 Vinaya Kumar Katneni1*
Vinaya Kumar Katneni1* Mudagandur S. Shekhar1
Mudagandur S. Shekhar1 Ashok Kumar Jangam1
Ashok Kumar Jangam1 Sudheesh K. Prabhudas1
Sudheesh K. Prabhudas1 Karthic Krishnan1
Karthic Krishnan1 Nimisha Kaikkolante1
Nimisha Kaikkolante1 Balasubramanian C. Paran2
Balasubramanian C. Paran2 Dushyant Singh Baghel3
Dushyant Singh Baghel3 Vijayan K. Koyadan1
Vijayan K. Koyadan1 Joykrushna Jena4
Joykrushna Jena4 Trilochan Mohapatra4
Trilochan Mohapatra4- 1Nutrition Genetics and Biotechnology Division, Indian Council of Agricultural Research-Central Institute of Brackishwater Aquaculture, Chennai, India
- 2Crustacean Culture Division, Indian Council of Agricultural Research-Central Institute of Brackishwater Aquaculture, Chennai, India
- 3Nucleome Informatics Private Limited, Hyderabad, India
- 4Indian Council of Agricultural Research, New Delhi, India
Introduction
Increasing the number of farmed species is one of the main strategies for enhancing aquaculture diversification toward sustainable production (Harvey et al., 2017). Global shrimp production is dominated by Penaeus vannamei and Penaeus monodon which accounted for 82.7 and 12.5% of production, respectively (FAO, 2020). Farmers' preference for these species is due to the availability of specific pathogen free (SPF) seed from genetically improved broodstock. Focussing on one or two species may be economically favorable in short-run owing to efficient technical advances made in limited number of species (Metian et al., 2020), however, dependence on single species might prove detrimental for long-term sustainable production. There is an urgent need to undertake research and genetic improvement programs for other region-specific shrimp species that have wide natural distribution. The Penaeus indicus is one such species with distribution ranging from East African Coast through South-East Asia to the Northern Australian Coast. Some field-level farming demonstrations of P. indicus along the Indian Coast indicated production performance at par with P. vannamei raising considerable interest among shrimp farmers and stakeholders (CIBA, 2019). Further, coordinated research efforts through functional studies and genetic improvement program are required to provide P. indicus as an additional and alternate species to farmers.
Though short-read based transcript assemblies were available for shrimp (Ghaffari et al., 2014; Powell et al., 2015; Sellars et al., 2015; Jones et al., 2017; Huerlimann et al., 2018; Shi et al., 2018), they are constrained in completeness in comparison to long-read based assemblies (Zeng et al., 2018; Zhang X. et al., 2019; Sittikankaew et al., 2020). It was further shown that full-length transcripts can be easily annotated than short transcripts (Zeng et al., 2018). Access to full-length transcript sequences is a mandatory resource for researchers to derive valuable results from functional studies. Therefore, for the first time, we have generated a transcriptome assembly for P. indicus to support the efforts of developing another shrimp species for aquaculture diversification and associated genetic improvement programs. We have used the latest Pacific Biosciences (Pacbio) Isoform Sequencing (Iso-Seq) approach on Sequel II instrument using 8M single molecule real-time (SMRT) sequencing cell and circular consensus sequence (CCS) technology to assemble transcriptome for P. indicus.
This is the first report of a comprehensive transcriptome generated from different tissues and larval stage of P. indicus with 238.98 Gb of sequence data from gills, hepatopancreas, muscle, and pooled post-larvae. The only related complete genome available for shrimp is that of P. vannamei (Zhang X. J. et al., 2019). In the absence of a genome for P. indicus, the full-length transcriptome generated in this study would be a valuable resource for conducting functional studies involving desired economic traits.
Methods
Sample Collection
Three tissues (gills, hepatopancreas and muscle) from an adult P. indicus shrimp and a pool of post-larvae (15 numbers at PL18 stage) being maintained at shrimp hatchery (Muttukadu Experimental Station of ICAR-CIBA, Muttukadu, Chennai, India) were used for iso-sequencing. The four samples were initially snap-frozen in liquid nitrogen and later stored at −80°C until RNA extraction.
RNA Extraction and Iso-Seq Library Preparation
Separately for each sample, the total RNA was extracted using RNAiso Plus (Takara Bio Inc., Japan) according to the manufacturer's instructions. The RNA quantification was carried out by Qubit3.0 (Invitrogen Life Technologies, USA) and the RNA integrity (RIN) was evaluated on an Agilent 2100 Bioanalyzer (Agilent, USA). The total RNA of each sample (RIN value, ≥7) was used for cDNA synthesis using NEBNext® Single Cell/Low Input cDNA Synthesis & Amplification Module (NEB Inc., France). With barcoded primers, the cDNA was amplified for 12 cycles and then amplicons were size-selected by Pronex beads following the standard workflow where the transcripts were primarily centered at approximately 2 Kb length. The sample-wise identity of the sequence data was retained due to unique barcodes. The sequencing libraries were prepared using Iso-Seq Express Oligo Kit and SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, USA). The libraries of four samples at a loading concentration of 85 pmol were sequenced together on PacBio Sequel II 8M SMRT cell runs (30 h movie, CCS/HiFi mode) at Nucleome Informatics, Hyderabad.
Transcript Assembly
About 238.98 Gb of sequence data in 120.7 million subreads were analyzed in SMRTLink v9.0 using Iso-Seq3 pipeline to obtain high-quality isoforms (Table 1 and Supplementary Table 1) with the following parameters: minLength = 50, maxLength = 15,000, minPasses = 1, minSnr = 2.5, Polish CCS = ON, minPredictedAccuracy = 0.8, and minimum accuracy for high quality isoforms = 0.99, while retaining the sample identity. Following the quality filters, CCS reads were generated for 89% (2.8 million) of zero-mode waveguides (ZMWs). Of these, about 2.6 million (90.6%) had 5′ primer, 3′ primer and polyA tails, thus qualifying to be full length non-concatemer (FLNC) reads. Further, the clustering and polishing of FLNC reads had generated 99,458 high quality transcripts with N50 length of 2,957 bases. The high quality isoforms were checked for contaminating sequences using Mash v2.2 (Ondov et al., 2019) with Refseq genomes sketch. Since no contaminant sequences were found, the transcripts were further analyzed with CPC2 (Kang et al., 2017), CNCI (Sun et al., 2013), and CPAT (Wang et al., 2013) to filter out 7,434 lncRNA sequences (Supplementary Table 2 and Supplementary Figure 1). The remaining coding transcripts (Figure 1A) were clustered (>99% similarity) with CDHIT V4.6 (Li and Godzik, 2006) to obtain a final set of 30,479 non-redundant coding transcripts.

Table 1. Iso-Seq data and transcriptome assembly statistics.
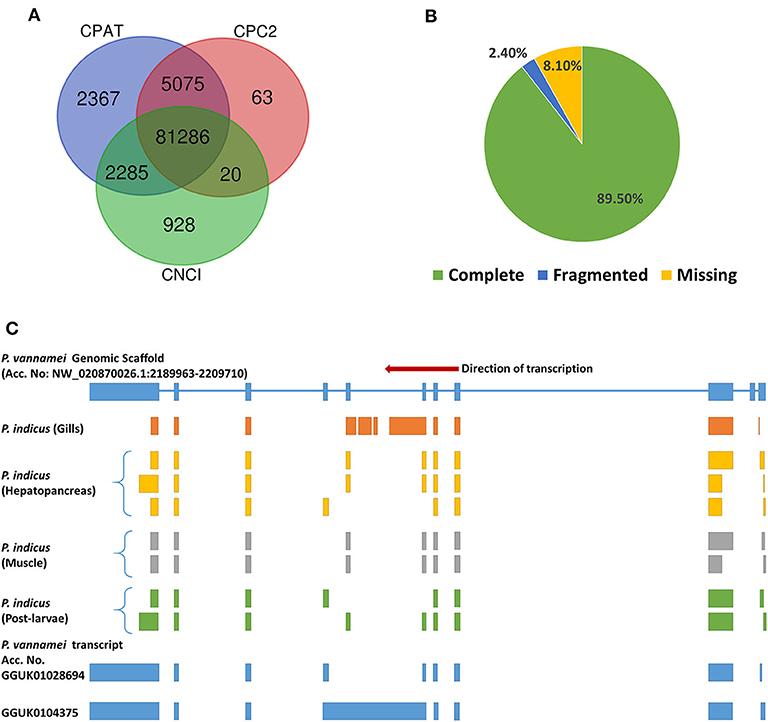
Figure 1. Transcriptome assembly of P. indicus. (A) The number of coding transcripts identified through analysis of high-quality transcripts (n = 99,458) in CPAT, CNCI and CPC2 tools. A high-quality transcript identified as ‘coding’ by atleast one of these 3 tools, is finally classified as “coding.” (B) Benchmarking the assembled transcripts against BUSCO orthologs for completeness. (C) Tissue-specific isoforms of choline phosphotransferase (CP) gene in P. indicus displaying isoform diversity. The transcripts of CP gene from each sample of P. indicus were aligned against the P. vannamei genomic scaffolds (ASM378908v1). The scaffold with positive hit (NW_020870026.1) has CP gene between bases 2189963 and 2209710. Each of the P. indicus CP transcripts was mapped to the NW_020870026.1 to identify the splice pattern. Similarly, the iso-seq transcripts of P. vannamei (Accession No. GGUK00000000) were also screened for CP gene to validate the exon junctions against the P. vannamei genomic scaffold. Each block in the figure represents an exon.
Transcript Annotation and Validation
The transcriptome assembly was checked for completeness against the eukaryota_odb10 (2019-11-20) dataset of BUSCO single-copy orthologs. The assembly has 228 (89.5%) complete orthologs out of 255 genes. About 6 (2.4%) and 21 (8.2%) orthologs were fragmented and missing, respectively, in the assembly (Figure 1B). Current transcriptome assembly had high number of complete genes when compared to isoseq-based assembly in tiger shrimp (80.86%, Pootakham et al., 2020). The inclusion of a sample from early development stage in the current study might have contributed to higher completeness.
The transcripts in the final assembly were annotated in OmicsBox v 1.3.11 (OmicsBox, 2019) which uses the high-quality GO terms obtained from GO mapping, InterProScan and EggNOG mapper, for annotation. While performing blastx search during annotation, Eukaryota subset of non-redundant protein database of Genbank was used. Of 30,479 transcripts, 25,156 (83%) could be annotated and 9,610 (31.5%) could be mapped with enzyme codes (Supplementary Figure 2 and Supplementary Table 3). The top 3 GO categories in the annotated assembly are organic substance metabolic process (biological processes), organelle (cellular component) and primary metabolic process (biological processes). The protein-binding is the major molecular function category in the assembly (Supplementary Figure 3). Of the enzyme code mapped transcripts, the hydrolases were predominant followed by transferases and oxidoreductases (Supplementary Figure 4). The EggNOG mapper gave GO annotations for 56% of transcripts (Supplementary Table 4) while assigning COG categories to information storage and processing (15.16%), cellular processes and signaling (34.71%) and metabolism (19.5%). The InterProScan gave protein family/domain information for 98.3% of transcripts and GO annotations for 61.8% of transcripts (Supplementary Table 3 and Supplementary Figures 5–10).
Further, SQUANTI3 (Tardaguila et al., 2018) was used to classify the high-quality isoforms and their junction sites, using the isoforms collapsed with CDHIT as input and P. vannamei genome as reference. About 19 and 23% of isoforms in P. indicus showed exact and incomplete matches, respectively with P. vannamei annotations. Additionally, the P. indicus transcriptome has 40% novel isoforms when compared to P. vannamei indicating high splice diversity between the two species (Supplementary Table 5). Among the four samples used for Iso-Seq study, the muscle tissue has got less isoform diversity with fewer genes having multiple isoforms (Supplementary Figure 11). The length of the majority transcripts was centered at 2 Kb which is in agreement with the size selection procedures followed during Iso-Seq library preparation (Supplementary Figure 12).
As tissue identity was also retained in the final set of transcripts, the assembled transcriptome resource would be highly valuable to study tissue-specific isoforms. For example, various isoforms of choline phosphotransferase mapped to the corresponding gene from P. vannamei genome (Zhang X. J. et al., 2019) indicated the isoform diversity between species and also between tissues within P. indicus (Figure 1C).
Re-Use Potential
For the first time, the study presents a full length transcriptome assembly for P. indicus based on long read sequence data generated using the latest sequencing platform (Pacific BioSciences, Sequel II, 8M chip) and analyzed with the latest software (SMRTLink v9.0). The transcriptome assembly has great value in annotation of P. indicus genome and to improve genome annotations for other related shrimp. The isoform-level full-length transcript resource aids researchers to derive meaningful results in functional studies. Finally, the transcript assembly would have potential to support the efforts of species diversification in introduction and propagation of the P. indicus as a sustainable culture species.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/ena, ERS4889090; https://www.ebi.ac.uk/ena, ERS4889091; https://www.ebi.ac.uk/ena, ERS4889092; https://www.ebi.ac.uk/ena, ERS4889093.
Author Contributions
VKo, MS, JJ, and TM conceived and designed the study and experiments. BP collected the samples. DB extracted RNA and prepared libraries for Iso-Seq. AJ, SP, KK, DB, VKa, and NK performed bioinformatics analysis. VKa wrote manuscript with inputs from all coauthors. All authors have read and approved the final manuscript.
Funding
The authors acknowledge the funding support from the ICAR-CRP on Genomics project, Whole genome sequencing of Indian white shrimp, Penaeus indicus.
Conflict of Interest
DB was employed by the company Nucleome Informatics Private Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.605098/full#supplementary-material
References
CIBA (2019). Annual Report 2019. ICAR-Central Institute of Brackishwater Aquaculture: Chennai, Tamil Nadu, India.
FAO (2020). Fishery and Aquaculture Statistics. Global Aquaculture Production 1950-2018 (FishstatJ). FAO Fisheries and Aquaculture Department: Rome. Available online at: www.fao.org/fishery/statistics/software/fishstatj/en (accessed August, 2020).
Ghaffari, N., Sanches-Flores, A., Doan, R., Garcia-Orozco, K. D., Chen, P. L., Ochoa-Levya, A., et al. (2014). Novel transcriptome assembly and improved annotation of the whiteleg shrimp (Litopenaeus vannamei), a dominant crustacean in global seafood mariculture. Sci. Rep. 4:7081. doi: 10.1038/srep07081
Harvey, B., Soto, D., Carolsfeld, J., Beveridge, M., and Bartley, D. M. (2017). Planning for Aquaculture Diversification: the Importance of Climate Change and Other Drivers. FAO Technical Workshop, 23–25 June 2016, FAO Rome. FAO Fisheries and Aquaculture Proceedings No. 47. Rome, FAO, 166.
Huerlimann, R., Wade, N. M., Gordon, L., Montenegro, J. D., Goodall, J., McWilliam, S., et al. (2018). De novo assembly, characterization, functional annotation and expression patterns of the black tiger shrimp (Penaeus monodon) transcriptome. Sci. Rep. 8:13553. doi: 10.1038/s41598-018-31148-4
Jones, D. B., Jerry, D. R., Khatkar, M. S., Raadsma, H. W., Steen, H. V. D., Prochaska, J., et al. (2017). A comparative integrated gene-based linkage and locus ordering by linkage disequilibrium map for the Pacific white shrimp, Litopenaeus vannamei. Sci. Rep. 7:10360. doi: 10.1038/s41598-017-10515-7
Kang, Y. J., Yang, D. C., Kong, L., Hou, M., Meng, Y. Q., Wei, L., et al. (2017). CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acid Res. 45, W12–W16. doi: 10.1093/nar/gkx428
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–9 doi: 10.1093/bioinformatics/btl158
Metian, M., Troell, M., Christensen, V., Steenbeek, J., and Pouil, S. (2020). Mapping diversity of species in global aquaculture. Rev. Aquac. 12, 1090–1100. doi: 10.1111/raq.12374
OmicsBox (2019). OmicsBox– Bioinformatics Made Easy. BioBam Bioinformatics. Available online at: https://www.biobam.com/omicsbox
Ondov, B. D., Starrett, G. J., Sappington, A., Kostic, A., Koren, S., Buck, C. B., et al. (2019). Mash Screen: high-throughput sequence containment estimation for genome discovery. Genome Biol. 20:232. doi: 10.1186/s13059-019-1841-x
Pootakham, W., Uengwetwanit, T., Sonthirod, C., Sittikankaew, K., and Karoonuthaisiri, N. (2020). A novel full-length transcriptome resource for black tiger shrimp (Penaeus monodon) developed using isoform sequencing (Iso-Seq). Front. Mar. Sci. 7:172. doi: 10.3389/fmars.2020.00172
Powell, D., Knibb, W., Remilton, C., and Elizur, A. (2015). De-novo transcriptome analysis of the banana shrimp (Fenneropenaeus merguiensis) and identification of genes associated with reproduction and development. Mar. Genomics. 22, 71–78. doi: 10.1016/j.margen.2015.04.006
Sellars, M. J., Trewin, C., McWilliam, S. M., Glaves, R., and Hertzler, P. L. (2015). Transcriptome profiles of Penaeus (Marsupenaeus) japonicus animal and vegetal half-embryos: identification of sex determination, germ line, mesoderm, and other developmental genes. Mar. Biotechnol. 17, 252–265. doi: 10.1007/s10126-015-9613-4
Shi, X., Meng, X., Kong, X., Luan, S., Luo, S., Cao, B., et al. (2018). Transcriptome analysis of ‘Huanghai No. 2′ fenneropenaeus chinensis response to WSSV using RNA-seq. Fish Shellfish Immunol. 75, 132–138. doi: 10.1016/j.fsi.2018.01.045
Sittikankaew, K., Pootakham, W., Sonthirod, C., Sangsrakru, D., Yoocha, T., Khudet, J., et al. (2020). Transcriptome analyses reveal the synergistic effects of feeding and eyestalk ablation on ovarian maturation in black tiger shrimp. Sci. Rep. 10:3239. doi: 10.1038/s41598-020-62221-6
Sun, L., Luo, H., Bu, D., Zhao, G., Yu, K., Zhang, C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acid Res. 41:e166. doi: 10.1093/nar/gkt646
Tardaguila, M., de la Fuente, L., Marti, C., Pereira, C., Pardo-Palacios, F. J., del Risco, H., et al. (2018). SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification Genome Res. 28, 396–411. doi: 10.1101/gr.222976.117
Wang, L., Park, H. J., Dasari, S., Wang, S., Kocher, J.-P., and Li, W. (2013). CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acid Res. 41:e74. doi: 10.1093/nar/gkt006
Zeng, D., Chen, X., Peng, J., Yang, C., Peng, M., Zhu, W., et al. (2018). Single-molecule long-read sequencing facilitates shrimp transcriptome research. Sci. Rep. 8:16920. doi: 10.1038/s41598-018-35066-3
Zhang, X., Li, G., Jiang, H., Li, L., Ma, J., Li, H., et al. (2019). Full-length transcriptome analysis of Litopenaeus vannamei reveals transcript variants involved in the innate immune system. Fish Shellfish Immunol. 87, 346–359. doi: 10.1016/j.fsi.2019.01.023
Keywords: lncRNA, transcriptome, Iso-Seq, Pacbio, isoform sequencing, Penaeus indicus, Indian white shrimp
Citation: Katneni VK, Shekhar MS, Jangam AK, Prabhudas SK, Krishnan K, Kaikkolante N, Paran BC, Baghel DS, Koyadan VK, Jena J and Mohapatra T (2020) Novel Isoform Sequencing Based Full-Length Transcriptome Resource for Indian White Shrimp, Penaeus indicus. Front. Mar. Sci. 7:605098. doi: 10.3389/fmars.2020.605098
Received: 11 September 2020; Accepted: 11 November 2020;
Published: 02 December 2020.
Edited by:
Andrew Stanley Mount, Clemson University, United StatesReviewed by:
Vittoria Roncalli, University of Naples Federico II, ItalyManuel Aranda, King Abdullah University of Science and Technology, Saudi Arabia (Arun Prasanna Nagarajan contributed to the review of Manuel Aranda)
Copyright © 2020 Katneni, Shekhar, Jangam, Prabhudas, Krishnan, Kaikkolante, Paran, Baghel, Koyadan, Jena and Mohapatra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinaya Kumar Katneni, dmluYXlAY2liYS5yZXMuaW4=; dmluYXluZHJpQHlhaG9vLmNvbQ==