 Anne Hilgendorff
Anne Hilgendorff Michael A. O’Reilly
Michael A. O’Reilly- 1Comprehensive Pneumology Center, Helmholtz Zentrum München, Member of the German Center for Lung Research (DZL), Munich, Germany
- 2Neonatology, Perinatal Center Grosshadern, Dr. von Hauner Children’s Hospital, Ludwig-Maximilians University, Munich, Germany
- 3Department of Pediatrics, School of Medicine and Dentistry, The University of Rochester, Rochester, NY, USA
Neonatal chronic lung disease, i.e., bronchopulmonary dysplasia, is characterized by impaired pulmonary development resulting from the impact of different risk factors including infections, hyperoxia, and mechanical ventilation on the immature lung. Remodeling of the extracellular matrix, apoptosis as well as altered growth factor signaling characterize the disease. The immediate consequences of these early insults have been studied in different animal models supported by results from in vitro approaches leading to the successful application of some findings to the clinical setting in the past. Nonetheless, existing information about long-term consequences of the identified early and most likely sustained changes to the developing lung is limited. Interesting results point towards a tremendous impact of these early injuries on the pulmonary repair capacity as well as aging related processes in the adult lung.
Clinical Background
The neonatal form of chronic lung disease (CLD), also known as bronchopulmonary dysplasia (BPD), is one of the most common forms of CLD in early infancy. The disease results from the impact of different risk factors on the undeveloped neonatal lung and is associated with a significantly increased risk for pulmonary and neurologic impairment persisting into adulthood in the cohort of formerly preterm infants (1). Defined by the need for supplemental oxygen and/or ventilatory support for >28 days, or beyond 36 weeks post-menstrual age (PMA), the disease can be classified into three different severity grades (mild, moderate, severe) (1).
The incidence of BPD is reported to be up to 77% in infants born at <32 weeks of gestation with a birth weight below 1 kg (2–4) but varies between newborn care centers, reflecting differences in patient population and infant management practices (2, 5–7). Even significant improvements in perinatal care including surfactant treatment, administration of antenatal corticosteroids, and improvement of invasive and non-invasive ventilation strategies could not significantly alter the incidence of long-term sequelae associated with the disease in the most immature infants (8).
Clinically, this form of CLD presents with hypoxemia leading to the need for supplemental O2 as well as hypercapnia, reflecting impaired respiratory gas exchange and alveolar hypoventilation, resulting in a mismatch of ventilation and perfusion (9). Lung function is characterized by diminished compliance, tachypnea, increased minute ventilation, and work of breathing and can be accompanied by an increase in lung microvascular filtration pressure leading to interstitial pulmonary edema (10). As a result of increased respiratory tract resistance and hyper-reactive airways, episodic bronchoconstriction and cyanosis can be observed (11), with early lung function impairment indicating more severe disease at term (12). The increased lung vascular resistance, typically associated with impaired responsiveness to inhaled nitric oxide and other vasodilators, can progress to reversible or sustained pulmonary hypertension and right heart failure (13, 14).
As a trigger for the onset of these pathophysiologic processes, large clinical trials have identified important risk factors (15–20). Besides postnatal infections, the requirement for prolonged assisted ventilation to treat acute respiratory failure caused by primary surfactant deficiency and the need for oxygen supplementation is known to injure the structural and functional immature lung (21–24). Here, the use of large tidal volumes and high inflation pressures, in concert with the magnitude and duration of exposure to supplemental oxygen, are major risk factors for disease development (25, 26). The occurence of pulmonary complications, e.g., air leaks, interstitial emphysema, and pneumothoraces, further increase the risk (25). With respect to postnatal growth and development, poor nutritional support, vitamin deficiency as well as insufficient adrenal and thyroid hormone release in the very premature infant is known to significantly contribute to adverse pulmonary outcome (27–29).
These postnatal stressors are known to act beyond the background of both prenatal as well as genetic risk factors influencing the capacity of the developing lung to respond to the indicated postnatal injuries:
Intrauterine growth retardation increases the risk of BPD three to fourfold (30–34), most likely through impaired alveolar and vascular growth associated with altered growth factor signaling (35). The prenatal impact of cytokines, in the presence, i.e., chorioamnionitis or absence, i.e., fetal inflammatory response syndrome of other signs of infection is known to prime the lung for a pathologic response to postnatal injury, thereby contributing to BPD development (36).
Regarding the impact of the genetic background, a study investigating twin preterm infants found genetic factors to account for 53% of the variance in liability for BPD (37). Several potential candidate genes have been associated with BPD, where genetic variants predisposing to BPD are usually polymorphisms, which are not causative, but have been shown to increase disease susceptibility. Genetic abnormalities include variations affecting the surfactant system or the innate immune response (38, 39). Such conclusions are consistent with a genome-wide association study involving more than 1700 very low birth weight infants that was not able to link specific genomic loci or pathways with BPD (40). Instead, gene linkage studies found that one of the corticotropin-releasing hormone receptors (CRHR1) or cytochrome P450 2E1 (CYP2E1) in the preterm infant, and ectonucleotide pyrophosphatase 1 (ENPP1), insulin-like growth factor binding protein 3 (IFGBP3), 7-dehydrocholesterol reductase (DHCR7), or tumor necrosis factor-associated factor 2 (TRAF2) in the mother were highly associated with preterm birth (41). Nonetheless, gestational age at birth still remains one of the most accurate ways to predict the incidence of BPD (42). In addition, male preterm infants are at a higher risk for the development of long-term impairment, including BPD (43), and premature changes to hormonal regulation have been discussed as an underlying cause (44). The effect of gender seems to be different with respect to the adult population, as female adult BPD patients are more severely affected with respect to developing long-term pulmonary impairment (45).
Animal models were instrumental in elucidating some of the underlying mechanisms by which the indicated risk factors resulted in profound and durable structural changes in the developing lung and will be discussed in the pathophysiologic context.
Pathophysiologic Characteristics
With respect to histopathology, neonatal CLD is characterized by impaired alveolarization and vascularization (1). Alveolarization in humans begins during late fetal development and continues into early childhood (46). Although continued growth of new alveoli was observed in rhesus monkeys up into adulthood (47), there is increasing evidence in both humans and rodents suggesting alveolar development to occur in two phases (46, 48, 49). The majority of alveoli develops in the early phase and occurs as double micro-capillaries mature into a single capillary network. This phase is followed by ongoing alveolar growth arising from existing alveoli. Knowledge about these two distinct phases in alveolar development is critical to design therapeutic approaches aiming to promote alveolar development in the injured neonatal lung. As a well-known example, postnatal corticosteroids have been used to promote lung function, facilitating extubation and thereby trying to prevent or ameliorate BPD development. Although their anti-inflammatory effects are potent (50), dexamethasone administered to newborn rats has been shown to cause alveolar thinning and structural simplification, presumably by inhibiting the early phase of alveolar development (51). Especially, the impact of postnatal steroid therapy on extracellular matrix (ECM) composition as observed in rats may account for long-term effects (52). Not only is the window of lung development a critical variable for the decision on postnatal steroid therapy, but the increased risk for gastrointestinal bleeding, cardiovascular disease, and cerebral palsy in infants that had received steroid therapy (53, 54), strongly limits dose, time-point, and length of treatment.
Besides the continuous changes in pre- and postnatal treatment strategies in preterm care, the altered histo- and pathophysiological picture of BPD over the last decades is mainly due to the degree of immaturity in the preterm cohort studied. As the disease was largely an atelectatic fibrosing disease attributed to persistent oxygen exposure in late preterm infants (>34–36 weeks) in the pre-surfactant era (55), the implementation of exogenous surfactant administration as well as improved ventilation strategies and the induction of lung maturation by prenatal steroids significantly increased the survival of infants as immature as 23–24 weeks of gestation. Although the amount of interstitial fibrosis is substantially less in these infants and tends to be more diffuse when compared to histopathology in the pre-surfactant era, extensive ECM remodeling together with increased smooth muscle in small pulmonary arteries and airways (56) remains a key finding in the diseased lung.
Many studies have shown that characteristic inflammatory changes and altered growth factor signaling precede and accompany these changes to the pulmonary scaffold that may not only hinder physiologic lung development but also transfer its long-term effects into adulthood. The characteristic degradation of lung elastin that accompanies its pathologic distribution pattern in infants who later acquire BPD manifests in increased urinary excretion of desmosin, a breakdown product of the mature elastic fiber, preceded and paralleled by an associated increase in protease activity (57–62). Nonetheless, studies in an inflammatory model of BPD have shown that a delicate balance of protease activity is critical for normal lung development, as complete matrix-metalloproteinase deficiency worsened lung injury (63). Experimental studies in rodents recapitulated the changes to the ECM and allowed further insight into disease relevant pathophysiology, linking ECM remodeling to apoptosis, inflammation, and growth factor signaling (64–67). First attempts were made in order to therapeutically prevent ECM degradation, thereby preserving lung growth in the presence of mechanical ventilation (68, 69).
Pulmonary inflammation induced by both non-infectious processes, such as positive-pressure ventilation or oxygen therapy aggravated by primary surfactant deficiency, or patent ductus arteriosus, as well as pre- or postnatal infections play an important role in the translation of different injury mechanisms to structural and functional changes as well as in the aggravation of ongoing pathologic processes in the developing lung (18, 22, 70, 71). The characteristic initial influx of neutrophils into the lung is accompanied and followed by increased numbers of macrophages in the course of the disease (32, 72, 73). Explaining the perpetuation of the inflammatory response, animal studies indicate that lung injury leading to ECM remodeling or early alveolar epithelial dysfunction further promotes lung inflammation (68, 74). In contrast, inflammation in fetal sheep infected with Ureaplasma parvum, a common microorganism present in chorioaminionitis, did not affect lung development (75), suggesting that inflammation acts in concert with other risk factors to provoke the development of BPD.
The release of cytokines and disturbance of growth factor signaling, [e.g., transforming growth factor (TGF)-β], leads to the activation of different transcription factors, and results in a characteristic increase in apoptosis affecting all different cell types (76). With relevance to the mesenchymal cells, the platelet derived growth factor-α, characterizing the myofibroblast driving secondary crest formation, and the fibroblast growth factor family [for review see Ref. (77)] are known to be of importance. The presence of dysmorphic capillaries is related to an altered pattern of angiogenic growth factors such as reduced expression of the vascular endothelial growth factor (VEGF) and its receptors (78–80) in the lung, accompanied by diminished endothelial nitric-oxide synthase (eNOS) and soluble guanylate cyclase (sGC) in lung blood vessels and airways (81, 82). These changes contribute to subsequent development of pulmonary hypertension and impaired lung lymphatic drainage (13, 14).
The growing evidence that prenatal factors impact on the incidence of childhood asthma (83) further underlines the importance of studies focusing on the role of prenatal variables in respiratory development following premature birth including the broad issue of maternal health. Suggested by a study in newborn mice exposed to hyperoxia after maternal exposure to LPS during pregnancy, the enhancement of cardiovascular disease in the animals points to an impact of chorioamnionitis not only on surfactant production but also on cardiovascular health in children born premature undergoing early oxygen treatment (84).
Relative deficiencies of anti-oxidants and inhibitors of proteolytic enzymes render the very immature lung, especially vulnerable to the effects of toxic oxygen metabolites and proteases released by the ECM, resident lung cells, or activated neutrophils and macrophages (85–88). Elevated urinary malondialdehyde concentrations in the first week of life, generated by peroxidation of lipid membranes after oxidant-mediated injury, were correlated with the risk for oxygen radical diseases including BPD (89). The association of genetic polymorphisms of the superoxide dismutase with the development of BPD underlines the importance of a balanced redox system (90).
To understand how oxygen affects lung development, non-human primates, preterm sheep, newborn guinea pigs, and newborn rodents have been exposed to excessive levels of oxygen [for review in Ref. (91)]. Early-life oxygen exposure leads to many characteristic pathologic features of the so called “new” BPD, including inhibition of microvascular development, alveolar simplification, inflammation, and mild interstitial thickening [Figure 1 and Ref. (92, 93)]. It also recapitulates many diseases in children who were born preterm, including altered host response to respiratory viral infections, mild cognitive changes, and cardiovascular disease. Despite the widespread use of hyperoxia as a tool to study BPD, a codified model of oxygen exposure has yet to be established, making the extrapolation of outcomes between different investigators using different doses and durations of hyperoxia on different developmental windows possible (91). Today, preterm infants are often exposed to excess oxygen during the saccular stage of lung development, and discharged breathing room air when entering the first phase of completion in alveolarization. In order to mimic these clinically relevant conditions, the influence of oxygen on saccular stages in lung development, i.e., mouse E17.5 to PN should be separated from its impact on the phase of alveolarization, i.e., mouse PN5–PN14 (94). Models using oxygen exposure during alveolar development may therefore better resume the characteristic picture of the so called “old BPD,” the scarring lung disease seen in infants born in late gestation in the pre-surfactant era. The process of organ maturation clearly is a modifier of the pulmonary response to oxygen exposure, as hyperoxia has been shown to reduce bone marrow, circulating and lung endothelial progenitor cells in the developing but not in the adult mouse lung (95). Additionally, different oxygen concentrations affected lung development and the host response to influenza A virus in neonatal mice (96). Hence, a better understanding of how dose and duration of the respective harmful agent interfere with a certain developmental window is important to make progress in the development of treatment strategies that could improve pulmonary health in preterm infants.
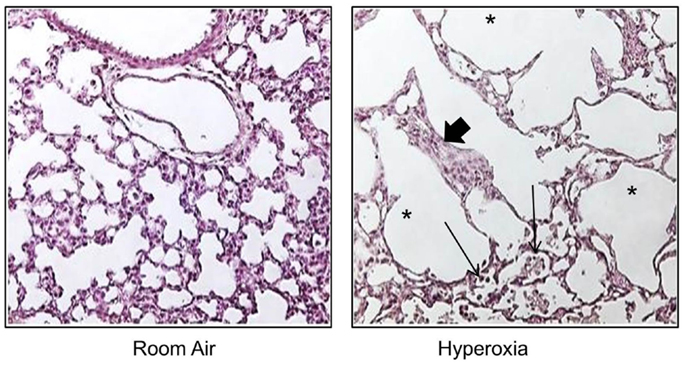
Figure 1. Neonatal hyperoxia disrupts postnatal alveolar development in the lung. Representative tissue slides (H&E stains) of newborn mouse lung exposed to room air or 100% oxygen from birth to PN10. Thickened alveolar septae (thick arrow), inflammatory cells (thin arrow), and simplified alveoli (asterisks).
Long-Term Consequences
Minimizing long-term pulmonary impairment, and neurologic complications associated with BPD has become the main focus of perinatal care (97, 98). Nonetheless, increasing evidence suggests that the early pathologic changes observed in BPD contribute to long-lasting consequences including premature aging of the lung.
Although only some affected infants remain oxygen dependent beyond 2 years of age, oxygen dependency for months or years is frequently described (99, 100) indicating the most severe lung disease. These infants require hospital readmission twice as often compared to infants who are not oxygen dependent. Even after having outgrown the need for oxygen supply, high readmission rates remain common in infants with BPD, with up to 70% requiring a hospital stay in the first 2 years of life (101). Here, infections with respiratory syncytial virus are the major cause for readmission among preterm infants regardless of BPD status (102). Furthermore, patients with moderate or severe BPD suffer more frequently from episodes of wheezing and need for inhalation therapies (12). Up to 80% of these infants demonstrate airway obstruction in early childhood and adolescence, with the majority being symptomatic (103–105). About 20–30% of infants with BPD suffer from those symptoms at 6 and 12 months of age (106, 107) and respiratory symptoms remain common at preschool and school age (99, 108). The most severely affected children remain symptomatic into adulthood (109). The described clinical symptoms are underlined by changes in pulmonary function as identified by clinical studies, suggesting that the presence of persistent airflow limitations and reduction in alveolar surface area as well as impaired lung growth, significantly lower peak oxygen consumption, forced expiratory volume at 1 s (FEV1), and gas transfer at school age after premature birth are accompanied by significantly lower peak workload, higher respiratory rates in combination with lower tidal volumes during peak exercise (110) as well as lower lung volumes and decreased gas mixing efficiency during infancy in BPD patients (111, 112).
As young adult BPD survivors present with a reduced maximal airway function and some even show a trend towards an early and steeper decline in lung function with age, the concern was raised that BPD may be a precursor of a COPD-like phenotype later in life (113) although the incidence has yet to be defined in this patient cohort (114).
With respect to the pathophysiologic concepts discussed, the effect of injury mechanism and treatment regimen has to be considered in the context of different stages in pulmonary development. Here, the inhibition of secondary alveolar formation in patients with BPD could result in premature lung structure degradation with age. Furthermore, the sustained and potentially irreversible reorganization of the ECM as observed after mechanical ventilation, oxygen exposure or steroid therapy may affect its function as a scaffold for lung development as well as its long-term repair potential, both resulting in long-term consequences. Latest studies have shown a “memory function” of the ECM as the fate of different pulmonary cell types re-populating the pulmonary scaffold was defined by its composition (115, 116). With respect to the lung vasculature, changes in angiogenic growth factor expression resemble the pattern observed in aged mice and are associated with reduced plasticity of the lung capillaries, potentially leading to sustained changes for lung development and injury response through life (117, 118). In line with this, oxygen exposure in the first week of life (FiO2 1.0) has been shown to increase mortality by inducing pulmonary vascular disease in mice (119). Again, maturational differences need to be considered and studied in more detail with respect to their initial impact and their consequences for aging related processes. Whereas oxygen enhances lung vascular and airway smooth muscle contraction and reduces nitric-oxide relaxation in the neonatal rat lung, the opposite occurs in the adult animal (120). These changes are associated with altered lung function and right ventricular hypertrophy at 10 months of age, indicating significant pulmonary hypertension. At this late stage, bone morphogenetic protein signaling is altered and may contribute to the cardiovascular phenotype observed in the adult lung. Furthermore, recent studies suggest that long-term respiratory abnormalities after preterm birth may be associated with a sustained alteration of the oxidative stress response. Here, adolescent BPD patients show evidence of heightened oxidative stress in the airways (121). Likewise, the early interference with different transcription factors disrupts normal lung morphogenesis in fetal life (122), subsequently resulting in pulmonary emphysema in adult mice preluded by severe chronic bronchial inflammation (123). Underlining these findings, studies showed that suppression of the nuclear factor kappa B (NF-κB) worsened pathophysiologic changes that potentially lead to BPD development, e.g., impaired alveolarization and vascularization (122, 124, 125). Studies focusing on the differential response to injury in the developing lung will help to broaden the understanding of identified targets and processes with a critical role in adult lung diseases.
With respect to early treatment regimen, different findings indicate the need for a careful investigation of their potential to induce long-term effects. As an example, the prenatal administration of betamethasone, although widely used to enhance lung maturation thereby preventing respiratory distress and reducing BPD rates (126, 127), has been shown to be associated with an increase in lipid membrane peroxidation (89). In addition, postnatal dexamethasone treatment in the neonate, besides its negative effects on neurologic and alveolar development, led to systolic dysfunction and reduced life expectancy in elderly rats (128, 129). Furthermore, the broad use of antibiotic treatment in the mother at risk for premature delivery leads to a sustained alteration of the bacterial flora of the child (130), affecting immune function long-term as shown in neonatal in contrast to adult-germ free mice (131).
Some therapeutic agents with suggested immediate effects on the redox system, displayed their treatment potential more with respect to middle and long-term effects (132, 133). Although improving oxygenation and pulmonary vascular development in preterm sheep, intra-tracheal administration of recombinant human CuZn superoxide dismutase did not diminish the incidence of BPD, but reduced respiratory morbidity at 1 year of age and the incidence of retinopathy (ROP) (134–136). As inflammatory cells recruited to the sites of oxidative damage or attracted during infections are very likely to contribute to disease development, a combination of anti-oxidant and anti-inflammatory therapies may be most efficacious for the treatment of the structural and functional immature lung in the preterm infant. Here, studies in newborn mice showed that blocking neutrophil influx using anti-CINC antibody in diminished hyperoxia-induced DNA damage and alveolar simplification (137, 138).
As oxygen is a leading risk factor but as well remains the main treatment option in infants suffering from impaired lung growth when diagnosed with BPD, the definition of the adequate dose for oxygen therapy will remain of critical importance. Several clinical trials have attempted to identify adequate therapeutic oxygen concentrations providing maximal benefit with minimal harm. Early trials found that the incidence of ROP correlated with unrestricted use of oxygen (139) and the benefits of oxygen-saturation targeting (BOOST) trial found that a higher oxygen-saturation range prolonged oxygen dependence (140). However, the surfactant, positive pressure, and pulse oximetry randomized trial (SUPPORT) showed in 1300 infants between 24 and 28 weeks gestation treated with low oxygen saturation (85–89%) to have reduced ROP but higher mortality compared to infants with high (91–95%) oxygen-saturation target (141). These findings were confirmed in the BOOST II trial, which enrolled 2400 infants at 54 hospitals in United Kingdom, Australia, and New Zealand (142). Finally, the supplemental therapeutic oxygen for pre-threshold retinopathy of prematurity (STOP-ROP) trial found high oxygen-saturation levels did not increase the severity of ROP in those infants with pre-threshold ROP (143). Taken together, preterm infants are likely to be treated with high oxygen saturations despite the increased risk for ROP. With respect to its effect on lung function, infants with the highest quartile of oxygen exposure as a cumulative dose of inhaled gas over the first 3 days of life were two to three times more likely to experience symptomatic airway dysfunction than infants in the lowest quartile (144). In line with this quantifying cumulative oxygen exposure in newborn mice can successfully predict an altered host response of adult mice infected with influenza A virus infection (145). These findings suggest that the quantification of FiO2 may be a better indicator of oxygen toxicity and risk for long-term respiratory morbidity than pAO2 or days on oxygen therapy.
Figure 2 summarizes some important pathophysiologic processes and long-term consequences following early pulmonary injury dicussed here.
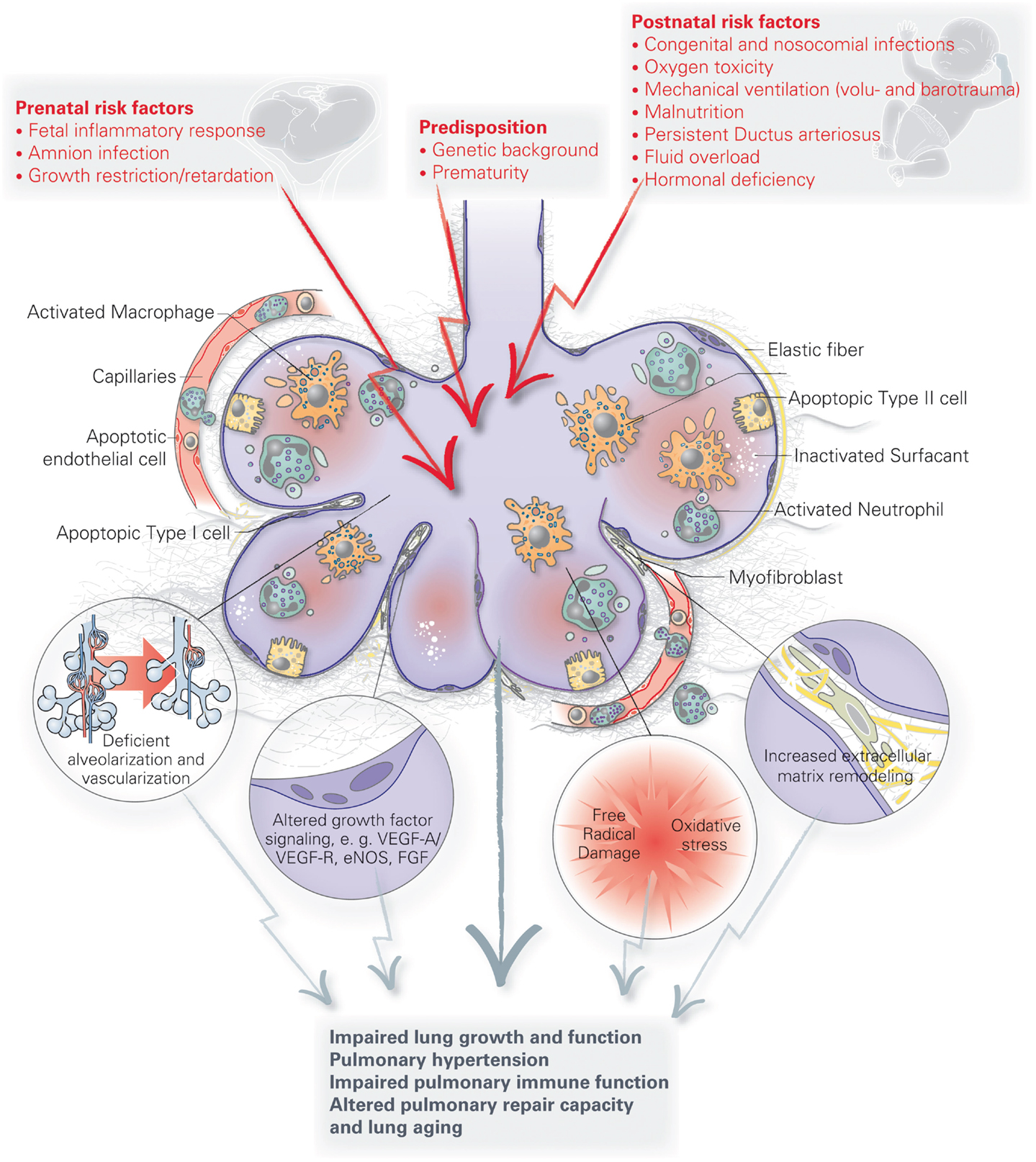
Figure 2. Summary of important pathophysiologic processes and long-term consequences following early pulmonary injury.
In order to consequently evaluate existing and further develop new treatment strategies, significant findings from disease models in rodents, allowing for the comprehensive investigation of pathophysiologic processes and long-term effects, need to be re-evaluated in animal models that very closely mimic clinical conditions in the preterm infant. Here, the premature baboon model, originally developed in the pre-surfactant era, has been shown to be very meaningful in the investigation of treatment strategies including antenatal exposure to maternal glucocorticoids, postnatal surfactant treatment, and assisted ventilation with more modest inflating pressures and concentrations of inspired oxygen (146). A similar strategy was followed in the studies performed in premature lambs (57, 81, 146–155). These studies in larger animals were integral in allowing mechanistic insight into the pathology of BPD, and permitting the evaluation of therapeutic strategies currently used in the care of premature infants, including surfactant replacement therapy (156, 157), high-frequency oscillatory ventilation (HFOV) (158), and nitric oxide (152). Further modification of these models to allow for the investigation of long-term effects as well as the evaluation of results obtained from clinical trials may provide more insight into the pros and cons of current and future treatment strategies.
To conclude, the unique response to injury observed in the immature lung including the effects on oxidative stress, ECM composition, growth factor signaling, and the sustained inflammatory response translate into a characteristic histophathologic picture. Furthermore, these changes rather contribute to a pulmonary “memory effect” than being erased over time. We now understand that early organ injury in the lung provokes pulmonary long-term consequences including alteration of physiologic aging processes as well as a characteristic response to challenges imposed on the adult lung. These considerations as well as the possible impact of an injured lung on the process of extra-pulmonary organ developmental, i.e., the brain have to be taken into account, when treatment strategies are designed and life-style issues are advocated to this patient population. Furthermore, the translation of the indicated findings into other fields of lung research and back holds great potential to inspire future ideas for the process of repair and re-programing in the diseased neonatal and adult lung.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the technical assistance of Min Yee who provided the histologic image of neonatal mouse lungs exposed to room air and hyperoxia. This work was supported in part by the National Institutes of Health grant HL091968.
References
1. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med (2001) 163:1723–9. doi:10.1164/ajrccm.163.7.2011060
2. Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, et al. Neonatal outcomes of extremely preterm infants from the nichd neonatal research network. Pediatrics (2010) 126:443–56. doi:10.1542/peds.2009-2959
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Johnson AH, Peacock JL, Greenough A, Marlow N, Limb ES, Marston L, et al. High-frequency oscillatory ventilation for the prevention of chronic lung disease of prematurity. N Engl J Med (2002) 347:633–42. doi:10.1056/NEJMoa020432
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Ehrenkranz RA, Walsh MC, Vohr BR, Jobe AH, Wright LL, Fanaroff AA, et al. Validation of the national institutes of health consensus definition of bronchopulmonary dysplasia. Pediatrics (2005) 116:1353–60. doi:10.1542/peds.2005-0249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Gortner L, Misselwitz B, Milligan D, Zeitlin J, Kollee L, Boerch K, et al. Rates of bronchopulmonary dysplasia in very preterm neonates in Europe: results from the mosaic cohort. Neonatology (2011) 99:112–7. doi:10.1159/000313024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Vachon E, Bourbonnais Y, Bingle CD, Rowe SJ, Janelle MF, Tremblay GM. Anti-inflammatory effect of pre-elafin in lipopolysaccharide-induced acute lung inflammation. Biol Chem (2002) 383:1249–56. doi:10.1515/BC.2002.138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Van Marter LJ, Pagano M, Allred EN, Leviton A, Kuban KC. Rate of bronchopulmonary dysplasia as a function of neonatal intensive care practices. J Pediatr (1992) 120:938–46. doi:10.1016/S0022-3476(05)81968-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Doyle LW. Evaluation of neonatal intensive care for extremely-low-birth-weight infants. Semin Fetal Neonatal Med (2006) 11:139–45. doi:10.1016/j.siny.2005.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Lopez E, Mathlouthi J, Lescure S, Krauss B, Jarreau PH, Moriette G. Capnography in spontaneously breathing preterm infants with bronchopulmonary dysplasia. Pediatr Pulmonol (2011) 46:896–902. doi:10.1002/ppul.21445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Petersson J, Glenny RW. Gas exchange and ventilation-perfusion relationships in the lung. Eur Respir J (2014) 44(4):1023–41. doi:10.1183/09031936.00037014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Hilgendorff A, Reiss I, Gortner L, Schuler D, Weber K, Lindemann H. Impact of airway obstruction on lung function in very preterm infants at term. Pediatr Crit Care Med (2008) 9:629–35. doi:10.1097/PCC.0b013e31818d17c8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. May C, Kennedy C, Milner AD, Rafferty GF, Peacock JL, Greenough A. Lung function abnormalities in infants developing bronchopulmonary dysplasia. Arch Dis Child (2011) 96:1014–9. doi:10.1136/adc.2011.212332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Kinsella JP, Greenough A, Abman SH. Bronchopulmonary dysplasia. Lancet (2006) 367:1421–31. doi:10.1016/S0140-6736(06)68615-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Steinhorn RH. Neonatal pulmonary hypertension. Pediatr Crit Care Med (2010) 11:S79–84. doi:10.1097/PCC.0b013e3181c76cdc
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Korhonen P, Tammela O, Koivisto AM, Laippala P, Ikonen S. Frequency and risk factors in bronchopulmonary dysplasia in a cohort of very low birth weight infants. Early Hum Dev (1999) 54:245–58. doi:10.1016/S0378-3782(98)00101-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Clyman R, Cassady G, Kirklin JK, Collins M, Philips JB III. The role of patent ductus arteriosus ligation in bronchopulmonary dysplasia: reexamining a randomized controlled trial. J Pediatr (2009) 154:873–6. doi:10.1016/j.jpeds.2009.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Oh W, Poindexter BB, Perritt R, Lemons JA, Bauer CR, Ehrenkranz RA, et al. Association between fluid intake and weight loss during the first ten days of life and risk of bronchopulmonary dysplasia in extremely low birth weight infants. J Pediatr (2005) 147:786–90. doi:10.1016/j.jpeds.2005.06.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Kramer BW. Antenatal inflammation and lung injury: prenatal origin of neonatal disease. J Perinatol (2008) 28(Suppl 1):S21–7. doi:10.1038/jp.2008.46
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Stevens TP, Harrington EW, Blennow M, Soll RF. Early surfactant administration with brief ventilation vs. Selective surfactant and continued mechanical ventilation for preterm infants with or at risk for respiratory distress syndrome. Cochrane Database Syst Rev (2007) 4:CD003063.
20. Mittendorf R, Covert R, Montag AG, elMasri W, Muraskas J, Lee KS, et al. Special relationships between fetal inflammatory response syndrome and bronchopulmonary dysplasia in neonates. J Perinat Med (2005) 33:428–34. doi:10.1515/JPM.2005.076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Jobe AH, Ikegami M. Mechanisms initiating lung injury in the preterm. Early Hum Dev (1998) 53:81–94. doi:10.1016/S0378-3782(98)00045-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Watterberg KL, Demers LM, Scott SM, Murphy S. Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics (1996) 97:210–5.
23. Hislop AA, Wigglesworth JS, Desai R, Aber V. The effects of preterm delivery and mechanical ventilation on human lung growth. Early Hum Dev (1987) 15:147–64. doi:10.1016/0378-3782(87)90003-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Brew N, Hooper SB, Allison BJ, Wallace MJ, Harding R. Injury and repair in the very immature lung following brief mechanical ventilation. Am J Physiol Lung Cell Mol Physiol (2011) 301:L917–26. doi:10.1152/ajplung.00207.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Wheeler KI, Klingenberg C, Morley CJ, Davis PG. Volume-targeted versus pressure-limited ventilation for preterm infants: a systematic review and meta-analysis. Neonatology (2011) 100:219–27. doi:10.1159/000326080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Saugstad OD, Aune D. In search of the optimal oxygen saturation for extremely low birth weight infants: a systematic review and meta-analysis. Neonatology (2011) 100:1–8. doi:10.1159/000322001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Biniwale MA, Ehrenkranz RA. The role of nutrition in the prevention and management of bronchopulmonary dysplasia. Semin Perinatol (2006) 30:200–8. doi:10.1053/j.semperi.2006.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Shenai JP, Chytil F, Stahlman MT. Vitamin a status of neonates with bronchopulmonary dysplasia. Pediatr Res (1985) 19:185–8. doi:10.1203/00006450-198502000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Watterberg KL, Scott SM. Evidence of early adrenal insufficiency in babies who develop bronchopulmonary dysplasia. Pediatrics (1995) 95:120–5.
30. Regev RH, Lusky A, Dolfin T, Litmanovitz I, Arnon S, Reichman B. Excess mortality and morbidity among small-for-gestational-age premature infants: a population-based study. J Pediatr (2003) 143:186–91. doi:10.1067/S0022-3476(03)00181-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Reiss I, Landmann E, Heckmann M, Misselwitz B, Gortner L. Increased risk of bronchopulmonary dysplasia and increased mortality in very preterm infants being small for gestational age. Arch Gynecol Obstet (2003) 269:40–4. doi:10.1007/s00404-003-0486-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Walsh MC, Yao Q, Horbar JD, Carpenter JH, Lee SK, Ohlsson A. Changes in the use of postnatal steroids for bronchopulmonary dysplasia in 3 large neonatal networks. Pediatrics (2006) 118:e1328–35. doi:10.1542/peds.2006-0359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Rieger-Fackeldey E, Schulze A, Pohlandt F, Schwarze R, Dinger J, Lindner W. Short-term outcome in infants with a birthweight less than 501 grams. Acta Paediatr (2005) 94:211–6. doi:10.1111/j.1651-2227.2005.tb01893.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Bose C, Van Marter LJ, Laughon M, O’Shea TM, Allred EN, Karna P, et al. Fetal growth restriction and chronic lung disease among infants born before the 28th week of gestation. Pediatrics (2009) 124:e450–8. doi:10.1542/peds.2008-3249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Rozance PJ, Seedorf GJ, Brown A, Roe GB, O’Meara MC, Gien J, et al. Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am J Physiol Lung Cell Mol Physiol (2011) 301(6):L860–71. doi:10.1152/ajplung.00197.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Kramer BW, Kallapur S, Newnham J, Jobe AH. Prenatal inflammation and lung development. Semin Fetal Neonatal Med (2009) 14:2–7. doi:10.1016/j.siny.2008.08.011
37. Bhandari V, Bizzarro MJ, Shetty A, Zhong X, Page GP, Zhang H, et al. Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics (2006) 117:1901–6. doi:10.1542/peds.2005-1414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Hallman M, Haataja R. Genetic influences and neonatal lung disease. Semin Neonatol (2003) 8:19–27. doi:10.1016/S1084-2756(02)00196-3
39. Hilgendorff A, Heidinger K, Pfeiffer A, Bohnert A, Konig IR, Ziegler A, et al. Association of polymorphisms in the mannose-binding lectin gene and pulmonary morbidity in preterm infants. Genes Immun (2007) 8:671–7. doi:10.1038/sj.gene.6364432
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Wang H, St Julien KR, Stevenson DK, Hoffmann TJ, Witte JS, Lazzeroni LC, et al. A genome-wide association study (gwas) for bronchopulmonary dysplasia. Pediatrics (2013) 132:290–7. doi:10.1542/peds.2013-0533
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Bream EN, Leppellere CR, Cooper ME, Dagle JM, Merrill DC, Christensen K, et al. Candidate gene linkage approach to identify DNA variants that predispose to preterm birth. Pediatr Res (2013) 73:135–41. doi:10.1038/pr.2012.166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Laughon MM, Langer JC, Bose CL, Smith PB, Ambalavanan N, Kennedy KA, et al. Prediction of bronchopulmonary dysplasia by postnatal age in extremely premature infants. Am J Respir Crit Care Med (2011) 183:1715–22. doi:10.1164/rccm.201101-0055OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Binet ME, Bujold E, Lefebvre F, Tremblay Y, Piedboeuf B. Role of gender in morbidity and mortality of extremely premature neonates. Am J Perinatol (2012) 29(3):159–66. doi:10.1055/s-0031-1284225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Trotter A, Maier L, Kron M, Pohlandt F. Effect of oestradiol and progesterone replacement on bronchopulmonary dysplasia in extremely preterm infants. Arch Dis Child Fetal Neonatal Ed (2007) 92:F94–8. doi:10.1136/adc.2006.097170
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Vrijlandt EJ, Gerritsen J, Boezen HM, Duiverman EJ. Gender differences in respiratory symptoms in 19-year-old adults born preterm. Respir Res (2005) 6:117. doi:10.1186/1465-9921-6-117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Zeltner TB, Burri PH. The postnatal development and growth of the human lung. II. Morphology. Respir Physiol (1987) 67:269–82. doi:10.1016/0034-5687(87)90057-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Hyde DM, Blozis SA, Avdalovic MV, Putney LF, Dettorre R, Quesenberry NJ, et al. Alveoli increase in number but not size from birth to adulthood in rhesus monkeys. Am J Physiol Lung Cell Mol Physiol (2007) 293:L570–9. doi:10.1152/ajplung.00467.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Schittny JC, Mund SI, Stampanoni M. Evidence and structural mechanism for late lung alveolarization. Am J Physiol Lung Cell Mol Physiol (2008) 294:L246–54. doi:10.1152/ajplung.00296.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Tschanz SA, Salm LA, Roth-Kleiner M, Barre SF, Burri PH, Schittny JC. Rat lungs show a biphasic formation of new alveoli during postnatal development. J Appl Physiol (2014) 117:89–95. doi:10.1152/japplphysiol.01355.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Jobe AH. Postnatal corticosteroids for bronchopulmonary dysplasia. Clin Perinatol (2009) 36:177–88. doi:10.1016/j.clp.2008.09.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Massaro D, Teich N, Maxwell S, Massaro GD, Whitney P. Postnatal development of alveoli. Regulation and evidence for a critical period in rats. J Clin Invest (1985) 76:1297–305. doi:10.1172/JCI112103
52. Roth-Kleiner M, Berger TM, Gremlich S, Tschanz SA, Mund SI, Post M, et al. Neonatal steroids induce a down-regulation of tenascin-c and elastin and cause a deceleration of the first phase and an acceleration of the second phase of lung alveolarization. Histochem Cell Biol (2014) 141:75–84. doi:10.1007/s00418-013-1132-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Doyle LW, Ehrenkranz RA, Halliday HL. Early (< 8 days) postnatal corticosteroids for preventing chronic lung disease in preterm infants. Cochrane Database Syst Rev (2014) 5:CD001146. doi:10.1002/14651858.CD001146.pub4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Halliday HL, Ehrenkranz RA, Doyle LW. Early (> 8 days) postnatal corticosteroids for preventing chronic lung disease in preterm infants. Cochrane Database Syst Rev (2010) 1:CD001146. doi:10.1002/14651858.CD001146.pub3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Northway WH Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. bronchopulmonary dysplasia. N Engl J Med (1967) 276:357–68. doi:10.1056/NEJM196702162760701
56. Husain AN, Siddiqui NH, Stocker JT. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol (1998) 29:710–7. doi:10.1016/S0046-8177(98)90280-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Pierce RA, Albertine KH, Starcher BC, Bohnsack JF, Carlton DP, Bland RD. Chronic lung injury in preterm lambs: disordered pulmonary elastin deposition. Am J Physiol (1997) 272:L452–60.
58. Thibeault DW, Mabry SM, Ekekezie II, Truog WE. Lung elastic tissue maturation and perturbations during the evolution of chronic lung disease. Pediatrics (2000) 106:1452–9. doi:10.1542/peds.106.6.1452
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Thibeault DW, Mabry SM, Ekekezie II, Zhang X, Truog WE. Collagen scaffolding during development and its deformation with chronic lung disease. Pediatrics (2003) 111:766–76. doi:10.1542/peds.111.4.766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Bruce MC, Schuyler M, Martin RJ, Starcher BC, Tomashefski JF Jr, Wedig KE. Risk factors for the degradation of lung elastic fibers in the ventilated neonate. Implications for impaired lung development in bronchopulmonary dysplasia. Am Rev Respir Dis (1992) 146:204–12. doi:10.1164/ajrccm/146.1.204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Bruce MC, Wedig KE, Jentoft N, Martin RJ, Cheng PW, Boat TF, et al. Altered urinary excretion of elastin cross-links in premature infants who develop bronchopulmonary dysplasia. Am Rev Respir Dis (1985) 131:568–72.
62. Merritt TA, Cochrane CG, Holcomb K, Bohl B, Hallman M, Strayer D, et al. Elastase and alpha 1-proteinase inhibitor activity in tracheal aspirates during respiratory distress syndrome. Role of inflammation in the pathogenesis of bronchopulmonary dysplasia. J Clin Invest (1983) 72:656–66. doi:10.1172/JCI111015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Lukkarinen H, Hogmalm A, Lappalainen U, Bry K. Matrix metalloproteinase-9 deficiency worsens lung injury in a model of bronchopulmonary dysplasia. Am J Respir Cell Mol Biol (2009) 41:59–68. doi:10.1165/rcmb.2008-0179OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Kroon AA, Wang J, Kavanagh BP, Huang Z, Kuliszewski M, van Goudoever JB, et al. Prolonged mechanical ventilation induces cell cycle arrest in newborn rat lung. PLoS One (2011) 6:e16910. doi:10.1371/journal.pone.0016910
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M, et al. Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol (2010) 298:L23–35. doi:10.1152/ajplung.00251.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Bland RD, Ertsey R, Mokres LM, Xu L, Jacobson BE, Jiang S, et al. Mechanical ventilation uncouples synthesis and assembly of elastin and increases apoptosis in lungs of newborn mice. Prelude to defective alveolar septation during lung development? Am J Physiol Lung Cell Mol Physiol (2008) 294:L3–14. doi:10.1152/ajplung.00362.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Bland RD, Mokres LM, Ertsey R, Jacobson BE, Jiang S, Rabinovitch M, et al. Mechanical ventilation with 40% oxygen reduces pulmonary expression of genes that regulate lung development and impairs alveolar septation in newborn mice. Am J Physiol Lung Cell Mol Physiol (2005) 293(5):L1099–110.
68. Hilgendorff A, Parai K, Ertsey R, Jain N, Navarro EF, Peterson JL, et al. Inhibiting lung elastase activity enables lung growth in mechanically ventilated newborn mice. Am J Respir Crit Care Med (2011) 184(5):537–46. doi:10.1164/rccm.201012-2010OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Hilgendorff A, Parai K, Ertsey R, Juliana Rey-Parra G, Thebaud B, Tamosiuniene R, et al. Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth. Am J Physiol Lung Cell Mol Physiol (2012) 303:L215–27. doi:10.1152/ajplung.00405.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Stoll BJ, Gordon T, Korones SB, Shankaran S, Tyson JE, Bauer CR, et al. Early-onset sepsis in very low birth weight neonates: a report from the national institute of child health and human development neonatal research network. J Pediatr (1996) 129:72–80. doi:10.1016/S0022-3476(96)70192-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Yoon BH, Romero R, Jun JK, Park KH, Park JD, Ghezzi F, et al. Amniotic fluid cytokines (interleukin-6, tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8) and the risk for the development of bronchopulmonary dysplasia. Am J Obstet Gynecol (1997) 177:825–30. doi:10.1016/S0002-9378(97)70276-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Speer CP. Pulmonary inflammation and bronchopulmonary dysplasia. J Perinatol (2006) 26(Suppl 1):S57–62 discussion S63–54. doi:10.1038/sj.jp.7211476
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Todd DA, Earl M, Lloyd J, Greenberg M, John E. Cytological changes in endotracheal aspirates associated with chronic lung disease. Early Hum Dev (1998) 51:13–22. doi:10.1016/S0378-3782(97)00069-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Atochina-Vasserman EN, Bates SR, Zhang P, Abramova H, Zhang Z, Gonzales L, et al. Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky-Pudlak syndrome. Am J Respir Crit Care Med (2011) 184:449–58. doi:10.1164/rccm.201011-1882OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Polglase GR, Dalton RG, Nitsos I, Knox CL, Pillow JJ, Jobe AH, et al. Pulmonary vascular and alveolar development in preterm lambs chronically colonized with Ureaplasma parvum. Am J Physiol Lung Cell Mol Physiol (2010) 299:L232–41. doi:10.1152/ajplung.00369.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Kunzmann S, Speer CP, Jobe AH, Kramer BW. Antenatal inflammation induced tgf-beta1 but suppressed ctgf in preterm lungs. Am J Physiol Lung Cell Mol Physiol (2007) 292:L223–31. doi:10.1152/ajplung.00159.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Jankov RP, Tanswell AK. Growth factors, postnatal lung growth and bronchopulmonary dysplasia. Paediatr Respir Rev (2004) 5:S265–75. doi:10.1016/S1526-0542(04)90050-4
78. Thebaud B. Angiogenesis in lung development, injury and repair: implications for chronic lung disease of prematurity. Neonatology (2007) 91:291–7. doi:10.1159/000101344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. De Paepe ME, Greco D, Mao Q. Angiogenesis-related gene expression profiling in ventilated preterm human lungs. Exp Lung Res (2010) 36:399–410. doi:10.3109/01902141003714031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. De Paepe ME, Mao Q, Powell J, Rubin SE, DeKoninck P, Appel N, et al. Growth of pulmonary microvasculature in ventilated preterm infants. Am J Respir Crit Care Med (2006) 173:204–11. doi:10.1164/rccm.200506-927OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Bland RD, Ling CY, Albertine KH, Carlton DP, MacRitchie AJ, Day RW, et al. Pulmonary vascular dysfunction in preterm lambs with chronic lung disease. Am J Physiol Lung Cell Mol Physiol (2003) 285(1):L76–85.
82. Vyas-Read S, Shaul PW, Yuhanna IS, Willis BC. Nitric oxide attenuates epithelial-mesenchymal transition in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol (2007) 293:L212–21. doi:10.1152/ajplung.00475.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Wark PA, Murphy V, Mattes J. The interaction between mother and fetus and the development of allergic asthma. Expert Rev Respir Med (2014) 8:57–66. doi:10.1586/17476348.2014.848795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Velten M, Hutchinson KR, Gorr MW, Wold LE, Lucchesi PA, Rogers LK. Systemic maternal inflammation and neonatal hyperoxia induces remodeling and left ventricular dysfunction in mice. PLoS One (2011) 6:e24544. doi:10.1371/journal.pone.0024544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Rose MJ, Stenger MR, Joshi MS, Welty SE, Bauer JA, Nelin LD. Inhaled nitric oxide decreases leukocyte trafficking in the neonatal mouse lung during exposure to >95% oxygen. Pediatr Res (2010) 67:244–9. doi:10.1203/PDR.0b013e3181ca0d93
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Bose CL, Dammann CE, Laughon MM. Bronchopulmonary dysplasia and inflammatory biomarkers in the premature neonate. Arch Dis Child Fetal Neonatal Ed (2008) 93:F455–61. doi:10.1136/adc.2007.121327
87. Vento G, Tirone C, Lulli P, Capoluongo E, Ameglio F, Lozzi S, et al. Bronchoalveolar lavage fluid peptidomics suggests a possible matrix metalloproteinase-3 role in bronchopulmonary dysplasia. Intensive Care Med (2009) 35:2115–24. doi:10.1007/s00134-009-1646-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Watterberg KL, Carmichael DF, Gerdes JS, Werner S, Backstrom C, Murphy S. Secretory leukocyte protease inhibitor and lung inflammation in developing bronchopulmonary dysplasia. J Pediatr (1994) 125:264–9. doi:10.1016/S0022-3476(94)70209-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Weinberger B, Anwar M, Henien S, Sosnovsky A, Hiatt M, Jochnowitz N, et al. Association of lipid peroxidation with antenatal betamethasone and oxygen radial disorders in preterm infants. Biol Neonate (2004) 85:121–7. doi:10.1159/000074968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Poggi C, Giusti B, Vestri A, Pasquini E, Abbate R, Dani C. Genetic polymorphisms of antioxidant enzymes in preterm infants. J Matern Fetal Neonatal Med (2012) 25(Suppl 4):131–4. doi:10.3109/14767058.2012.714976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Buczynski BW, Maduekwe ET, O’Reilly MA. The role of hyperoxia in the pathogenesis of experimental bpd. Semin Perinatol (2013) 37:69–78. doi:10.1053/j.semperi.2013.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol (1998) 275:L110–7.
93. Bonikos DS, Bensch KG, Ludwin SK, Northway WH Jr. Oxygen toxicity in the newborn. The effect of prolonged 100 per cent o2 exposure on the lungs of newborn mice. Lab Invest (1975) 32:619–35.
94. Maeda Y, Dave V, Whitsett JA. Transcriptional control of lung morphogenesis. Physiol Rev (2007) 87:219–44. doi:10.1152/physrev.00028.2006
95. Balasubramaniam V, Mervis CF, Maxey AM, Markham NE, Abman SH. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol (2007) 292:L1073–84. doi:10.1152/ajplung.00347.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Buczynski BW, Yee M, Paige Lawrence B, O’Reilly MA. Lung development and the host response to influenza a virus are altered by different doses of neonatal oxygen in mice. Am J Physiol Lung Cell Mol Physiol (2012) 302:L1078–87. doi:10.1152/ajplung.00026.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Jobe AH, Ikegami M. Prevention of bronchopulmonary dysplasia. Curr Opin Pediatr (2001) 13:124–9. doi:10.1097/00008480-200104000-00006
98. Newman JB, Debastos AG, Batton D, Raz S. Neonatal respiratory dysfunction and neuropsychological performance at the preschool age: a study of very preterm infants with bronchopulmonary dysplasia. Neuropsychology (2011) 25:666–78. doi:10.1037/a0023895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Greenough A, Alexander J, Burgess S, Bytham J, Chetcuti PA, Hagan J, et al. Preschool healthcare utilisation related to home oxygen status. Arch Dis Child Fetal Neonatal Ed (2006) 91:F337–41. doi:10.1136/adc.2005.088823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Greenough A, Alexander J, Burgess S, Chetcuti PA, Cox S, Lenney W, et al. Home oxygen status and rehospitalisation and primary care requirements of infants with chronic lung disease. Arch Dis Child (2002) 86:40–3. doi:10.1136/adc.86.1.40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Greenough A, Cox S, Alexander J, Lenney W, Turnbull F, Burgess S, et al. Health care utilisation of infants with chronic lung disease, related to hospitalisation for RSV infection. Arch Dis Child (2001) 85:463–8. doi:10.1136/adc.85.6.463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Broughton S, Roberts A, Fox G, Pollina E, Zuckerman M, Chaudhry S, et al. Prospective study of healthcare utilisation and respiratory morbidity due to RSV infection in prematurely born infants. Thorax (2005) 60:1039–44. doi:10.1136/thx.2004.037853
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Broughton S, Thomas MR, Marston L, Calvert SA, Marlow N, Peacock JL, et al. Very prematurely born infants wheezing at follow-up: lung function and risk factors. Arch Dis Child (2007) 92:776–80. doi:10.1136/adc.2006.112623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Yuksel B, Greenough A. Relationship of symptoms to lung function abnormalities in preterm infants at follow-up. Pediatr Pulmonol (1991) 11:202–6. doi:10.1002/ppul.1950110304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Pelkonen AS, Hakulinen AL, Turpeinen M. Bronchial lability and responsiveness in school children born very preterm. Am J Respir Crit Care Med (1997) 156:1178–84. doi:10.1164/ajrccm.156.4.9610028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Greenough A, Limb E, Marston L, Marlow N, Calvert S, Peacock J. Risk factors for respiratory morbidity in infancy after very premature birth. Arch Dis Child Fetal Neonatal Ed (2005) 90:F320–3. doi:10.1136/adc.2004.062018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Baraldi E, Carraro S, Filippone M. Bronchopulmonary dysplasia: definitions and long-term respiratory outcome. Early Hum Dev (2009) 85:S1–3. doi:10.1016/j.earlhumdev.2009.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Gross SJ, Iannuzzi DM, Kveselis DA, Anbar RD. Effect of preterm birth on pulmonary function at school age: a prospective controlled study. J Pediatr (1998) 133:188–92. doi:10.1016/S0022-3476(98)70219-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Northway WH Jr, Moss RB, Carlisle KB, Parker BR, Popp RL, Pitlick PT, et al. Late pulmonary sequelae of bronchopulmonary dysplasia. N Engl J Med (1990) 323:1793–9. doi:10.1056/NEJM199012273232603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Welsh L, Kirkby J, Lum S, Odendaal D, Marlow N, Derrick G, et al. The epicure study: maximal exercise and physical activity in school children born extremely preterm. Thorax (2010) 65:165–72. doi:10.1136/thx.2008.107474
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Greenough A, Dimitriou G, Bhat RY, Broughton S, Hannam S, Rafferty GF, et al. Lung volumes in infants who had mild to moderate bronchopulmonary dysplasia. Eur J Pediatr (2005) 164(9):583–6. doi:10.1007/s00431-005-1706-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Hjalmarson O, Sandberg KL. Lung function at term reflects severity of bronchopulmonary dysplasia. J Pediatr (2005) 146:86–90. doi:10.1016/j.jpeds.2004.08.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Doyle LW, Faber B, Callanan C, Freezer N, Ford GW, Davis NM. Bronchopulmonary dysplasia in very low birth weight subjects and lung function in late adolescence. Pediatrics (2006) 118:108–13. doi:10.1542/peds.2005-2522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Filippone M, Carraro S, Baraldi E. From bpd to copd? The hypothesis is intriguing but we lack lung pathology data in humans. Eur Respir J (2010) 35:1419–20 author reply 1420. doi:10.1183/09031936.00013310
115. Bonvillain RW, Danchuk S, Sullivan DE, Betancourt AM, Semon JA, Eagle ME, et al. A nonhuman primate model of lung regeneration: detergent-mediated decellularization and initial in vitro recellularization with mesenchymal stem cells. Tissue Eng Part A (2012) 18(23–24):2437–52. doi:10.1089/ten.TEA.2011.0594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Jensen T, Roszell B, Zang F, Girard E, Matson A, Thrall R, et al. A rapid lung de-cellularization protocol supports embryonic stem cell differentiation in vitro and following implantation. Tissue Eng Part C Methods (2012) 18:632–46. doi:10.1089/ten.TEC.2011.0584
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Ito Y, Betsuyaku T, Nagai K, Nasuhara Y, Nishimura M. Expression of pulmonary VEGF family declines with age and is further down-regulated in lipopolysaccharide (LPS)-induced lung injury. Exp Gerontol (2005) 40:315–23. doi:10.1016/j.exger.2005.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol (2006) 290:H560–76. doi:10.1152/ajpheart.00133.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Yee M, White RJ, Awad HA, Bates WA, McGrath-Morrow SA, O’Reilly MA. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am J Pathol (2011) 178:2601–10. doi:10.1016/j.ajpath.2011.02.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Belik J, Jankov RP, Pan J, Yi M, Chaudhry I, Tanswell AK. Chronic O2 exposure in the newborn rat results in decreased pulmonary arterial nitric oxide release and altered smooth muscle response to isoprostane. J Appl Physiol (1985) 96(2):725–30.
121. Filippone M, Bonetto G, Corradi M, Frigo AC, Baraldi E. Evidence of unexpected oxidative stress in airways of adolescents born very pre-term. Eur Respir J (2012) 40:1253–9. doi:10.1183/09031936.00185511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Iosef C, Alastalo TP, Hou Y, Chen C, Adams ES, Lyu SC, et al. Inhibiting NF-kappaB in the developing lung disrupts angiogenesis and alveolarization. Am J Physiol Lung Cell Mol Physiol (2012) 302:L1023–36. doi:10.1152/ajplung.00230.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Didon L, Roos AB, Elmberger GP, Gonzalez FJ, Nord M. Lung-specific inactivation of ccaat/enhancer binding protein alpha causes a pathological pattern characteristic of copd. Eur Respir J (2010) 35:186–97. doi:10.1183/09031936.00185008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Alvira CM, Abate A, Yang G, Dennery PA, Rabinovitch M. Nuclear factor-kappab activation in neonatal mouse lung protects against lipopolysaccharide-induced inflammation. Am J Respir Crit Care Med (2007) 175:805–15. doi:10.1164/rccm.200608-1162OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Yang G, Abate A, George AG, Weng YH, Dennery PA. Maturational differences in lung nf-kappab activation and their role in tolerance to hyperoxia. J Clin Invest (2004) 114:669–78. doi:10.1172/JCI200419300
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Hennessy EM, Bracewell MA, Wood N, Wolke D, Costeloe K, Gibson A, et al. Respiratory health in pre-school and school age children following extremely preterm birth. Arch Dis Child (2008) 93:1037–43. doi:10.1136/adc.2008.140830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Merritt TA, Deming DD, Boynton BR. The ‘new’ bronchopulmonary dysplasia: challenges and commentary. Semin Fetal Neonatal Med (2009) 14:345–57. doi:10.1016/j.siny.2009.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Bal MP, de Vries WB, van Oosterhout MF, Baan J, van der Wall EE, van Bel F, et al. Long-term cardiovascular effects of neonatal dexamethasone treatment: hemodynamic follow-up by left ventricular pressure-volume loops in rats. J Appl Physiol (2008) 104:446–50. doi:10.1152/japplphysiol.00951.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Kamphuis PJ, de Vries WB, Bakker JM, Kavelaars A, van Dijk JE, Schipper ME, et al. Reduced life expectancy in rats after neonatal dexamethasone treatment. Pediatr Res (2007) 61:72–6. doi:10.1203/01.pdr.0000249980.95264.dd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, et al. Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. N Engl J Med (2002) 347:240–7. doi:10.1056/NEJMoa012657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science (2012) 336:489–93. doi:10.1126/science.1219328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Davis JM, Auten RL. Maturation of the antioxidant system and the effects on preterm birth. Semin Fetal Neonatal Med (2010) 15:191–5. doi:10.1016/j.siny.2010.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Wright CJ, Kirpalani H. Targeting inflammation to prevent bronchopulmonary dysplasia: can new insights be translated into therapies? Pediatrics (2011) 128:111–26. doi:10.1542/peds.2010-3875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Davis JM, Parad RB, Michele T, Allred E, Price A, Rosenfeld W. North American Recombinant Human CuZn SODSG. Pulmonary outcome at 1 year corrected age in premature infants treated at birth with recombinant human cuzn superoxide dismutase. Pediatrics (2003) 111:469–76. doi:10.1542/peds.111.3.469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Kinsella JP, Parker TA, Davis JM, Abman SH. Superoxide dismutase improves gas exchange and pulmonary hemodynamics in premature lambs. Am J Respir Crit Care Med (2005) 172:745–9. doi:10.1164/rccm.200501-146OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Parad RB, Allred EN, Rosenfeld WN, Davis JM. Reduction of retinopathy of prematurity in extremely low gestational age newborns treated with recombinant human cu/zn superoxide dismutase. Neonatology (2012) 102:139–44. doi:10.1159/000336639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Auten RL Jr, Mason SN, Tanaka DT, Welty-Wolf K, Whorton MH. Anti-neutrophil chemokine preserves alveolar development in hyperoxia-exposed newborn rats. Am J Physiol Lung Cell Mol Physiol (2001) 281(2):L336–44.
138. Auten RL, Whorton MH, Nicholas Mason S. Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed newborn rat lung. Am J Respir Cell Mol Biol (2002) 26:391–7. doi:10.1165/ajrcmb.26.4.4708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Patz A, Hoeck LE, De La Cruz E. Studies on the effect of high oxygen administration in retrolental fibroplasia. I. Nursery observations. Am J Ophthalmol (1952) 35:1248–53. doi:10.1016/0002-9394(52)91140-9
140. Askie LM, Henderson-Smart DJ, Irwig L, Simpson JM. Oxygen-saturation targets and outcomes in extremely preterm infants. N Engl J Med (2003) 349:959–67. doi:10.1056/NEJMoa023080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Carlo WA, Finer NN, Walsh MC, Rich W, Gantz MG, Laptook AR, et al. Target ranges of oxygen saturation in extremely preterm infants. N Engl J Med (2010) 362:1959–69. doi:10.1056/NEJMoa0911781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. BOOST II United Kingdom Collaborative Group, BOOST II Australia Collaborative Group, BOOST II New Zealand Collaborative Group; Stenson BJ, Tarnow-Mordi WO, Darlow BA, et al. Oxygen saturation and outcomes in preterm infants. N Engl J Med (2013) 368:2094–104. doi:10.1056/NEJMoa1302298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Supplemental therapeutic oxygen for prethreshold retinopathy of prematurity (STOP-ROP), a randomized, controlled trial. I: primary outcomes. Pediatrics (2000) 105(2):295–310. doi:10.1542/peds.105.2.295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Stevens TP, Dylag A, Panthagani I, Pryhuber G, Halterman J. Effect of cumulative oxygen exposure on respiratory symptoms during infancy among VLBW infants without bronchopulmonary dysplasia. Pediatr Pulmonol (2010) 45:371–9. doi:10.1002/ppul.21199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Maduekwe ET, Buczynski BW, Yee M, Rangasamy T, Stevens TP, Lawrence BP, et al. Cumulative neonatal oxygen exposure predicts response of adult mice infected with influenza a virus. Pediatr Pulmonol (2014). doi:10.1002/ppul.23063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med (1999) 160:1333–46. doi:10.1164/ajrccm.160.4.9810071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Awasthi S, Coalson JJ, Yoder BA, Crouch E, King RJ. Deficiencies in lung surfactant proteins a and d are associated with lung infection in very premature neonatal baboons. Am J Respir Crit Care Med (2001) 163:389–97. doi:10.1164/ajrccm.163.2.2004168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Yoder BA, Siler-Khodr T, Winter VT, Coalson JJ. High-frequency oscillatory ventilation: effects on lung function, mechanics, and airway cytokines in the immature baboon model for neonatal chronic lung disease. Am J Respir Crit Care Med (2000) 162:1867–76. doi:10.1164/ajrccm.162.5.9912145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Maniscalco WM, Watkins RH, Pryhuber GS, Bhatt A, Shea C, Huyck H. Angiogenic factors and alveolar vasculature: development and alterations by injury in very premature baboons. Am J Physiol Lung Cell Mol Physiol (2002) 282(4):L811–23.
150. Afshar S, Gibson LL, Yuhanna IS, Sherman TS, Kerecman JD, Grubb PH, et al. Pulmonary no synthase expression is attenuated in a fetal baboon model of chronic lung disease. Am J Physiol Lung Cell Mol Physiol (2003) 284(5):L749–58.
151. Yoder BA, Coalson JJ, Winter VT, Siler-Khodr T, Duffy LB, Cassell GH. Effects of antenatal colonization with Ureaplasma urealyticum on pulmonary disease in the immature baboon. Pediatr Res (2003) 54:797–807. doi:10.1203/01.PDR.0000091284.84322.16
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. McCurnin DC, Pierce RA, Chang LY, Gibson LL, Osborne-Lawrence S, Yoder BA, et al. Inhaled no improves early pulmonary function and modifies lung growth and elastin deposition in a baboon model of neonatal chronic lung disease. Am J Physiol Lung Cell Mol Physiol (2005) 288:L450–9. doi:10.1152/ajplung.00347.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Thomson MA, Yoder BA, Winter VT, Martin H, Catland D, Siler-Khodr TM, et al. Treatment of immature baboons for 28 days with early nasal continuous positive airway pressure. Am J Respir Crit Care Med (2004) 169:1054–62. doi:10.1164/rccm.200309-1276OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho SC, et al. Chronic lung injury in preterm lambs. Disordered respiratory tract development. Am J Respir Crit Care Med (1999) 159:945–58. doi:10.1164/ajrccm.159.3.9804027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Bland RD, Albertine KH, Carlton DP, Kullama L, Davis P, Cho SC, et al. Chronic lung injury in preterm lambs: abnormalities of the pulmonary circulation and lung fluid balance. Pediatr Res (2000) 48:64–74. doi:10.1203/00006450-200007000-00013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Maeta H, Raju TN, Vidyasagar D, Bhat R, Esterly J, Matsuda H, et al. Effect of exogenous surfactant on the development of bronchopulmonary dysplasia in a baboon hyaline membrane disease model. Crit Care Med (1990) 18:403–9. doi:10.1097/00003246-199004000-00011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Shimada S, Raju TN, Vidyasagar D, Maeta H, Bhat R. Chest radiographic course after exogenous surfactant therapy in baboons with respiratory distress syndrome. Crit Care Med (1990) 18:969–73. doi:10.1097/00003246-199009000-00013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Kinsella JP, Gerstmann DR, Clark RH, Null DM Jr, Morrow WR, Taylor AF, et al. High-frequency oscillatory ventilation versus intermittent mandatory ventilation: early hemodynamic effects in the premature baboon with hyaline membrane disease. Pediatr Res (1991) 29:160–6. doi:10.1203/00006450-199102000-00012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: bronchopulmonary dysplasia, neonatal chronic lung disease, lung development, long-term consequences, hyperoxia, mechanical ventilation, inflammation, animal models
Citation: Hilgendorff A and O’Reilly MA (2015) Bronchopulmonary dysplasia early changes leading to long-term consequences. Front. Med. 2:2. doi: 10.3389/fmed.2015.00002
Received: 14 September 2014; Accepted: 05 January 2015;
Published online: 12 February 2015.
Edited by:
Benjamin John Marsland, Centre Hospitalier Universitaire Vaudois, SwitzerlandReviewed by:
Christophe Guignabert, INSERM UMR 999, FranceMatthias Roth-Kleiner, Clinic of Neonatology, Switzerland
Copyright: © 2015 Hilgendorff and O’Reilly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne Hilgendorff, Comprehensive Pneumology Center, Ludwig-Maximilians-Universität, Asklepios Klinik Gauting, Helmholtz Zentrum München, Max-Lebsche-Platz 31, München 81377, Germany e-mail:YW5uZS5oaWxnZW5kb3JmZkBoZWxtaG9sdHotbXVlbmNoZW4uZGU=