 Linlin Zhao
Linlin Zhao Shouqiang Wang
Shouqiang Wang Fangrui Lou2
Fangrui Lou2 Tianxiang Gao
Tianxiang Gao Zhiqiang Han
Zhiqiang Han- 1First Institute of Oceanography, Ministry of Natural Resources, Qingdao, China
- 2School of Ocean, Yantai University, Yantai, China
- 3Fishery College, Zhejiang Ocean University, Zhoushan, China
The evolutionary relationships of lungfish can provide crucial information on the transition from Sarcopterygii to tetrapods. Phylogenomics is necessary to explore accurate internal phylogenetic relationships among all lungfish species. In the context of the lack of genome-wide genetic information for Protopterus amphibious, we are the first to systematically report the transcriptome of P. amphibius and these sequences can be used to enrich the genome-wide genetic information of lungfish. Meanwhile, we also found significant differences in the expression levels of 3,189 genes between the lung and heart of P. amphibious. Based on phylogenomics, 1,094 shared orthologous genes were identified and then applied to reconstruct the internal phylogenetic structure of lungfish species. The reconstructed phylogenetic relationships provide evidence that lungfish is the sister group of terrestrial vertebrates and that Neoceratodus forsteri is the most primitive lungfish. Moreover, the divergence time between the most primitive lungfish and other lungfish species is between 186.11 and 195.36 MYA. Finally, 43 protein metabolism-related, stress response-related, and skeletogenesis-related genes were found to have undergone positive selection and fast evolution in N. forsteri. We suspected that these genes possibly helped ancient fish adapt to the new terrestrial environment and ultimately contributed to its spreading to land.
Introduction
Approximately 400 million years ago, the ancient Sarcopterygii that appeared in coastal environments near the equator gradually colonized the land, which eventually became a major event in tetrapod and even human evolution (Dial et al., 2015). For the initial landing, the terrestrial environments created more serious selective pressures on Sarcopterygii. The pressures resulted in a number of physiological, morphological and behavioral adaptations and evolution of these fishes, including the appearance of limbs and lungs, as well as more developed musculoskeletal and nervous systems, and others than other aquatic organisms (Clack, 2002). A previous study suspected that the amphibious behavior of Sarcopterygii evolved repeatedly many times, possibly independently at least 30 times during the landing process (Ord and Cooke, 2016). However, how the ancient Sarcopterygii conquered and adapted to the terrestrial environments and eventually evolved into tetrapods remains a mystery.
The coelacanth and lungfish are recognized as the only two extant Sarcopterygii groups, characterized by the presence of lungs and lobed fins (Liang et al., 2013). Although phylogenetic relationships based on morphology, paleontology, and molecular levels have been extensively studied, the relationships among coelacanths, lungfish, and tetrapods have remained contentious during the last three decades (Figure 1; Fritzsch, 1987; Meyer, 1995; Zardoya and Meyer, 1996; Hedges, 2009; Christensen-Dalsgaard et al., 2011; Shan and Gras, 2011). It is worth noting that the subjectiveness of traditional analysis methods, the different analysis methods and the amounts of molecular markers used may be the main reasons for the above disagreements (Takezaki et al., 2004). However, a large number of researchers have recently suggested that lungfish are the closest living relative of tetrapods (Meyer and Wilson, 1990; Hedges et al., 1993; Brinkmann et al., 2004; Panchen and Smithson, 2008). In fact, genome-wide phylogenetic relationships of molecular markers also confirmed this hypothesis (Meyer et al., 2021). Such as, Irisarri and Meyer (2016) and Irisarri et al. (2017) have accurate phylogenomic datasets from RNA sequencing and reconstructed a robust and strongly supported timetree of coelacanths, lungfish, and tetrapods. Furthermore, Amemiya et al. (2013) have reported the genome sequence of the African coelacanth, Latimeria chalumnae and then reconstructed a phylogenomic relationship among coelacanths, lungfish, and tetrapods. It is worth noting that these results confirmed that the lungfish, and not the coelacanth, is the closest living relative of tetrapods. Despite extensive molecular and morphological research on the relationships among coelacanths, lungfish, and tetrapods, the phylogenetic relationships within lungfish have not been reported. This implies that more complete genetic information is necessary to explore accurate internal evolutionary relationships among all lungfish species. More accurate lungfish phylogenetic relationships will also help us understand how ancient Sarcopterygii colonized the land and eventually evolved into tetrapods.
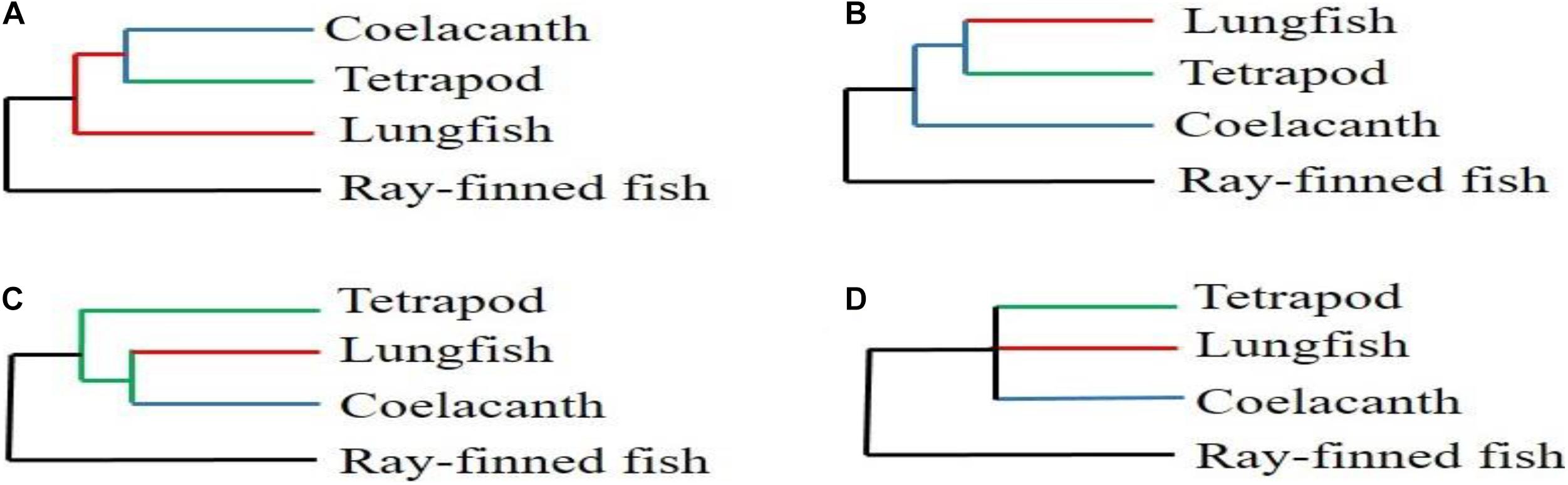
Figure 1. The relationships among coelacanth, lungfish, and tetrapod. (A; Fritzsch, 1987; Meyer, 1995; Christensen-Dalsgaard et al., 2011) The coelacanth is the closest living relative of tetrapods. (B; Meyer and Wilson, 1990; Hedges et al., 1993; Brinkmann et al., 2004; Panchen and Smithson, 2008) The lungfish is the closest living relative of tetrapod. (C; Zardoya and Meyer, 1996; Shan and Gras, 2011) and (D; Zardoya and Meyer, 1996; Shan and Gras, 2011) The lungfish and coelacanth are equally related to tetrapods.
Phylogenomics provides an opportunity to explore the accurate internal phylogenetic relationships among all lungfish species. Considering that lungfish may also have provided crucial information for the transition from Sarcopterygii to tetrapod, it is essential to investigate the complete genetic information of lungfish. Unfortunately, there is only one genome sequencing project for Neoceratodus forsteri (Meyer et al., 2021). This may be because lungfish have the largest genomes of all vertebrates (Liang et al., 2013). Because whole-genome information for lungfish is not available, modern RNA-Seq may solve the issues mentioned above because it can effortlessly capture genome-wide protein-coding sequences (Lou et al., 2020). Transcriptome datasets can also avoid the large computational costs required to analyze for whole-genome datasets (Hughes et al., 2018). Therefore, we believe that transcriptome datasets are probably the most effective method to explore accurate internal evolutionary relationship among all lungfish species. Moreover, we can also calculate and compare the evolutionary rates of genes based on transcriptome datasets, and then infer the potential terrestrial adaptation characteristics of the ancient lungfish.
Currently, only six lungfish species have been found in the world, including N. forsteri, Lepidosiren paradoxa, Protopterus aethiopicus, P. amphibius, P. dolloi, and P. annectens (Brinkmann et al., 2004). It is worth noting that whole-genome datasets have been produced for all but P. amphibius (Biscotti et al., 2016). In the present study, we sequenced a P. amphibius reference transcriptome that can be used by the scientific community to solve biological issues on the transition from the Sarcopterygii to the tetrapods. In detail, clean reads produced by RNA-seq were first applied to assemble the relatively integrated transcriptome of P. amphibius. Subsequently, phylogenomics was applied to detect the protein-coding orthologous genes. The identified orthologous genes were used to construct the phylogenetic relationship among all lungfish species. Moreover, we investigated the potential genome-wide adaptation signatures of N. forsteri. The aim of the present study was to determine the accurate internal evolutionary relationships among all lungfish species and then provide valuable information on the ancient Sarcopterygii landing processes.
Materials and Methods
Ethics Approval and Consent to Participate
P. amphibius is not an endangered or protected species in China. In addition, frost anesthesia was performed to minimize suffering of P. amphibius. All experimental protocols and procedures were performed in strict accordance with the Laboratory Animal Management Principles of China.
Sample Collection, RNA Extraction and Illumina Sequencing
One healthy female P. amphibius individual were obtained from an aquaculture farm (Guangzhou Health Aquarium Company) in Guangzhou (China) in October 2019. P. amphibius was immediately anesthetized by freezing, and the heart and lung tissues were rapidly clipped, snap-frozen in liquid nitrogen and stored at −80°C prior to RNA extraction. The total RNA of each tissue was extracted using a standard TRIzol Reagent Kit (Huayueyang Biotech Co., Ltd., Beijing, China) following the manufacturer’s protocol. After the quantitative evaluation of total RNA was completed using the Agilent 2,100 Bioanalyzer (Agilent Technologies, Santa Clara, United States), we purified mRNA by depleting rRNA from total RNA using RNA Purification Beads (Illumina, San Diego, CA, United States). The purified mRNA was cut into fragments of appropriate size, and fragmented mRNA was further used to construct the paired-end cDNA library. Paired-end cDNA libraries were generated using NEBNext®UltraTM RNA Library Prep Kit for Illumina® (New England Biolabs, Beijing, China) following manufacturer’s recommendations and index codes were added to attribute sequences to each tissue. Subsequently, we diluted two paired-end cDNA libraries and the quality of two libraries were assessed on the Agilent Bioanalyzer 2,100 system. The clustering of the index-coded tissues was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina, San Diego, CA, United States) according to the manufacturer’s instructions. After cluster generation, two tagged paired-end cDNA libraries were further sequenced on the Illumina HiSeq 2,500 platform and 150 bp paired-end reads were generated.
Transcriptome Data Analysis
All the raw reads in FASTQ format generated by the sequencing platform were first quality controlled through in-house Perl scripts. In this process, clean reads were obtained by removing reads containing adaptors and unknown nucleotides (N ratio > 10%) and low quality reads (quality scores < 20) from raw reads. At the same time, Q20, Q30, GC-content and sequence duplication level of all the clean reads were calculated. All downstream analyses were based on the remaining high-quality clean reads. Transcriptome assembly was accomplished based on left.fq and right.fq using Trinity software (version 5.0.0; Grabherr et al., 2011) with the parameter: min_kmer_cov 2. The redundant transcripts were removed and the longest unigenes were further spliced.
All unigenes were annotated using BLAST (version 2.2.31; Altschul et al., 1997), KOBAS (version 2.0; Xie et al., 2011) and HMMER (version 3.1b2; Wheeler and Eddy, 2013) software with the following databases: NR (NCBI non-redundant protein sequences); Pfam (Protein family); KOG/COG/eggNOG (Clusters of Orthologous Groups of proteins); Swiss-Prot (A manually annotated and reviewed protein sequence database); KEGG (Kyoto Encyclopedia of Genes and Genomes); and GO (Gene Ontology).
Tissue-Specific Expression Analyses
Gene expression levels were estimated by mapping all clean reads to the unigenes based on RSEM software (version 1.2.19; Li and Colin, 2011). Prior to differential expression analysis, the read counts of each cDNA library were adjusted by EBSeq software (version 1.6.0; Leng et al., 2013) through an empirical Bayesian approach. Differential expression analysis of two tissues was performed using EBSeq software (version 1.6.0; Leng et al., 2013), and a q-value < 0.005 and fold change (FC) > 2 were set as the threshold for significantly differential expression. Furthermore, the functional categories and biochemical metabolic pathways of differentially expressed genes (DEGs) were predicted based on Blast2GO software (version 2.5; Conesa et al., 2005), and the threshold for a significant difference was Q ≤ 0.05.
Phylogenomics Analyses
To investigate the more accurate internal phylogenetic relationships among all lungfish species, we performed an extensive orthologous gene comparison among P. amphibius and other vertebrates with transcriptome or genome datasets. First, we obtained the coding sequences (CDSs) of Gallus gallus, L. chalumnae, L. paradoxa, N. forsteri, P. aethiopicus, P. annectens, and P. dolloi from the Ensembl genome database. The CDS of P. amphibius transcriptome were extracted using TransDecoder (version 5.0.0; Grabherr et al., 2011) with the parameter: m 200. We further converted all CDSs into amino acid sequence files according to the codon table. All amino acid sequence files were translated into orthomcl-compatible FASTA files, and the low-quality sequences (<200 bp and <20% identity among taxa) in each FASTA file were then eliminated (Li et al., 2003). All-vs.-All BLASTP was conducted for all high-quality amino acid sequences with a cutoff E-value of 1E−5. The single-copy orthologous genes among all species were extracted using OrthoMCL software (Li et al., 2003). The obtained high-quality single-copy orthologous genes of each species were aligned and spliced into single amino acid sequences using MAFFT software (version 7.0; Katoh and Standley, 2013). Conserved sequences were extracted from each amino acid sequence using Gblocks software (v.0.91b; Castresana, 2000) with parameter: −t = p. Finally, we performed 1,000 non-parametric bootstrap replicates for the optimal GTRGAMMA substitution model of all concatenated amino acid sequences in RAxML software (Stamatakis, 2006), and then a maximum likelihood (ML) tree was constructed. Furthermore, the divergence time was estimated using the r8s software (Sanderson, 2003) and molecular clock data from the divergence time between G. gallus and L. chalumnae (403–423 MYA) from the TimeTree database (Kumar et al., 2017).
It is well known that N. forsteri is the most primitive lungfish (Kemp, 1986). In the present study, the codeml program in PAML software (version 4.9; Yang, 2007) was applied to identify the genome-wide selection signatures of the N. forsteri. First, we reconstructed a phylogenetic relationship among L. paradoxa, P. amphibious and N. forsteri based on concatenated amino acid sequences. Furthermore, we set the N. forsteri as the foreground branch in the tree files. The branch-site model (model = 2, NSsites = 2) was further used to identify the positively selected genes (PSGs) of the N. forsteri. The null model assumed that the foreground branch was under purifying selection and those sites were evolved neutrally (non-synonymous (dN)/synonymous (dS) = 1, modelA1, fix_omega = 1, and omega = 1.5), and the alternative model assumed that those sites on the foreground branch were under positive selection (dN/dS > 1, modelA2, fix_omega = 0, and omega = 1.5). A likelihood ratio test (LRT) was applied to calculate the log-likelihood values (2△ln) between the null model and alternative model of each single-copy orthologous gene. After a chi-square statistical analysis, a gene was considered as a PSG of the lungfish if the FDR-adjusted p-value was < 0.01. Moreover, we used the branch model (model = 1; NSsites = 0) to estimate the fast evolving genes (FEGs). The null model assumed that all branches evolved at the same rate, and the alternative model assumed that the foreground branch could evolve at a different rate. An LRT with df = 1 was constructed to calculate the 2△ln between the null model and the alternative model. A gene that satisfied the following two conditions was considered as a FEG: (i) the ratio of the non-synonymous nucleotide substitution rate to the synonymous nucleotide substitution rate (ω) of the foreground branch was higher than that of the background branch; (ii) FDR-adjusted p-value < 0.05. Finally, the union of FEGs and PSGs of N. forsteri were assumed to be the adaptive genes (AGs) of N. forsteri. Blast2GO software (version 2.5; Conesa et al., 2005) was applied to predict the functional categories of these AGs.
Results
P. amphibius Transcriptome Assembly and Annotation
We obtained raw reads from the heart and lung tissues of P. amphibius with Illumina HiSeq 2,500 platform and deposited them into the NCBI database under accession numbers SRR13486841–SRR13486842 under BioProject PRJNA692649 and BioSample SAMN17359469. After filtering, 14.07 Gb of clean reads were generated and the evaluation results of clean reads were listed in Table 1. All high-quality clean reads were assembled to produce 71,592 transcripts with an average length of 1,353 bp and an N50 length of 2,319 bp. All transcripts were subjected to redundancy analysis and spliced into 55,177 unigenes with an average length of 1,168 bp and an N50 length of 1,963 bp.

Table 1. The evaluation results of clean reads from two tissues.
All unigenes were considered to analyze the gene ontology and orthologous classifications based on protein databases. The results showed that a total of 23,155 unigenes were annotated in all databases (Table 2). Of all annotated unigenes, 5,551, 15,170, 14,045, 15,023, 16,507, 13,078, 21,333, and 22,754 unigenes had significant matches with sequences in the COG, GO, KEGG, KOG, Pfam, Swiss-Prot, eggNOG, and NR database, respectively.

Table 2. The annotation information of all unigenes blasted to protein databases.
Tissue-Specific Gene Expression Pattern of P. amphibius
We analyzed the number and function of DEGs to explore tissue-specific expression pattern of P. amphibius. The results showed that there were significant differences in the expression levels of 3,189 genes between lung and heart of P. amphibius (Figure 2).
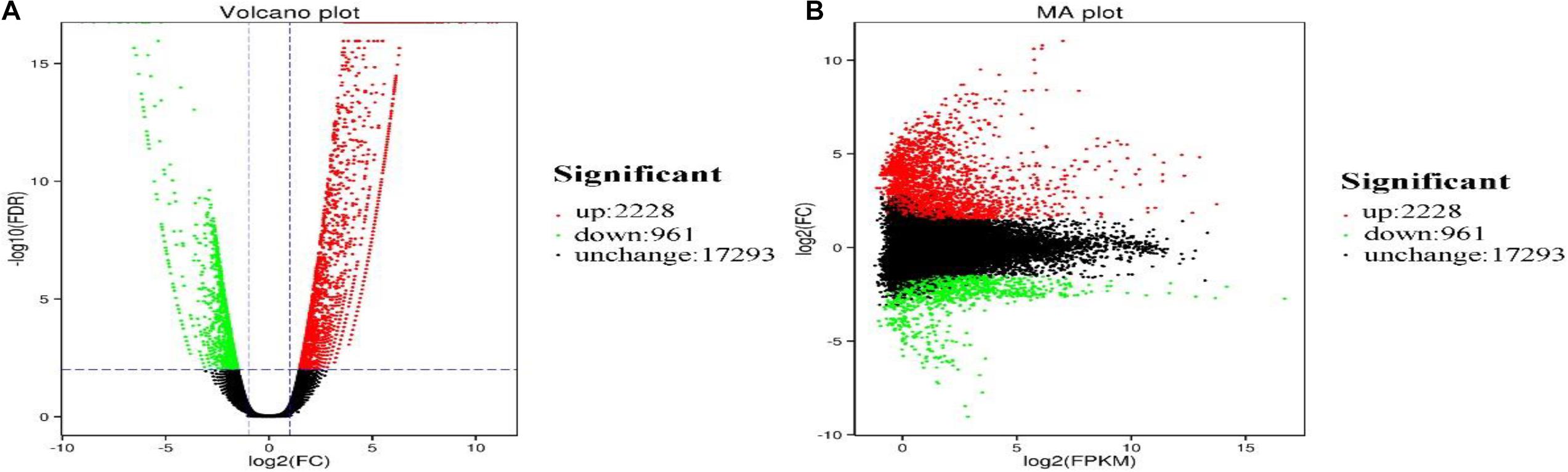
Figure 2. Volcano plot (A) and MA plot (B) showing gene expression profiles in the lung and heart libraries of P. amphibius. Red and green dots indicate upregulated and downregulated genes, respectively. Black dots represent genes with no significant difference.
Enrichment analyses of 3,189 DEGs in GO terms and KEGG pathways were further performed to explore the potential functions of these DEGs and their products. The results showed that a total of 1,440 DEGs were successfully mapped to 58 GO terms (Figure 3). Among these GO terms, “cellular process,” “binding,” and “cell” were dominant in “biological process,” “molecular function,” and “cellular component,” respectively.
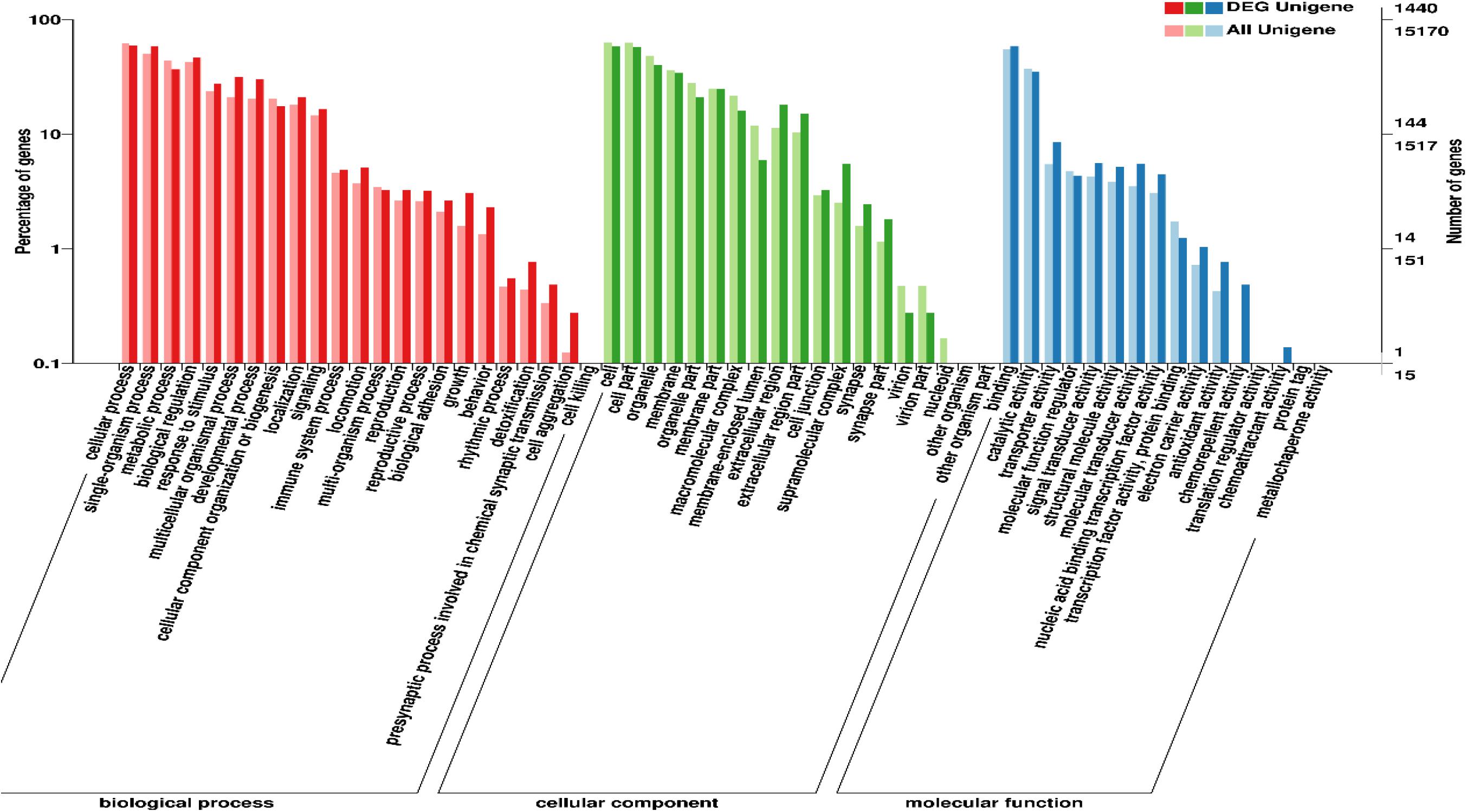
Figure 3. Results of the GO enrichment analysis of DEGs.
Additionally, 1,305 DEGs were assigned to 235 metabolic pathways. The results also indicated that 7 pathways were significantly enriched, and the statistics for the top 20 enriched pathways are shown in Figure 4. DEGs between lung and heart were predominately associated with the pathways neuroactive ligand-receptor interaction (ko04080; Q = 1.26 × 10–7), cardiac muscle contraction (ko04260; Q = 8.13 × 10–7), biosynthesis of amino acids (ko01230; Q = 9.37 × 10–6), adrenergic signaling in cardiomyocytes (ko04261; Q = 1.82 × 10–3), glycine, serine and threonine metabolism (ko00260; Q = 4.04 × 10–3), glycolysis/Gluconeogenesis (ko00010; Q = 7.82 × 10–3), and tight junction (ko04530; Q = 1.82 × 10–2).
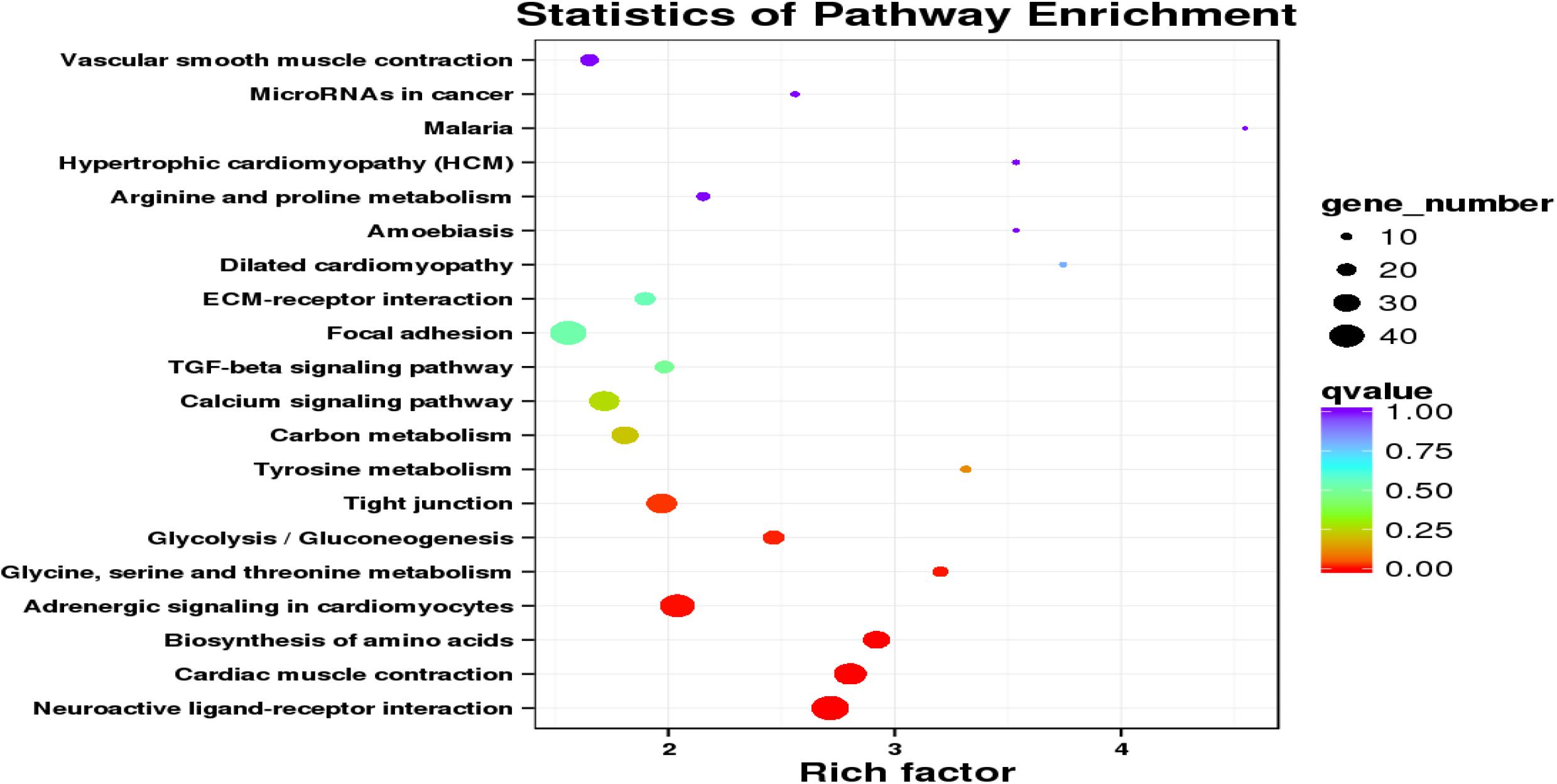
Figure 4. Statistics for the top 20 enriched pathways associated with DEGs.
Orthologous Gene Identification and the Internal Phylogenetic Relationship of the Lungfish
Using OrthoMCL software, we identified a set of 1,094 single-copy orthologous genes longer than 200 bp among all species. After alignment, all single-copy orthologous genes of each species were concatenated and then used to generate a data matrix. RAxML software was used to construct the phylogenetic tree of the lungfishes and other species based on the data matrix of the concatenated amino acids (Figure 5). According to the phylogenetic analysis, P. amphibius clustered together with five other lungfish species, which further clustered with G. gallus belonging to the terrestrial vertebrates, and then clustered with L. chalumnae belonging to the coelacanth. This result also confirms the hypothesis that N. forsteri is the most primitive lungfish. Moreover, the differentiation time range between N. forsteri and other lungfishes ranged from 186.11 to 195.36 MYA.
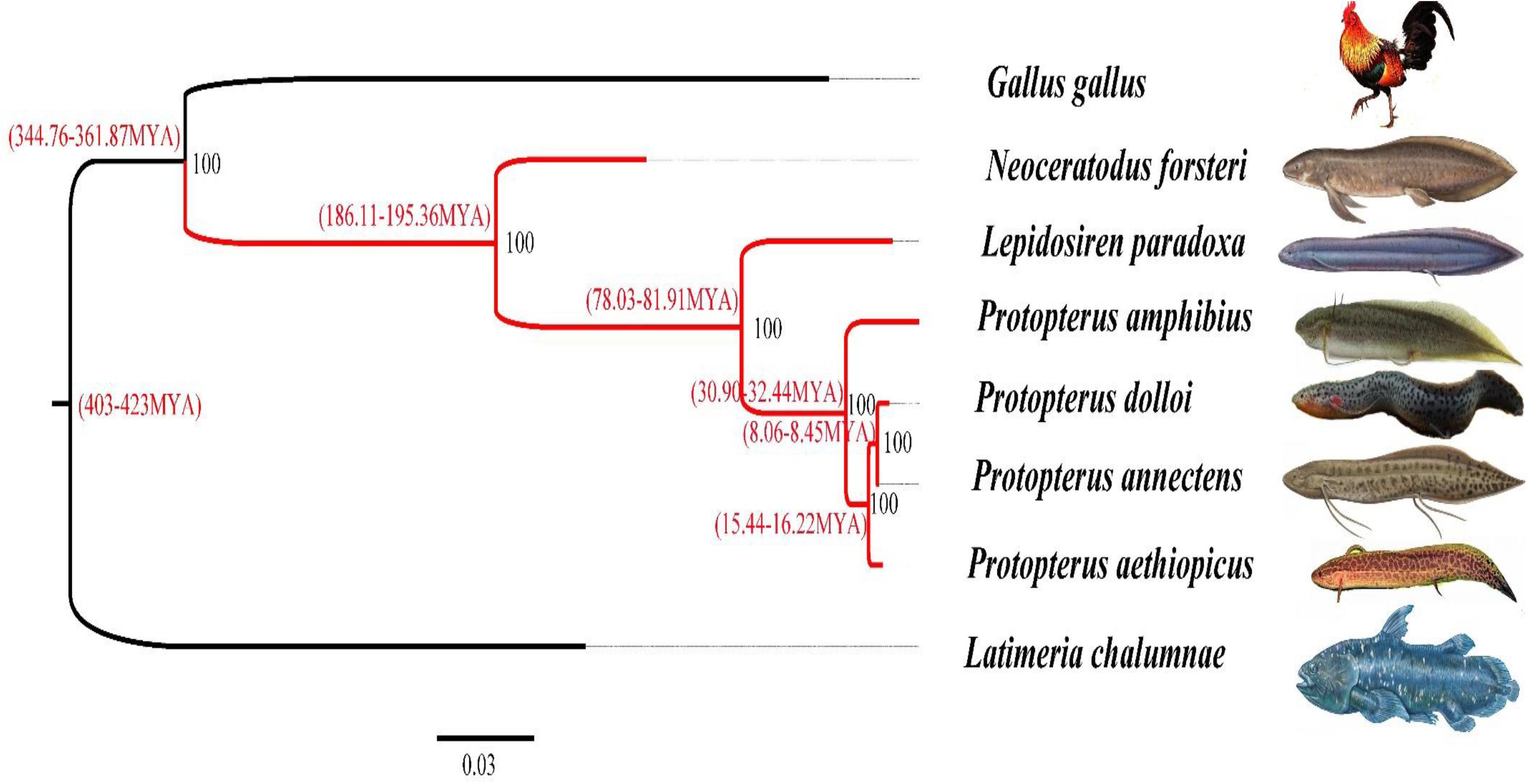
Figure 5. Inferred phylogenetic relationships of Sarcopterygii and the tetrapods based on the concatenated amino acid sequences. Note: The red lines represents the lungfish and 0.03 represents the difference degree of all sequences.
AGs Representative of N. forsteri
We set N. forsteri as the foreground branch in the tree files and then calculated the 2△ln values between the null model and alternative model for 1,094 orthologous genes. After chi-square statistical analyses, a total of 981 and 43 genes were identified as PSGs and FEGs, respectively. The intersection (43 genes) of PSGs and FEGs was identified as the AGs of the N. forsteri (Table 3).
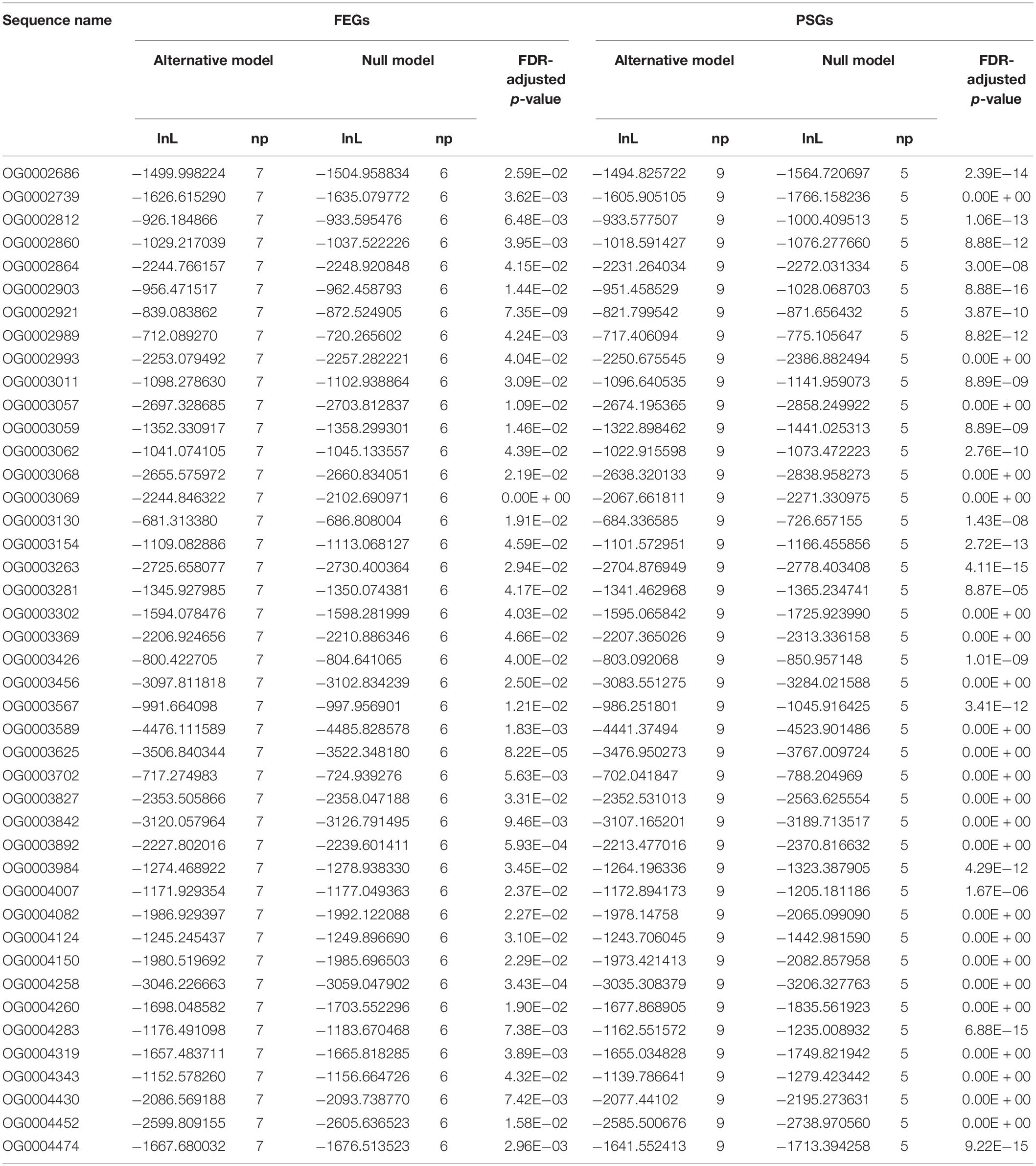
Table 3. The lnL and FDR-adjusted p-value of 43 AGs of N. forsteri.
By combining annotation information, we found that these AGs are potentially associated with protein metabolism, stress response, skeletogenesis and others (Table 4). Moreover, GO annotation results showed that these AGs were primarily involved in “cellular process,” “cell part,” “binding,” and other functions (Figure 6).
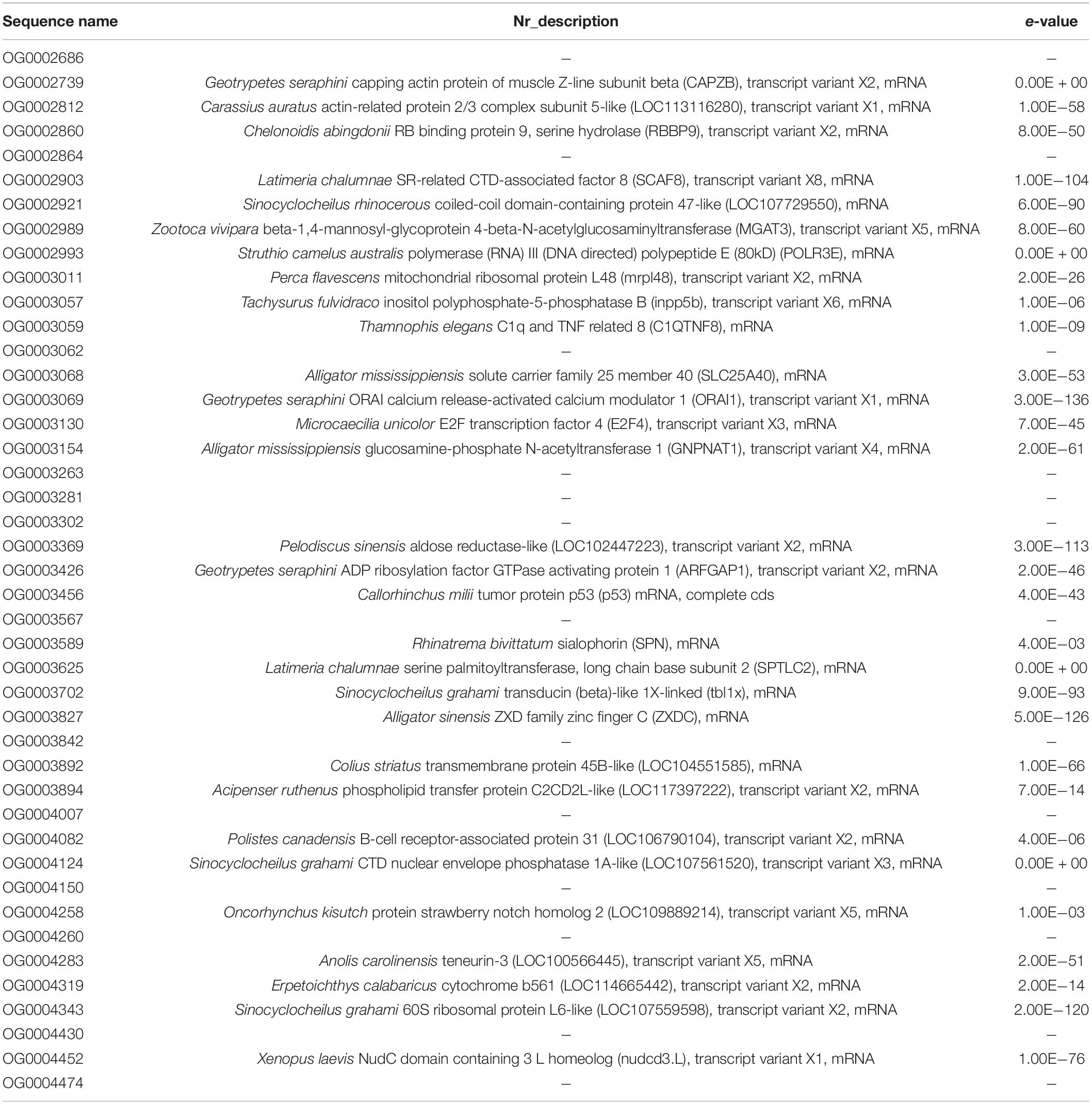
Table 4. AGs representative of N. forsteri.
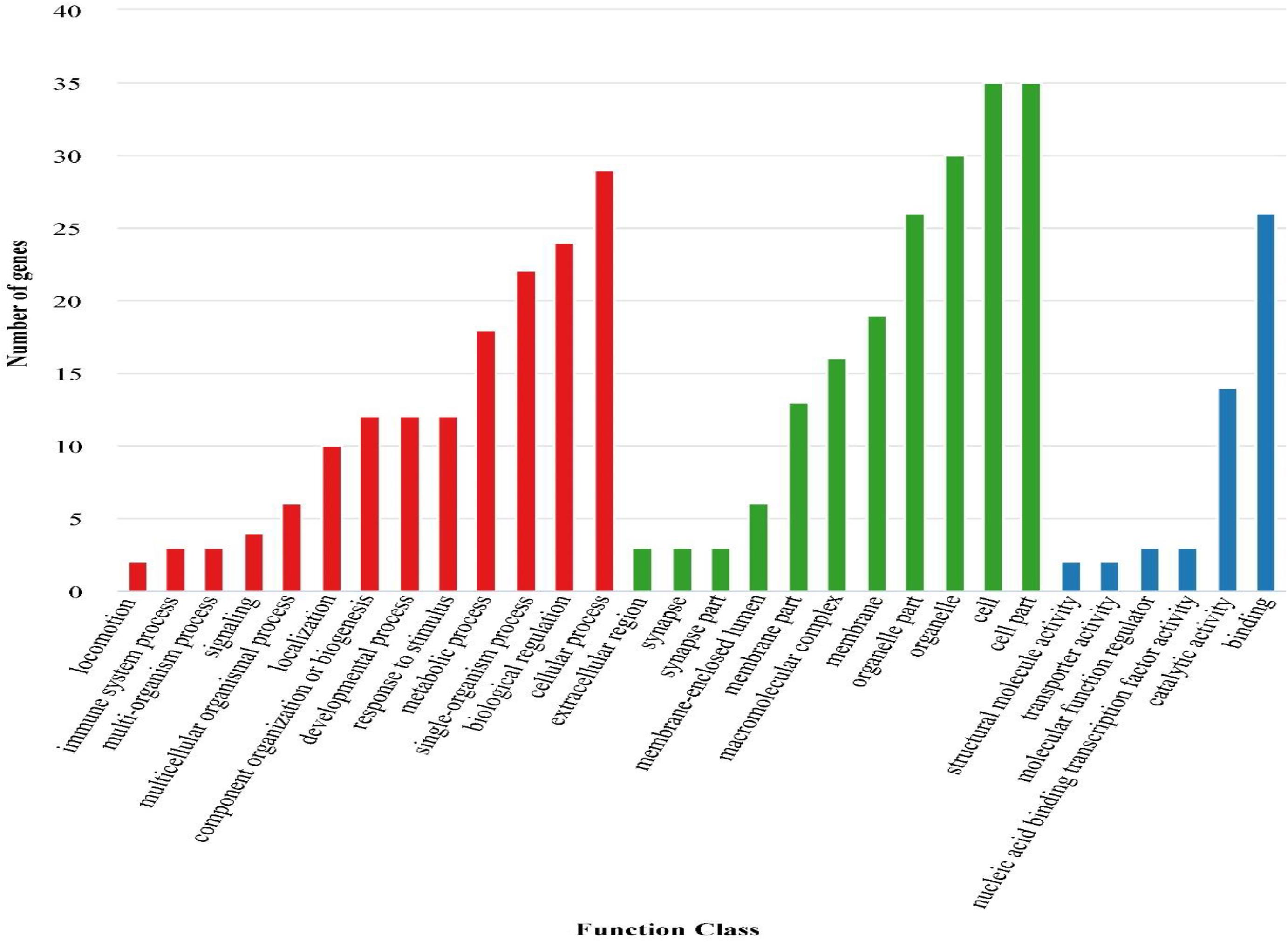
Figure 6. Results of the enrichment analysis of GO terms for AGs of N. forsteri.
Discussion
As the only two extant Sarcopterygii groups, the coelacanths and lungfish have provided ideal research sources for investigating tetrapod landing. More accurate identification of the internal phylogenetic relationship of lungfish is essential to our understanding of how ancient Sarcopterygii originated and colonized the land (Ord and Cooke, 2016). Despite extensive molecular and morphological research on Sarcopterygii during the last three decades, the evolutionary relationships of among all lungfish species remain unresolved (Liang et al., 2013). Limited evidence has been used to evaluate the crucial genes responsible for Sarcopterygii landing processes. Genome-wide genetic information provides an opportunity to solve the above problems effectively (Amemiya et al., 2013). To date, genome-wide genetic information has been obtained by RNA-seq for all the six remaining lungfish species except P. amphibius. In the present study, we first sequenced and assembled the reference transcriptome of P. amphibius. Then, we reconstructed the internal phylogenetic relationships among all lungfish species and inferred the AGs of the most primitive lungfish based on orthologous genes. In brief, we believe that this research can provide new perspectives for investigating ancient Sarcopterygii landing processes.
RNA-Seq Processing
In the present study, we systematically describe the genome-wide genetic information of P. amphibius for the first time based on RNA-seq. The results showed that a total of 14.07 Gb of clean transcriptomic reads were obtained from heart and lung tissues of P. amphibius and the N50 length of clustered unigenes was 1,963 bp. This means that the transcriptome information of P. amphibius has high integrity and credibility. Additionally, it is worth noting that only 41.96% (23,155/55,177) of unigenes were annotated across the eight databases. This may be due to the lack of a lungfish reference genome influences the final assembly efficiency (Liang et al., 2013). However, it is undeniable that the transcriptomic data for P. amphibius obtained in this study still expand the currently available genomic resources for lungfish.
Tissue-Specific Gene Expression Levels
Considering that the gene expression level may be tissue-specific and ultimately lead to different tissues having different physiological functions (Lou et al., 2019), we analyzed DEGs in the heart and lung of P. amphibius. As predicted, numerous (3,189) DEGs were detected between the two tissues. For the DEGs in the two tissues, GO terms were primarily (top 6) related to cellular process, binding, single-organism process, cell, cell part, and biological regulation. The KEGG enrichment results varied slightly in each tissue and included the following pathways: neuroactive ligand-receptor interaction, cardiac muscle contraction, biosynthesis of amino acids, adrenergic signaling in cardiomyocytes, glycine, serine, and threonine metabolism, glycolysis/gluconeogenesis, and tight junctions. These results showed that genes had different biological functions in different tissues based on their unique expression levels and that the gene expression levels with tissue specificity might lead to various roles via different pathways (Li et al., 2018).
Internal Phylogenetic Relationships Among All Lungfish Species
Genome-wide genetic information obtained by high-throughput sequencing technologies can provide large amounts of orthologous genes for the exploration of the phylogenetic relationships mentioned above (Delsuc et al., 2005). In fact, orthologous genes are more suitable for phylogenetic tree construction, because the differentiation of orthologous genes directly leads to species differentiation (Warnock et al., 2012). Moreover, it can also avoid conflicting gene trees caused by different genetic markers (Delsuc et al., 2005). In the present study, we performed an extensive gene comparison among coelacanths, lungfish, and terrestrial vertebrates with transcriptome or genome datasets and 1,094 single-copy orthologous genes were obtained. This is probably because the screening of Blast pairwise comparison may be more rigorous (Li et al., 2003). We hypothesized that RNA-seq could not obtain complete genomic information and further resulted in a small number of orthologous genes. Although the number of gene numbers was small, we still believe that those genes can help us obtain a more complete phylogenetic relationship among all lungfish species. The reconstructed phylogenetic structure in the present study provided evidence that lungfishes are the closest living relatives of terrestrial vertebrates (Liang et al., 2013). Unsurprisingly, the phylogenetic tree is significantly congruent with the prevailing morphological and molecular biological view of lungfish species. We verified that N. forsteri is the most primitive lungfish species. A previous study considered that the morphology of lungfish barely changed over millions of years (Kemp, 1986). In fact, the morphological characteristics (such as body shape, large scales, and paddle-shaped fins) of N. forsteri make them more similar to their ancestors than to other lungfish species. Moreover, the fins of all lungfish species (especially the African lungfish species) except N. forsteri have almost completely disappeared and the fin morphology has been reduced to filaments (Gunther, 1870; Kemp, 1986). This result further confirms the findings of the present study that N. forsteri is the most primitive lungfish species. There is no denying the fact that high statistical support for a given topology does not imply completely accurate phylogenetic inference (Amemiya et al., 2013; Liang et al., 2013). In fact, we also suspect that the species selection and number of orthologous genes used for phylogenetic analysis may also contribute to differences in phylogenetic tree shape (Edwards et al., 2017). Accordingly, future studies will need to sequence the genomes of all lungfish species to obtain the complete genetic information for constructing a more comprehensive evolutionary relationship.
AGs Information Supports N. forsteri as the Most Primitive Lungfish Species and Reveals the Sarcopterygii Landing Processes
As the most primitive lungfish, N. forsteri has only a single swim bladder and cannot live under dry conditions for long periods of time. In the present study, only 43 genes showed positive selection and rapid evolution in N. forsteri. The GO annotation showed that AGs mainly participate in cell part and cellular processes, and their functions mainly involve protein binding.
The most primitive Sarcopterygii transitioned to the land, and a new terrestrial environment is a major physiological challenge (including stronger ultraviolet radiation, evaporation, and others) for the landing Sarcopterygii. In the present study, we suspect that some genes (RBBP9, MGAT3, inpp5b, and nudcd3.L) may be involved in the maintenance of the cytoskeleton (Lu, 2011; Zhang et al., 2018). The cytoskeleton plays an important role in the maintenance of cell morphology, movement under deformation and the transport of intracellular substances (Pontaini et al., 2009; Lee et al., 2010). The evaporation of terrestrial environment will inevitably cause the loss of water in the cells of landing Sarcopterygii and eventually damage the cytoskeleton. Therefore, rapid evolution or positive selection of cytoskeleton related-genes in N. forsteri may be beneficial to cellular defense. Additionally, although mucus glands all over the body can help landing Sarcopterygii reduce water evaporation, landing Sarcopterygii still needs to improve their humoral regulation mechanism to cope with evaporation stress. In fact, we found that some ion-exchange related genes (such SLC25A40) underwent rapid evolution and positive selection. The solute carrier superfamily has been shown to mediate the transmembrane transport of various solutes between cells and the outside environment or within cells (Hoglund et al., 2011). Amemiya et al. (2013) considered that early landing Sarcopterygii may be more vulnerable to ammonia in terrestrial environments because ammonia cannot be quickly diluted by water (Amemiya et al., 2013). It is possible that stimulation from multiple noxious elements in the terrestrial environment might contribute to oxidative stress in landing Sarcopterygii and eventually lead to DNA damage and even apoptosis (Kantidze et al., 2016; Morales et al., 2016). In the present study, some genes associated with stress response (such as ORAI1 and ZXDC) were found to have undergone positive selection and fast evolution in the N. forsteri. Previous study considered that ORAI1 plays a role in endoplasmic reticulum stress (Zhang et al., 2020). In fact, endoplasmic reticulum stress caused by environmental stress usually results in misfolded or unfolded proteins, and ultimately leads to the disruption of normal cell function (Ron and Walter, 2007). The ZXDC gene has been shown to be associated with inflammation (Ramsey and Fontes, 2013). Therefore, such results indicated that rapid evolution and positive selection of stress-related genes helped landing Sarcopterygii survive in harsher terrestrial environments. In fact, these genes have been shown to have a range of protective effects, including antioxidation, antiapoptosis and DNA damage repair. It is worth noting that genetic changes may y manifest in both locus variation and quantitative changes during ancient Sarcopterygii landing processes (Amemiya et al., 2013). We agree that critical characteristics in the morphological transition (fin-to-limb transition, etc.) during the ancient Sarcopterygii landing processes are associated with changes in some gene deletions (Navratilova et al., 2009; Jones et al., 2012). Interestingly, we also found that some AGs were involved in the skeletogenesis (such CAPZB). Although the present study cannot determine which genes were lost during the Sarcopterygii landing processes due to the lack of genome sequences of Sarcopterygii, it provides the first steps toward new studies on the field. Therefore, sequencing genomes of all Sarcopterygii is probably the next step on determining the crucial mechanisms of the ancient Sarcopterygii landing processes through comparative genomics.
Conclusion
The present study is the first systematic report on the transcriptome of P. amphibius and we believe that these sequences can be used to enrich the genome-wide genetic information of lungfishes. We obtained 1,094 single-copy orthologous genes shared by Sarcopterygii and tetrapods through comparative genomics, and the phylogenetic structure was indicated that the lungfishes are the closest living relatives of the terrestrial vertebrates. We first reconstructed the internal phylogenetic relationship of all lungfish species based on phylogenomics. In addition, AGs information supports N. forsteri as the most primitive lungfish and reveals the pivotal genes associated with Sarcopterygii landing processes. In conclusion, the present study is one a small step in how the ancient Sarcopterygii transitioned to the land. Future studies will require sequencing of all lungfish genomes to construct a more comprehensive internal phylogenetic relationship and to determine the crucial mechanisms for the ancient Sarcopterygii landing processes.
Data Availability Statement
The data presented in this study are publicly accessible at NCBI under accession numbers: SRR13486841–SRR13486842.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of First Institute of Oceanography, Ministry of Natural Resources.
Author Contributions
LZ and ZH conceptualized the study and conducted the analyses. LZ, SW, FL, TG, and ZH analyzed the data and wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (LR21D060003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Altschul, S. F., Madden, T. L., Schffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Amemiya, C. T., Alföldi, J., Lee, A. P., Fan, S., Philippe, H., Maccallum, I., et al. (2013). The African coelacanth genome provides insights into tetrapod evolution. Nature 496, 311–316. doi: 10.1038/nature12027
Biscotti, M. A., Gerdol, M., Canapa, A., Forconi, M., Olmo, E., Pallavicini, A., et al. (2016). The lungfish transcriptome: a glimpse into molecular evolution events at the transition from water to land. Sci. Rep. 6:21571. doi: 10.1038/srep2157
Brinkmann, H., Venkatesh, B., Brenner, S., and Meyer, A. (2004). Nuclear protein-coding genes support lungfish and not the coelacanth as the closest living relatives of land vertebrates. Proc. Natl. Acad. Sci. U.S.A. 101, 4900–4905. doi: 10.1073/pnas.0400609101
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Christensen-Dalsgaard, J., Brandt, C., Wilson, M., Wahlberg, M., and Madsen, P. (2011). Hearing in the African lungfish (Protopterus annectens): preadaptation for pressure hearing in tetrapods? Biol. Lett. 7, 139–141. doi: 10.1098/rsbl.2010.0636
Clack, J. A. (2002). Gaining Ground: The Origin and Early Evolution of Tetrapods. Bloomington, IN: Indiana University Press.
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Delsuc, F., Brinkmann, H., and Philippe, H. (2005). Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 6, 361–375. doi: 10.1038/nrg1603
Dial, K. P., Shubin, N., and Brainerd, E. L. (2015). Great Transformations in Vertebrate Evolution. Chicago, IL: The University of Chicago Press, 114–115. doi: 10.1093/sysbio/syv117
Edwards, S. V., Cloutier, A., and Baker, A. J. (2017). Conserved nonexonic elements: a novel class of marker for phylogenomics. Syst. Biol. 66, 1028–1044. doi: 10.1093/sysbio/syx058
Fritzsch, B. (1987). Inner ear of the coelacanth fish Latimeria has tetrapod affinities. Nature 327, 153–154. doi: 10.1038/327153a0
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J., Thompson, D., Amit, I., et al. (2011). Full length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gunther, A. (1870). Description of Ceratodus, a genus of ganoid fishes, recently discovered in rivers of Queensland, Australia. Philos. Proc. R. Soc. Lond. 161, 377–379. doi: 10.1098/rspl.1870.0056
Hedges, S. B., Hass, C. A., and Maxson, L. R. (1993). Relations of fish and tetrapods. Nature 363, 501–502. doi: 10.1038/363501b0
Hoglund, P. J., Nordstrom, K. J., Schioth, H. B., and Fredriksson, R. (2011). The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of Bilaterian species. Mol. Biol. Evol. 28, 1531–1541. doi: 10.1093/molbev/msq350
Hughes, L. C., Ortía, G., Huang, Y., Sun, Y., Baldwin, C., Thompson, A., et al. (2018). Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. Proc. Natl. Acad. Sci. U.S.A. 115, 6249–6254. doi: 10.1073/pnas.1719358115
Irisarri, I., and Meyer, A. (2016). The identification of the closest living relative(s) of tetrapods: phylogenomic lessons for resolving short ancient internodes. Syst. Biol. 65, 1057–1075. doi: 10.1093/sysbio/syw057
Irisarri, I., Baurain, D., Brinkmann, H., Delsuc, F., Sire, J., Kupfer, A., et al. (2017). Phylotranscriptomic consolidation of the jawed vertebrate timetree. Nat. Ecol. Evol. 1, 1370–1378. doi: 10.1038/s41559-017-0240-5
Jones, F. C., Grabherr, M. G., Chan, Y. F., Russell, P., Mauceli, E., Johnson, J., et al. (2012). The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61. doi: 10.1038/nature10944
Kantidze, O. L., Velichko, A. K., Luzhin, A. V., and Razin, S. V. (2016). Heat stress-induced DNA damage. Acta Nat. 8, 75–78.
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kemp, A. (1986). The biology of the Australian lungfish, Neoceratodus forsteri (Krefft 1870). J. Morphol. 190, 181–198. doi: 10.1002/jmor.1051900413
Kumar, S., Stecher, G., Suleski, M., and Hedges, S. B. (2017). TimeTree: a resource for timelines, timetrees, and divergence times. Bioinformatics 34, 1812–1819. doi: 10.1093/molbev/msx116
Lee, H., Ferrer, J. M., Nakamura, F., Lang, M., and Kamm, R. (2010). Passive and active microrheology for cross-linked F-actin networks in vitro. Acta Biomater. 6, 1207–1218. doi: 10.1016/j.actbio.2009.10.044
Leng, N., Dawson, J. A., Thomson, J. A., Ruotti, V., Rissman, A. I, Smits, B., et al. (2013). EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043. doi: 10.1093/bioinformatics/btt087
Li, B., and Colin, N. D. (2011). RSEM: accurate transcript quantification from RNA Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189.
Li, Y., Fang, C. C., Fu, Y. H., Hu, A., Li, C., Zou, C., et al. (2018). A survey of transcriptome complexity in Sus scrofa using single-molecule long-read sequencing. DNA Res. 25, 421–437. doi: 10.1093/dnares/dsy014
Liang, D., Shen, X. X., and Zhang, P. (2013). One thousand two hundred ninety nuclear genes from a genome-wide survey support lungfishes as the sister group of tetrapods. Mol. Biol. Evol. 30, 1803–1807. doi: 10.1093/molbev/mst072
Lou, F. R., Han, Z. Q., and Gao, T. X. (2019). Transcriptomic responses of two ecologically divergent populations of Japanese mantis shrimp (Oratosquilla oratoria) under thermal stress. Animals 9, 399. doi: 10.3390/ani9070399
Lou, F. R., Zhang, Y., Song, N., Ji, D., and Gao, T. (2020). Comprehensive transcriptome analysis reveals insights into phylogeny and positively selected genes of Sillago species. Animals 10:633. doi: 10.3390/ani10040633
Lu, Y. Q. (2011). The Function and Mechanism of NudC in Cell Migration. Hangzhou: Zhejiang University, 34–35.
Meyer, A. (1995). Molecular evidence on the origin of tetrapods and the relationships of the coelacanth. Trends Ecol. Evol. 10, 111–116.
Meyer, A., and Wilson, A. C. (1990). Origin of tetrapods inferred from their mitochondrial DNA affiliation to lungfish. J. Mol. Evol. 31, 359–364. doi: 10.1007/BF02106050
Meyer, A., Schloissnig, S., and Franchini, P. (2021). Giant lungfish genome elucidates the conquest of land by vertebrates. Nature 590, 284–289.
Morales, M. E., Derbes, R. S., Ade, C. M., Ortego, J. C., Stark, J., Deininger, P., et al. (2016). Heavy metal exposure influences double strand break DNA repair outcomes. PLoS One 11:e0151367. doi: 10.1371/journal.pone.0151367
Navratilova, P., Fredman, D., Hawkins, T. A., Turner, K., Lenhard, B., and Becker, T. S. (2009). Systematic human/zebrafish comparative identification of cis-regulatory activity around vertebrate developmental transcription factor genes. Dev. Biol. 327, 526–540. doi: 10.1016/j.ydbio.2008.10.044
Ord, T. J., and Cooke, G. M. (2016). Repeated evolution of Amphibius behavior in fish and its implications for the colonization of novel environments. Evolution 70, 1747–1759. doi: 10.1111/evo.12971.2016
Panchen, A. L., and Smithson, T. S. (2008). Character diagnosis, fossils and the origin of tetrapods. Biol. Rev. 62, 341–438. doi: 10.1111/j.1469-185X.1987.tb01635.x
Pontaini, L. L., Jasper, V., Salbreux, G., Heuvingh, J., Joanny, J., and Sykes, C. (2009). Reconstitution of an actin cortex inside a liposome. Biophys. J. 96, 192–198. doi: 10.1016/j.bpj.2008.09.029
Ramsey, J. E., and Fontes, J. D. (2013). The zinc finger transcription factor ZXDC activates CCL2 gene expression by opposing BCL6-mediated repression. Mol. Immunol. 56, 768–780. doi: 10.1016/j.molimm.2013.07.001
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Sanderson, M. (2003). r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302. doi: 10.1093/bioinformatics/19.2.301
Shan, Y. F., and Gras, R. (2011). 43 genes support the lungfish-coelacanth grouping related to the closest living relative of Tetrapods with the Bayesian method under the coalescence model. BMC Res. Notes 4:49. doi: 10.1186/1756-0500-4-49
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Takezaki, N., Figueroa, F., Zaleska-Rutczynska, Z., Takahata, N., and Klein, J. (2004). The phylogenetic relationship of tetrapod, coelacanth, and lungfish revealed by the sequences of forty-Four nuclear genes. Mol. Biol. Evol. 21, 1512–1524.
Warnock, R. C., Yang, Z., and Donoghue, P. C. (2012). Exploring uncertainty in the calibration of the molecular clock. Biol. Lett. 8, 156–159. doi: 10.1098/rsbl.2011.0710
Wheeler, T. J., and Eddy, S. R. (2013). nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489. doi: 10.1093/bioinformatics/btt403
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, 316–322. doi: 10.1093/nar/gkr483
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Zardoya, R., and Meyer, A. (1996). Evolutionary relationships of the coelacanth, lungfish, and tetrapods based on the 28S ribosomal RNA gene. Proc. Natl. Acad. Sci. U.S.A. 93, 5449–5454. doi: 10.1073/pnas.93.11.5449
Zhang, B. B., Li, M., Yang, W., Loor, J. J., Wang, S., Zhao, Y., et al. (2020). Orai calcium release-activated calcium modulator 1 (ORAI1) plays a role in endoplasmic reticulum stress in bovine mammary epithelial cells challenged with physiological levels of ketone bodies. J. Dairy Sci. 103, 4691–4701.
Keywords: Protopterus amphibious, phylogenomics, terrestrial environment adaption, phylogenetic relationships, lungfish
Citation: Zhao L, Wang S, Lou F, Gao T and Han Z (2021) Phylogenomics Based on Transcriptome Data Provides Evidence for the Internal Phylogenetic Relationships and Potential Terrestrial Evolutionary Genes of Lungfish. Front. Mar. Sci. 8:724977. doi: 10.3389/fmars.2021.724977
Received: 14 June 2021; Accepted: 26 July 2021;
Published: 16 August 2021.
Edited by:
Jin Sun, Ocean University of China, ChinaReviewed by:
Kai Zhang, Hong Kong University of Science and Technology, Hong Kong, SAR ChinaNadayca Mateussi, UNESP, Brazil
Ruth Morona, Complutense University of Madrid, Spain
Copyright © 2021 Zhao, Wang, Lou, Gao and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiqiang Han, ZDYzMzkxMjRAMTYzLmNvbQ==