 Tomas Israel Grijalva-Rodriguez1†
Tomas Israel Grijalva-Rodriguez1† Alexandra V. Turchyn2
Alexandra V. Turchyn2 Sharon Albin1
Sharon Albin1 Efrat Eliani-Russak1
Efrat Eliani-Russak1 Alen Manashirov1†Irina Zweig1
Alen Manashirov1†Irina Zweig1 Alexey Kamyshny Jr.1*
Alexey Kamyshny Jr.1*- 1Department of Earth and Environmental Sciences, Faculty of Natural Sciences, Ben-Gurion University of the Negev, Beer Sheva, Israel
- 2Department of Earth Sciences, University of Cambridge, Cambridge, United Kingdom
Cryptic sulfur cycling, characterized by nearly quantitative oxidation of hydrogen sulfide, is characteristic for sediments with low organic matter and high reactive iron contents. A common source of reactive iron in sediments of marine systems that are located in arid environments is aeolian dry deposition of desert dust. The presence of such a cryptic sulfur cycle has previously been documented in sediments of enclosed basins such as the Red Sea and, especially, in its north-eastern extension, the Gulf of Aqaba. Here we present speciation data for iron, manganese and sulfur as well as the isotopic composition of sulfate, and demonstrate cryptic sulfur cycling in sediments from the north-east Atlantic Ocean situated in the vicinity of the Sahara. These sediments are characterized by high reactive-to-total-iron ratios and extremely low sulfur-bound iron contents. In the near-shore sediments (e.g., sediments which were sampled at < 550 km from the coast) highly reactive iron contents are higher than in the sediments, which were retrieved at higher distances from the continent. In the near-shore sediments, the most abundant highly reactive iron fraction is dithionite-extractable iron, which comprises goethite, hematite, and akaganéite. This is the most abundant group of minerals in the Saharan dust collected above the Red Sea. On the other hand, contents and speciation of highly reactive iron does not significantly affect sedimentary sulfur-bound iron contents. We suggest that cryptic sulfur cycling is a common phenomenon for sediments in the ocean situated near arid environments.
1 Introduction
Understanding the mechanisms of microbial metabolism beneath the seafloor in anoxic sediments remains a widely studied research area due to its importance in understanding planetary evolution, astrobiology, and life in extreme environments. In marine sediments, organic carbon is the dominant electron donor due to its availability and high metabolic energy yield (Orcutt et al., 2011). The oxygen penetration depth in marine sediments is usually only a few millimeters or centimeters (Glud, 2008; Boyko et al., 2018), while oxygen can penetrate meters into the sediments of extremely oligotrophic marine systems such as the South Pacific gyre (Fischer et al., 2009). Nitrate is consumed immediately below this depth and in the deeper anoxic sediments sulfate, Fe(III) (hyrdr)oxides and Mn(IV) oxides are the quantitatively-important terminal electron acceptors utilized for microbial metabolism (Thamdrup, 2000; Jørgensen, 2021). Mineralization of organic matter coupled to the reduction of Mn(IV) phases produces the highest amount of metabolic energy in anoxic zones, but sulfate and Fe(III) phases are more abundant in modern marine sediments. Although reduction of poorly crystallized Fe(III) (hydr)oxides produces more metabolic energy than reduction of sulfate, reduction of more stable Fe(III) phases often yields less metabolic energy than sulfate reduction (Bethke et al., 2011; Postma and Jakobsen, 1996). Depending on the content and reactivity of sedimentary organic matter and terminal electron acceptors, biogeochemical zones which are characterized by the consumption of various electron acceptors, often partially overlap (Postma and Jakobsen, 1996; Blonder et al., 2017).
The sulfur cycle in marine sediments starts with microbial sulfate reduction to hydrogen sulfide coupled to the oxidation of organic matter (Jørgensen, 1982). The fate of this produced hydrogen sulfide includes re-oxidation (Yao and Millero, 1993, 1996; Avetisyan et al., 2021), precipitation (Berner, 1970), and incorporation into organic matter (Amrani, 2014 and references therein). The oxidation of H2S in natural aquatic systems results in the formation of elemental sulfur and sulfate, as well as polysulfides, sulfite and thiosulfate (Yao and Millero, 1993, 1996; Zopfi et al., 2004; Kamyshny and Ferdelman, 2010; Findlay and Kamyshny, 2017). The sulfur and iron cycles in sediments are coupled through the oxidation of hydrogen sulfide by reactive iron(III) (hydr)oxides (Yao and Millero, 1996; Zopfi et al., 2004); and Mn(IV) oxides (Burdige and Nealson, 1986; Yao and Millero, 1993; Avetisyan et al., 2021) as well as the formation of pyrite (FeS2) (Berner, 1970; Raiswell and Canfield, 1998). Iron(III) (hydr)oxides minerals also represent the most bioavailable form of iron, although there is no direct correlation between the reactivity of iron minerals toward hydrogen sulfide and their bioavailability (Laufer et al., 2020).
In the case where the flux of iron to a sedimentary environment is higher than the flux of reduced sulfur, two situations may arise. First, microbial sulfate reduction may be - to some extent - suppressed due to ecological competition with iron-reducing microorganisms (Lovley and Phillips, 1987). Secondly, hydrogen sulfide, which is formed by microbial sulfate reduction, will be immediately oxidized by the Fe(III) phases (Yao and Millero, 1996). Combination of these processes may lead to formation of so-called “cryptic” sulfur cycling, e.g., the absence of hydrogen sulfide or a decrease in sulfate concentrations in the pore waters despite the presence of microbial sulfate reduction (Wehrmann et al., 2014, 2017; Mills et al., 2016; Blonder et al., 2017; Boyko et al., 2022). One of the lines of evidence for a cryptic sulfur cycle is the presence of sulfide oxidation intermediates. Isotopic composition of sulfur species, especially oxygen isotopes of sulfate, may also serve as evidence for cryptic sulfur cycling in the sediments (Mills et al., 2016; Blonder et al., 2017). Trace amounts of pyrite in the sediments may provide an additional line of evidence for the existence of microbial sulfate reduction (Blonder et al., 2017). The ultimate proof of presence of the active sulfur cycling is a direct quantification of microbial sulfate reduction (Holmkvist et al., 2011).
Measurements of the sulfur isotope composition of sulfate provide only very limited information on transformations of sulfur species during the cryptic sulfur cycling (Mills et al., 2016; Blonder et al., 2017). In such systems, the oxygen isotopic composition of sulfate (δ18OSO4) may serve as the best isotopic tool for understanding redox transformations of sulfur. The enrichment of sulfate in 18O isotope during microbial sulfate reduction is a result of incorporation of oxygen isotopes from water into sulfate when it is in intracellular intermediate valence state sulfur species (Turchyn et al., 2006; Antler et al., 2013; Blonder et al., 2017). Cryptic sulfur cycling in iron-rich sediments has been reported in various environments including salt marshes (Mills et al., 2016) and coastal sediments influenced by high iron input from fluvial and aeolian sources (Wehrmann et al., 2014, 2017; Blonder et al., 2017; Boyko et al., 2018, 2019, 2022).
One sedimentary system that hosts a cryptic sulfur cycle due to high fluxes of highly reactive iron to the sediments is Svalbard fjords. A source of iron to the sediments in the Svalbard fjords is glacial runoff and the source of organic matter is primary productivity in the water column (Michaud et al., 2020). According to Wehrmann et al. (2017), in these sediments low concentrations of “easily” reducible particulate iron phases coincides with low porewater Fe2+(aq) and Mn2+(aq), and high concentrations of reduced sulfur. In contrast at high concentrations Fe2+(aq) (> 850 μM) and Mn+2(aq) (>650 μM), FeS and FeS2 sedimentary contents are low, and sulfur cycle may be defined as cryptic. Another sedimentary system with a similar sedimentary cryptic sulfur cycle is the Red Sea and especially its North-Eastern extension, the Gulf of Aqaba (Blonder et al., 2017; Boyko et al., 2018, 2019, 2022). Two sources of reactive iron to these environments include direct deposition of dust from adjacent deserts, especially the Sahara, and seasonal flash floods transferring dust that was deposited on the shores to the gulf (Katz et al., 2015; Torfstein et al., 2017; Blonder et al., 2017; Boyko et al., 2019). At 700 m water depth, the concentrations of hydrogen sulfide, its oxidation intermediates and pyrite are extremely low, the presence of active sulfur cycling is evident from the oxygen isotope composition of sulfate (Blonder et al., 2017).
The Red Sea and especially the Gulf of Aqaba are enclosed basins with limited water exchange with the open ocean. Thus, it is not clear if the presence of a cryptic sulfur cycle in the sediments, which are strongly affected by aeolian dust deposition from Sahara, is a common phenomenon or rather restricted to the specific condition of high inputs of highly reactive iron, which prevail in the Red Sea and the Gulf of Aqaba (Boyko et al., 2022). The main goal of this research was to clarify if the sediments of the north-eastern Atlantic Ocean which are strongly affected by the aeolian deposition of Saharan dust are also hosting a cryptic sulfur cycling. In order to answer this question, we studied sulfur, iron, and manganese speciation as well as the isotopic composition of sulfur species in the ODP and IODP cores from the Atlantic coast of Africa in sediment during the last ~100 ka.
2 Sites description and cores information
The Sahara Desert is the largest source of dust on Earth and accounts for up to half of all aeolian material deposited at the surface of the world’s oceans (Goudie and Middleton, 2001 and references therein). Up to 700 Tg of crustal material is mobilized from the Sahara per year and about 1/3 of this amount is deposited in the North Atlantic Ocean (Duce et al., 1991). Total iron content in the Saharan dust is 240–330 mmol kg-1 (Goudie and Middleton, 2001 and references therein). Sediments in the North Atlantic primarily originate from hemipelagic particle flux due to coastal upwelling and dust input, with limited fluvial contribution in northern Morocco (Henrich et al., 2010; Holz et al., 2004). The area between 18°N and 20°N along the NW-African margin features well-developed canyon systems and gullies that influence sediment distribution and transport on the continental slope by serving as path for turbidity currents (Henrich et al., 2010).
Over the last 100 ka, the period with the highest amount of precipitation in Sahara occurred at c.a. 5.5 – 15.0 ka and is known as the “Green Sahara” period, due to the abundant vegetation (Cheddadi et al., 2021). Between 15.0–100 ka, the amount of precipitation was lower than during the Green Sahara period with periods of enhanced dust transport to the Atlantic Ocean (Skonieczny et al., 2019).
We suggest that the distance from shore and latitude are potential controls on the source of iron, transport and delivery to this area west of the Sahara. Thus, the sediments studied in this work can be divided into three groups (Table 1; Figure 1). Group 1: cores from DSDP 79 (Site 546), ODP 157 (Site 954), and DSDP 41 (Site 368) are located < 550 km from the African shore and in the 17°N to 34°N area, e.g. at the same latitude as the Sahara Desert (12°N to 36°N). Group two: core ODP 157 (Site 952) is in the same latitude interval but is 1114 km from the African shore. Group 3: cores DSDP 41 (Site 366) and ODP 108 (Site 665) are located 790–930 km from the shore of Africa, but to the south of Sahara, at 2°N to 6°N. Sediments lithology and composition are summarized in Table 2.
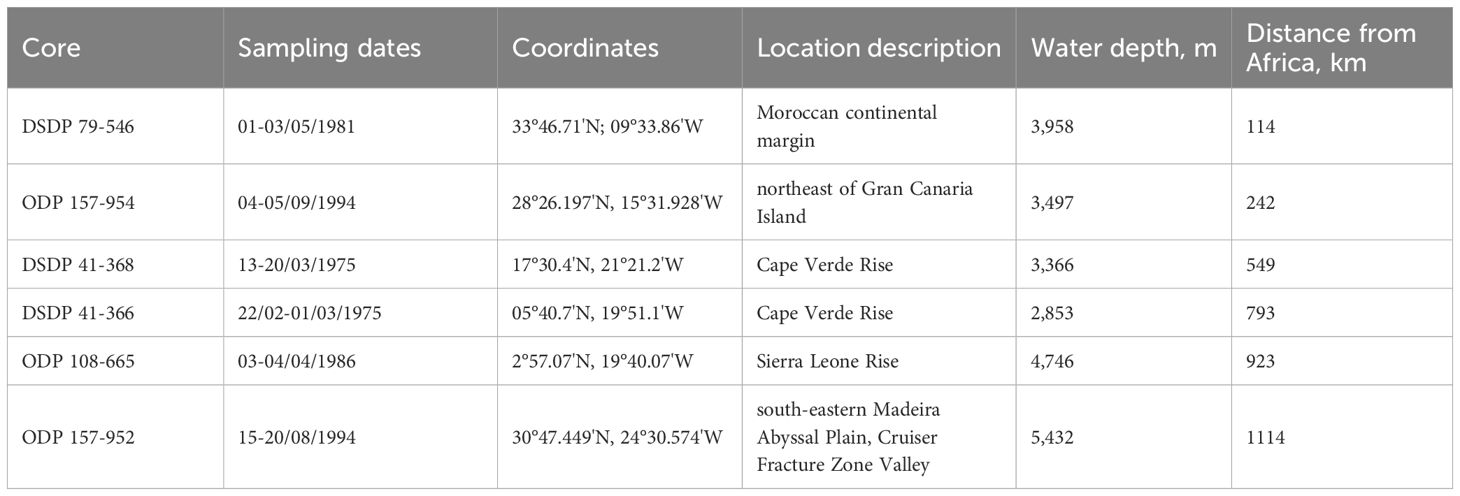
Table 1. List of sampling dates, sampling locations, and water depths.
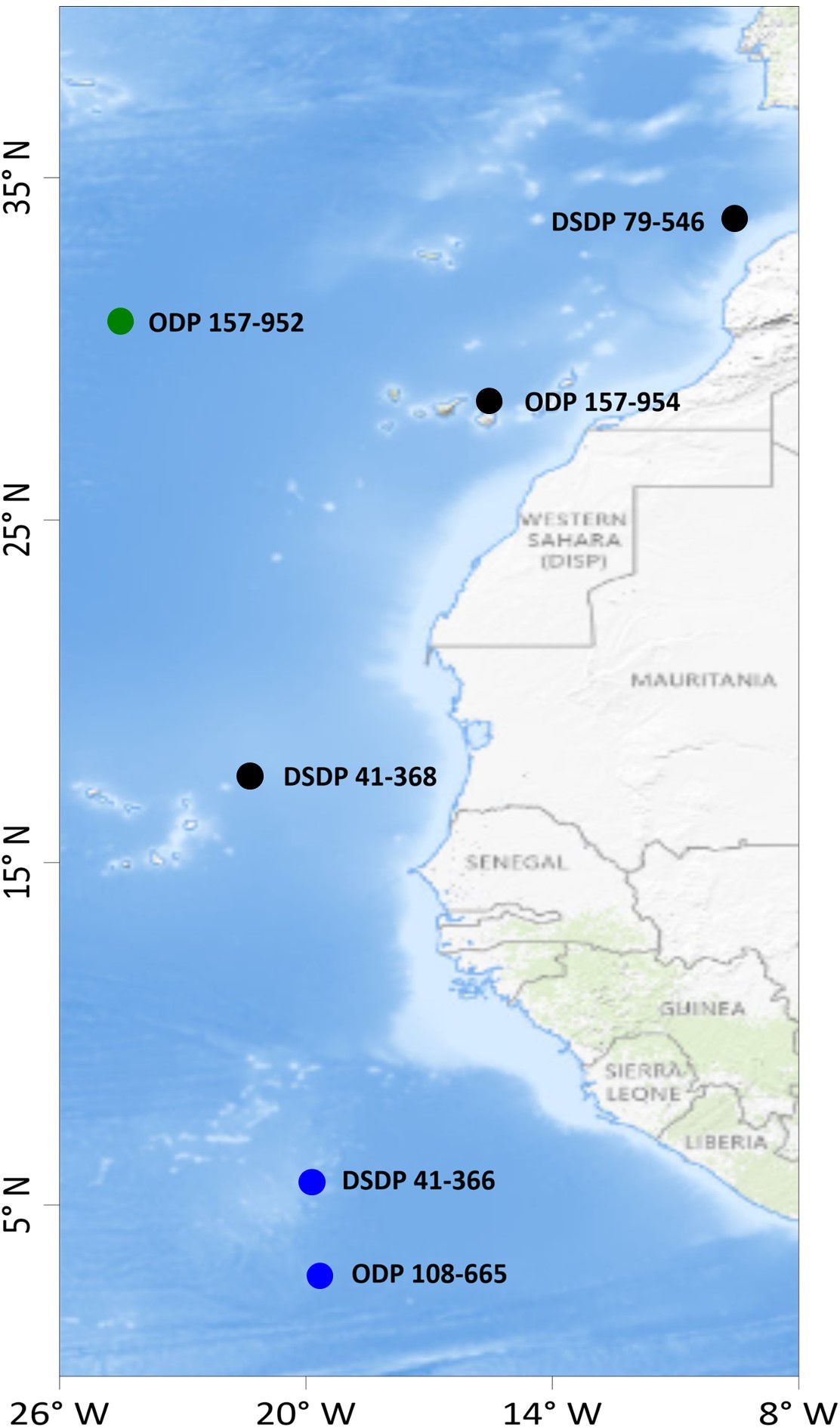
Figure 1. Sampling locations. DSDP stands for “Deep Sea Drilling Project”, ODP stands for “Ocean Drilling Program”. The first number stands for the leg, and the second number for the core hole. Black, green and blue circles stand for locations of the cores from groups 1, 2, and 3, respectively (see explanation in the text).
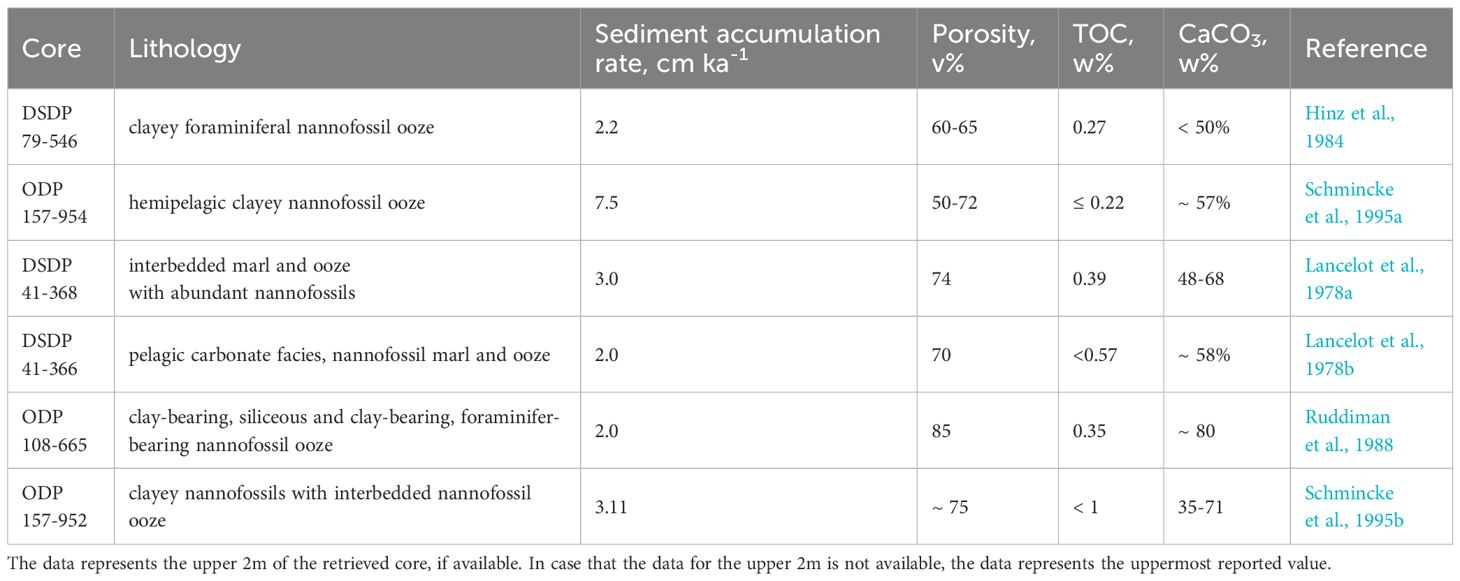
Table 2. Sediments lithology and composition.
Although we did not measure the ages of the individual samples in this study, a combination of the reported sediment accumulation rates and an assumption that up to one meter of surface sediment was lost during sampling, allows us to place all our samples in the interval between 0 and 115 ka (Lancelot et al., 1978a, b; Hinz et al., 1984; Ruddiman et al., 1988; Schmincke et al., 1995a, b). This period corresponds to two types of climatic conditions: (1) the “Green Sahara” period, 14.5 to 5.5 ka before present and (2) more arid periods.
3 Methods
The samples were received from the IODP Bremen Core Repository (MARUM) in sealed plastic bags. The samples were weighed, dried to the constant weight at 50°C for 5 days and stored in hermetically closed vials at ambient conditions prior to analysis.
The same sub-samples were used for analysis of zero-valent sulfur (ZVS), acid volatile sulfide (AVS), and chromium-reducible sulfur (CRS). Around 1.0 g of sediment was transferred into a 50 mL plastic centrifuge tube. Methanol (HPLC-grade) was added to the sample at a sample-to-methanol ratio of 1:20 [w/v], following the methodology of Zopfi et al. (2004). Extraction of ZVS was performed on a rotary shaker at 120 rpm for 16 hours. Quantification of ZVS was carried out on the supernatant after centrifugation of the sample. Determination of ZVS in the methanol extract was done with HPLC with UV-VIS detector and a reverse-phase column. An Agilent Technologies 1260 Infinity High-Performance Liquid Chromatography (HPLC) system was utilized for chromatographic analyses, employing a Grace Prevail C18 reversed-phase column (5µm, 250 x 4.6 mm I.D). This setup was complemented with an Agilent G1365D UV-VIS detector and an Agilent G1321B fluorescence detector (FLD). The mobile phase consisted of HPLC-grade methanol and the flow rate was 1 mL min-1. Spectrophotometric quantification was performed at a 230 nm wavelength. Spectrometric analyses were carried out using the Lambda 365 UV-vis spectrometer by PerkinElmer®. The method detection limit (MDL) was 10 µmol kg-1 of wet sediment. Following the methanol extraction, analyses of AVS and CRS were performed on the same sample by distillation with HCl and with an acidic solution of CrCl2, respectively, as described by Fossing and Jørgensen (1989), with a 15 mL of 50 g L-1 zinc acetate as the trapping solution. The quantification of zinc sulfide in the trapping solution was performed spectrophotometrically at 665 nm. The MDL for the method was 10 µmol kg-1.
Total manganese (MnTOT) and total iron contents (FeTOT) were quantified in c.a. 0.5 g of sediment sample, which was ashed at 450°C for 8 hours, followed by a 24-hour digestion in near-boiling 6 mol L-1 HCl as described by Aller et al. (1986). Iron contents were determined by the ferrozine method outlined by Stookey (1970) with one amendment: an ascorbic acid solution (20 mg per 10 mL) was added, and the solution was shaken for 1 min by hand in order to convert all iron to the Fe2+ form. Total manganese contents were measured by the photometric (2-pyridylazo)-2-naphthol (PAN) technique according to Goto et al. (1977) after adjusting pH of the sample to 7 with a 6 mol L-1 NaOH solution. Both manganese and total iron were quantified spectrophotometrically at a wavelength of 562 nm, with the MDLs 20 µmol kg-1 for both metals.
The determination of highly reactive iron (FeHR) in ~0.5 g of sediment samples was performed following the procedure published by Poulton and Canfield (2005), in which FeHR is defined as the iron extracted in the five steps of sequential extraction and sulfur-bound iron (FeSB), which is calculated as a sum of AVS and CRS divided by 2. The first step of the sequential procedure was extraction of ion-exchangeable iron (Fe2+) using a magnesium chloride solution (Fechl). No iron was released in this step from all analyzed samples. This step was followed by the treatment with a sodium acetate solution to extract carbonate-associated iron (Feac) typically in the form of siderite and ankerite. Subsequently, “easily reducible” iron (hydr)oxides (Fehydr) including ferrihydrite and lepidocrocite were extracted with hydroxylamine hydrochloride solution. The fourth step employed a sodium dithionite solution (Fedith) to remove “reducible” oxides, which include goethite, hematite, and akaganéite. The final extraction step involved the use of ammonium oxalate solution (Feox) to dissolve poorly crystalline iron oxides, e.g., magnetite. Concentrations of the dissolved iron were measured by spectrophotometric technique according to Stookey (1970). The MDL was 20 µmol kg-1 for each fraction.
For the analysis of isotopic composition of sulfur in CRS, zinc sulfide formed during hydrogen sulfide distillation was converted to silver sulfide by the addition of an excess of silver nitrate solution. Silver sulfide samples were stored for at least one week in the dark, washed three times with 10 mL of Milli-Q water, stored overnight in 1 mL of 1 mol L-1 NH4OH solution, and washed three times with 10 mL of Milli-Q water. The samples were dried at 50°C to a constant weight.
For the oxygen isotope analysis of sulfate, 2 g of the sediment was shaken with 20 mL of 10% NaCl solution for 24 hours. After centrifugation and filtration through a 0.22 µm filter, 3 mL of solution was mixed with 1 mL of 1 mol L-1 BaCl2 solution stored at 4°C for 48 hours, centrifuged, washed with MQ water and dried at 70°C for five days. Barium sulfate was purified according to Bao (2006), e.g., 14 mg of the sample was dissolved in 7 mL of diethylenetriaminepentaacetic acid solution (40 g NaOH and 19.7 g diethylenetriaminepentaacetic acid in 1 L solution) followed by acidification with 10.2 M HCl to pH 3-4. After one hour, the samples were centrifuged and decantated to remove excess liquid. The samples were washed three times with Milli-Q water and dried at 70°C for five days.
Approximately 400 μg of barite was crushed and placed in tin capsules for analysis. The sulfur isotope composition of the barite was measured after combustion at 1020°C in an EA Isolink (Thermo Fisher) and continuous helium flow to a Delta V mass spectrometer in the Godwin Laboratory for Paleoclimate Research at the University of Cambridge. The samples were bracketed with blank boats due to extensive memory effects in the EA Isolink. Samples were bracketed with repeat analysis of NBS127 (20.3‰) and SO-6 (-34‰) which was used for drift correction. Analytical precision based on repeat analysis of the standards and replicates of several samples was 0.2‰ (2σ).
The oxygen isotope composition of sulfate (as barite) was measured with a high temperature elemental analyzer coupled to Sercon 2022 isotope ratio mass spectrometer. Analytical standards IAEA-601 (23‰ VSMOW), IAEA-SO-5 (12‰ VSMOW), and IAEA-SO-6 (-11‰ VSMOW) were used for calibration, and the analytical error was less than 1.0‰ for δ18O values.
The prolonged storage of sediment cores at the Bremen Core Repository, at temperatures 4-6°C, can lead to the oxidation of pyrite and Fe(II) minerals, resulting in the formation of fresh, highly reactive Fe(III) (hydr)oxides. In order to quantify fresh reactive ferrihydrite and to evaluate the quality of the samples a method proposed by Boyko et al. (2022) was applied. Dry samples (0.5 g) were mixed with 10 mL of a 0.5 mol L-1 HCl solution, a condition noted for neither reducing ferric ions nor oxidizing ferrous ions (Lovley and Phillips, 1986). The samples were shaken for one hour followed by centrifugation at 4000 RPM for 10 minutes. Two 1 mL subsamples of the supernatant were used for iron quantification: the first subsample was treated with 100 µL of ferrozine, and the second was treated with both 100 µL of ferrozine and 100 µL of 1.4 mol L-1 hydroxylamine hydrochloride in 2 mol L-1 HCl, for Fe2+ and total dissolved iron quantification, respectively. The concentration of “fresh” ferrihydrite in the samples was calculated by subtracting the iron content measured in the first subsample from that in the second. However, it is crucial to acknowledge, as noted by Boyko et al. (2022), that this methodology does not distinguish between ferrihydrite which was present in the core during its retrieval and ferrihydrite which could be developed during its subsequent storage.
4 Results
The contents of AVS in the sediments were extremely low, <0.22 mmol kg-1 of dry sediment in all samples (Figures 2A, B) with the exception of one sample at Site 546 from DSDP Leg 79 which had a content of 0.55 mmol kg-1 of dry sediment. The CRS contents were low as well. In all samples, the CRS content was <3 mmol kg-1 of dry sediment except in the 1.33-1.45 m interval of ODP 157–954 in which the CRS content was up to 13 mmol kg-1 (Figures 2C, D). The average MnTOT was relatively low in the four cores which were sampled closer to the shore, 52–86 mmol kg-1 of dry sediment for DSDP 79-546, ODP 157-954, DSDP 41-368, and DSDP 41-366. For the two sites further from shore, ODP 108–665 and ODP 157-952, the average total MnTOT content was >100 mmol kg-1 of dry sediment (Figures 2E, F). The average total iron (FeTOT) content was relatively high (>400 mmol kg-1 of dry sediment) in three cores, which were sampled at distances closer to shore as well as ODP 108-665 (Figures 2G, H). In the two other cores, the average FeTOT content was <330 mmol kg-1 of dry sediment. The average FeHR contents were higher in the cores with high FeTOT contents: >150 mmol kg-1 of dry sediment for DSDP 79-546, ODP 157-954, DSDP 41-368, ODP 108-665, and < 128 mmol kg-1 dry sediment in two other cores (Figures 2I, J). The FeSB pool consisted mostly of CRS (Figures 2C, D, K, L).
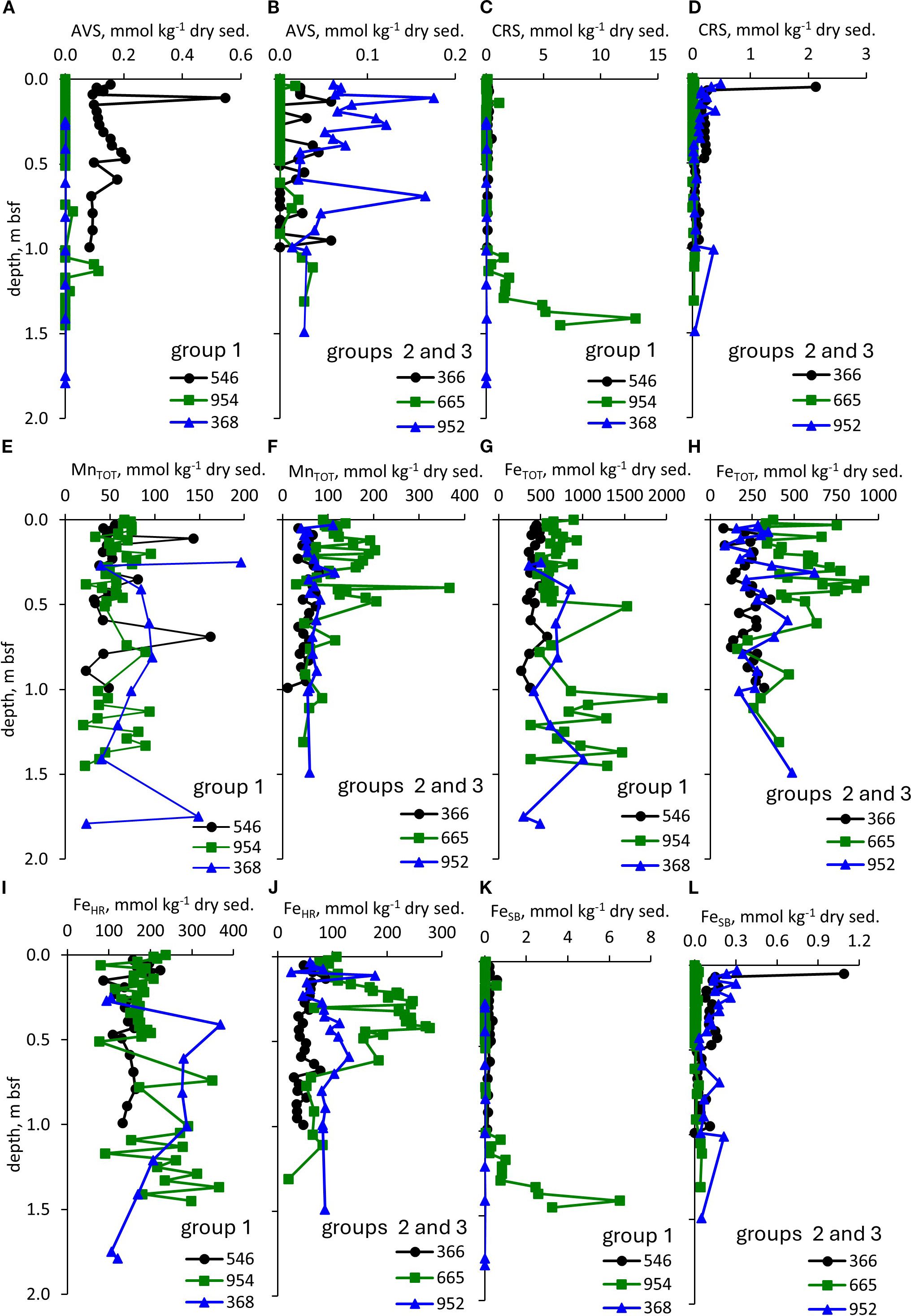
Figure 2. Speciation of sulfur, iron and manganese. Profiles of: (A, B) – AVS, (C, D) – CRS, (E, F) – MnTOT, (G, H) – FeTOT, (I, J) – FeHR, (K, L) – FeSB. The numbers stand for the core hole.
The most abundant of the FeHR pools in four of the six cores (ODP 157-954, DSDP 41-368, DSDP 41-366, ODP 108-665) was Fedith, which comprises goethite, hematite, and akaganéite. This is the most abundant group of minerals in the Saharan dust collected at the Gulf of Aqaba and in its sediments (Boyko et al., 2019, 2022) (Figure 3). On the other hand, in some sections of these cores, the contents of Fehydr, which accounts for the most reactive Fe(III) phases, ferrihydrite and lepidocrocite, were higher than those of Fedith. In cores DSDP 79–546 and ODP 157-952, Fehydr was the most abundant of the FeHR pools in the studied core. The third most abundant of the FeHR pools (and even the second in cores DSDP 79–546 and ODP 157-952) was the least reactive Feox. In core ODP 157-952, sampled at the site with the furthest distance from the shore of Africa, Feox fraction comprised 35 ± 11% (± 1σ) of FeHR.
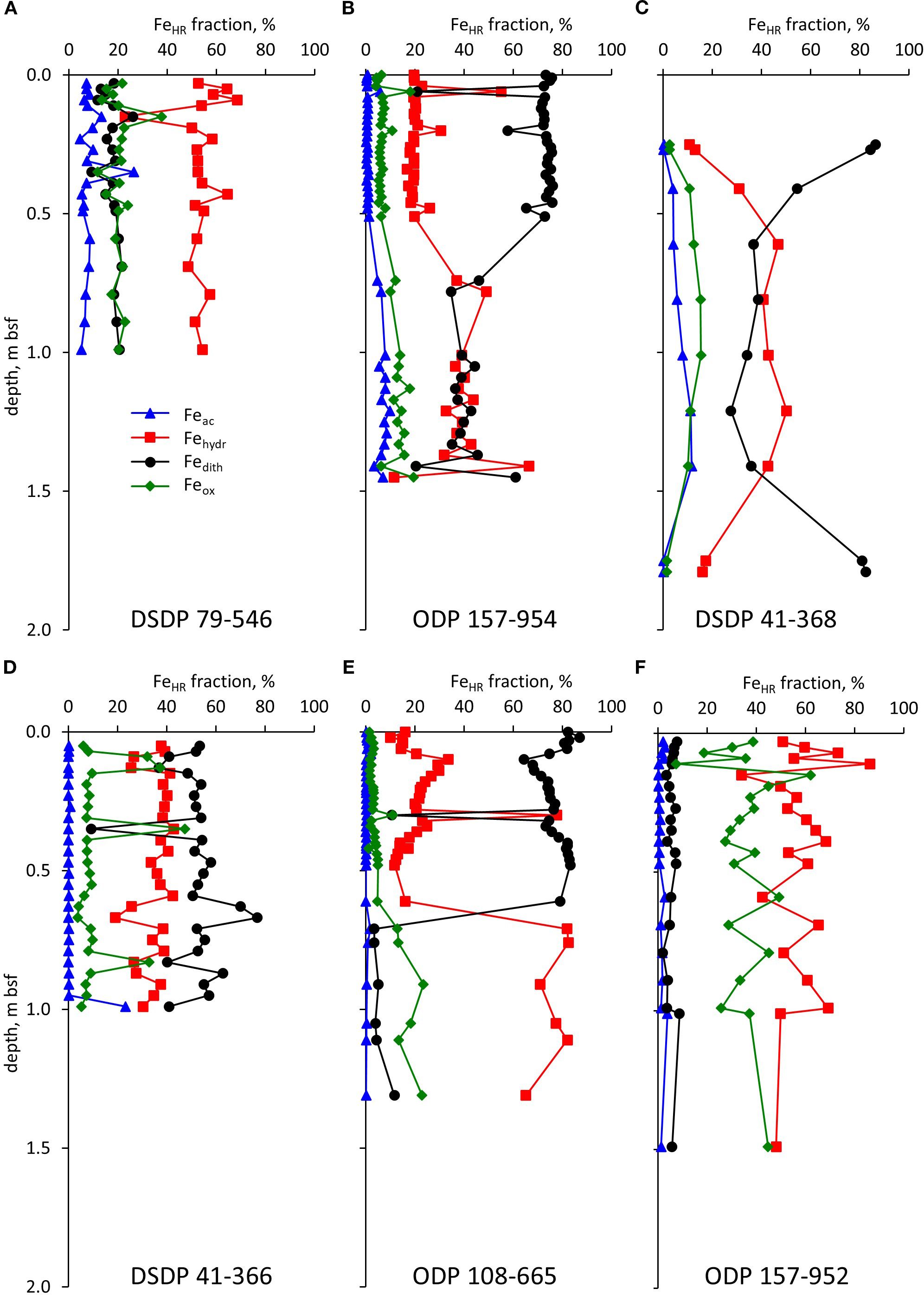
Figure 3. Speciation of the FeHR. Profiles for cores DSDP 79-546 (A), ODP 157-954 (B), DSDP 41-368 (C), DSDP 41-366 (D), ODP 108-665 (E), and ODP 157-952 (F).
The pyrite contents in all samples were too low for isotopic analysis. On the other hand, valuable information is gained by looking at the sulfur and oxygen isotopic composition of pore fluid sulfate. In Figures 4A, B, the sulfur and oxygen isotopic composition of sulfate are presented, and δ18OSO4 vs. δ34SSO4 are plotted in Figure 4C, with a schematic of microbial sulfate reduction in Figure 4C. The isotopic composition of sulfate does not show significant variability with depth for cores DSDP 41–366 and DSDP 79-546. In these cores, the δ34SSO4 is in the 19-22‰ range, close to the seawater value of 21.15 ± 0.15 ‰ (Johnston et al., 2014). In ODP 157–954 core, the δ34SSO4 is slightly lower and more variable – in the 15 to 20‰ range. The oxygen isotope composition of sulfate reveals the following trends: DSDP 41–366 and DSDP 79–546 cores do not show significant variability, with δ18OSO4 ranging between 8-11‰ range, close to the value for the Atlantic Ocean, 9.06‰ (Longinelli and Craig, 1967). In ODP 157-954, the δ18OSO4 are in the range of 12-15‰.
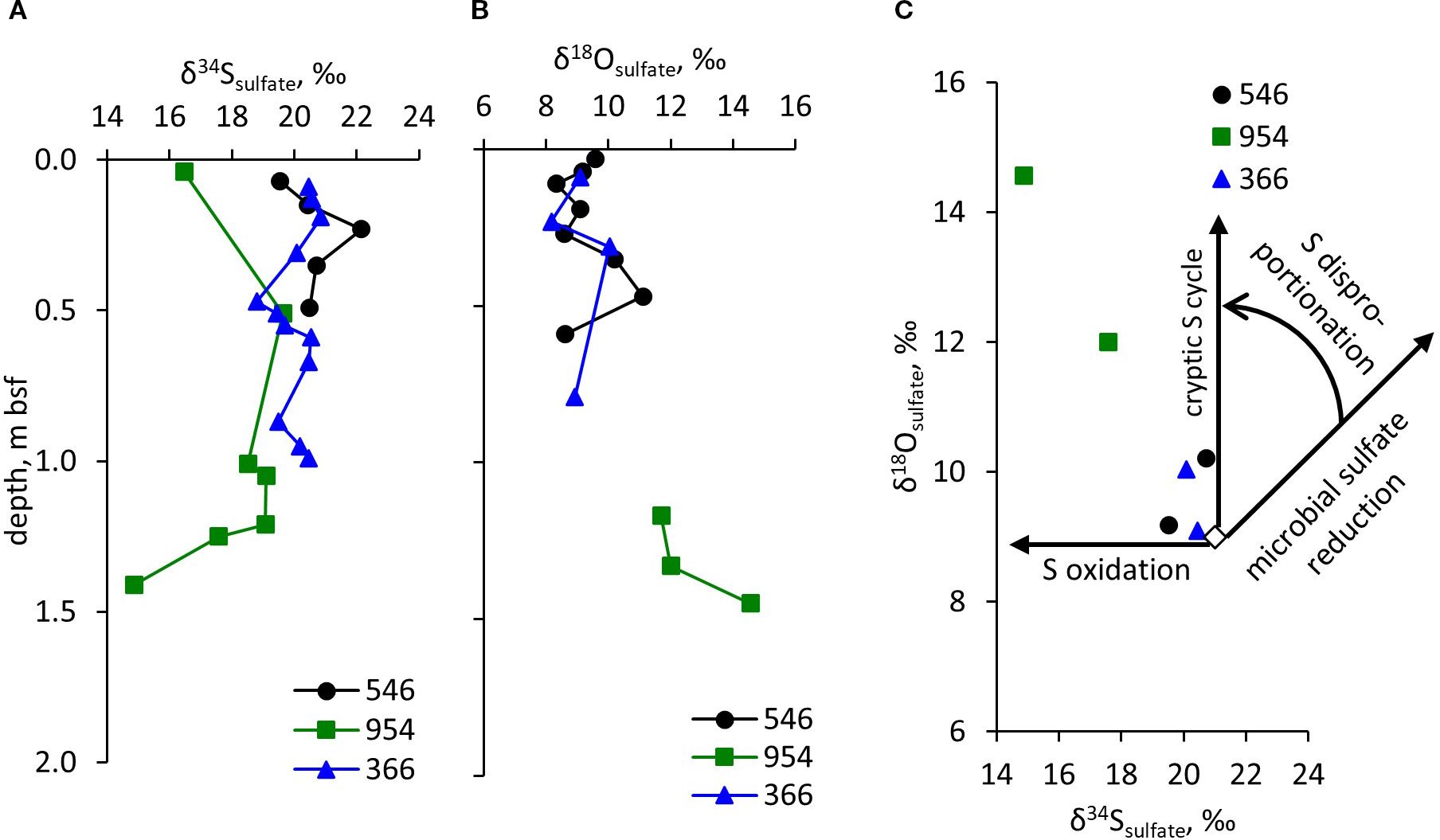
Figure 4. Isotopic composition of sulfate. Profiles of: (A) δ34S of sulfate, (B) δ18O of sulfate, and (C) δ18O vs. δ34S values of sulfate as well as trends of sulfur isotope fractionation by microbial sulfur transformation processes. The empty diamond in (C) represents the isotopic composition of sulfate in the northern Atlantic Ocean (Longinelli and Craig, 1967).
5 Discussion
5.1 Evaluation of the effect of oxidation during storage on iron and sulfur speciation
The iron speciation in the cores, which were stored under oxic conditions, may be compromised by the oxidation of FeSB and Fe(II) minerals to ferrihydrite (Boyko et al., 2022 and references therein). To evaluate any possible changes in iron speciation in the samples, we measured the freshly precipitated Fe(III) (hydr)oxides (Fe (III)fresh), which represent the most reactive part of the Fehydr pool, in 200 samples from all six cores. The possible impacts of the oxidation of iron species during storage are presented in Supplementary Table 1 while a short summary of the results is presented in Table 3. Our results show that the possible artifacts of oxidation are not quantitatively important to impact our results for the speciation of highly reactive iron. As both Fe(II) carbonate minerals and FeSB represent parts of the FeHR pool, their oxidation to Fe(III)fresh does not affect the total FeHR content. Our results show that the oxidation of these species has a relatively small effect on the distribution of iron pools within FeHR. The Fe(III)fresh represents <4.6% of the total FeHR pool in all analyzed samples, and the average value for each core does not exceed 0.31%. Even the most reactive Fe(III) fraction, Fehydr, was possibly impacted by oxic storage by less than 7.1% for all samples, and the average value for each core did not exceed 0.47%. In four of the six analyzed cores Fe(III)fresh was detected in less than a half of the samples. On the other hand, in the samples with low FeSB, the impact of oxidation on the quantification of FeSB may be high. For samples with FeSB >0.04 mmol kg-1, the possible bias is <10%. For cores with low FeSB 88-100% of the FeSB may be oxidized during storage. Despite this, low Fe(III)fresh/FeHR values suggest that even if FeSB is the only source of Fe(III)fresh, the original FeSB/FeHR ratios were still low (see discussion below).
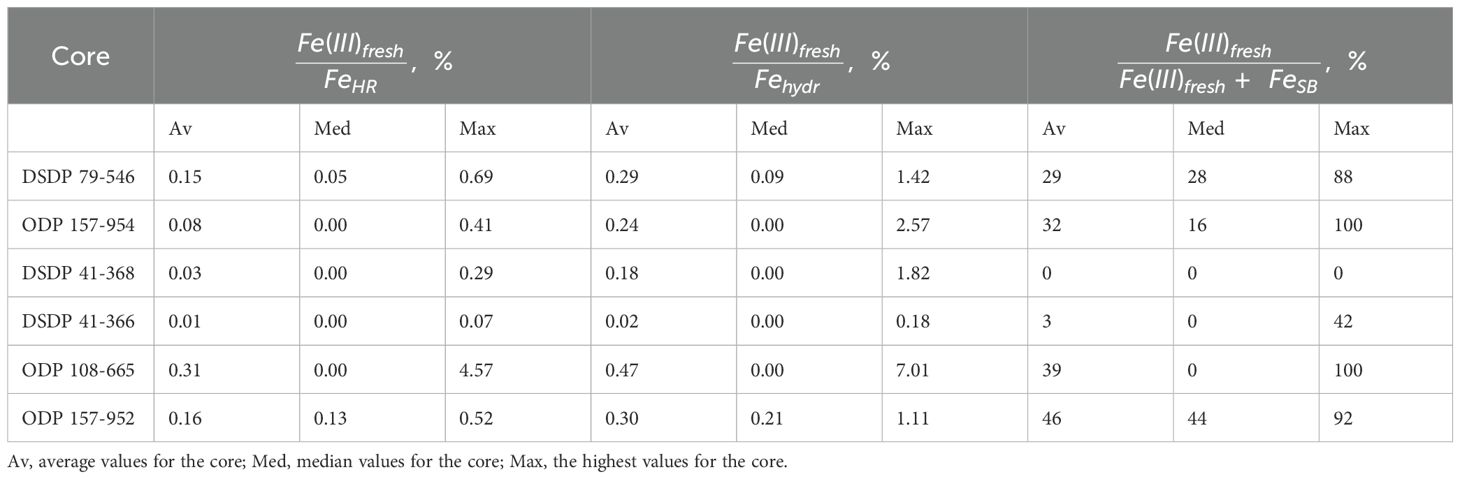
Table 3. Estimation of the impact of the oxidation of pyrite and Fe(II) during storage of the core on the speciation of iron and sulfur.
5.2 Evidence for the cryptic sulfur cycling in the sediments
In all cores, sulfur speciation is consistent with the presence of cryptic sulfur cycling. In the absence of direct measurements of sulfate reduction rates or porefluid concentrations of hydrogen sulfide and its oxidation intermediates, the presence of cryptic sulfur cycling may be confirmed by three lines of evidence (Blonder et al., 2017; Boyko et al., 2022). The first line of evidence for cryptic sulfur cycling is the presence of trace amounts of sedimentary pyrite (Luther, 1991; Rickard and Luther, 1997), the second line of evidence is the presence of the sulfide oxidation intermediate, ZVS, in the solid phase (Zopfi et al., 2004; Findlay and Kamyshny, 2017), and the third line of evidence is the isotopic composition of the sulfur species (Harrison and Thode, 1958; Berner, 1970; Antler et al., 2013).
First line of evidence.
In all analyzed samples from four cores, the AVS content in the cores are <0.62 mmol kg-1 (Figures 2A, B), while CRS content was somewhat higher, up to 4.5 mmol kg-1, in all samples, except four samples in core ODP 157–954 at 1.33-1.45 m bsf depth (Figures 2C, D). The presence of even trace amounts of pyrite (in the form of CRS) in the sediments requires that there was microbial sulfate reduction to produce hydrogen sulfide which is required for pyrite formation.
Second line of evidence.
Concentrations of ZVS were found to be below the detection limit in all samples. Low content of the reduced sulfur species in the sediments is most likely a result of fast hydrogen sulfide oxidation in the sediments, which are rich in FeHR (Poulton et al., 2004).
Third line of evidence.
Strong evidence for the presence of cryptic sulfur cycling in the studied sediments is also reflected in the isotopic composition of sulfate (Figure 4). The combination of δ18OSO4 values, which are higher than seawater values, and δ34SSO4 values, which are similar to or lower than seawater values, is diagnostic of cryptic sulfur cycling (Figure 4C). Although microbial sulfate reduction results in the formation of hydrogen sulfide with lower δ34S while residual sulfate becomes increasingly enriched in the 34S isotope (“microbial sulfate reduction” arrow in Figure 4C), nearly complete re-oxidation of hydrogen sulfide results in the absence of difference between sulfur isotopic composition of sulfate in seawater and in sedimentary porewaters because the sulfur isotope ratio in sulfate remains unchanged. However, the oxygen atoms in sulfate that is formed due to the re-oxidation of hydrogen sulfide or disproportionation of elemental sulfur derive from water. During the oxidation of sulfur to sulfate, sulfite is formed as an intermediate species. Oxygen isotope exchange between sulfite and water is fast and, in equilibrium with water, oxygen in sulfite is enriched in 18O isotope compared to water. The combination of these factors results in formation of sulfate with higher δ18O values than the seawater sulfate values (Böttcher et al., 2005; Antler et al., 2013) (“S disproportionation” arrow in Figure 4C). In the case where reoxidation of 32S-enriched pyrite, formed earlier, occurs, δ34S values of sulfate may be even lower than those measured in seawater and the increase in δ18O values as compared to the seawater sulfate may vary from zero to the values produced as the result of cryptic sulfur cycling (“S oxidation” arrow in Figure 4C).
A similar pattern in the sulfur and oxygen isotopic composition of sulfate was reported in the sediments of the Gulf of Aqaba by Blonder et al. (2017). Three lines of evidence suggest that, although δ34SSO4 values are lower than those of seawater, pyrite oxidation during core storage is not the sole reason for these anomalously low δ34SSO4 values. First, no significant dependence of δ34SSO4 values on the Fe(III)fresh contents was detected (p = 0.07). Second, for only two samples with δ34SSO4 < 20‰ (ODP 157-954, 1.21 m bsf and 1.25 m bsf), a mass balance calculation suggests that the observed low δ34SSO4 values could be attributed solely to pyrite oxidation during storage (assuming that the formation of Fe(III)fresh resulted only from pyrite oxidation and microbial sulfate reduction occurred with ϵ = 70‰). Third, for five samples in which no Fe(III)fresh was detected, δ34SSO4 values were found to be below 20‰ (Figure 4A; Supplementary Table 1).
A combination of the first and the third lines of evidence strongly supports the presence of cryptic sulfur cycling in these Atlantic Ocean sediments, which are affected by aeolian dry deposition of dust from the Saharan desert. On the other hand, direct measurements of sulfate reduction rates and concentrations of sulfur, iron and manganese species in the pore-waters of the freshly recovered cores are required for the understanding of the controls of cycling of the redox-sensitive elements.
5.3 Differences and similarities between the cores
Average contents of FeTOT in the cores from group 1 are somewhat higher than in the cores from groups 2 and 3 (Figures 2G, H). We suggest that this is a result of the high fluxes of dust from Sahara. This explanation is supported by the higher sedimentary silicate content (calculated as 100% minus percent of carbonate content) in the sediments located between 17°N to 34°N than in the sediments located at 2°N to 6°N (group 3). Another support for a similar source of iron in all analyzed cores comes from the analysis of FeHR contents (Figures 2I, J). The highest average contents of FeHR, 151–202 mmol kg-1 of dry sediment were detected in the cores from group 1 (Figure 5). On the other hand, the differences in speciation of FeHR suggests that the sources of dust to the sediments of the Atlantic Ocean are heterogeneous and vary with latitude (Figure 3). As was shown above (see Part 5.1), high amounts of the most reactive fraction of FeHR are unlikely to be an artifact introduced during the storage of the sample under oxic conditions. Contents of FeSB are low in all cores and the average content of FeSB does not depend either on the core group or on the average FeHR content (Figure 5). We interpret this observation as further support for the presence of cryptic sulfur cycling in the sediments of all studied sites, e.g., flux of highly reactive iron to the sediments at all sites is sufficient to reoxidize nearly all hydrogen sulfide, which is produced by microbial sulfate reduction.
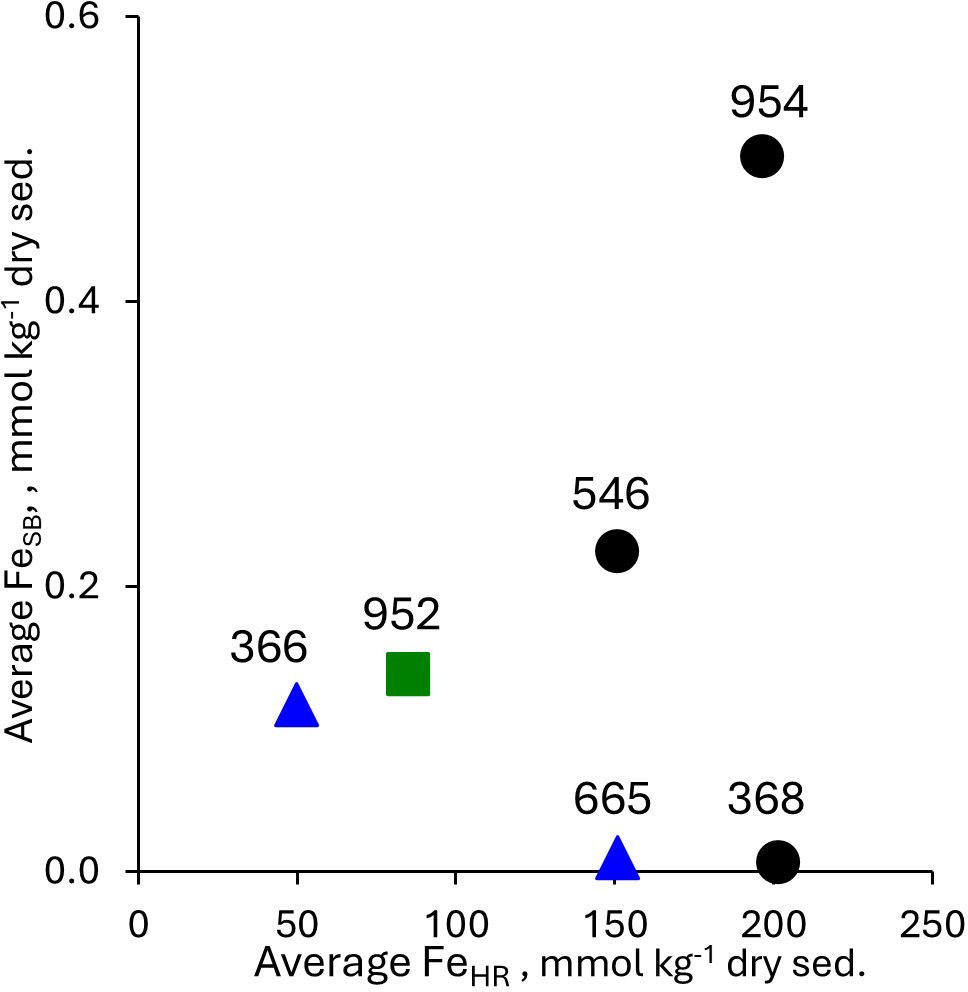
Figure 5. Average FeSB as a function of FeHR, the numbers stand for the core hole. Black circles – cores from group 1, green rectangle – group 2, blue triangles – group 3.
The ratio between FeHR and FeTOT is one of the most commonly applied paleoproxies for the redox state of the water column. In this application, values for this ratio below 0.22 and above 0.38 are interpreted to reflect deposition under oxic and anoxic water columns, respectively (Raiswell and Canfield, 1998; Lyons and Severmann, 2006). However, interpretation of iron proxies may be complicated by other environmental factors (Raiswell et al., 2018). These factors include enhanced sedimentation rates, the remobilization of sedimentary iron (Lyons and Severmann, 2006) as well as external iron inputs to the ocean, such as glacial and aeolian deposition (Wehrmann et al., 2014; Raiswell et al., 2018; Boyko et al., 2019). Iron speciation in the local sources was suggested to play an important role in controlling iron speciation in the sediments (Pasquier et al., 2022). In the Atlantic Ocean samples analyzed in this work at least one sample in each core had a FeHR/FeTOT value above 0.38 (Figures 6A, B). For all cores, the average FeHR/FeTOT ratio was in the range of 0.22-0.38 (Figure 6C). For three cores, the average FeHR/FeTOT ratio was in the range of 0.35-0.37. All three of these cores, DSDP 79-546, DSDP 41-368, ODP 157–952 were retrieved north of 15°N, and thus we hypothesize were strongly affected by the dry deposition of Saharan dust. For the cores sampled south of the Sahara, e.g., DSDP 41–366 and ODP 108-665, the average FeHR/FeTOT ratio was lower, 0.26 and 0.31, respectively. These observations provide further support for the hypothesis that the source of highly reactive iron in sediments of the north-eastern Atlantic Ocean is aeolian dry deposition of Saharan dust. Evaluation of the entire dataset rather than the average core values shows that FeSB depends, to some extent, on the FeHR values: at FeHR/FeTOT values above 0.27, FeSB/FeHR values do not exceed 0.0039, while at lower FeHR/FeTOT values, FeSB/FeHR values may be up to 0.0109 (except for one sample from ODP 157–954 with FeHR/FeTOT 0.48 and FeSB/FeHR 0.0360) (Figures 6A, B) This observation suggests that although cryptic sulfur cycling is common, occurring in organic carbon-poor sediments with a wide range of iron and manganese contents, at lower FeHR/FeTOT ratios, higher amounts of hydrogen sulfide may escape reoxidation and be preserved in the form of FeSB.
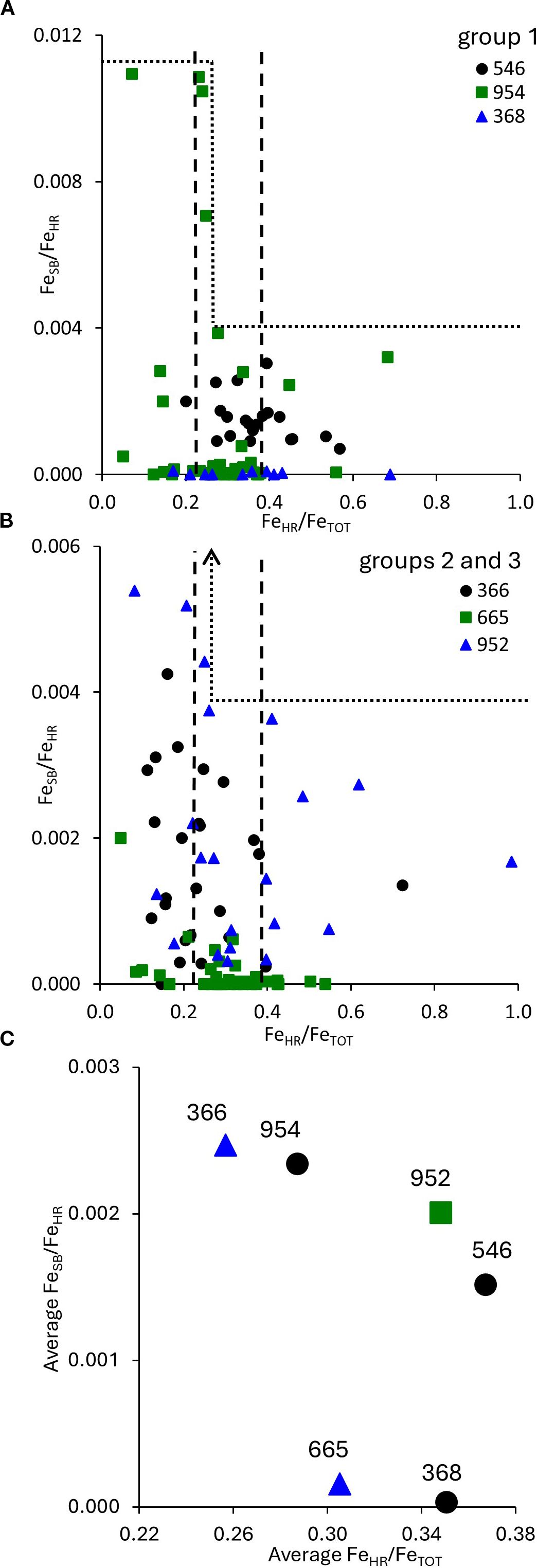
Figure 6. (A) and (B) – plots of FeSB/FeHR vs. FeHR/FeTOT ratios (in panel (A) one outliner point is omitted from the plot). Vertical dashed lines stand for the boundaries of oxic (FeHR/FeTOT <0.22) and ferruginous (FeHR/FeTOT >0.38) conditions (see text for details). Dotted lines in (A) and (B) represent the upper limit of the FeSB/FeHR values. (C) – plot of the average FeHR/FeSB vs. median FeHR/FeTOT ratios. The numbers stand for the core hole. Black circles represent cores from group 1, green rectangle from group 2, blue triangles from group 3.
5.4 Trends in the individual cores
In four of the six studied cores there is no large variability in FeHR content. Only in cores DSDP 41–368 and ODP 108-665, peaks of FeHR content were found at 40–42 cm depth (Figures 2I, J). This implies that sediments on the southern edge of Sahara and south of Sahara were subject to some local influences, the sources of which cannot be detected based on this dataset. Interestingly, these fluctuations have no effect on the sedimentary FeSB content, which was extremely low in both cores (Figures 2K, L). This observation implies that cryptic sulfur existed in the studied sediments during both dryer and wetter climatic conditions in Sahara and is a ubiquitous mode of cycling of the redox-sensitive elements in the oligotrophic marine systems situated in the arid regions and affected by high fluxes of aeolian dust deposition.
5.5 Comparison to the Red Sea
Until now, Red Sea was the only system in which sedimentary cryptic sulfur cycling was linked to the dry deposition of highly reactive iron-rich Sharan dust (Blonder et al., 2017; Boyko et al., 2019, 2022). The Atlantic Ocean and the Red Sea are affected by similar sources of highly reactive iron. Comparison between the speciation of iron and sulfur in the Gulf of Aqaba, Red Sea, and Atlantic Ocean sediments, which were analyzed in this work, are presented in Table 4. TOC contents in the Atlantic Ocean sediments are low and similar or lower than in the Gulf of Aqaba and the Red Sea. The FeTOT contents are similar between the studied basins, although the variability is quite high. On the other hand, the upper limits of FeHR contents are similar in the Gulf of Aqaba and Atlantic Ocean sediments. Sedimentary manganese content in the Atlantic Ocean cores is similar or higher than in the Gulf of Aqaba and the Red Sea cores. It is likely that in these sediments, Mn(IV) phases play an important quantitative role in oxidation of hydrogen sulfide. Concentrations of the reduced inorganic sulfur species are extremely low in all basins. Sedimentary contents of AVS are always < 0.8 mmol kg-1. The content of the most abundant reduced inorganic sulfur pool, CRS, is also similar in the deep-water sites of the Gulf of Aqaba and Atlantic Ocean sediments: 0–11 mmol kg-1 and 0–13 mmol kg-1, respectively. The value of 13 mmol kg-1 of pyrite sulfur corresponds to only 0.16 w% of pyrite.
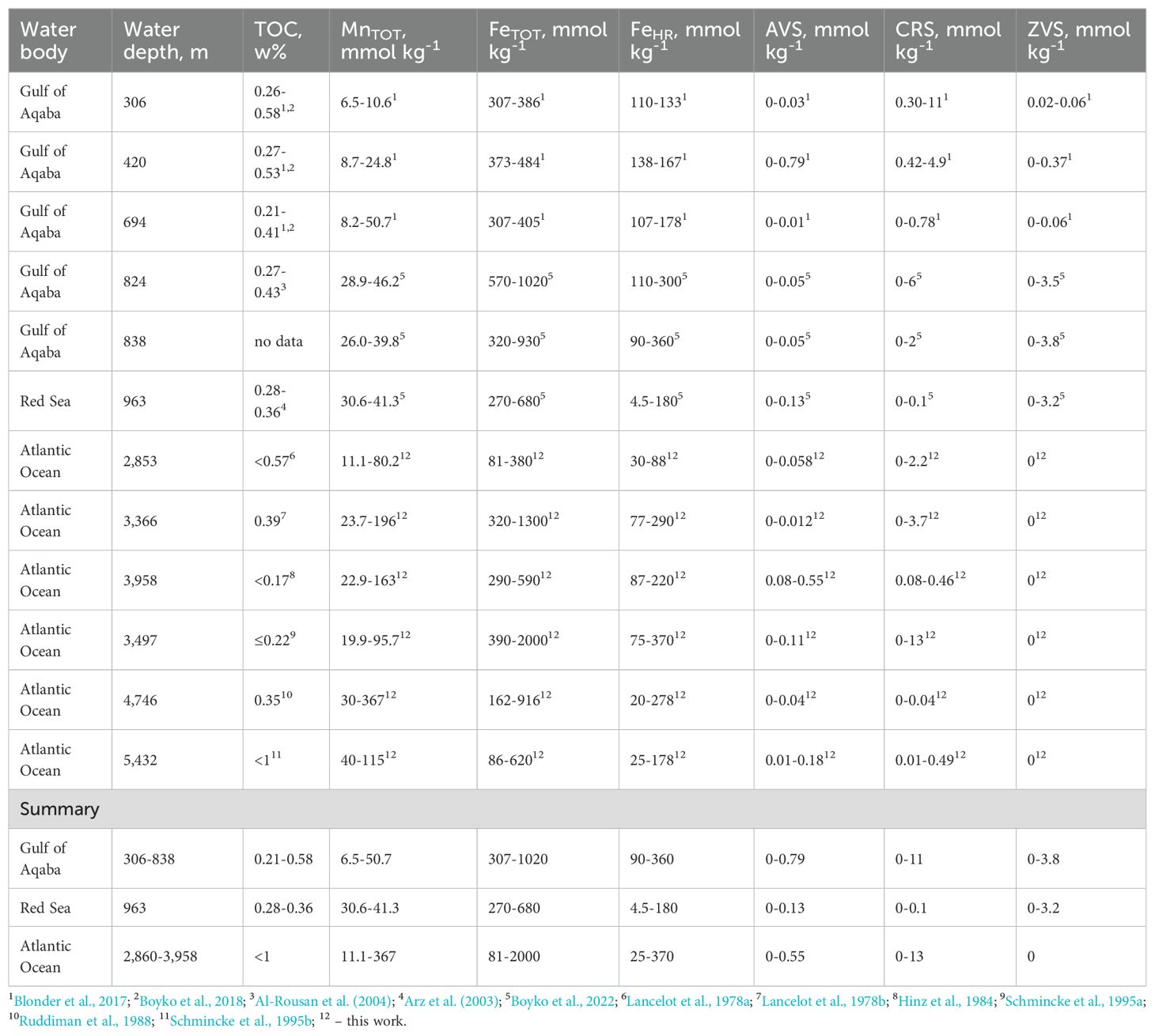
Table 4. Comparison of TOC contents as well as manganese, iron and sulfur speciation in the solid phase of the sediments from the Gulf of Aqaba, Red Sea, and Atlantic Ocean.
These observations provide evidence for the presence of similar modes of sulfur and iron cycling in the sediments of oligotrophic marine systems, which are affected by the aeolian dust deposition from Sahara on the Western and Eastern shores of the African continent. In order to further prove the presence of the cryptic sulfur cycling in the sediments of the Atlantic Ocean near the African shore, sampling of the fresh sediments followed by detection of the solid phase and pore-water composition as well as evaluation of the rates of transformations of the redox-sensitive elements are required.
In summary, the results of this work point to cryptic sulfur cycle as a ubiquitous mode of transformations of the redox-sensitive elements in the marine sediments characterized by a combination of low organic matter content and high contents of the reactive Fe(III) and Mn(IV) species. Cryptic sulfur cycle is not limited to the enclosed water bodies such as the Red Sea and the Gulf of Aqaba. Although in this work we clarified mechanistic constraints on the sulfur cycling in the sediments of the Atlantic Ocean, understanding of the quantitative constraints requires analyses and incubations of the fresh sediments immediately upon their retrieval.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
TG-R: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. AT: Data curation, Funding acquisition, Investigation, Methodology, Resources, Writing – review & editing. SA: Investigation, Methodology, Writing – review & editing. EE-R: Investigation, Methodology, Writing – review & editing. AM: Investigation, Methodology, Writing – review & editing. IZ: Investigation, Methodology, Writing – review & editing. AK: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was funded by ISF, Grant No. 1670/19 and NE/T006838/1 (to AT).
Acknowledgments
This research used samples provided by International Ocean Discovery Program Core Repository at MARUM, Bremen, Germany. We are grateful to Keren Solomovich for assistance with analyses of the samples. This work was funded by ISF, Grant No. 1670/19 and NE/T006838/1 (to AT).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1663305/full#supplementary-material
References
Aller R. C., Mackin J. E., and Cox R. T. (1986). Diagenesis of Fe and S in Amazon inner shelf muds: apparent dominance of Fe reduction and implications for the genesis of ironstones. Cont Shelf Res. 6, 263–289. doi: 10.1016/0278-4343(86)90064-6
Al-Rousan S., Pätzold J., Al-Moghrabi S., and Wefer G. (2004). Invasion of anthropogenic CO2 recorded in planktonic foraminifera from the northern Gulf of Aqaba. Int. J. Earth Sci. 93, 1066–1076. doi: 10.1007/s00531-004-0433-4
Amrani A. (2014). Organosulfur compounds: molecular and isotopic evolution from biota to oil and gas. Annu. Rev. Earth Planet Sci. 42, 733–768. doi: 10.1146/annurev-earth-050212-124126
Antler G., Turchyn A. V., Rennie V., Herut B., and Sivan O. (2013). Coupled sulfur and oxygen isotope insight into bacterial sulfate reduction in the natural environment. Geochim Cosmochim Acta 118, 98–117. doi: 10.1016/j.gca.2013.05.005
Arz H. W., Pätzold J., Müller P. J., and Moammar M. O. (2003). Influence of Northern Hemisphere climate and global sea level rise on the restricted Red Sea marine environment during termination I. Paleoceanography 18, 1053. doi: 10.1029/2002PA000864
Avetisyan K., Zweig I., Luther G. W., and Kamyshny A. (2021). Kinetics and mechanism of polysulfides and elemental sulfur formation by a reaction between hydrogen sulfide and δ-MnO2. Geochim Cosmochim Acta 313, 21–37. doi: 10.1016/j.gca.2021.08.022
Bao H. (2006). Purifying barite for oxygen isotope measurement by dissolution and reprecipitation in a chelating solution. Anal. Chem. 78, 304–309. doi: 10.1021/ac051568z, PMID: 16383341
Bethke C. M., Sanford R. A., Kirk M. F., Jin Q., and Flynn T. M. (2011). The thermodynamic ladder in geomicrobiology. Am. J. Sci. 311, 183–210. doi: 10.2475/03.2011.01
Blonder B., Boyko V., Turchyn A. V., Antler G., Sinichkin U., Knossow N., et al. (2017). Impact of aeolian dry deposition of reactive iron minerals on sulfur cycling in sediments of the gulf of aqaba. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.01131, PMID: 28676799
Böttcher M. E., Thamdrup B., Gehre M., and Theune A. (2005). 34S/32S and 18O/16O fractionation during sulfur disproportionation by Desulfobulbus propionicus. Geomicrobiol J. 22, 219–226. doi: 10.1080/01490450590947751
Boyko V., Blonder B., and Kamyshny A. (2019). Sources and transformations of iron in the sediments of the Gulf of Aqaba (Red Sea). Mar. Chem. 216, 103691. doi: 10.1016/j.marchem.2019.103691
Boyko V., Pätzold J., and Kamyshny A. (2022). Iron and sulfur speciation and cycling in the sediments of marine systems located in arid environments: the northern Red Sea. J. Geol Soc. London 179, 103691. doi: 10.1144/jgs2021-027
Boyko V., Torfstein A., and Kamyshny A. (2018). Oxygen consumption in permeable and cohesive sediments of the gulf of aqaba. Aquat Geochem 24, 165–193. doi: 10.1007/s10498-018-9338-x
Burdige D. J. and Nealson K. H. (1986). Chemical and microbiological studies of sulfide-mediated manganese reduction. Geomicrobiol J. 4, 361–387. doi: 10.1080/01490458609385944
Cheddadi R., Carré M., Nourelbait M., François L., Rhoujjati A., Manay R., et al. (2021). Early Holocene greening of the Sahara requires Mediterranean winter rainfall. Proc. Natl. Acad. Sci. 118, e2024898118. doi: 10.1073/pnas.2024898118, PMID: 34074769
Duce R. A., Liss P. S., Merrill J. T., Atlas E. L., Buat-Menard P., Hicks B. B., et al. (1991). The atmospheric input of trace species to the world ocean. Global Biogeochem Cycles 5, 193–259. doi: 10.1029/91GB01778
Findlay A. J. and Kamyshny A. (2017). Turnover rates of intermediate sulfur species (Sx2-, S0, S2O32-, S4O62-, SO32-) in anoxic freshwater and sediments. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02551, PMID: 29312234
Fischer J. P., Ferdelman T. G., D`Hoindt S., Røy H., and Wenzhöfer F. (2009). Oxygen penetration deep into the sediment of the South Pacific gyre. Biogeosciences 6, 1467–1478. doi: 10.5194/bg-6-1467-2009
Fossing H. and Jørgensen B. B. (1989). Measurement of bacterial sulfate reduction in sediments: Evaluation of a single-step chromium reduction method. Biogeochemistry 8, 205–222. doi: 10.1007/BF00002889
Glud R. N. (2008). Oxygen dynamics of marine sediments. Mar. Biol. Res. 4, 243–289. doi: 10.1080/17451000801888726
Goto K., Taguchi S., Fukue Y., Ohta K., and Watanabe H. (1977). Spectrophotometric determination of manganese with 1-(2-pyridylazo)-2-naphthol and a non-ionic surfactant. Talanta 24, 752–753. doi: 10.1016/0039-9140(77)80206-3, PMID: 18962190
Goudie A. S. and Middleton N. J. (2001). Saharan dust storms: Nature and consequences. Earth Sci. Rev. 56, 179–204. doi: 10.1016/S0012-8252(01)00067-8
Harrison A. G. and Thode H. G. (1958). Mechanism of the bacterial reduction of sulphate from isotope fractionation studies. Trans. Faraday Soc. 54, 84. doi: 10.1039/tf9585400084
Henrich R., Hanebuth T. J. J., Cherubini Y., Krastel S., Pierau R., and Zühlsdorff C. (2010). “Climate-Induced Turbidity Current Activity in NW-African Canyon Systems,” in Submarine Mass Movements and Their Consequences (Springer Netherlands, Dordrecht), 447–459. doi: 10.1007/978-90-481-3071-9_37
Hinz K., Winterer E. L., Baumgartner P. O., Bradshaw M. J., Channell J. E. T., Jaffrezo M., et al. (1984). “Site 546,” in Initial Reports of the Deep Sea Drilling Project (Washington: U.S Government Printing Office), 79, 179–221. doi: 10.2973/dsdp.proc.79.104.1984
Holmkvist L., Kamyshny A., Vogt C., Vamvakopoulos K., Ferdelman T. G., and Jørgensen B. B. (2011). Sulfate reduction below the sulfate-methane transition in Black Sea sediments. Deep Sea Res. 1 Oceanogr Res. Pap 58, 493–504. doi: 10.1016/j.dsr.2011.02.009
Holz C., Stuut J. W., and Henrich R. (2004). Terrigenous sedimentation processes along the continental margin off NW Africa: implications from grain-size analysis of seabed sediments. Sedimentology 51, 1145–1154. doi: 10.1111/j.1365-3091.2004.00665.x
Johnston D. T., Gill B. C., Masterson A., Beirne E., Casciotti K. L., Knapp A. N., et al. (2014). Placing an upper limit on cryptic marine sulphur cycling. Nature 513, 530–533. doi: 10.1038/nature13698, PMID: 25209667
Jørgensen B. B. (1982). Mineralization of organic matter in the sea bed—the role of sulphate reduction. Nature 296, 643–645. doi: 10.1038/296643a0
Jørgensen B. B. (2021). Sulfur biogeochemical cycle of marine sediments. Geochem Perspect. 10, 145–307. doi: 10.7185/geochempersp.10.2
Kamyshny A. and Ferdelman T. G. (2010). Dynamics of zero-valent sulfur species including polysulfides at seep sites on intertidal sand flats (Wadden Sea, North Sea). Mar. Chem. 121, 17–26. doi: 10.1016/j.marchem.2010.03.001
Katz T., Ginat H., Eyal G., Steiner Z., Braun Y., Shalev S., et al. (2015). Desert flash floods form hyperpycnal flows in the coral-rich Gulf of Aqaba, Red Sea. Earth Planet Sci. Lett. 417, 87–98. doi: 10.1016/j.epsl.2015.02.025
Lancelot Y., Seibold E., Cepek P., Dean W., Eremeev V., Gardner J., et al. (1978a). “Site 368: Cape Verde Rise,” in Initial Reports of the Deep Sea Drilling Project, (La Jolla: U.S Government Printing Office) 41, 233–326. doi: 10.2973/dsdp.proc.41.104.1978
Lancelot Y., Seibold E., Cepek P., Dean W. E., Eremeev V., Gardner J., et al. (1978b). “Site 366: Sierra Leone Rise,” in Initial Reports of the Deep Sea Drilling Project, (La Jolla: U.S. Government Printing Office), 21–161. doi: 10.2973/dsdp.proc.41.102.1978
Laufer K., Michaud A. B., Røy H., and Jørgensen B. B. (2020). Reactivity of iron minerals in the seabed toward microbial reduction–A comparison of different extraction techniques. Geomicrobiol J. 37, 170–189. doi: 10.1080/01490451.2019.1679291
Longinelli A. and Craig H. (1967). Oxygen-18 variations in sulfate ions in sea water and saline lakes. Science 156, 56–59. doi: 10.1126/science.156.3771.56, PMID: 6020038
Lovley D. R. and Phillips E. J. P. (1986). Organic matter mineralization with reduction of ferric iron in anaerobic sediments. Appl. Environ. Microbiol. 51, 683–689. doi: 10.1128/aem.51.4.683-689.1986, PMID: 16347032
Lovley D. R. and Phillips E. J. P. (1987). Competitive mechanisms for inhibition of sulfate reduction and methane production in the zone of ferric iron reduction in sediments. Appl. Environ. Microbiol. 53, 2636–2641. doi: 10.1128/aem.53.11.2636-2641.1987, PMID: 16347483
Luther G. W. (1991). Pyrite synthesis via polysulfide compounds. Geochim Cosmochim Acta 55, 2839–2849. doi: 10.1016/0016-7037(91)90449-F
Lyons T. W. and Severmann S. (2006). A critical look at iron paleoredox proxies: New insights from modern euxinic marine basins. Geochim Cosmochim Acta 70, 5698–5722. doi: 10.1016/j.gca.2006.08.021
Michaud A. B., Laufer K., Findlay A., Pellerin A., Antler G., Turchyn A. V., et al. (2020). Glacial influence on the iron and sulfur cycles in Arctic fjord sediments (Svalbard). Geochim Cosmochim Acta 280, 423–440. doi: 10.1016/j.gca.2019.12.033
Mills J. V., Antler G., and Turchyn A. V. (2016). Geochemical evidence for cryptic sulfur cycling in salt marsh sediments. Earth Planet Sci. Lett. 453, 23–32. doi: 10.1016/j.epsl.2016.08.001
Orcutt B. N., Sylvan J. B., Knab N. J., and Edwards K. J. (2011). Microbial Ecology of the Dark Ocean above, at, and below the Seafloor. Microbiol. Mol. Biol. Rev. 75, 361–422. doi: 10.1128/mmbr.00039-10, PMID: 21646433
Pasquier V., Fike D. A., Révillon S., and Halevy I. (2022). A global reassessment of the controls on iron speciation in modern sediments and sedimentary rocks: A dominant role for diagenesis. Geochim Cosmochim Acta 335, 211–230. doi: 10.1016/j.gca.2022.08.037
Postma D. and Jakobsen R. (1996). Redox zonation: Equilibrium constraints on the Fe(III)/SO4-reduction interface. Geochim Cosmochim Acta 60, 3169–3175. doi: 10.1016/0016-7037(96)00156-1
Poulton S. W. and Canfield D. E. (2005). Development of a sequential extraction procedure for iron: Implications for iron partitioning in continentally derived particulates. Chem. Geol 214, 209–221. doi: 10.1016/j.chemgeo.2004.09.003
Poulton S. W., Krom M. D., and Raiswell R. (2004). A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide. Geochim Cosmochim Acta 68, 3703–3715. doi: 10.1016/j.gca.2004.03.012
Raiswell R. and Canfield D. E. (1998). Sources of iron for pyrite formation in marine sediments. Am. J. Sci. 298, 219–245. doi: 10.2475/ajs.298.3.219
Raiswell R., Hardisty D. S., Lyons T. W., Canfield D. E., Owens J. D., Planavsky N. J., et al. (2018). The iron paleoredox proxies: A guide to the pitfalls, problems and proper practice. Am. J. Sci. 318, 491–526. doi: 10.2475/05.2018.03
Rickard D. and Luther G. W. (1997). Kinetics of pyrite formation by the H2S oxidation of iron (II) monosulfide in aqueous solutions between 25 and 125 °C: The mechanism. Geochim Cosmochim Acta 61, 135–147. doi: 10.1016/S0016-7037(96)00322-5
Ruddiman W., Sarnthein M., and Baldauf J. (1988). “Site 665,” in Proceedings of the Ocean Drilling Program, 108 Initial Reports (Ocean Drilling Program). Texas A&M University. doi: 10.2973/odp.proc.ir.108.110.1988
Schmincke H. U., Weaver P. P. E., Firth J. V., Baraza J., Bristow J. F., Brunner C., et al. (1995a). “Site 954,” in Proceedings of the Ocean Drilling Program, 157 Initial Reports (Ocean Drilling Program), 395–431. College Station, TX. doi: 10.2973/odp.proc.ir.157.108.1995
Schmincke H. U., Weaver P. P. E., Firth J. V., Baraza J., Bristow J. F., Brunner C., et al. (1995b). “Site 952,” in Proceedings of the Ocean Drilling Program, 157 Initial Reports (Ocean Drilling Program), 135–178. College Station, TX. doi: 10.2973/odp.proc.ir.157.106.1995
Skonieczny C., McGee D., Winckler G., Bory A., Bradtmiller L. I., Kinsley C. W., et al. (2019). Monsoon-driven Saharan dust variability over the past 240,000 years. Sci. Adv. 5, eaav1887. doi: 10.1126/sciadv.aav1887, PMID: 30613782
Stookey L. L. (1970). Ferrozine - A new spectrophotometric reagent for iron. Anal. Chem. 42, 779–781. doi: 10.1021/ac60289a016
Thamdrup B. (2000). “Bacterial Manganese and Iron Reduction in Aquatic Sediments,” in Advances in Microbial Ecology 16, 41–84. New York: Springer Science. doi: 10.1007/978-1-4615-4187-5_2
Torfstein A., Teutsch N., Tirosh O., Shaked Y., Rivlin T., Zipori A., et al. (2017). Chemical characterization of atmospheric dust from a weekly time series in the north Red Sea between 2006 and 2010. Geochim Cosmochim Acta 211, 373–393. doi: 10.1016/j.gca.2017.06.007
Turchyn A. V., Sivan O., and Schrag D. P. (2006). Oxygen isotopic composition of sulfate in deep sea pore fluid: Evidence for rapid sulfur cycling. Geobiology 4, 191–201. doi: 10.1111/j.1472-4669.2006.00079.x
Wehrmann L. M., Formolo M. J., Owens J. D., Raiswell R., Ferdelman T. G., Riedinger N., et al. (2014). Iron and manganese speciation and cycling in glacially influenced high-latitude fjord sediments (West Spitsbergen, Svalbard): Evidence for a benthic recycling-transport mechanism. Geochim Cosmochim Acta 141, 628–655. doi: 10.1016/j.gca.2014.06.007
Wehrmann L. M., Riedinger N., Brunner B., Kamyshny A., Hubert C. R. J., Herbert L. C., et al. (2017). Iron-controlled oxidative sulfur cycling recorded in the distribution and isotopic composition of sulfur species in glacially influenced fjord sediments of west Svalbard. Chem. Geol 466, 678–695. doi: 10.1016/j.chemgeo.2017.06.013
Yao W. and Millero F. J. (1993). The rate of sulfide oxidation by δMnO2 in seawater. Geochim Cosmochim Acta 57, 3359–3365. doi: 10.1016/0016-7037(93)90544-7
Yao W. and Millero F. J. (1996). Oxidation of hydrogen sulfide by hydrous Fe(III) oxides in seawater. Mar. Chem. 52, 1–16. doi: 10.1016/0304-4203(95)00072-0
Keywords: Atlantic ocean sediments, Saharan dust, dry aeolian deposition, iron, sulfur, manganese, cryptic sulfur cycling
Citation: Grijalva-Rodriguez TI, Turchyn AV, Albin S, Eliani-Russak E, Manashirov A, Zweig I and Kamyshny A Jr. (2025) Evidence for cryptic sulfur cycling in Atlantic ocean sediments affected by aeolian dry deposition of Saharan dust. Front. Mar. Sci. 12:1663305. doi: 10.3389/fmars.2025.1663305
Received: 10 July 2025; Accepted: 01 September 2025;
Published: 01 October 2025.
Edited by:
Yan Li, China University of Geosciences, ChinaReviewed by:
Dirk De Beer, Max Planck Society, GermanyWilliam Patrick Gilhooly III, Indiana University Indianapolis, United States
Copyright © 2025 Grijalva-Rodriguez, Turchyn, Albin, Eliani-Russak, Manashirov, Zweig and Kamyshny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexey Kamyshny Jr., a2FteXNobnlAYmd1LmFjLmls
†Present addresses: Tomas Israel Grijalva-Rodriguez, Departamento Académico de Ciencias de La Tierra, Universidad Autónoma de Baja California Sur, La Paz, México
Alen Manashirov, Department of Earth and Planetary Sciences, Weizmann Institute of Science, Rehovot, Israel