 Ashley M. Dungan
Ashley M. Dungan Madeleine J. H. van Oppen
Madeleine J. H. van Oppen Linda L. Blackall
Linda L. Blackall- 1School of BioSciences, The University of Melbourne, Melbourne, VIC, Australia
- 2Australian Institute of Marine Science, Townsville, QLD, Australia
The global decline of coral reefs heightens the need to understand how corals may persist under changing environmental conditions. Restructuring of the coral-associated bacterial community, either through natural or assisted strategies, has been suggested as a means of adaptation to climate change. A low complexity microbial system would facilitate testing the efficacy of microbial restructuring strategies. We used the model organism for corals, Exaiptasia diaphana, and determined that short-term (3 weeks) exposure to filter-sterilized seawater conditions alone reduced the complexity of the microbiome. Metabarcoding of the V5–V6 region of the bacterial 16S rRNA gene revealed that alpha diversity was approximately halved in anemones reared in filter-sterilized seawater compared to controls reared in unfiltered seawater and that the composition (beta diversity) differed significantly between the two. By reducing the complexity of the E. diaphana microbiome, the development of a system for testing assisted strategies such as probiotics, is more feasible.
Introduction
Corals are colonized by microbes, including bacteria, eukaryotes, archaea, and viruses (Blackall et al., 2015; Ainsworth et al., 2017; Huggett and Apprill, 2019), that contribute to the overall health of this complex host-microbe association, or holobiont (Rohwer et al., 2002). A noted eukaryote in the coral holobiont is the algal endosymbiont, a dinoflagellate of the family Symbiodiniaceae, which actively interacts with other coral-associated microbes (Camp et al., 2020; Matthews et al., 2020). Coral-associated bacteria, hereafter referred to as the “microbiome,” likely consist of a combination of commensals, transients, and long-term stable members, and combined with the host, form a mutually beneficial, stable symbiosis. Evolution of the coral microbiome to confer benefits to the holobiont may occur naturally (Ziegler et al., 2017) or be directed through assisted evolution strategies, in which the microbiome of cnidarians is manipulated (van Oppen et al., 2015, 2018; Epstein et al., 2019a). Manipulation of the coral microbiome could be achieved through the addition of a bacterial consortium with specific purposes such as degradation of oil (dos Santos et al., 2015), nutrient cycling, disease prevention (Alagely et al., 2011), or mitigation of bleaching (Rosado et al., 2018). These consortia, also known as probiotics, are developed to target specific aspects of holobiont health and functioning and should be based on the management of the already complex ménage à trois of host-bacteria-algae interactions.
One strategy to unravel the functions of and interactions between host-associated bacteria is to identify and synthetically recreate the minimal microbiome (MM). The MM is defined as the fewest microbes and/or microbial functions needed to maintain health and functioning of the holobiont (de Vos, 2013). The concept of a MM has been explored in plant science (Qin et al., 2016) and the human gut microbiome (de Vos, 2013; Clavel et al., 2017), with suggestions for its application in coral science (van Oppen and Blackall, 2019). While the ability to achieve the MM is important for functional microbiome studies, realizing this with antibiotics is difficult for many marine invertebrates (D’agostino, 1975), including the model organism for corals, the sea anemone Exaiptasia diaphana (Hartman, 2020).
As an alternative approach to creating the MM, we have explored a community reduction strategy of the E. diaphana microbiome by manipulating the external environment (Clavel et al., 2017), instead of the application of antibiotics. Studies on the microbiome of E. diaphana indicate a similar phylum level diversity to corals (Blackall et al., 2015) for anemones sourced from the Great Barrier Reef (GBR) (Hartman et al., 2019, 2020; Dungan et al., 2020a), Hawaii (Herrera et al., 2017), Pacific and Caribbean (Brown et al., 2017), Atlantic (Röthig et al., 2016), and Red Sea (Ahmed et al., 2019). We hypothesize that selective forces acting on E. diaphana and its microbiome, when placed in sterile seawater, will reduce the complexity of the microbiome with the loss of some (e.g., transient) members. This hypothesis is driven by existing evidence that environment has a significant influence on the cnidarian microbiome (Zhang et al., 2015; Webster and Reusch, 2017; Hartman, 2020), and that autoclave-sterilized seawater is correlated with a reduction in alpha diversity for GBR-sourced E. diaphana (Hartman et al., 2019). If their microbiomes were successfully reduced, we sought to identify bacterial taxa that persisted in anemones reared in sterile seawater as these taxa may be critical to holobiont health.
Methods
Sample Collection
Anemones and the surrounding seawater from each of four GBR-sourced genotypes (Dungan et al., 2020b) were sampled once from each of two conditions:
1) control anemones—maintained in Red Sea SaltTM (R11065, Red Sea, United States; RSS) seawater prepared with reverse osmosis water at ∼34 parts per thousand (ppt) salinity, or
2) “sterile” anemones—defined as anemones removed from stock conditions (see 1), maintained in sterilized glass jars with 300 mL of 0.2 μm filtered RSS (fRSS), and sampled after 3 weeks.
Before fRSS was added to the anemones, the water was plated on Marine Agar (DifcoTM Marine Agar 2216; MA); the absence of any bacterial growth on agar plates after 7 days confirmed sterility. In both conditions, anemones were fed ad libitum with freshly hatched Artemia salina (brine shrimp, Salt Creek, UT, United States) nauplii twice weekly. A. salina feedstock was not sterile prior to feeding (see Hartman et al., 2020 for details on feedstock bacterial community composition). Control and “sterile” anemone tanks were cleaned each week after feeding with complete RSS or fRSS water changes, respectively. Each anemone (n = 6 for control anemones and n = 15 for “sterile” anemones from each genotype) was individually homogenized in a sterile glass homogenizer in 1 mL fRSS, flash frozen in liquid nitrogen, and stored at –20°C for later DNA extraction. Immediately following anemone acquisition, RSS and fRSS was collected from each anemone culture container (n = 3 for RSS and n = 5 for fRSS for each genotype container). The collected RSS and fRSS had hosted the anemones for 7 days since the last water change. Briefly, 250 mL of RSS or fRSS was filtered through a 0.2 μm Supor hydrophilic polyethersulfone membrane (Pall, New York) with a Sentino peristaltic pump (Pall). The membrane was flash frozen then stored at –20°C until genomic DNA extraction.
Anemone Health Metrics
Health metrics were measured in control and “sterile” anemones. Photosynthetic efficiency, according to maximum quantum yield (Fv/Fm) was measured on the intracellular algae by pulse-amplitude-modulated (PAM) fluorometry with an imaging-PAM system (IMAG-MAX/L, Walz Heinz, Germany) weekly, and immediately prior to sampling for both control and “sterile” anemones. Additionally, a subset (n = 5 per genotype for both control and “sterile” anemones) were sacrificed for host protein and Symbiodiniaceae cell density determinations as described in Dungan et al. (2020b). Additionally, feeding behaviors, anemone body and tentacle position, and asexual propagation were qualitatively documented throughout the experiment.
DNA Extraction and MiSeq Library Preparation
DNA was extracted (Wilson et al., 2002; Hartman et al., 2019) from the anemones and RSS/fRSS samples for bacterial community analysis by metabarcoding of the 16S rRNA genes. Extraction blanks (n = 7) were processed simultaneously to identify contaminants introduced during DNA extractions. Extracted DNA along with no template negative PCR controls (n = 2) were amplified by PCR in triplicate using bacterial primers with Illumina adapters (underlined) targeting the V5–V6 regions of the 16S rRNA genes: 784F [5′ GTGACCTATGAACTCAGGAGTCAGGATTAGATACCCTGG TA 3′]; 1061R [5′ CTGAGACTTGCACATCGCAGCCRRCACGA GCTGACGAC 3′] (Andersson et al., 2008). Each PCR contained 12.5 μL MyTaq HS Mix polymerase (Bioline, Australia), 2 μL of DNA template, 0.4 μM of each primer, and nuclease-free water up to 25 μL. PCR conditions were: 1 cycle × 95°C for 3 min; 18 cycles × 95°C for 15 s, 55°C for 30 s, and 72°C for 30 s; 1 cycle × 72°C for 7 min; hold at 4°C. Triplicates were not pooled and instead treated as technical replicates in library preparation and sequencing.
To prepare DNA sequencing libraries, 20 μL of each PCR product was purified by size-selection using Ampure XP magnetic beads (Agencourt, Beckman Coulter, Australia). Indexing PCRs were created by combining 10 μL of purified DNA with 10 μL 2x Taq master mix (M0270S, New England BioLabs, Australia) and 0.25 μM of forward and reverse indexing primers. PCR conditions were: 1 cycle × 95°C for 3 min; 24 cycles × 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s; 1 cycle × 72°C for 7 min; hold 4°C. A subset of samples, chosen at random, were checked for product size and quantity (2200 TapeStation, Agilent Technologies, Australia); sequencing libraries were created by pooling 5 μL from each reaction and performing a final bead clean-up on 50 μL. Each library was checked for quality and quantity to guide pool normalization (2200 TapeStation), then sequenced on a single Illumina MiSeq run using v3 (2 × 300 bp) reagents at the Walter and Eliza Hall Institute, Melbourne, Australia.
Metabarcoding Data Processing and Analysis
Raw 16S rRNA gene sequences were imported into QIIME2 v2019.10.0 (Bolyen et al., 2019) and demultiplexed. Primers were removed with cutadapt v2.6 (Martin, 2011). Filtering, denoising, and chimera checking was performed using DADA2 (Callahan et al., 2016) in the QIIME2 environment to correct sequencing errors, remove low quality bases (mean Qscore < 30), and generate bacterial amplicon sequence variants (ASVs). Taxonomy for each ASV was assigned against a SILVA database (version 132) trained with a naïve Bayes classifier against the same V5–V6 region targeted for sequencing (Bokulich et al., 2018). A phylogenetic tree was produced in QIIME2 by aligning ASVs using the PyNAST method (Caporaso et al., 2010) with mid-point rooting.
All data were analyzed in R (v3.6.2; R Core Team, 2019). Statistical tests were considered significant at α = 0.05. ASV, taxonomy, metadata and phylogenetic tree files were imported into R and combined into a phyloseq object (McMurdie and Holmes, 2013). Potential contaminant ASVs were identified and removed sequentially from the dataset according to their abundance in the extraction and PCR negative controls relative to the anemone and seawater samples using the prevalence method in the R package decontam with p = 0.1 (Davis et al., 2018).
Alpha diversity of the bacteria was investigated to assess the impact of RSS type on the bacterial communities. Alpha diversity metrics were calculated in the R package vegan (Oksanen et al., 2018) after rarefying the samples to 3,718 reads per sample. Alpha diversity data were then analyzed for overall differences using linear mixed effects models (LME) against the variables genotype, maintenance in RSS or fRSS, and sample type (anemone or seawater) with culture container specified as a random effect, in the R package nlme (Pinheiro et al., 2019). Post hoc pairwise comparisons were performed by genotype using Tukey’s honestly significant difference test (Tukey, 1949) in the R package emmeans (Searle et al., 1980) with Tukey’s adjustment for multiple comparisons. Standard deviations from the LME model were used to determine the within and between sample variation for triplicate sequencing data. Bar charts were created by agglomerating taxa at the family level based on relative abundances using the R package phyloseq.
Differences in the bacterial community compositions of each genotype were visualized in principal coordinates analysis (PCoA) ordinations based on a weighted UniFrac distance matrix. To compare bacterial community compositions, we applied a non-parametric permutational multivariate analysis of variance (PERMANOVA) using the weighted UniFrac distance matrix as implemented in the vegan function adonis (permutation = 999) (Oksanen et al., 2018). Data were center log ratio transformed (Morton et al., 2017) prior to PERMANOVA and all data were evaluated for homogeneity of dispersion among groups as an assumption for adonis. Genera that were significantly associated with treatment groups were identified using the indicspecies function multipatt (Cáceres and Legendre, 2009) and changes in their relative abundances were visualized in bubble plots for each genotype.
Results
Anemone Health
Throughout the 3-week period, all E. diaphana maintained good health based on the density and photophysiological performance of their in hospite Symbiodiniaceae, tentacle extension, active feeding on A. salina nauplii, and asexual propagation. Average Symbiodiniaceae cell densities (normalized to host protein) were not significantly different between control and “sterile” anemones by genotype (p > 0.05, Supplementary File S1). After an initial drop in Fv/Fm following the transfer of anemones to fRSS and in a different incubator, Fv/Fm values returned to control levels.
Metabarcoding Data Processing
Sequencing produced 8.8 M reads across E. diaphana (n = 84), RSS/fRSS (n = 32), extraction blanks (n = 7), and negative control samples (n = 2) sequenced as triplicate technical replicates for a total of 375 samples. After merging, denoising and chimera filtering, 6.2 M reads remained; one triplicate from each of three samples was removed as they had <20 reads. Decontam identified 138 putative contaminant ASVs constituting 4.25% relative abundance of the bacterial communities. Upon review of these putative contaminants, 20 were found to be common marine bacteria. To be conservative, these 20 were removed from the decontam output and thus included in the subsequent data analyses. The remaining 118 ASVs (3.91% relative abundance) were removed from the data set prior to all statistical analyses (Supplementary File S2). After all filtering steps, there were 1171 ASVs across the remaining samples.
Triplicate Technical Replicates
For amplicon sequencing of the bacterial 16S rRNA gene V5–V6 region, the standard error within the three replicates of a single sample was greater than the error between biological replicates for the observed number of ASVs (13.12 and 9.35, respectively). For both Shannon’s and Simpson’s diversity metrics, the error between biological replicates and within a biological replicate was approximately the same. Given that these technical replicates all stem from one biological sample, the triplicates were merged for visualization and exploration of the bacterial microbial communities.
Bacterial Community Shifts
For all alpha diversity metrics, a one-way analysis of variance (ANOVA) reported a significant interaction of sample type (anemone vs. seawater) and environment (RSS vs. fRSS) [FObsASVs(1, 81) = 62.35, p < 0.0001; FSimp(1, 81) = 12.26, p = 0.0008; FShan(1, 81) = 51.67, p < 0.0001]. Substantial decreases in bacterial alpha diversity were observed in all “sterile” anemone genotypes (Figure 1A and Supplementary File S3) with the average number of observed ASVs dropping 55% from (mean ± SE) 134 ± 6 in control anemones to 74 ± 3 [t(18) = 8.85, p < 0.0001] in “sterile” anemones. Simpson’s and Shannon’s bacterial diversity also decreased in all anemones, regardless of genotype, with significant reductions in AIMS2 (Shannon’s only), AIMS3 (both Shannon’s and Simpson’s), and AIMS4 (Shannon’s only) (Figures 1B,C and Supplementary File S3). A total of 13 bacterial families were found in at least three genotypes of control anemones and absent in at least three genotypes of “sterile” anemones (Supplementary File S4). These families were all part of the rare bacterial biosphere (Sogin et al., 2006) with the most abundant, Microtrichaceae, having an average of 0.12% relative abundance across all control genotypes. Conversely, there were five families that were present in the “sterile” anemones, yet absent in control anemones (Supplementary File S4). The most abundant of these was Nisaeaceae at 0.18% relative abundance in “sterile” anemones.
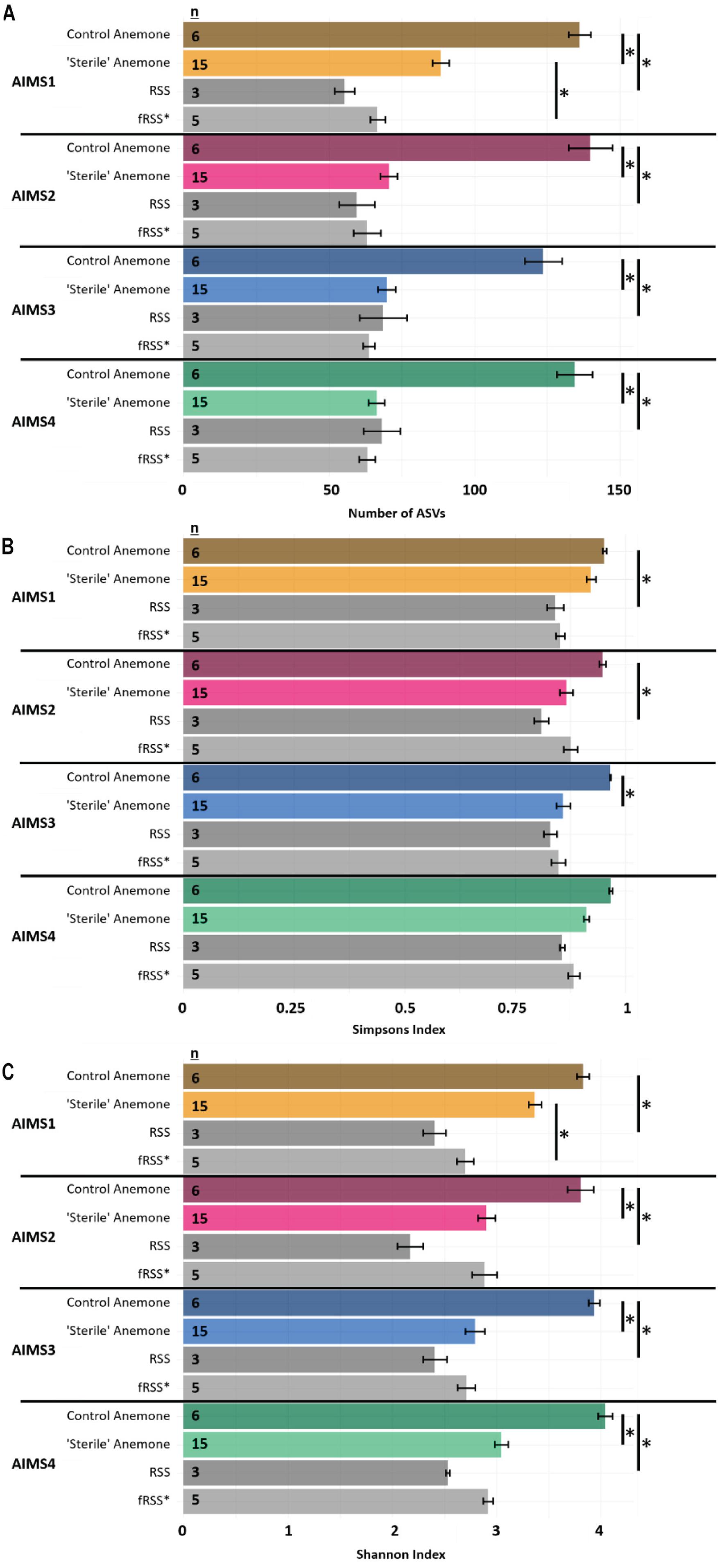
Figure 1. Observed ASVs (A), Simpson’s Index (B), and Shannon’s Index (C) for AIMS1-4 anemones (orange, purple, blue, green) and seawater (gray) samples in control (unfiltered seawater) or sterilized seawater (SW). The data set was rarefied to 16,993 reads per sample with technical triplicates pooled. Asterisks indicate significant pairwise differences between sample types by genotype. Averages ± SE are shown. Statistical output for pairwise comparisons is in Supplementary File S3. ∗fRSS was collected after hosting anemones for 7 days.
Principal coordinate analyses (PCoA) showed a change in composition of the bacterial communities within each genotype (Figure 2A). There was clear clustering of data points by sample type (anemone vs. fRSS/RSS) and environmental condition. For all genotypes, homogeneity of dispersion was >0.05 for seawater treatment and sample type. PERMANOVA results by genotype confirmed that the bacterial community structure (Figure 2B) varied significantly with the interaction of sample type and environment [FAIMS1(1, 86) = 6.59, p = 0.001; FAIMS2(1, 83) = 5.82, p = 0.001; FAIMS3(1, 85) = 6.18, p = 0.001; FAIMS4(1, 86) = 5.64, p = 0.001]. The indicator species analysis highlighted genera that are characteristic of the microbiomes in these distinct groups (Figure 3 and Supplementary Files S5–S8). Erythrobacter, Winogradskyella, Phaeodactylibacter, Muricauda, and unresolved Rhodobacteraceae genera were significantly more abundant in control anemones compared to “sterile” anemones (Supplementary File S9). Few genera were significantly more abundant in “sterile” anemones (only Nisaea, Thalassobaculum, and Alteromonas in AIMS3 and Tropicibacter in AIMS4). Groups that were significantly more abundant in the anemones, regardless of environment or genotype, compared to seawater were Alteromonadaceae, Terasakiellaceae, Coxiella, Labrenzia, Nannocystaceae, Sphingobacteriales, and Fulvivirga. In the RSS Roseibacterium and Pseudohongiella were consistently more abundant while Algiphilus was in greater abundance in fRSS. Saprospiraceae was the only group that was significantly more abundant in all seawater samples, regardless of genotype or environment (Supplementary File S9).
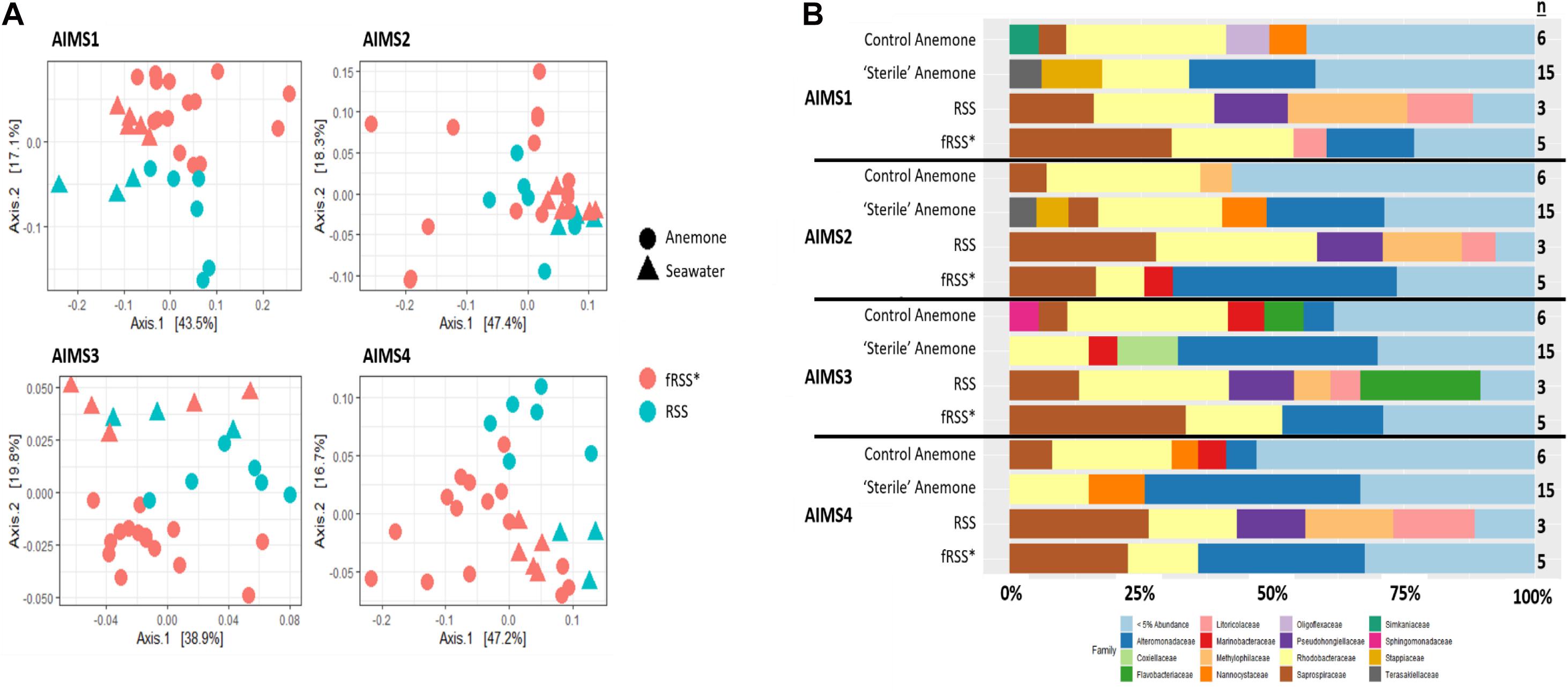
Figure 2. Principle coordinate analysis (PCoA) (A) for AIMS1-4 using a weighted UniFrac distance matrix for the anemone (circles) and seawater (triangles) samples in the control (blue symbols) and sterile (red symbols) treatments. Each point is an individual sample with technical triplicates merged. Relative abundance of bacterial families (B) for anemone and seawater samples in RSS and fRSS treatments between GBR-sourced anemone genotypes AIMS1-4. Families whose relative abundance was less than 5% across the whole data set were pooled into a single category. ∗fRSS was collected after hosting anemones for 7 days.
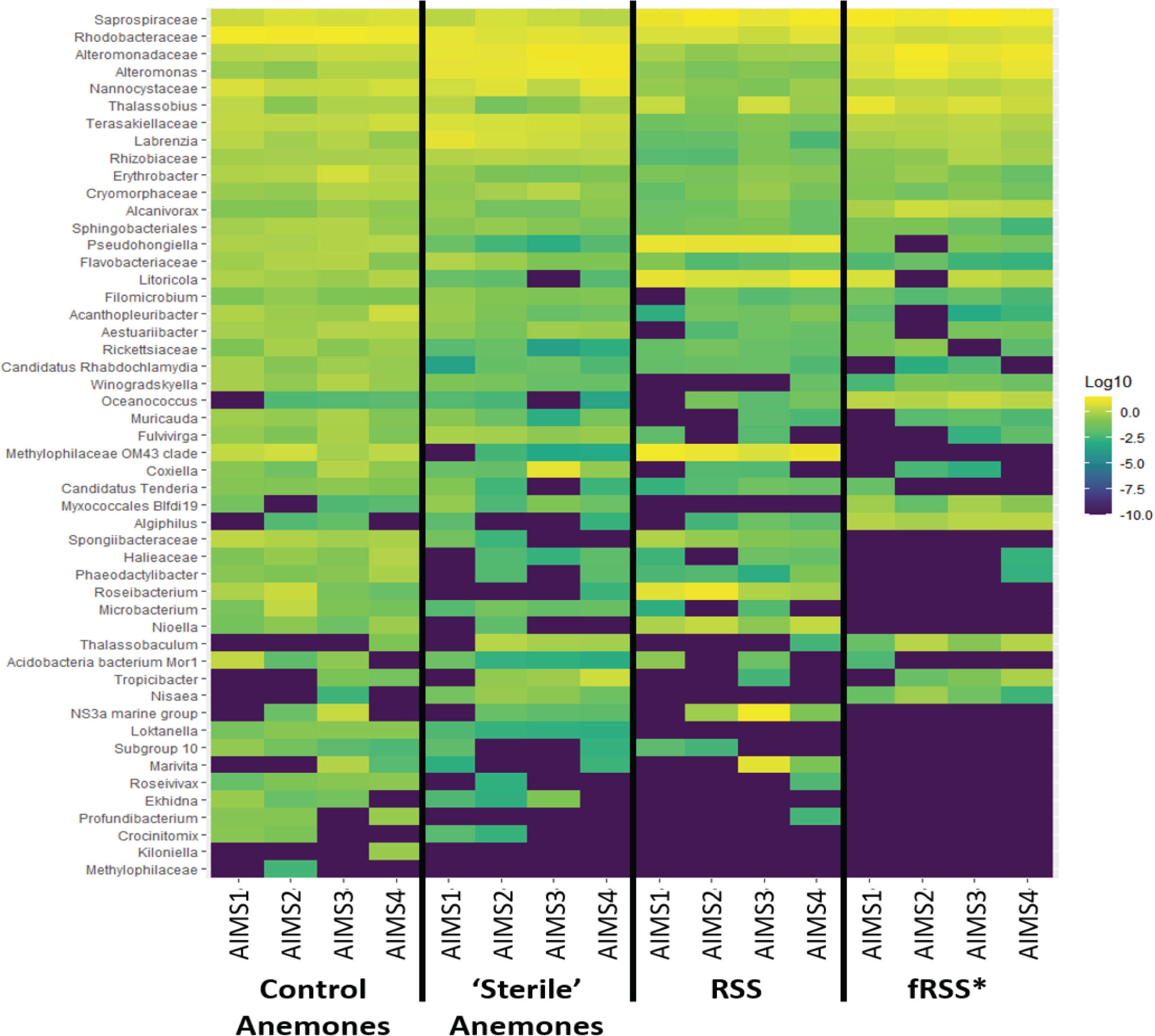
Figure 3. Heat map showing the relative abundance of bacterial taxa for each treatment group (control and “sterile” anemones, RSS and fRSS) for AIMS1-4 anemones. The color of each square corresponds with the relative abundance, which was log10 normalized. Taxa were selected from an indicator species analysis with parameters At = 0.8 and Bt = 0.8, where A is the probability that a sample belongs to its target group or specificity and B is the probability of finding the genus in samples belonging to the sample group or fidelity. ∗fRSS was collected after hosting anemones for 7 days.
Discussion
The coral holobiont has the capacity to restructure its bacterial component, either through natural (Ziegler et al., 2017) or assisted strategies (Damjanovic et al., 2017), which has been suggested as a means of holobiont adaptation to climate change (Ziegler et al., 2019). We found that the bacterial diversity and community structure for each of four anemone genotypes was significantly different with a reduction in alpha diversity when anemones were moved from RSS to fRSS and maintained in fRSS for 3 weeks. Several taxa, including Alteromonadaceae, Terasakiellaceae, Coxiella, Labrenzia, Nannocystaceae, Sphingobacteriales, and Fulvivirga, maintained high abundances in “sterile” anemones despite changes in other aspects of community structure. We suggest that these groups have a key role in holobiont health. Our results have implications for future manipulation of the E. diaphana holobiont, as these anemones can be manipulated to reduce the diversity of their microbiome while remaining healthy and thus are good models. The loss of bacterial diversity in the “sterile” anemones and likely passage of these microbes into the seawater hints at the interaction of E. diaphana with their seawater environment.
Exaiptasia diaphana Microbiome Can Be Reduced With Sterile Seawater
From this study, the average number of ASVs from GBR-sourced E. diaphana was 134, which is substantially lower than >750 ASVs, which was previously reported for these anemones (Hartman et al., 2020). After anemones were maintained for 3 weeks in fRSS, the number of observed bacterial ASVs was significantly reduced in all genotypes while maintaining holobiont health. This has been previously shown with GBR-sourced E. diaphana (Hartman et al., 2019). A common approach to reduce the microbiome of a complex system is the application of antibiotics, which has been successful in marine invertebrates, including corals (Sweet et al., 2011), but often at the expense of holobiont health (D’agostino, 1975; Gilbert et al., 2012; Hartman, 2020). Even when there are no visible adverse effects of antibiotic treatment, the artificial shift in or depletion of the holobiont microbiome may have unintended consequences such as perturbations of native microbial communities and proliferation of pathogens (Raymann et al., 2017), or the development of antibiotic resistance. A disruption to the bacterial community of coral via antibiotic treatment has resulted in diminished resilience to thermal stress (Gilbert et al., 2012), bleaching, and necrosis (Glasl et al., 2016). We have demonstrated that the microbial diversity of E. diaphana is rapidly (in 3 weeks) reduced in sterile seawater with an average of 55% of ASVs lost, critically, with no changes in Symbiodiniaceae photophysiology or anemone heath. A total of 13 bacterial families were found in at least three genotypes of control anemones and absent in at least three genotypes of “sterile” anemones (Supplementary File S3). The capacity for horizontal transmission (recruitment of microorganisms from the environment) has been proposed for several marine organisms (Bright and Bulgheresi, 2010), including corals (Damjanovic et al., 2019b) and sponges (Taylor et al., 2007). It is plausible that horizontal transmission of bacteria in a marine environment is an active process with some host-associated bacteria capable of leaving if the conditions are favorable. Here, by filter sterilizing the seawater, we may have removed competitive bacterial taxa that prevented some host-associated taxa from proliferating. Alternatively, filtering of RSS could have removed non-soluble organic compounds that supported the growth of facultative taxa. Thus, fRSS might not have been a favorable environment for a diverse set of bacteria to persist.
Potential Key Microbiome Members of Exaiptasia
Bacterial genera were identified as indicators that reflect the treatment (RSS or fRSS) of the anemones and seawater (Figure 3). Erythrobacter, Winogradskyella, Phaeodactylibacter, Muricauda, and unresolved Rhodobacteraceae genera were significantly more abundant in control anemones compared to “sterile” anemones. At least three of these groups (Erythrobacter, Winogradskyella, and Muricauda) are common members of the Symbiodiniaceae microbiome (Lawson et al., 2017; Nitschke et al., 2020) and some are known carotenoid producers (Prabhu et al., 2014; Zhang et al., 2016; Setiyono et al., 2019), which could serve as potent antioxidants protecting the algal symbiont during periods of oxidative stress (Schmitt et al., 2014). Erythrobacter produces enzymes that can inactivate common antibacterial compounds (Jiang et al., 2018), while Winogradskyella produces antifouling compounds (Dash et al., 2009). Muricauda has an increased competitive advantage during stressful conditions due to their production of the signal molecule indole (Wang et al., 2017). Given their absence in healthy “sterile” anemones and ability to outcompete other bacteria, they might be considered opportunistic colonizers. The only bacterial genera that were significantly more abundant in “sterile” anemones were Nisaea, Thalassobaculum, and Alteromonas (AIMS3) and Tropicibacter (AIMS4). These four genera are all Alphaproteobacteria, with the former two being members of the order Thalassobaculales and the latter two being Rhodobacterales members.
Several taxa were significantly more represented in the anemones vs. seawater, regardless of genotype or seawater filtration status. These included Alteromonadaceae, Labrenzia, Terasakiellaceae, Coxiella, Nannocystaceae, Sphingobacteriales, and Fulvivirga. These potentially obligate anemone-associated taxa likely contribute to an important anemone feature like health or metabolism. The taxa we identified as stably associated with E. diaphana can be divided into those that are frequently found in cnidarians or their algal symbionts, including Alteromonadaceae (Rothig et al., 2017; Ahmed et al., 2019; Damjanovic et al., 2019a; Epstein et al., 2019b), Labrenzia (Lawson et al., 2017), Fulvivirga (Glasl et al., 2016; Ziegler et al., 2017; Epstein et al., 2019b; Pootakham et al., 2019; Damjanovic et al., 2020) and Sphingobacteriales (Meron et al., 2012; Kellogg et al., 2013; Li et al., 2014; van de Water et al., 2017; Bonthond et al., 2018), and those that occur much more infrequently, including Terasakiellaceae, Coxiella, and Nannocystaceae. Labrenzia is part of the core microbiome of the anemones’ Symbiodiniaceae (Lawson et al., 2017) and may have a role in the production of antioxidants such as dimethylsulfoniopropionate (DMSP) and its breakdown products (Sunda et al., 2002). The DMSP biosynthesis (Curson et al., 2017) and degradation genes, were found in the whole genome sequences of E. diaphana-sourced Labrenzia isolates (Dungan et al., 2020a). Sphingobacteriales in soil systems are believed to be copiotrophic, referring to their ability to metabolize a wide array of carbon sources (Hester et al., 2018).
Terasakiellaceae (Rhodospirallales) were reported in the cold water coral Paragorgia arborea (Weiler et al., 2018) where they were implicated in nitrogen cycling, and they could have a similar role in the anemone holobiont. Coxiella are members of the order Legionellales, which are described as facultative or obligate intracellular parasites known to infect invertebrate and vertebrate species (Garrity and Brenner, 2005), including marine animals; their role in anemones should be determined. While there are few references to Nannocystaceae in the literature (i.e., Gignoux-Wolfsohn et al., 2020), the order Myxococcales has been reported in coral metabarcoding studies (Godoy-Vitorino et al., 2017; Ziegler et al., 2017; Pollock et al., 2018; Rosales et al., 2019). Myxococcales has been associated with disease resistant corals (Rosales et al., 2019) and could have some importance in pathogen control.
Mitigating Metabarcoding Biases
Recent evidence suggests that pooling triplicate PCRs prior to sequencing is redundant and that a single replicate is sufficient as modern amplicon data analysis is robust enough to reduce the influence of artifacts introduced during the PCR (Marotz et al., 2019). We compared triplicate technical replicates from single reactions for amplicon sequencing of the bacterial 16S rRNA gene V5–V6 region and found that the standard error within was greater than the error between biological samples from a single sampling replicate for the observed number of ASVs. Because each sample was sequenced in the same MiSeq run, we would expect less variation between the technical replicates than the biological samples. However, in the case of observed numbers of ASVs, there was a strong, consistent effect of technical replicate. The variation between technical replicates is likely an artifact of errors that arise during the early stages of PCR, which are then amplified exponentially, and chimera formation (Polz and Cavanaugh, 1998), thus these results support the continued practice of pooling triplicate PCRs prior to the indexing step as outlined in the Earth’s Microbiome Project protocol (Gilbert et al., 2014). Like the Earth’s Microbiome Project, having standard procedures for cnidarian sampling and processing, as well as the establishment of a mock community standard comprised of coral-associated taxa, could mitigate some of these potential biases.
Conclusion
The results from this study have implications for future manipulation of the E. diaphana microbiome and perhaps that of corals. Recent studies have shown that cnidarians can acquire bacteria introduced into their external environment (Damjanovic et al., 2017, 2019b, 2020) and, with the addition of probiotic bacteria, may have the ability to mitigate the effects of climate change (Rosado et al., 2018). Future studies should test the ability of E. diaphana to acquire bacteria from their environment with a focus on microbes that have potentially beneficial traits for the holobiont.
The use of probiotics or synthetic bacterial communities has been effective in treating disease in humans (de Vos, 2013; Shetty et al., 2019) and aquaculture (Verschuere et al., 2000; Ringø, 2020) as well as promoting plant growth (Qin et al., 2016). Following these systems, modifying or engineering the microbiome of corals with probiotics may be an option to enhance coral recovery from bleaching or disease (Peixoto et al., 2017; Rosado et al., 2018). These types of interventions will likely require extensive experimentation, thus, the ability to work with a reduced microbiome in a healthy anemone holobiont is important. We found that culturing E. diaphana in fRSS vs. RSS was able to halve the microbiome diversity while still maintaining holobiont health and function. This finding will be invaluable for bacterial bioengineering approaches to increase the climate resilience of the coral holobiont.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA576556).
Author Contributions
AD, MO, and LB designed and conceived the experiments. AD conducted the experiments, collected, analyzed, and interpreted the data. MO and LB contributed reagents, materials, and analysis tools. AD wrote the first draft of the manuscript. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication and read and approved the final manuscript.
Funding
This research was supported by the Australian Research Council Discovery Project grant DP160101468 (to MO and LB). MO acknowledges Australian Research Council Laureate Fellowship FL180100036.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Leon Hartman for help with figure design and review. We also thank Cameron Patrick and the Melbourne Statistical Consulting Platform for support with data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.599314/full#supplementary-material
References
Ahmed, H. I., Herrera Sarrias, M., Liew, Y. J., and Aranda, M. (2019). Long-term temperature stress in the coral model Aiptasia supports the “Anna Karenina principle” for bacterial microbiomes. Front. Microbiol. 10:975. doi: 10.3389/fmicb.2019.00975
Ainsworth, T. D., Fordyce, A. J., and Camp, E. F. (2017). The other microeukaryotes of the coral reef microbiome. Trends Microbiol. 25, 980–991. doi: 10.1016/j.tim.2017.06.007
Alagely, A., Krediet, C. J., Ritchie, K. B., and Teplitski, M. (2011). Signaling-mediated cross-talk modulates swarming and biofilm formation in a coral pathogen Serratia marcescens. ISME J. 5, 1609–1620. doi: 10.1038/ismej.2011.45
Andersson, A. F., Lindberg, M., Jakobsson, H., Bäckhed, F., Nyrén, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 3:e2836. doi: 10.1371/journal.pone.0002836
Blackall, L. L., Wilson, B., and van Oppen, M. J. H. (2015). Coral-the world’s most diverse symbiotic ecosystem. Mol. Ecol. 24, 5330–5347. doi: 10.1111/mec.13400
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90.
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857.
Bonthond, G., Merselis, D. G., Dougan, K. E., Graff, T., Todd, W., Fourqurean, J. W., et al. (2018). Inter-domain microbial diversity within the coral holobiont Siderastrea siderea from two depth habitats. PeerJ 6:e4323. doi: 10.7717/peerj.4323
Bright, M., and Bulgheresi, S. (2010). A complex journey: transmission of microbial symbionts. Nat. Rev. Microbiol. 8, 218–230. doi: 10.1038/nrmicro2262
Brown, T., Otero, C., Grajales, A., Rodriguez, E., and Rodriguez-Lanetty, M. (2017). Worldwide exploration of the microbiome harbored by the cnidarian model, Exaiptasia pallida (Agassiz in Verrill, 1864) indicates a lack of bacterial association specificity at a lower taxonomic rank. PeerJ 5:e3235. doi: 10.7717/peerj.3235
Cáceres, M. D., and Legendre, P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Camp, E. F., Kahlke, T., Nitschke, M. R., Varkey, D., Fisher, N. L., Fujise, L., et al. (2020). Revealing changes in the microbiome of Symbiodiniaceae under thermal stress. Environ. Microbiol. 22, 1294–1309. doi: 10.1111/1462-2920.14935
Caporaso, J. G., Bittinger, K., Bushman, F. D., Desantis, T. Z., Andersen, G. L., and Knight, R. (2010). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Clavel, T., Gomes-Neto, J. C., Lagkouvardos, I., and Ramer-Tait, A. E. (2017). Deciphering interactions between the gut microbiota and the immune system via microbial cultivation and minimal microbiomes. Immunol. Rev. 279, 8–22. doi: 10.1111/imr.12578
Curson, A. R., Liu, J., Bermejo Martinez, A., Green, R. T., Chan, Y., Carrion, O., et al. (2017). Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nat. Microbiol. 2:17009.
D’agostino, A. (1975). “Antibiotics in cultures of invertebrates,” in Culture of Marine Invertebrate Animals, eds M. H. Chanley and W. L. Smith (Berlin: Springer), 109–133. doi: 10.1007/978-1-4615-8714-9_7
Damjanovic, K., Blackall, L. L., Webster, N. S., and van Oppen, M. J. H. (2017). The contribution of microbial biotechnology to mitigating coral reef degradation. Microb. Biotechnol. 10, 1236–1243. doi: 10.1111/1751-7915.12769
Damjanovic, K., Menéndez, P., Blackall, L. L., and van Oppen, M. J. H. (2019a). Early life stages of a common broadcast spawning coral associate with specific bacterial communities despite lack of internalized bacteria. Microb. Ecol. 79, 706–719. doi: 10.1007/s00248-019-01428-1
Damjanovic, K., Menéndez, P., Blackall, L. L., and van Oppen, M. J. H. (2020). Mixed-mode bacterial transmission in the common brooding coral Pocillopora acuta. Environ. Microbiol. 22, 397–412. doi: 10.1111/1462-2920.14856
Damjanovic, K., van Oppen, M. J. H., Menéndez, P., and Blackall, L. L. (2019b). Experimental inoculation of coral recruits with marine bacteria indicates scope for microbiome manipulation in Acropora tenuis and Platygyra daedalea. Front. Microbiol. 10:1702. doi: 10.3389/fmicb.2019.01702
Dash, S., Jin, C., Lee, O. O., Xu, Y., and Qian, P.-Y. (2009). Antibacterial and antilarval-settlement potential and metabolite profiles of novel sponge-associated marine bacteria. J. Ind. Microbiol. Biotechnol. 36, 1047–1056. doi: 10.1007/s10295-009-0588-x
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A., and Callahan, B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6:226.
de Vos, W. M. (2013). Fame and future of faecal transplantations–developing next-generation therapies with synthetic microbiomes. Microb. Biotechnol. 6, 316–325. doi: 10.1111/1751-7915.12047
dos Santos, F. A. H., Duarte, G. A., Rachid, C. T., Chaloub, R. M., Calderon, E. N., Marangoni, L. F., et al. (2015). Impact of oil spills on coral reefs can be reduced by bioremediation using probiotic microbiota. Sci. Rep. 5:18268.
Dungan, A. M., Bulach, D., Lin, H., van Oppen, M. J. H., and Blackall, L. L. (2020a). Development of a free radical scavenging probiotic to mitigate coral bleaching. bioRxiv [Preprint]. doi: 10.1101/2020.07.02.185645
Dungan, A. M., Hartman, L. M., Tortorelli, G., Belderok, R., Lamb, A. M., Pisan, L., et al. (2020b). Exaiptasia diaphana from the great barrier reef: a valuable resource for coral symbiosis research. Symbiosis 80, 195–206. doi: 10.1007/s13199-020-00665-0
Epstein, H. E., Smith, H. A., Torda, G., and van Oppen, M. J. H. (2019a). Microbiome engineering: enhancing climate resilience in corals. Front. Ecol. Environ. 17, 100–108. doi: 10.1002/fee.2001
Epstein, H. E., Torda, G., and van Oppen, M. J. H. (2019b). Relative stability of the Pocillopora acuta microbiome throughout a thermal stress event. Coral Reefs 38, 373–386. doi: 10.1007/s00338-019-01783-y
Garrity, G., and Brenner, D. (2005). Bergey’s Manual of Systematic Bacteriology: The Gammaproteobacteria. Cham: Springer.
Gignoux-Wolfsohn, S. A., Precht, W. F., Peters, E. C., Gintert, B. E., and Kaufman, L. S. (2020). Ecology, histopathology, and microbial ecology of a white-band disease outbreak in the threatened staghorn coral Acropora cervicornis. Dis. Aquat. Organ 137, 217–237. doi: 10.3354/dao03441
Gilbert, J. A., Hill, R., Doblin, M. A., and Ralph, P. J. (2012). Microbial consortia increase thermal tolerance of corals. Mar. Biol. 159, 1763–1771. doi: 10.1007/s00227-012-1967-9
Gilbert, J. A., Jansson, J. K., and Knight, R. (2014). The Earth Microbiome project: successes and aspirations. BMC Biol. 12:69. doi: 10.1186/s12915-014-0069-1
Glasl, B., Herndl, G. J., and Frade, P. R. (2016). The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 10, 2280–2292. doi: 10.1038/ismej.2016.9
Godoy-Vitorino, F., Ruiz-Diaz, C. P., Rivera-Seda, A., Ramirez-Lugo, J. S., and Toledo-Hernandez, C. (2017). The microbial biosphere of the coral Acropora cervicornis in Northeastern Puerto Rico. PeerJ 5:e3717. doi: 10.7717/peerj.3717
Hartman, L. M. (2020). Manipulation of prokaryotic communities in the coral model organism, Exaiptasia diaphana. Ph.D. thesis, Swinburne University of Technology, Hawthorn.
Hartman, L. M., van Oppen, M. J. H., and Blackall, L. L. (2019). The effect of thermal stress on the bacterial microbiome of Exaiptasia diaphana. Microorganisms 8:20. doi: 10.3390/microorganisms8010020
Hartman, L. M., van Oppen, M. J. H., and Blackall, L. L. (2020). Microbiota characterization of Exaiptasia diaphana from the Great Barrier Reef. Anim. Microb. 2, 1–14.
Herrera, M., Ziegler, M., Voolstra, C. R., and Aranda, M. (2017). Laboratory-cultured strains of the sea anemone exaiptasia reveal distinct bacterial communities. Front. Mar. Sci. 4:115. doi: 10.3389/fmars.2017.00115
Hester, E. R., Harpenslager, S. F., Van Diggelen, J. M. H., Lamers, L. L., Jetten, M. S. M., Lüke, C., et al. (2018). Linking nitrogen load to the structure and function of wetland soil and rhizosphere microbial communities. mSystems 3, e00214–e00217.
Huggett, M. J., and Apprill, A. (2019). Coral microbiome database: integration of sequences reveals high diversity and relatedness of coral-associated microbes. Environ. Microbiol. Rep. 11, 372–385. doi: 10.1111/1758-2229.12686
Jiang, X.-W., Cheng, H., Huo, Y.-Y., Xu, L., Wu, Y.-H., Liu, W.-H., et al. (2018). Biochemical and genetic characterization of a novel metallo-β-lactamase from marine bacterium Erythrobacter litoralis HTCC 2594. Sci. Rep. 8:803.
Kellogg, C. A., Piceno, Y. M., Tom, L. M., Desantis, T. Z., Gray, M. A., Zawada, D. G., et al. (2013). Comparing bacterial community composition between healthy and white plague-like disease states in Orbicella annularis using PhyloChip G3 microarrays. PLoS One 8:e79801. doi: 10.1371/journal.pone.0079801
Lawson, C. A., Raina, J. B., Kahlke, T., Seymour, J. R., and Suggett, D. J. (2017). Defining the core microbiome of the symbiotic dinoflagellate, Symbiodinium. Environ. Microbiol. Rep. 10, 7–11. doi: 10.1111/1758-2229.12599
Li, J., Chen, Q., Long, L. J., Dong, J. D., Yang, J., and Zhang, S. (2014). Bacterial dynamics within the mucus, tissue and skeleton of the coral Porites lutea during different seasons. Sci. Rep. 4:7320.
Marotz, C., Sharma, A., Humphrey, G., Gottel, N., Daum, C., Gilbert, J. A., et al. (2019). Triplicate PCR reactions for 16S rRNA gene amplicon sequencing are unnecessary. BioTechniques 67, 29–32. doi: 10.2144/btn-2018-0192
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 17, 10–12. doi: 10.14806/ej.17.1.200
Matthews, J. L., Raina, J. B., Kahlke, T., Seymour, J. R., van Oppen, M. J. H., and Suggett, D. J. (2020). Symbiodiniaceae-bacteria interactions: rethinking metabolite exchange in reef-building corals as multi-partner metabolic networks. Environ. Microbiol. 22, 1675–1687. doi: 10.1111/1462-2920.14918
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: x10.1371/journal.pone.0061217
Meron, D., Rodolfo-Metalpa, R., Cunning, R., Baker, A. C., Fine, M., and Banin, E. (2012). Changes in coral microbial communities in response to a natural pH gradient. ISME J. 6, 1775–1785. doi: 10.1038/ismej.2012.19
Morton, J. T., Toran, L., Edlund, A., Metcalf, J. L., Lauber, C., and Knight, R. (2017). Uncovering the horseshoe effect in microbial analyses. mSystems 2:e00166-16.
Nitschke, M. R., Fidalgo, C., Simões, J., Brandão, C., Alves, A., Serôdio, J., et al. (2020). Symbiolite formation: a powerful in vitro model to untangle the role of bacterial communities in the photosynthesis-induced formation of microbialites. ISME J. 14, 1533–1546. doi: 10.1038/s41396-020-0629-z
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’hara, R. B., et al. (2018). ”vegan: Community Ecology Package”. 2.5.
Peixoto, R. S., Rosado, P. M., Leite, D. C., Rosado, A. S., and Bourne, D. G. (2017). Beneficial microorganisms for corals (BMC): proposed mechanisms for coral health and resilience. Front. Microbiol. 8:341. doi: 10.3389/fmicb.2017.00341
Pinheiro, J., Bates, D., Debroy, S., Sarkar, D., and R Core Team (2019). ”nlme: Linear and Nonlinear Mixed Effects Models”. 3.1, 141 Edn.
Pollock, F. J., Mcminds, R., Smith, S., Bourne, D. G., Willis, B. L., Medina, M., et al. (2018). Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat. Commun. 9:4921.
Polz, M. F., and Cavanaugh, C. M. (1998). Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 64, 3724–3730. doi: 10.1128/aem.64.10.3724-3730.1998
Pootakham, W., Mhuantong, W., Yoocha, T., Putchim, L., Jomchai, N., Sonthirod, C., et al. (2019). Heat-induced shift in coral microbiome reveals several members of the Rhodobacteraceae family as indicator species for thermal stress in Porites lutea. MicrobiologyOpen 8:e935.
Prabhu, S., Rekha, P., and Arun, A. (2014). Zeaxanthin biosynthesis by members of the genus Muricauda. Pol. J. Microbiol. 63, 115–119. doi: 10.33073/pjm-2014-017
Qin, Y., Druzhinina, I. S., Pan, X., and Yuan, Z. (2016). Microbially mediated plant salt tolerance and microbiome-based solutions for saline agriculture. Biotechnol. Adv. 34, 1245–1259. doi: 10.1016/j.biotechadv.2016.08.005
R Core Team (2019). A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Raymann, K., Shaffer, Z., and Moran, N. A. (2017). Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLoS Biol. 15:e2001861. doi: 10.1371/journal.pbio.2001861
Ringø, E. (2020). Probiotics in shellfish aquaculture. Aquac. Fish. 5, 1–27. doi: 10.1016/j.aaf.2019.12.001
Rohwer, F., Seguritan, V., Azam, F., and Knowlton, N. (2002). Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243:10.
Rosado, P. M., Leite, D. C. A., Duarte, G. A. S., Chaloub, R. M., Jospin, G., Nunes Da Rocha, U., et al. (2018). Marine probiotics: increasing coral resistance to bleaching through microbiome manipulation. ISME J. 13, 921–936. doi: 10.1038/s41396-018-0323-6
Rosales, S. M., Miller, M. W., Williams, D. E., Traylor-Knowles, N., Young, B., and Serrano, X. M. (2019). Microbiome differences in disease-resistant vs. susceptible Acropora corals subjected to disease challenge assays. Sci. Rep. 9, 1–11.
Röthig, T., Costa, R. M., Simona, F., Baumgarten, S., Torres, A. F., Radhakrishnan, A., et al. (2016). Distinct bacterial communities associated with the coral model aiptasia in aposymbiotic and symbiotic states with symbiodinium. Front. Mar. Sci. 3:234. doi: 10.3389/fmars.2016.00234
Rothig, T., Yum, L. K., Kremb, S. G., Roik, A., and Voolstra, C. R. (2017). Microbial community composition of deep-sea corals from the Red Sea provides insight into functional adaption to a unique environment. Sci. Rep. 7:44714.
Schmitt, F.-J., Renger, G., Friedrich, T., Kreslavski, V. D., Zharmukhamedov, S. K., Los, D. A., et al. (2014). Reactive oxygen species: re-evaluation of generation, monitoring and role in stress-signaling in phototrophic organisms. Biochim. Biophys. Acta Bioenerget. 1837, 835–848. doi: 10.1016/j.bbabio.2014.02.005
Searle, S. R., Speed, F. M., and Milliken, G. A. (1980). Population marginal means in the linear model: an alternative to least squares means. Am. Stat. 34, 216–221. doi: 10.2307/2684063
Setiyono, E., Pringgenies, D., Shioi, Y., Kanesaki, Y., Awai, K., and Brotosudarmo, T. H. P. (2019). Sulfur-containing carotenoids from a marine coral symbiont Erythrobacter flavus Strain KJ5. Mar. Drugs 17:349. doi: 10.3390/md17060349
Shetty, S. A., Smidt, H., and de Vos, W. M. (2019). Reconstructing functional networks in the human intestinal tract using synthetic microbiomes. Curr. Opin. Biotechnol. 58, 146–154. doi: 10.1016/j.copbio.2019.03.009
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Sunda, W., Kieber, D. J., Kiene, R. P., and Huntsman, S. (2002). An antioxidant function for DMSP and DMS in marine algae. Nature 418, 317–320. doi: 10.1038/nature00851
Sweet, M., Croquer, A., and Bythell, J. (2011). Dynamics of bacterial community development in the reef coral Acropora muricata following experimental antibiotic treatment. Coral Reefs 30:1121. doi: 10.1007/s00338-011-0800-0
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/mmbr.00040-06
Tukey, J. (1949). Comparing individual means in the analysis of variance. Biometrics 5, 99–144. doi: 10.2307/3001913
van de Water, J. A., Melkonian, R., Voolstra, C. R., Junca, H., Beraud, E., Allemand, D., et al. (2017). Comparative assessment of mediterranean gorgonian-associated microbial communities reveals conserved core and locally variant bacteria. Microb. Ecol. 73, 466–478. doi: 10.1007/s00248-016-0858-x
van Oppen, M. J. H., and Blackall, L. L. (2019). Coral microbiome dynamics, functions and design in a changing world. Nat. Rev. Microbiol. 17, 557–567. doi: 10.1038/s41579-019-0223-4
van Oppen, M. J. H., Bongaerts, P., Frade, P., Peplow, L. M., Boyd, S. E., Nim, H. T., et al. (2018). Adaptation to reef habitats through selection on the coral animal and its associated microbiome. Mol. Ecol. 27, 2956–2971. doi: 10.1111/mec.14763
van Oppen, M. J. H., Oliver, J. K., Putnam, H. M., and Gates, R. D. (2015). Building coral reef resilience through assisted evolution. Proc. Natl. Acad. Sci. U.S.A. 112, 2307–2313. doi: 10.1073/pnas.1422301112
Verschuere, L., Rombaut, G., Sorgeloos, P., and Verstraete, W. (2000). Probiotic bacteria as biological control agents in aquaculture. Microbiol. Mol. Biol. Rev. 64:17.
Wang, Y., Li, H., Cui, X., and Zhang, X.-H. (2017). A novel stress response mechanism, triggered by indole, involved in quorum quenching enzyme MomL and iron-sulfur cluster in Muricauda olearia Th120. Sci. Rep. 7:4252.
Webster, N. S., and Reusch, T. B. H. (2017). Microbial contributions to the persistence of coral reefs. ISME J. 11, 2167–2174. doi: 10.1038/ismej.2017.66
Weiler, B. A., Verhoeven, J. T. P., and Dufour, S. C. (2018). Bacterial communities in tissues and surficial mucus of the cold-water coral Paragorgia arborea. Front. Mar. Sci. 5:378. doi: 10.3389/fmars.2018.00378
Wilson, K., Li, Y., Whan, V., Lehnert, S., Byrne, K., Moore, S., et al. (2002). Genetic mapping of the black tiger shrimp Penaeus monodon with amplified fragment length polymorphism. Aquaculture 204, 297–309. doi: 10.1016/s0044-8486(01)00842-0
Zhang, D.-C., Liu, Y.-X., Huang, H.-J., Weber, K., and Margesin, R. (2016). Winogradskyella sediminis sp. nov., isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 66, 3157–3163. doi: 10.1099/ijsem.0.001157
Zhang, Y.-Y., Ling, J., Yang, Q.-S., Wang, Y.-S., Sun, C.-C., Sun, H.-Y., et al. (2015). The diversity of coral associated bacteria and the environmental factors affect their community variation. Ecotoxicology 24, 1467–1477. doi: 10.1007/s10646-015-1454-4
Ziegler, M., Grupstra, C. G. B., Barreto, M. M., Eaton, M., Baomar, J., Zubier, K., et al. (2019). Coral bacterial community structure responds to environmental change in a host-specific manner. Nat. Commun. 10:3092.
Keywords: metabarcoding, microbiome manipulation, Exaiptasia pallida, bacteria, cnidarian
Citation: Dungan AM, van Oppen MJH and Blackall LL (2021) Short-Term Exposure to Sterile Seawater Reduces Bacterial Community Diversity in the Sea Anemone, Exaiptasia diaphana. Front. Mar. Sci. 7:599314. doi: 10.3389/fmars.2020.599314
Received: 26 August 2020; Accepted: 28 December 2020;
Published: 21 January 2021.
Edited by:
Stefano Goffredo, University of Bologna, ItalyReviewed by:
Matthew R. Nitschke, University of Aveiro, PortugalSilvia Franzellitti, University of Bologna, Italy
Copyright © 2021 Dungan, van Oppen and Blackall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashley M. Dungan, YWR1bmdhbjMxQGdtYWlsLmNvbQ==