as haplotype diversity (h), nucleotide diversity (π), polymorphic sites, haplotypes distribution, transversions, transitions, and indels (Excoffier and Laval, 2007).
We used the software of MrBayes 3.1.2 to construct the phylogenetic relationships among haplotypes (Ronquist and Huelsenbeck, 2003) [44]. We also used the software of MrModeltest 2.3 combined with PAUP 4.0b10 to obtain the best fit model (HKY + I + G) for the MrBayes setting (BIC; Swofford, 2002; Nylander, 2004). Bayesian searches included four chains. Each chain was run for thirty million generations with a tree sampling frequency of every 100 generations, and the first 25% of the resulting trees were discarded as burn-ins. The haplotype tree was visualized by FigTree (ver.1.3.1; http://tree.bio.ed.ac.uk/software/figtree/). Genealogical structures among haplotypes for the two molecular markers were further evaluated based on the minimum spanning tree (MST) analysis calculated by ARLEQUIN. We also used homologous mtDNA Cyt b sequences of C. nasus and C. grayii to construct the MST to verify the relationships between them (Ma et al., 2010).
We employed the quation μ = dA/2T to estimate the nucleotide substitution rate for grenadier anchovy, where T is an estimate of 3.6 Myr divergence between C. nasus and C. ectenes [this sample should actually be a specimen of C. grayii from Lavoué et al. (2013)], and dA was the net average genetic distance estimated using the homologous sequences of mtDNA D-loop. The genetic distance between lineages originated from the equation dA = dXY – (dX + dY)/2 was calculated based on the Tamura and Nei model using MEGA 3.1 (Kumar et al., 2004). Using this method, the lineage-specific divergence rate for mtDNA D-loop was estimated at about 1.34%/Myr. A 10-fold faster mutation rate was applied for population-level estimates for two reasons: first, the divergence rate with 13.4%/Myr is near the high end of estimates reported for sardines fishes (Bowen et al., 2006); second, Ho et al. (2008) reported up to 10 × elevated rates on short time scales, so an order-of-magnitude range of rate estimates should bracket the most extreme plausible cases.
We used the AMOVA analysis to estimate the population structure of C. nasus throughout the distribution of species (Excoffier et al., 1992). According to the geographic pattern, three groups, including the middle reaches of the Yangtze River group, the low reaches of the Yangtze River and the Chinese coastal group, and the Ariake Sea group, were designed for the AMOVA analyses. Ten thousands permutations were used to test the significance of covariances component from different assembly levels. Pairwise different values among the populations were also carried out in the examined ranges (Excoffier et al., 1992). Ten thousands permutations were also used to test the significance of the ΦST for each pairwise comparison. All the parameters of the population structure were calculated using the ARLEQUIN software.
We used the Google Earth (Version 6) to obtain the accurate geographical distances along with coast lines among localities. Then, we used Mantel tests implemented in GeneAlex to calculate the corelations between ΦST/(1-ΦST) and standardized geographical distance (Peakall and Smouse, 2006). We used the neutrality test, mismatch distribution analyses implemented in the ARLEQUIN to check population historical demography for C. nasus based on the mtDNA D-loop (Fu, 1997). The Bayesian skyline analyses were used to estimate the fluctuations of effective population sizes (ESS) for C. nasus (Drummond et al., 2005). We used the beast 1.7.5 software to create the Bayesian skyline plot (BSP) based on the mtDNA D-loop datasets (Drummond et al., 2012). Genealogies were combined from 1 × 108 steps with a burn-in of 1 × 107 lineages. To obtain the effective convergence, we performed multiple analyses that were run for 5 × 108 iterations with a burn-in of 107 under the HKY + I + G model, a strict molecular clock and stepwise skyline model. The results of 10 times runs for C. nasus were integrated using LogCombiner software, and the values of ESS for each parameter all exceeded 200. The skyline plot was generated by Tracer 1.5 (Rambaut and Drummond, 2007).
Results
Genetic Diversity of C. nasus
The mtDNA D-loop (624–703 bp) of C. nasus contained one to four copies of tandemly repeated 38 bp sequence units. After the removal of the tandemly repeated sequences and a conserved 10 bp portion of the tRNAPro gene, nucleotide sequence with length of 465 bp mtDNA D-loop was determined in 424 individuals. The mtDNA D-loop alignment consisted of 108 polymorphic sites, of which 88 were transition sites, 19 were transversion sites, and 7 were indels, yielding 171 haplotypes. One hundred and twenty-four haplotypes (72.5%) were represented by a single sequence. Forty-one of the remaining haplotypes were shared by more than one population and the last remaining 6 haplotypes were shared by at least two individuals in the same population. The average values of intrapopulation genetic diversity indices showed that a pattern with both high levels of haplotype diversity (h = 0.97) and nucleotide diversity (π = 0.02) were detected in the examined range.
The mtDNA Cyt b data set, with an aligned fragment length of 402 bp, was obtained from 139 individual from 8 localities. A total of 9 substitutions were detected in the mtDNA Cyt b fragment with 8 transitions and 1 transversions, which defined 10 haplotypes. Seven of the ten haplotypes were represented by a single individual. The average values of haplotype diversity (h) and nucleotide diversity (π) for all the examined populations were 0.455 and 0.001, respectively.
Phylogenetic Relationships and Four Lineages
Four genealogical clades (labeled A, B, C, and D) were detected in the phylogenetic tree under the HKY + I + G model based on the mtDNA D-loop data set (Figure 2). The MST analysis also indicated that four distinct lineages with three or six mutational steps corresponding to the phylogenetic tree were existed in the examined range (Figure 3). The MST was characterized by star-like structure with common haplotypes distributing in the center of the structure (Figure 3). The genetic distance among four clades were also calculated as follows: A/B, 0.0270; A/C, 0.0236; A/D, 0.0296; B/C, 0.0145; B/D, 0.0252; and C/D, 0.0220. According to the population-level divergence rate (13.4% per Myr), the divergence events among these genealogical clades happened at about 110,000–220,000 years ago.
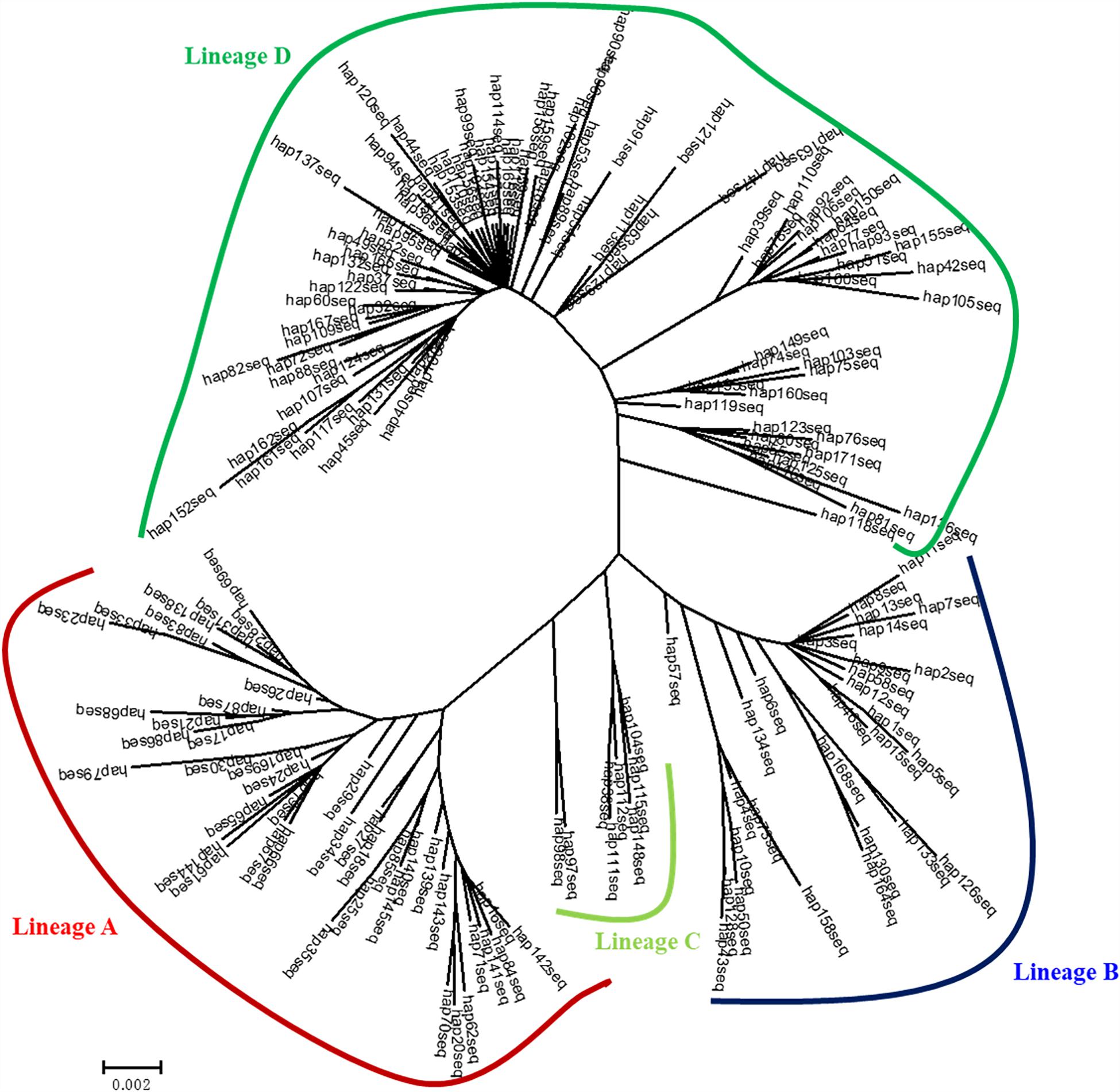
Figure 2. Bayesian tree for mtDNA D-loop haplotypes for C. nasus constructed under the HKY + I + G model using MRBAYES. Numbers above branches show posterior probabilities of nodes.
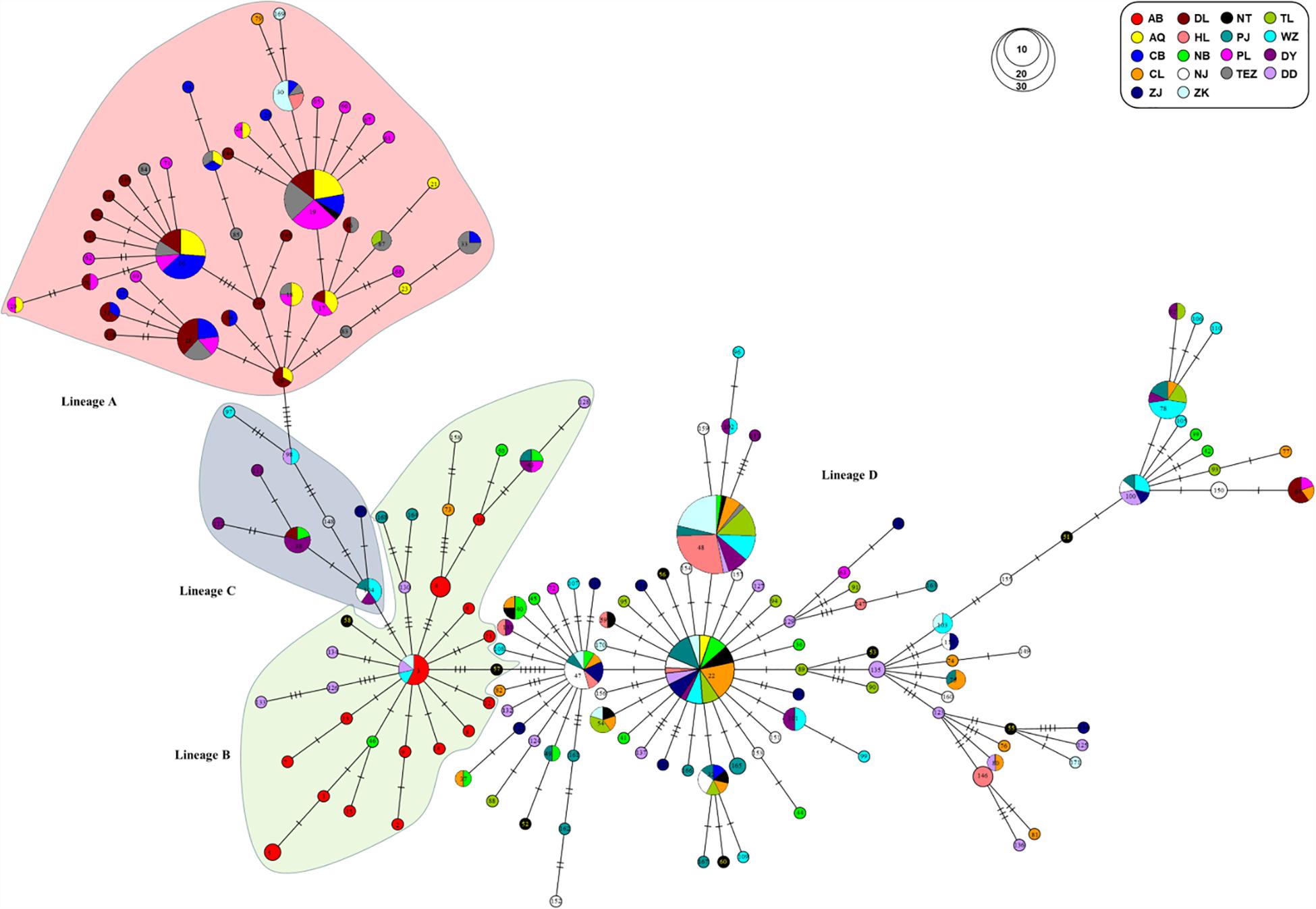
Figure 3. MST showed genetic relationship among mtDNA D-loop haplotypes for four lineages of C. nasus.
The distribution of haplotype frequencies for the four genealogical clades showed significantly geographic differentiation in the examined ranges. The populations along the middle reaches of the Yangtze River were mainly occupied by the lineage A, which included 44 haplotypes with 132 individuals. In addition, low frequencies of the haplotypes from lineage A were also detected in the six populations from the lower reaches of the Yangtze River and the Huai River (Table 1 and Figure 1). Additionally, two haplotypes of lineages A (hap97 and hap98) were also found in Dandong and Wenzhou. Five mutational steps existed between these two haplotypes and other haplotypes of lineage A (Figure 3). The frequency of lineage B (29 haplotypes, 41 individuals), which was sympatric with lineage D (92 haplotypes, 237 individuals) in the populations from the coast of China, declined along the coast from Dandong to Wenzhou. The lineage B haplotypes were also observed at low frequency in those populations from the middle and lower reaches of the Yangtze River and the Huai River system, such as Poyang Lake, Chao Lake, Nanjing, Nantong, and Zhoukou. Only lineage B haplotypes were detected in the Ariake Bay. The frequency of lineage D was higher than that of lineage B in the examined populations. Lineage D also was found at low frequency in populations from the middle reaches of the Yangtze River. The haplotypes of lineage C (6 haplotypes, 14 individuals) were distributed in populations from the coast of China except Dandong, and with a high frequency (33.3%) in Dongying. The lineage C haplotypes also were found at a low frequency in the Yangtze River basin, such as in Dongting Lake, Nanjing and Zhenjiang.
For the mtDNA Cyt b sequences, the MST of haplotypes revealed the C. nasus and C. grayii were separated from each other by 10 mutational steps (d = 2.5%). The MST for mtDNA Cyt b haplotypes was also characterized by star-like structure for C. nasus and C. grayii (Figure 4). For C. nasus, two main haplotypes (hap1 and hap3) were separated from each other only by one mutational step (Figure 4). For C. grayii, 8 haplotypes were also divergent from one to another by one mutational step. Furthermore, the MST of C. grayii also suggested the sequences of C. nasus from Ma et al. (2010) and Qiao et al. (2013) were within the lineage of C. grayii (Figure 4). As a consequence, these sequences reported by Ma et al. (2010) and Qiao et al. (2013) should belong to the specimen of C. grayii.
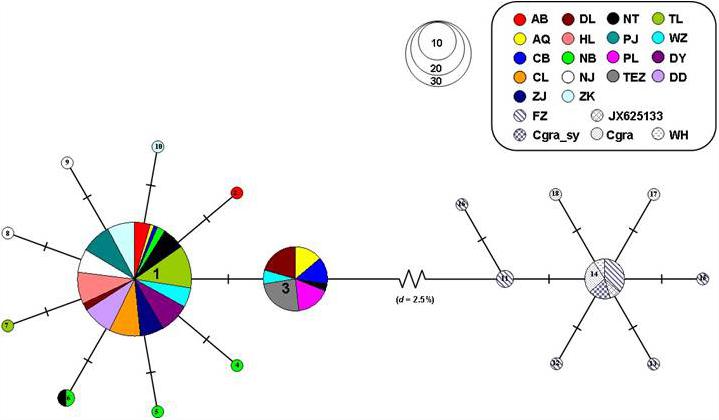
Figure 4. MST showed genetic relationship among mtDNA Cyt b haplotypes for C. nasus.
Population Structure
AMOVAs based on the geographic scale were carried out and revealed 50.92% variation among groups (P < 0.01) and 1.32% differentiation among populations (P < 0.01). The values of ΦCT and ΦSC were calculated as follows: 0.558 (P < 0.01) and 0.022 (P < 0.01). The pairwise ΦST values showed that non-significant differentiations were checked among populations from the middle reaches of the Yangtze River group (Table 3). Within the lower reaches of the Yangtze River and the Chinese coastal group, the genetic differentiation was significant between Dandong and other most of the populations from the Huai River, the lower reaches of the Yangtze and Wenzhou. The genetic structure between the Huai River and the Yangtze River system was demonstrated by three significant ΦST values (HL & NJ, ZK & NJ, and ZK & TL; ΦST = 0.05583∼0.06663; P < 0.05). Significant ΦST values were also found between population pairs such as DY & TL, DY & CL, HL & NB, HL & WZ, and ZK & WZ (ΦST = 0.6671∼0.11248; P < 0.05).

Table 3. Pairwise ΦST values among populations of Coilia nasus.
A significant low-level signal of isolation-by-distance (IBD) was observed among all populations (R2 = 0.2175; P = 0.001; Figure 5A). The significant low-level IBD pattern (R2 = 0.1978; P = 0.018) was also found among all populations except the five populations of the middle reaches of the Yangtze River group (R2 = 0.0161; P = 0.338; Figures 5B,C). No IBD pattern was detected for the twelve populations from the lower reaches of the Yangtze and the coast of China group (R2 = 0.056, P = 0.123; Figure 5D).
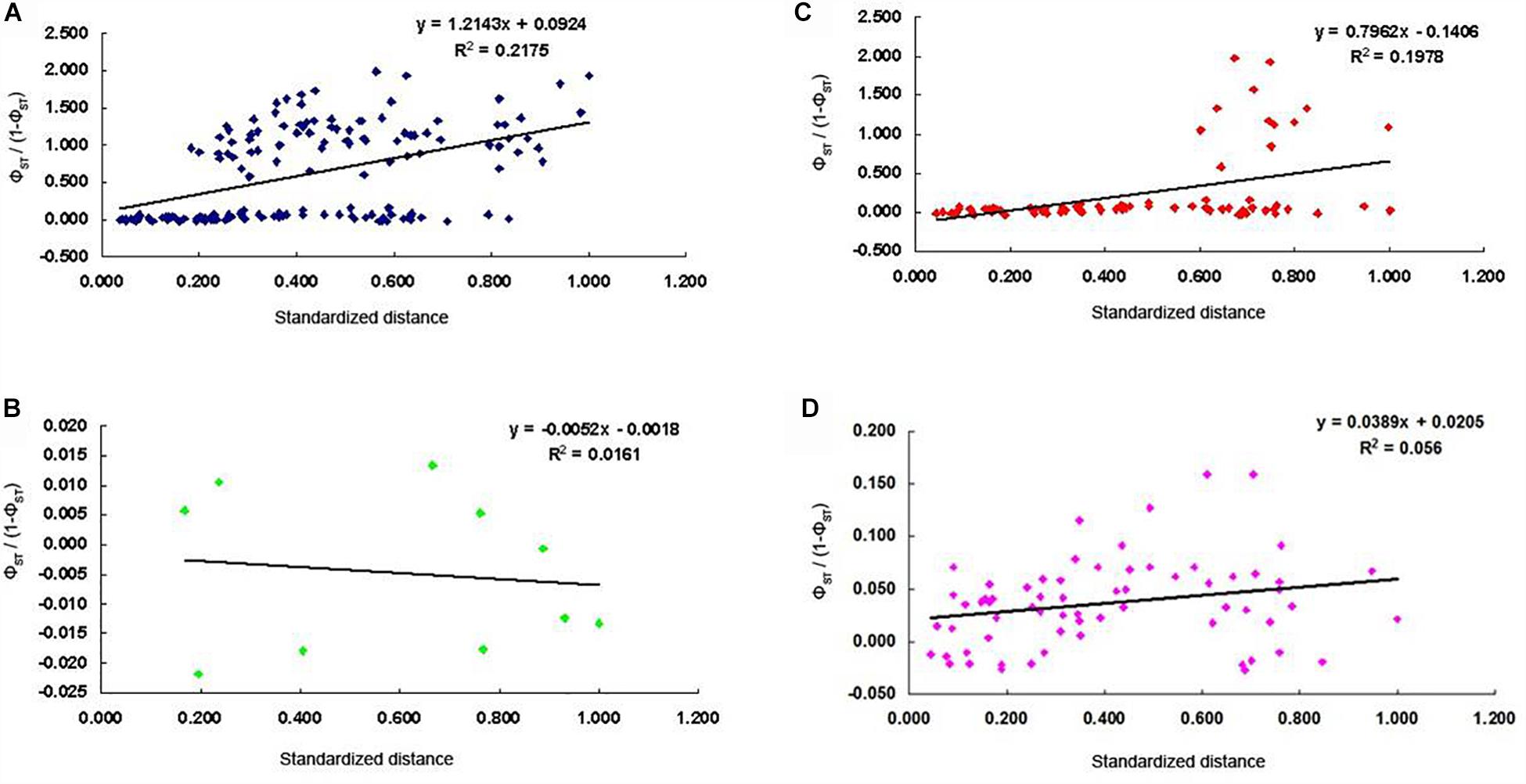
Figure 5. Corelation between pairwise differentiation (ΦST) and standardized geographic distance for C. nasus; (A), all populations (R2 = 0.2175, P = 0.001; B), five populations in the middle reaches of the Yangtze River (R2 = 0.0161, P > 0.05; C), all populations except the five populations from the middle reaches of the Yangtze River group (R2 = 0.1978, P = 0.018; D), twelve populations from the lower reaches of the Yangtze and the coast of China group (R2 = 0.056, P = 0.123).
Population Demography
The FS tests for all the samples and the four lineages were significantly negative (Table 4), indicating a possible population expansion event for the C. nasus. The unimodal mismatch distributions were detected in each lineage A, B, and C, which further demonstrated that the population expansion event had happened in the species (Figure 6). The bimodal mismatch distribution was detected in the lineage D, one of which corresponded to the differences among subclades, and the other of which corresponded to the differences among individuals within subclades (Figure 6). Based on τ values and 10-fold faster rate (13.4% per Myr), the expansion time for the three clades was dated back to the late Pleistocene period. Bayesian skyline plots demonstrated demographic expansion in each of the four lineages (Figure 7). Applying the 10 × faster mutation rate (13.4%/Myr), the population expansion time of lineage A occurred at about 20,000–30,000 years ago. For lineages C and D, the demographic expansion was approximately 20,000–25,000 and 25,000–27,000 years ago, respectively. Furthermore, the expansion time for lineage B was much higher than those of the left lineages happened at about 55,000–75,000 years ago.
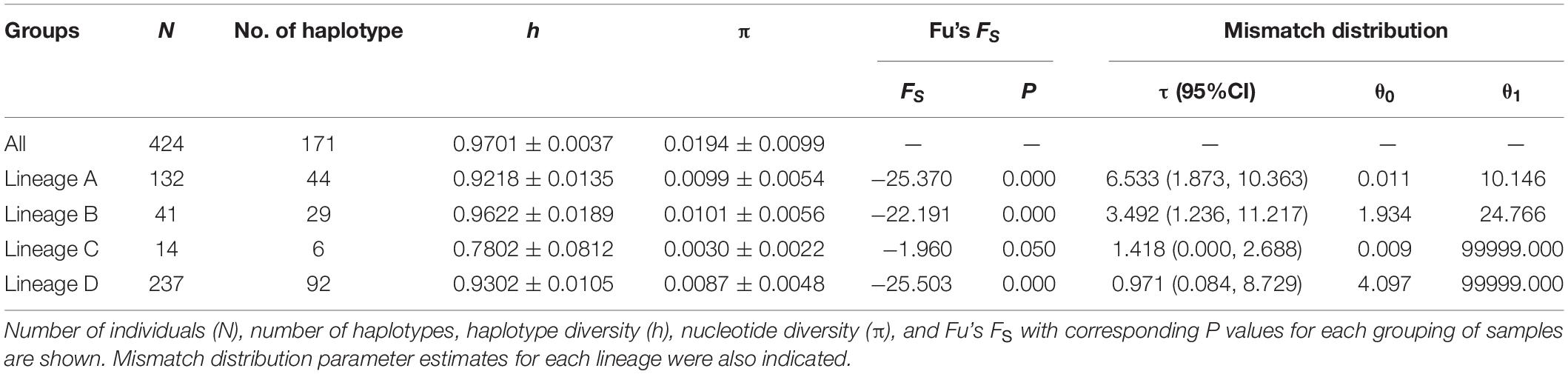
Table 4. Summary of molecular diversity of mtDNA D-loop for Coilia nasus.
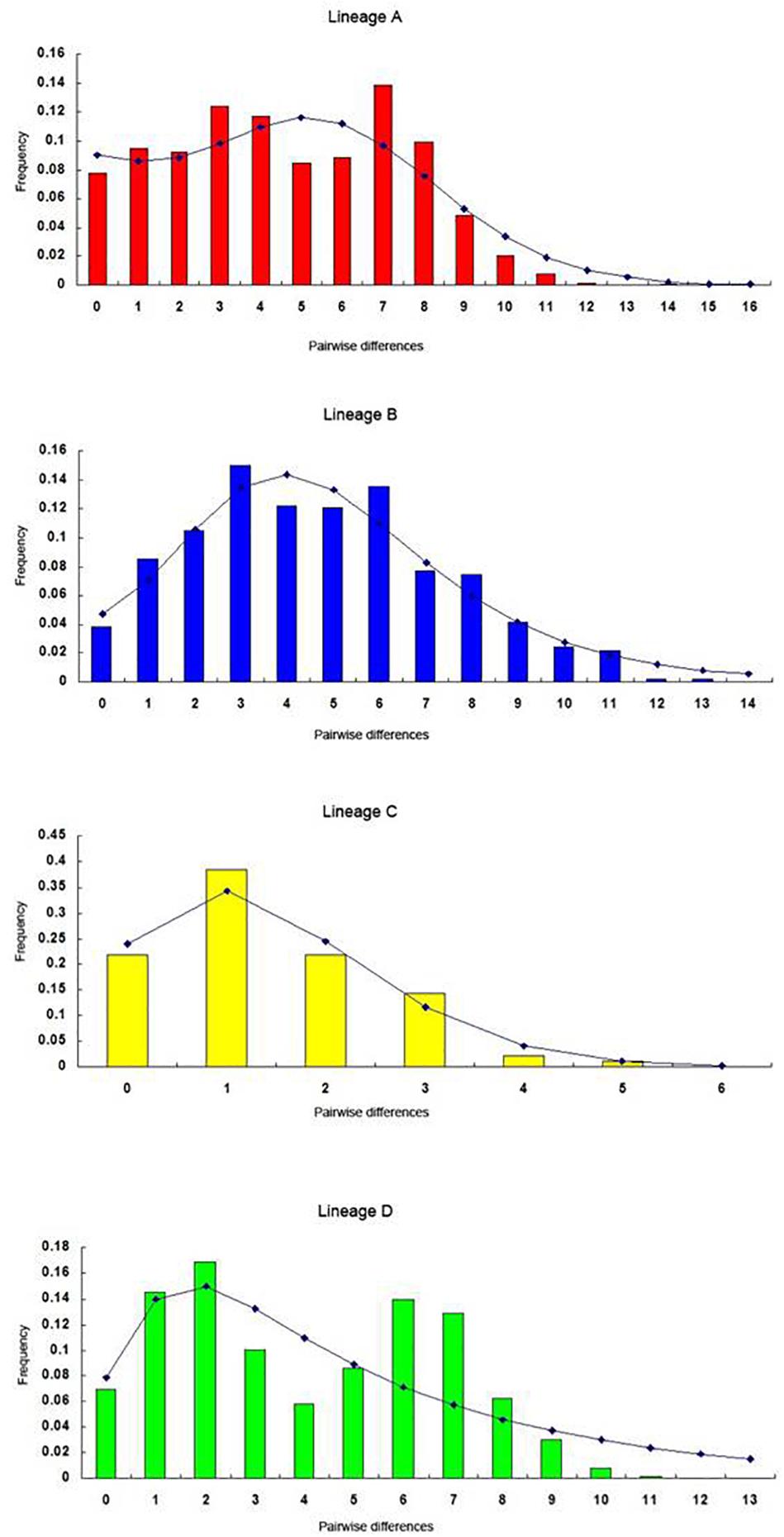
Figure 6. The observed pairwise differences (bars), and the expected mismatch distributions under the sudden expansion model (solid line) for the mtDNA D-loop haplotypes in lineages A, B, C, and D.
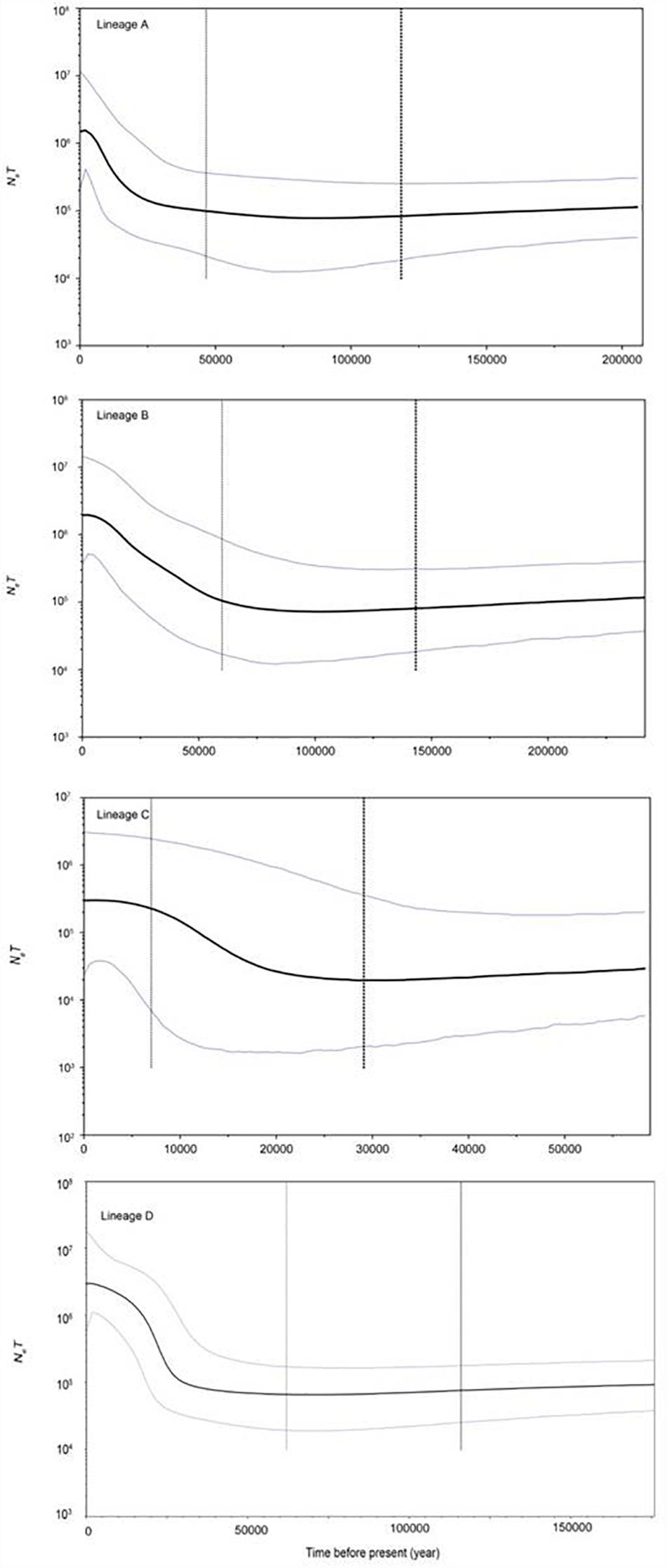
Figure 7. BSP results revealing changes of NeT (Ne = effective population size; T = generation time) through time inferred by BEAST software based on mtDNA D-loop data set. Blue lines represent the upper and lower 95% highest posterior density (HPD) limits of NeT; and the vertical dotted line represents the median estimate of time to most recent common ancestor (TMRCA). The y-axis is plotted on a logarithmic scale.
Discussion
The fossil records were always used as calibration points in the determination of the divergence rate, paleogeologic range, and paleogeological environment of the organisms (Lavoué et al., 2012, 2013). Lavoué et al. (2013) investigated 73 clupeoid taxa collected from 5 families and used fossils as the calibration to estimate the node age. According to their analyses, we could identify the divergence time between the C. nasus and C. ectenes was as approximately 3.6 Myr. The lineage-specific divergence rate for mtDNA D-loop sequence was approximately 1.34%/Myr for the Japanese grenadier anchovies based on the comparison results of mtDNA Cty b.
Recently, Ho et al. (2005) argued that the phylogenetic substitution rates could not be directly applied to population-level analyses. The substitution rates should be ensured based on the calibration point age (Emerson, 2007; Burridge et al., 2008). To account to some extent for such concerns, Liu et al. (2011b) applied two different alternative rate calibrations (“phylogenetic” and 10-fold faster rates) to the analyses of Pleistocene lineage diversification for Pacific herring. However, the substitution rates for mtDNA D-loop was uncertain and seemed vary among taxonomic groups (Bowen et al., 2006). Various molecular rates were used in the previous researches of Clupeoidei as follows: 7.2%/MY for California anchovy (Engraulidae), 5–10%/MY for Japanese anchovy and Australian anchovy (Engraulidae), and 15–20%/MY for Indo-Pacific sardines (Clupeidae; Bowen and Grant, 1997; Liu et al., 2006; Díaz-Viloria et al., 2012). Thus, the “phylogenetic” rate (1.34%/Myr) based on the presumed split between C. nasus and C. grayii was slower and inappropriate for population-level analyses of C. nasus. In this study, we applied a substitution rate (13.4%/Myr) that is 10-fold higher than the phylogenetic rate and is close to the estimates reported for sardines (Bowen et al., 2006).
Four major genealogical clades were detected in C. nasus based on the mtDNA D-loop analyses. The freshwater form of C. nasus mainly existed in the Yangtze River under the effects of geographic isolation, which strongly supported by the geographical distribution of lineage A. The lineage B of C. nasus was originated from the geographic isolation between Ariake Bay and Northwestern Pacific during the late Pleistocene. The lineage D of C. nasus was mainly composed of populations from the Chinese coast waters and the lower reaches of the Yangtze River. Lineage C may be constituted by some ancient haplotypes remained in the East China Sea following the last glacial in accordance with a low frequency distribution along the Chinese coast populations and the low reaches of the Yangtze River. According to the population-level divergence rates utilized in the present study, lineages A, B, C and D separated from one another at approximately 110,000–220,000 years ago.
Additionally, two dominant haplotypes were found in analysis of the partial mtDNA cytochrome b sequences. One was mainly shared by the five populations in the middle reaches of the Yangtze River and the other was shared by the populations along coast and the lower reaches of the Yangtze River. The results of phylogenetic analysis for both the mtDNA D-loop and Cyt b sequences were consistent with the allocation of the freshwater form and anadromous form reported by Cheng (2011).
The genetic variation of marine species has been shaped by the periodic climatic oscillations of the Pleistocene in East Asia, which have raised range contractions, secondary contact between different basins (Zhang et al., 2008). Strong genetic divergence between populations in the middle and lower reaches of the Yangtze River have also been reported for Macrobrachium nipponense, Neosalanx taihuensis, Hypophthalmichthys molitrix, Ctenopharyngodon piceus, Aristichthys nobilis, and Mylopharyngodon piceus (Lu et al., 1997; Li and Lu, 1998; Feng et al., 2008; Zhao et al., 2008). These concordant results showed that paleogeographic factors had greatly influenced the evolutionary processes of species in the middle and lower reaches of the Yangtze River. The Pleistocene glacial-interglacial cycles was characterized by the intervals of ∼100 kyr over the past ∼800 kyr (Lambeck et al., 2002). During the glaciations periods, the sea level reduced to the 120–140 m and the basin in the middle of the Yangtze River formed deeply incised river valleys (Faure et al., 2002). During the inter-glaciations periods, water level of the Yangtze River rose and the lakes expanded after heavy rainfall (Yang, 1986). The lower reaches of the Yangtze River could be inundated by the seawater during the sea level rise.
The Yangtze River was an inland river derived from the impacts of arid climate, reduced rainfall and desertification during the LGM, which isolated with East China Sea (Xiao et al., 2003). Thus, some populations of C. nasus inhabited along in the deep-water channel of the Yangtze River during the glaciation periods and became the freshwater form under the effect of geographic isolation. The glacial refugium for the freshwater population was inferred to be located in the middle reaches of the Yangtze River. However, the existence of special sediment proves that Dongting Lake and Poyang Lake were formed less than 10,000 years ago (Guan and Cai, 1986). Therefore, Dongting Lake and Poyang Lake were unlikely to have been the glacial refuges for these freshwater populations. The deficiency of gene flow between the migratory and non-migratory populations caused the differentiations of morphology, behavior, and genetics, and further gave rise to incipient speciation of island freshwater fish (Michel et al., 2008). Freshwater resident individuals of C. nasus have short maxilla and small body size, and their main breeding season is different from the individuals in the lower reaches of the Yangtze River (Yuan, 1986). The anadromous populations in the lower reaches of the Yangtze River mainly spawn from April to May, whereas the freshwater populations in the middle reaches of the Yangtze River spawns from May to August (Zhang et al., 2005; Luo, 2006; Liu, 2008). The incongruence of the breeding season probably accelerates the genetic divergence between the freshwater form and the anadromous form.
Strong genetic differentiations between the Ariake Sea populations and the Chinese coast populations have been observed in some marine fishes, such as Pennahia argentata, Verasper variegatus, Salanx ariakensis, and Mugil cephalus, marine invertebrates and marine mammal (Han et al., 2008; Yang et al., 2008; Hua et al., 2009; Mao et al., 2011; Sekino et al., 2011; Shen et al., 2011). The Ariake Bay is a semi-closed basin, whose geographical environment has been greatly influenced by the sea-level fluctuations during the Pleistocene glacial-interglacial periods. The population genetic compositions of marine species in the Ariake Bay have been proved to be isolated with the other populations distributed along the extension of the bay (Shimoyama et al., 1996; Kikuchi, 2000; Sato, 2001; Kojima et al., 2004). In the present study, the results of the distribution of lineages B and D strongly supported the hypothesis that the population genetic composition of C. nasus between Chinese coast and Ariake Bay of Japan had been dramatically shaped by the Pleistocene climatic oscillations. The results of lineages B and C showed deeply separated mtDNA lineages coexisted in the examined ranges, however, no significantly geographical isolation could be responsible for the sympatric differentiations. Although it was reported that enough population size with stable or grew throughout long history would accumulate numerous sequence differences (Avise, 2004), we suggested that the secondary contacts of different lineages originated from the isolations among different glacial-refugia (e.g., the Ariake Bay, the East China Sea, the Huang River, and the Huai River) during the Pleistocene period should be responsible for the geographic distribution of the lineages in C. nasus.
The current analyses do not support the interspecific genetic divergence as reported by Ma et al. (2010). Their conclusions suggested that there were some highly diverged mtDNA cytochrome b haplotypes (29 substitutions) in Fuzhou (FZ) and Wuhan (WH) populations. But from our analyses, we can conclude the special haplotypes in Fuzhou and Wuhan populations belong to the specimen of C. grayii, not C. nasus (Ma et al., 2010). Gray’s grenadier anchovy, C. grayii are widely distributed in the East and South China Sea (from Fuzhou City to Guangxi Province; Whitehead et al., 1988; Zhang, 2001). The Gray’s grenadier anchovy doesn’t inhabit along the reaches of Yangtze River. Thus, Ma et al. misidentified the specimen and confused the sample sites of C. nasus and C. grayii.
Strong gene frequency changes of the three dominant lineages (A, B, and D) in the examined ranges showed highly limited genetic exchange existed among groups of C. nasus. Our results also showed that the presence of lineage A (freshwater individuals) occupied low frequencies of haplotypes in the low reaches of the Yangtze implied extremely limited gene flow existed between the anadromous group and freshwater group. Our result was also supported by the phylogenetic analysis of representative individuals clearly corresponding to the anadromous group and freshwater group of C. nasus based on the entire genome SNP set (Xu et al., 2020). The middle reaches of the Yangtze River harbors a large number of lakes with different sizes (e.g., Tianezhou, Poyang Lake, and Dongting Lake), which would support optimum living and spawning conditions (e.g., fresh water, water temperature (15∼27.5°C), depth (0.5∼3.0 m), suitable current (0.057∼0.075 m/s), and abundant zooplankton required for spawning) for freshwater group of C. nasus. The optimum living and spawning conditions for freshwater group of C. nasus, especially high quality fresh water, gentle currents and abundant zooplankton, could not be found in the low reaches of the Yangtze River and the China coast (Xu et al., 2020). The effect of isolation (the middle reaches of the Yangtze River vs. the low reaches of the Yangtze River and the China coast) and adaptation to the new freshwater habitats of the middle reaches of the Yangtze River for C. nasus freshwater form might be responsible for the module of finitely asymmetric gene flow from freshwater to anadromous populations (Zhang, 2001; Schluter and Conte, 2009). The conclusions were also further supported by the module with high gene exchanges among anadromous form populations for C. nasus (Tonteri et al., 2007; Bourret et al., 2013a,b; Perrier et al., 2013). The lineage A was also found in the populations (ZK and HL) from the system of the Huai River, which might be derived from the gene flow channel (Hongze Lake) connecting the Huai River and the Yangtze River since 1851 (Lu and Wang, 2009). Therefore, individuals of lineage A might have dispersed into the system of the Huai River by the connection between the two rivers. Then, the freshwater form (lineage A) was amplified in the Huai River to bring about the high frequency of lineage A existed in the Zhoukou population, which can be explained by the “transporter” hypothesis. Moreover, the presence of lineages B, C, and D at low frequencies within the five freshwater populations may suggest hybridization between the two forms, or upstream spawning migration of anadromous individuals. High frequencies variations of haplotypes were detected in the lineages B and D between the Ariake Bay and the coast of China, which suggested highly limited gene flow existed among populations. During the Pleistocene ice ages, the East China Sea was pushed to the East to be a huge enclosed sea because of exposed continental shelves (Liu et al., 2007). Therefore, the populations of C. nasus in the Ariake Sea were separated from those populations in the East China Sea. The isolation of marginal seas and the contact and mix among populations during the Pleistocene glacial and inter-glacial epoch would be responsible for the current distribution of lineages B, C and D in the East China Sea.
All the ΦST values of the comparisons among five populations of the middle reaches of the Yangtze River group were not statistically significant, which indicated the genetic structures among the freshwater forms were lacked. The result was further supported by the IBD pattern (R2 = 0.0161, P > 0.05) in the middle reaches of the Yangtze River (Figure 5B). A limited range of activities and current coupled with pelagic eggs of C. nasus could be responsible for the frequent gene flow among populations in the middle reaches of the Yangtze River (Yuan et al., 1976; Ni et al., 1990; Xu et al., 2020). The similar results with no significant IBD pattern was also detected among populations from the lower reaches of the Yangtze and the coast of China group (R2 = 0.056, P = 0.123), which might be derived from integrations of the upstream spawning migration behavior of anadromous individuals and transportation of juveniles by China Coastal Current (Figure 5D; Yuan et al., 1976; Yuan et al., 1980; Liu et al., 2007; Cheng, 2011; Xu et al., 2020). The Hongze Lake and Zhoukou populations belonged to the Huai River system which was connected by the Yangtze River via a human-made channel. However, the dams located the human-made channel could limit the Yangtze River populations to enter into the Huai River. Thus, the existence of genetic substructure between the Huai River and the Yangtze system was supported by the significant ΦST values of three comparisons between populations (HL & NJ, ZK & NJ, and ZK & TL). Significant ΦST values were observed between the Dandong (the estuary of the Yalu River) population and other three populations (NT, TL, and CL) from the lower reaches of the Yangtze River. This level of subdivision may be reflecting the homing behavior of adults (Campton and Utter, 1987; McDowall, 2001; Keefer and Caudill, 2014). Furthermore, the significant ΦST values of the comparisons were also found between populations (DD & WZ, DY & TL, DY & CL, HL & NB, HL & WZ, and ZK & WZ) reflecting the homing behavior of different populations. Therefore, although no IBD pattern was detected for populations from the lower reaches of the Yangtze and the coast of China group, different homing behavioral mechanisms may bring about the weak genetic structure between the ZK population and other two estuary populations (DY and PJ).
Neutrality test, mismatch distribution and network analyses combined with Bayesian skyline plots consistently showed that all the four lineages (lineages A, B, C, and D) had populations with relatively stable over time and then subjected to sudden expansion during the late-Pleistocene era (c. 20,000–75,000 years ago). According to the divergence rate of 13.4%/Myr, the initial population expansion of lineages A, C and D occurred 20,000–30,000 years ago that largely coincided with the onset of the current Pleistocene interglacial period. During the last interglacial period, the transgression has happened in the East China Sea Shelf, which caused the coastline to migrate from the Okinawa Trough to the coast of the Bohai bay by 1200 km (Wang, 1999; Xu and Oda, 1999). Under this scenario, populations of C. nasus from the China coast would expand their range along the coastal areas following the glacial retreat. Moreover, water level of the Yangtze River rose and the lakes expanded after heavy rainfall during the last deglaciation. Freshwater populations in the Yangtze River might also expand their range.
Our results showed that the phylogeographic and demographic history of C. nasus has been greatly influenced by the Pleistocene glacial-interglacial cycles. With respect to phylogeography, the genetic divergences between four lineages were observed in the middle reaches of Yangtze River, Chinese coastal and Japanese coastal populations. Geographic isolation resulted from climatic fluctuations of the Pleistocene has a great influence on the distribution of the four lineages. The glacial refugium for the freshwater population of C. nasus was proved to be located in the middle reaches of the Yangtze River. With respect to the historical demography, our result showed that C. nasus had experienced dramatic population expansion event during the glacial period of the Pleistocene. But beyond all that, additional nuclear markers are needed to be used for further studies to test our conclusion of the biogeographical pattern for C. nasus.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics Statement
The animal study was reviewed and approved by Institutional Ethics Committee of Center for Ocean Mega-Science.
Author Contributions
TG, QY, and YX conceived and designed research. TG, QY, NS, YY, and YX conducted experiments, analyzed data, and wrote the manuscript. Authors critically reviewed and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key R&D Program of China (2019YFD0901301) and National Natural Science Foundation of China (41776171).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Koji Yokogawa, Cunyin Dou, Yuting Wang, Yancui Chen, Xifu Sun, and Xiao Chen for their assistance in collecting samples. We would like to give sincere thanks to Jinxian Liu for his contributions to paper analysis and sample sequencing.
References
Aldenhoven, J. T., Miller, M. A., Corneli, P. S., and Shapiro, M. D. (2010). Phylogeography of ninespine sticklebacks (Pungitius pungitius) in North America: glacial refugia and the origins of adaptive traits. Mol. Ecol. 19, 4061–4076. doi: 10.1111/j.1365-294x.2010.04801.x
Avise, J. C. (2000). Phylogeography: The History and Formation of Species. Cambridge, MA: Harvard University Press, 25–65.
Avise, J. C. (2004). Molecular Markers, Natural History, and Evolution, 2nd Edn. Sunderland, MA: Sinauer Associates, 1–30.
Bell, M. A., and Foster, S. A. (1994). “Introduction to the evolutionary biology of the threespine stickleback,” in The Evolutionary Biology of the Threespine Stickleback, eds M. A. Bell and S. A. Foster (Oxford: Oxford University Press), 1–26.
Bermingham, E., and Martin, A. P. (1998). Comparative phylogeography of neotropical freshwater fish: testing shared history to infer the evolutionary landscape of Central America. Mol. Ecol. 7, 499–517. doi: 10.1046/j.1365-294x.1998.00358.x
Bernatchez, L., and Wilson, L. C. (1998). Comparative phylogeography of Nearctic and Palearctic fishes. Mol. Ecol. 7, 431–452. doi: 10.1046/j.1365-294x.1998.00319.x
Bourret, V., Dionne, M., Kent, M. P., Lien, S., and Bernatchez, L. (2013a). Landscape genomics in Atlantic salmon (Salmo salar): searching for gene-environment interactions driving local adaptation. Evolution 67, 3469–3487. doi: 10.1111/evo.12139
Bourret, V., Kent, M. P., Primmer, C. R., Vasemägi, A., Karlsson, S., Hindar, K., et al. (2013b). SNP-array reveals genome-wide patterns of geographical and potential adaptive divergence across the natural range of Atlantic salmon (Salmosalar). Mol. Ecol. 22, 532–551. doi: 10.1111/mec.12003
Bowen, B. W., and Grant, W. S. (1997). Phylogeography of the sardines (Sardinops spp.): assessing biogeographic models and population histories in temperate upwelling zones. Evolution 51, 1601–1610. doi: 10.2307/2411212
Bowen, B. W., Muss, A., Rocha, L. A., and Grant, W. S. (2006). Shallow mtDNA coalescence in Atlantic pygmy angelfishes (Genus Centropyge) indicates a recent invasion from the Indian Ocean. J. Hered. 97, 1–12. doi: 10.1093/jhered/esj006
Burridge, C. P., Craw, D., Fletcher, D., and Waters, J. M. (2008). Geological dates and molecular rates: fish DNA sheds light on time dependency. Mol. Biol. Evol. 25, 624–633. doi: 10.1093/molbev/msm271
Cai, D., Ji, D., and Zhou, C. (1980). An ecological investigation and fishery of the anchovy (Coilia ectenes Jordan et Seale) in the lower Yellow River. J. Shandong Univ. 2, 97–107.
Campton, D. E., and Utter, F. M. (1987). Genetic structure of anadromous cutthroat trout (Salmo clarkiclarki) populations in the Puget Sound area: evidence for restricted gene flow. Can. J. Fish. Aquat. Sci. 44, 573–582. doi: 10.1139/f87-070
Cheng, W. (2011). The Research of Some Phenotypic Differences from Different Ecotypes of Coilia nasus from Yangtze River, Master’s thesis, Shanghai Ocean University, Shanghai, 1–25.
Consuegra, S., García de Leániz, C., Serdio, A., González Morales, M., Straus, L. G., Verspoor, E., et al. (2002). Mitochondrial DNA variation in Pleistocene and modern Atlantic salmon from the Iberian glacial refugium. Mol. Ecol. 11, 2037–2048. doi: 10.1046/j.1365-294x.2002.01592.x
Díaz-Viloria, N., Sánchez-Velasco, L., and Pérez-Enríquez, R. (2012). Recent population expansion in the evolutionary history of the Californian anchovy Engraulis mordax. Hidrobioloìgica 22, 258–266.
Dodson, J. J., Tremblay, S., Colombani, F., Carscadden, J. E., and Lecomte, F. (2007). Trans-Arctic dispersals and the evolution of a circumpolar marine fish species complex, the capelin (Mallotus villosus). Mol. Ecol. 16, 5030–5043. doi: 10.1111/j.1365-294x.2007.03559.x
Drummond, A. J., Rambaut, A., Shapiro, B., and Pybus, O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192. doi: 10.1093/molbev/msi103
Drummond, A. J., Suchard, M. A., and Xie, D. A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Duan, J., Zhang, H., Liu, K., Xu, D., Zhang, M., and Sui, W. (2012). An overview of Coilia ectenes in Jiangsu section of the Yangtze River. Agric. Sci. Technol. 13, 1950–1954.
Emerson, B. C. (2007). Alarm bells for the molecular clock? No support for Ho et al.’s model of time-dependent molecular rate estimates. Syst. Biol. 56, 337–345. doi: 10.1080/10635150701258795
Excoffier, L., and Laval, G. (2007). Schneider, S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinformatics 1, 47–50.
Excoffier, L., Smouse, P. E., and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491.
Faure, H., Walter, R. C., and Grant, D. R. (2002). The coastal oasis: ice age springs on emerged continental shelves. Glob. Planet. Change 33, 47–56. doi: 10.1016/s0921-8181(02)00060-7
Feng, J., Sun, Y., Cheng, X., and Li, J. (2008). Sequence analysis of mitochondrial COI gene of Macrobrachium nipponense from the five largest freshwater lakes in China. J. Fish. China 32, 517–723.
Fu, Y. X. (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 14, 915–925.
Guan, Z., and Cai, S. (1986). The formation and evolution of the Dongting Lake. J. Sediment. Res. 1, 70–72.
Han, Z. Q., Gao, T. X., Yanagimoto, T., and Sakurai, Y. (2008). Deep phylogeographic break among white croaker Pennahiaargentata (Sciaenidae, Perciformes) populations in North-western Pacific. Fish. Sci. 74, 770–780. doi: 10.1111/j.1444-2906.2008.01588.x
Hewitt, G. (2000). The genetic legacy of the Quaternary ice ages. Nature 405, 907–913. doi: 10.1038/35016000
Hewitt, G. M. (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 58, 247–276. doi: 10.1006/bijl.1996.0035
Hewitt, G. M. (1999). Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 68, 87–112. doi: 10.1111/j.1095-8312.1999.tb01160.x
Hewitt, G. M. (2004). The structure of biodiversity—insights from molecular phylogeography. Front. Zool. 1:4. doi: 10.1186/1742-9994-1-4
Ho, S. Y., Phillips, M. J., Cooper, A., and Drummond, A. J. (2005). Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 22, 1561–1568. doi: 10.1093/molbev/msi145
Ho, S. Y., Saarma, U., Barnett, R., Haile, J., and Shapiro, B. (2008). The Effect of inappropriate calibration: three case studies in molecular ecology. PLoS One 3:e1615. doi: 10.1371/journal.pone.0001615
Ho, S. Y. W., and Shapiro, B. (2011). Skyline-plot methods for estimating demographic history from nucleotide sequences. Mol. Ecol. Resour. 11, 423–434. doi: 10.1111/j.1755-0998.2011.02988.x
Hua, X., Wang, W., Yin, W., He, Q., Jin, B., Li, J., et al. (2009). Phylogeographical analysis of an estuarine fish, Salanxari akensis (Osmeridae: Salanginae) in the north-western Pacific. J. Fish Biol. 75, 354–367. doi: 10.1111/j.1095-8649.2009.02323.x
Jordan, D. S., and Seale, A. (1905). List of fishes collected by DrBashford Dean on the island of Negros, Philippines. Proc. Natl. Acad. Sci. U.S.A. 28, 769–803. doi: 10.5479/si.00963801.28-1407.769
Keefer, M. L., and Caudill, C. (2014). Homing and straying by Anadromous salmonids a review of mechanisms and rates. Rev. Fish Biol. Fish. 24, 333–368. doi: 10.1007/s11160-013-9334-6
Kikuchi, T. (2000). “Significance of conservation of tidal flats and their adjacent waters,” in Life in Ariake Sea: Biodiversity in Tidal Flats and Estuaries, ed. M. Sato (Tokyo: Kaiyu-Sha), 306–317.
Kitamura, A., Takano, O., Takata, H., and Omote, H. (2000). Late Pliocene–early Pleistocene paleoceanographic evolution of the Japan Sea. Palaeoecology 172, 81–98. doi: 10.1016/s0031-0182(01)00272-3
Kojima, S., Hayashi, I. D., Kim, S., Iijima, A., and Furota, T. (2004). Phylogeography of an intertidal direct-developing gastropod Batillaria cumingi around the Japanese Islands. Mar. Ecol. Prog. Ser. 276, 161–172. doi: 10.3354/meps276161
Kumar, S., Tamura, K., and Nei, M. (2004). MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinformatics 5, 150–163. doi: 10.1093/bib/5.2.150
Lambeck, K., Esat, T. M., and Potter, E. K. (2002). Links between climate and sea levels for the past three million years. Nature 419, 199–206. doi: 10.1038/nature01089
Lavoué, S., Miya, M., Arnegard, M. E., Sullivan, J. P., Hopkins, C. D., and Nishida, M. (2012). Comparable ages for the independent origins of electrogenesis in African and South American weakly electric fishes. PLoS One 7:e36287. doi: 10.1371/journal.pone.0036287
Lavoué, S., Miya, M., Musikasinthorn, P., Chen, W. J., and Nishida, M. (2013). Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One 8:e56485. doi: 10.1371/journal.pone.0056485
Li, S. F., and Lu, G. Q. (1998). Diversity of mitochondrial DNA in the populations of silver carp, bighead carp, grass carp and black carp in the middle and lower reaches of the Yangtze River. Acta Zool. Sin. 44, 82–93.
Liu, J., Li, Q., Kong, L. F., and Zheng, X. D. (2011a). Cryptic diversity in the pen shell Atrina pectinata (Bivalvia: Pinnidae): high divergence and hybridization revealed by molecular and morphological data. Mol. Ecol. 20, 4332–4345. doi: 10.1111/j.1365-294x.2011.05275.x
Liu, J. X., Gao, T. X., Wu, S. F., and Zhang, Y. P. (2007). Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Mol. Ecol. 16, 275–288. doi: 10.1111/j.1365-294x.2006.03140.x
Liu, J. X., Gao, T. X., Zhuang, Z. M., Jin, X. S., Yokogawa, K., and Zhang, Y. P. (2006). Late Pleistocene divergence and subsequent population expansion of two closely related fish species, Japanese anchovy (Engraulis japonicus) and Australian anchovy (Engraulis australis). Mol. Phylogenet. Evol. 40, 712–723. doi: 10.1016/j.ympev.2006.04.019
Liu, J. X., Tatarenkov, A., Beacham, T. D., Gorbachev, V., Wildes, S., and Avise, J. C. (2011b). Effects of Pleistocene climatic fluctuations on the phylogeographic and demographic histories of Pacific herring (Clupea pallasii). Mol. Ecol. 19, 4535–4553.
Liu, Y. (2008). Species Identification and Biological Characteristics of Coilia brachynathus in the Poyang Lake. Master’s thesis, Nanchang University, Nanchang, 35–82.
Lu, G., Li, S., and Bernatchez, L. (1997). Mitochondrial DNA diversity, population structure, and conservation genetics of four native carps within the Yangtze River, China. Can. J. Fish. Aquat. Sci. 54, 47–58. doi: 10.1139/f96-266
Lu, Y., and Wang, S. (2009). Ming and Qing Dynasties to the south of Huai River and the surrounding environment evolution. Chin. Agric. Sci. Bull. 5, 494–499.
Luo, H. (2006). Age, Growth and Gonadal Development of Coilia brachynathus in Swan Oxbow of the Yangtze River. Mater’s thesis, Southwest University, Chongqing, 1–35.
Ma, C., Cheng, Q., Zhang, Q., Zhuang, P., and Zhao, Y. (2010). Genetic variation of Coilia ectenes (Clupeiformes: Engraulidae) revealed by the complete cytochrome b sequences of mitochondrial DNA. J. Exp. Mar. Biol. Ecol. 385, 14–19. doi: 10.1016/j.jembe.2010.01.015
Mäkinen, H. S., and Merilä, J. (2008). Mitochondrial DNA phylogeography of the three-spined stickleback (Gasterosteus aculeatus) in Europe—Evidence for multiple glacial refugia. Mol. Phylogenet. Evol. 46, 167–182. doi: 10.1016/j.ympev.2007.06.011
Mao, Y., Gao, T., Yanagimoto, T., and Xiao, Y. (2011). Molecular phylogeography of Ruditapes philippinarum in the Northwestern Pacific Ocean based on COI gene. J. Exp. Mar. Biol. Ecol. 407, 171–181. doi: 10.1016/j.jembe.2011.06.002
McDowall, R. M. (2001). Anadromy and homing two life-history traits with adaptive synergies in salmonid fishes? Fish Fish. 2, 78–85. doi: 10.1046/j.1467-2979.2001.00036.x
Michel, C., Hicks, B. J., Stölting, K. N., Clarke, A. C., Stevens, M. I., Tana, R., et al. (2008). Distinct migratory and non-migratory ecotypes of an endemic New Zealand eleotrid (Gobiomorphus cotidianus) – implications for incipient speciation in island freshwater fish species. BMC Evol. Biol. 8:49. doi: 10.1186/1471-2148-8-49
Ni, Y., Wang, Y., Xu, C., Xiong, G., and Zhan, H. (1990). Fishes of Shanghai. Shanghai: Shanghai Science and Technology Press, 1–30.
Nylander, J. A. (2004). MrModeltest v2. Program Distributed by the Author. Evolutionary Biology Centre. Uppsala: Uppsala University, 1–20.
Peakall, R., and Smouse, P. E. (2006). GENEALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol. Ecol. Note 6, 288–295. doi: 10.1111/j.1471-8286.2005.01155.x
Perrier, C., Bourret, V., Kent, M. P., and Bernatchez, L. (2013). Parallel and nonparallel genome-wide divergence among replicate population pairs of freshwater and anadromous Atlantic salmon. Mol. Ecol. 22, 5577–5593. doi: 10.1111/mec.12500
Qiao, H., Cheng, Q., Chen, Y., Chen, W., and Zhu, Y. (2013). The complete mitochondrial genome sequence of Coilia ectenes (Clupeiformes: Engraulidae). Mitochondrial DNA 24, 123–125. doi: 10.3109/19401736.2012.731405
Rambaut, A., and Drummond, A. J. (2007). Tracer 1.5. Available at: http://beast.bio.ed.ac.uk/Tracer (accessed August, 2019).
Ronquist, F., and Huelsenbeck, J. P. (2003). MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Sato, S. (2001). Temporal changes of bivalves and gastropods in Isahaya bay shut off from Ariake Sea, western Kyushu, Japan. Quat. Res. 40, 85–95. doi: 10.4116/jaqua.40.85
Schluter, D., and Conte, G. L. (2009). Genetics and ecological speciation. Proc. Natl. Acad. Sci. U.S.A. 106, 9955–9962.
Schmitt, T. (2007). Molecular biogeography of Europe: pleistocene cycles and postglacial trends. Front. Zool. 4:11. doi: 10.1186/1742-9994-4-11
Sekino, M., Saitoh, K., Shimizu, D., Wada, T., Kamiyama, K., Gambe, S., et al. (2011). Genetic structure in species with shallow evolutionary lineages a case study of the rare flatfish Verasper variegates. Conserv. Genet. 12, 139–159. doi: 10.1007/s10592-010-0128-2
Shen, K. N., Jamandre, B. W., Hsu, C. C., Tzeng, W. N., and Durand, J. D. (2011). Plio-Pleistocene sea level and temperature fluctuations in the northwestern Pacific promoted speciation in the globally-distributed flathead mullet Mugil cephalus. BMC Evol. Biol. 11:83.
Shimoyama, S., Kosugi, M., Matsuoka, K., Kataoka, H., and Sato, N. (1996). Integral analysis for the paleoenvironment of the coastal lowland around the innermost Ariake Bay, West Japan. Kanto Heiya 4, 53–76.
Swofford, D. L. (2002). PAUP∗: Phylogenetic Analysis Using Parsimony (∗and Other Methods), version 4.0b10. Sunderland, MA: Sinauer Associates, 10–21.
Takita, T. (1967). The spawning and the early life history of the Engraulid fish Coilia sp. distributed in Ariake Sound. Bull. Fac. Fish. Nagasaki Univ. 23, 107–122.
Tang, W., Hu, X., and Yang, J. (2007). Species validities of Coilia brachygnathus and C. nasustaihuensis based on sequence variations of complete mtDNA control region. Biodivers. Sci. 15, 224–231. doi: 10.1360/biodiv.060263
Teacher, A. G., Shikano, T., Karjalainen, M. E., and Merilä, J. (2011). Phylogeography and Genetic Structuring of European Nine-Spined Sticklebacks (Pungitius pungitius) — Mitochondrial DNA Evidence. PLoS One 6:e19476. doi: 10.1371/journal.pone.0019476
Tonteri, A., Titov, S., Veselov, A., Zubchenko, A., Koskinen, M. T., Lesbarrères, D., et al. (2005). Phylogeography of anadromous and non-anadromous Atlantic salmon (Salmo salar) from northern Europe. Ann. Zool. Fenn. 42, 1–22. doi: 10.1111/j.1095-8649.1981.tb03753.x
Tonteri, A., Veselov, A. J., Titov, S., Lumme, J., and Primmer, C. R. (2007). The effect of migratory behaviour on genetic diversity and population divergence: a comparison of anadromous and freshwater Atlantic salmon Salmo salar. J. Fish Biol. 70, 381–398. doi: 10.1111/j.1095-8649.2007.01519.x
Wang, P. X. (1999). Response of Western Pacific marginal seas to glacial cycles: paleoceanographic and sedimentological features. Mar. Geol. 156, 5–39. doi: 10.1016/s0025-3227(98)00172-8
Whitehead, P. J., Nelson, G. J., and Wongratana, T. (1988). Clupeoid Fishes of the World (Suborder Clupeoidei): An Annotated and Illustrated Catalogue of the Herrings, Sardines, Pilchards, Sprats, Shads, Anchovies and Wolf Herrings. Available online at: http://www.fao.org/3/a-ac482e.pdf (accessed March, 2019).
Wilson, A. B., and Veraguth, I. E. (2010). The impact of Pleistocene glaciation across the range of a wide spread European coastal species. Mol. Ecol. 19, 4535–4553. doi: 10.1111/j.1365-294x.2010.04811.x
Xiao, X., Huang, P., Wan, S., and Li, A. (2003). Entering sea history of the Yangtze River during the last pleniglacial stage. J. Univ. Petrol. China 27, 125–130.
Xu, G. C., Bian, C., Nie, Z. J., Li, J., Wang, Y. Y., Xu, D. P., et al. (2020). Genome and population sequencing of a chromosome-level genome assembly of the Chinese tapertail anchovy (Coilia nasus) provides novel insights into migratory adaptation. Gigascience 9:giz157.
Xu, J., Chan, T. Y., Tsang, L. M., and Chu, K. H. (2009). Phylogeography of the mitten crab Eriocheir sensu stricto in East Asia: pleistocene isolation, population expansion and secondary contact. Mol. Phylogenet. Evol. 52, 45–56. doi: 10.1016/j.ympev.2009.02.007
Xu, X., and Oda, M. (1999). Surface-water evolution of the eastern East China Sea during the last 36,000 years. Mar. Geol. 156, 285–304. doi: 10.1016/s0025-3227(98)00183-2
Yang, D. (1986). The paleoenvironment of the mid-lower regions of Changjiang in the full-glacial period of late Pleistocene. Acta Geograph. Sin. 41, 302–310.
Yang, G., Guo, L., Bruford, M. W., Wei, F., and Zhou, K. (2008). Mitochondrial phylogeography and population history of finless porpoises in Sino-Japanese waters. Biol. J. Linn. Soc. 95, 193–204. doi: 10.1111/j.1095-8312.2008.0932.x
Yang, J., Hou, L., Wu, T., Ren, J., and Ba, L. (2012). The analysis for the quaternary sediments from the southwest area of the Huai River Plain. Earth Sci. Technol. 1, 12–17. doi: 10.1016/j.quaint.2014.08.053
Yang, J., Wu, T., Ren, J., Zhang, Y., and Zhao, H. (2010a). The geologic age of OSL and 14C for the quaternary sediments from the Xinyang area in Henan Province. Earth Sci. Bull. Henan Prov. 2, 250–254.
Yang, Q., Gao, T., and Miao, Z. (2011). Differentiation between populations of Japanese grenadier anchovy (Coilia nasus) in Northwestern Pacific based on ISSR markers: Implications for biogeography. Biochem. Syst. Ecol. 39, 286–296. doi: 10.1016/j.bse.2011.07.019
Yang, Q., Han, Z., Sun, D., Xie, S., Lin, L., and Gao, T. (2010b). Genetics and phylogeny of genus Coilia in China based on AFLP markers. Chin. J. Oceanol. Limnol. 28, 795–801. doi: 10.1007/s00343-010-9093-3
Yuan, C., Lin, J., Qin, A., and Liu, R. (1976). Historical and present taxonomic status about the genus Coilia in China. J. Nanjing Univ. 2, 1–12.
Yuan, C., Qin, A., Liu, R., and Lin, J. (1980). On the classification of the anchovies, Coilia, from the lower Yangtze River and the southeast coast of China. J. Nanjing Univ. 3, 67–122.
Zhang, H., Yan, J., Zhang, G., and Zhou, K. (2008). Phylogeography and demographic history of Chinese black-spotted frog populations (Pelophylax nigromaculata): evidence for independent refugia expansion and secondary contact. BMC Evol. Biol. 8:21. doi: 10.1186/1471-2148-8-21
Zhang, J., Gao, T., Han, Z., Guo, Y., and Dong, C. (2007). Sequence analysis of partial cytochrome b and 16S rRNA genes of three Coregonus species. J. Fish. Sci. China 14, 9–14.
Zhang, M., Xu, D., Liu, K., and Shi, W. (2005). Studies on biological characteristics and change of resource of Coilia nasus Schlegel in the lower reaches of the Yangtze River. Resour. Environ. Yangtze Basin 14, 694–698.
Zhang, S. (2001). Fauna Sinica: Osteichthyes, Acipenseriformes, Elopiformes, Clupeiformes, Gonorhynchiformes. Beijing: Science Press, 137–147.
Keywords: climatic oscillations, genetic differentiation, last glacial maximum, phylogeography, anadromous form, demographic expansion
Citation: Gao T, Ying Y, Yang Q, Song N and Xiao Y (2020) The Mitochondrial Markers Provide New Insights Into the Population Demographic History of Coilia nasus With Two Ecotypes (Anadromous and Freshwater). Front. Mar. Sci. 7:576161. doi: 10.3389/fmars.2020.576161
Received: 25 June 2020; Accepted: 07 October 2020;
Published: 02 November 2020.
Edited by:
XueWei Xu, Ministry of Natural Resources, ChinaReviewed by:
Qiong Shi, Beijing Genomics Institute (BGI), ChinaDongsheng Zhang, Ministry of Natural Resources, China
Copyright © 2020 Gao, Ying, Yang, Song and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongshuang Xiao, ZGFoYWlzaHVhbmcxOTgyQDE2My5jb20=