 Jaeyoung Yu
Jaeyoung Yu Marika Kawahisa
Marika Kawahisa Abdullah Adham Zulmajdi
Abdullah Adham Zulmajdi Tetsushi Mori
Tetsushi Mori- Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, Koganei-shi, Japan
Marine bacteria have been targeted by industry and pharmaceutics as genetic resources for highly active enzymes or novel lead compounds. Although numerous techniques have been introduced to isolate useful bacteria from the environment, we are still highly dependent on the conventional direct cultivation method to attain pure cultures. However, efficient bacterial isolation is hindered by several factors, including the presence of impurities. In this work, to demonstrate the significance of removing impurities and their impact on bacterial isolation, we employed two approaches: dielectrophoresis (DEP) and fluorescent D-amino acids (FDAA). We successfully attained clean bacterial fractions applicable for downstream processing using these approaches, uniquely designed to identify bacteria based on their characteristics and features. The diversity of bacteria attained by both approaches was investigated using 16S rRNA sequencing and compared to that attained by the standard differential centrifugation method. In addition, the viability of the isolates was also determined via direct cultivation. As a result, the separation of bacteria from impurities allowed for the identification of novel and useful bacteria unique to each approach. Successful cultivation also suggested that both approaches were applicable for attaining viable bacteria. In conclusion, removing impurities to attain clean bacterial fractions promotes the isolation of novel bacteria and thus could aid in the successful isolation of useful bacteria within complex environmental samples.
1 Introduction
The ocean covers over 70% of the Earth’s surface and harbors numerous unique habitats distinctly different from terrestrial architecture. Marine bacteria have been discovered and isolated from these unique habitats, ranging from open water environments, seabed sediments, hydrothermal vents, and even as symbionts from specified marine organisms (Di Donato et al., 2018). Their adaptation to these habitats allows them to exhibit various functions and characteristics unique from their terrestrial counterparts (Alex et al., 2023). Due to these features, marine bacteria have been frequently targeted by industry and pharmaceutics as genetic resources for highly active enzymes or novel lead compounds (Lindequist, 2016). Recent advancements in sequencing technologies have also allowed for more comprehensive bacterial diversity analyses, leading to the discovery of countless novel bacteria that have not been characterized or exploited (Escudeiro et al., 2022). Thus, the demand for more useful marine bacteria is much higher now and needs to be addressed.
For the exploitation of useful bacteria, it is important for the target bacteria to be cultivated since it allows users to metabolically engineer and optimize them for the efficient and mass production of important compounds (Jung et al., 2021). Over the years, much effort has been placed into establishing techniques to isolate novel and useful bacteria from the environment (Rodrigues and de Carvalho, 2022). In addition to the conventional Fluorescence In Situ Hybridization (FISH) approach, where specific bacteria can be fluorescently stained, identified, and isolated using flow cytometry or micromanipulation, recent efforts for large-scale screening and high-throughput systems using microfluidics and in combination with single-cell analytical techniques have allowed for a comprehensive understanding of bacterial communities (Moon et al., 2023), construction of near-complete draft genomes of novel and unique bacteria (Kogawa et al., 2018), and elucidation of highly efficient enzymes applicable for downstream processes (Baysoy et al., 2023). However, originally designed for nucleic acid analyses, most of these techniques have incorporated cell fixation or lysis, resulting in many isolated bacteria to be inapplicable for subsequent cultivation.
Thus, there is still a high dependency on conventional direct cultivation in which researchers try to establish culture media and mimic the environmental conditions in standard laboratory settings. Nevertheless, this approach is tedious and time-consuming, and, in many cases, since cultivation conditions are hard to replicate, many bacteria still remain uncultivable. Factors that have been speculated to majorly contribute to this difficulty include undefined growth conditions (Kapinusova et al., 2023) or the dependency of the bacteria on specific scaffolds, metabolites, and nutrients only available from its host or surroundings (Zengler and Zaramela, 2018). Thus, to promote the cultivation of environmental bacteria, an approach to cultivate bacteria in their original environment has been introduced (Chaudhary et al., 2019). Co-cultivation has also been introduced as an alternative approach based on the necessity of some bacteria to depend on other bacteria within their community (Watterson et al., 2020). Although these approaches have been successful in the cultivation of several bacteria, it is still challenging to cultivate environmental bacteria as pure cultures. However, among these factors, one other possible factor that is often missed and could provide a large impact to the direct cultivation of bacteria is the presence of impurities within the bacterial fractions prepared from environmental samples. Environmental samples can contain various organic and inorganic materials, including soil particles, organic debris, and minerals, creating complex matrices that may interfere with bacterial growth (Wilpiszeski et al., 2019) or particles that have a similar size or density to bacteria, making it challenging to distinguish between them (Fatoyinbo et al., 2014).
Currently, the preparation of the bacterial fractions for use with the bacterial isolation techniques is generally performed using conventional differential centrifugation or filtration, but these methods can still result in fractions that contain commendable amounts of impurities (Vaishampayan et al., 2010; Nnadozie et al., 2015). In this work, to demonstrate the significance of removing impurities and their impact on bacterial isolation, we employed two approaches, dielectrophoresis (DEP) and fluorescent D-amino acids (FDAA), to attain clean bacterial fractions from environmental samples. Using the marine sponge Halichondria okadai as a model, clean bacterial fractions were attained, and the distribution and diversity of bacterial populations were determined using nanopore sequencing and compared with differential centrifugation. Finally, the viability of bacteria attained using these approaches was evaluated using direct cultivation. Through our findings, we believe that DEP and FDAA can be applied to prepare clean bacterial fractions for the further exploration and investigation of diverse bacteria from complex environmental samples.
2 Materials and methods
2.1 Sample collection and preparation of the bacterial fraction
Bacteria associated with the H. okadai were used as the sample. H. okadai, attached to large rocks found along the shoreline of Mabori Beach (35.263101° N, 139.721886° E), Kanagawa Prefecture, Japan, was collected at low tide. The collected H. okadai specimens were submerged in prechilled filter-sterilized seawater (FSW) and quickly transported to the nearest laboratory. The samples were processed approximately one hour after collection. To begin with, the samples were rinsed 2 times with FSW to remove surface contaminants. The rinsed specimens were cut into small pieces averaging 2 cm in size and placed in 25 mL centrifuge tubes. FSW was added to the sponge pieces, and they were further diced with a scalpel. The diced samples were vortexed at maximum speed for 30 s, and debris was settled for 15 min to allow bacteria to dissociate from the sponge tissues. Adding FSW, vortexing, and settling the samples were repeated 3 times. The supernatant containing the dissociated bacteria was collected after each settling and filtered through a 32 μm nylon mesh. A 32 µm mesh was used to compensate for the possible presence of large bacteria within the sample. The filtered flowthrough was collected in 50 mL centrifuge tubes and initially centrifuged 2 times at 100 × g for 10 min to remove any carried over debris. Upon centrifugation, the final supernatant, comprised of the filtered dissociated bacteria, was collected in new 50 mL centrifuge tubes and centrifuged 2 times at 7,500 × g for 10 min, in which bacterial pellets attained at each centrifugation step were collected. The bacterial pellets were resuspended in FSW, pooled, and adjusted to OD660 = 1.0 to attain the final H. okadai bacterial fraction. The final H. okadai bacterial fraction was kept at 4°C until use and was used within one week upon collection.
2.2 Attaining clean bacterial fractions via DEP
500 µL of the final H. okadai bacterial fraction was transferred to a 1.5 mL centrifuge tube, centrifuged at 7,500 × g for 5 min, and resuspended in 1 mL of ELESTA-PBS (EP) buffer at room temperature (RT, 23°C) to set the conductivity of the sample to 100 μS/cm. EP buffer was prepared by mixing ELESTA buffer (ELB100N; AFI Corporation, Kyoto, Japan) with 1 × Phosphate-Buffered Saline (PBS; Thermo Fisher Scientific, Tokyo, Japan) at a ratio of 200:1. Resuspension was repeated 3 times to ensure complete replacement of the final H. okadai bacterial fraction in EP buffer. Solution conductivity was measured with a LAQUAtwin EC-33 conductivity meter (HORIBA Advanced Techno Co. Ltd., Tokyo, Japan). The bacterial capture via DEP was performed using PixeeMo® (AFI Corporation) based on the manufacturer’s guidelines and recommendations. Briefly, microchannels within the microchip (Supplementary Figure 1; D32L210; AFI Corporation) designed for the device were first equilibrated with EP buffer at a flow rate of 60 µL/min for 5 min prior to use. Subsequently, the prepared final H. okadai bacterial fraction was loaded into the inlet of the microchip at a flow rate of 8 μL/min, and a frequency of 3,000 kHz and 20 Vpp of voltage were applied to capture viable bacteria at electrodes located within the entrapment region. The first flowthrough (waste fluid), comprised mostly of debris and non-viable bacteria, was collected from the outlet to evaluate bacterial capture efficiency. Upon bacteria capture, the frequency and voltage were turned off, and bacteria captured on the electrodes were released and collected as the second flowthrough (DEP-captured bacteria sample) at the outlet by loading freshly prepared EP buffer through the microchip at 60 μL/min for 10 min. To ensure efficient bacteria capture, the waste fluid was re-loaded into the microchip, and the procedure for collecting the DEP-captured bacteria sample was repeated. The collected DEP-captured bacteria sample was centrifuged at 7,500 × g for 5 min, resuspended in 500 µL of 1 × PBS, subsequently checked for viability, and processed for nanopore sequencing. The efficiency of bacterial capture and viability of the DEP-captured bacteria sample were evaluated using live/dead bacterial staining and subsequent fluorescence microscopy. The results were compared with the final H. okadai bacterial fraction and waste fluid.
2.3 Bacterial staining with FDAA
In this work, we used sBADA (green sulfonated BODIPY-FL 3-amino-D-alanine; maximum λex/em = 490/510 nm; bio-techne, Minneapolis, MN, USA) as the FDAA. The sBADA stock solution was prepared by dissolving sBADA in dimethyl sulfoxide (DMSO; FUJIFILM Wako Pure Chemical Co., Osaka, Japan) upon arrival and stored as 10 mM aliquots at -20°C. 100 μM working solutions diluted in 1 × PBS were prepared immediately prior to use. Staining of bacteria using sBADA was performed based on a previously published protocol (Hsu et al., 2017). Four individual 1.5 mL centrifuge tubes, each aliquoted with 500 μL of the final H. okadai bacterial fraction, were prepared, and each sample was washed 2 times by centrifugation at 7,500 × g for 5 min and resuspension in 1 × PBS. After washing, each sample was centrifuged at 7,500 × g for 5 min, resuspended in 500 μL of 100 μM sBADA solution, and incubated for 5, 10, 15, and 24 h at 25°C, respectively. sBADA stained cells were collected at the designated time points, washed 3 times by centrifugation at 7,500 × g for 5 min and resuspension in cold 1 × PBS (Kuru et al., 2015), and immediately observed using fluorescence microscopy. sBADA uptake efficiency was determined by flow cytometry using the cells incubated for 24 h (FDAA-stained bacteria sample).
For bacterial diversity analysis using nanopore sequencing, the FDAA-stained bacteria sample was sorted using flow cytometry into 15 mL centrifuge tubes containing 5 mL of 1 × PBS and collected by membrane filtration onto an Omnipore membrane filter (0.2 µm pore size; 47 mm diameter; Merck, Tokyo, Japan). A membrane filter containing the FDAA-stained bacteria sample was stored at -20°C until use. For direct cultivation, the FDAA-stained bacteria sample was similarly sorted using flow cytometry into 15 mL centrifuge tubes containing 5 mL of 1 × PBS, collected by centrifugation at 7,500 × g for 5 min, and resuspended in 500 μL of 1 × PBS.
2.4 Nanopore sequencing and data analysis
The membrane filter containing the FDAA-stained bacteria sample stored at -20°C was thawed at RT. Using sterilized surgical scissors (autoclaved prior to use), the membrane filter was cut to small pieces, approximately 1 × 1 mm in size, for bacterial genomic DNA extraction. For comparison, 500 μL of the final H. okadai bacterial fraction was centrifuged at 7,500 × g for 5 min (centrifuged bacteria sample) and used as a control. Genomic DNAs from 3 samples, the centrifuged bacteria sample, DEP-captured bacteria sample, and membrane filter-bound FDAA-stained bacteria sample, were extracted using the DNeasy UltraClean Microbial Kit (QIAGEN, Tokyo, Japan). A 16S rRNA gene library was prepared using the 16S Barcoding Kit (SQK-16S024; Oxford Nanopore Technologies (ONT), Oxford, UK), and sequencing was carried out on a MinION Mk1B (ONT) with a FLO-MIN106D flow cell (ONT). Genome extraction and nanopore sequencing were performed according to the manufacturer’s protocol. Sequencing was stopped when sufficient reads were obtained. The raw data was base-called with Guppy v6.4.6 (ONT) using the high-accuracy mode (Zulmajdi et al., 2022). The quality score and length of the sequenced reads were obtained using NanoPlot v1.41.0 and trimmed (Q-score, >10; length, 1,200 – 1,800 bp) using NanoFilt v2.8.0 (De Coster et al., 2018). The FASTQ file was converted to a FASTA file using seqkit v2.3.0 (Shen et al., 2016). The finalized reads were determined using BLASTn v2.13.0 (Camacho et al., 2009) and the NCBI 16S rRNA gene database (O'Leary et al., 2016). MEGAN v6.24.23 was used to generate the bacterial distribution data (Huson et al., 2016). Upon evaluation of the bacterial diversity and abundance within our samples, bacteria showing distributions below 0.1% were considered contaminants and removed from subsequent analysis. The nanopore sequencing raw data has been submitted to the DDBJ Sequence Read Archive (DRA) with the accession number DRA017584.
2.5 Bacterial viability test
The viability of each sample collected using DEP and FDAA staining was evaluated using the LIVE/DEAD™ BacLight™ Bacterial Viability Kit (Thermo Fisher Scientific) or direct cultivation. The former was carried out according to the manufacturer’s protocol. For direct cultivation, 3 samples, the centrifuged bacteria sample, DEP-captured bacteria sample, and FDAA-stained bacteria sample, were diluted in 1 x PBS to 104, 104, and 103 times, respectively, spread onto marine broth 2216 (MB; BD Biosciences) agar plates, and cultivated at 25°C for 48 h. Single colonies with distinct morphology were identified, selected, and subjected to direct colony PCR using the 16S rRNA universal primers 27F (5’ – AKWGTTTGATCMTGGCTCAG) and 1492R (5’ – GGHTACCTTGTTACGACTT). The PCR reaction was prepared using AmpliTaq Gold 360 Master Mix (Thermo Fisher Scientific) based on the manufacturer’s protocols, and amplification was conducted using the C1000 Touch Thermal Cycler (Bio-Rad, Hercules, CA, USA). Thermocycling conditions were as follows: 10 min at 95°C for initial denaturation, followed by 25 cycles of 30 s at 95°C, 30 s at 54°C and 90 s at 72°C, and 7 min at 72°C for a final extension. DNA sequencing of the PCR products was performed by FASMAC Co., Ltd., Kanagawa, Japan, and the attained sequencing results were analyzed using Geneious Prime v2020.2.5 (Geneious; AKL, New Zealand).
2.6 Flow cytometric analysis and fluorescence microscopy
Fluorescing cells were detected and sorted using flow cytometry (BD FACSAria™ II cell sorter; BD Biosciences, Tokyo, Japan) equipped with a 488-nm blue laser. Green fluorescence was detected using the 530/30-nm FITC filters. Fluorescence compensation and data analysis were performed using the BD FACSDiva™ software. Thresholds to determine fluorescing cells were set based on non-FDAA-stained bacteria.
Microscopic images were captured with a BX53 upright fluorescence microscope equipped with a 100x objective lens (Olympus, Tokyo, Japan) and a DP74 digital camera. Fluorescing cells were observed using the U-FGW and U-FBWA mirror units for red and green fluorescence, respectively.
2.7 Data presentation and image editing
All graphs were generated using Prism8 (GraphPad Software Inc., San Diego, CA, USA). Microscopic images were minimally modified and cropped to the desired sizes using Adobe Photoshop CC 2018 v19.1.3 (Adobe System, Tokyo, Japan).
3 Results
3.1 Clean bacterial fraction via DEP
The device, PixeeMo®, was originally designed to capture and count bacteria in drinking water and food samples (Wakizaka et al., 2020; Ogawa et al., 2021). However, it has not been tested against environmental samples comprised of more diverse bacteria. Thus, prior to employing this device on the H. okadai bacterial fraction, we evaluated the ability of PixeeMo® to capture phylogenetically distinct bacteria and its efficiency with several bacteria when pooled together in a mixed population. For the former evaluation, we used the model bacteria, including Bacillus megaterium, Acinetobacter radioresistens, Escherichia coli, Synechocystis sp. PCC6803, and Vibrio alginolyticus. In this evaluation, we showed that all bacteria could be efficiently captured on the electrode within the entrapment region of the microchip (Supplementary Figure 2A). For the later evaluation, we used E. coli, Edwardsiella ictalurid, Erwinia persicina, Pantoea agglomerans, and Yersinia bercovieri as the model bacteria. Upon collection of the mixed bacterial population after capture and release within the microchip, DNA bands from the extracted genomic DNA corresponding to each bacteria were confirmed by PCR and agarose gel electrophoresis (Supplementary Table 2, Supplementary Figure 2B). Having proven that PixeeMo® is applicable to capturing diverse bacteria and distinct morphology, we subsequently applied it to capture bacteria from the final H. okadai bacterial fraction.
We showed that bacteria were successfully captured within the microchip when the final H. okadai bacterial fraction was applied (Figure 1A). Upon confirming bacterial capture and collection, we evaluated bacterial viability against the DEP-captured bacteria sample, the waste fluid, and the final H. okadai bacterial fraction prior to DEP treatment (centrifuged bacteria sample) (Figure 1B). In the centrifuged bacteria sample, we first confirmed that viable and non-viable bacteria, each showing green and red fluorescence, respectively, were present. Impurities were also observed to be present but showed low autofluorescence. Next, we confirmed that most impurities from the sample were present within the waste fluid via bright field microscopy, while non-viable bacteria were also confirmed by the high presence of red-fluorescing cells. In the DEP-captured bacteria sample, on the other hand, most of the cells showed green fluorescence, indicating the capture of viable bacteria, while the presence of impurities was low. Based on this result, we confirmed that DEP was applicable in capturing and enriching viable bacteria from environmental samples.
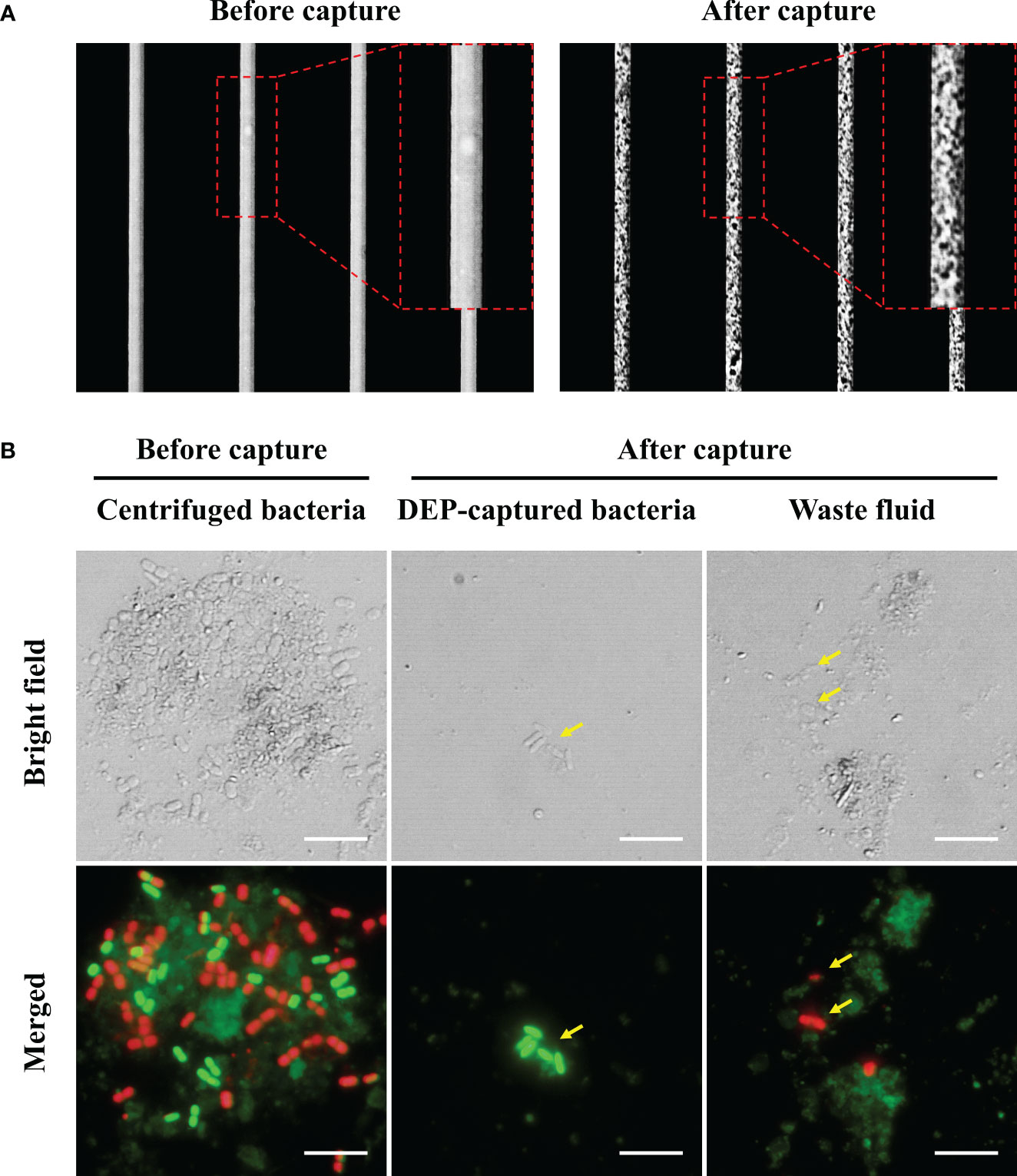
Figure 1 (A) The images of the electrodes before and after bacteria capture (B) The results of the bacterial viability test before and after capture using DEP. Cell viability was determined using the LIVE/DEAD™ BacLight™ Bacterial Viability Kit. Green and red fluorescence indicate viable and non-viable cells, respectively. The yellow arrows indicate bacteria. Scale bars are 5 μm.
3.2 FDAA for attaining viable bacteria
Having evaluated the ability of DEP to separate viable bacteria from environmental samples, we subsequently employed another approach for attaining viable bacteria using FDAA. Using sBADA as the FDAA molecule, when we evaluated the staining conditions of sBADA with the final H. okadai bacterial fraction, we observed the presence of green-fluorescing cells by fluorescence microscopy after 10 h of incubation, but it was most prominent after 24 h (Figure 2A). The presence of green-fluorescing cells indicated the uptake of sBADA by viable bacteria within the sample and was observed to be mostly cocci and bacilli in morphology. Due to the nature of sBADA, no autofluorescence was observed from the impurities present within the sample. Quantitative analysis of the sBADA-stained sample at 24 h was performed by flow cytometry, and 34.8% of the population were the FDAA-stained bacteria sample (Figure 2B). 6.0 × 106 cells were subsequently sorted by flow cytometry for nanopore sequencing and bacterial viability test.
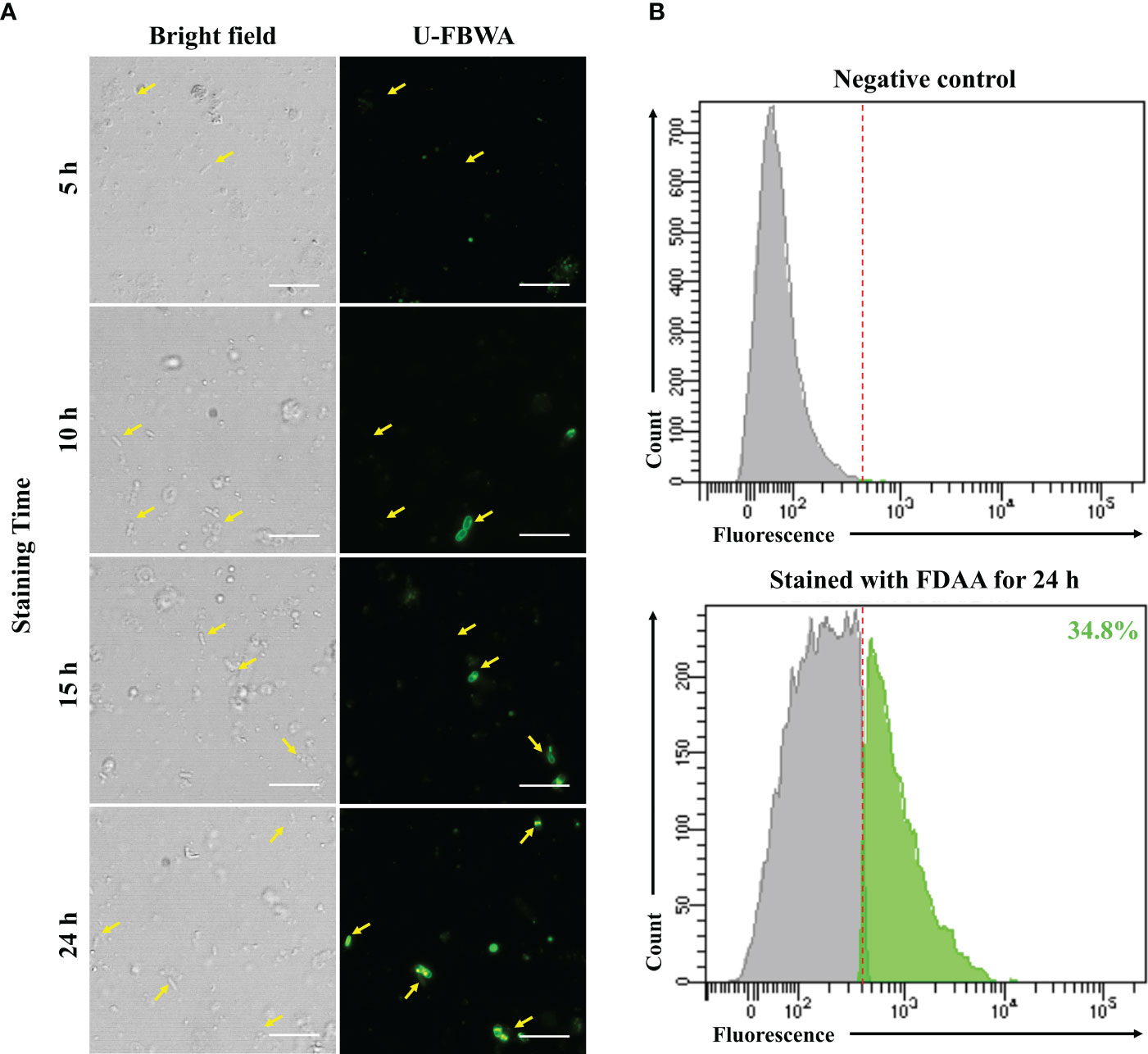
Figure 2 (A) Microscopic images of sBADA stained H okadai bacterial fraction samples at 4 time points for 24 h. The yellow arrows indicate the presence of green fluorescing bacteria. Scale bars are 5 μm. (B) Flow cytometry results of FDAA-stained bacteria sample after 24 h. A non-FDAA-stained bacteria sample was used as the negative control.
3.3 Comparative analysis of bacterial distribution via nanopore sequencing
Nanopore sequencing was performed to compare the diversity and composition of bacteria attained using the DEP and FDAA approaches. After quality check and trimming the raw sequence reads to the designated size (Supplementary Table 1), the final read count and bacterial genera identified from each sample via BLAST search were 122,481 and 11 for the centrifuged bacteria sample, 114,708 and 7 for the DEP-captured bacteria sample, and 79,511 and 10 for the FDAA-stained bacteria sample, respectively (Table 1). Interestingly, 2 and 4 genera, not detected in the centrifuged bacteria sample, were detected using DEP and FDAA, respectively. Distribution analysis of the major genera (>5% population) between the samples showed that Psychrobacter accounted for 58.7%, Pseudomonas 26.2%, and Clostridium 5.6% within the centrifuged bacteria sample, Pseudomonas accounted for 61.3% and Psychrobacter 37.4% within the DEP-captured bacteria sample, and Clostridium accounted for 73.5%, Saccharospirillum 9.3%, and Psychrobacter 6.8%, within the FDAA-stained bacteria sample.
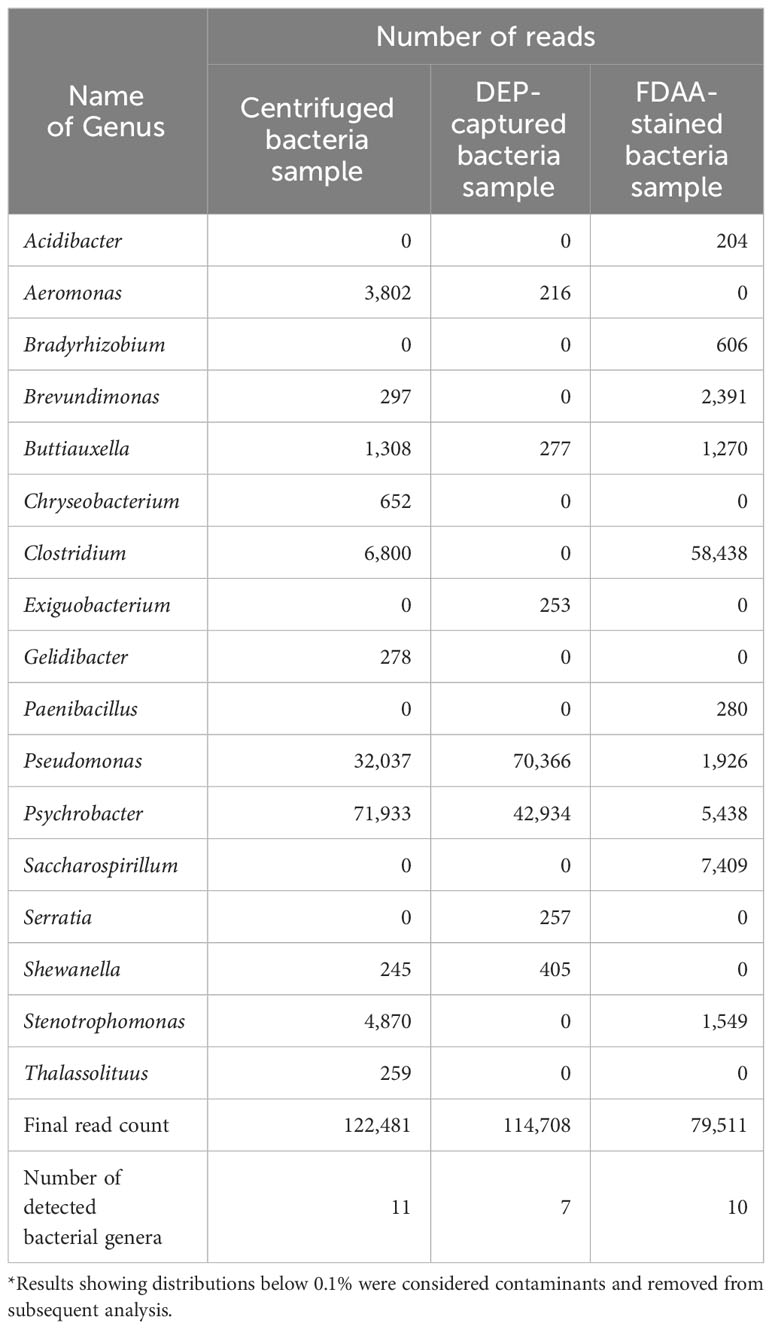
Table 1 Distribution of bacteria from nanopore sequencing results.
3.4 Viability conformation of DEP-captured and FDAA-stained bacteria sample
Finally, we confirmed the viability of bacteria attained using DEP and FDAA through direct cultivation. When we identified the bacterial colonies grown on MB agar plates, there were no morphological differences other than the size of the colonies. Therefore, 30 colonies were randomly selected for each sample and subjected to 16S rRNA gene identification via PCR. As a result, from the centrifuged bacteria sample, Psychrobacter accounted for 88.9%, while Pseudomonas was 5.3%. From the DEP-captured bacteria sample, Pseudomonas accounted for 92.9% and Exiguobacterium 7.1%, whereas in the FDAA-stained bacteria, Pseudomonas accounted for 58.3% and Stenotrophomonas 41.7% (Figure 3). These results showed that bacteria not cultivated from the centrifuged bacteria sample were attained via DEP and FDAA. Specifically, in our case, from the DEP-captured bacteria sample, it was Exiguobacterium, and from the FDAA-stained bacteria sample, it was Stenotrophomonas. We also validated that the bacteria attained from cultivation were not contaminants by cross-referencing with the bacteria identified by nanopore sequencing (Table 1).
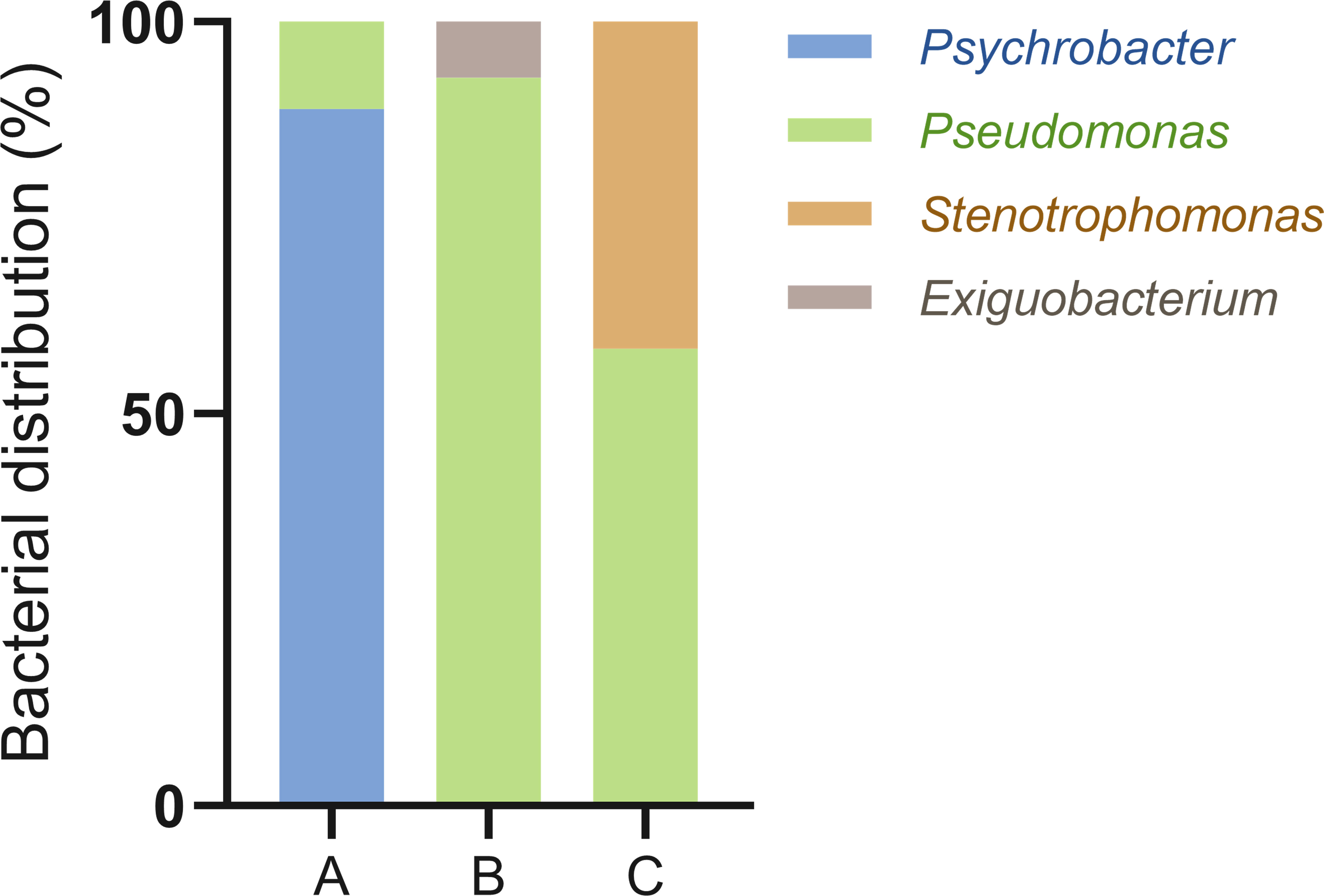
Figure 3 Bacterial viability results through direct cultivation. MB agar plates were used as the growth media in this analysis. (A) centrifuged bacteria sample, (B) DEP-captured bacteria sample, (C) FDAA-stained bacteria sample.
4 Discussion
Attaining clean bacterial fractions with less or no impurities from environmental samples is crucial as this greatly influences the possibility of isolating diverse and useful bacteria. Therefore, to attain such clean bacterial fractions, we needed approaches that could distinctly identify bacteria based on their unique characteristics and features. Having searched for possible solutions, we found two approaches that fulfilled this criterion. DEP employs generating non-uniform electric fields via predesigned electrodes that allow for manipulating and capturing polarized particles within microfluidic systems (Wakizaka et al., 2020). Viable bacteria with intact cellular membranes have intracellular ion homeostasis, resulting in them being highly polarized and, therefore, applicable for manipulation with DEP. As a result, DEP can be used to specifically detect, separate, and characterize viable bacteria from impurities present within environmental samples. FDAA, on the other hand, is D-amino acid residues artificially labeled with fluorophores and is designed to be incorporated into the peptidoglycan (PG) layer during cell membrane synthesis for specific labeling of actively growing bacteria (García-Heredia et al., 2018). Hence, we employed DEP and FDAA and tested their applicability in attaining clean bacterial fractions. From our results, where we used the bacterial fraction from the marine sponge H. okadai, in addition to attaining relatively clean bacterial fractions comprised of viable bacteria (Figures 1, 2), our bacterial distribution and diversity analysis via nanopore sequencing also showed differences in bacterial populations attained using both approaches (Table 1).
From the bacterial distribution and diversity analysis of the DEP-captured bacteria sample, we found that Pseudomonas and Psychrobacter were the most abundant, covering 98.7% of the total bacterial population (Table 1). This correlated with the bacterial distribution from the centrifuged bacteria sample, where Pseudomonas and Psychrobacter were also most dominant at 84.9%. Interestingly, we observed that the populations of Pseudomonas and Psychrobacter were inverted in the DEP-captured bacteria sample, where Pseudomonas was at 61.3%, followed by Psychrobacter at 37.4%. We found this as an interesting observation because the bacterial genera captured by DEP, excluding Psychrobacter, all had motile characteristics. Referring to the principle of DEP and the design of PixeeMo®, we speculated that it was easier to capture motile bacteria with this system since the probability of these bacteria approaching the electrodes is higher in comparison to non-motile bacteria. Psychrobacter, however, still covered a high percentage of the DEP-captured bacteria sample population simply because they were at high abundance in the final H. okadai bacterial fraction. Being a non-motile bacteria, Psychrobacter may also have the tendency to attach themselves to some of the impurities rather than being in suspension (Staloch et al., 2022), resulting in them to be also removed during the DEP capture process. In addition to Pseudomonas and Psychrobacter, although covering an abundance of only 0.3% in average, motile bacteria such as Aeromonas, Shewanella, Buttiauxella, Serratia, and Exiguobacterium were also captured by DEP.
In contrast to the bacteria captured by DEP, from the FDAA-stained bacteria sample, we found that Clostridium was most abundant, accounting for 73.5% of the total bacterial population (Table 1). The dominance of Clostridium within the FDAA-stained bacteria sample was intriguing, as this genus consists of obligate anaerobes (Maier et al., 2014). As described, the FDAA is incorporated into the PG layer of bacteria during cell growth. Since sBADA staining of the bacteria was performed for 24 h not in anaerobic conditions, it is hard to assume that growth occurred. However, bacteria classified as Clostridium are known to have spore-forming characteristics (Talukdar et al., 2015). The non-optimal growth conditions for Clostridium could have possibly triggered the formation of spores, hence promoting the incorporation of sBADA. The large abundance of Clostridium in the FDAA-stained bacteria sample compared to the centrifuged bacteria sample could be explained by selective sorting during flow cytometry. Among the bacteria from the FDAA-stained bacteria sample, one other spore-forming genus, Paenibacillus, was also identified (Grady et al., 2016). Although not showing high abundance compared to Clostridium, this genus was also only identified from the FDAA-stained bacteria sample. The low abundance of Paenibacillus in the FDAA-stained bacteria sample could be due to their initial abundance within the centrifuged bacteria sample (Table 1). The distribution of other genera within the FDAA-stained bacteria sample comprised mainly of gram-negative bacteria but was at much lower abundance compared to the centrifuged and the DEP-captured bacteria samples. Particularly for Psychrobacter and Pseudomonas, this is probably due to the staining conditions where the samples were stained with sBADA in 1 x PBS. Although we risk the possibility of bacteria losing their viability when incubating in PBS, we avoided using culture media since we wanted to avoid bias growth but instead attain a more diverse bacterial population.
Next, we tested the viability of the bacteria attained from each approach by plating the samples on MB agar plates (Figure 3). Here, we used MB as the culture medium as it is considered a rich media commonly used to isolate marine bacteria. From the centrifuged bacteria sample, upon performing 16S rRNA analysis, we found that they represented the two dominant bacteria, Psychrobacter and Pseudomonas (Figure 3). This was expected as both genera can grow on MB and cover more than 80% of the total bacterial population (Table 1). Likewise, in the DEP-captured bacteria sample, the presence of Pseudomonas was confirmed. However, in contrast to the centrifuged bacteria sample, although representing the second most abundant genus, Psychrobacter was not identified. Surprisingly, Psychrobacter was also not identified from the FDAA-stained bacteria sample. Even though it is hard to determine the exact reason why the growth of Psychrobacter from the DEP-captured and FDAA-stained bacteria samples was not observed, the removal and absence of impurities including possible metabolites, proteins or compounds from the centrifuged bacteria sample during DEP and FDAA treatment may suggest that Psychrobacter from H. okadai could be dependent on such constituents present within the marine sponge to support growth (Staloch et al., 2022). Besides Pseudomonas, the growth of Exiguobacterium was also confirmed from the DEP-captured bacteria sample (Figure 3). Since the abundance of this bacterium was very low (Table 1) and all the other low-abundant genera are also motile and can grow on MB, we assumed that the identification of Exiguobacterium was random.
In the direct cultivation of the FDAA-stained bacteria sample, the growth of two genera, Pseudomonas and Stenotrophomonas, was confirmed (Figure 3). Although, both these bacteria were not highly abundant, cultivation of these bacteria suggests that bacteria isolated by FDAA were viable (Table 1). Clostridium, which represents the most abundant genus, was not identified since we knew that bacteria from this genus are mostly obligate anaerobes, and the growth conditions were not ideal for their cultivation. Subsequent attempts to further cultivate the Clostridium with the proper growth conditions were performed but were not successful. Here, we speculated that the cultivation of Clostridium could have been hindered by other factors rather than viability. Subsequent to Clostridium, from the nanopore sequencing analysis, Saccharospirillum, Psychrobacter, and Brevundimonas were the next most abundant, but no colonies representing neither of these bacteria was identified. As described, we speculated that the growth of Psychrobacter could be its dependency on organic compounds or molecules present within H. okadai. Looking at the characteristics of Saccharospirllum and Brevundimonas, where some bacteria from both genera have been reported to be non-motile (Choi et al., 2011) or adopts a sessile lifestyle (Sperling et al., 2019), we hypothesized that both these bacteria in H. okadai also have similar features to Psychrobacter, hence, no colonies were identified.
H. okadai has brought much interest since unique natural compounds, including halichondrin B, have been identified (Hirata and Uemura, 1986), yet only a few reports on the bacterial distribution in H. okadai (Abe et al., 2012; Jeong et al., 2015) have been published. Thus, in addition to evaluating the distribution and viability of bacteria attained using DEP and FDAA, we also highlight the ability of these approaches to identify unique and useful bacteria. From the DEP-captured bacteria sample, Exiguobacterium and Serratia were newly identified (Table 1). Both genera have been reported from marine sponges, where they harbor enzymes applicable for industrial applications (Shanthakumar et al., 2015; Jadhav et al., 2020). The FDAA-stained bacteria sample, on the other hand, showed the presence of 4 unique genera which were Acidibacter, Bradyrhizobium, Paenibacillus, and Saccharospirillum, among which, as far as we know, Acidibacter, Bradyrhizobium, and Saccharospirillum have not been reported from marine sponges. All 3 genera are commonly found in freshwater and terrestrial environments and have been reported to be associated with wastewater bioremediation (Oren, 2015; Ratnasari et al., 2021) or plants (Saranraj et al., 2021). However, several genera have recently been reported from marine environments. Bradyrhizobium and Saccharospirillum, for example, have recently been found abundant in bacteriocytes of a nudibranch (Zhukova et al., 2022) and associated with a marine dinoflagellate (Yang et al., 2020), respectively, indicating they do serve as possible candidates for harboring novel natural compounds. In contrast, Paenibacillus has been identified in marine sponges and has been associated with antibacterial compounds (Grady et al., 2016). With the employment of DEP and FDAA, we believe these approaches could assist in isolating and discovering novel, unique, and useful bacteria.
5 Conclusions
In summary, we showed that DEP and FDAA can be used to prepare clean bacterial fractions for the investigation and exploration of viable and useful bacteria from complex environmental samples. Both approaches have been designed to capture or stain bacteria based on their distinct characteristics, allowing for the identification of novel bacteria unique to each approach apart from conventional differential centrifugation. The differences in bacterial populations between the centrifuged bacteria sample and samples attained via both approaches strongly indicate that the removal of impurities from bacterial fractions does indeed have an impact on bacteria isolation. Although further optimization of the DEP and FDAA approaches may lead to the discovery of more unique bacteria, they still have their limitations. Thus, in addition to further optimization of these approaches, other new approaches designed for targeted isolation of bacteria should also be explored.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: DDBJ Sequence Read Archive; BioProject: PRJDB17192 (PSUB021998); BioSample: SAMD00664446-SAMD00664448 (SSUB027844); Experiment: DRX502901-DRX502903 (moritets-0003_Experiment_0001-0003); Run: DRR519015-DRR519017 (moritets-0003_Run_0001-0003). The nanopore sequencing raw data has been submitted to the DDBJ Sequence Read Archive (DRA) with the accession number DRA017584.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
JY: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. MK: Data curation, Investigation, Methodology, Writing – original draft. AK: Investigation, Methodology, Writing – original draft. AZ: Investigation, Methodology, Writing – original draft. TM: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Grant-in-Aid for Scientific Research (B) (JSPS KAKENHI Grant number JP21H02280), Grant-in-Aid for Challenging Exploratory Research (JSPS KAKENHI Grant number JP21K19137), and by The Japan Science and Technology Agency (Fusion Oriented Research for disruptive Science and Technology program) all awarded to TM.
Acknowledgments
We would like to extend our deepest gratitude to Dr. Rei Suo for assisting in the collection of the H. okadai specimens, Dr. Satoshi Morita of AFI Corporation for providing technical support in the handling and optimization of their PixeeMo® system, and Prof. Tomoko Yoshino from the Tokyo University of Agriculture and Technology for allowing us to use their flow cytometry facility.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1354979/full#supplementary-material
References
Abe T., Sahin F. P., Akiyama K., Naito T., Kishigami M., Miyamoto K., et al. (2012). Construction of a metagenomic library for the marine sponge Halichondria okadai. Biosci. biotechnol. Biochem. 76, 633–639. doi: 10.1271/bbb.110533
Alex R. A., Augustine J., Abraham J. (2023). “Industrial Aspect of Marine Bioprocessing,” in Microbial products for future industrialization (Singapore: Springer), 55–76. doi: 10.1007/978-981-99-1737-2_4
Baysoy A., Bai Z., Satija R., Fan R. (2023). The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 24, 695–713. doi: 10.1038/s41580-023-00615-w
Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10, 1–9. doi: 10.1186/1471-2105-10-421
Chaudhary D. K., Khulan A., Kim J. (2019). Development of a novel cultivation technique for uncultured soil bacteria. Sci. Rep. 9, 1–11. doi: 10.1038/s41598-019-43182-x
Choi A., Oh H.-M., Cho J.-C. (2011). Saccharospirillum aestuarii sp. nov., isolated from tidal flat sediment, and an emended description of the genus Saccharospirillum. Int. J. Syst. Evol. Microbiol. 61, 487–492. doi: 10.1099/ijs.0.022996-0
De Coster W., D’hert S., Schultz D. T., Cruts M., Van Broeckhoven C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Di Donato P., Buono A., Poli A., Finore I., Abbamondi G. R., Nicolaus B., et al. (2018). Exploring marine environments for the identification of extremophiles and their enzymes for sustainable and green bioprocesses. Sustainability 11, 149. doi: 10.3390/su11010149
Escudeiro P., Henry C. S., Dias R. P. (2022). Functional characterization of prokaryotic dark matter: the road so far and what lies ahead. Curr. Res. Microb. Sci. 3, 100159. doi: 10.1016/j.crmicr.2022.100159
Fatoyinbo H. O., Mcdonnell M. C., Hughes M. P. (2014). Dielectrophoretic sample preparation for environmental monitoring of microorganisms: Soil particle removal. Biomicrofluidics, 8. doi: 10.1063/1.4892036
García-Heredia A., Pohane A. A., Melzer E. S., Carr C. R., Fiolek T. J., Rundell S. R., et al. (2018). Peptidoglycan precursor synthesis along the sidewall of pole-growing mycobacteria. Elife 7, e37243. doi: 10.7554/eLife.37243.039
Grady E. N., Macdonald J., Liu L., Richman A., Yuan Z.-C. (2016). Current knowledge and perspectives of Paenibacillus: a review. Microb. Cell Factories 15, 1–18. doi: 10.1186/s12934-016-0603-7
Hirata Y., Uemura D. (1986). Halichondrins-antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 58, 701–710. doi: 10.1351/pac198658050701
Hsu Y.-P., Rittichier J., Kuru E., Yablonowski J., Pasciak E., Tekkam S., et al. (2017). Full color palette of fluorescent d-amino acids for in situ labeling of bacterial cell walls. Chem. Sci. 8, 6313–6321. doi: 10.1039/C7SC01800B
Huson D. H., Beier S., Flade I., Górska A., El-Hadidi M., Mitra S., et al. (2016). MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PloS Comput. Biol. 12, e1004957. doi: 10.1371/journal.pcbi.1004957
Jadhav S. B., Shah N., Rathi A., Rathi V., Rathi A. (2020). Serratiopeptidase: Insights into the therapeutic applications. Biotechnol. Rep. 28, e00544. doi: 10.1016/j.btre.2020.e00544
Jeong J.-B., Kim K.-H., Park J.-S. (2015). Sponge-specific unknown bacterial groups detected in marine sponges collected from Korea through barcoded pyrosequencing. J. Microbiol. Biotechnol. 25, 1–10. doi: 10.4014/jmb.1406.06041
Jung D., Liu L., He S. (2021). Application of in situ cultivation in marine microbial resource mining. Mar. Life Sci. Technol. 3, 148–161. doi: 10.1007/s42995-020-00063-x
Kapinusova G., Lopez Marin M. A., Uhlik O. (2023). Reaching unreachables: Obstacles and successes of microbial cultivation and their reasons. Front. Microbiol. 14, 1089630. doi: 10.3389/fmicb.2023.1089630
Kogawa M., Hosokawa M., Nishikawa Y., Mori K., Takeyama H. (2018). Obtaining high-quality draft genomes from uncultured microbes by cleaning and co-assembly of single-cell amplified genomes. Sci. Rep. 8, 2059. doi: 10.1038/s41598-018-20384-3
Kuru E., Tekkam S., Hall E., Brun Y. V., Van Nieuwenhze M. S. (2015). Synthesis of fluorescent D-amino acids and their use for probing peptidoglycan synthesis and bacterial growth in situ. Nat. Protoc. 10, 33–52. doi: 10.1038/nprot.2014.197
Lindequist U. (2016). Marine-derived pharmaceuticals–challenges and opportunities. Biomol. Ther. 24, 561. doi: 10.4062/biomolther.2016.181
Maier E., Anderson R. C., Roy N. C. (2014). Understanding how commensal obligate anaerobic bacteria regulate immune functions in the large intestine. Nutrients 7, 45–73. doi: 10.3390/nu7010045
Moon J.-H., Roh D.-H., Kwack K. H., Lee J.-H. (2023). Bacterial single-cell transcriptomics: Recent technical advances and future applications in dentistry. Jpn Dent. Sci. Rev. 59, 253–262. doi: 10.1016/j.jdsr.2023.08.001
Nnadozie C. F., Lin J., Govinden R. (2015). Selective isolation of bacteria for metagenomic analysis: impact of membrane characteristics on bacterial filterability. Biotechnol. Prog. 31, 853–866. doi: 10.1002/btpr.2109
O'Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., Mcveigh R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Ogawa U., Koyama K., Koseki S. (2021). Rapid detection and enumeration of aerobic mesophiles in raw foods using dielectrophoresis. J. Microbiol. Methods 186, 106251. doi: 10.1016/j.mimet.2021.106251
Oren A. (2015). Halophilic microbial communities and their environments. Curr. Opi Biotech. 33, 119–124. doi: 10.1016/j.copbio.2015.02.005
Ratnasari A., Zaidi N. S., Syafiuddin A., Boopathy R., Kueh A. B. H., Amalia R., et al. (2021). Prospective biodegradation of organic and nitrogenous pollutants from palm oil mill effluent by acidophilic bacteria and archaea. Bioresour. Technol. Rep. 15, 100809. doi: 10.1016/j.biteb.2021.100809
Rodrigues C. J., de Carvalho C. C. (2022). Cultivating marine bacteria under laboratory conditions: Overcoming the “unculturable” dogma. Front. Bioeng. Biotechnol. 10, 964589. doi: 10.3389/fbioe.2022.964589
Saranraj P., Sivasakthivelan P., Al-Tawaha A., Sudha A., Al-Tawaha A., Sirajuddin S. (2021). “Diversity and evolution of Bradyrhizobium communities relating to Soybean cultivation: A review,” in IOP Conference Series: Earth and Environmental Science, The 3rd International Conference of Animal Science and Technology, 3-4 November 2020, Makassar, Indonesia (IOP Publishing) 788, 012208. doi: 10.1088/1755-1315/788/1/012208
Shanthakumar S., Duraisamy P., Vishwanath G., Selvanesan B. C., Ramaraj V., David B. V. (2015). Broad spectrum antimicrobial compounds from the bacterium Exiguobacterium mexicanum MSSRFS9. Microbiol. Res. 178, 59–65. doi: 10.1016/j.micres.2015.06.007
Shen W., Le S., Li Y., Hu F. (2016). SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PloS One 11, e0163962. doi: 10.1371/journal.pone.0163962
Sperling L., Mulero Alegría M. D., Kaever V., Curtis P. D. (2019). Analysis of Brevundimonas subvibrioides developmental signaling systems reveals inconsistencies between phenotypes and c-di-GMP levels. J. Bacteriol. 201.20, 10–1128. doi: 10.1128/JB.00447-19
Staloch B. E. K., Niero H., De Freitas R. C., Ballone P., Rodrigues-Costa F., Trivella D. B. B., et al. (2022). Draft genome sequence of Psychrobacter nivimaris LAMA 639 and its biotechnological potential. Data Br. 41, 107927. doi: 10.1016/j.dib.2022.107927
Talukdar P. K., Olguín-Araneda V., Alnoman M., Paredes-Sabja D., Sarker M. R. (2015). Updates on the sporulation process in Clostridium species. Res. Microbiol. 166, 225–235. doi: 10.1016/j.resmic.2014.12.001
Vaishampayan P. A., Kuehl J. V., Froula J. L., Morgan J. L., Ochman H., Francino M. P. (2010). Comparative metagenomics and population dynamics of the gut microbiota in mother and infant. Genome Biol. Evol. 2, 53–66. doi: 10.1093/gbe/evp057
Wakizaka Y., Itoi T., Takano M., Kato E., Sato Y., Morita S., et al. (2020). AFI Corp. PixeeMo™ for enumeration of aerobic bacteria in drinking water: AOAC performance tested methodSM 012002. J. AOAC Int. 103, 1610–1618. doi: 10.1093/jaoacint/qsaa070
Watterson W. J., Tanyeri M., Watson A. R., Cham C. M., Shan Y., Chang E. B., et al. (2020). Droplet-based high-throughput cultivation for accurate screening of antibiotic resistant gut microbes. Elife 9, e56998. doi: 10.7554/eLife.56998.sa2
Wilpiszeski R. L., Aufrecht J. A., Retterer S. T., Sullivan M. B., Graham D. E., Pierce E. M., et al. (2019). Soil aggregate microbial communities: towards understanding microbiome interactions at biologically relevant scales. Appl. Environ. Microbiol. 85, e00324–e00319. doi: 10.1128/AEM.00324-19
Yang Q., Jiang Z., Zhou X., Xie Z., Wang Y., Wang D., et al. (2020). Saccharospirillum alexandrii sp. nov., isolated from the toxigenic marine dinoflagellate Alexandrium catenella LZT09. Int. J. Syst. Evol. Microbiol. 70, 820–826. doi: 10.1099/ijsem.0.003832
Zengler K., Zaramela L. S. (2018). The social network of microorganisms—how auxotrophies shape complex communities. Nat. Rev. Microbiol. 16, 383–390. doi: 10.1038/s41579-018-0004-5
Zhukova N. V., Eliseikina M. G., Balakirev E. S., Ayala F. J. (2022). Multiple bacterial partners in symbiosis with the nudibranch mollusk Rostanga alisae. Sci. Rep. 12, 169. doi: 10.1038/s41598-021-03973-7
Keywords: bacterial isolation, bacterial fraction purification, impurities, dielectrophoresis, fluorescent D-amino acids
Citation: Yu J, Kawahisa M, Kinoshita A, Zulmajdi AA and Mori T (2024) Approaches for attaining clean bacterial fractions from complex environmental samples. Front. Mar. Sci. 11:1354979. doi: 10.3389/fmars.2024.1354979
Received: 13 December 2023; Accepted: 11 March 2024;
Published: 27 March 2024.
Edited by:
Mathew Cumming, The New Zealand Institute for Plant and Food Research Ltd, New ZealandReviewed by:
Noora Barzkar, King Mongkut’s University of Technology North Bangkok, ThailandZongze Shao, Third Institute of Oceanography, China
Copyright © 2024 Yu, Kawahisa, Kinoshita, Zulmajdi and Mori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tetsushi Mori, bW9yaXRldHNAZ28udHVhdC5hYy5qcA==