 Shichao Wei
Shichao Wei Wenliang Zhou
Wenliang Zhou Huizhong Fan
Huizhong Fan Zhiwei Zhang
Zhiwei Zhang Weijian Guo1
Weijian Guo1 Fuwen Wei
Fuwen Wei- 1Center for Evolution and Conservation Biology, Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Guangzhou, China
- 2CAS Key Laboratory of Animal Ecology and Conservation Biology, Institute of Zoology, Chinese Academy of Sciences, Beijing, China
- 3University of Chinese Academy of Sciences, Beijing, China
- 4Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming, China
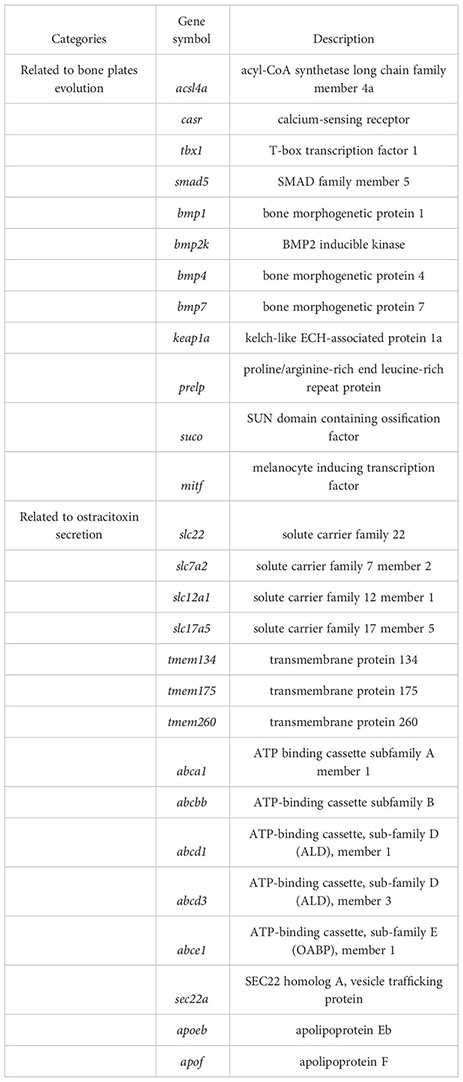
The Ostracion cubicus, commonly known as the yellow boxfish, is a remarkable species with a body encased in a bone plate and the ability to produce an ostracitoxin from their skin when under stress. However, the genetic basis of those effective defense traits is still largely unknown due to the lack of genomic resources. Here, we assembled the first chromosome-level genome of O. cubicus with 867.50 Mb in genome size and 34.86 Mb N50 scaffold length by HiFi and Hi-C sequencing. Twenty-five pseudo-chromosomes, numbered according to size, covered 94.13% of the total assembled sequences. A total of 23,224 protein-coding genes were predicted, with a BUSCO completeness of 98.6%. Positive selection or rapid evolution was observed in genes related to scale and bone development (acsl4a, casr, keap1a, tbx1), and up-regulation of transcription was found in the skin of boxfish (bmp1, bmp2k, bmp4, bmp7, smad5, suco, prelp, mitf), likely associated with the bone plates evolution in the yellow boxfish. An expansion of the solute carrier family 22, a cluster of genes in solute carrier (SLCs) family, transmembrane protein family (TMEMs), vesicle trafficking (SECs), ATP-binding cassette (ABCs) and apolipoproteins (APOs) were identified under positive selection, rapid evolution, or up-regulated in the skin of boxfish, likely associated with the ostracitoxin secretion in the yellow boxfish. Our study not only presents a high-quality boxfish genome but also provides insights into bone plates evolution and ostracitoxin secretion of O. cubicus.
Introduction
Tetraodontiformes, a group of approximately 430 extant fishes, can be divided into 10 families, including boxfish (Ostraciidae), pufferfish (Tetraodontidae), porcupinefish (Diodontidae), triggerfish (Balistidae), trunkfish (Ostraciidae), and ocean sunfish (Molidae) (Tyler and Santini, 2002; Santini and Tyler, 2003). These fishes are mainly found in shallow tropical or warm-temperate water coral reefs and exhibit a remarkable diversity in defense strategies, body size, and ecology (Alfaro et al., 2007). Pufferfish and boxfish species are known for adopting toxin-defense strategies to avoid predation by accumulating lethal amounts of tetrodotoxin and boxfish toxin (Thomson, 1964; Noguchi et al., 2006). Porcupinefish are covered with long, hard spines, and when attacked, their stomachs puff up into prickly balls (Wainwright and Turingan, 1997). The triggerfish species are aggressive, while the ocean sunfish are sluggish due to their large size and defunct tail fins (Dornburg et al., 2011). All these special behaviors and characteristics make tetraodontiformes an ideal group for examining the genomic basis the evolution of complex traits in marine teleost.
Boxfishes of the family Ostraciidae are characterized by having a body encased in bone plates, which covers most of the head and body, with gaps only for the mouth, nostrils, gill opening, anus, caudal peduncle, and fins (Yang et al., 2015). The formation of closed bone plates is one of the most important defense strategies of boxfishes. Although the boxy shape and bone plates considerably restrict the movement of boxfish, it eventually provides effective protection against predators (Gordon et al., 2000). Thus, it is clear that the evolution of the bone plates of boxfish is an adaptation to predatory pressures and should be under natural selection, which may leaving its marks on the genomic level as well. Because that these bone plates are mainly composed of dermal scutes and derived from the skin’s dermis with a highly mineralized surface plate and a compliant collagen base (Yang et al., 2015). This suggests that genomic changes may have occurred in gene regions associated with bone formation and scales keratinization during the evolution of boxfish bone plates.
Another key defense strategy of boxfish is that it quickly secretes a toxin called ostracitoxin (also known as pahutoxin) through club cells in the epidermis in response to external stress, such as preying (Thomson, 1964). Ostracitoxin has a wide variety of effects on biological systems, the most notable being its high toxicity to marine fishes and its hemolytic-agglutinating action on fish erythrocytes (Thomson, 1964; Thomson, 1969). When caught or touched, boxfish often release this toxic substance, which can kill other fish that swimming together (Thomson, 1964; Thomson, 1969). However, it is still unclear how boxfish form boxfish toxins and how they are secreted quickly in response to danger.
A high-quality genome is the key to understanding the molecular mechanisms of species adaptation (Hu et al., 2017; Fan et al., 2019; Hu et al., 2023). Due to the lack of genetic information, the genetic basis underlying the two remarkable anti-predator adaptation characteristic traits of the boxfish remains unknown. Herein, the yellow boxfish (Ostracion cubicus Linnaeus, 1758), belonging to the family Ostraciidae with widely distributed across the Indo-Pacific region (Froese and Pauly, 2023), was chosen as a model species to investigate the genetic basis of boxfishes’ two characteristic traits, bone plates and ostracitoxin secretion. First, we used long high-fidelity (HiFi) sequencing data and high‐throughput chromosome conformation capture (Hi-C) technique to obtain a chromosome-level assembly genome of O. cubicus. And then, we conducted comparative genomic and transcriptomic analyses to identify the genes that associated with the bone plates evolution and ostracitoxin secretion. Our results provide potential clues to interpret the genetic basis of the bone plates and ostracitoxin secretion in the yellow boxfish, and the availability of genomic and transcriptomic resources will be valuable for elucidating the complex traits evolution and ecology of tetraodontiformes.
Materials and methods
Sampling and sequencing
One adult male yellow boxfish was sampled in December 2021 from offshore Sanya, China with a body weight of 186.1 g and a body length of 16.6 cm. Genome DNA from the fresh muscle was extracted and used to construct PacBio, paired-end (PE) MGI and Hi-C libraries. Additionally, the skin from five adult boxfishes (two yellow boxfish and three longhorn cowfish (Lactoria cornuta)) and the skin tissues from three adult fugu (Takifugu rubripes) were collected from offshore Sanya, China, and extracted for RNA sequencing. Furthermore, the RNA sequencing data of the skin tissue of two adult T. flavidus individuals from NCBI (SRR accession numbers: SRR18358223 and SRR18358224) were downloaded. All sampled tissues were stored frozen at -80°C.
Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA), according to the standard operating procedure provided by the manufacturer. DNA degradation and contamination was checked on 1% agarose gels with lambda DNA standard. DNA purity was then assessed using NanoDrop™ One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA), of which OD260/280 ranging from 1.8 to 2.0 and OD 260/230 is between 2.0-2.2. DNA concentration was further measured by Qubit® 4.0 Fluorometer (Invitrogen, USA). Then, A total amount of 15 μg DNA was fragmented into approximately 15-kb fragments for PacBio sequencing according to PacBio’s standard protocol (Pacific Biosciences, CA, USA). Single-strand overhangs were then removed, followed by damage repair, end-repair and A-tailing. DNA fragments were ligated to blunt hairpins and DNA polymerase was bound to the annealed SMRTbell templates. Sequencing was performed on a PacBio Sequel II instrument with Sequencing Primer V2 and Sequel II Binding Kit 2.0 in Grandomics.
A PE library with 300-bp insert sizes was prepared with the MGIEasy FS DNA library preparation set (MGI Tech) for the whole genome resequencing. The library was sequenced using a DNBSEQ-T7 platform (MGI Tech, China).
The fresh muscle was cut into 2-cm pieces and vacuum infiltrated in nuclei isolation buffer supplemented with 2% formaldehyde for Hi-C sequencing. Glycine was added to stop the crosslinking reaction. The crosslinked DNA was extracted and digested with 100 units of Dpnll restriction enzyme. The sticky ends of the digested products were marked with biotin and ligated. After the removal of the biotin from non-ligated DNA ends owing to the exonuclease activity of T4 DNA polymerase, the ligated DNA was sheared into 300-600 bp fragments. The Hi-C libraries were quantified and sequenced using the MGI DNBSEQ.
The four tissues were used for RNA extraction using TRIzol reagents for the RNA sequencing. The cDNA libraries were constructed by following the manufacturer’s recommendations and paired-end sequenced with 150 bp using the MGI DNBSEQ-T7 sequencing platform.
Genome assembly
The PE libraries sequencing data was used to estimate the yellow boxfish genome size and heterozygosity rate. The k-mer spectrum was calculated using Jellyfish v2.3.0 (Marçais and Kingsford, 2011). And then, the genome size and heterozygosity rate were measured using 21 k-mer spectrum by Genome Characteristics Estimation (GCE) v1.0.2 (Liu et al., 2013).
We first produced a high-quality contig genome assembly using Hifiasm v0.16.1 (Cheng et al., 2021) to assemble the yellow boxfish’s contig-level genome. And then, the redundant sequences in the assembled genome were removed by Purge_dups v1.2.5 (Guan et al., 2020). The purged contigs were subsequently polished by the PE libraries sequencing data. Briefly, the PE libraries sequencing data were firstly aligned to the genome using BWA v0.7.17 (Li, 2013), and polished with default parameters using Pilon v1.23 (Walker et al., 2014). After that, high-accuracy yellow boxfish contigs had been obtained.
The assembled contigs were aligned to the Hi-C sequencing data with default parameters using Juicer v1.6 (Durand et al., 2016) to improve the level of genome from the contig to the chromosome. After that, the chromosome-level genome assembly was performed with default parameters using the 3D-DNA pipeline (Dudchenko et al., 2017). Whole-genome sequence collinearity alignments were performed between O. cubicus and T. rubripes using LastZ v1.03.54 (Harris, 2007). The syntenic relationships among chromosomes were visualized using Circos v0.69 (Krzywinski et al., 2009).
Genome completeness was evaluated by searching against the actinopterygii_odb10 database using the BUSCO v4.1.4 (Seppey et al., 2019). The accuracy of the genome was further assessed by mapping the PE libraries sequencing data to the assembled chromosome-level genome using BWA v0.7.17 (Li, 2013), and counting the mapping rate and depth using SAMtools v1.12 (Danecek et al., 2011).
Genome annotation
We initially employed RepeatModeler v2.0 (Flynn et al., 2020) to classify repeats for the genome. Next, to find known and novel transposable elements (TEs), we used RepeatMasker v4.1.1 (Chen, 2004) to map the yellow boxfish genome sequences against the de novo repeat library and Repbase TE library v16.02 (Bao et al., 2015). Subsequently, to find TE-relevant proteins, we applied the RepeatProteinMask v.4.0.6 (Chen, 2004). In addition, to find tandem repeats, we initially employed Tandem Repeats Finder (TRF) v.4.07 (Benson, 1999).
We also used three methods to annotate genes in the genome, including the de novo prediction, homology-based prediction, and transcriptome-based prediction were combined using EvidenceModeler (EVM) v1.1.1 (Haas et al., 2008) and PASA v2.4.0 (Haas et al., 2008). For de novo gene predictions, we used Augustus v3.4.0 (Stanke et al., 2008), Genscan v3.1 (Burge and Karlin, 1998), and GlimmerHMM v3.0.1 (Majoros and Salzberg, 2004) to analyze the repeat-masked genome. For homology-based predictions, the protein sequences of Cynoglossus semilaevis, Danio rerio, Gasterosteus aculeatus, Gadus morhua, Larimichthys crocea, Oryzias latipes, Oreochromis niloticus, T. rubripes, T. bimaculatus and Tetraodon nigroviridis obtained from NCBI were aligned to the yellow boxfish genome by following the pipeline of Chen et al. (Chen et al., 2019). For transcriptome-based prediction, RNA-seq data were aligned to the genome with default parameters by Hisat v2.1.0 (Kim et al., 2015). And then, transcripts were reconstructed with default parameters using StringTie v2.0 (Pertea et al., 2015).
We aligned the protein or nucleotide format gene sequences to the National Center for Biotechnology Information nonredundant protein (NR), the National Center for Biotechnology Information nonredundant nucleotide sequence (NT) and SwissProt (Bairoch and Apweiler, 2000) databases using Blastp and Blastn (-e 1e-5) to annotate the genes’ function. Gene ontology (GO) terms were retrieved and assigned to the yellow boxfish query sequences, and enzyme codes (EC) corresponding to GO were retrieved and mapped to KEGG pathway annotations.
Comparative genomics and phylogenetic reconstruction
The annotated protein-coding sequences of the 12 related Clupeocephala fishes (C. semilaevis, D. rerio, G. aculeatus, G. morhua, L. crocea, Mola Leptocephali, O. latipes, O. niloticus, T. rubripes, T. bimaculatus, T. nigroviridis and T. palembangensis) were downloaded from NCBI to cluster gene family. We filtered the genes either with frameshifts, less than 50 amino acids, or redundant copies, and only keep the longest transcripts to ensure the accuracy of downstream comparative genomics analysis. We aligned the protein sequences of the yellow fish with the 12 Clupeocephala fishes based on sequence identity to call orthologous by the OrthoFinder v2.3.8 (Emms and Kelly, 2019).
Protein-coding gene sequences of single-copy orthogroups genes shared by the 13 Clupeocephala fishes were aligned by Macse v2.06 (Ranwez et al., 2011) at the codon level. Gaps and nonhomologous fragments were filtered out using Gblocks v0.91b (Castresana, 2000) with strict parameters (“-t = c, -b5 = n”). After excluding the datasets with less than 150-bp nucleotide sites, high-quality multiple sequence alignments (MSAs) were obtained for subsequent analysis. The alignments were then concatenated into a super-gene alignment by in-house Perl script. This super-gene was used to construct a maximum-likelihood phylogeny tree by RAxML (Stamatakis, 2006) with 1000 bootstrap replicates. GTR + G models for each amino acid and nucleotide partition were set as substitution models. The divergence times between fishes were estimated by the MCMCtree program in Paml v4.9j (Yang, 2007), using a Bayesian relaxed-molecular clock model calibrated with three calibrated nodes by fossil records (ancestral node of fugu and Tetraodon: 32.2-56.0 Mya (million years ago); ancestral node of gasterosteiform and tetraodontiform: 96.9-150.9 Mya; ancestral node of zebrafish and medaka: 149.9-165.2 Mya) (Benton and Donoghue, 2007).
We used Cafe v3.1 (De Bie et al., 2006) to explore the gene family evolution by construct the gene families that underwent expansion or contraction across the 13 fish’s phylogeny. The significantly expanded gene families were classified through R package clusterProfiler v3.14.2 (Yu et al., 2012) based on the gene annotation database.
We used the branch model of CodeML in Paml by setting yellow boxfish as the foreground branch to identify potential rapidly evolving genes (REGs). The null hypothesis assumes that the omega (ω) value of each branch was equal, the alternative hypothesis assumes that the omega value of the foreground branch was not equal to those of the background branches. A likelihood ratio test was performed after correcting P values using the FDR test with Bonferroni correction. Genes were identified to REGs when the ω value on the foreground branch was larger than those on the background branches and had a corrected P < 0.05.
We employed the branch-site model of CodeML in PAML with yellow boxfish as the foreground branch to identify potential positively selected genes (PSGs). The null hypothesis was that the ω value of each site on the foreground branch was ≤ 1, and the alternative hypothesis was that ω values were > 1. A likelihood ratio test was performed, with the null distribution set to a 50:50 mixture of χ2 distributions with 1 degree of freedom and a point mass of zero. To account for multiple testing, FDR testing with Bonferroni correction was performed.
Gene transcription quantification
We investigated the differences in gene transcription profiles between the skin of boxfish and fugu by align RNA-seq clean reads from the respective tissues of five boxfishes (two yellow boxfish and three longhorn cowfish) to the yellow boxfish genome and five fugu (three T. rubripes and two T. flavidus) to T. rubripes reference genome (fTakRub1.2) using Hisat2 v2.2.1 (Kim et al., 2019). Read counts were calculated with Stringtie v2.2.1 (Pertea et al., 2015), and gene transcription was quantified as fragments per kilobase of gene per million mapped reads (FPKM) using prepDE.py in Stringtie. Differentially expressed genes (DEGs) were identified using edgeR v3.40.1 (Robinson et al., 2010), and adjusted P values were calculated by Benjamini-Hochberg false discovery correction. Genes with adjusted P values less than 0.05 were considered as DEGs.
Results
Genome sequencing and assembly
A total of 18.5 Gb HiFi, 112.1 Gb Hi-C and 27.0 Gb PE library sequencing data were generated from PacBio SequelII, DNBSEQ-T7 and DNBSEQ-T7 platform, respectively (Table 1). We estimated the genome size and heterozygosity of the preliminary assembly using GCE software, which yielded 881.46 Mb and 0.48%, respectively, indicating a relatively low complexity. Subsequently, we used PacBio subreads to generate a contig-level assembly of 867.42 Mb, with an N50 contig length of 1.61 Mb (Table 2). Further, a scaffolded genome was constructed using Hi-C sequencing, with an 867.50 Mb assembly size and an N50 scaffold length of 34.86 Mb (Table 2).
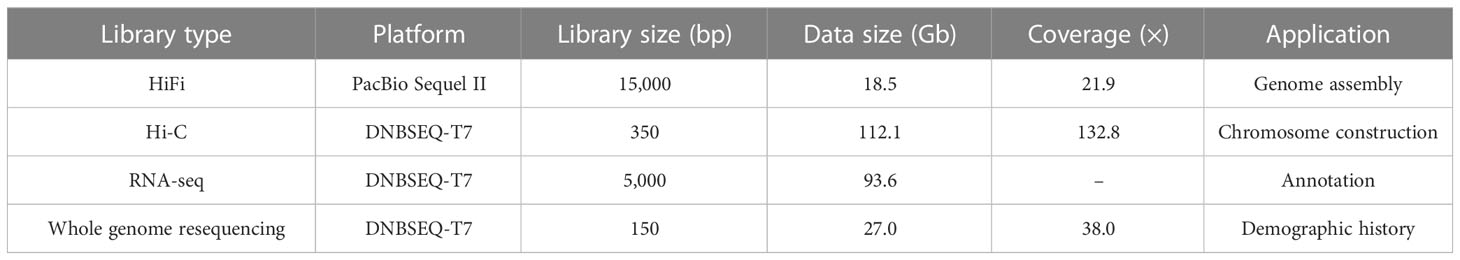
Table 1 Statistics of the sequencing data generated for O. cubicus genome assembly.
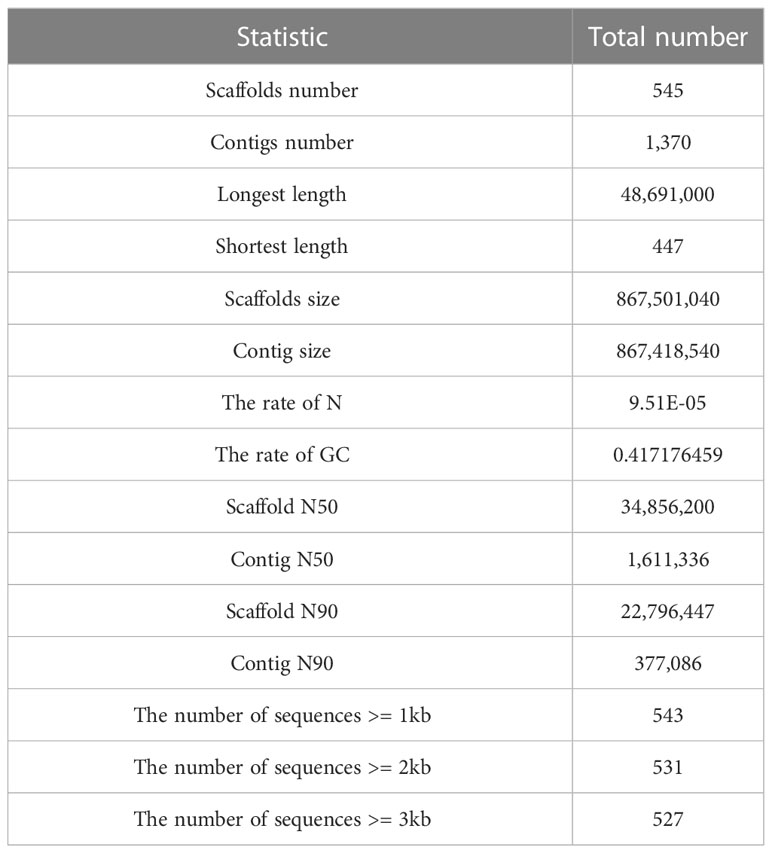
Table 2 Statistics of the O. cubicus genome assembly.
Twenty-five pseudo-chromosomes numbered according to size and covered 94.13% of the 867.50 Mb assembly (Figure 1A, Table 3). Twenty-five pseudo-chromosomes, numbered according to size and covering 94.13% of the 867.50 Mb assembly, were generated (Figure 1A, Table 3). The 25 pseudo-chromosomes were directly aligned to the 22 chromosomes of the fugu (T. rubripes) genome (Figures 1B, S1), indicating a highly contiguous assembly compared to other pufferfish genomes.
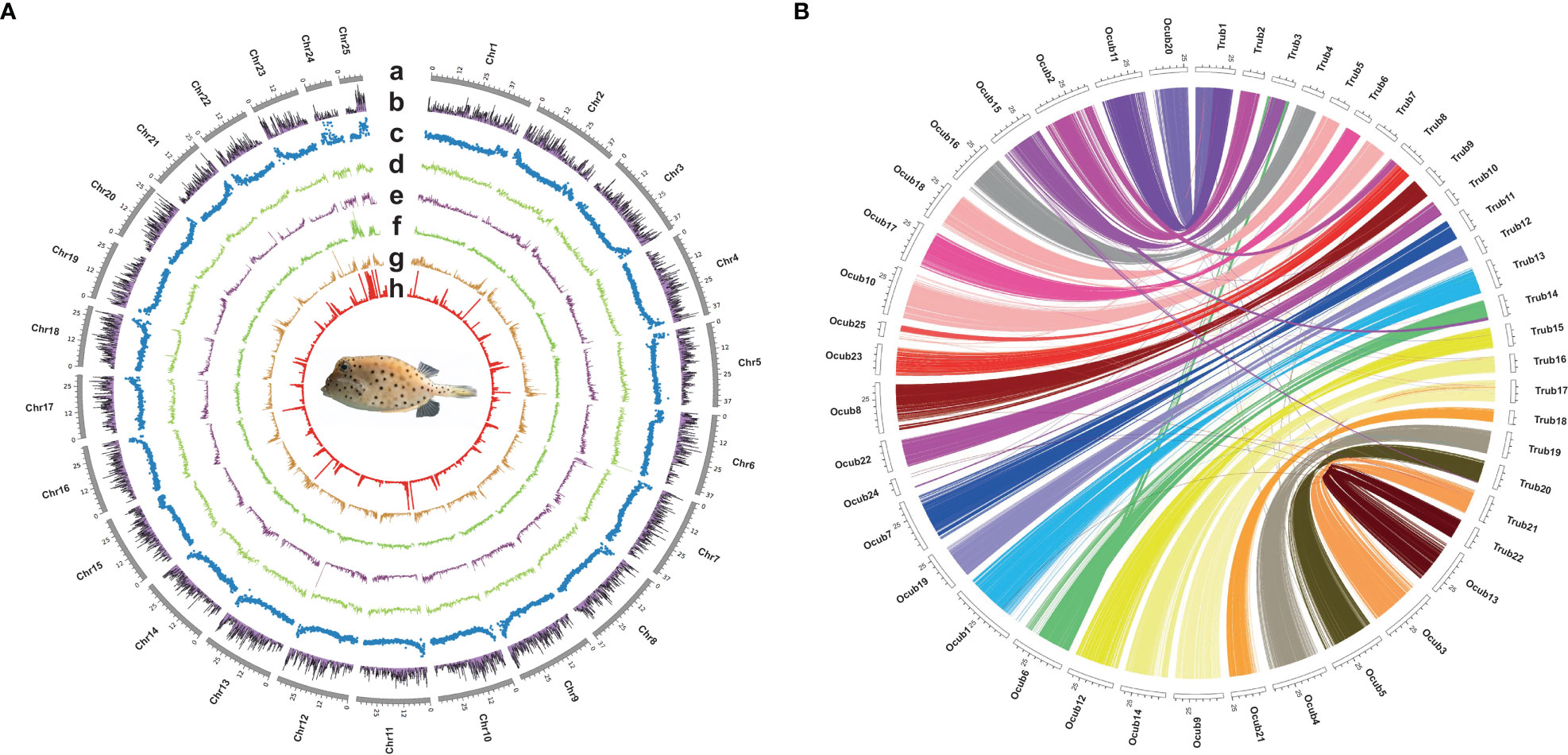
Figure 1 Circos plot basic characteristics of O. cubicus and T. rubripes genomes. (A) Circos view of the genome assembly of O. cubicus. (A) Tracks depict a circular representation of haplotype chromosomes in megabases (Mb). (B) GC content, (C) protein-coding genes density, (D–H) repeat sequence density (DNA, LINE, LTR, SINE and TRF, respectively); all statistics use the 100-kb window. (B) Collinear gene blocks between 25 pseudo-chromosomes O. cubicus and 22 chromosomes of T. rubripes.
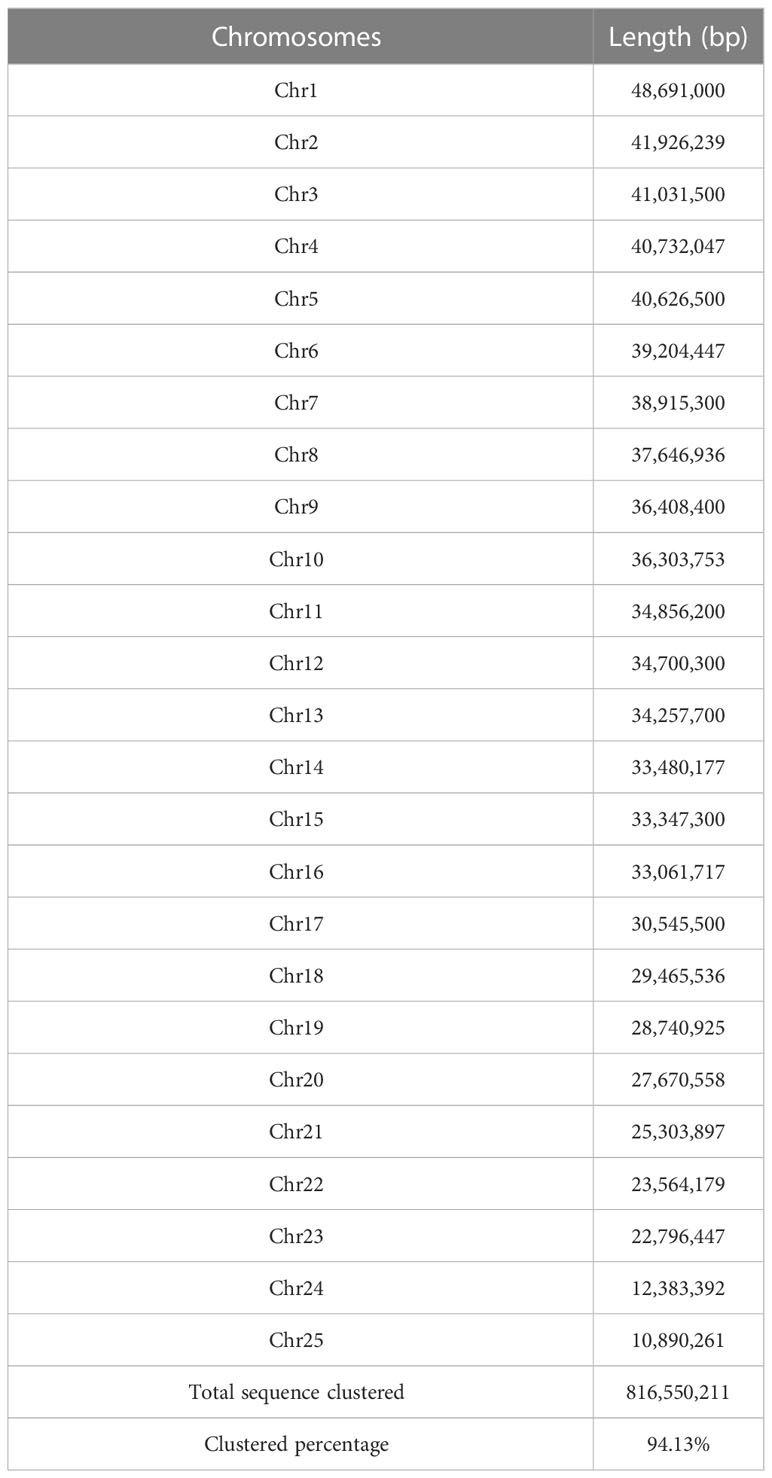
Table 3 Summary of assembled 25 chromosomes of O. cubicus.
Completeness of the assembled genome
To assess the completeness and accuracy of the assembly, we employed three approaches. First, BUSCO analysis revealed 97.8% complete actinopterygii BUSCOs, including 96.7% classified as “complete and single-copy”, 1.1% as “complete and duplicated”, 0.4% as “fragmented”, and 1.8% as “missing” (Figure 2B; Table S1). Second, 99.9% of the PE short-read sequences could be aligned to the genome sequences. Third, over 99.85% of the genomic regions had a coverage depth larger than 5. Collectively, these results suggest that we obtained a high-quality yellow boxfish genome resource.
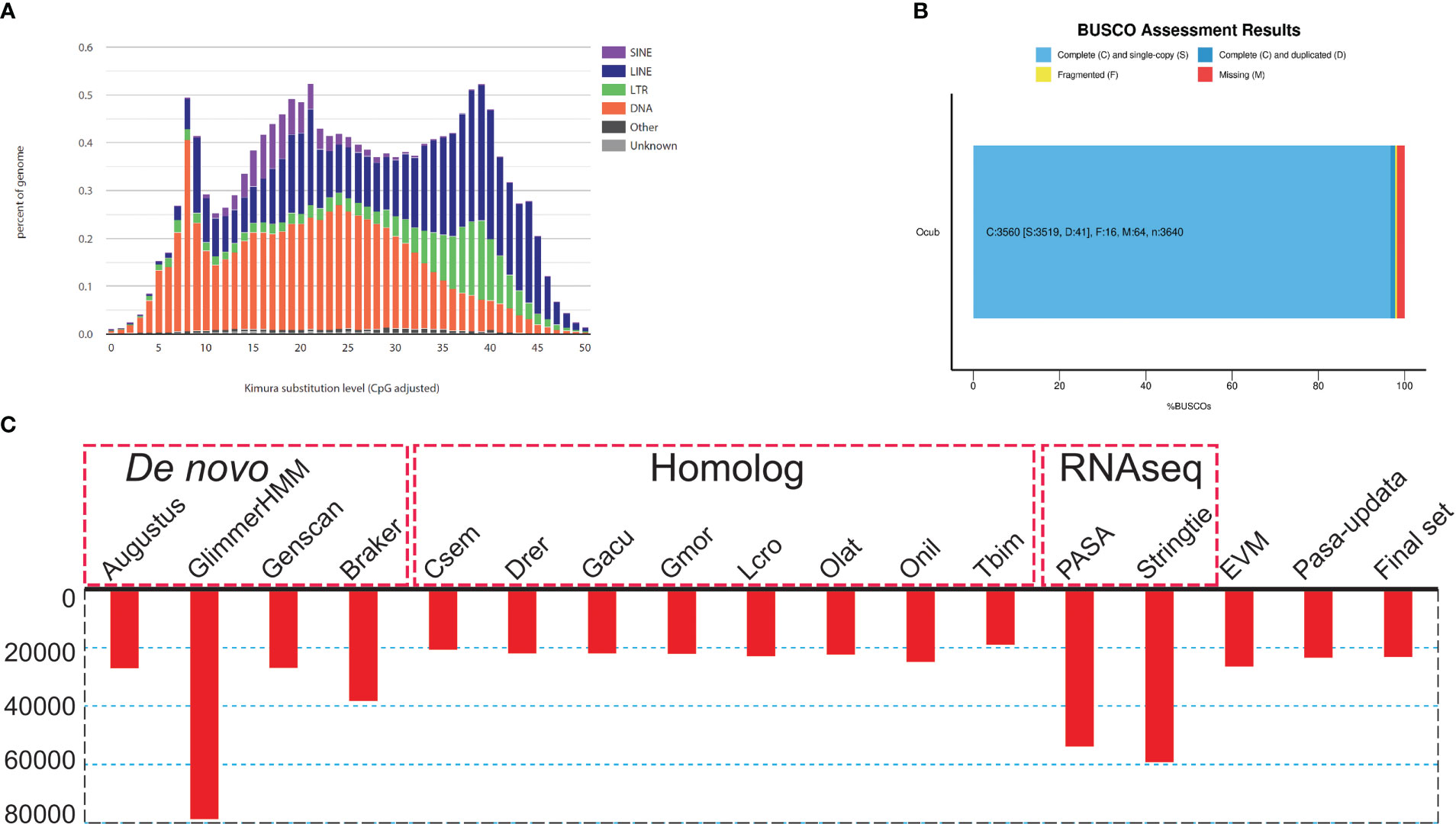
Figure 2 Genome assembly and annotation information. (A) Kimura distance-based copy divergence analysis of transposable elements in O. cubicus genome. Graphs represent genome coverage (Y-axis) for each type of TE (SINE, LINE, LTR retrotransposons and DNA transposons) in the genome analyzed, clustered to its corresponding consensus sequence according to Kimura distances (X-axis, K-value from 0 to 50). (B) BUSCO evaluation results, 97.3% classified as “complete and single-copy”, 1.3% as “complete and duplicated” and 0.2% as “fragmented” BUSCOs were present in the O. cubicus assembly genome. (C) Summary statistics of the identified protein-coding genes by using different strategies, including de novo prediction, homology-based prediction, and transcriptome-based prediction.
Repeat annotation and gene structure annotation
For repeat annotation, we found that 14.87% (128.92 Mb) of the yellow boxfish genome was composed of TEs (Figures 1A, 2A). The top three categories of repetitive elements were DNA transposons (6.16%), long interspersed nuclear elements (LINEs, 5.74%), and long terminal repeats (LTRs, 1.73%).
For the gene structure annotation, we employed a combination of de novo prediction, homology-based prediction, and transcript evidence, resulting in a total of 23,224 predicted protein-coding genes (Figure 2C; Table S2). The BUSCO analysis showed that yellow boxfish gene predictions recovered 98.6% of the highly conserved orthologues (97.3% classified as “complete and single-copy”, 1.3% as “complete and duplicated” and 0.2% as “fragmented”), while 1.2% of the conserved orthologs were missing from the gene prediction (Table S1). Of the 23,224 predicted genes, 88.5% (20,543), 88.8% (20,622) and 77.8% (18,058) could be functionally annotated using the NR, NT and Swissprot databases, respectively (Table S3). In total, 96.11% were successfully annotated with putative functions. Of these annotated genes, 90.1% had GO annotations and 78.3% could be assigned to KEGG pathways.
Genome expansion and contraction
Analysis of the 13 Clupeocephala fishes’ genomes revealed 22,269 orthogroups, 296,436 genes, 1,548 species-specific orthogroups, 3,150 single-copy orthogroups and 3704 single-copy orthogroups genes. Using the 3704 single-copy orthogroups genes shared by the 13 Clupeocephala fishes, a maximum-likelihood tree was constructed using RaxML. As shown in the phylogenetic tree, boxfish and sunfish had a closer evolutionary relationship and shared a common ancestor at 83.29 Mya (confidence interval: 65.58-100.24 Mya) (Figure 3A).
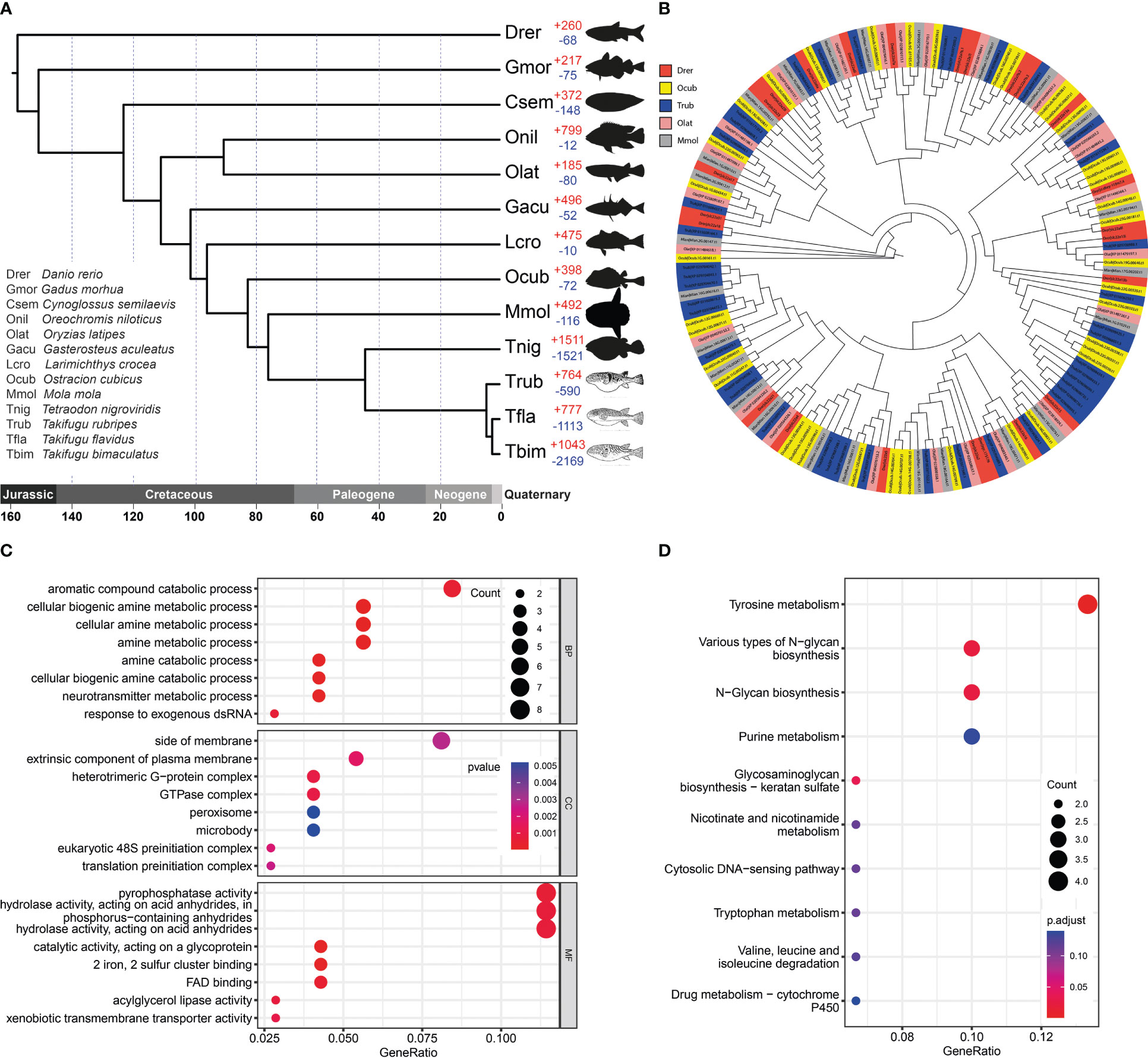
Figure 3 Gene family evolution and thesolute carrier family 22 gene clusters comparison. (A) Phylogenetic analysis of O. cubicus within the Clupeocephala lineage and gene family gain-and-loss analysis, including the number of gained gene families (+) and lost gene families (-). Abbreviations corresponding to the Latin name of species were listed in the bottom left corner. (B) The phylogeny of the solute carrier family 22 in ray finned fishes. Drer, Ocub, Trub, Olat and Mmol are presented in the zebrafish, yellow boxfish, fugu, medaka and sunfish, respectively. (C) GO enrichment analysis of expanded gene families. (D) KEGG enrichment analysis of expanded gene families.
A total of 398 significantly expanded and 72 significantly contracted gene families were identified and annotated in the yellow boxfish (Figure 3A). Gene Ontology (GO) enrichment analysis of the expanded gene families showed significant enrichment in the oxidation-reduction process and other processes related to aromatic and amine compound secretion, including the “aromatic compound catabolic process”, “cellular biogenic amine metabolic process”, “cellular amine metabolic process”, “amine metabolic process”, “amine catabolic process” and “cellular biogenic amine catabolic process” (Figure 3C; Table S4). KEGG enrichment analysis of the expanded gene families demonstrated that they were mainly assigned to the “Tyrosine secretion”, “Various types of N-glycan biosynthesis”, “N-Glycan biosynthesis” and “Purine secretion” pathways, which are related to secretion (Figure 3D; Table S5).
Evolution of the bone plates
For gene evolution analysis, we identified a set of 79 genes that are evolving significantly faster in the yellow boxfish lineage compared with other branches (Table S6). One of these is the extracellular calcium-sensing receptor (casr), which activates the BMP/Smad signaling pathway by increasing transcription of bmp2 and plays an essential role in fish bone development (Herberger and Loretz, 2013). In addition, using the branch-site model, we found 49 genes that contain positively selected sites specifically in the yellow boxfish (Table S7). Three of the genes identified (acsl4a, casr, tbx1) play a role in activating the BMP/Smad signaling pathway, which is crucial for regulating osteogenesis and promoting new bone formation. Additionally, one of the genes (keap1a) triggers the KEAP1/NRF2 signaling pathway, which primarily mediates keratinization. (Figure 4) (Ishitsuka et al., 2020).
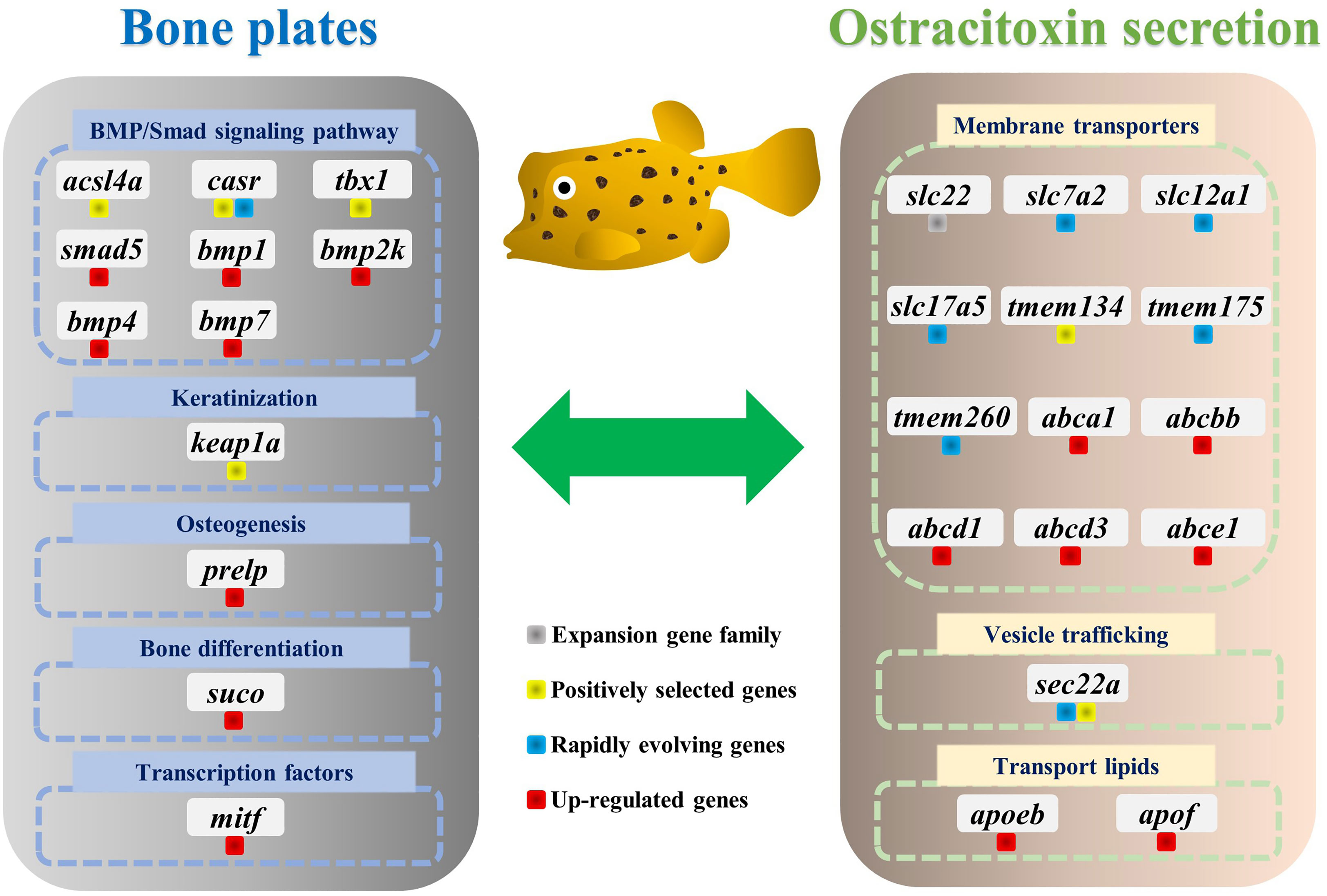
Figure 4 Genes involved in the evolution of bone plates and ostracitoxin secretion in the yellow boxfish. Genes are labeled with different colors to indicate expansion gene family, positively selected genes (PSGs), rapidly evolving genes and up-regulated genes in the skin in boxfish.
We analyzed differentially expressed genes (DEGs) between the skin of five boxfishes and five fugu. A total of 1608 DEGs were identified, 883 of which were up-regulated and the remaining 725 were down-regulated in the boxfish group (Figure 5A; Table S8). Gene Ontology (GO) enrichment analysis of the up-regulated genes revealed enrichment in biological processes potentially related to bone plates evolution, such as “embryonic organ morphogenesis”, “skeletal system development”, “embryonic cranial skeleton morphogenesis”, “skeletal myofibril assembly”, “embryonic skeletal system morphogenesis” (Figure 5B; Table S9). No enriched categories were identified in the KEGG enrichment analysis of the up-regulated genes. Notably, several genes involved in bone development were up-regulated in the skin in boxfish, including genes (bmp1, bmp2k, bmp4, bmp7, smad5), bone differentiation (suco), SLRP gene family (prelp) and transcription factors (mitf) (Figures 4, 5A; Table S8).
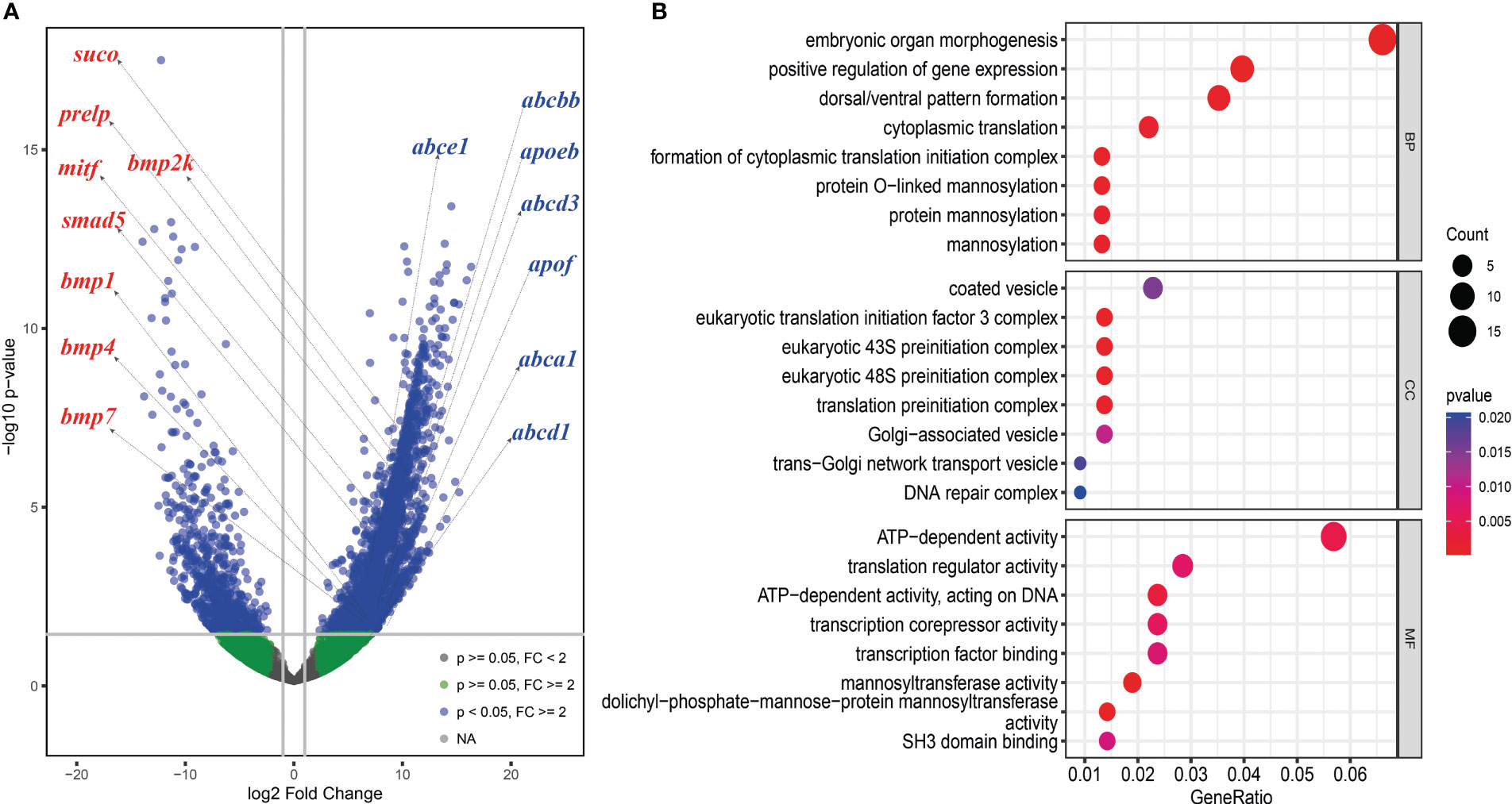
Figure 5 Differently expressed genes (DEGs) analysis between the skin of boxfish and fugu. (A) Volcano map of DEGs showing the threshold line of the screening criteria. Several up-regulated genes related to bone plates (left with red color) and ostracitoxin secretion (right with blue color) are shown on the map. (B) GO enrichment analysis of up-regulated genes.
Ostracitoxin secretion and genome characteristics
Gene family evolution analysis revealed a large expansion of the solute carrier family 22 (slc22) in the boxfish and fugu genomes, with 39 and 41 genes, respectively, compared to 25 in sunfish, 24 in medaka, and 24 in zebrafish (Figures 3B, 4). Gene Ontology (GO) enrichment analysis of the up-regulated genes revealed enrichment in biological processes potentially related to ostracitoxin secretion, such as “cytosolic transport”, “regulation of cellular amide metabolic process”, “regulation of translation”, “membrane lipid biosynthetic process” (Figure 5B; Table S9). In addition, several genes associated with membrane transporters were found to be under rapid evolution (slc7a2, slc12a1, slc17a5, tmem175, tmem260), positive selection (tmem134) and up-regulated in the skin in boxfish (abca1, abcbb, abcd1, abcd3, abce1) (Figure 4; Tables S6–S8). Furthermore, one of the vesicle trafficking genes (sec22a) was found to be under rapid evolution and positive selection (Figure 4; Tables S6, S7). Two apolipoprotein (APO) genes, which are involved in lipid transport, were identified as up-regulated genes (Figures 4, 5A; Table S8).
Discussion
In this study, we successfully assembled a chromosome-level yellow boxfish genome using HiFi and Hi-C sequencing data. The assembly size (867.50 Mb) corresponded well with the genome size estimation based on the analysis of k-mer distribution of MGI short reads (881.46 Mb). Twenty-five pseudo-chromosomes were easily distinguished and corresponded to the karyotype of this species (Arai, 1983). We observed that 98.6% of complete actinopterygii BUSCOs were present in the assembled genome and 99.9% of the MGI short reads could be aligned to the genome sequences, indicating that our genome assembly was of high completeness and accuracy. The assembled genome contained only 545 scaffolds, with a scaffold N50 of 34.86 Mb, which will be one of the most contiguous fishes’ genome assemblies with a high scaffold N50. Among the genome sequenced tetraodontiform fishes, due to the expansion of TEs the genome size of the yellow boxfish was the largest, when compared to Takifugu, Tetraodon, filefish (Thamnaconus septentrionalis) and sunfish (M. mola) (Aparicio et al., 2002; Jaillon et al., 2004; Gao et al., 2014; Pan et al., 2016; Bian et al., 2020). This result provided further evidence that DNA transposons and LINEs have been active and strongly contributed to the evolution of tetraodontiform’ genomes (Brainerd et al., 2001; Kang et al., 2020). In addition, we obtained a total of 23,224 protein-coding genes, which was comparable to the average number of genes (23,475) analyzed in 22 fish species (Lehmann et al., 2019). The yellow boxfish gene predictions recovered 98.6% of the highly conserved orthologues, suggesting the gene annotations of yellow boxfish were of high quality.
The dermal scutes of boxfishes are composed of a highly mineralized surface plate and a compliant collagen base (Yang et al., 2015), which should be due to the evolution of genes associated with bone formation or scale keratinization in teleost. In this study, we identified several genes that exhibited signatures of positive selection (acsl4a, casr, keap1a, tbx1) and rapid evolution (casr), which play a key role in the bone formation or scale keratinization of fish. Acsl4a, an LC-PUFA activating enzyme, is thought to modulate Bmp-Smad signaling by influencing the activity of Smad transcription factors (Miyares et al., 2013). Loss of acsl4a results in dorsalized embryos due to attenuated bone morphogenetic protein signaling (Miyares et al., 2013). We speculated that the positive selection of acsl4a might enhance skeletal formation through increasing the ability to inhibit critical inhibitors of Smad activity, such as p38 mitogen-activated protein kinase and the Akt-mediated inhibition of glycogen synthase kinase 3. Casr transcription causes increase transcription of bmp2 and further activation of BMP/Smad signaling pathway, and play an essential role in bone development in fish (Herberger and Loretz, 2013). Keap1a induced the KEAP1/NRF2 signaling pathway, which regulates primarily mediates keratinization (Ishitsuka et al., 2020). Tbx1 encodes a T-box transcription factor, modulated negatively Smad1-dependent transactivation by interfering with Smad1-Smad4 interaction, and regulates scale and bone development in zebrafish (Zhang et al., 2022b). Here, we speculated that the positively selection on the tbx1 may reduce binding with smad1, thus increasing the activity of BMP/Smad signaling pathway and facilitating bone plate formation in the yellow boxfish. The evidence of five BMP pathway genes (bmp1, bmp2k, bmp4, bmp7, smad5) with high transcription pattern in the skin in boxfish support this claim.
Our results showed that changes in gene transcription patterns also contributed to the evolution of bone plates in boxfish. Gene Ontology enrichment analysis of the up-regulated genes revealed several enrichments in biological processes potentially related to bone plates evolution. In addition, five BMP pathway genes and three bone formation related genes (suco, prelp, and mitf) were identified as up-regulated DEGs. Mutagenesis of suco has been shown to lead to failure of osteoblast maturation by decreasing the synthesis of type I collagen, and eventually catastrophic defects in skeletal development (Sohaskey et al., 2010). Prelp, a leucine-rich repeat protein present in connective tissue extracellular matrix, has been shown to reduce ALP activity, mineralization and transcription of osteogenic marker gene runx2 upon down-regulation (Li et al., 2016). Mitf, a transcription factor, has been shown to regulate hematopoietic stem cell differentiation into osteoclasts precursors (Amarasekara et al., 2018). These genes may have been rewired and regulated for the formation of bone plates of boxfish, suggesting a hypothesis that can be tested experimentally in the future.
In our study, comparative genomics analysis identified a set of genes belonging to two major groups of membrane transporters (the solute carriers (SLCs) and transmembrane protein family (TMEMs)), and vesicle trafficking (SECs), which may be involved in the enhanced secretion of ostracitoxin observed in boxfish compared to the outgroup. In detail, we revealed an expansion of the SLC22 gene family in the boxfish (with ostracitoxin secretion) and fugu (with TTX secretion) genomes, with three solute carrier (SLCs) family genes (slc7a2, slc12a1 and slc17a5) under positive selection or rapid evolution in the yellow boxfish genome. These genes encode transmembrane transporters for moving small molecule endogenous metabolites, drugs, and toxins (exogenous and endogenous) between tissues and interfacing body fluids in fishes (Saito, 2010), and play critical roles in TTX accumulation and translocation in T. rubripes (Zhang et al., 2022a). Furthermore, TMEMs is constituted by a large number of proteins that span the lipid bilayer, which are wildly expressed in various types of tissues and are supposed to act as channels to transport different substances (Beasley et al., 2021). Sec22a complex with SNARE and is thought to play a role in the ER-Golgi protein trafficking (Adnan et al., 2019), and may play a key role in the secretion of macromolecular materials in ostracitoxin secretion. We hypothesize that these genes have evolved to encode proteins or regulate the function of down-stream genes, leading to enhanced skin secretion in boxfish.
In addition, our transcriptome analysis showed that gene transcription pattern changes may also contribute to the ostracitoxin secretion. Five ATP-binding cassettes (ABCs) genes (abca1, abcbb, abcd1, abcd3, abce1) and two apolipoprotein (APOs) genes (apoeb, apof) were upregulated in the skin in the yellow boxfish. These transporters are involved in substrate translocation across biological membranes and have been previously reported to be upregulated in the blood and liver of T. rubripes, which are involved in tetrodotoxin (TTX) accumulation and translocation (Matsumoto et al., 2011; Luckenbach et al., 2014; Zhang et al., 2022a).
Conclusion
In the present study, we released a high-quality chromosome-level genome assembly of the yellow boxfish (O. cubicus). The final size of the genome assembly was 867.50 Mb, with an N50 scaffold length of 34.86 Mb. The assembled sequences were clustered into 25 pseudo-chromosomes by using Hi-C data and covered 94.13% of the total assembled sequences. A total of 23,224 protein-coding genes were predicted, with a BUSCO completeness of 98.6%, suggesting a high-quality genome annotation. This high-quality assembled genome and annotation information provides important information for exploring the evolution of complex traits such as bone plate formation and toxin secretion in boxfish. Positively selection or rapidly evolution was observed in genes related to scale and bone development (acsl4a, casr, keap1a, tbx1), and up-regulation of transcription was found in the skin of boxfish (bmp1, bmp2k, bmp4, bmp7, smad5, suco, prelp, mitf), likely associated with the evolution of bone plates. An expansion of the solute carrier family 22, three solute carrier family genes (slc7a2, slc12a1, slc17a5), two transmembrane protein family genes (tmem134, tmem175, tmem260) and one vesicle trafficking gene (sec22a) were also found to be under positive selection or rapid evolution. Five ATP-binding cassettes genes (abca1, abcbb, abcd1, abcd3, abce1) and two apolipoprotein genes (apoeb, apof) were up-regulated in the skin of boxfish, which may have contributed to ostracitoxin secretion in the yellow boxfish.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: China National GeneBank DataBase (CNGBdb) with accession number CNP0004153.
Ethics statement
The animal study was reviewed and approved by Committee for Animal Experiments of the Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou).
Author contributions
FW and WZ conceived and managed the project. SW, HF, ZZ, WG and ZP collected the sequencing samples. SW performed the analysis and wrote the manuscript. FW, SW and WZ revised the manuscript. All authors reviewed and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Ministry of Science and Technology of China (2021YFF0502802), National Natural Science Foundation of China (32222014), Science and Technology Department of Guangdong Province (2021QN02H103), the PI Project of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2020GD0804, GML2022GD0804) and Postdoctoral Research Foundation of Guangzhou (GML2022BH0905).
Acknowledgments
We express sincere thanks to Xin Du, Xin Huang, Lin Yang and Dengfeng Guan for their technical assistance. We are also grateful to Mingpan Huang for their kind assistance in the samples collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1170704/full#supplementary-material
References
Adnan M., Islam W., Zhang J., Zheng W., Lu G.-D. (2019). Diverse role of SNARE protein Sec22 in vesicle trafficking, membrane fusion, and autophagy. Cells 8, 337. doi: 10.3390/cells8040337
Alfaro M. E., Santini F., Brock C. D. (2007). Do reefs drive diversification in marine teleosts? evidence from the pufferfish and their allies (Order tetraodontiformes). Evolution: Int. J. Organic Evol. 61, 2104–2126. doi: 10.1111/j.1558-5646.2007.00182.x
Amarasekara D. S., Yun H., Kim S., Lee N., Kim H., Rho J. (2018). Regulation of osteoclast differentiation by cytokine networks. Immune Netw. 18:e8. doi: 10.4110/in.2018.18.e8
Aparicio S., Chapman J., Stupka E., Putnam N., Chia J., Dehal P., et al. (2002). Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297, 1301–1310. doi: 10.1126/science.1072104
Arai R. (1983). Karyological and osteological approach to phylogenetic systematics of tetraodontiform fishes. Bull. Natn. Sci. Mus. Tokyo Ser. A. 9, 175–210.
Bairoch A., Apweiler R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45
Bao W., Kojima K. K., Kohany O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 1–6. doi: 10.1186/s13100-015-0041-9
Beasley H. K., Rodman T. A., Collins G. V., Hinton A. Jr., Exil V. (2021). TMEM135 is a novel regulator of mitochondrial dynamics and physiology with implications for human health conditions. Cells 10, 1750. doi: 10.3390/cells10071750
Benson G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Benton M. J., Donoghue P. C. (2007). Paleontological evidence to date the tree of life. Mol. Biol. Evol. 24, 26–53. doi: 10.1093/molbev/msl150
Bian L., Li F., Ge J., Wang P., Chang Q., Zhang S., et al. (2020). Chromosome-level genome assembly of the greenfin horse-faced filefish (Thamnaconus septentrionalis) using Oxford nanopore PromethION sequencing and Hi-c technology. Mol. Ecol. Resour. 20, 1069–1079. doi: 10.1111/1755-0998.13183
Brainerd E. L., Slutz S. S., Hall E. K., Phillis R. W. (2001). Patterns of genome size evolution in tetraodontiform fishes. Evolution 55, 2363–2368. doi: 10.1111/j.0014-3820.2001.tb00750.x
Burge C. B., Karlin S. (1998). Finding the genes in genomic DNA. Curr. Opin. Struct. Biol. 8, 346–354. doi: 10.1016/S0959-440X(98)80069-9
Castresana J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen N. (2004). Using repeat masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 5, 4.10. doi: 10.1002/0471250953.bi0410s05
Chen L., Qiu Q., Jiang Y., Wang K., Lin Z., Li Z., et al. (2019). Large-Scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 364, eaav6202. doi: 10.1126/science.aav6202
Cheng H., Concepcion G. T., Feng X., Zhang H., Li H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., Depristo M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
De Bie T., Cristianini N., Demuth J. P., Hahn M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Dornburg A., Sidlauskas B., Santini F., Sorenson L., Near T. J., Alfaro M. E. (2011). The influence of an innovative locomotor strategy on the phenotypic diversification of triggerfish (Family: Balistidae). Evolution 65, 1912–1926. doi: 10.1111/j.1558-5646.2011.01275.x
Dudchenko O., Batra S. S., Omer A. D., Nyquist S. K., Hoeger M., Durand N. C., et al. (2017). De novo assembly of the Aedes aegypti genome using Hi-c yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Durand N. C., Shamim M. S., Machol I., Rao S. S., Huntley M. H., Lander E. S., et al. (2016). Juicer provides a one-click system for analyzing loop-resolution Hi-c experiments. Cell Syst. 3, 95–98. doi: 10.1016/j.cels.2016.07.002
Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14. doi: 10.1186/s13059-019-1832-y
Fan H., Wu Q., Wei F., Yang F., Ng B. L., Hu Y. (2019). Chromosome-level genome assembly for giant panda provides novel insights into Carnivora chromosome evolution. Genome Biol. 20, 1–12. doi: 10.1186/s13059-019-1889-7
Flynn J. M., Hubley R., Goubert C., Rosen J., Clark A. G., Feschotte C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Froese R., Pauly D. (2023) FishBase. Available at: www.fishbase.org.
Gao Y., Gao Q., Zhang H., Wang L., Zhang F., Yang C., et al. (2014). Draft sequencing and analysis of the genome of pufferfish Takifugu flavidus. DNA Res. 21, 627–637. doi: 10.1093/dnares/dsu025
Gordon M. S., Hove J. R., Webb P. W., Weihs D. (2000). Boxfishes as unusually well-controlled autonomous underwater vehicles. Physiol. Biochem. Zool. 73, 663–671. doi: 10.1086/318098
Guan D., Mccarthy S. A., Wood J., Howe K., Wang Y., Durbin R. (2020). Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898. doi: 10.1093/bioinformatics/btaa025
Haas B. J., Salzberg S. L., Zhu W., Pertea M., Allen J. E., Orvis J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, 1–22. doi: 10.1186/gb-2008-9-1-r7
Harris R. S. (2007). Improved pairwise alignment of genomic DNA. Pennsylvania State University, University Park, Pennsylvania.
Herberger A. L., Loretz C. A. (2013). Morpholino oligonucleotide knockdown of the extracellular calcium-sensing receptor impairs early skeletal development in zebrafish. Comp. Biochem. Physiol. Part A: Mol. Integr. Physiol. 166, 470–481. doi: 10.1016/j.cbpa.2013.07.027
Hu Y., Wang X., Xu Y., Yang H., Tong Z., Tian R., et al. (2023). Molecular mechanisms of adaptive evolution in wild animals and plants. Sci. China Life Sci. 66, 453–495. doi: 10.1007/s11427-022-2233-x
Hu Y., Wu Q., Ma S., Ma T., Shan L., Wang X., et al. (2017). Comparative genomics reveals convergent evolution between the bamboo-eating giant and red pandas. Proc. Natl. Acad. Sci. 114, 1081–1086. doi: 10.1073/pnas.1613870114
Ishitsuka Y., Ogawa T., Roop D. (2020). The KEAP1/NRF2 signaling pathway in keratinization. Antioxidants 9, 751. doi: 10.3390/antiox9080751
Jaillon O., Aury J., Brunet F., Petit J., Stangethomann N., Mauceli E., et al. (2004). Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431, 946–957. doi: 10.1038/nature03025
Kang S., Kim J. H., Jo E., Lee S. J., Jung J., Kim B. M., et al. (2020). Chromosomal-level assembly of Takifugu obscurus (Abe 1949) genome using third-generation DNA sequencing and Hi-c analysis. Mol. Ecol. Resour. 20, 520–530. doi: 10.1111/1755-0998.13132
Kim D., Langmead B., Salzberg S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim D., Paggi J. M., Park C., Bennett C., Salzberg S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lehmann R., Lightfoot D. J., Schunter C., Michell C. T., Ohyanagi H., Mineta K., et al. (2019). Finding nemo’s genes: A chromosome-scale reference assembly of the genome of the orange clownfish Amphiprion percula. Mol. Ecol. Resour. 19, 570–585. doi: 10.1111/1755-0998.12939
Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997.
Li H., Cui Y., Luan J., Zhang X., Li C., Zhou X., et al. (2016). PRELP (proline/arginine-rich end leucine-rich repeat protein) promotes osteoblastic differentiation of preosteoblastic MC3T3-E1 cells by regulating the β-catenin pathway. Biochem. Biophys. Res. Commun. 470, 558–562. doi: 10.1016/j.bbrc.2016.01.106
Liu B., Shi Y., Yuan J., Hu X., Zhang H., Li N., et al. (2013). Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv, 1308.2012. doi: 10.48550/arXiv.1308.2012
Luckenbach T., Fischer S., Sturm A. (2014). Current advances on ABC drug transporters in fish. Comp. Biochem. Physiol. Part C: Toxicol. Pharmacol. 165, 28–52. doi: 10.1016/j.cbpc.2014.05.002
Majoros W. H., Salzberg S. L. (2004). An empirical analysis of training protocols for probabilistic gene finders. BMC Bioinf. 5, 1–12. doi: 10.1186/1471-2105-5-206
Marçais G., Kingsford C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Matsumoto T., Ishizaki S., Nagashima Y. (2011). Differential gene expression profile in the liver of the marine puffer fish Takifugu rubripes induced by intramuscular administration of tetrodotoxin. Toxicon 57, 304–310. doi: 10.1016/j.toxicon.2010.12.007
Miyares R. L., Stein C., Renisch B., Anderson J. L., Hammerschmidt M., Farber S. A. (2013). Long-chain acyl-CoA synthetase 4A regulates smad activity and dorsoventral patterning in the zebrafish embryo. Dev. Cell 27, 635–647. doi: 10.1016/j.devcel.2013.11.011
Noguchi T., Arakawa O., Takatani T. (2006). TTX accumulation in pufferfish. Comp. Biochem. Physiol. Part D: Genomics Proteomics 1, 145–152. doi: 10.1016/j.cbd.2005.10.006
Pan H., Yu H., Ravi V., Li C., Lee A. P., Lian M. M., et al. (2016). The genome of the largest bony fish, ocean sunfish (Mola mola), provides insights into its fast growth rate. Gigascience 5, 36. doi: 10.1186/s13742-016-0144-3
Pertea M., Pertea G. M., Antonescu C. M., Chang T.-C., Mendell J. T., Salzberg S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Ranwez V., Harispe S., Delsuc F., Douzery E. J. (2011). MACSE: Multiple alignment of coding squences accounting for frameshifts and stop codons. PloS One 6, e22594. doi: 10.1371/journal.pone.0022594
Robinson M. D., Mccarthy D. J., Smyth G. K. (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Saito H. (2010). Pathophysiological regulation of renal SLC22A organic ion transporters in acute kidney injury: pharmacological and toxicological implications. Pharmacol. Ther. 125, 79–91. doi: 10.1016/j.pharmthera.2009.09.008
Santini F., Tyler J. C. (2003). A phylogeny of the families of fossil and extant tetraodontiform fishes (Acanthomorpha, tetraodontiformes), upper Cretaceous to recent. Zool. J. Linn. Soc. 139, 565–617. doi: 10.1111/j.1096-3642.2003.00088.x
Seppey M., Manni M., Zdobnov E. M. (2019). “BUSCO: assessing genome assembly and annotation completeness,” in Gene prediction, ed. M. Kollmar (New York, NY: Springer), 227–245. doi: 10.1007/978-1-4939-9173-0_14
Sohaskey M. L., Jiang Y., Zhao J. J., Mohr A., Roemer F., Harland R. M. (2010). Osteopotentia regulates osteoblast maturation, bone formation, and skeletal integrity in mice. J. Cell Biol. 189, 511–525. doi: 10.1083/jcb.201003006
Stamatakis A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Stanke M., Diekhans M., Baertsch R., Haussler D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Thomson D. A. (1964). Ostracitoxin: an ichthyotoxic stress secretion of the boxfish, Ostracion lentiginosus. Science 146, 244–245. doi: 10.1126/science.146.3641.244
Thomson D. A. (1969). Toxic stress secretions of the boxfish Ostracion meleagris. Copeia 2, 335–352. doi: 10.2307/1442084
Tyler J. C., Santini F. (2002). Review and reconstructions of the tetraodontiform fishes from the Eocene of Monte bolca, Italy, with comments on related tertiary taxa. Studi e Ricerche sui giacimenti Terziari di Bolca 9, 47–119.
Wainwright P. C., Turingan R. G. (1997). Evolution of pufferfish inflation behavior. Evolution 51, 506–518. doi: 10.2307/2411123
Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9, e112963. doi: 10.1371/journal.pone.0112963
Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang W., Naleway S. E., Porter M. M., Meyers M. A., Mckittrick J. (2015). The armored carapace of the boxfish. Acta Biomater. 23, 1–10. doi: 10.1016/j.actbio.2015.05.024
Yu G., Wang L., Han Y., He Q. (2012). clusterProfiler: an r package for comparing biological themes among gene clusters. Omics: J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
Zhang Z., Ji F., Jiang S., Wu Z., Xu Q. (2022b). Scale development-related genes identified by transcriptome analysis. Fishes 7, 64. doi: 10.3390/fishes7020064
Zhang H., Li P., Wu B., Hou J., Ren J., Zhu Y., et al. (2022a). Transcriptomic analysis reveals the genes involved in tetrodotoxin (TTX) accumulation, translocation, and detoxification in the pufferfish Takifugu rubripes. Chemosphere 303, 134962. doi: 10.1016/j.chemosphere.2022.134962
Glossary
Keywords: boxfish, genome assembly, anti-predation traits, bony carapace, ostracitoxin secretion
Citation: Wei S, Zhou W, Fan H, Zhang Z, Guo W, Peng Z and Wei F (2023) Chromosome-level genome assembly of the yellow boxfish (Ostracion cubicus) provides insights into the evolution of bone plates and ostracitoxin secretion. Front. Mar. Sci. 10:1170704. doi: 10.3389/fmars.2023.1170704
Received: 21 February 2023; Accepted: 27 March 2023;
Published: 06 April 2023.
Edited by:
Taewoo Ryu, Okinawa Institute of Science and Technology Graduate University, JapanReviewed by:
Sonia Andrade, University of São Paulo, BrazilQiong Shi, BGI Academy of Marine Sciences, China
Copyright © 2023 Wei, Zhou, Fan, Zhang, Guo, Peng and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenliang Zhou, emhvdXdsQGdtbGFiLmFjLmNu