 Grith L. Sorensen
Grith L. Sorensen- Institute of Molecular Medicine, University of Southern Denmark, Odense, Denmark
Surfactant protein D (SP-D) is a multimeric collectin that is involved in innate immune defense and expressed in pulmonary, as well as non-pulmonary, epithelia. SP-D exerts antimicrobial effects and dampens inflammation through direct microbial interactions and modulation of host cell responses via a series of cellular receptors. However, low protein concentrations, genetic variation, biochemical modification, and proteolytic breakdown can induce decomposition of multimeric SP-D into low-molecular weight forms, which may induce pro-inflammatory SP-D signaling. Multimeric SP-D can decompose into trimeric SP-D, and this process, and total SP-D levels, are partly determined by variation within the SP-D gene, SFTPD. SP-D has been implicated in the development of respiratory diseases including respiratory distress syndrome, bronchopulmonary dysplasia, allergic asthma, and chronic obstructive pulmonary disease. Disease-induced breakdown or modifications of SP-D facilitate its systemic leakage from the lung, and circulatory SP-D is a promising biomarker for lung injury. Moreover, studies in preclinical animal models have demonstrated that local pulmonary treatment with recombinant SP-D is beneficial in these diseases. In recent years, SP-D has been shown to exert antimicrobial and anti-inflammatory effects in various non-pulmonary organs and to have effects on lipid metabolism and pro-inflammatory effects in vessel walls, which enhance the risk of atherosclerosis. A common SFTPD polymorphism is associated with atherosclerosis and diabetes, and SP-D has been associated with metabolic disorders because of its effects in the endothelium and adipocytes and its obesity-dampening properties. This review summarizes and discusses the reported genetic associations of SP-D with disease and the clinical utility of circulating SP-D for respiratory disease prognosis. Moreover, basic research on the mechanistic links between SP-D and respiratory, cardiovascular, and metabolic diseases is summarized. Perspectives on the development of SP-D therapy are addressed.
Surfactant Protein D (SP-D)
Surfactant protein D is a pattern-recognition molecule belonging to the collectin family, a group of collagen-containing C-type lectins. Human collectins also include surfactant protein A (SP-A), which has a tissue distribution and functions partially overlapping with those of SP-D. One main effect of SP-D is the aggregation and enhancement of phagocytosis of microbes and dying host cells. An additional classical member of the collectin family is the serum protein, mannan-binding lectin (MBL), which evokes complement activation through the lectin pathway in a complex with MBL-associated serine proteases (MASPs) (1). Moreover, three novel human defense collagens: collectin-10 (CL-10) [collectin liver 1, CL-L1, or CL-10], collectin-11 (CL-11) (collectin kidney 1, CL-K1, or CL-11), and collectin-12 (CL-12) (collectin placenta 1, CL-P1, or CL-12) have been identified (2–4). CL-10 and CL-11, partly in heterocomplexes with one another, are found in the circulation associated with MASPs and can induce complement activation (5). Different from the other collectins, CL-12 is a type II membrane protein with a fluid phase variant that has scavenger receptor, as well as complement activation, functions (6, 7).
Pulmonary surfactant is a multimolecular complex consisting of phospholipids and cholesterol (total 90%) and surfactant proteins (10%). The surfactant proteins consist of the high-molecular weight (HMW) hydrophilic proteins, SP-A and SP-D, and the low-molecular weight (LMW) and extraordinarily lipophilic surfactant protein B (SP-B) and surfactant protein C (SP-C), which are essential for the biophysical properties of surfactant phospholipids (8).
Sites of SP-D Synthesis
Surfactant protein D has been localized to both the lung and non-pulmonary tissues. The protein is associated with external or luminal surfaces of the respiratory, digestive, glandular, reproductive tract, urinary, and vascular epithelia and glands (Table 1). This is consistent with the role of SP-D in pattern recognition, as the majority of locations in which it is expressed are at interfaces with the external milieu or with plasma, urine, tears, cerebrospinal fluid, and amniotic fluid, where the maintenance of a sterile milieu is critical.

Table 1. Detection of SP-D immunoreactivity or SP-D gene (SFTPD) expression in human organ systems.
Some SP-D expressing non-pulmonary sites may produce surfactant-like materials and phospholipidic lubrication is present at numerous distinct sites. A major function of SP-D in the lung is as regulator of pulmonary surfactant lipid levels and, although not investigated, it has been speculated that SP-D may also participate in phospholipid homeostasis at extrapulmonary sites (52). Moreover, SP-D is expressed in the muscle cells and endothelium of the cardiovascular system, where it is suggested to function as an inhibitor of inflammatory signaling (47, 48). The expression patterns of SP-D have frequently been validated by different observers or by the use of diverse techniques (Table 1). SP-D immunostaining has also been observed in infiltrating white blood cells; for example, in the lung (9) and placenta (32). Moreover, SP-D immune-staining has been detected in the phagolysosome compartment or as granular staining of the cell membrane (9). Ultrastructural studies have demonstrated the presence of SP-D in the endocytic compartment of rat alveolar macrophages, but not in biosynthetic organelles (53), indicating that SP-D is not produced by these inflammatory cells, but rather is taken up by endocytosis.
Regulation of SP-D Expression
The proximal promoter of SP-D mediates cell type-restricted, basal, and glucocorticoid-stimulated promoter activities as demonstrated in vitro (54). The SP-D promoter was originally identified containing multiple potential cis-regulatory elements including half-site glucocorticoid response elements, a canonical AP-1 consensus, several AP-1-like sequences, E-box sequences, several C/EBP and PEA3 motifs, putative interferon response elements, FoxA-bindings sites, and a GT-containing regulatory element and regulatory roles for AP-1 (junB, junD, c-Jun, and c-Fos), FoxA1/2 and GT-box binding proteins were identified by mutational studies (55–57). It was suggested that the permissive glucocorticoid regulation of SP-D expression is caused by increased promoter occupancy of C/EBPβ (58). Furthermore, retinoblastoma protein is demonstrated to stimulate SFTPD gene activation by forming a complex with C/EBPs bound to the C/EBPβ consensus site in the SFTPD promoter (59). Moreover, the calcineurin/NFAT pathway was demonstrated to be active in vitro resulting in assembly of NFATs, AP-1, and TFF-1 in a transcriptional complex in the proximal promoter of mouse SFTPD (60). Mitogen-activated protein kinase (MAPK)-mediated upregulation of SP-D expression has been reported in human corneal epithelial cells (61) and in human lung epithelial cells, where the expressional regulation was mediated via signaling through JNK, a MAPK (62). The expression of SP-D in corneal epithelium was further inhibited by pharmacological inhibitors of toll-like receptor (TLR)4 and myeloid differentiation primary response gene 88 (MyD88) signaling (44). Tumor necrosis factor-α (TNF-α) significantly augmented the level of SP-D expression in primary coronary endothelial cells. Moreover, the basal level SP-D was reduced by nitric oxide (NO) synthase inhibitor l-NAME, inhibitor of phosphoinositide 3-kinases (PI3Ks) Wortmannin and inhibitor of MEK1 activation and the MAP kinase cascade PD 98059. Inversely, SP-D expression could be increased by DETA NONOate (donor of NO) or insulin (activator of PI3K/Akt) (63).
Surfactant protein D expression is developmentally regulated and further regulated by epigenetic allele-specific expression outside the lung (64). Dexamethasone treatment during culture of fetal lung explants increased SP-D mRNA and protein (54), maternal steroid treatment increased fetal serum SP-D (65), and in vitro and in vivo studies have confirmed regulation of SP-D expression by glucocorticoids and shown a dramatic increase prior to birth (66–69). Fetal lung maturation occurs on exposure to glucocorticoids with a simultaneous increase in expression of SP-D by lung epithelial cells (70, 71). In vivo studies have further demonstrated an increase in SP-D mRNA after pharmacological inhibition of dipeptidyl peptidase activity (72) and both mRNA and protein after a brief 95% oxygen exposure in rats (73), and mRNA and protein was markedly increased following mouse exposure to the cytokines interleukin (IL)-4 (74, 75), IL-13 (76), and TNF-α (77), whereas insulin is reported to inhibit SP-D expression in lung epithelial cell line (78).
In addition, estrogen positively regulates expression of SP-D in the mouse uterus (79). Progesterone, along with estrogen synergizes SP-D expression, however, when administered alone results in negative regulation (80). SP-D transcript levels increased sevenfold in the prostate of castrated rats suggesting negative regulation by testosterone (81), while testosterone suppression downregulated transcript levels of SP-D in murine testis (38). Moreover, serum SP-D levels increase in Turner syndrome patients treated with growth hormone (82).
Effects of SP-D
The primary reported effects of SP-D include binding of bacteria, viruses, fungi, and, recently, helminthic parasites, for clearance via opsonization for phagocyte recognition (83–90). A detailed review of the numerous interactions of SP-D with pathogenic microbes was provided by Nayak et al. (91). SP-D can also bind to other biological or abiotic particles and participate in their clearance from the airways and potential additional sites. Hence, SP-D is known to aggregate allergens and aid in their removal (92, 93), to enhance clearance of genomic DNA and apoptotic material (94), to aggregate and remove particulate material (95), and has the capacity to affect the mouse intestinal microbiota under certain experimental conditions (96, 97). The lectin activity of human SP-D favors its interactions with microbial ligands glycosylated with a variety of saccharides, including N-acetylmannosamine (ManNAc) > mannose > fucose (36, 98); however, SP-D is also recognized to bind a wide range of inhaled pathogens and can bind saccharides as well as lipids and nucleic acids, with broad specificity, to initiate phagocytosis. The diversity of its ligand interactions were recently reviewed by Jakel et al. (99).
In addition to opsonization for phagocytosis, the antimicrobial effects of SP-D include aggregation (100–102), which may enhance the efficiency of neutrophil extracellular traps (103), bacterial and fungal cell-membrane lysis (104–106), neutralization of infectivity (107–109), or dampening of innate signaling evoked by microbe-derived ligands (110).
Surfactant protein D enhanced phagocytosis and additional antimicrobial activity is beneficial to the host; however, in some rare cases, SP-D binding to pathogens can be a risk factor contributing to deterioration of (murine) disease and increased pathogen burden, for example, of hypocapsular Cryptococcus neoformans (111, 112) and Pneumocystis carinii (113). Initial studies of SP-D interactions with Mycobacterium tuberculosis demonstrated reduced bacterial uptake by macrophages, whereas in vivo studies suggest that SP-D is dispensable for immune control of infection (114–116).
SP-D-Mediated Cellular Activation
A wealth of data from in vitro studies demonstrate that SP-D modulates immune cell, epithelial cell, fibrocyte, and smooth muscle cell functions (Table 2).
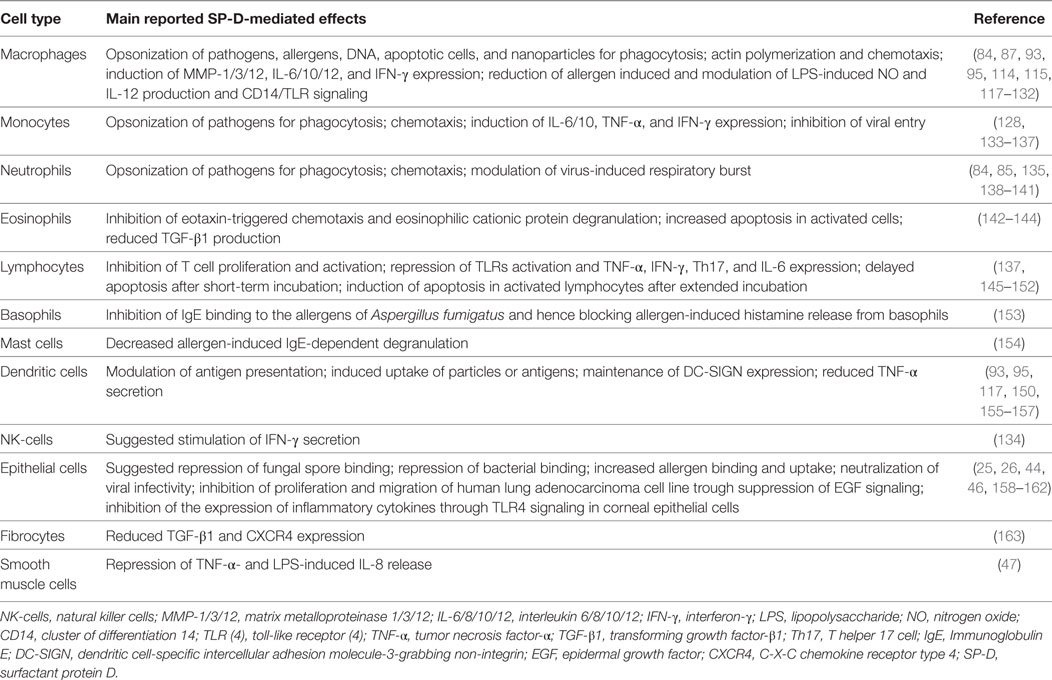
Table 2. SP-D-mediated cellular effects.
SP-D Receptors
The identification of SP-D receptors is important for understanding its immune-regulatory and homeostatic functions in different cell types. A series of SP-D receptors, or receptor candidates, have been identified; however, the cellular effects of SP-D via some of these receptors have yet to be determined mechanistically and validated by independent research.
Cluster of Differentiation 14 (CD14)/TLR/Myeloid Differentiation Factor 2 (MD-2)
Initially, SP-D was demonstrated to interact in a Ca2+-dependent manner with glycosylated CD14 via its C-type lectin domain (CTLD), thereby inhibiting lipopolysaccharide (LPS) binding (164). Subsequently, allergen-induced activation of macrophages and dendritic cells by SP-D mediated by suppression of the CD14/TLR signaling pathway was discovered (117). Furthermore, SP-D can inhibit the cell surface binding of LPS to TLR4/MD-2-expressing cells and attenuate MD-2 binding to LPS through the CTLD (118, 165, 166). Moreover, SP-D modulation of epithelial responses to additional microbial stimuli was recently documented as dependent on TLR4 and MyD88 (44). Such observations may partially explain the results of in vivo studies indicating that SP-D inhibits inflammation caused by bacterial LPS (167–169). SP-D can also bind to TLR2 (165).
Signal-Regulatory Protein-α (SIRP-α)/Calreticulin/CD91
In a highly cited study from 2003, it was suggested that free SP-D binds to the cellular receptor, SIRP-α, through its CTLD, resulting in an inhibitory signal preventing activation of mononuclear phagocytes, nuclear factor-κB activation, and secretion of inflammatory cytokines through phospho-p38-dependent signaling. By contrast, when the CTLD is occupied by a ligand, SP-D was suggested to interact with a cell surface receptor complex consisting of CD91 and calreticulin, through its collagen-like domain, promoting inflammatory cell activation (170). However, the main focus of the study was SP-A interactions. Consequently, the evidence for direct SP-D/SIRP-α binding was weak and CD91/calreticulin interactions were only demonstrated for SP-A. The study inspired further research, which validated the binding of SP-D to N-glycosylated sites in the membrane-proximal domain of SIRPα, and to SIRPβ, a related SIRP (171). Moreover, in the absence of inflammation, SP-D can suppress the phagocytic function of alveolar macrophages by binding to SIRPα, thereby altering the activity of its downstream signaling effectors. By contrast, during LPS-induced inflammation, recruited mononuclear phagocytes partly escape SP-D-mediated inhibition and contribute to cell clearance (119) while IL-12p40 production is suppressed (120). Glucocorticoid treatment further relieves SP-D-driven suppression of apoptotic cell uptake through downregulation of SIRPα (172).
At the same time that SP-D interaction with SIRPα was validated, it was demonstrated that an interaction of SP-D with macrophage calreticulin appeared to be dependent on biochemical modification of SP-D. S-nitrosothiol (SNO)-SP-D, formed by nitrosylation of N-terminal cysteines in SP-D, but not native SP-D, was chemoattractive for macrophages, inducing downstream p38 phosphorylation. The authors suggested that SP-D acts to integrate the status of the lung lining, initiating inflammatory responses under various pathological conditions, through calreticulin-mediated signaling, while maintaining a quiescent state through SIRPα signaling in the absence of stress (173).
Leukocyte-Associated Immunoglobulin-Like Receptor 1 (LAIR-1)
Leukocyte-associated immunoglobulin-like receptor 1, which is a receptor expressed on most immune cells, and for which collagens are high-affinity ligands, is an inhibitory SP-D receptor. The collagen stalk of SP-D is essential for the interaction with LAIR-1, which results in functional reduction of reactive oxygen species (ROS) signaling in a neutrophilic cell line (174). The authors therefore suggested that a lack of SP-D/LAIR-1 interaction could be responsible for the increased production of hydrogen peroxide in lung homogenates previously observed in SP-D deficient (Sftpd−/−) mice (175).
Osteoclast-Associated Receptor (OSCAR)
Another collagen receptor, OSCAR, expressed in inflammatory C-C chemokine receptor type 2 (CCR2) + monocytes and macrophages, can functionally interact with multimeric (cruciform) SP-D, resulting in a pro-inflammatory response. This interaction leads to TNF-α release from CCR2+ monocytes and apparent internalization of the SP-D/OSCAR complex in alveolar macrophages (133). OSCAR is also expressed in osteoclasts, dendritic cells, and endothelial cells (176).
Fc Receptor γII (FcγRII/CD32)
Surfactant protein D binding to FcγRII (CD32) on eosinophils has been detected by flow cytometric analysis and may explain the inhibitory effect of SP-D on IgG and serum-triggered eosinophilic cationic protein degranulation by eosinophils (142).
NKp46
Sftpd−/− mice have reduced expression of pulmonary interferon-γ (IFN-γ) when exposed to ozone; therefore, it was hypothesized that IFN-γ-producing natural killer (NK) cells interact with SP-D through the glycosylated membrane receptor, NKp46. Indirect evidence for such an interaction came from the reduced binding between SP-D and NK cells obtained from NKp46−/− mice, relative to those from NKp46+/+ mice; the authors of this study suggested that this interaction may be involved in the IFN-γ-dependent impaired dendritic cell homing to lymphoid tissue in Sftpd−/− mice (134).
G Protein-Coupled Receptor 116 (GPR116)
The phenotype of GPR116 (Ig-Hepta) deficient mice is very similar to that of Sftpd−/− mice, including accumulation of surfactant lipids, enlarged alveoli, hypertrophy of type II alveolar (AT-II) cells, decreased surfactant uptake by type II alveolar cells (AT-II cells), accumulation of enlarged foamy macrophages, and enhanced expression of the matrix metalloproteinase 12 (Mmp12) gene. Therefore, it was hypothesized that this adhesion class of G protein-coupled receptor may interact with SP-D. GPR116 is highly expressed in type II pneumocytes and immunoprecipitation of flag-tagged recombinant proteins supports SP-D as a likely ligand of this receptor (177).
Uroplakin Ia (UPIa)
Uroplakin Ia is a glycoprotein expressed on bladder urothelium that serves as a receptor for FimH, a lectin in bacterial pili, and this interaction initiates uropathogenic Escherichia coli (UPEC) infection. SP-D binds directly to UPIa, which is rich in high mannose glycans, and thereby inhibits the adherence and cytotoxicity of UPEC in a human bladder epithelial cell line. These in vitro observations were supported by the results of experiments demonstrating that exogenous administration of SP-D inhibited UPEC adherence to the bladder and dampened UPEC-induced inflammation in mice (26).
Epidermal Growth Factor Receptor (EGFR)
Surfactant protein D binds directly to high mannose-type N-glycans in EGFR and the interaction blocks the binding of epidermal growth factor (EGF) to EGFR, suppressing EGF signaling and inhibiting the proliferation and migration of two human lung adenocarcinoma epithelial cell lines, indicating that lung cancer cells are regulated by SP-D via autocrine mechanisms (158, 178).
Additional receptor candidates, Jäkel and Sim, demonstrated that SP-D can bind to a 20- to 22-kDa structure on macrophages and dendritic cells in a calcium-dependent manner; however, they were unable to identify the nature of the structure (179). Interaction of SP-D with dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin reduces SP-D-mediated HIV-1 capture and transfer to CD4+ T cells (180).
Additional Interacting Host Molecules
Secreted host molecules, reported to bind SP-D include decorin (181); the protease inhibitor, alpha(2)-macroglobulin, which enhances bacterial agglutination and protects SP-D against elastase-mediated degradation (182); deleted in malignant brain tumor 1/gp340, which enhances SP-D-mediated viral aggregation (183, 184); and defensins, which, according to subtype, may cause SP-D to precipitate out of bronchoalveolar lavage (BAL) fluid or have additive viral neutralizing activity when combined with SP-D (185, 186). Moreover, various classes of immunoglobulin, including IgG, IgM, IgE, and secretory IgA, bind SP-D. SP-D aggregates immunoglobulin-coated beads and enhances their phagocytosis and IgM–SP-D complexes effectively opsonize late apoptotic cells and enhance their clearance by alveolar macrophages in the lungs (121, 187).
SP-D Structure, Decomposition, and Proteolytic Degradation
The amino acid sequence (375 aa) of the mature SP-D monomer consists of four structural domains: (1) an N-terminal domain involved in intermolecular disulfide bond formation; (2) a collagen domain, important for spacing of the CTLDs; (3) an α-helical neck region involved in protein trimerization and spacing of the CTLDs; and (4) a globular C-terminal CTLD (Figure 1A), responsible for Ca2+-dependent binding of microbial ligands (188). The SP-D protein structure is stabilized by assembly into trimers and multimers via two conserved cysteine residues in the N-terminal domain (33, 189).
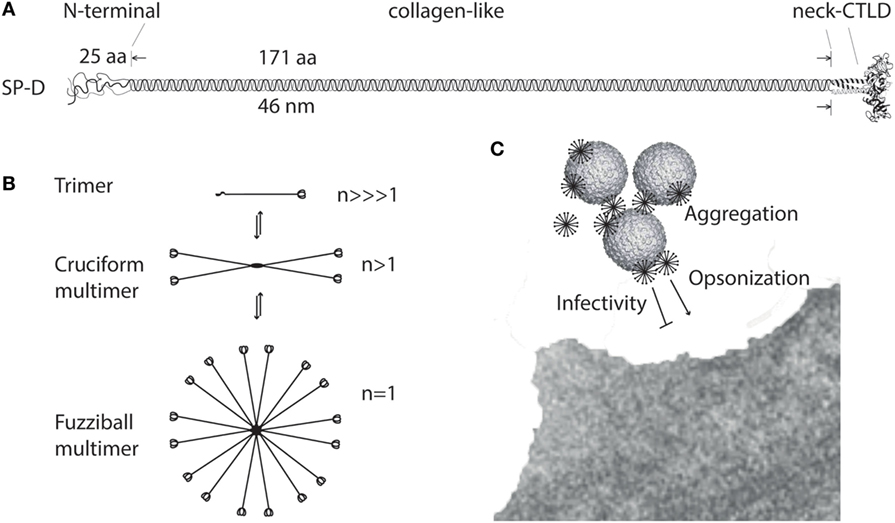
Figure 1. Multimerization of surfactant protein D (SP-D). (A) Regions of the trimeric SP-D subunit. The subunit structure has been drawn to the approximate dimensions of the protein domains. Adapted with permission from Ref. (190). (B) Multimerization of the trimeric SP-D subunit (3 chains) into 4-subunit cruciform (12 chains) or fuzziball >4-subunit (>12 chains) structures of SP-D. (C) Schematic overview of how multimeric SP-D is implicated in antimicrobial defense. Binding of multimeric SP-D to microbe-associated glycans may block interaction of the microbe with its receptors, aggregate the microbes, or SP-D may act as an opsonin, enhancing endocytic uptake of the microbe in host cells. Only fuzziball SP-D multimers are shown for simplicity. CTLD, C-type lectin domain.
The individual “arm” length of an SP-D monomer is approximately 46 nm (188), making it a molecule with dimensions the same order of magnitude as certain viruses, as shown by electron microscopy (191). The molecular mass of SP-D molecules ranges from <100 to >1,000 kDa, as a consequence of different degrees of multimerization. Disulfide-bridge dependent trimerization of SP-D into subunits and further multimerization into cruciform or fuzziball structures (astral bodies), which can contain 32 (or more) trimeric subunits (17, 192) (Figure 1B), provides the spatial arrangement required for high-avidity interaction of the CTLDs with multivalent microbial ligands (85, 100). The effects of SP-D thus depend on the degree of multimerization for the binding of ligands (108, 115) (Figure 1C) or the cellular receptors evoking major SP-D antimicrobial effects.
HMW and LMW SP-D
High-molecular weight SP-D multimers are only partly dependent on disulfide crosslinking of the N-termini, and a proportion of SP-D subunits are non-covalently associated. This allows interconversion between HMW SP-D and LMW SP-D trimers, as demonstrated using size permeation chromatography (36) (Figure 1B). The HMW/LMW ratio depends on the concentration of the protein in solution, with low-protein concentrations favoring the decomposition of multimers into trimers. In addition, the HMW/LMW ratio increases with affinity purification of SP-D, suggesting that ligand-binding facilitates assembly of SP-D trimers into multimers (36).
A single-nucleotide polymorphism (SNP), rs721917, in the SP-D gene (SFTPD) results in expression of either methionine or threonine at position 11 (Met11Thr) in the mature protein. The HMW/LMW ratio in body fluids appears to vary according to the amino acid at this position, with 1:(1–1.6) for methionine 11 (Met11) and 1:(3–5) for threonine 11 (Thr11) allelic variants (36, 37), as assessed by monoclonal immunodetection of SP-D CTLDs after size permeation chromatography. HMW SP-D exhibits markedly increased binding to a majority of microbial ligands and microbes (36, 37), which calls into question the role of LMW SP-D, other than as a large reservoir of subunits available for assembly of HMW SP-D when enriched on microbial surfaces; however, whereas HMW SP-D binds preferentially to intact influenza A virus and bacteria, trimeric SP-D favors Ca2+-independent binding to isolated bacterial LPS (37). Use of ManNAc-affinity chromatography, in place of traditional maltose-affinity chromatography, allowed preparation of enriched natural trimeric human SP-D, facilitating investigation of this molecule. Natural LMW SP-D also binds to endogenous circulating ligands including low-density lipoprotein (LDL), oxidized LDL (oxLDL), and high-density lipoprotein (HDL), in a partially Ca2+-independent manner, whereas HMW SP-D does not bind lipoproteins (36).
In Vivo Studies of SP-D Size Variants
Transgenic Sftpd−/− mice expressing either the human SP-D Met11 or Thr11 allelic variants were generated to test the hypothesis that this allelic variation is implicated in disease; however, the expression of the allelic variants was under the control of a promoter for ubiquitous expression. Consequently, the distribution of SP-D levels in the lung and serum appeared to differ from that of endogenous SP-D. This made comparisons of effects with normal mice difficult, despite sustained allele-dependent HMW/LMW SP-D distribution. The low transgene expression levels in the lung generated pulmonary phenotypes in both transgenic mice somewhat resembling Sftpd deficiency, which is characterized by mild emphysema and the presence of foam cell-like macrophages (193–197). Alternative studies made point mutations affecting the N-terminal cysteines involved in the stabilization of SP-D HMW multimers, or deletions of the collagen region and/or the N-terminal region. Such studies demonstrated that trimeric SP-D subunits have the same saccharide selectivity as multimers, but appear to have a weaker and more restricted range of antimicrobial activity (85, 100, 198, 199). In vivo studies, where the N-terminal cysteine mutated SP-D was overexpressed, or an SP-D collagen deletion mutant protein expressed, in Sftpd−/− or wild-type mice demonstrated that native SP-D is essential for pulmonary phospholipid homeostasis and prevention of airspace enlargement (189, 200). Collectively, those studies suggested that multimerization is important; however, in vivo administration of repeated high doses of a 60-kDa recombinant trimeric fragment of SP-D [60-kDa recombinant trimeric fragment of SP-D lacking the N-terminal but retaining a part of the collagen region (rfhSP-D)], lacking the N-terminus but retaining part of the collagen region, appeared to have similar effects to native SP-D in reducing lipidosis, apoptotic macrophages, alveolar type II cell numbers, and airspace enlargement in mice (201, 202), suggesting that the homeostatic effects of SP-D are predominantly mediated by the CTLD, and that high therapeutic levels of trimeric human SP-D may compensate for a relatively low target avidity. Trimeric rfhSP-D has subsequently been used successfully in antimicrobial or anti-inflammatory therapy in a series of in vivo studies (110, 203–206) and can also induce apoptosis of activated immune cells (143, 145). However, rfhSP-D has also failed to demonstrate effects in some contexts (92) and the extent to which the effects of full-length multimeric SP-D can be mimicked by high levels of trimeric SP-D is not yet entirely clear.
Genetic Variation Affecting SP-D Structure
According to a study of adult twins, genetic factors explain an estimated 83% of variation in constitutive serum SP-D levels. Moreover, the rs721917 SFTPD SNP is associated with serum levels of SP-D (207) and explains 39% of phenotypic variation (36), with the Met11 allelic variant associated with the highest levels (37). Both allelic variants were detected at relatively high frequencies (Thr11/Thr11 = 0.18, Met11/Thr11 = 0.43, Met11/Met11 = 0.36) in a North European population (208). Moreover, the rs721917 allelle frequency distribution is highly similar in different populations (209, 210), although ethnic differences are documented (211).
Consistent with genetic determination of SP-D levels, the constitutive distribution of HMW and LMW SP-D in human body fluids is also genetically determined, with individuals homozygous for the Met11 allele having a relative predominance of HMW SP-D and Thr11 allele homozygotes more LMW SP-D. The dependency of SP-D molecular size on rs721917 genetic variation is supported by the size distribution of recombinant SP-D expressed from the two allelic variants in a human cell line (37). The varying abilities of the allelic variants to assemble into multimers is postulated to be attributable to the different hydrophobic properties of Met and Thr, or partial O-linked glycosylation of the Thr11-residue, which may affect close-proximity disulfide bonding, thus limiting the stability of Thr11-variant multimers (34, 37).
Several studies have linked rs721917 with clinical pathologies (Table 3), which may indicate involvement of SP-D size variation in pathogenesis. Some rs721917 association studies have demonstrated a strong interaction with smoking, including in preclinical cardiovascular disease (CVD) (212–214). Moreover, some data are contradictory; for example, SNP-analysis associated rs721917 variation with chronic obstructive pulmonary disease (COPD) (215), whereas genome-wide association (GWA) analysis did not confirm this association, although several other SFTPD SNPs were identified as associated with COPD (216). Recently, an association of rs721917 variation with SP-D size variation was reported in respiratory disease (217); however, the overall conclusion from studies of rs721917 variant associations is that both allelic variants may be deleterious in different disease contexts, although the majority of studies suggest disease associations with the Thr11 allele.
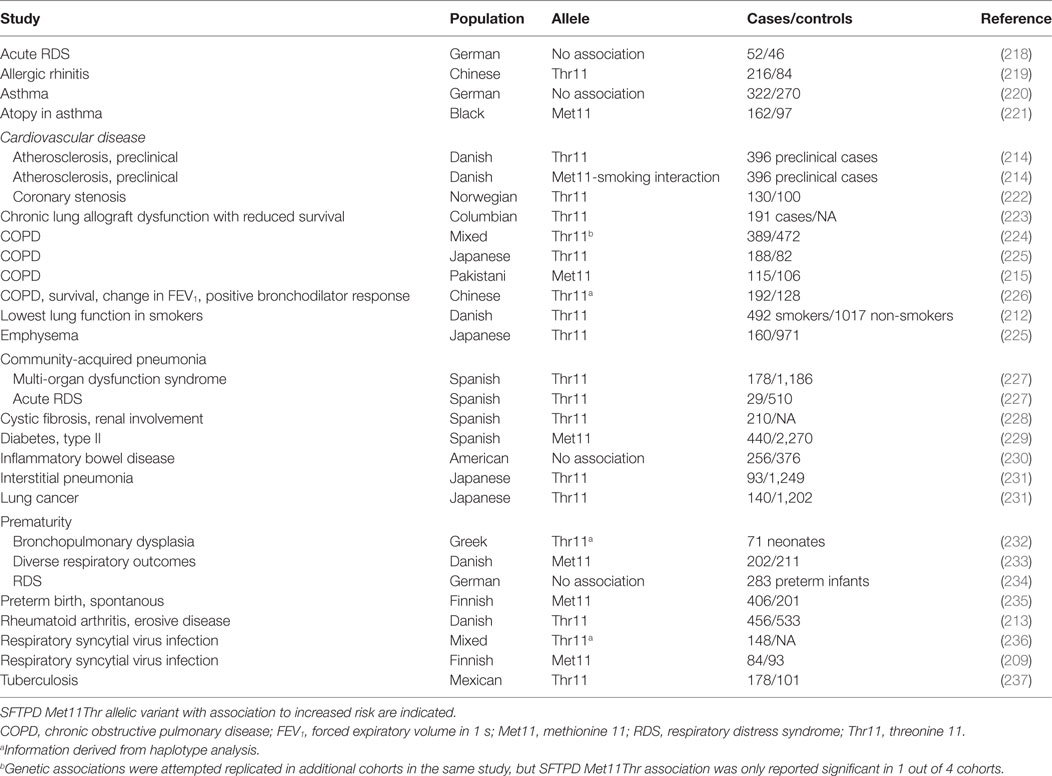
Table 3. SFTPD rs721917 (Met11Thr) variation in disease.
Biochemical Modification of SP-D
Posttranslational modifications of SP-D include partial hydroxylation of proline and lysine residues in the collagen-like region. Furthermore, SP-D can undergo glycosyl-galactosyl O-linked glycosylation of hydroxylated lysine residues and O-linked glycosylation of N-terminal threonines. N-glycosylation of SP-D also occurs within the collagen-like region. These modifications are partial, hence the molecular weight of resulting molecules varies from 37 to 50 kDa, as assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and mass spectrometry (34, 188, 238–241). Some posttranslational SP-D modifications may alter its propensity for multimerization and a fraction of LMW SP-D purified from normal body fluid (late amniotic fluid) is unable to assemble into multimers (36). This fraction probably contains modified or partly degraded SP-D and appears to be enriched under inflammatory conditions.
Glycosylation Variants
Surfactant protein D can undergo O-linked glycosylation of Thr11 and this variant is essentially present in the trimeric form (34). Moreover, endothelial SP-D appear with lower molecular weight band pattern in SDS-PAGE and may represent immature intracellular protein, or a posttranslationally modified version of SP-D, different from the forms produced by lung cells or cells that generate SP-D in amniotic fluid (214); however, this remains to be explored.
One type of disease-induced modification of SP-D is suggested to be increased levels of core fucose in the SP-D N-glycan. The N-glycan is not expected to affect the quaternary structure of SP-D and normal SP-D N-glycan comprises core fucose; however, the glycosylation is only partial (240) and depends on fucosyltransferase activity. Glycomic analysis demonstrated that the levels of core-fucosylated N-glycan in SP-D are increased, relative to total SP-D, in smoking COPD subjects, but not in non-smokers (241).
Nitrosylation
The two N-terminal cysteines at positions 15 and 20 of the SP-D N-terminal region are implicated in inducible nitric oxide synthase (iNOS)- and NO-mediated control of multimerization of SP-D through formation of S-nitrosothiol SP-D (SNO-SP-D) (173). SNO-SP-D levels increase during inflammation, and its formation results in decomposition of SP-D multimers (173, 242–245). Radiation-induced lung injury is thus reported to result in iNOS-dependent disruption of SP-D multimers in mouse BAL (246). As described earlier, SNO-SP-D, but not SP-D (neither multimeric nor trimeric), is chemoattractive for macrophages and can induce cellular p38 phosphorylation, indicating that SNO-SP-D reacts differently with cellular receptors compared with SP-D (173).
Oxidative Damage
Non-reducible crosslinking of HMW SP-D occurs in normal late amniotic fluid (36), in BAL from alveolar proteinosis (17), and in asthma patient BAL after provocation with an allergen (242). Formation of tyrosine-dependent covalent crosslinking within the neck/CTLD of SP-D is a result of reactive oxidant species (ROS), including peroxynitrite activity, and reduces SP-D ligand aggregation (247).
Additional oxidative damage is caused by neutrophil myeloperoxidase and its specific reactive oxidant product, hypochlorous acid. Hypochlorous acid can cause abnormal but reducible N-terminal disulfide crosslinking of SP-D, and although the mechanism is not fully elucidated, these modifications result in loss of the aggregating activity of SP-D in vitro and in the context of acute inflammation in vivo (248).
Proteolytic Degradation
Surfactant protein D is subjected to diverse types of proteolytic degradation, resulting in the release of LMW breakdown products (<30 kDa). Various studies have demonstrated that SP-D can be fragmented in the human lung (249–252), and relevant enzymes include host proteases such as neutrophil elastase, cathepsin G, protease 3, and MMP-9, along with elastase produced by Pseudomonas aeruginosa, and house dust mite protease (253–261).
Clinically relevant proteolytic degradation of SP-D is observed in different settings, including acute lung injury (ALI) (262) and cystic fibrosis (CF) BAL. SP-D degradation by CF relevant proteases is well described; neutrophil elastase appears to be the most important contributor and reduces the CTLD lectin activity of SP-D in vitro (249, 250, 254, 263); however, the functional consequences of this have been questioned, as physiological concentrations of calcium can delay or abolish SP-D proteolysis (250, 254). Some studies have shown that, although present, the proteolytic fragmentation of SP-D does not appear to be responsible for reduced SP-D lectin activity in CF, which is instead suggested to be mediated by oxidative modifications (18). Nevertheless, SP-D levels are decreased in elastase-positive CF BAL samples, and the depressed levels are generally suggested to result from proteolytic activity (249, 250, 264). Clinical SP-D deficiency is not documented; however, BAL levels of SP-D may decrease during disease to levels that prevent immunodetection (264–266).
Surfactant protein D breakdown products are also detected in samples obtained from patients with severe asthma. In one recent study, no breakdown products were detected by western blotting (WB) in BAL, whereas the authors succeeded in identifying breakdown products in serum after a StrataClean Resin™ incubation step. The detection of breakdown products correlated with increased enzyme-linked immunosorbent assay (ELISA)-based detection of serum SP-D, at the expense of BAL SP-D. Hence, the ELISA readout may represent a mixture of complete and degraded SP-D forms (19). LMW SP-D breakdown products, together with trimeric SP-D, and non-reducible SP-D, can be separated by size permeation chromatography and WB, as demonstrated using BAL obtained from children with gastroesophageal reflux (267) and serum from patients with asthma (217).
Determinants of SP-D Levels
Surfactant protein D is a hydrophilic molecule and a variety studies using disease selected cohorts have demonstrated that variation in levels of BAL or circulatory SP-D may be associated with pulmonary disease, as previously reviewed (16). Moreover, sex, smoking, adiposity, and age were reported as important determinants of constitutional levels of circulating SP-D in a homogenous Caucasian population (208); however, studies of age-induced changes in SP-D levels are contradictory. Rat studies have demonstrated an association between reduced alveolar SP-D levels with increased oxidative damage (268). By contrast, studies of human alveolar SP-D levels demonstrated no detectable change in its levels with aging (269), neither was induction of human nor mouse alveolar SP-D observed during aging, alongside induction of cytokines and oxidants (270). Although a clear age-related induction of circulating SP-D was reported in Danes (208), this relationship has not been identified in all ethnicities (271), and may not occur in the presence of disease (272, 273).
Translocation of SP-D from Lung to Blood
Loss of air–blood barrier integrity is responsible for the outward intravascular leakage of secreted lung proteins and inward edematous flooding in the interstitium and air spaces (274). A concentration gradient of SP-D thus allows SP-D synthesized in the respiratory tract to leak into the bloodstream in acute and chronic lung injury following cigarette smoke exposure, as demonstrated using mice (77, 275, 276) and shown in human subjects (277). Thus, in some settings, including acute cigarette smoke exposure, SP-D may be decreased in BAL while being simultaneously enriched in serum (16, 252, 277). Smoking status is a strong predictor of such translocation (208, 277, 278). Examples include investigations of SP-D variation in COPD (252, 279, 280), asthma (19), and CF (249, 250, 264, 265, 281, 282). Studies in rabbits and humans have provided evidence for molecular size-dependent clearance of proteins from the air spaces of the lung (274); however, although highly anticipated, this study did not conclusively confirmed that LMW SP-D can translocate from the lung into the blood more easily than HMW SP-D (Figure 2). Herbein and Wright (283) reported that SP-D clearance from lavage was elevated in LPS-treated lungs compared with control lungs, due to the increased SP-D uptake in tissue neutrophils, and such clearance may also contribute to decreased alveolar SP-D levels in disease.
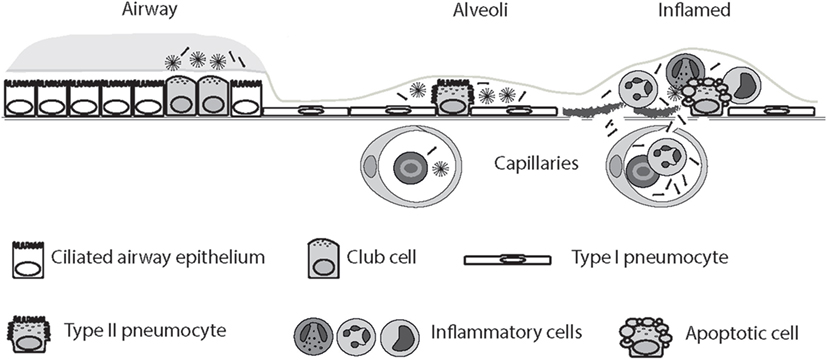
Figure 2. Circulatory spill-over of pulmonary surfactant protein D (SP-D) in inflammatory disease. SP-D is synthesized by Club cells, type II alveolar cells, and endothelial cells, and the levels of SP-D multimers and trimers in the serum are highly genetically determined. In the inflamed lung, the production of trimeric SP-D is increased, due to various chemical modifications and proteolytic breakdown of the protein, and loss of air–blood barrier integrity allows spill-over of pulmonary SP-D into the circulation. For simplicity, only alveolar damage is illustrated. Moreover, only fuzziball SP-D multimers are depicted.
Interpretation of the quantitation of SP-D in alveolar fluid and serum in various pulmonary diseases may be hampered because anti-SP-D antibodies may have varying affinities for oxidized and/or crosslinked species, as recently discussed by Atochina-Vasserman (284). HMW and LMW SP-D structural variants have been examined in BAL by native gel electrophoresis in various clinical pulmonary diseases and, as described above, different studies have demonstrated that levels of trimeric SP-D increase in BAL in inflammatory disease, and that trimeric SP-D is composed of both partially degraded, oxidized, and inactive SP-D, as well as a Thr11-glycosylation variant (50 kDa) (18, 19, 242, 249, 252, 262, 267, 285).
Identification of Modified or Degraded SP-D in Clinical Samples
Variation in SNO-SP-D
The presence of SNO-SP-D in BAL from clinically relevant samples has recently been demonstrated. SNO-SP-D was indirectly identified in BAL from patients with Hermansky–Pudlak syndrome type 1 (122) and directly detected in samples from asthmatic patients after segmental challenge with allergen (242). Hence, a role for SNO-SP-D as a disease marker is conceivable, as it is enriched in BAL from patients with pulmonary disease (122, 242). An ELISA-based method to measure circulating SNO-SP-D in COPD patients has been developed using anti-SP-D capture antibodies and antibodies reacting with S-nitrosylated groups for detection; however, this approach did not identify an association with disease severity and found only a weak correlation with radiologist score of emphysema, while possible associations with disease activity have not been verified (286). Nevertheless, detection of circulating SNO-SP-D in additional pulmonary diseases is warranted.
Variation in Fucosylated SP-D
A similar ELISA-based approach has been applied for measurement of fucosylated SP-D, demonstrating the presence of a core-fucose in N-glycans groups on serum SP-D, and an association of circulating levels of fucosylated SP-D with COPD outcomes. This modification represents a promising circulating disease-associated SP-D biomarker candidate, although in initial experiments measured levels of fucosylated SP-D could not adequately separate never-smokers, COPD smokers, and COPD (241).
Variation in SP-D Degradation Products
An alternative approach to simple SDS-PAGE-based measurement of SP-D degradation products in BAL or serum is permeation chromatography-based size separation; however, both techniques may be biased by genetic size variants (encoded by rs721917) and by oxidative crosslinking. Moreover, enzymatic neutrophil elastase degradation of SP-D and consecutive production of monoclonal antibodies against proteolysis products did not appear to result in immunological recognition of disease-specific SP-D breakdown products (286). Although various proteases are known to degrade SP-D, this approach has not yet been developed sufficiently to enable detection of disease-induced SP-D neoepitopes.
The Role of SP-D in Respiratory Disease
Circulating SP-D levels and genetic variants in SFTPD are associated with the development, progression, and severity of various pulmonary diseases. The variation of constitutional serum SP-D levels spans a >30-fold range (208); therefore, disease-induced serum levels may exhibit considerable overlap with control levels. Nevertheless, the significance of disease-induced levels in prognosis is underscored by association of serum SP-D with mortality in pulmonary disorders, including COPD (287), idiopathic pulmonary fibrosis (288), and ALI/acute respiratory distress syndrome (ARDS) (289). Genetic or phenotypic SP-D variation is associated with ALI/ARDS (289–293), lung injury in critically ill mechanically ventilated patients (294), respiratory distress syndrome (RDS)/bronchopulmonary dysplasia (BPD) (233, 234, 295), community-acquired pneumonia (227, 296), viral infection (297–300), asthma (19, 242), lung cancer (178, 301, 302), pulmonary aspergillosis (281), interstitial lung disease (15, 288, 303–305), and COPD (215, 224, 280). This review is concerned with the role of SP-D in RDS/BPD, asthma, and COPD.
RDS and BPD
Genetic Association
Fetuses carrying the SFTPD rs721917 Met11 allele have been associated with spontaneous preterm birth (235), and an initial genetic association of SP-D with pulmonary outcomes in premature infants was suggested by the observations that 2-marker SP-D/SP-A haplotypes including the Met11 allelic SP-D variant were protective against the development of RDS (306) and harmful in BPD (232), respectively. Subsequently, the rs1923537 polymorphism, located downstream of SFTPD, was demonstrated as associated with RDS and the requirement for oxygen supplementation at day 28 (a proxy of mild BPD) in very early preterm birth infants (234), whereas other SFTPD SNPs, including rs721917 (Met11Thr), were not associated with either RDS or BPD in that study or a subsequent investigation (307). Similar tendencies were observed in another study, although the genetic Met11 SP-D variant appeared to be protective for BPD defined radiologically and with requirement for supplemental oxygen at a gestational age of 36 weeks (295). In the most recent study, several SNPs in SFTPD, including the Met11 variant, were found to be positively associated with circulating SP-D levels and harmful in respiratory distress, the requirement for oxygen supplementation at day 28, and respiratory support (233).
Although they generated conflicting data, the above studies support a contribution of SP-D genetic variation to pulmonary outcomes in prematurity, and the discrepancies between studies may be explained by differences in statistical power, different mean gestational ages and the associated variation in requirement for oxygen supplementation at day 28/36, or the investigated SNPs or haplotypes may reflect variation at other, causative variants. No association between SFTPD alleles and neonatal mortality has been identified, and this may explain the persistence of high-frequency SNPs associated with respiratory outcomes in prematurity. No association was identified between SP-D SNP variation and diffuse lung disease enriched for genetic surfactant dysfunction (308).
Phenotypic Association
Surfactant protein D expression in the fetal distal airways increases with advancing gestation and is evident in 10-week-old fetuses; however, in lungs from infants with RDS and BPD, only open terminal airways were bordered with SP-D expression. Injured areas lined with hyaline membranes, or alveoli filled with hemorrhage, infection, or edema fluid, were lightly stained or unstained, whereas serous cells of bronchial and tracheal glands were consistently stained, particularly in infants with lung inflammation (10).
The percentage of multimeric SP-D in neonatal BAL, which is capable of binding microbial compounds, appeared to be lower in preterm than term infants (309), and alveolar SP-D may essentially be absent in the presence of RDS, increasing after surfactant treatment (310, 311). However, an increase in preterm BAL SP-D the first day (day 1) after birth has been demonstrated (309, 312), in parallel with a transient increase in capillary SP-D levels. This transient, circulatory increase was predominantly observed in infants homozygous for the rs721917 Met11 allele and was more apparent in infants with respiratory distress or receiving respiratory support. Such observations suggest that genetic SP-D variation determines the magnitude of induction of respiratory support-dependent systemic SP-D levels. Serum SP-D levels, which were not significantly affected by antenatal steroids, were positively associated with gestational age, mode of delivery, risk of later septicemia, and risk of respiratory distress (65, 233, 313). A recent study correlated serum SP-D measured at later time points (day 3 and day 7 after birth) in preterm infants and found no relation with the requirement for mechanical ventilation or oxygen, or with the development of BPD using that approach (314).
Basic Research
Mouse fetal pulmonary SP-D expression increases with advancing gestation, and levels predominantly elevate shortly before birth, stimulated by vascular endothelial growth factor signaling and glucocorticoid treatment (54, 66–69, 71, 315, 316). Pulmonary SP-D expression is therefore very low in experimental prematurity (317); however, various studies of the Sftpd−/− lung phenotype do not support a role for SP-D in normal fetal or postnatal lung development, although the vast accumulation of phospholipids in the Sftpd−/− lung have effects on surfactant homeostasis. Rather, these studies support a role for SP-D in emphysema and fibrosis development during airway remodeling processes later in life, as recently reviewed by Bersani et al. (318).
An early study of baboons showed that both the expression and protein secretion of pulmonary SP-D precede that of SP-A in normal gestational development in the baboon, and they are comparable to, or exceed, adult levels during advancing gestational age. In line with observations of the induction of circulating SP-D with respiratory support, data from premature baboons receiving 100% oxygen for 10 days to produce chronic lung injury indicated that lavage concentrations of SP-A reached a low percentage of that of normal adults, while those of SP-D equaled the amounts present in normal adults. The combined lavage SP-A/SP-D pool reached a low percentage of that of normal adults and the authors concluded that the combined decreased concentration of surfactant host-defense proteins may augment proclivity to infection and worsening injury (319). This suggestion was partly supported by a subsequent experiment demonstrating an increased tendency for lung infection in the same model; however, the major experimental effects were related to massive depletion of SP-A (320). Surprisingly, Sftpd−/− mice are resistant to hyperoxia, which may be partly explained by phospholipid and SP-B-mediated induction of surfactant resistance to inactivation (321).
Consistent with clinical lung maturation, the major effects of experimental chorioamnionitis are fetal lung inflammation, increased airway surfactants, and increased lung volumes (322–324), while the pro-inflammatory stimulus of chorioamnionitis is also commonly associated with preterm delivery and subsequent RDS (325, 326). A clinical study did not support an association of increased SP-D in amniotic fluid with intra-amniotic infection (327). In contrast, studies of mice and lambs have demonstrated a clear induction of SP-D expression in the fetal lung after LPS treatment (328, 329), although an earlier study indicated greater fluctuations in SP-D expression (330). Furthermore, studies using transgenic mice overexpressing rat SP-D under the SP-C promoter demonstrated that SP-D enhances cytokine production in the fetal and maternal compartments on maternal LPS exposure. Moreover, a significantly higher proportion of the pups born to dams overexpressing SP-D were stillborn after LPS treatment compared with those from wild-type mice (331). Moreover, mice that are doubly deficient in both SP-D and SP-A had delayed parturition and decreased expression of inflammatory and contractile genes (332). A recent study extended these findings, demonstrating that Sftpd−/− female mice have fertility defects, evidenced by smaller litter size, increased pre-implantation embryo loss, and elevated uterine inflammation, when mated with wild-type males; however, in support of previous findings, maternal LPS administration did not result in increased embryo loss or pro-inflammatory responses in Sftpd−/− females (333).
In contrast to the surprising association between high intra-amniotic SP-D and preterm birth, addition of recombinant human SP-D to commercial surfactant containing SP-B and SP-C alone improved surfactant function by protecting the premature lung from ventilation-induced inflammation and by increasing its resistance to protein inhibition of surfactant function and changing its biophysical properties and structure when tested in lambs shortly after birth (334). Although pulmonary inflammation was not blocked by SP-D, exogenous SP-D was effective in a model of endotoxin shock in newborn preterm lambs, where intratracheal administration prevented systemic inflammation and decreased cytokine expression in the spleen and liver (169).
Asthma
Genetic Association
Although structural SP-D polymorphisms are not associated with allergic bronchial asthma (220), an association with decreased atopy was identified in black subjects (221).
Phenotypic Association
Several studies have documented induction of BAL or systemic SP-D levels in asthma, which may be attributable to a combination of induced SP-D synthesis in airway epithelia (335) and increased air–blood barrier integrity, as described ealier.
Levels of SP-D are increased in BAL samples from allergic asthma patients (336), may further increase after segmental allergen challenge, and be correlated with those of BAL eosinophils, which in turn are correlated with NO content in BAL and oxidized SP-D species (242). Baseline SP-D levels are elevated in serum from patients with allergic asthma, and further elevated after allergen challenge, which is predictive for the late asthmatic response and for eosinophil cationic protein concentrations post-challenge (337).
Recent studies have supported these initial observations and demonstrated that sputum SP-D is increased in severe asthma or severity of exacerbation (338, 339), that serum SP-D increases stepwise in mild to moderate and severe disease, and correlates inversely with lung function and directly with small airway resistance (273). Mackay et al. (19) demonstrated that serum SP-D was increased in severe asthma with mixed eosinophilic and neutrophilic inflammation and enriched for SP-D breakdown products. The latter observation was supported by the recent findings of Fakih et al. (217) of a significantly decreased HMW/LMW serum SP-D ratio in asthmatic patients. In the studies of Mackay et al., the BAL/serum SP-D ratio was reported to be decreased, implicating depletion of SP-D from BAL due to leakage of degraded SP-D to the serum. Furthermore, serum SP-D levels were inversely associated with alveolar neutrophil infiltration and alveolar endotoxin levels (19).
Investigations that did not support a relationship between asthma and SP-D variation include one of the earliest studies of serum SP-D (340), and a recent study of serum SP-D by Akiki et al. (341). These discrepancies may be explained by differences in the distributions of mild and severe asthma between investigations. Serum SP-D enrichment is observed in pulmonary allergies (342–346) and allergic rhinitis (219), which is considered to be a manifestation of the same underlying disease processes as asthma (347), and basic research has demonstrated a link between SP-D and T helper 2 cell (Th2)-mediated inflammation. Thus, differences in clinical observations may also be due to a lack of stratification for different distributions of allergic asthma, or asthma characterized by a Th2-high profile, which accounts for approximately 50% of patients with steroid naïve asthma (348), and the impact of neutrophilic inflammation or pathogenic load, as implicated by the observations of Mackay et al. (19).
Basic Research
Allergic asthma models have provided insight into the physiological influence of SP-D in the development of Th2 type allergic asthma. The use of Sftpd−/− mice as allergic asthma models has provided both clear and subtle allergic asthma phenotypes; however, uniform data have been obtained from studies using exogenous administration of SP-D (110, 197, 349). A large body of studies have used rfhSP-D in treatment protocols and, although rfhSP-D did not efficiently aggregate and opsonize pollen grains relative to native SP-D (92), both SP-D and rfhSP-D appear to dampen the majority of aspects of the allergic phenotype. Intranasal administration of SP-D/rfhSP-D in murine models of pulmonary hypersensitivity induced by diverse allergens and antigens suppresses specific IgE levels in serum, reduces peripheral and pulmonary eosinophilia, and causes T helper 1 cell polarization from the allergic Th2-mediated inflammation to varying degrees (110, 123, 144, 203, 204, 206, 221, 349–353). In one model, beneficial effects of exogenous SP-D were observed when it was administered 6 h after, but not 24 h before, allergen challenge. A single application of rfhSP-D to allergen-sensitized mice led to a dampening of the allergic airway response equilibrium, similar to the effect of budesonide (352).
Moreover, allergen exposure induced SP-D protein levels in an IL-4/IL-13-dependent manner, resulting in increased murine alveolar SP-D levels (353, 354), and this negative feedback loop appears to protect the airways from inflammatory damage after allergen inhalation, as described in a recent review (355). SP-D in lavage and tissue is derived from AT-II cells and Club cells; however, it is also synthesized in hyperplastic goblet cells of inflamed lungs (356).
Additional beneficial SP-D effects in asthma may be anticipated in virus-induced asthma, because of the anti-viral effects of SP-D; however, deleterious effects may also be expected due to disease-induced formation of SNO-SP-D. Disease-related proteolysis is suspected to render a fraction of SP-D malfunctioning or deleterious (260), nevertheless, the overall effects of SP-D appear to be beneficial in the context of allergic asthma. The main reported individual steps leading to SP-D-dependent experimental phenotypes are illustrated in Figure 3.
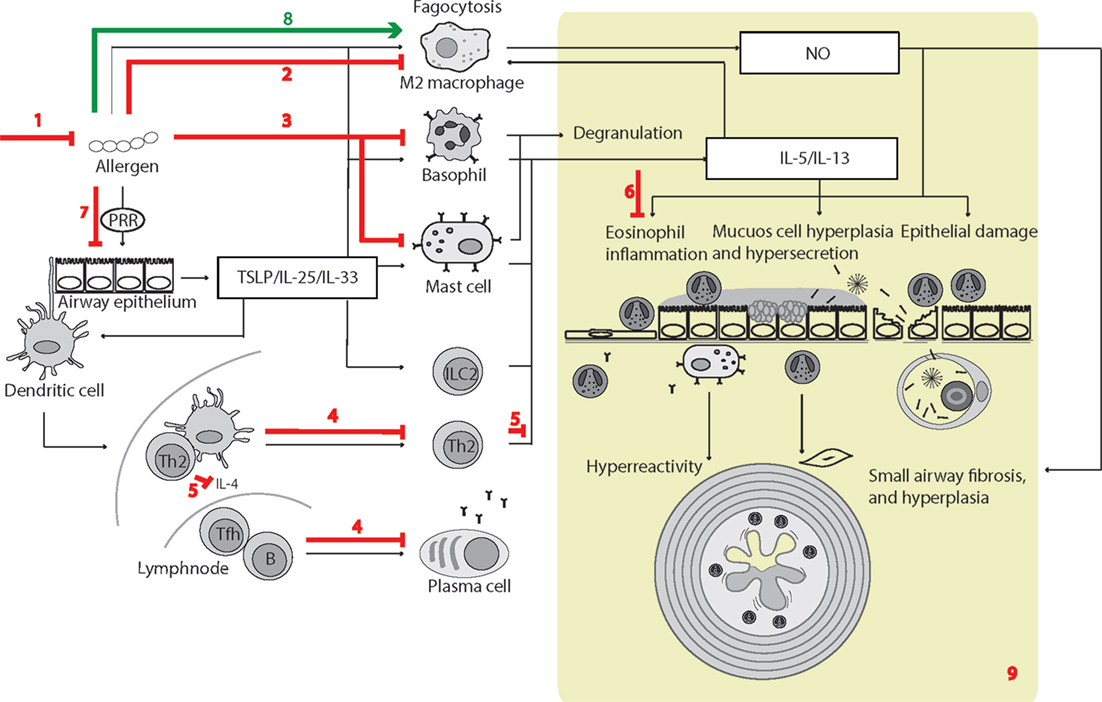
Figure 3. Surfactant protein D (SP-D)-mediated effects in experimental allergic asthma. The overview of cellular functions in allergic asthma was inspired by Lambrecht and Hammad (347) and Fahy (357). The multiple effects of SP-D include (1) removal of allergens by induction of aggregation and accelerating their binding and uptake by alveolar macrophages (92, 154, 358); (2) suppression of M2 macrophage polarization and allergen-stimulated macrophage NO production (123, 352); (3) inhibition of IgE binding to allergens, blocking allergen-induced histamine release by basophils and degranulation by mast cells (153, 154); (4) suppression of peripheral blood mononuclear cell interleukin (IL)-2 secretion (146), lymphocyte proliferation (358), and cytotoxic T-lymphocyte-associated protein 4 (CTLA4)-dependent induction of apoptosis (145). SP-D-mediated T-cell responses are CTLA4 dependent (149); (5) decreased lymphocyte IL-4 and IL-13 release (221); (6) suppression of eosinophil chemotaxis and degranulation, and induction of apoptosis (142, 143); (7) SP-D increases allergen interaction with respiratory epithelium, yet dampens epithelial chemotactic signaling (161); (8) SP-D increases uptake and removal of allergens in macrophages (93); (9) the overall effects of SP-D in allergic asthma in vivo include dampening of eosinophilia, alveolar macrophage accumulation, increased specific antibody levels, airway hyperreactivity, subepithelial fibrosis, and mucous metaplasia. These are features, which have either been observed in Sftpd−/− mice or that are subjected to phenotype rescue by endogenous SP-D, or administration of recombinant SP-D/60-kDa recombinant trimeric fragment of SP-D lacking the N-terminal but retaining a part of the collagen region (110, 123, 144, 203, 204, 206, 221, 349–353, 359). Leakage of pulmonary SP-D to the circulation in allergic asthma has been demonstrated in clinical samples (19). Only trimeric SP-D and fuzziball SP-D multimers are shown for simplicity.
Some of the effects illustrated in Figure 3 remain unclear. SP-D can increase pollen starch granule (PSG)-positive cells in vitro and accelerate PSG binding/uptake in vivo; however, studies by Winkler et al. (93) demonstrated that it did not affect total clearance of PSGs from the mouse lung nor enhance T-cell proliferation induced by PSG-positive dendritic cells. Hence, the different results obtained using human cell cultures or clinically isolated cells, compared with those from mice models, require further investigation.
A wider role for SP-D in Th2 immunity was recently proposed in a study of infection with the helminth Nippostrongylus brasiliensis. Elevated SP-D production in Th2 immunity is partly driven by IL-4 and IL-13. In turn, SP-D can exert negative feedback control of Th2 responses (90, 353); however, whereas allergic asthma studies identified increased type 2 immunity in Sftpd−/− mice, similar effects were not observed in N. brasiliensis infections. The results of these investigations demonstrate that elevated SP-D can enhance type 2 immunity and suggest that SP-D is an important modulator of protective IL-13 producing type 2 innate lymphoid cell-mediated and alveolar macrophage responses against N. brasiliensis (90).
Chronic Obstructive Pulmonary Disease
Genetic Associations
Associations of polymorphisms or haplotypes in, or flanking, SFTPD with COPD, emphysema, and COPD survival have been identified in various populations and by GWA studies (215, 216, 224–226, 360–362). As reviewed by Lock-Johansson et al. (363), specific SNP associations have not been validated in all investigated populations (363). The SNPs associated with COPD and circulating SP-D levels differed within some investigations, suggesting distinct genetic influences on COPD susceptibility and SP-D levels. In addition, two coding SNPs in SFTPD were associated with expiratory lung function in a study of preclinical smoke-induced lung injury (212), suggesting that structural variants of SP-D affect the susceptibility of COPD development in smokers. Recently, Mendelian randomization analyses were used to analyze serum SP-D-associated genetic variants and their association to COPD in the largest study executed until today. Variants were tested for association with COPD risk in 11,157 cases and 36,699 controls and with 11 years decline of lung function in 4,061 individuals. This study concluded that variants associated with increased serum SP-D levels decreased the risk of COPD and slowed the lung function decline (364).
Effects of Smoking
The majority of studies of smokers have reported reduced alveolar levels of SP-D (252, 269, 278, 365), and alveolar epithelial injury after LPS instillation was more severe in smokers than non-smokers, with increased circulatory SP-D and decreased BAL SP-D (277). These clinical observations are supported by in vitro observations that nicotine can cause reduced levels of SP-D in human airway epithelial cells (366). Additional studies have demonstrated increased circulating SP-D with tobacco smoking or other types of noxious exposure, including data from a twin study showing that smoking monozygotic twins had markedly increased serum SP-D relative to their non-smoking twins, despite the very high heritability of serum SP-D levels (208, 252, 280, 340, 367, 368); however, smoke-induced serum levels may not be clearly evident in mixed populations of respiratory patients, where SP-D levels may be influenced by other processes (369). As described earlier, cigarette smoke disrupts the quaternary structure of SP-D molecules (252). However, an initial attempt to construct an immunoassay for proteolytic SP-D breakdown products in serum did not provide additional information regarding COPD (286).
Differentiation between COPD, Smoking, and Other Respiratory Diseases
Whether SP-D levels can differentiate COPD from smoking or other respiratory diseases is uncertain. Some studies have noted significant decreases in SP-D in BAL samples from COPD patients compared with current smokers (252, 279), while others have not (269, 278). In a multicenter study, “Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints” (ECLIPSE), of approximately 2,000 individuals with COPD, higher serum SP-D levels were reported among COPD patients relative to current and former smokers without respiratory obstruction. As the largest difference in serum SP-D levels occurred between non-smokers and current/former smokers, it was concluded that SP-D is a powerful biomarker for smoking; however, there was no difference in serum SP-D levels in individuals with COPD, or in smoker controls with chronic bronchitis, compared with those who did not have this symptom (280). In addition, there are examples of smaller studies that have not shown significant differences between COPD smokers and control smokers (226, 370, 371).
Chronic obstructive pulmonary disease is a heterogeneous disease with several features that overlap with asthma, which has important therapeutic implications for some patients. Data from a randomized control trial of inhaled glucocorticoid therapy in COPD (Groningen Leiden universities chronic obstructive pulmonary disease) confirmed that alterations in airway gene expression may coexist in asthma and COPD, and suggested that Th2 inflammation is important in a subset of patients with COPD who have no history of asthma (372). Despite the vast body of experimental evidence for a role of SP-D in eosinophil-mediated allergy/asthmatic disease, there is no reported evidence linking changes in SP-D expression/function and eosinophil activity in COPD; however, independent studies have provided evidence that supports elevation of circulating SP-D in COPD compared with asthma patients; thus, SP-D may differentiate these two diseases. Yet, these studies may not have controlled sufficiently for the effects of smoking or the severity of asthma (340, 341). Size separation of HMW and LMW serum SP-D appeared to provide a clear distinction between controls and COPD subjects, with the HMW/LMW ratio significantly decreased in COPD after controlling for smoking and additional confounders; however, a similar association was evident for asthmatic patients (217). Additional studies have suggested that smoking-related SP-D variation is prognostic in lung cancer (301, 302).
Phenotypic Associations
Severity
In the ECLIPSE study, serum SP-D levels were not associated with COPD disease severity, as defined by the Global Initiative for chronic Obstructive lung Disease status, and there was no association with forced expiratory lung function (280). Some smaller studies have reported similar findings (272, 341, 373, 374); however, Ju et al. (375) reported an association with the BODE (Body-mass index, airflow Obstruction, Dyspnea, and Exercise) index of severity (375).
There are contradicting data regarding the association between circulating SP-D and expiratory lung function. The discrepancy indicates that there may be an inverse correlation between serum SP-D and forced expiratory lung function only in smokers with and without COPD (212, 226, 252, 370, 376–379). Such correlations may be difficult to identify in cohorts including both smokers and non-smokers (380), because the two groups exhibit opposite associations with SP-D variation (212). In addition, the correlation may also be affected by differences in COPD treatment.
A recent analysis of data from independent longitudinal population-based cohorts suggested that a large proportion of individuals who develop airflow limitation exhibit a rapid decline in forced expiratory volume in 1 s (FEV1) from a normal level of lung function in early adulthood, whereas another fraction had a relatively gradual rate of FEV1 decline, but started from a low initial FEV1 value (381). In this regard, circulating SP-D has been suggested to provide the means to identify those smokers with low initial lung function who are at highest risk of COPD development and thus eligible for early intervention with pharmacological treatment, in addition to recommendation of tobacco-smoking cessation (212). Nevertheless, longitudinal studies are warranted to validate the use of SP-D measurements as information complementary to spirometric testing for early detection.
Treatment
Surfactant protein D is a rather stable marker when measured repeatedly within a few months (252, 280, 382). Nevertheless, Sin et al. (376) reported a significant association between serum SP-D decline and dyspnea improvement within a 3-month study (376). Inhaled corticosteroids (ICS), ICS/long-acting beta(2)-agonist combinations, or prednisolone were also reported to significantly reduce plasma concentrations of SP-D and, in some reports, changes in SP-D levels were associated with symptom scores (279, 280, 373, 383). By contrast, ICS use was independently associated with higher SP-D levels in BAL, and AT-II cells isolated from adult rat lungs responded to dexamethasone treatment by significantly increasing SP-D (279). However, some studies have not validated SP-D as a marker of COPD treatment (374).
Activity
Increased levels of circulating SP-D have been reported during exacerbation of COPD compared with stable COPD (384, 385). The observations from these cross-sectional studies were extended by longitudinal studies demonstrating that the level of circulating SP-D decreases in the weeks after hospitalization, due to exacerbation, but ultimately increases some months after hospitalization (375, 386, 387). The variation in levels after the onset of exacerbation appeared to mirror the white blood cell count and was ascribed to the effects of glucocorticoid treatment (386).
Chronic obstructive pulmonary disease subjects from the ECLIPSE cohort, as well as other cohorts, who had the highest serum SP-D concentrations at baseline also had increased risk of exacerbation during the follow-up period (226, 280, 370). The ECLIPSE consortium subsequently performed a more detailed analysis and concluded that the association with SP-D levels did not persist after adjustment for history of exacerbation (388). Overall, the reported data indicate that SP-D is a robust marker for treatment effects, but that the utility of circulating SP-D as marker for prediction of COPD exacerbation may be limited, despite induction of serum SP-D in the acute phase.
Emphysema and Mortality
Further data analyses from the ECLIPSE cohort have demonstrated that baseline serum SP-D levels are associated with baseline lung density and its decline over time (389); however, the correlation between SP-D and emphysema was recently challenged by observations of serum SP-D variation in smoking, pulmonary emphysema, and combined pulmonary fibrosis and emphysema (CPFE), where serum SP-D was clearly induced only in the CPFE group compared with the other groups (390). Moreover, a study of indium-exposed workers demonstrated that increased serum SP-D was more closely associated with progression of interstitial changes than with progression of emphysema (391). The ECLIPSE consortium subsequently confirmed that COPD is a highly heterogeneous disease with poor correlations between FEV1, symptoms, quality of life, functional outcomes, and biomarkers in general (392), which highlighted the need for patient subgrouping. The consortium then included serum SP-D into a multi-marker cluster analysis of COPD subgroups and found that it was reduced among individuals with more severe emphysema, but higher among those with progressive emphysema. The resulting data suggest that high-circulating SP-D levels could indicate highly active disease and progression, whereas low levels may reflect loss of lung tissue, and thus more severe emphysema. The authors further concluded that the implications of SP-D appeared to differ from cluster to cluster (393). Furthermore, the ECLIPSE and COPDGene multicenter cohort studies both indicated that combinations of multiple biomarkers, including SP-D, are much more strongly predictive of airflow limitation, emphysema, and mortality than any individual biomarker; however, the amount of variance explained by the multiple biomarkers was lower than that of clinical variables (394).
Together with the observation that circulating SP-D is associated with the hard endpoint “all-cause mortality” in COPD (287), its relationship with treatment effects and inverse correlation with FEV1 in smokers indicates that SP-D is a useful COPD marker within specified prognostic subgroups and in multi-marker analyses. Multi-marker analyses including SP-D may be useful to limit clinical trials to subgroups of patients likely to benefit from a given intervention or serve as surrogate endpoints.
Basic Research
Semi-quantitative studies on aging Sftpd−/− mice (395) have indicated that they develop progressive emphysema, increasing septal wall thickness (395–397), and subpleural fibrosis (194), which may indicate a phenotype that combines emphysematous and fibrotic changes in the lungs. Highly detailed stereological investigations of the long-term progression of lung parenchymal remodeling, destruction, and septal wall thickening in aging Sftpd−/− mice were recently undertaken by Schneider et al. (398). These investigations concluded that the unchallenged aging Sftpd−/− pulmonary phenotype was characterized by airspace enlargement only, without septal wall thickening (fibrosis) or destruction (emphysema). The age-induced airway enlargement was suggested to result from unexplained altered airway mechanics. The authors further suggested that the discrepancies in observations with previous studies might partly result from the different genetic backgrounds of the mouse strains used and ongoing alveolarization in C57BL6 mice (398).
Although the role of SP-D in spontaneous airway remodeling was recently questioned, several studies of Sftpd−/− mice challenged with noxious stimuli have provided clear evidence for a role of SP-D in pulmonary inflammation and emphysema development, which are essential components of COPD pathophysiology. These results support a role for SP-D in modulating alveolar macrophage activation, oxidant production, and MMP activity, leading to emphysema-like and fibrotic changes of the lung, and histological and morphometric studies have demonstrated airspace enlargement and/or emphysema development associated with Sftpd deficiency. Inflammatory and structural changes may be reversed or prevented by local treatment with rfhSP-D or full-length SP-D (200, 202, 399, 400). These changes appear to predominantly affect the alveolar compartment, and there is no apparent SP-D-dependent effect on vascular physiology after cigarette smoke exposure (401).
In contrast to findings in humans with COPD, in Sftpd−/− mice neutrophil infiltration was not associated with lung remodeling during emphysema development (175). Moreover, alveolar levels of SP-D in mice and rats may increase after pulmonary exposure to noxious stimuli (276, 402–405), in contrast to the majority of findings in humans; however, reductions of SP-D in rat BAL after exposure to noxious stimuli have been reported (406). Viral and bacterial respiratory tract infections amplify the chronic inflammation in COPD by triggering pathogen-recognition receptors, as recently reviewed by Brusselle et al. (407). Additional beneficial SP-D effects in COPD development or progression may be anticipated due to the anti-viral and anti-bacterial effects of SP-D. The main individual steps contributing to the SP-D-depressed experimental phenotypes relevant to COPD are illustrated in Figure 4.
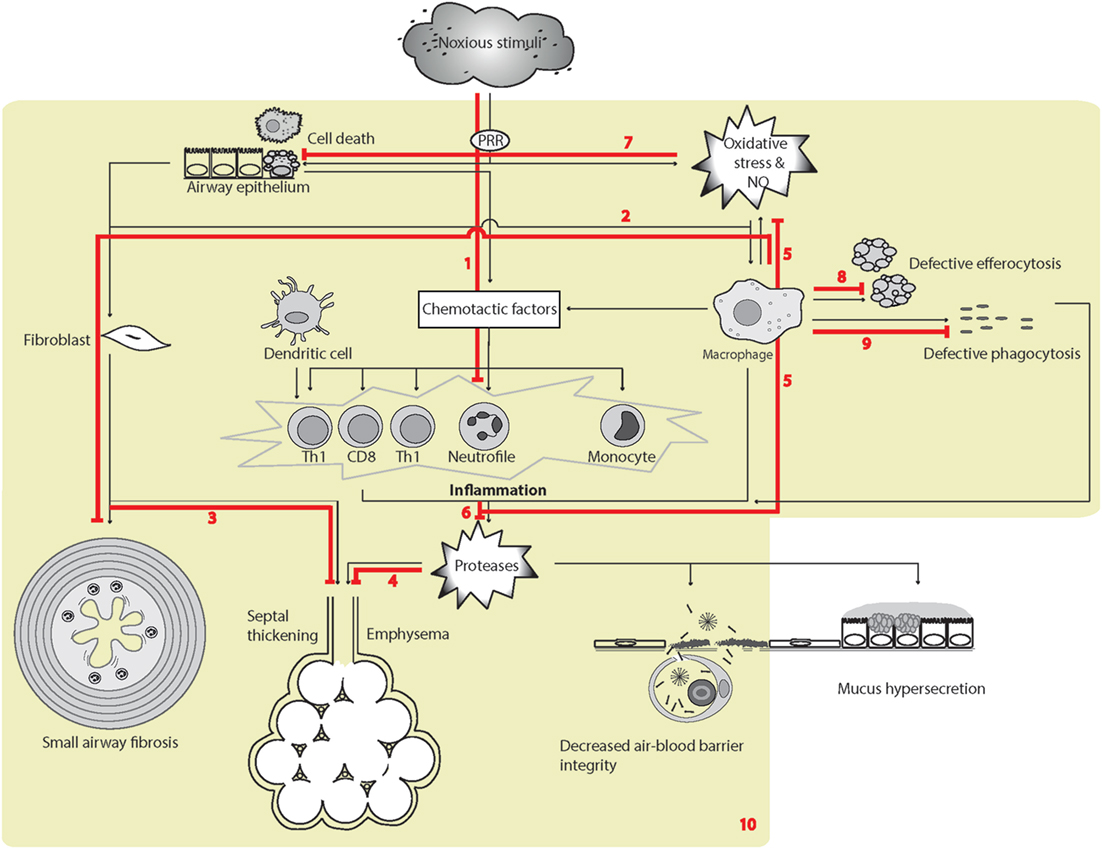
Figure 4. Surfactant protein D (SP-D)-mediated effects in experimental pulmonary inflammation and airspace enlargement in chronic obstructive pulmonary disease (COPD). The overview of cellular functions in COPD was inspired by Barnes (408) and Brusselle et al. (407). Multiple effects of endogenous or exogenous SP-D include (1) suppression of inflammation elicited by noxious stimuli (395, 400, 402, 409). The SP-D-mediated mechanisms includes SP-Ds interaction with immune-regulatory receptors (117, 134, 170, 174, 177), depression of oxidative stress, including iNOS activity, protection of phospholipid oxidation (397, 410–413); (2) decrease of macrophage transforming growth factor-β (TGF-β) production and fibrocyte recruitment (163). These SP-D effects may partly enable suppression of age-induced influences; (3) increased septal wall thickening by fibrotic deposition (194, 397); (4) airspace enlargement and loss of surface area of alveolar epithelia (194, 202, 397, 414); (5) suppression of the production of ROS and NO by macrophages, and possibly additional cell types (125, 194, 395, 409, 411). Fibrotic and emphysematic changes in the lung may also depend on inhibition of iNOS by SP-D (397, 413). (5/6) SP-D suppresses metalloproteinase production in alveolar macrophages (125, 194) and putatively additional cell types. The result is an overall decrease in pulmonary protease activity via oxidant-sensitive pathways (125, 194); (7) prolonged alveolar epithelial cell and macrophage survival after cigarette smoke extract exposure or oxidative stress (276, 410); (8) increased efferocytosis (94, 121, 170, 201, 415); (9) opsonization of microbes for phagocytosis (91). (10) The resulting effects of SP-D relevant for COPD-like phenotypes in vivo include dampening of chronic low-level pulmonary inflammation predominantly mediated by macrophages and correlated with reduced oxidative stress and protease activity (125, 194), which can prevent changes in pulmonary elastance due to both tissue breakdown and fibrotic build up that occur sequentially with increasing age and exposure to noxious stimuli (395, 397, 409, 413). Leakage of pulmonary SP-D to the circulation in COPD has been demonstrated using clinical samples (280). Only trimeric SP-D and fuzziball SP-D multimers are shown for simplicity.
The phenotype of mice deficient for the proposed SP-D receptor, GPR116, is highly similar to that reported for Sftpd−/− mice, including alveolar enlargement, enhanced ROS production, accumulation of foamy macrophages, and enhanced expression of Mmp12. The surfactant lipid changed induced by SP-D may be attributable to its effects on alveolar type II cell activity (177, 193, 414). Although GPR116 deficiency also causes an emphysema-like phenotype that is associated with alveolar macrophage activation (177, 416), the phenotypes of neither GPR116 nor Sftpd-deficient mice fully reflect those of human COPD. Mouse alveolar macrophages become foamy with lipid-laden phagosomes due to extensive uptake of surfactant lipids (177, 417, 418); however, in human pathology, foamy macrophages are found in diverse disorders, including diffuse pan-bronchiolitis or bronchiolitis associated with bronchiecstasis (419), diseases of surfactant homeostasis (420), extrinsic allergic alveolitis (421), and tuberculosis, rather than in COPD. Moreover, the alveolar accumulation of surfactant lipids in the two types of gene deficiency appears to reflect aspects of pulmonary alveolar lipoproteinosis, derived from defective granulocyte macrophage colony-stimulating factor signaling (124, 396, 422), or other types of macrophage exhaustion. Thus, the appearance of foamy macrophages in the Sftpd−/− lung may not be specific to COPD-related processes, although induction of MMP-12-synthesizing foamy macrophages results after exposure of mice to cigarette smoke (276), and foamy macrophages provide a source of ROS and inflammatory signaling in mouse models as a result of Sftpd deficiency (125).
SP-D in Non-Respiratory Diseases
Genetic SP-D variation, altered local protein expression, and serum variation have been reported in extrapulmonary diseases involving autoimmune disorders [including rheumatoid disease (213, 423–426) and diabetes (229, 427)] and in diseases involving specific organs [including the intestines (230, 428), skin (20), brain (41), and large arteries (214, 429)]. In some cases, the association between circulatory SP-D and extrapulmonary disease may be partly explained by the coexistence of respiratory disease or the pulmonary effects of disease (430–434). The antimicrobial or anti-inflammatory effects of SP-D at extrapulmonary sites have been demonstrated using animal models or cell culture, including disease models of the eye (258, 261, 435), pancreas (436), kidney/urinary tract (437, 438), gastric mucosa (439–441), intestine (442), gestational tissue (27, 331, 443), and large arteries (48). Inflammation-modulatory effects of SP-D at extrapulmonary sites is, to a certain degree, similar to SP-D-mediated effects in the pulmonary compartment, yet unique extrapulmonary effects are emerging in studies of CVD, such as atherosclerosis, and also in metabolic disease.
Cardiovascular Disease
Association with All-Cause Mortality
A recent study of constitutional circulating SP-D measurements in an elderly twin population, where subclinical disease was likely to be present, demonstrated a relationship between the highest SP-D levels and increased all-cause mortality. Female twins with the highest SP-D levels had a significantly increased risk of dying before their co-twin during the study follow-up period. Adjustment of this analysis for intrapair differences in smoking pack-years did not affect the association, indicating that SP-D is not merely a proxy for smoking in reflecting mortality (444). A study of dementia in the elderly reached a similar conclusion about the association with all-cause mortality, without a gender-bias (445). Hence, it is possible that SP-D is associated with CVD, which is the number one cause of death globally.1
The Relationship between Pulmonary Impairment and CVD
The sources of circulating SP-D are not entirely clear, and it is possible that spill-over of SP-D from the diseased arterial wall into the circulation may affect total serum levels, in addition to the lung, as a main contributor. COPD and coronary artery disease (CAD) frequently coexist, COPD is an independent risk factor for CAD and increased risk for cardiovascular mortality, and patients with diagnosed and treated COPD are at increased risk for hospitalization and death due to CVD (432, 446–451). These correlations suggest that the association of SP-D with all-cause mortality may be rather complex. Some recent studies have demonstrated that SP-D levels correlate with alveolar leakage in heart failure (433), and with the presence of submassive pulmonary embolism (434), supporting the hypothesis that variation in circulatory SP-D results from disease-mediated lung damage in some types of CVD.
Genetic Association
Recently obtained data indicated nominal associations between SFTPD coding variants, including the Met11Thr polymorphism, and subclinical atherosclerosis, and demonstrated that the direction of the effects was dependent on tobacco smoking, but independent of circulating SP-D levels (214). An additional study identified a borderline significant association of the Met11Thr polymorphism with advanced atherosclerosis, and this variation was observed to significantly contribute to the risk of arterial stenosis when included in a complex model with other inflammatory gene variants (222).
Phenotypic Association
As suggested above, innate immune factors, including SP-D, may provide a link between respiratory and vascular morbidity and mortality. In this context, circulating SP-D was positively associated with both all-cause mortality and cardiovascular mortality in a large study of the Vancouver Coronary Angiography Cohort by Hill et al. (429), and inclusion of SP-D into a multi-marker analysis improved risk prediction across all levels of risk. There was also a significant positive association of SP-D with triple vessel disease and a borderline association with total cholesterol; however, no associations with other serum lipid fractions or diabetes mellitus were observed in that study. SP-D was also associated with all-cause mortality in the ECLIPSE COPD cohort (287), as described above, and in a study of the relationships among COPD, coronary artery calcification, and mortality using the same cohort, circulatory SP-D was correlated with Agatston score (432). The relationship between SP-D and CVD is further supported by the observation of Hu et al. (452) of positive associations between circulatory SP-D levels and carotid artery intima-media thickness and severe coronary artery calcification, which persisted after multivariate adjustment. Hill et al. (429) and Hu et al. (452) did not report BAL SP-D or other means of monitoring the presence of lung disease and, therefore, did not address the question of the organ source of circulatory SP-D.
Basic Research into the Role of SP-D in Atherosclerosis
Surfactant protein D expression, as well as expression of the SP-D-receptor, OSCAR, which induces secretion of TNF-α by CCR2 + inflammatory monocytes exposed to SP-D, was localized to the tunica intima and tunica media of clinical atherosclerotic plaques (48, 133). Arterial SP-D expression is documented both in endothelial cells (48) and coronary smooth muscle cells (47), where the pattern of expression regulation implicates SP-D in modulation of inflammation, and TNF-α induces SP-D expression in both cell types (47, 63). Direct interaction between LMW, but not HMW, SP-D and HDL, LDL, and oxLDL was demonstrated using ELISA-based assays (36). These observations suggest that the dominant effects of SP-D in the vasculature may not be the multivalent interactions with microbes, but rather may include specialized LMW SP-D-mediated effects on lipid metabolism.
The SP-D receptor, OSCAR, is expressed by cell types relevant for atherogenesis, including monocytic cells (133), neutrophils (453), dendritic cells (454, 455), and endothelial cells (456). Sftpd deficiency is shown to be protective in a diet-induced atherosclerosis model in C57BL/6 mice. Endogenous SP-D can influence systemic plasma lipids and inflammation, reducing HDL cholesterol and triglyceride levels while increasing those of TNF-α in mice maintained long term on a cholate-rich diet. Intravenous treatment with rfhSP-D for 5 days resulted in reduction in HDL cholesterol, but also, in parallel, in LDL cholesterol and total cholesterol, while there was no effect on triglycerides. The role of SP-D-mediated modulation of plasma lipids in atherogenesis is thus uncertain. However, the long-term effects of these events were not studied (48). A subsequent study using double-deficient apolipoprotein E (Apoe)−/−Sftpd−/− mice showed similar quantitative effects in decreasing lesion size. Moreover, Sftpd deficiency resulted in less macrophage accumulation, and more smooth muscle cell coverage of lesions (457). In that study, endogenous SP-D reduced several plasma lipid fractions, including total cholesterol and induced circulatory IL-6, rather than TNF-α. In contrast to the two previous studies, a study using double-deficient LDL-receptor (Ldlr)−/−Sftpd−/− mice did not provide clear evidence for a role of SP-D in atherogenesis (Sorensen, unpublished data).
Bridges et al. (410) reported that SP-D was a potent inhibitor of oxidation of LDL to oxLDL in in vitro assays, but later ascribed the majority of the effects to residual EDTA in the SP-D vehicle in a published erratum. However, the consequences of Sftpd deficiency in atherogenesis may also result from the antioxidant effects of SP-D and the effects on the formation of ROS and in NO-mediated signaling, although these aspects have not been investigated in detail in the vascular system.
A potential link between low SP-D levels and the sequela of atherosclerosis was recently demonstrated using cerebrospinal fluid samples from patients with cerebral infarction (41). Almost simultaneously, a mouse model of cerebral ischemia was analyzed, but showed no effect of Sftpd deficiency; however, that model was based on permanent occlusion of the middle cerebral artery and did not involve atherosclerosis (49).
The main reported individual steps leading to SP-D-enhanced experimental outcomes relevant for atherosclerosis are illustrated in Figure 5.
Figure 5. Surfactant protein D (SP-D)-mediated effects in atherogenesis. The simplified overview of cellular functions in atherogenesis was inspired by Skaggs et al. (458). Effects of SP-D in a long-term diet-induced model of atherosclerosis include (1) repression of high-density lipoprotein (HDL)-cholesterol levels (48) and also additional plasma lipid levels in a genetic model (457); (2) model-dependent induction of tumor necrosis factor-α (TNF-α) in vivo (48, 457) and TNF-α induction in monocytes in vitro, dependent on osteoclast-associated receptor (OSCAR) signaling (133); (3) induction of macrophage proliferation (457); (4) induction of circulating monocytes; (5) decreased coverage of smooth muscle in plaques (457); (6) the result is an overall increase in atherosclerotic plaque formation with accumulation of foam cells and cholesterol crystals, accompanied by disturbed plasma lipid levels in Sftpd−/− mice compared with Sftpd+/+ mice (48, 457). Moreover, TNF-α and NO increase endothelial SP-D expression (63) and circulating SP-D is increased in clinical cardiovascular disease (CVD) (25, 429). Only trimeric SP-D and fuzziball SP-D multimers are shown for simplicity.
Metabolic Disease
Relationship between Pulmonary Impairment and Metabolic Disease
In addition to the association of SP-D with CVD, possible causes of the association between circulating SP-D and all-cause mortality may include the role of SP-D in metabolic homeostasis. As described earlier, the brain, pancreas, gut, and endothelium express SP-D, and these locations suggest potential effects in systemic metabolism. Diabetes and metabolic syndrome are frequent comorbidities in COPD patients (459–462). COPD, diabetes, and the metabolic syndrome are all related to systemic inflammation, which may explain their cooccurrence (463). Moreover, pulmonary function impairment is well recognized in diabetes and may be attributable to increased pulmonary bacterial colonization due to hyperglycemia and exacerbation of COPD, among the additional causes recently discussed by Kinney et al. (464). By contrast, a reduced ability to eradicate pathogens may cause frequent respiratory tract infections, and chronic inflammation resulting in insulin resistance and type 2 diabetes, as reviewed by Fernández-Real and Pickup (465). Moreover, cigarette smoking is an independent predictor of type 2 diabetes, although the link between lower vital capacity and diabetes risk is independent of cigarette exposure and stronger in never-smokers (466).
Obesity is the most frequent cause of insulin resistance (467) and, although impaired lung function is present before the development of metabolic syndrome or diabetes, the associations between pulmonary and metabolic impairment may, at least partly, result from the effects of obesity, as recently shown in the longitudinal Strong Heart Study (468). Multiple cellular and molecular mechanisms of the innate immune system may contribute to this relationship, as reviewed by Lackey and Olefsky (469). Several studies have independently identified positive associations between lung function impairment and full metabolic syndrome or obesity (470–473), and metabolic biomarkers are predictors of impaired lung function in response to particulate inhalation (474).
Overall, the close relationship between metabolic and respiratory impairment suggests that lung innate immunity, as inferred from genetic or phenotypic SP-D variation, could reside at the intersect of inflammation, obesity, and insulin resistance.
Genetic Association
Multivariate genetic analysis was used to investigate genetic associations of serum SP-D with factors involved in metabolic syndrome in a twin study (GEMINAKAR). The study found that genetic correlations between traits differed, but were close to zero for SP-D versus body mass index (BMI), which may indicate that the common genetic contribution to the regulation of the traits (i.e., pleiotropy) is significant but limited. There was no significant genetic correlation between serum SP-D and HDL cholesterol or total cholesterol, and the study did not investigate possible genetic associations between SP-D and diabetes (208). A subsequent study by Ortega et al. (51) demonstrated that a common SNP variant in the SFTPD promotor, which is positively associated with circulating SP-D levels, was enriched in individuals with lower BMI and decreased percentage fat mass. A study by Pueyo et al. (229) showed that the presence of the variant SP-D allele, Met11, increased the odds of developing insulin resistance and type 2 diabetes, especially among women. Moreover, this SNP, and others in the SFTPD genomic region, were significantly associated with glucose homeostasis, insulin sensitivity, and type 2 diabetes, according to GWA datasets, and these associations were independent of circulating SP-D concentrations (229). A borderline significant inverse association was observed between free LPS concentration in plasma and circulating SP-D in subjects homozygous for Thr11, but not those homozygous for Met11, in a small subpopulation of the study. This observation indicates that the different genotypes may predispose to differing buffering capacity in metabolic endotoxemia (229), consistent with observations by Leth-Larsen et al. (37), showing that Thr11 SP-D bound preferentially to LPS compared with Met11 SP-D (37).
Phenotypic Association
Fernández-Real et al. (427) evaluated serum SP-D in four different cohorts and found that SP-D concentrations were significantly decreased in non-obese populations with type 2 diabetes, and that they were correlated with several metabolic variables, including BMI, and fasting and post-load glucose levels. Obese subjects showed significantly decreased serum SP-D concentrations, and weight loss led to further significantly decreased serum SP-D concentrations, while serum SP-D concentrations were not significantly associated with lung function parameters in these studies. When multiple linear regressions were performed using data from subjects with altered glucose tolerance, fasting glucose and fasting triglycerides were identified as independently contributing to 33% of SP-D variance (427). However, a recent study by López-Cano et al. (475) demonstrated increased serum SP-D in diabetes in obese patients with type 2 diabetes. Moreover, serum SP-D in this population was independently and inversely associated with FEV1 in the obese diabetes patients and the authors concluded serum SP-D might be a useful biomarker for detecting lung impairment in obese patients with type 2 diabetes.
The findings of Ortega et al. (51) supported previous observations that SFTPD was expressed in human adipose tissue and adipocytes, and this expression was decreased in both omental and subcutaneous adipose tissue from obese subjects with and without type 2 diabetes, compared with the control group. Omental SP-D expression was inversely associated with several metabolic variables, including BMI, percentage fat mass, waist circumference, and fasting glucose; and positively associated with the expression of insulin receptor substrate 1 and fatty acid synthase, suggesting that SP-D may effect glucose tolerance in fat tissue or vice versa.
At least two previous studies demonstrated decreased constitutional circulating SP-D in obese or overweight subjects after correction for gender, smoking, and age, which may support an initial influence of the processes leading to accumulation of fat tissues on the correlations between SP-D and metabolic impairment. As a part of the Danish population-based twin study on metabolic syndrome, GEMINAKAR, serum SP-D was identified as significantly and inversely associated with weight and waist circumference in men, and with BMI in both sexes (476). Similar findings were obtained from a Chinese population-based cohort (271). Moreover, the relationship was also observed in patients who were obese and had ALI (477).
Basic Research into the Role of SP-D in Metabolism
Three independent studies have demonstrated a link between Sftpd deficiency and obesity (457, 476, 478). According to a study by Stidsen et al. (478), body mass development in Sftpd-deficient mice was significantly increased compared with that of corresponding wild-type mice receiving both ad libitum and fixed energy intake diets. The study also identified a significant redistribution of body fat in parallel to obesity-related insulin resistance, as well as increased metabolic endotoxemia associated with the Sftpd deficiency in this model (478). Relative insulin resistance in Sftpd-deficient mice was later validated by Hirano et al. (457). Additional links between SP-D and body fat mass include the observation of decreased SP-D expression in human adipose tissue with increasing BMI (51), and direct SP-D interactions with fatty acids (479).
Furthermore, SP-D was linked to metabolic homeostasis in two independent studies seeking to identify genes involved in β cell adaptation during mouse pregnancy or in rat perinatal regenerating pancreas. The studies demonstrated that Sftpd mRNA and protein levels in β cells exhibited large but transient perinatal increases (480, 481). TNF-α- and IL-1β-induced expression of SP-D in islets was demonstrated (480), suggesting that TNF-α induction of SP-D is a general effect for many cell types.
No direct role of SP-D in experimental diabetes development has been demonstrated; however, the development of diabetes in mother rats is accompanied by delayed pulmonary histogenesis and decreased SP-D expression in the pups (482). Moreover, viral neutralization by SP-D in vitro was abolished in the presence of glucose at levels commonly found in diabetic mice (483), which may contribute to reduced anti-viral protection in diabetes.
Summary, Including Unresolved Questions
SP-D Effects and Variants
Widespread SP-D expression has been documented in a multitude of studies and underscores the fact that, although the lungs remain a major site of SP-D synthesis, the essential effects of SP-D in innate immunity can be expected at numerous additional sites, and various examples of such extrapulmonary effects have now been investigated and documented. For many years, research into SP-D-mediated immune-modulation has primarily focused on its roles in phagocytosis and efferocytosis, opsonization, and oxidative signaling in phagocytes. However, analyses of SP-D interactions with various white blood cells, epithelial cells, fibrocytes, and smooth muscle cells have provided more detailed insights into SP-D-modulated pathologies, and imply that additional cellular SP-D-mediated effects remain unexplored. Further examples include endothelial cells and adipocytes, where SP-D expression is reported, but where the effects of its expression remain largely unknown. Gaps in knowledge regarding the role of SP-D in non-immune cells and the levels of expression of SP-D during homeostasis and disease have the potential to hamper the design and interpretation of both in vivo experiments and clinical biomarker studies.
In support of observations of SP-D-induced cellular signaling, more than 10 different SP-D receptors or receptor complexes have been identified; however, the majority of studies suggesting new SP-D receptors have yet to be validated by independent researchers. In the majority of reports, the pulmonary effect of SP-D signaling is the dampening of inflammation. Pulmonary suppression of inflammation is thus the documented outcome when applying SP-D or rfhSP-D treatment, despite the potential inflammatory side effects of SP-D variants, which are unable to form HMW SP-D due to modifications, or which may represent proteolytic breakdown products.
Low-molecular weight SP-D is characterized by preferential binding to LPS and circulating lipoproteins, compared with HMW SP-D; however, the effects on pro- or anti-inflammatory cellular signaling resulting from the recognition of such complexes have not been explored. Moreover, it is unknown to what degree interaction of SP-D with LPS and lipoproteins mediates assembly of LMW into HMW forms in vivo, as observed with immobilized ManNAc ligands in vitro. However, observations suggest different effects of LMW SP-D compared with HMW SP-D and studies of a previously proposed role for collectins in quenching the immune-activating effects of hydrophobic polymers, such as lipids or cholesterols from lipoprotein particles (484), are warranted, particularly for LMW SP-D.
In contrast to the primarily anti-inflammatory effect of SP-D in the lung, during development of mouse atherosclerosis the major impact of SP-D appears to be enhancement of inflammation (48, 457) and this is also the case during fetoplacental development (331). Such observations could potentially be explained by site-specific expression of SP-D receptors. Determination of the contribution of SP-D-mediated OSCAR signaling to respiratory and non-respiratory disease may help to clarify the reasons underlying conflicting observations of pro- and anti-inflammatory effects of SP-D.
There is a paucity of in vivo data confirming native SP-D structural size variation-dependent effects in disease, although initial attempts have been made using Sftpd−/− mice overexpressing the common human SFTPD Met11Thr variants to determine their effects on the degree of SP-D multimerization. By contrast, various studies have demonstrated that treatment with the LMW rfhSP-D molecule may rescue histogenesis in Sftpd−/− mice and resolve inflammation both in Sftpd−/− and Sftpd+/+ mice. This suggests that this particular fragment may be capable of signaling through the same set of cellular receptors as endogenous SP-D, or that some in vivo inflammatory effects may be ameliorated by high local concentrations of SP-D, regardless of the distribution of HMW and LMW SP-D. However, rfhSP-D has also failed to demonstrate effects in several settings and the question of the extent to which the effects of full-length multimeric SP-D may be mimicked by high levels of trimeric SP-D has yet to be fully answered.
The degree to which SFTPD variants affect SP-D structural size in disease also remains to be elucidated. Nevertheless, the Met11Thr polymorphism, which affects the degree of multimerization in non-diseased populations, has also been identified as associated with numerous respiratory diseases, as well as with CVD and metabolic diseases. Such SFTPD Met11Thr associations with disease may indicate that, not only the expressional levels of SP-D but also its ability to form multimers, are essential SP-D-mediated effects beyond interaction with multivalent ligands and clearance thereof. Overall, the majority of data from genetic studies of this particular polymorphism indicate associations of the Thr11 allele with diverse clinical conditions. However, this allele has also been reported as protective in some diseases, including severe respiratory syncytial virus infection, which can cause infant death, and in which the allele is observed at a relatively high frequency.
Phenotypic SP-D Variation Induced by Disease-Related Processes
The lungs are among the five largest organs of the human body, and SP-D concentrations in the alveolar lining fluid appear to be higher relative to circulating levels. Thus, loss of air–blood barrier integrity in respiratory disease may cause pulmonary SP-D to translocate, leading to an increase in systemic levels, despite the huge variation in constitutional levels. Clinical SP-D deficiency is not documented; however, BAL levels of SP-D may decrease during respiratory disease to a level that does not allow immunodetection, while circulatory levels rise. However, it has not been conclusively confirmed that LMW SP-D will translocate from the lung into blood more easily than HMW SP-D, and just two studies (19, 217) have confirmed increased levels of circulating LMW SP-D in respiratory disease.
The translocation of SP-D appears to be reflected in a change of the ratio between BAL SP-D and circulating SP-D. This ratio is effective for differentiation of respiratory cases from controls and smokers, which is not always possible using isolated serum or BAL measurements (252); however, due to the invasive nature of sampling, SP-D measurements in BAL do not provide useful means for disease monitoring. Few attempts to monitor LMW SP-D, or breakdown products of SP-D, have been reported, and these primarily rely on the use of native gel electrophoresis, which may not be applicable to large-scale analysis. The development of immunoassay-based methods, which have previously been used to detect circulating extracellular matrix epitopes (485, 486), is warranted for studies of SP-D as disease biomarker. Whether particular LMW SP-D species are enriched in the lung or other organs requires further exploration.
Biomarker Utility
This review has focused on SP-D variation as prognostic marker for infant respiratory distress syndrome and BPD, moderate and severe asthma, and COPD. The involvement of SP-D in these clinical respiratory diseases is partly supported by genetic analyses. The data regarding SFTPD associations with RDS and BPD are conflicting, and an association of SFTPD with atopy has been demonstrated, but not with asthma. Moreover, associations of polymorphisms within or flanking SFTPD have been identified in various populations of COPD patients and by GWA, although the overlap between observations is not complete.
By contrast, circulating SP-D levels appear to be uniformly increased with disease severity, particularly in RDS and asthma, and are associated with further progression or exacerbation of disease, and with mortality from COPD. Clinical measurements of SP-D primarily support that the total circulating SP-D can be used to correct for the background variation in SP-D in interstitial lung diseases or to facilitate differentiation of idiopathic pulmonary fibrosis from other idiopathic interstitial pneumonias when included into a multi-marker array (15, 304, 487). Nevertheless, measurement of total circulating SP-D may also prove to be valuable in multi-marker-based identification of disease subtypes for pharmacological testing in other types of respiratory disease, including COPD (393).
RDS and BPD
Important information has been obtained about SP-D variation in premature infants, including the normal range of SP-D in term and preterm newborns and the influence of perinatal factors; however, there is a paucity of independent studies validating the observed association between the highest serum SP-D levels measured day 1 after birth and the most severe outcomes in premature infants, which warrants further validation.
Allergic Asthma
Recent research findings support increased circulating SP-D in moderate or severe asthma and in addition to allergic rhinitis. Yet, SP-D breakdown products were also enriched in the blood system in severe asthma, indicating that such fragments may provide a more specific measure of disease activity.
Chronic Obstructive Pulmonary Disease
Although SP-D levels are highly increased in response to smoking, the highest circulating SP-D levels appear to be not only proxies for smoking but also related to disease outcomes after adjustment for smoking, hence providing a robust marker for treatment effects. Association with mortality has been identified in CVD in cohorts selected, and not selected, for COPD. The presence of CVD is thus anticipated to be a significant contributor to induction of circulating SP-D levels in COPD. The authors of the ECLIPSE study stressed that division of the COPD population into more homogenous subgroups with similar prognoses provides the potential to test interventions. That study further underscored that monitoring of biomarker arrays including SP-D is useful as an indicator of disease prognosis, or for prediction and monitoring of clinical responses to an intervention; however, this strategy is yet to be validated in randomized clinical trials.
In general, there appears to be an untapped potential for monitoring circulating LMW SP-D or SP-D neoepitopes, with the aim of separating disease cases from controls, as well as for use in disease prognosis. Specific proteases are known to degrade SP-D, and this aspect has not yet been pursued sufficiently to identify disease-induced SP-D neoepitopes. More elaborate immunoassay-based methods, which have been previously used to detect circulating extracellular matrix epitopes, are warranted for studies of SP-D breakdown product variation in respiratory, as well as non-respiratory, diseases and experimental data support the presence of such circulating breakdown products. This approach has the potential to provide measures of disease and organ specific SP-D breakdown products.
It remains to be determined whether those subjects presenting with the highest circulating SP-D levels are prone to the development of clinical disease due to the presence of preclinical lung injury or CVD and if monitoring of SP-D may facilitate early intervention.
Circulating SP-D is confounded by adiposity and diabetes, and experimental and genetic links have been established to these disorders; however, these associations have not been explained mechanistically. The correlation between circulating SP-D and adiposity is inverse and may, therefore, counteract phenotypic associations with both respiratory disease and CVD.
Perspectives: Recombinant SP-D Treatment
Alveolar levels of endogenous SP-D may be vastly decreased in respiratory disease, particularly RDS, but also in severe asthma and COPD, and preclinical tests of treatment by local SP-D enrichment have been undertaken in animal models. Alveolar SP-D may be further modified, cleaved, or tightly bound to other components.
RDS and BPD
Pulmonary instillation of SP-D is beneficial, for example, in preterm ventilated lambs representing a clinically relevant model for preterm surfactant needs, and also in allergic asthma models and smoke-induced pulmonary injury. While currently used surfactant therapeutic formulations lacking SP-D have greatly reduced mortality from RDS, a significant proportion of patients develop oxygen dependency and ventilatory requirements extending beyond the first week of life, especially among survivors of very low-birth weight and extreme prematurity. Using the preterm lamb model, treatment effects were more pronounced in animals treated with surfactant (Survanta®) enriched with SP-D versus those receiving surfactant alone. This suggests that there is a therapeutic potential for the use of SP-D as a supplement to current surfactant formulations for the treatment of neonatal RDS or development of BPD, which merits clinical investigation.
Allergic Asthma
A multitude of in vivo data support the beneficial effects of exogenous SP-D treatment for allergic asthma and pulmonary infections. Treatment with exogenous SP-D may have dual effects in asthma, targeting both inflammation and fibrotic development, and induced anti-inflammatory effects comparable with those obtained by treatment with budesonide. By contrast, Survanta® did not have a suppressive effect in a mouse model of a house dust mite-induced inflammatory asthma process. In addition, exogenous SP-D could reduce fibrotic depositions in house dust mite allergen challenged mice, or when administered throughout the experimental period of bleomycin treatment for the induction of pulmonary fibrosis. However, it remains to be seen whether exogenous administration of SP-D can attenuate fibrosis in wild-type mice and when it is administered only during the fibrotic phase. Such experiments will be crucial to add weight to the potential of SP-D as a therapeutic intervention in allergic asthma or other respiratory diseases characterized by fibrotic deposition, including BPD, COPD, and idiopathic pulmonary fibrosis.
COPD and Atherosclerosis
The effects of exogenous SP-D in models of COPD and atherosclerosis require validation by independent laboratories and further mechanistic studies. However, observations from basic cardiovascular studies and studies of metabolism, suggest that systemic side effects from exogenous SP-D treatment include disturbance of circulating lipoprotein levels and whole-body fat distribution. Such observations imply that local administration of SP-D for the treatment of respiratory disease may be preferred over systemic induction of SP-D expression. However, the understanding that SP-D may also be harmful in atherogenesis and in infection during fetoplacental development suggests that the development of pharmaceutical means to induce tissue or cell type-specific inhibition of SP-D is warranted.
Additional Potential Types of Treatment and Applications
Additional potential treatments that have been suggested include the use of covalently bound complexes, comprising antibody crosslinked to point mutated SP-D (299, 488), or introduction of specific N-linked glycans (489), in enhanced anti-viral or anti-bacterial therapies (490); for example, genetically engineered SP-D, which is resistant to proteolysis and relevant for the treatment of P. aeruginosa (250, 260); reduction of lung inflammation and injury after allogeneic hematopoietic stem cell transplantation (491); inclusion of SP-D in nasal spray for chronic rhinosinusitis with polyps (13); alleviation of pneumonia severity and intestinal injury in Staphylococcus aureus pneumonia (441) or pulmonary aspergillosis (205). Moreover, therapeutic approaches have been suggested to increase transcription of SP-D (62, 492).
Development of Therapies
No clinical trials treating patients with recombinant SP-D have yet been undertaken, despite the body of preclinical data supporting significant treatment effects. Nevertheless, the early observation that recombinant SP-D prevented endotoxin shock in preterm lambs paved the way for the formation of a spin-out company from Cincinnati Children’s Hospital Medical Center, “Airway Therapeutics”2 in 2011, with the goal of producing full-length recombinant SP-D as a replacement protein therapy for the prevention of BPD in premature infants, and this product has received orphan designation status for BPD in the US and European Union. According to the website, weare.techohio.ohio.gov, AT-100 is currently in an advanced preclinical development stage. The GMP-production process is in development and investigational new drug-enabling preclinical experiments are being conducted.
Although airway therapeutics has focused on producing full-length SP-D and may be close to conducting trials, an alternative approach would be to produce rfhSP-D, which can be synthesized by Escherichia coli, and an rfhSP-D-based treatment has secured funding for continued development (Professor Howard Clark, University of Southampton and Honorary Consultant in Pediatrics at the Southampton University Hospitals Trust, personal communication).
In addition to the intellectual property rights for production of full-length SP-D or rfhSP-D for treatment and prophylaxis of respiratory disease (493, 494), more patents have been obtained to facilitate the commercial development of SP-D-based monitoring of respiratory disease or pharmaceutical treatments based on recombinant SP-D, or fragments thereof. A non-exhaustive list of patents and patent applications cover SP-D as marker for dyspnea (495), as a marker of steroid response in asthma and COPD (496), glycyrrhetinic acid for reduction of SP-D expression (497), SP-D modulation of eosinophil activity (498), combined administration with lysozyme and SP-D in respiratory prophylaxis (499), and recombinant chimeric SP-D-antibody-fragment proteins for enhanced neutrophilic uptake of pathogens (500).
The critical step for the future development of SP-D-based therapy will be the outcomes of well-performed clinical trials confirming the hypothesis that exogenous SP-D will improve neonatal RDS or BPD. The results of such trials have the potential to facilitate further development or testing of SP-D treatment in additional diverse settings, including fibrosis of the lung, allergic asthma, and COPD.
Author Contributions
GS conceived, drafted, and approved the final version of the manuscript and is accountable for all aspects of the work.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by the Danish Strategic Research Council (the Centre of COPD Research, http://www.cekol.dk) (09-066945) and the Danish Lung Association.
Abbreviations
AHR, airway hyperreactivity; ALI, acute lung injury; ARDS, acute respiratory distress syndrome; AT-II cells, type II alveolar cells; BAL, bronchoalveolar lavage; BMI, body mass index; BPD, bronchopulmonary dysplasia; CAD, coronary artery disease; CCR2, C-C chemokine receptor type 2; CD14, cluster of differentiation 14; CF, cystic fibrosis; CL-10, collectin-10; CL-11, collectin-11; CL-12, collectin-12; COPD, chronic obstructive pulmonary disease; CTLA4, cytotoxic T-lymphocyte-associated protein 4; CTLD, C-type lectin domain; CVD, cardiovascular disease; DC-SIGN, dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin; ECLIPSE, evaluation of COPD longitudinally to identify predictive surrogate endpoints; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; ELISA, enzyme-linked immunosorbent assay; FcγRII/εRI, Fc receptor γII/εI; FEV1, forced expiratory volume in 1 s; GLUCOLD, Groningen Leiden universities chronic obstructive pulmonary disease; GM-CSF, granulocyte macrophage colony-stimulating factor; GOLD, Global initiative for chronic Obstructive Lung Disease; GPR116, G protein-coupled receptor 116; GWA, genome-wide association; HDL, high-density lipoprotein; HMW, high-molecular weight; ICS, inhaled corticosteroids; IFN-γ, interferon-γ; IgA/E/G/M, immunoglobulin A/E/G/M; IHC, immunohistochemistry; IL-, interleukin-; iNOS, inducible nitric oxide synthase; LAIR-1, leukocyte-associated immunoglobulin-like receptor 1; LDL, low-density lipoprotein; LMW, low-molecular weight; LPS, lipopolysaccharide; ManNAc, N-acetylmannosamine; MASP, MBL-associated serine protease; MBL, mannan-binding lectin; MD-2, myeloid differentiation factor 2; Met11, methionine 11; Met11Thr, methionine11threonine; MMP-, matrix metalloproteinase-; NO, nitric oxide; OSCAR, osteoclast-associated receptor; oxLDL, oxidized LDL; PRR, pathogen-recognition receptor; RDS, respiratory distress syndrome; rfhSP-D, 60 kDa recombinant trimeric fragment of SP-D lacking the N-terminal but retaining a part of the collagen region; ROS, reactive oxygen species; RT-PCR, reverse transcription polymerase chain reaction; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SFTPD/Sftpd, SP-D gene; SIRP-α/β, signal-regulatory protein-α/β; SNO-SP-D, S-nitrosothiol SP-D; SNP, single-nucleotide polymorphism; SP-A/B/C/D, surfactant protein A/B/C/D; TGF-β1, transforming growth factor-β1; Th1/2/17, T helper 1/2/17 cell; Thr11, threonine 11; TLR (2/4), toll-like receptor (2/4); TNF-α, tumor necrosis factor-α; UPEC, uropathogenic Escherichia coli; UPIa, uroplakin Ia; WB, western blotting.
Footnotes
References
1. Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol (2003) 21:547–78. doi:10.1146/annurev.immunol.21.120601.140954
2. Ohtani K, Suzuki Y, Eda S, Kawai T, Kase T, Yamazaki H, et al. Molecular cloning of a novel human collectin from liver (CL-L1). J Biol Chem (1999) 274:13681–9. doi:10.1074/jbc.274.19.13681
3. Nakamura K, Funakoshi H, Miyamoto K, Tokunaga F, Nakamura T. Molecular cloning and functional characterization of a human scavenger receptor with C-type lectin (SRCL), a novel member of a scavenger receptor family. Biochem Biophys Res Commun (2001) 280:1028–35. doi:10.1006/bbrc.2000.4210
4. Keshi H, Sakamoto T, Kawai T, Ohtani K, Katoh T, Jang SJ, et al. Identification and characterization of a novel human collectin CL-K1. Microbiol Immunol (2006) 50:1001–13. doi:10.1111/j.1348-0421.2006.tb03868.x
5. Henriksen ML, Brandt J, Andrieu JP, Nielsen C, Jensen PH, Holmskov U, et al. Heteromeric complexes of native collectin kidney 1 and collectin liver 1 are found in the circulation with MASPs and activate the complement system. J Immunol (2013) 191:6117–27. doi:10.4049/jimmunol.1302121
6. Ohtani K, Suzuki Y, Eda S, Kawai T, Kase T, Keshi H, et al. The membrane-type collectin CL-P1 is a scavenger receptor on vascular endothelial cells. J Biol Chem (2001) 276:44222–8. doi:10.1074/jbc.M103942200
7. Ma YJ, Hein E, Munthe-Fog L, Skjoedt MO, Bayarri-Olmos R, Romani L, et al. Soluble collectin-12 (CL-12) is a pattern recognition molecule initiating complement activation via the alternative pathway. J Immunol (2015) 195:3365–73. doi:10.4049/jimmunol.1500493
8. Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol (2005) 5:58–68. doi:10.1038/nri1528
9. Madsen J, Kliem A, Tornoe I, Skjodt K, Koch C, Holmskov U. Localization of lung surfactant protein D on mucosal surfaces in human tissues. J Immunol (2000) 164:5866–70. doi:10.4049/jimmunol.164.11.5866
10. Stahlman MT, Gray ME, Hull WM, Whitsett JA. Immunolocalization of surfactant protein-D (SP-D) in human fetal, newborn, and adult tissues. J Histochem Cytochem (2002) 50:651–60. doi:10.1177/002215540205000506
11. Kankavi O. Immunodetection of surfactant proteins in human organ of corti, eustachian tube and kidney. Acta Biochim Pol (2003) 50:1057–64.
12. Woodworth BA, Lathers D, Neal JG, Skinner M, Richardson M, Young MR, et al. Immunolocalization of surfactant protein A and D in sinonasal mucosa. Am J Rhinol (2006) 20:461–5. doi:10.2500/ajr.2006.20.2892
13. Schicht M, Knipping S, Hirt R, Beileke S, Sel S, Paulsen F, et al. Detection of surfactant proteins A, B, C, and D in human nasal mucosa and their regulation in chronic rhinosinusitis with polyps. Am J Rhinol Allergy (2013) 27:24–9. doi:10.2500/ajra.2013.27.3838
14. Woodworth BA, Neal JG, Newton D, Joseph K, Kaplan AP, Baatz JE, et al. Surfactant protein A and D in human sinus mucosa: a preliminary report. ORL J Otorhinolaryngol Relat Spec (2007) 69:57–60. doi:10.1159/000096718
15. Honda Y, Kuroki Y, Matsuura E, Nagae H, Takahashi H, Akino T, et al. Pulmonary surfactant protein D in sera and bronchoalveolar lavage fluids. Am J Respir Crit Care Med (1995) 152:1860–6. doi:10.1164/ajrccm.152.6.8520747
16. Sorensen GL, Husby S, Holmskov U. Surfactant protein A and surfactant protein D variation in pulmonary disease. Immunobiology (2007) 212:381–416. doi:10.1016/j.imbio.2007.01.003
17. Crouch E, Persson A, Chang D. Accumulation of surfactant protein D in human pulmonary alveolar proteinosis. Am J Pathol (1993) 142:241–8.
18. Kotecha S, Doull I, Davies P, McKenzie Z, Madsen J, Clark HW, et al. Functional heterogeneity of pulmonary surfactant protein-D in cystic fibrosis. Biochim Biophys Acta (2013) 1832:2391–400. doi:10.1016/j.bbadis.2013.10.002
19. Mackay RA, Grainge CL, Lau LC, Barber C, Clark HW, Howarth PH. Airway surfactant protein D (SP-D) deficiency in adults with severe asthma. Chest (2016) 149(5):1165–72. doi:10.1016/j.chest.2015.11.012
20. Hohwy T, Otkjaer K, Madsen J, Soerensen G, Nielsen O, Vestergaard C, et al. Surfactant protein D in atopic dermatitis and psoriasis. Exp Dermatol (2006) 15:168–74. doi:10.1111/j.1600-0625.2006.00406.x
21. Mo YK, Kankavi O, Masci PP, Mellick GD, Whitehouse MW, Boyle GM, et al. Surfactant protein expression in human skin: evidence and implications. J Invest Dermatol (2007) 127:381–6. doi:10.1038/sj.jid.5700561
22. Brauer L, Moschter S, Beileke S, Jager K, Garreis F, Paulsen FP. Human parotid and submandibular glands express and secrete surfactant proteins A, B, C and D. Histochem Cell Biol (2009) 132:331–8. doi:10.1007/s00418-009-0609-x
23. Schicht M, Stengl C, Sel S, Heinemann F, Gotz W, Petschelt A, et al. The distribution of human surfactant proteins within the oral cavity and their role during infectious diseases of the gingiva. Ann Anat (2015) 199:92–7. doi:10.1016/j.aanat.2014.05.040
24. Saka R, Yanagihara I, Sasaki T, Nose S, Takeuchi M, Nakayama M, et al. Immunolocalization of surfactant protein D in the liver from infants with cholestatic liver disease. J Pediatr Surg (2015) 50:297–300. doi:10.1016/j.jpedsurg.2014.11.020
25. Hu F, Liang W, Ren Z, Wang G, Ding G. Surfactant protein D inhibits lipopolysaccharide-induced monocyte chemoattractant protein-1 expression in human renal tubular epithelial cells: implication for tubulointerstitial fibrosis. Clin Exp Immunol (2012) 167:514–22. doi:10.1111/j.1365-2249.2011.04521.x
26. Kurimura Y, Nishitani C, Ariki S, Saito A, Hasegawa Y, Takahashi M, et al. Surfactant protein D inhibits adherence of uropathogenic Escherichia coli to the bladder epithelial cells and the bacterium-induced cytotoxicity: a possible function in urinary tract. J Biol Chem (2012) 287:39578–88. doi:10.1074/jbc.M112.380287
27. Oberley RE, Goss KL, Ault KA, Crouch EC, Snyder JM. Surfactant protein D is present in the human female reproductive tract and inhibits Chlamydia trachomatis infection. Mol Hum Reprod (2004) 10:861–70. doi:10.1093/molehr/gah117
28. Leth-Larsen R, Floridon C, Nielsen O, Holmskov U. Surfactant protein D in the female genital tract. Mol Hum Reprod (2004) 10:149–54. doi:10.1093/molehr/gah022
29. Yadav AK, Chaudhari H, Warke H, Shah PK, Dodagatta-Marri E, Kishore U, et al. Differential expression of collectins in human placenta and role in inflammation during spontaneous labor. PLoS One (2014) 9:e108815. doi:10.1371/journal.pone.0108815
30. Miyamura K, Malhotra R, Hoppe HJ, Reid KB, Phizackerley PJ, Macpherson P, et al. Surfactant proteins A (SP-A) and D (SP-D): levels in human amniotic fluid and localization in the fetal membranes. Biochim Biophys Acta (1994) 1210:303–7. doi:10.1016/0005-2760(94)90233-X
31. Madhukaran SP, Kishore U, Jamil K, Choolani M, Lu J. Decidual expression and localization of human surfactant protein SP-A and SP-D, and complement protein C1q. Mol Immunol (2015) 66:197–207. doi:10.1016/j.molimm.2015.03.001
32. Sati L, Seval-Celik Y, Demir R. Lung surfactant proteins in the early human placenta. Histochem Cell Biol (2010) 133:85–93. doi:10.1007/s00418-009-0642-9
33. Lu J, Willis AC, Reid KB. Purification, characterization and cDNA cloning of human lung surfactant protein D. Biochem J (1992) 284:795–802. doi:10.1042/bj2840795
34. Mason RJ, Nielsen LD, Kuroki Y, Matsuura E, Freed JH, Shannon JM. A 50-kDa variant form of human surfactant protein D. Eur Respir J (1998) 12:1147–55. doi:10.1183/09031936.98.12051147
35. Inoue T, Matsuura E, Nagata A, Ogasawara Y, Hattori A, Kuroki Y, et al. Enzyme-linked immunosorbent assay for human pulmonary surfactant protein D. J Immunol Methods (1994) 173:157–64. doi:10.1016/0022-1759(94)90295-X
36. Sorensen GL, Hoegh SV, Leth-Larsen R, Thomsen TH, Floridon C, Smith K, et al. Multimeric and trimeric subunit SP-D are interconvertible structures with distinct ligand interaction. Mol Immunol (2009) 46:3060–9. doi:10.1016/j.molimm.2009.06.005
37. Leth-Larsen R, Garred P, Jensenius H, Meschi J, Hartshorn K, Madsen J, et al. A common polymorphism in the SFTPD gene influences assembly, function, and concentration of surfactant protein D. J Immunol (2005) 174:1532–8. doi:10.4049/jimmunol.174.3.1532
38. Rokade S, Madan T. Testicular expression of SP-A, SP-D and MBL-A is positively regulated by testosterone and modulated by lipopolysaccharide. Immunobiology (2016) 221(9):975–85. doi:10.1016/j.imbio.2016.05.005
39. Beileke S, Claassen H, Wagner W, Matthies C, Ruf C, Hartmann A, et al. Expression and localization of lung surfactant proteins in human testis. PLoS One (2015) 10:e0143058. doi:10.1371/journal.pone.0143058
40. Oberley RE, Goss KL, Dahmoush L, Ault KA, Crouch EC, Snyder JM. A role for surfactant protein D in innate immunity of the human prostate. Prostate (2005) 65:241–51. doi:10.1002/pros.20292
41. Schob S, Schicht M, Sel S, Stiller D, Kekule AS, Paulsen F, et al. The detection of surfactant proteins A, B, C and D in the human brain and their regulation in cerebral infarction, autoimmune conditions and infections of the CNS. PLoS One (2013) 8:e74412. doi:10.1371/journal.pone.0074412
42. Schob S, Lobsien D, Friedrich B, Bernhard MK, Gebauer C, Dieckow J, et al. The cerebral surfactant system and its alteration in hydrocephalic conditions. PLoS One (2016) 11:e0160680. doi:10.1371/journal.pone.0160680
43. Brauer L, Kindler C, Jager K, Sel S, Nolle B, Pleyer U, et al. Detection of surfactant proteins A and D in human tear fluid and the human lacrimal system. Invest Ophthalmol Vis Sci (2007) 48:3945–53. doi:10.1167/iovs.07-0201
44. Wu X, Zhao G, Lin J, Jiang N, Li C, Hu L, et al. The production mechanism and immunosuppression effect of pulmonary surfactant protein D via toll like receptor 4 signaling pathway in human corneal epithelial cells during Aspergillus fumigatus infection. Int Immunopharmacol (2015) 29:433–9. doi:10.1016/j.intimp.2015.10.018
45. Ni M, Evans DJ, Hawgood S, Anders EM, Sack RA, Fleiszig SM. Surfactant protein D is present in human tear fluid and the cornea and inhibits epithelial cell invasion by Pseudomonas aeruginosa. Infect Immun (2005) 73:2147–56. doi:10.1128/IAI.73.4.2147-2156.2005
46. Che CY, Jia WY, Xu Q, Li N, Hu LT, Jiang N, et al. The roles of surfactant protein D during Aspergillus fumigatus infection in human corneal epithelial cells. Int J Ophthalmol (2012) 5:13–7. doi:10.3980/j.issn.2222-3959.2012.01.03
47. Snyder GD, Oberley-Deegan RE, Goss KL, Romig-Martin SA, Stoll LL, Snyder JM, et al. Surfactant protein D is expressed and modulates inflammatory responses in human coronary artery smooth muscle cells. Am J Physiol Heart Circ Physiol (2008) 294:H2053–9. doi:10.1152/ajpheart.91529.2007
48. Sorensen GL, Madsen J, Kejling K, Tornoe I, Nielsen O, Townsend P, et al. Surfactant protein D is proatherogenic in mice. Am J Physiol Heart Circ Physiol (2006) 290:H2286–94. doi:10.1152/ajpheart.01105.2005
49. Lambertsen KL, Ostergaard K, Clausen BH, Hansen S, Stenvang J, Thorsen SB, et al. No effect of ablation of surfactant protein-D on acute cerebral infarction in mice. J Neuroinflammation (2014) 11:123. doi:10.1186/1742-2094-11-123
50. Christensen AF, Sorensen GL, Junker K, Revald PH, Varnum C, Sorensen FB, et al. Localization of surfactant protein-D in the rheumatoid synovial membrane. APMIS (2017) 126(1):9–13. doi:10.1111/apm.12785
51. Ortega FJ, Pueyo N, Moreno-Navarrete JM, Sabater M, Rodriguez-Hermosa JI, Ricart W, et al. The lung innate immune gene surfactant protein-D is expressed in adipose tissue and linked to obesity status. Int J Obes (Lond) (2013) 37:1532–8. doi:10.1038/ijo.2013.23
52. Bourbon JR, Chailley-Heu B. Surfactant proteins in the digestive tract, mesentery, and other organs: evolutionary significance. Comp Biochem Physiol A Mol Integr Physiol (2001) 129:151–61. doi:10.1016/S1095-6433(01)00312-9
53. Voorhout WF, Veenendaal T, Kuroki Y, Ogasawara Y, van Golde LM, Geuze HJ. Immunocytochemical localization of surfactant protein D (SP-D) in type II cells, Clara cells, and alveolar macrophages of rat lung. J Histochem Cytochem (1992) 40:1589–97. doi:10.1177/40.10.1527377
54. Dulkerian SJ, Gonzales LW, Ning Y, Ballard PL. Regulation of surfactant protein D in human fetal lung. Am J Respir Cell Mol Biol (1996) 15:781–6. doi:10.1165/ajrcmb.15.6.8969273
55. Rust K, Bingle L, Mariencheck W, Persson A, Crouch EC. Characterization of the human surfactant protein D promoter: transcriptional regulation of SP-D gene expression by glucocorticoids. Am J Respir Cell Mol Biol (1996) 14:121–30. doi:10.1165/ajrcmb.14.2.8630261
56. He Y, Crouch EC, Rust K, Spaite E, Brody SL. Proximal promoter of the surfactant protein D gene: regulatory roles of AP-1, forkhead box, and GT box binding proteins. J Biol Chem (2000) 275:31051–60. doi:10.1074/jbc.M003499200
57. Besnard V, Wert SE, Kaestner KH, Whitsett JA. Stage-specific regulation of respiratory epithelial cell differentiation by Foxa1. Am J Physiol Lung Cell Mol Physiol (2005) 289:L750–9. doi:10.1152/ajplung.00151.2005
58. He Y, Crouch E. Surfactant protein D gene regulation. Interactions among the conserved CCAAT/enhancer-binding protein elements. J Biol Chem (2002) 277:19530–7. doi:10.1074/jbc.M201126200
59. Charles A, Tang X, Crouch E, Brody JS, Xiao ZX. Retinoblastoma protein complexes with C/EBP proteins and activates C/EBP-mediated transcription. J Cell Biochem (2001) 83:414–25. doi:10.1002/jcb.1239
60. Dave V, Childs T, Whitsett JA. Nuclear factor of activated T cells regulates transcription of the surfactant protein D gene (Sftpd) via direct interaction with thyroid transcription factor-1 in lung epithelial cells. J Biol Chem (2004) 279:34578–88. doi:10.1074/jbc.M404296200
61. Ni M, Tam C, Verma A, Ramphal R, Hawgood S, Evans DJ, et al. Expression of surfactant protein D in human corneal epithelial cells is upregulated by Pseudomonas aeruginosa. FEMS Immunol Med Microbiol (2008) 54:177–84. doi:10.1111/j.1574-695X.2008.00461.x
62. Kamio K, Usuki J, Azuma A, Matsuda K, Ishii T, Inomata M, et al. Nintedanib modulates surfactant protein-D expression in A549 human lung epithelial cells via the c-Jun N-terminal kinase-activator protein-1 pathway. Pulm Pharmacol Ther (2015) 32:29–36. doi:10.1016/j.pupt.2015.03.001
63. Lee MY, Sorensen GL, Holmskov U, Vanhoutte PM. The presence and activity of SP-D in porcine coronary endothelial cells depend on Akt/PI(3)K, Erk and nitric oxide and decrease after multiple passaging. Mol Immunol (2008) 46(6):1050–7. doi:10.1016/j.molimm.2008.09.027
64. Lin Z, Floros J. Heterogeneous allele expression of pulmonary SP-D gene in rat large intestine and other tissues. Physiol Genomics (2002) 11:235–43. doi:10.1152/physiolgenomics.00061.2002
65. Dahl M, Juvonen PO, Holmskov U, Husby S. Surfactant protein D in newborn infants: factors influencing surfactant protein D levels in umbilical cord blood and capillary blood. Pediatr Res (2005) 58:908–12. doi:10.1203/01.PDR.0000181379.72900.EC
66. Deterding RR, Shimizu H, Fisher JH, Shannon JM. Regulation of surfactant protein D expression by glucocorticoids in vitro and in vivo. Am J Respir Cell Mol Biol (1994) 10:30–7. doi:10.1165/ajrcmb.10.1.8292379
67. Crouch E, Rust K, Marienchek W, Parghi D, Chang D, Persson A. Developmental expression of pulmonary surfactant protein D (SP-D). Am J Respir Cell Mol Biol (1991) 5:13–8. doi:10.1165/ajrcmb/5.1.13
68. Ogasawara Y, Kuroki Y, Tsuzuki A, Ueda S, Misaki H, Akino T. Pre- and postnatal stimulation of pulmonary surfactant protein D by in vivo dexamethasone treatment of rats. Life Sci (1992) 50:1761–7. doi:10.1016/0024-3205(92)90059-X
69. Wong CJ, Akiyama J, Allen L, Hawgood S. Localization and developmental expression of surfactant proteins D and A in the respiratory tract of the mouse. Pediatr Res (1996) 39:930–7. doi:10.1203/00006450-199606000-00002
70. Wang JY, Yeh TF, Lin YC, Miyamura K, Holmskov U, Reid KB. Measurement of pulmonary status and surfactant protein levels during dexamethasone treatment of neonatal respiratory distress syndrome. Thorax (1996) 51:907–13. doi:10.1136/thx.51.9.907
71. Mariencheck W, Crouch E. Modulation of surfactant protein D expression by glucocorticoids in fetal rat lung. Am J Respir Cell Mol Biol (1994) 10:419–29. doi:10.1165/ajrcmb.10.4.8136157
72. Schmiedl A, Grutzner D, Hoffmann T, von Horsten S, Stephan M. DPP4 inhibitors increase differentially the expression of surfactant proteins in Fischer 344 rats. Acta Physiol (Oxf) (2014) 212:248–61. doi:10.1111/apha.12350
73. Aderibigbe AO, Thomas RF, Mercer RR, Auten RL Jr. Brief exposure to 95% oxygen alters surfactant protein D and mRNA in adult rat alveolar and bronchiolar epithelium. Am J Respir Cell Mol Biol (1999) 20:219–27. doi:10.1165/ajrcmb.20.2.3348
74. Jain-Vora S, LeVine AM, Chroneos Z, Ross GF, Hull WM, Whitsett JA. Interleukin-4 enhances pulmonary clearance of Pseudomonas aeruginosa. Infect Immun (1998) 66:4229–36.
75. Ikegami M, Whitsett JA, Chroneos ZC, Ross GF, Reed JA, Bachurski CJ, et al. IL-4 increases surfactant and regulates metabolism in vivo. Am J Physiol Lung Cell Mol Physiol (2000) 278:L75–80. doi:10.1152/ajplung.2000.278.1.L75
76. Homer RJ, Zheng T, Chupp G, He S, Zhu Z, Chen Q, et al. Pulmonary type II cell hypertrophy and pulmonary lipoproteinosis are features of chronic IL-13 exposure. Am J Physiol Lung Cell Mol Physiol (2002) 283:L52–9. doi:10.1152/ajplung.00438.2001
77. Fujita M, Shannon JM, Ouchi H, Voelker DR, Nakanishi Y, Mason RJ. Serum surfactant protein D is increased in acute and chronic inflammation in mice. Cytokine (2005) 31:25–33. doi:10.1016/j.cyto.2005.02.006
78. Rucka Z, Vanhara P, Koutna I, Tesarova L, Potesilova M, Stejskal S, et al. Differential effects of insulin and dexamethasone on pulmonary surfactant-associated genes and proteins in A549 and H441 cells and lung tissue. Int J Mol Med (2013) 32:211–8. doi:10.3892/ijmm.2013.1363
79. Oberley RE, Goss KL, Hoffmann DS, Ault KA, Neff TL, Ramsey KH, et al. Regulation of surfactant protein D in the mouse female reproductive tract in vivo. Mol Hum Reprod (2007) 13:863–8. doi:10.1093/molehr/gam074
80. Kay S, Metkari SM, Madan T. Ovarian hormones regulate SP-D expression in the mouse uterus during estrous cycle and early pregnancy. Am J Reprod Immunol (2015) 74:77–88. doi:10.1111/aji.12369
81. Oberley RE, Goss KL, Quintar AA, Maldonado CA, Snyder JM. Regulation of surfactant protein D in the rodent prostate. Reprod Biol Endocrinol (2007) 5:42. doi:10.1186/1477-7827-5-42
82. Gravholt CH, Leth-Larsen R, Lauridsen AL, Thiel S, Hansen TK, Holmskov U, et al. The effects of GH and hormone replacement therapy on serum concentrations of mannan-binding lectin, surfactant protein D and vitamin D binding protein in Turner syndrome. Eur J Endocrinol (2004) 150:355–62. doi:10.1530/eje.0.1500355
83. Pikaar JC, Voorhout WF, van Golde LM, Verhoef J, Van Strijp JA, van Iwaarden JF. Opsonic activities of surfactant proteins A and D in phagocytosis of Gram-negative bacteria by alveolar macrophages. J Infect Dis (1995) 172:481–9. doi:10.1093/infdis/172.2.481
84. Madan T, Eggleton P, Kishore U, Strong P, Aggrawal SS, Sarma PU, et al. Binding of pulmonary surfactant proteins A and D to Aspergillus fumigatus conidia enhances phagocytosis and killing by human neutrophils and alveolar macrophages. Infect Immun (1997) 65:3171–9.
85. Hartshorn KL, Crouch E, White MR, Colamussi ML, Kakkanatt A, Tauber B, et al. Pulmonary surfactant proteins A and D enhance neutrophil uptake of bacteria. Am J Physiol (1998) 274:L958–69.
86. Restrepo CI, Dong Q, Savov J, Mariencheck WI, Wright JR. Surfactant protein D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. Am J Respir Cell Mol Biol (1999) 21:576–85. doi:10.1165/ajrcmb.21.5.3334
87. Ofek I, Mesika A, Kalina M, Keisari Y, Podschun R, Sahly H, et al. Surfactant protein D enhances phagocytosis and killing of unencapsulated phase variants of Klebsiella pneumoniae. Infect Immun (2001) 69:24–33. doi:10.1128/IAI.69.1.24-33.2001
88. LeVine AM, Elliott J, Whitsett JA, Srikiatkhachorn A, Crouch E, DeSilva N, et al. Surfactant protein-d enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am J Respir Cell Mol Biol (2004) 31:193–9. doi:10.1165/rcmb.2003-0107OC
89. Tecle T, White MR, Sorensen G, Gantz D, Kacak N, Holmskov U, et al. Critical role for cross-linking of trimeric lectin domains of surfactant protein D in antiviral activity against influenza A virus. Biochem J (2008) 412:323–9. doi:10.1042/BJ20071663
90. Thawer S, Auret J, Schnoeller C, Chetty A, Smith K, Darby M, et al. Surfactant protein-D is essential for immunity to helminth infection. PLoS Pathog (2016) 12:e1005461. doi:10.1371/journal.ppat.1005461
91. Nayak A, Dodagatta-Marri E, Tsolaki AG, Kishore U. An insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Front Immunol (2012) 3:131. doi:10.3389/fimmu.2012.00131
92. Erpenbeck VJ, Malherbe DC, Sommer S, Schmiedl A, Steinhilber W, Ghio AJ, et al. Surfactant protein D increases phagocytosis and aggregation of pollen-allergen starch granules. Am J Physiol Lung Cell Mol Physiol (2005) 288:L692–8. doi:10.1152/ajplung.00362.2004
93. Winkler C, Huper K, Wedekind AC, Rochlitzer S, Hartwig C, Muller M, et al. Surfactant protein D modulates pulmonary clearance of pollen starch granules. Exp Lung Res (2010) 36:522–30. doi:10.3109/01902141003790148
94. Palaniyar N, Clark H, Nadesalingam J, Hawgood S, Reid KB. Surfactant protein D binds genomic DNA and apoptotic cells, and enhances their clearance, in vivo. Ann N Y Acad Sci (2003) 1010:471–5. doi:10.1196/annals.1299.085
95. Kendall M, Ding P, Mackay RM, Deb R, McKenzie Z, Kendall K, et al. Surfactant protein D (SP-D) alters cellular uptake of particles and nanoparticles. Nanotoxicology (2013) 7:963–73. doi:10.3109/17435390.2012.689880
96. Sarashina-Kida H, Negishi H, Nishio J, Suda W, Nakajima Y, Yasui-Kato M, et al. Gallbladder-derived surfactant protein D regulates gut commensal bacteria for maintaining intestinal homeostasis. Proc Natl Acad Sci U S A (2017) 114:10178–83. doi:10.1073/pnas.1712837114
97. Barfod KK, Roggenbuck M, Al-Shuweli S, Fakih D, Sorensen SJ, Sorensen GL. Alterations of the murine gut microbiome in allergic airway disease are independent of surfactant protein D. Heliyon (2017) 3:e00262. doi:10.1016/j.heliyon.2017.e00262
98. Crouch EC, Smith K, McDonald B, Briner D, Linders B, McDonald J, et al. Species differences in the carbohydrate binding preferences of surfactant protein D. Am J Respir Cell Mol Biol (2006) 35:84–94. doi:10.1165/rcmb.2005-0462OC
99. Jakel A, Qaseem AS, Kishore U, Sim RB. Ligands and receptors of lung surfactant proteins SP-A and SP-D. Front Biosci (Landmark Ed) (2013) 18:1129–40. doi:10.2741/4168
100. Hartshorn K, Chang D, Rust K, White M, Heuser J, Crouch E. Interactions of recombinant human pulmonary surfactant protein D and SP-D multimers with influenza A. Am J Physiol (1996) 271:L753–62.
101. Jounblat R, Kadioglu A, Iannelli F, Pozzi G, Eggleton P, Andrew PW. Binding and agglutination of Streptococcus pneumoniae by human surfactant protein D (SP-D) vary between strains, but SP-D fails to enhance killing by neutrophils. Infect Immun (2004) 72:709–16. doi:10.1128/IAI.72.2.709-716.2004
102. Hartshorn KL, White MR, Crouch EC. Contributions of the N- and C-terminal domains of surfactant protein d to the binding, aggregation, and phagocytic uptake of bacteria. Infect Immun (2002) 70:6129–39. doi:10.1128/IAI.70.11.6129-6139.2002
103. Douda DN, Jackson R, Grasemann H, Palaniyar N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J Immunol (2011) 187:1856–65. doi:10.4049/jimmunol.1004201
104. Wu H, Kuzmenko A, Wan S, Schaffer L, Weiss A, Fisher JH, et al. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest (2003) 111:1589–602. doi:10.1172/JCI16889
105. Kuzmenko AI, Wu H, McCormack FX. Pulmonary collectins selectively permeabilize model bacterial membranes containing rough lipopolysaccharide. Biochemistry (2006) 45:2679–85. doi:10.1021/bi0522652
106. McCormack FX, Gibbons R, Ward SR, Kuzmenko A, Wu H, Deepe GS Jr. Macrophage-independent fungicidal action of the pulmonary collectins. J Biol Chem (2003) 278:36250–6. doi:10.1074/jbc.M303086200
107. Meschi J, Crouch EC, Skolnik P, Yahya K, Holmskov U, Leth-Larsen R, et al. Surfactant protein D binds to human immunodeficiency virus (HIV) envelope protein gp120 and inhibits HIV replication. J Gen Virol (2005) 86:3097–107. doi:10.1099/vir.0.80764-0
108. Hartshorn KL, White MR, Tecle T, Tornoe I, Sorensen GL, Crouch EC, et al. Reduced influenza viral neutralizing activity of natural human trimers of surfactant protein D. Respir Res (2007) 8:9. doi:10.1186/1465-9921-8-9
109. Goh BC, Rynkiewicz MJ, Cafarella TR, White MR, Hartshorn KL, Allen K, et al. Molecular mechanisms of inhibition of influenza by surfactant protein D revealed by large-scale molecular dynamics simulation. Biochemistry (2013) 52:8527–38. doi:10.1021/bi4010683
110. Fakih D, Pilecki B, Schlosser A, Jepsen CS, Thomsen LK, Ormhoj M, et al. Protective effects of surfactant protein D treatment in 1,3-beta-glucan-modulated allergic inflammation. Am J Physiol Lung Cell Mol Physiol (2015) 309:L1333–43. doi:10.1152/ajplung.00090.2015
111. Geunes-Boyer S, Oliver TN, Janbon G, Lodge JK, Heitman J, Perfect JR, et al. Surfactant protein D increases phagocytosis of hypocapsular Cryptococcus neoformans by murine macrophages and enhances fungal survival. Infect Immun (2009) 77:2783–94. doi:10.1128/IAI.00088-09
112. Holmer SM, Evans KS, Asfaw YG, Saini D, Schell WA, Ledford JG, et al. Impact of surfactant protein D, interleukin-5, and eosinophilia on cryptococcosis. Infect Immun (2014) 82:683–93. doi:10.1128/IAI.00855-13
113. Yong SJ, Vuk-Pavlovic Z, Standing JE, Crouch EC, Limper AH. Surfactant protein D-mediated aggregation of Pneumocystis carinii impairs phagocytosis by alveolar macrophages. Infect Immun (2003) 71:1662–71. doi:10.1128/IAI.71.4.1662-1671.2003
114. Ferguson JS, Voelker DR, McCormack FX, Schlesinger LS. Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J Immunol (1999) 163:312–21.
115. Ferguson JS, Voelker DR, Ufnar JA, Dawson AJ, Schlesinger LS. Surfactant protein D inhibition of human macrophage uptake of Mycobacterium tuberculosis is independent of bacterial agglutination. J Immunol (2002) 168:1309–14. doi:10.4049/jimmunol.168.3.1309
116. Lemos MP, McKinney J, Rhee KY. Dispensability of surfactant proteins A and D in immune control of Mycobacterium tuberculosis infection following aerosol challenge of mice. Infect Immun (2011) 79:1077–85. doi:10.1128/IAI.00286-10
117. Liu CF, Rivere M, Huang HJ, Puzo G, Wang JY. Surfactant protein D inhibits mite-induced alveolar macrophage and dendritic cell activations through TLR signalling and DC-SIGN expression. Clin Exp Allergy (2010) 40:111–22. doi:10.1111/j.1365-2222.2009.03367.x
118. Yamazoe M, Nishitani C, Takahashi M, Katoh T, Ariki S, Shimizu T, et al. Pulmonary surfactant protein D inhibits lipopolysaccharide (LPS)-induced inflammatory cell responses by altering LPS binding to its receptors. J Biol Chem (2008) 283:35878–88. doi:10.1074/jbc.M807268200
119. Janssen WJ, McPhillips KA, Dickinson MG, Linderman DJ, Morimoto K, Xiao YQ, et al. Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am J Respir Crit Care Med (2008) 178:158–67. doi:10.1164/rccm.200711-1661OC
120. Yamaguchi R, Sakamoto A, Yamamoto T, Ishimaru Y, Narahara S, Sugiuchi H, et al. Surfactant protein D inhibits interleukin-12p40 production by macrophages through the SIRPalpha/ROCK/ERK signaling pathway. Am J Med Sci (2017) 353:559–67. doi:10.1016/j.amjms.2017.03.013
121. Litvack ML, Djiadeu P, Renganathan SD, Sy S, Post M, Palaniyar N. Natural IgM and innate immune collectin SP-D bind to late apoptotic cells and enhance their clearance by alveolar macrophages in vivo. Mol Immunol (2010) 48:37–47. doi:10.1016/j.molimm.2010.09.014
122. Atochina-Vasserman EN, Bates SR, Zhang P, Abramova H, Zhang Z, Gonzales L, et al. Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky-Pudlak syndrome. Am J Respir Crit Care Med (2011) 184:449–58. doi:10.1164/rccm.201011-1882OC
123. Takeda K, Miyahara N, Rha YH, Taube C, Yang ES, Joetham A, et al. Surfactant protein D regulates airway function and allergic inflammation through modulation of macrophage function. Am J Respir Crit Care Med (2003) 168:783–9. doi:10.1164/rccm.200304-548OC
124. Douda DN, Farmakovski N, Dell S, Grasemann H, Palaniyar N. SP-D counteracts GM-CSF-mediated increase of granuloma formation by alveolar macrophages in lysinuric protein intolerance. Orphanet J Rare Dis (2009) 4:29. doi:10.1186/1750-1172-4-29
125. Yoshida M, Korfhagen TR, Whitsett JA. Surfactant protein D regulates NF-kappaB and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol (2001) 166:7514–9. doi:10.4049/jimmunol.166.12.7514
126. Tino MJ, Wright JR. Surfactant proteins A and D specifically stimulate directed actin-based responses in alveolar macrophages. Am J Physiol (1999) 276:L164–74.
127. Trask BC, Malone MJ, Lum EH, Welgus HG, Crouch EC, Shapiro SD. Induction of macrophage matrix metalloproteinase biosynthesis by surfactant protein D. J Biol Chem (2001) 276:37846–52. doi:10.1074/jbc.M102524200
128. Keisari Y, Wang H, Mesika A, Matatov R, Nissimov L, Crouch E, et al. Surfactant protein D-coated Klebsiella pneumoniae stimulates cytokine production in mononuclear phagocytes. J Leukoc Biol (2001) 70:135–41.
129. Kudo K, Sano H, Takahashi H, Kuronuma K, Yokota S, Fujii N, et al. Pulmonary collectins enhance phagocytosis of Mycobacterium avium through increased activity of mannose receptor. J Immunol (2004) 172:7592–602. doi:10.4049/jimmunol.172.12.7592
130. Palaniyar N, Clark H, Nadesalingam J, Shih MJ, Hawgood S, Reid KB. Innate immune collectin surfactant protein D enhances the clearance of DNA by macrophages and minimizes anti-DNA antibody generation. J Immunol (2005) 174:7352–8. doi:10.4049/jimmunol.174.11.7352
131. Liu CF, Chen YL, Chang WT, Shieh CC, Yu CK, Reid KB, et al. Mite allergen induces nitric oxide production in alveolar macrophage cell lines via CD14/toll-like receptor 4, and is inhibited by surfactant protein D. Clin Exp Allergy (2005) 35:1615–24. doi:10.1111/j.1365-2222.2005.02387.x
132. Atochina-Vasserman EN, Abramova EV, Tomer Y, Scott P, Nazarov VA, Kruglov SV, et al. SP-D-dependent regulation of NO metabolism in lipopolysaccharide-stimulated peritoneal macrophages. Bull Exp Biol Med (2009) 147:415–20. doi:10.1007/s10517-009-0525-z
133. Barrow AD, Palarasah Y, Bugatti M, Holehouse AS, Byers DE, Holtzman MJ, et al. OSCAR is a receptor for surfactant protein D that activates TNF-alpha release from human CCR2+ inflammatory monocytes. J Immunol (2015) 194:3317–26. doi:10.4049/jimmunol.1402289
134. Ge MQ, Kokalari B, Flayer CH, Killingbeck SS, Redai IG, MacFarlane AW IV, et al. Cutting edge: role of NK cells and surfactant protein D in dendritic cell lymph node homing: effects of ozone exposure. J Immunol (2016) 196:553–7. doi:10.4049/jimmunol.1403042
135. Crouch EC, Persson A, Griffin GL, Chang D, Senior RM. Interactions of pulmonary surfactant protein D (SP-D) with human blood leukocytes. Am J Respir Cell Mol Biol (1995) 12:410–5. doi:10.1165/ajrcmb.12.4.7695920
136. Bufler P, Schmidt B, Schikor D, Bauernfeind A, Crouch EC, Griese M. Surfactant protein A and D differently regulate the immune response to nonmucoid Pseudomonas aeruginosa and its lipopolysaccharide. Am J Respir Cell Mol Biol (2003) 28:249–56. doi:10.1165/rcmb.4896
137. Pandit H, Gopal S, Sonawani A, Yadav AK, Qaseem AS, Warke H, et al. Surfactant protein D inhibits HIV-1 infection of target cells via interference with gp120-CD4 interaction and modulates pro-inflammatory cytokine production. PLoS One (2014) 9:e102395. doi:10.1371/journal.pone.0102395
138. Hartshorn KL, Crouch EC, White MR, Eggleton P, Tauber AI, Chang D, et al. Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J Clin Invest (1994) 94:311–9. doi:10.1172/JCI117323
139. Hartshorn KL, Reid KB, White MR, Jensenius JC, Morris SM, Tauber AI, et al. Neutrophil deactivation by influenza A viruses: mechanisms of protection after viral opsonization with collectins and hemagglutination-inhibiting antibodies. Blood (1996) 87:3450–61.
140. White MR, Crouch E, Vesona J, Tacken PJ, Batenburg JJ, Leth-Larsen R, et al. Respiratory innate immune proteins differentially modulate the neutrophil respiratory burst response to influenza A virus. Am J Physiol Lung Cell Mol Physiol (2005) 289:L606–16. doi:10.1152/ajplung.00130.2005
141. White M, Kingma P, Tecle T, Kacak N, Linders B, Heuser J, et al. Multimerization of surfactant protein D, but not its collagen domain, is required for antiviral and opsonic activities related to influenza virus. J Immunol (2008) 181:7936–43. doi:10.4049/jimmunol.181.11.7936
142. von Bredow C, Hartl D, Schmid K, Schabaz F, Brack E, Reinhardt D, et al. Surfactant protein D regulates chemotaxis and degranulation of human eosinophils. Clin Exp Allergy (2006) 36:1566–74. doi:10.1111/j.1365-2222.2006.02598.x
143. Mahajan L, Madan T, Kamal N, Singh VK, Sim RB, Telang SD, et al. Recombinant surfactant protein-D selectively increases apoptosis in eosinophils of allergic asthmatics and enhances uptake of apoptotic eosinophils by macrophages. Int Immunol (2008) 20:993–1007. doi:10.1093/intimm/dxn058
144. Ogawa H, Ledford JG, Mukherjee S, Aono Y, Nishioka Y, Lee JJ, et al. Surfactant protein D attenuates sub-epithelial fibrosis in allergic airways disease through TGF-beta. Respir Res (2014) 15:143. doi:10.1186/s12931-014-0143-9
145. Pandit H, Thakur G, Koippallil Gopalakrishnan AR, Dodagatta-Marri E, Patil A, Kishore U, et al. Surfactant protein D induces immune quiescence and apoptosis of mitogen-activated peripheral blood mononuclear cells. Immunobiology (2016) 221:310–22. doi:10.1016/j.imbio.2015.10.004
146. Borron PJ, Crouch EC, Lewis JF, Wright JR, Possmayer F, Fraher LJ. Recombinant rat surfactant-associated protein D inhibits human T lymphocyte proliferation and IL-2 production. J Immunol (1998) 161:4599–603.
147. Borron PJ, Mostaghel EA, Doyle C, Walsh ES, McHeyzer-Williams MG, Wright JR. Pulmonary surfactant proteins A and D directly suppress CD3+/CD4+ cell function: evidence for two shared mechanisms. J Immunol (2002) 169:5844–50. doi:10.4049/jimmunol.169.10.5844
148. Fisher JH, Larson J, Cool C, Dow SW. Lymphocyte activation in the lungs of SP-D null mice. Am J Respir Cell Mol Biol (2002) 27:24–33. doi:10.1165/ajrcmb.27.1.4563
149. Lin KW, Jen KY, Suarez CJ, Crouch EC, Perkins DL, Finn PW. Surfactant protein D-mediated decrease of allergen-induced inflammation is dependent upon CTLA4. J Immunol (2010) 184:6343–9. doi:10.4049/jimmunol.0901947
150. Madsen J, Gaiha GD, Palaniyar N, Dong T, Mitchell DA, Clark HW. Surfactant protein D modulates HIV infection of both T-cells and dendritic cells. PLoS One (2013) 8:e59047. doi:10.1371/journal.pone.0059047
151. Djiadeu P, Kotra LP, Sweezey N, Palaniyar N. Surfactant protein D delays Fas- and TRAIL-mediated extrinsic pathway of apoptosis in T cells. Apoptosis (2017) 22:730–40. doi:10.1007/s10495-017-1348-4
152. Djiadeu P, Farmakovski N, Azzouz D, Kotra LP, Sweezey N, Palaniyar N. Surfactant protein D regulates caspase-8-mediated cascade of the intrinsic pathway of apoptosis while promoting bleb formation. Mol Immunol (2017) 92:190–8. doi:10.1016/j.molimm.2017.10.016
153. Madan T, Kishore U, Shah A, Eggleton P, Strong P, Wang JY, et al. Lung surfactant proteins A and D can inhibit specific IgE binding to the allergens of Aspergillus fumigatus and block allergen-induced histamine release from human basophils. Clin Exp Immunol (1997) 110:241–9. doi:10.1111/j.1365-2249.1997.tb08323.x
154. Malherbe DC, Erpenbeck VJ, Abraham SN, Crouch EC, Hohlfeld JM, Wright JR. Surfactant protein D decreases pollen-induced IgE-dependent mast cell degranulation. Am J Physiol Lung Cell Mol Physiol (2005) 289:L856–66. doi:10.1152/ajplung.00009.2005
155. Brinker KG, Martin E, Borron P, Mostaghel E, Doyle C, Harding CV, et al. Surfactant protein D enhances bacterial antigen presentation by bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol (2001) 281:L1453–63. doi:10.1152/ajplung.2001.281.6.L1453
156. Hansen S, Lo B, Evans K, Neophytou P, Holmskov U, Wright JR. Surfactant protein D augments bacterial association but attenuates major histocompatibility complex class II presentation of bacterial antigens. Am J Respir Cell Mol Biol (2007) 36:94–102. doi:10.1165/rcmb.2006-0195OC
157. Hortobagyi L, Kierstein S, Krytska K, Zhu X, Das AM, Poulain F, et al. Surfactant protein D inhibits TNF-alpha production by macrophages and dendritic cells in mice. J Allergy Clin Immunol (2008) 122:521–8. doi:10.1016/j.jaci.2008.05.002
158. Hasegawa Y, Takahashi M, Ariki S, Asakawa D, Tajiri M, Wada Y, et al. Surfactant protein D suppresses lung cancer progression by downregulation of epidermal growth factor signaling. Oncogene (2015) 34:838–45. doi:10.1038/onc.2014.20
159. Yang Z, Jaeckisch SM, Mitchell CG. Enhanced binding of Aspergillus fumigatus spores to A549 epithelial cells and extracellular matrix proteins by a component from the spore surface and inhibition by rat lung lavage fluid. Thorax (2000) 55:579–84. doi:10.1136/thorax.55.7.579
160. Schleh C, Erpenbeck VJ, Winkler C, Lauenstein HD, Nassimi M, Braun A, et al. Allergen particle binding by human primary bronchial epithelial cells is modulated by surfactant protein D. Respir Res (2010) 11:83. doi:10.1186/1465-9921-11-83
161. Schleh C, Rothen-Rutishauser BM, Blank F, Lauenstein HD, Nassimi M, Krug N, et al. Surfactant protein D modulates allergen particle uptake and inflammatory response in a human epithelial airway model. Respir Res (2012) 13:8. doi:10.1186/1465-9921-13-8
162. McKenzie Z, Kendall M, Mackay RM, Tetley TD, Morgan C, Griffiths M, et al. Nanoparticles modulate surfactant protein A and D mediated protection against influenza A infection in vitro. Philos Trans R Soc Lond B Biol Sci (2015) 370:20140049. doi:10.1098/rstb.2014.0049
163. Aono Y, Ledford JG, Mukherjee S, Ogawa H, Nishioka Y, Sone S, et al. Surfactant protein-D regulates effector cell function and fibrotic lung remodeling in response to bleomycin injury. Am J Respir Crit Care Med (2012) 185:525–36. doi:10.1164/rccm.201103-0561OC
164. Sano H, Chiba H, Iwaki D, Sohma H, Voelker DR, Kuroki Y. Surfactant proteins A and D bind CD14 by different mechanisms. J Biol Chem (2000) 275:22442–51. doi:10.1074/jbc.M001107200
165. Ohya M, Nishitani C, Sano H, Yamada C, Mitsuzawa H, Shimizu T, et al. Human pulmonary surfactant protein D binds the extracellular domains of toll-like receptors 2 and 4 through the carbohydrate recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry (2006) 45:8657–64. doi:10.1021/bi060176z
166. Nie X, Nishitani C, Yamazoe M, Ariki S, Takahashi M, Shimizu T, et al. Pulmonary surfactant protein D binds MD-2 through the carbohydrate recognition domain. Biochemistry (2008) 47:12878–85. doi:10.1021/bi8010175
167. LeVine AM, Whitsett JA, Gwozdz JA, Richardson TR, Fisher JH, Burhans MS, et al. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol (2000) 165:3934–40. doi:10.4049/jimmunol.165.7.3934
168. Ikegami M, Scoville EA, Grant S, Korfhagen T, Brondyk W, Scheule RK, et al. Surfactant protein-D and surfactant inhibit endotoxin-induced pulmonary inflammation. Chest (2007) 132:1447–54. doi:10.1378/chest.07-0864
169. Ikegami M, Carter K, Bishop K, Yadav A, Masterjohn E, Brondyk W, et al. Intratracheal recombinant surfactant protein D prevents endotoxin shock in the newborn preterm lamb. Am J Respir Crit Care Med (2006) 173(12):1342–7. doi:10.1164/rccm.200509-1485OC
170. Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell (2003) 115:13–23. doi:10.1016/S0092-8674(03)00758-X
171. Fournier B, Andargachew R, Robin AZ, Laur O, Voelker DR, Lee WY, et al. Surfactant protein D (Sp-D) binds to membrane-proximal domain (D3) of signal regulatory protein alpha (SIRPalpha), a site distant from binding domain of CD47, while also binding to analogous region on signal regulatory protein beta (SIRPbeta). J Biol Chem (2012) 287:19386–98. doi:10.1074/jbc.M111.324533
172. McCubbrey AL, Sonstein J, Ames TM, Freeman CM, Curtis JL. Glucocorticoids relieve collectin-driven suppression of apoptotic cell uptake in murine alveolar macrophages through downregulation of SIRPalpha. J Immunol (2012) 189:112–9. doi:10.4049/jimmunol.1200984
173. Guo CJ, Atochina-Vasserman EN, Abramova E, Foley JP, Zaman A, Crouch E, et al. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol (2008) 6:e266. doi:10.1371/journal.pbio.0060266
174. Olde Nordkamp MJ, van Eijk M, Urbanus RT, Bont L, Haagsman HP, Meyaard L. Leukocyte-associated Ig-like receptor-1 is a novel inhibitory receptor for surfactant protein D. J Leukoc Biol (2014) 96:105–11. doi:10.1189/jlb.3AB0213-092RR
175. Yoshida M, Whitsett JA. Alveolar macrophages and emphysema in surfactant protein-D-deficient mice. Respirology (2006) 11(Suppl):S37–40. doi:10.1111/j.1440-1843.2006.00806.x
176. Nemeth K, Schoppet M, Al-Fakhri N, Helas S, Jessberger R, Hofbauer LC, et al. The role of osteoclast-associated receptor in osteoimmunology. J Immunol (2011) 186:13–8. doi:10.4049/jimmunol.1002483
177. Fukuzawa T, Ishida J, Kato A, Ichinose T, Ariestanti DM, Takahashi T, et al. Lung surfactant levels are regulated by Ig-Hepta/GPR116 by monitoring surfactant protein D. PLoS One (2013) 8:e69451. doi:10.1371/journal.pone.0069451
178. Umeda Y, Hasegawa Y, Otsuka M, Ariki S, Takamiya R, Saito A, et al. Surfactant protein D inhibits activation of non-small cell lung cancer-associated mutant EGFR and affects clinical outcomes of patients. Oncogene (2017) 36(46):6432–45. doi:10.1038/onc.2017.253
179. Jäkel A, Sim RB. The human lung surfactant proteins A (SP-A) and D (SP-D) share similar binding mechanisms and common lignads on macrophages and dendritic cells. J Unsolved Quest (2012) 2:12–8.
180. Dodagatta-Marri E, Mitchell DA, Pandit H, Sonawani A, Murugaiah V, Idicula-Thomas S, et al. Protein-protein interaction between surfactant protein D and DC-SIGN via C-type lectin domain can suppress HIV-1 transfer. Front Immunol (2017) 8:834. doi:10.3389/fimmu.2017.00834
181. Nadesalingam J, Bernal AL, Dodds AW, Willis AC, Mahoney DJ, Day AJ, et al. Identification and characterization of a novel interaction between pulmonary surfactant protein D and decorin. J Biol Chem (2003) 278:25678–87. doi:10.1074/jbc.M210186200
182. Craig-Barnes HA, Doumouras BS, Palaniyar N. Surfactant protein D interacts with alpha2-macroglobulin and increases its innate immune potential. J Biol Chem (2010) 285:13461–70. doi:10.1074/jbc.M110.108837
183. Holmskov U, Mollenhauer J, Madsen J, Vitved L, Gronlund J, Tornoe I, et al. Cloning of gp-340, a putative opsonin receptor for lung surfactant protein D. Proc Natl Acad Sci U S A (1999) 96:10794–9. doi:10.1073/pnas.96.19.10794
184. White MR, Crouch E, van Eijk M, Hartshorn M, Pemberton L, Tornoe I, et al. Cooperative anti-influenza activities of respiratory innate immune proteins and neuraminidase inhibitor. Am J Physiol Lung Cell Mol Physiol (2005) 288:L831–40. doi:10.1152/ajplung.00365.2004
185. Hartshorn KL, White MR, Tecle T, Holmskov U, Crouch EC. Innate defense against influenza A virus: activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J Immunol (2006) 176:6962–72. doi:10.4049/jimmunol.176.11.6962
186. Doss M, White MR, Tecle T, Gantz D, Crouch EC, Jung G, et al. Interactions of alpha-, beta-, and theta-defensins with influenza A virus and surfactant protein D. J Immunol (2009) 182:7878–87. doi:10.4049/jimmunol.0804049
187. Nadesalingam J, Reid KB, Palaniyar N. Collectin surfactant protein D binds antibodies and interlinks innate and adaptive immune systems. FEBS Lett (2005) 579:4449–53. doi:10.1016/j.febslet.2005.07.012
188. Crouch E, Persson A, Chang D, Heuser J. Molecular structure of pulmonary surfactant protein D (SP-D). J Biol Chem (1994) 269:17311–9.
189. Zhang L, Ikegami M, Crouch EC, Korfhagen TR, Whitsett JA. Activity of pulmonary surfactant protein-D (SP-D) in vivo is dependent on oligomeric structure. J Biol Chem (2001) 276:19214–9. doi:10.1074/jbc.M010191200
190. Hansen S, Holmskov U. Structural aspects of collectins and receptors for collectins. Immunobiology (1998) 199:165–89. doi:10.1016/S0171-2985(98)80025-9
191. Perino J, Thielens NM, Crouch E, Spehner D, Crance JM, Favier AL. Protective effect of surfactant protein d in pulmonary vaccinia virus infection: implication of A27 viral protein. Viruses (2013) 5:928–53. doi:10.3390/v5030928
192. Crouch EC. Surfactant protein-D and pulmonary host defense. Respir Res (2000) 1:93–108. doi:10.1186/rr19
193. Botas C, Poulain F, Akiyama J, Brown C, Allen L, Goerke J, et al. Altered surfactant homeostasis and alveolar type II cell morphology in mice lacking surfactant protein D. Proc Natl Acad Sci U S A (1998) 95:11869–74. doi:10.1073/pnas.95.20.11869
194. Wert SE, Yoshida M, LeVine AM, Ikegami M, Jones T, Ross GF, et al. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc Natl Acad Sci U S A (2000) 97:5972–7. doi:10.1073/pnas.100448997
195. Atochina EN, Beers MF, Hawgood S, Poulain F, Davis C, Fusaro TT, et al. Surfactant protein-D, A mediator of innate lung immunity, alters the products of NO metabolism. Am J Respir Cell Mol Biol (2004) 30(3):271–9.
196. Knudsen L, Ochs K, Boxler L, Tornoe I, Lykke-Sorensen G, Mackay RM, et al. Surfactant protein D (SP-D) deficiency is attenuated in humanised mice expressing the Met(11)Thr short nucleotide polymorphism of SP-D: implications for surfactant metabolism in the lung. J Anat (2013) 223:581–92. doi:10.1111/joa.12120
197. Winkler C, Bahlmann O, Viereck J, Knudsen L, Wedekind D, Hoymann HG, et al. Impact of a Met(11)Thr single nucleotide polymorphism of surfactant protein D on allergic airway inflammation in a murine asthma model. Exp Lung Res (2014) 40:154–63. doi:10.3109/01902148.2014.891062
198. Ogasawara Y, Voelker DR. The role of the amino-terminal domain and the collagenous region in the structure and the function of rat surfactant protein D. J Biol Chem (1995) 270:19052–8. doi:10.1074/jbc.270.32.19052
199. Brown-Augsburger P, Hartshorn K, Chang D, Rust K, Fliszar C, Welgus HG, et al. Site-directed mutagenesis of Cys-15 and Cys-20 of pulmonary surfactant protein D. Expression of a trimeric protein with altered anti-viral properties. J Biol Chem (1996) 271:13724–30. doi:10.1074/jbc.271.23.13724
200. Kingma PS, Zhang L, Ikegami M, Hartshorn K, McCormack FX, Whitsett JA. Correction of pulmonary abnormalities in Sftpd-/- mice requires the collagenous domain of surfactant protein D. J Biol Chem (2006) 281(34):24496–505. doi:10.1074/jbc.M600651200
201. Clark H, Palaniyar N, Strong P, Edmondson J, Hawgood S, Reid KB. Surfactant protein D reduces alveolar macrophage apoptosis in vivo. J Immunol (2002) 169:2892–9. doi:10.4049/jimmunol.169.6.2892
202. Knudsen L, Ochs M, Mackay R, Townsend P, Deb R, Muhlfeld C, et al. Truncated recombinant human SP-D attenuates emphysema and type II cell changes in SP-D deficient mice. Respir Res (2007) 8:70. doi:10.1186/1465-9921-8-70
203. Strong P, Reid KB, Clark H. Intranasal delivery of a truncated recombinant human SP-D is effective at down-regulating allergic hypersensitivity in mice sensitized to allergens of Aspergillus fumigatus. Clin Exp Immunol (2002) 130:19–24. doi:10.1046/j.1365-2249.2002.01968.x
204. Strong P, Townsend P, Mackay R, Reid KB, Clark HW. A recombinant fragment of human SP-D reduces allergic responses in mice sensitized to house dust mite allergens. Clin Exp Immunol (2003) 134:181–7. doi:10.1046/j.1365-2249.2003.02281.x
205. Singh M, Madan T, Waters P, Sonar S, Singh SK, Kamran MF, et al. Therapeutic effects of recombinant forms of full-length and truncated human surfactant protein D in a murine model of invasive pulmonary aspergillosis. Mol Immunol (2009) 46:2363–9. doi:10.1016/j.molimm.2009.03.019
206. Erpenbeck VJ, Ziegert M, Cavalet-Blanco D, Martin C, Baelder R, Glaab T, et al. Surfactant protein D inhibits early airway response in Aspergillus fumigatus-sensitized mice. Clin Exp Allergy (2006) 36:930–40. doi:10.1111/j.1365-2222.2006.02524.x
207. Heidinger K, Konig IR, Bohnert A, Kleinsteiber A, Hilgendorff A, Gortner L, et al. Polymorphisms in the human surfactant protein-D (SFTPD) gene: strong evidence that serum levels of surfactant protein-D (SP-D) are genetically influenced. Immunogenetics (2005) 57:1–7. doi:10.1007/s00251-005-0775-5
208. Sorensen GL, Hjelmborg JB, Kyvik KO, Fenger M, Hoj A, Bendixen C, et al. Genetic and environmental influences of surfactant protein D serum levels. Am J Physiol Lung Cell Mol Physiol (2006) 290:L1010–7. doi:10.1152/ajplung.00487.2005
209. Lahti M, Lofgren J, Marttila R, Renko M, Klaavuniemi T, Haataja R, et al. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr Res (2002) 51:696–9. doi:10.1203/00006450-200206000-00006
210. Liu W, Bentley CM, Floros J. Study of human SP-A, SP-B and SP-D loci: allele frequencies, linkage disequilibrium and heterozygosity in different races and ethnic groups. BMC Genet (2003) 4:13. doi:10.1186/1471-2156-4-13
211. Horimasu Y, Hattori N, Ishikawa N, Tanaka S, Bonella F, Ohshimo S, et al. Differences in serum SP-D levels between German and Japanese subjects are associated with SFTPD gene polymorphisms. BMC Med Genet (2014) 15:4. doi:10.1186/1471-2350-15-4
212. Johansson SL, Tan Q, Holst R, Christiansen L, Hansen NC, Hojland AT, et al. Surfactant protein D is a candidate biomarker for subclinical tobacco smoke-induced lung damage. Am J Physiol Lung Cell Mol Physiol (2014) 306:L887–95. doi:10.1152/ajplung.00340.2013
213. Kristiansen M, Frisch M, Madsen HO, Garred P, Jacobsen S. Smoking and polymorphisms of genes encoding mannose-binding lectin and surfactant protein-D in patients with rheumatoid arthritis. Rheumatol Int (2014) 34:373–80. doi:10.1007/s00296-013-2904-z
214. Sorensen GL, Bladbjerg EM, Steffensen R, Tan Q, Madsen J, Drivsholm T, et al. Association between the surfactant protein D (SFTPD) gene and subclinical carotid artery atherosclerosis. Atherosclerosis (2016) 246:7–12. doi:10.1016/j.atherosclerosis.2015.12.037
215. Shakoori TA, Sin DD, Bokhari SN, Ghafoor F, Shakoori AR. SP-D polymorphisms and the risk of COPD. Dis Markers (2012) 33:91–100. doi:10.3233/DMA-2012-0909
216. Kim DK, Cho MH, Hersh CP, Lomas DA, Miller BE, Kong X, et al. Genome-wide association analysis of blood biomarkers in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2012) 186:1238–47. doi:10.1164/rccm.201206-1013OC
217. Fakih D, Akiki Z, Junker K, Medlej-Hashim M, Waked M, Salameh P, et al. Surfactant protein D (SP-D) levels, polymorphisms and multimerization in COPD and asthma. Respirology (2017). doi:10.1111/resp.13193
218. Lin Z, Pearson C, Chinchilli V, Pietschmann SM, Luo J, Pison U, et al. Polymorphisms of human SP-A, SP-B, and SP-D genes: association of SP-B Thr131Ile with ARDS. Clin Genet (2000) 58:181–91. doi:10.1034/j.1399-0004.2000.580305.x
219. Deng YQ, Tao ZZ, Kong YG, Xiao BK, Chen SM, Xu Y, et al. Association between single nucleotide polymorphisms of surfactant protein D and allergic rhinitis in Chinese patients. Tissue Antigens (2009) 73:546–52. doi:10.1111/j.1399-0039.2009.01232.x
220. Krueger M, Puthothu B, Gropp E, Heinze J, Braun S, Heinzmann A. Amino acid variants in surfactant protein D are not associated with bronchial asthma. Pediatr Allergy Immunol (2006) 17:77–81. doi:10.1111/j.1399-3038.2005.00353.x
221. Brandt EB, Mingler MK, Stevenson MD, Wang N, Khurana Hershey GK, Whitsett JA, et al. Surfactant protein D alters allergic lung responses in mice and human subjects. J Allergy Clin Immunol (2008) 121:1140–7.e1142. doi:10.1016/j.jaci.2008.02.011
222. Berg KK, Madsen HO, Garred P, Wiseth R, Gunnes S, Videm V. The additive contribution from inflammatory genetic markers on the severity of cardiovascular disease. Scand J Immunol (2009) 69:36–42. doi:10.1111/j.1365-3083.2008.02187.x
223. Aramini B, Kim C, Diangelo S, Petersen E, Lederer DJ, Shah L, et al. Donor surfactant protein D (SP-D) polymorphisms are associated with lung transplant outcome. Am J Transplant (2013) 13:2130–6. doi:10.1111/ajt.12326
224. Foreman MG, Kong X, Demeo DL, Pillai SG, Hersh CP, Bakke P, et al. Polymorphisms in surfactant protein D are associated with COPD. Am J Respir Cell Mol Biol (2010) 44(3):316–22. doi:10.1165/rcmb.2009-0360OC
225. Ishii T, Hagiwara K, Kamio K, Ikeda S, Arai T, Mieno MN, et al. Involvement of surfactant protein D in emphysema revealed by genetic association study. Eur J Hum Genet (2012) 20:230–5. doi:10.1038/ejhg.2011.183
226. Ou CY, Chen CZ, Hsiue TR, Lin SH, Wang JY. Genetic variants of pulmonary SP-D predict disease outcome of COPD in a Chinese population. Respirology (2015) 20:296–303. doi:10.1111/resp.12427
227. Garcia-Laorden MI, Rodriguez de Castro F, Sole-Violan J, Rajas O, Blanquer J, Borderias L, et al. Influence of genetic variability at the surfactant proteins A and D in community-acquired pneumonia: a prospective, observational, genetic study. Crit Care (2011) 15:R57. doi:10.1186/cc10030
228. Soto-Cardenas MJ, Gandia M, Brito-Zeron P, Arias MT, Armiger N, Bove A, et al. Etiopathogenic role of surfactant protein d in the clinical and immunological expression of primary Sjogren syndrome. J Rheumatol (2015) 42:111–8. doi:10.3899/jrheum.140394
229. Pueyo N, Ortega FJ, Mercader JM, Moreno-Navarrete JM, Sabater M, Bonas S, et al. Common genetic variants of surfactant protein-D (SP-D) are associated with type 2 diabetes. PLoS One (2013) 8:e60468. doi:10.1371/journal.pone.0060468
230. Lin Z, John G, Hegarty JP, Berg A, Yu W, Wang Y, et al. Genetic variants and monoallelic expression of surfactant protein-D in inflammatory bowel disease. Ann Hum Genet (2011) 75:559–68. doi:10.1111/j.1469-1809.2011.00662.x
231. Ishii T, Hagiwara K, Ikeda S, Arai T, Mieno MN, Kumasaka T, et al. Association between genetic variations in surfactant protein d and emphysema, interstitial pneumonia, and lung cancer in a Japanese population. COPD (2012) 9:409–16. doi:10.3109/15412555.2012.676110
232. Pavlovic J, Papagaroufalis C, Xanthou M, Liu W, Fan R, Thomas NJ, et al. Genetic variants of surfactant proteins A, B, C, and D in bronchopulmonary dysplasia. Dis Markers (2006) 22:277–91. doi:10.1155/2006/817805
233. Sorensen GL, Dahl M, Tan Q, Bendixen C, Holmskov U, Husby S. Surfactant protein-D-encoding gene variant polymorphisms are linked to respiratory outcome in premature infants. J Pediatr (2014) 165:683–9. doi:10.1016/j.jpeds.2014.05.042
234. Hilgendorff A, Heidinger K, Bohnert A, Kleinsteiber A, Konig IR, Ziegler A, et al. Association of polymorphisms in the human surfactant protein-D (SFTPD) gene and postnatal pulmonary adaptation in the preterm infant. Acta Paediatr (2009) 98:112–7. doi:10.1111/j.1651-2227.2008.01014.x
235. Karjalainen MK, Huusko JM, Tuohimaa A, Luukkonen A, Haataja R, Hallman M. A study of collectin genes in spontaneous preterm birth reveals an association with a common surfactant protein D gene polymorphism. Pediatr Res (2012) 71:93–9. doi:10.1038/pr.2011.2
236. Thomas NJ, DiAngelo S, Hess JC, Fan R, Ball MW, Geskey JM, et al. Transmission of surfactant protein variants and haplotypes in children hospitalized with respiratory syncytial virus. Pediatr Res (2009) 66:70–3. doi:10.1203/PDR.0b013e3181a1d768
237. Floros J, Lin HM, Garcia A, Salazar MA, Guo X, DiAngelo S, et al. Surfactant protein genetic marker alleles identify a subgroup of tuberculosis in a Mexican population. J Infect Dis (2000) 182:1473–8. doi:10.1086/315866
238. Persson A, Chang D, Rust K, Moxley M, Longmore W, Crouch E. Purification and biochemical characterization of CP4 (SP-D), a collagenous surfactant-associated protein. Biochemistry (1989) 28:6361–7. doi:10.1021/bi00441a031
239. Crouch E, Chang D, Rust K, Persson A, Heuser J. Recombinant pulmonary surfactant protein D. Post-translational modification and molecular assembly. J Biol Chem (1994) 269:15808–13.
240. Leth-Larsen R, Holmskov U, Hojrup P. Structural characterization of human and bovine lung surfactant protein D. Biochem J (1999) 343(Pt 3):645–52. doi:10.1042/0264-6021:3430645
241. Ito E, Oka R, Ishii T, Korekane H, Kurimoto A, Kizuka Y, et al. Fucosylated surfactant protein-D is a biomarker candidate for the development of chronic obstructive pulmonary disease. J Proteomics (2015) 127:386–94. doi:10.1016/j.jprot.2015.07.011
242. Atochina-Vasserman EN, Winkler C, Abramova H, Schaumann F, Krug N, Gow AJ, et al. Segmental allergen challenge alters multimeric structure and function of surfactant protein d in humans. Am J Respir Crit Care Med (2011) 183:856–64. doi:10.1164/rccm.201004-0654OC
243. Atochina-Vasserman EN, Guo CJ, Abramova E, Golden TN, Sims M, James ML, et al. Surfactant dysfunction and lung inflammation in the female mouse model of lymphangioleiomyomatosis. Am J Respir Cell Mol Biol (2015) 53:96–104. doi:10.1165/rcmb.2014-0224OC
244. Guo C, Atochina-Vasserman E, Abramova H, George B, Manoj V, Scott P, et al. Role of NOS2 in pulmonary injury and repair in response to bleomycin. Free Radic Biol Med (2016) 91:293–301. doi:10.1016/j.freeradbiomed.2015.10.417
245. Atochina-Vasserman EN, Gow AJ, Abramova H, Guo CJ, Tomer Y, Preston AM, et al. Immune reconstitution during Pneumocystis lung infection: disruption of surfactant component expression and function by S-nitrosylation. J Immunol (2009) 182:2277–87. doi:10.4049/jimmunol.0802775
246. Malaviya R, Gow AJ, Francis M, Abramova EV, Laskin JD, Laskin DL. Radiation-induced lung injury and inflammation in mice: role of inducible nitric oxide synthase and surfactant protein D. Toxicol Sci (2015) 144:27–38. doi:10.1093/toxsci/kfu255
247. Matalon S, Shrestha K, Kirk M, Waldheuser S, McDonald B, Smith K, et al. Modification of surfactant protein D by reactive oxygen-nitrogen intermediates is accompanied by loss of aggregating activity, in vitro and in vivo. FASEB J (2009) 23:1415–30. doi:10.1096/fj.08-120568
248. Crouch EC, Hirche TO, Shao B, Boxio R, Wartelle J, Benabid R, et al. Myeloperoxidase-dependent inactivation of surfactant protein D in vitro and in vivo. J Biol Chem (2010) 285:16757–70. doi:10.1074/jbc.M109.097048
249. Cooley J, McDonald B, Accurso FJ, Crouch EC, Remold-O’Donnell E. Patterns of neutrophil serine protease-dependent cleavage of surfactant protein D in inflammatory lung disease. J Leukoc Biol (2008) 83:946–55. doi:10.1189/jlb.1007684
250. Duvoix A, Mackay RM, Henderson N, McGreal E, Postle A, Reid K, et al. Physiological concentration of calcium inhibits elastase-induced cleavage of a functional recombinant fragment of surfactant protein D. Immunobiology (2011) 216:72–9. doi:10.1016/j.imbio.2010.03.006
251. Alcorn JF, Wright JR. Degradation of pulmonary surfactant protein D by Pseudomonas aeruginosa elastase abrogates innate immune function. J Biol Chem (2004) 279:30871–9. doi:10.1074/jbc.M400796200
252. Winkler C, Atochina-Vasserman EN, Holz O, Beers MF, Erpenbeck VJ, Krug N, et al. Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir Res (2011) 12:29. doi:10.1186/1465-9921-12-29
253. Mariencheck WI, Alcorn JF, Palmer SM, Wright JR. Pseudomonas aeruginosa elastase degrades surfactant proteins A and D. Am J Respir Cell Mol Biol (2003) 28:528–37. doi:10.1165/rcmb.2002-0141OC
254. von Bredow C, Wiesener A, Griese M. Proteolysis of surfactant protein D by cystic fibrosis relevant proteases. Lung (2003) 181:79–88. doi:10.1007/s00408-003-1008-z
255. Hirche TO, Crouch EC, Espinola M, Brokelman TJ, Mecham RP, DeSilva N, et al. Neutrophil serine proteinases inactivate surfactant protein D by cleaving within a conserved subregion of the carbohydrate recognition domain. J Biol Chem (2004) 279:27688–98. doi:10.1074/jbc.M402936200
256. Malloy JL, Veldhuizen RA, Thibodeaux BA, O’Callaghan RJ, Wright JR. Pseudomonas aeruginosa protease IV degrades surfactant proteins and inhibits surfactant host defense and biophysical functions. Am J Physiol Lung Cell Mol Physiol (2005) 288:L409–18. doi:10.1152/ajplung.00322.2004
257. Deb R, Shakib F, Reid K, Clark H. Major house dust mite allergens Dermatophagoides pteronyssinus 1 and Dermatophagoides farinae 1 degrade and inactivate lung surfactant proteins A and D. J Biol Chem (2007) 282:36808–19. doi:10.1074/jbc.M702336200
258. Mun JJ, Tam C, Kowbel D, Hawgood S, Barnett MJ, Evans DJ, et al. Clearance of Pseudomonas aeruginosa from a healthy ocular surface involves surfactant protein D and is compromised by bacterial elastase in a murine null-infection model. Infect Immun (2009) 77:2392–8. doi:10.1128/IAI.00173-09
259. Bratcher PE, Weathington NM, Nick HJ, Jackson PL, Snelgrove RJ, Gaggar A. MMP-9 cleaves SP-D and abrogates its innate immune functions in vitro. PLoS One (2012) 7:e41881. doi:10.1371/journal.pone.0041881
260. Stolley JM, Gong D, Farley K, Zhao P, Cooley J, Crouch EC, et al. Increased surfactant protein D fails to improve bacterial clearance and inflammation in serpinB1-/- mice. Am J Respir Cell Mol Biol (2012) 47:792–9. doi:10.1165/rcmb.2012-0145OC
261. Zhang Z, Abdel-Razek O, Hawgood S, Wang G. Protective role of surfactant protein D in ocular Staphylococcus aureus infection. PLoS One (2015) 10:e0138597. doi:10.1371/journal.pone.0138597
262. Todd DA, Marsh MJ, George A, Henderson NG, Barr H, Sebastian S, et al. Surfactant phospholipids, surfactant proteins, and inflammatory markers during acute lung injury in children. Pediatr Crit Care Med (2010) 11:82–91. doi:10.1097/PCC.0b013e3181ae5a4c
263. Griese M, Wiesener A, Lottspeich F, von Bredow C. Limited proteolysis of surfactant protein D causes a loss of its calcium-dependent lectin functions. Biochim Biophys Acta (2003) 1638:157–63. doi:10.1016/S0925-4439(03)00063-2
264. Noah TL, Murphy PC, Alink JJ, Leigh MW, Hull WM, Stahlman MT, et al. Bronchoalveolar lavage fluid surfactant protein-A and surfactant protein-D are inversely related to inflammation in early cystic fibrosis. Am J Respir Crit Care Med (2003) 168:685–91. doi:10.1164/rccm.200301-005OC
265. Postle AD, Mander A, Reid KB, Wang JY, Wright SM, Moustaki M, et al. Deficient hydrophilic lung surfactant proteins A and D with normal surfactant phospholipid molecular species in cystic fibrosis. Am J Respir Cell Mol Biol (1999) 20:90–8. doi:10.1165/ajrcmb.20.1.3253
266. Griese M, Steinecker M, Schumacher S, Braun A, Lohse P, Heinrich S. Children with absent surfactant protein D in bronchoalveolar lavage have more frequently pneumonia. Pediatr Allergy Immunol (2008) 19:639–47. doi:10.1111/j.1399-3038.2007.00695.x
267. Griese M, Maderlechner N, Ahrens P, Kitz R. Surfactant proteins A and D in children with pulmonary disease due to gastroesophageal reflux. Am J Respir Crit Care Med (2002) 165:1546–50. doi:10.1164/rccm.2107147
268. Umstead TM, Freeman WM, Chinchilli VM, Phelps DS. Age-related changes in the expression and oxidation of bronchoalveolar lavage proteins in the rat. Am J Physiol Lung Cell Mol Physiol (2009) 296:L14–29. doi:10.1152/ajplung.90366.2008
269. Betsuyaku T, Kuroki Y, Nagai K, Nasuhara Y, Nishimura M. Effects of ageing and smoking on SP-A and SP-D levels in bronchoalveolar lavage fluid. Eur Respir J (2004) 24:964–70. doi:10.1183/09031936.04.00064004
270. Moliva JI, Rajaram MV, Sidiki S, Sasindran SJ, Guirado E, Pan XJ, et al. Molecular composition of the alveolar lining fluid in the aging lung. Age (Dordr) (2014) 36:9633. doi:10.1007/s11357-014-9633-4
271. Zhao XM, Wu YP, Wei R, Cai HX, Tornoe I, Han JJ, et al. Plasma surfactant protein D levels and the relation to body mass index in a chinese population. Scand J Immunol (2007) 66:71–6. doi:10.1111/j.1365-3083.2007.01943.x
272. Ilumets H, Mazur W, Toljamo T, Louhelainen N, Nieminen P, Kobayashi H, et al. Ageing and smoking contribute to plasma surfactant proteins and protease imbalance with correlations to airway obstruction. BMC Pulm Med (2011) 11:19. doi:10.1186/1471-2466-11-19
273. Benfante A, Battaglia S, Principe S, Di Mitri C, Paterno A, Spatafora M, et al. Asthmatics with high levels of serum surfactant protein D have more severe disease. Eur Respir J (2016) 47(6):1864–7. doi:10.1183/13993003.02142-2015
274. Hastings RH, Grady M, Sakuma T, Matthay MA. Clearance of different-sized proteins from the alveolar space in humans and rabbits. J Appl Physiol (1985) (1992) 73:1310–6. doi:10.1152/jappl.1992.73.4.1310
275. Gaunsbaek MQ, Rasmussen KJ, Beers MF, Atochina-Vasserman EN, Hansen S. Lung surfactant protein D (SP-D) response and regulation during acute and chronic lung injury. Lung (2013) 191:295–303. doi:10.1007/s00408-013-9452-x
276. Hirama N, Shibata Y, Otake K, Machiya J, Wada T, Inoue S, et al. Increased surfactant protein-D and foamy macrophages in smoking-induced mouse emphysema. Respirology (2007) 12:191–201. doi:10.1111/j.1440-1843.2006.01009.x
277. Moazed F, Burnham EL, Vandivier RW, O’Kane CM, Shyamsundar M, Hamid U, et al. Cigarette smokers have exaggerated alveolar barrier disruption in response to lipopolysaccharide inhalation. Thorax (2016) 71(12):1130–6. doi:10.1136/thoraxjnl-2015-207886
278. More JM, Voelker DR, Silveira LJ, Edwards MG, Chan ED, Bowler RP. Smoking reduces surfactant protein D and phospholipids in patients with and without chronic obstructive pulmonary disease. BMC Pulm Med (2010) 10:53. doi:10.1186/1471-2466-10-53
279. Sims MW, Tal-Singer RM, Kierstein S, Musani AI, Beers MF, Panettieri RA, et al. Chronic obstructive pulmonary disease and inhaled steroids alter surfactant protein D (SP-D) levels: a cross-sectional study. Respir Res (2008) 9:13. doi:10.1186/1465-9921-9-13
280. Lomas DA, Silverman EK, Edwards LD, Locantore NW, Miller BE, Horstman DH, et al. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur Respir J (2009) 34(1):95–102. doi:10.1183/09031936.00156508
281. Krane M, Griese M. Surfactant protein D in serum from patients with allergic bronchopulmonary aspergillosis. Eur Respir J (2003) 22:592–5. doi:10.1183/09031936.03.00060603
282. Olesen HV, Holmskov U, Schiotz PO, Sorensen GL. Serum-surfactant SP-D correlates inversely to lung function in cystic fibrosis. J Cyst Fibros (2010) 9:257–62. doi:10.1016/j.jcf.2010.03.011
283. Herbein JF, Wright JR. Enhanced clearance of surfactant protein D during LPS-induced acute inflammation in rat lung. Am J Physiol Lung Cell Mol Physiol (2001) 281:L268–77. doi:10.1152/ajplung.2001.281.1.L268
284. Atochina-Vasserman EN. S-nitrosylation of surfactant protein D as a modulator of pulmonary inflammation. Biochim Biophys Acta (2012) 1820:763–9. doi:10.1016/j.bbagen.2011.12.006
285. Starosta V, Griese M. Oxidative damage to surfactant protein D in pulmonary diseases. Free Radic Res (2006) 40:419–25. doi:10.1080/10715760600571248
286. Duvoix A, Miranda E, Perez J, Sorensen GL, Holmskov U, Trapnell BC, et al. Evaluation of full-length, cleaved and nitrosylated serum surfactant protein D as biomarkers for COPD. COPD (2011) 8:79–95. doi:10.3109/15412555.2011.558542
287. Celli BR, Locantore N, Yates J, Tal-Singer R, Miller BE, Bakke P, et al. Inflammatory biomarkers improve clinical prediction of mortality in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2012) 185:1065–72. doi:10.1164/rccm.201110-1792OC
288. Barlo NP, van Moorsel CH, Ruven HJ, Zanen P, van den Bosch JM, Grutters JC. Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis (2009) 26:155–61.
289. Eisner MD, Parsons P, Matthay MA, Ware L, Greene K; Acute Respiratory Distress Syndrome Network. Plasma surfactant protein levels and clinical outcomes in patients with acute lung injury. Thorax (2003) 58:983–8. doi:10.1136/thorax.58.11.983
290. Cheng IW, Ware LB, Greene KE, Nuckton TJ, Eisner MD, Matthay MA. Prognostic value of surfactant proteins A and D in patients with acute lung injury. Crit Care Med (2003) 31:20–7. doi:10.1097/00003246-200301000-00003
291. Ware LB, Koyama T, Billheimer DD, Wu W, Bernard GR, Thompson BT, et al. Prognostic and pathogenetic value of combining clinical and biochemical indices in patients with acute lung injury. Chest (2010) 137:288–96. doi:10.1378/chest.09-1484
292. Ware LB, Koyama T, Zhao Z, Janz DR, Wickersham N, Bernard GR, et al. Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit Care (2013) 17:R253. doi:10.1186/cc13080
293. King BA, Kingma PS. Surfactant protein D deficiency increases lung injury during endotoxemia. Am J Respir Cell Mol Biol (2011) 44:709–15. doi:10.1165/rcmb.2009-0436OC
294. Determann RM, Royakkers AA, Haitsma JJ, Zhang H, Slutsky AS, Ranieri VM, et al. Plasma levels of surfactant protein D and KL-6 for evaluation of lung injury in critically ill mechanically ventilated patients. BMC Pulm Med (2010) 10:6. doi:10.1186/1471-2466-10-6
295. Ryckman KK, Dagle JM, Kelsey K, Momany AM, Murray JC. Genetic associations of surfactant protein D and angiotensin-converting enzyme with lung disease in preterm neonates. J Perinatol (2012) 32:349–55. doi:10.1038/jp.2011.104
296. Leth-Larsen R, Nordenbaek C, Tornoe I, Moeller V, Schlosser A, Koch C, et al. Surfactant protein D (SP-D) serum levels in patients with community-acquired pneumonia. Clin Immunol (2003) 108:29–37. doi:10.1016/S1521-6616(03)00042-1
297. Jambo KC, French N, Zijlstra E, Gordon SB. AIDS patients have increased surfactant protein D but normal mannose binding lectin levels in lung fluid. Respir Res (2007) 8:42. doi:10.1186/1465-9921-8-42
298. Wu YP, Liu ZH, Wei R, Pan SD, Mao NY, Chen B, et al. Elevated plasma surfactant protein D (SP-D) levels and a direct correlation with anti-severe acute respiratory syndrome coronavirus-specific IgG antibody in SARS patients. Scand J Immunol (2009) 69:508–15. doi:10.1111/j.1365-3083.2009.02245.x
299. Hartshorn KL, White MR, Tecle T, Sorensen G, Holmskov U, Crouch EC. Viral aggregating and opsonizing activity in collectin trimers. Am J Physiol Lung Cell Mol Physiol (2010) 298:L79–88. doi:10.1152/ajplung.00223.2009
300. Boonarkart CH, Suptawiwat O, Uiprasertkul M, Kongchanagul A, Rodpothong P, Bunthi CH, et al. A reduced expression of surfactant protein D in the lungs of fatal influenza H1N1 cases in 2009. Acta Virol (2012) 56:253–5. doi:10.4149/av_2012_03_253
301. Sin DD, Man SF, McWilliams A, Lam S. Surfactant protein D and bronchial dysplasia in smokers at high risk of lung cancer. Chest (2008) 134:582–8. doi:10.1378/chest.08-0600
302. Shiels MS, Chaturvedi AK, Katki HA, Gochuico BR, Caporaso NE, Engels EA. Circulating markers of interstitial lung disease and subsequent risk of lung cancer. Cancer Epidemiol Biomarkers Prev (2011) 20:2262–72. doi:10.1158/1055-9965.EPI-11-0326
303. Ichiyasu H, Ichikado K, Yamashita A, Iyonaga K, Sakamoto O, Suga M, et al. Pneumocyte biomarkers KL-6 and surfactant protein D reflect the distinct findings of high-resolution computed tomography in nonspecific interstitial pneumonia. Respiration (2012) 83:190–7. doi:10.1159/000326924
304. White ES, Xia M, Murray S, Dyal R, Flaherty CM, Flaherty KR, et al. Plasma surfactant protein-D, matrix metalloproteinase-7, and osteopontin index distinguishes idiopathic pulmonary fibrosis from other idiopathic interstitial pneumonias. Am J Respir Crit Care Med (2016) 194(10):1242–51. doi:10.1164/rccm.201505-0862OC
305. Nakamura K, Kato M, Shukuya T, Mori K, Sekimoto Y, Ihara H, et al. Surfactant protein-D predicts prognosis of interstitial lung disease induced by anticancer agents in advanced lung cancer: a case control study. BMC Cancer (2017) 17:302. doi:10.1186/s12885-017-3285-6
306. Thomas NJ, Fan R, Diangelo S, Hess JC, Floros J. Haplotypes of the surfactant protein genes A and D as susceptibility factors for the development of respiratory distress syndrome. Acta Paediatr (2007) 96:985–9. doi:10.1111/j.1651-2227.2007.00319.x
307. Chang HY, Li F, Li FS, Zheng CZ, Lei YZ, Wang J. Genetic polymorphisms of SP-A, SP-B, and SP-D and risk of respiratory distress syndrome in preterm neonates. Med Sci Monit (2016) 22:5091–100. doi:10.12659/MSM.898553
308. Gower WA, Nogee LM. Candidate gene analysis of the surfactant protein D gene in pediatric diffuse lung disease. J Pediatr (2013) 163:1778–80. doi:10.1016/j.jpeds.2013.06.063
309. Kotecha S, Davies PL, Clark HW, McGreal EP. Increased prevalence of low oligomeric state surfactant protein D with restricted lectin activity in bronchoalveolar lavage fluid from preterm infants. Thorax (2013) 68:460–7. doi:10.1136/thoraxjnl-2012-202729
310. Nakamura T, Shimizu H, Nishitani C, Yoshioka T. Remarkably delayed occurrence of normal surfactant composition in an extremely preterm infant. Pediatr Int (2013) 55:790–2. doi:10.1111/ped.12202
311. Bae YM, Bae CW, Oh MH, Lee SH, Woo KM, Jung KB. Effect of exogenous surfactant therapy on levels of pulmonary surfactant proteins A and D in preterm infants with respiratory distress syndrome. J Perinat Med (2009) 37:561–4. doi:10.1515/JPM.2009.100
312. Beresford MW, Shaw NJ. Bronchoalveolar lavage surfactant protein a, B, and d concentrations in preterm infants ventilated for respiratory distress syndrome receiving natural and synthetic surfactants. Pediatr Res (2003) 53:663–70. doi:10.1203/01.PDR.0000054653.89527.F8
313. Dahl M, Holmskov U, Husby S, Juvonen PO. Surfactant protein D levels in umbilical cord blood and capillary blood of premature infants. The influence of peri-natal factors. Pediatr Res (2006) 59(6):806–10. doi:10.1203/01.pdr.0000219122.81734.03
314. Vinod S, Gow A, Weinberger B, Potak D, Hiatt M, Chandra S, et al. Serum surfactant protein D as a marker for bronchopulmonary dysplasia. J Matern Fetal Neonatal Med (2017) 26:1–5. doi:10.1080/14767058.2017.1392506
315. Ogasawara Y, Kuroki Y, Shiratori M, Shimizu H, Miyamura K, Akino T. Ontogeny of surfactant apoprotein D, SP-D, in the rat lung. Biochim Biophys Acta (1991) 1083:252–6. doi:10.1016/0005-2760(91)90079-W
316. Chen CM, Wang LF. High-dose vascular endothelial growth factor increases surfactant protein gene expressions in preterm rat lung. Early Hum Dev (2007) 83:581–4. doi:10.1016/j.earlhumdev.2006.12.005
317. Lykkedegn S, Sorensen GL, Beck-Nielsen SS, Pilecki B, Duelund L, Marcussen N, et al. Vitamin D depletion in pregnancy decreases survival time, oxygen saturation, lung weight and body weight in preterm rat offspring. PLoS One (2016) 11:e0155203. doi:10.1371/journal.pone.0155203
318. Bersani I, Speer CP, Kunzmann S. Surfactant proteins A and D in pulmonary diseases of preterm infants. Expert Rev Anti Infect Ther (2012) 10:573–84. doi:10.1586/eri.12.34
319. Awasthi S, Coalson JJ, Crouch E, Yang F, King RJ. Surfactant proteins A and D in premature baboons with chronic lung injury (bronchopulmonary dysplasia). Evidence for an inhibition of secretion. Am J Respir Crit Care Med (1999) 160:942–9. doi:10.1164/ajrccm.160.3.9806061
320. Awasthi S, Coalson JJ, Yoder BA, Crouch E, King RJ. Deficiencies in lung surfactant proteins A and D are associated with lung infection in very premature neonatal baboons. Am J Respir Crit Care Med (2001) 163:389–97. doi:10.1164/ajrccm.163.2.2004168
321. Jain D, Atochina-Vasserman E, Kadire H, Tomer Y, Inch A, Scott P, et al. SP-D-deficient mice are resistant to hyperoxia. Am J Physiol Lung Cell Mol Physiol (2007) 292:L861–71. doi:10.1152/ajplung.00145.2006
322. Kallapur SG, Moss TJ, Ikegami M, Jasman RL, Newnham JP, Jobe AH. Recruited inflammatory cells mediate endotoxin-induced lung maturation in preterm fetal lambs. Am J Respir Crit Care Med (2005) 172:1315–21. doi:10.1164/rccm.200506-1007OC
323. Jobe AH, Newnham JP, Willet KE, Sly P, Ervin MG, Bachurski C, et al. Effects of antenatal endotoxin and glucocorticoids on the lungs of preterm lambs. Am J Obstet Gynecol (2000) 182:401–8. doi:10.1016/S0002-9378(00)70231-6
324. Bry K, Lappalainen U. Intra-amniotic endotoxin accelerates lung maturation in fetal rabbits. Acta Paediatr (2001) 90:74–80. doi:10.1111/j.1651-2227.2001.tb00259.x
325. Alexander JM, Gilstrap LC, Cox SM, McIntire DM, Leveno KJ. Clinical chorioamnionitis and the prognosis for very low birth weight infants. Obstet Gynecol (1998) 91:725–9. doi:10.1097/00006250-199805000-00016
326. Andrews WW, Goldenberg RL, Faye-Petersen O, Cliver S, Goepfert AR, Hauth JC. The Alabama Preterm Birth study: polymorphonuclear and mononuclear cell placental infiltrations, other markers of inflammation, and outcomes in 23- to 32-week preterm newborn infants. Am J Obstet Gynecol (2006) 195:803–8. doi:10.1016/j.ajog.2006.06.083
327. Chaiworapongsa T, Hong JS, Hull WM, Romero R, Whitsett JA. Amniotic fluid concentration of surfactant proteins in intra-amniotic infection. J Matern Fetal Neonatal Med (2008) 21:663–70. doi:10.1080/14767050802215664
328. Schmiedl A, Behrens J, Zscheppang K, Purevdorj E, von Mayersbach D, Liese A, et al. Lipopolysaccharide-induced injury is more pronounced in fetal transgenic ErbB4-deleted lungs. Am J Physiol Lung Cell Mol Physiol (2011) 301:L490–9. doi:10.1152/ajplung.00131.2010
329. Kuypers E, Collins JJ, Kramer BW, Ofman G, Nitsos I, Pillow JJ, et al. Intra-amniotic LPS and antenatal betamethasone: inflammation and maturation in preterm lamb lungs. Am J Physiol Lung Cell Mol Physiol (2012) 302:L380–9. doi:10.1152/ajplung.00338.2011
330. Salminen A, Paananen R, Vuolteenaho R, Metsola J, Ojaniemi M, Autio-Harmainen H, et al. Maternal endotoxin-induced preterm birth in mice: fetal responses in toll-like receptors, collectins, and cytokines. Pediatr Res (2008) 63:280–6. doi:10.1203/PDR.0b013e318163a8b2
331. Salminen A, Vuolteenaho R, Paananen R, Ojaniemi M, Hallman M. Surfactant protein D modulates levels of IL-10 and TNF-alpha in intrauterine compartments during lipopolysaccharide-induced preterm birth. Cytokine (2012) 60:423–30. doi:10.1016/j.cyto.2012.07.021
332. Montalbano AP, Hawgood S, Mendelson CR. Mice deficient in surfactant protein A (SP-A) and SP-D or in TLR2 manifest delayed parturition and decreased expression of inflammatory and contractile genes. Endocrinology (2013) 154:483–98. doi:10.1210/en.2012-1797
333. Kay S, Madan T. Fertility defects in surfactant associated protein D knockout female mice: altered ovarian hormone profile. Mol Immunol (2016) 71:87–97. doi:10.1016/j.molimm.2016.01.002
334. Sato A, Whitsett JA, Scheule RK, Ikegami M. Surfactant protein-d inhibits lung inflammation caused by ventilation in premature newborn lambs. Am J Respir Crit Care Med (2010) 181:1098–105. doi:10.1164/rccm.200912-1818OC
335. Xu J, Singhera GK, Dorscheid DR. Expression of surfactant protein D in airways of asthmatics and interleukin-13 modulation of surfactant protein D in human models of airway epithelium. Respir Res (2015) 16:26. doi:10.1186/s12931-015-0177-7
336. Cheng G, Ueda T, Numao T, Kuroki Y, Nakajima H, Fukushima Y, et al. Increased levels of surfactant protein A and D in bronchoalveolar lavage fluids in patients with bronchial asthma. Eur Respir J (2000) 16:831–5. doi:10.1183/09031936.00.16583100
337. Koopmans JG, van der Zee JS, Krop EJ, Lopuhaa CE, Jansen HM, Batenburg JJ. Serum surfactant protein D is elevated in allergic patients. Clin Exp Allergy (2004) 34:1827–33. doi:10.1111/j.1365-2222.2004.02083.x
338. Emmanouil P, Loukides S, Kostikas K, Papatheodorou G, Papaporfyriou A, Hillas G, et al. Sputum and BAL Clara cell secretory protein and surfactant protein D levels in asthma. Allergy (2015) 70:711–4. doi:10.1111/all.12603
339. Okazaki S, Murai H, Kidoguchi S, Nomura E, Itoh N, Hashimoto N, et al. The biomarker salivary SP-D may indicate small airway inflammation and asthma exacerbation. J Investig Allergol Clin Immunol (2017) 27:305–12. doi:10.18176/jiaci.0174
340. Mutti A, Corradi M, Goldoni M, Vettori MV, Bernard A, Apostoli P. Exhaled metallic elements and serum pneumoproteins in asymptomatic smokers and patients with COPD or asthma. Chest (2006) 129:1288–97. doi:10.1378/chest.129.5.1288
341. Akiki Z, Fakih D, Jounblat R, Chamat S, Waked M, Holmskov U, et al. Surfactant protein D, a clinical biomarker for chronic obstructive pulmonary disease with excellent discriminant values. Exp Ther Med (2016) 11:723–30. doi:10.3892/etm.2016.2986
342. Tanaka H, Sugawara H, Saikai T, Tsunematsu K, Takahashi H, Abe S. Mushroom worker’s lung caused by spores of Hypsizygus marmoreus (Bunashimeji): elevated serum surfactant protein D levels. Chest (2000) 118:1506–9. doi:10.1378/chest.118.5.1506
343. Saikai T, Tanaka H, Fuji M, Sugawara H, Takeya I, Tsunematsu K, et al. Hypersensitivity pneumonitis induced by the spore of Pleurotus eryngii (Eringi). Intern Med (2002) 41:571–3. doi:10.2169/internalmedicine.41.571
344. Higashi A, Higashi N, Tsuburai T, Takeuchi Y, Taniguchi M, Mita H, et al. Involvement of eicosanoids and surfactant protein D in extrinsic allergic alveolitis. Eur Respir J (2005) 26:1069–73. doi:10.1183/09031936.05.00106104
345. Tsushima K, Fujimoto K, Yoshikawa S, Kawakami S, Koizumi T, Kubo K. Hypersensitivity pneumonitis due to Bunashimeji mushrooms in the mushroom industry. Int Arch Allergy Immunol (2005) 137:241–8. doi:10.1159/000086337
346. Inase N, Ohtani Y, Sumi Y, Umino T, Usui Y, Miyake S, et al. A clinical study of hypersensitivity pneumonitis presumably caused by feather duvets. Ann Allergy Asthma Immunol (2006) 96:98–104. doi:10.1016/S1081-1206(10)61047-2
347. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol (2015) 16:45–56. doi:10.1038/ni.3049
348. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med (2012) 18:716–25. doi:10.1038/nm.2678
349. Madan T, Reid KB, Singh M, Sarma PU, Kishore U. Susceptibility of mice genetically deficient in the surfactant protein (SP)-A or SP-D gene to pulmonary hypersensitivity induced by antigens and allergens of Aspergillus fumigatus. J Immunol (2005) 174:6943–54. doi:10.4049/jimmunol.174.11.6943
350. Madan T, Kishore U, Singh M, Strong P, Clark H, Hussain EM, et al. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest (2001) 107:467–75. doi:10.1172/JCI10124
351. Schaub B, Westlake RM, He H, Arestides R, Haley KJ, Campo M, et al. Surfactant protein D deficiency influences allergic immune responses. Clin Exp Allergy (2004) 34:1819–26. doi:10.1111/j.1365-2222.2004.02068.x
352. Liu CF, Chen YL, Shieh CC, Yu CK, Reid KB, Wang JY. Therapeutic effect of surfactant protein D in allergic inflammation of mite-sensitized mice. Clin Exp Allergy (2005) 35:515–21. doi:10.1111/j.1365-2222.2005.02205.x
353. Haczku A, Cao Y, Vass G, Kierstein S, Nath P, Atochina-Vasserman EN, et al. IL-4 and IL-13 form a negative feedback circuit with surfactant protein-D in the allergic airway response. J Immunol (2006) 176:3557–65. doi:10.4049/jimmunol.176.6.3557
354. Cao Y, Tao JQ, Bates SR, Beers MF, Haczku A. IL-4 induces production of the lung collectin surfactant protein-D. J Allergy Clin Immunol (2004) 113:439–44. doi:10.1016/j.jaci.2003.11.031
355. Qaseem AS, Sonar S, Mahajan L, Madan T, Sorensen GL, Shamji MH, et al. Linking surfactant protein SP-D and IL-13: implications in asthma and allergy. Mol Immunol (2013) 54:98–107. doi:10.1016/j.molimm.2012.10.039
356. Kasper M, Sims G, Koslowski R, Kuss H, Thuemmler M, Fehrenbach H, et al. Increased surfactant protein D in rat airway goblet and Clara cells during ovalbumin-induced allergic airway inflammation. Clin Exp Allergy (2002) 32:1251–8. doi:10.1046/j.1365-2745.2002.01423.x
357. Fahy JV. Type 2 inflammation in asthma – present in most, absent in many. Nat Rev Immunol (2015) 15:57–65. doi:10.1038/nri3807
358. Wang JY, Shieh CC, You PF, Lei HY, Reid KB. Inhibitory effect of pulmonary surfactant proteins A and D on allergen-induced lymphocyte proliferation and histamine release in children with asthma. Am J Respir Crit Care Med (1998) 158:510–8. doi:10.1164/ajrccm.158.2.9709111
359. Atochina EN, Beers MF, Tomer Y, Scanlon ST, Russo SJ, Panettieri RA Jr, et al. Attenuated allergic airway hyperresponsiveness in C57BL/6 mice is associated with enhanced surfactant protein (SP)-D production following allergic sensitization. Respir Res (2003) 4:15. doi:10.1186/1465-9921-4-15
360. van Diemen CC, Postma DS, Aulchenko YS, Snijders PJ, Oostra BA, van Duijn CM, et al. Novel strategy to identify genetic risk factors for COPD severity: a genetic isolate. Eur Respir J (2010) 35:768–75. doi:10.1183/09031936.00054408
361. Issac MS, Ashur W, Mousa H. Genetic polymorphisms of surfactant protein D rs2243639, interleukin (IL)-1beta rs16944 and IL-1RN rs2234663 in chronic obstructive pulmonary disease, healthy smokers, and non-smokers. Mol Diagn Ther (2014) 18:343–54. doi:10.1007/s40291-014-0084-5
362. Perez-Rubio G, Silva-Zolezzi I, Fernandez-Lopez JC, Camarena A, Velazquez-Uncal M, Morales-Mandujano F, et al. Genetic variants in IL6R and ADAM19 are associated with COPD severity in a Mexican Mestizo population. COPD (2016) 13(5):610–5. doi:10.3109/15412555.2016.1161017
363. Lock-Johansson S, Vestbo J, Sorensen G. Surfactant protein D, club cell protein 16, pulmonary and activation-regulated chemokine, C-reactive protein, and fibrinogen biomarker variation in chronic obstructive lung disease. Respir Res (2014) 15:147. doi:10.1186/s12931-014-0147-5
364. Obeidat M, Li X, Burgess S, Zhou G, Fishbane N, Hansel NN, et al. Surfactant protein D is a causal risk factor for COPD: results of Mendelian randomisation. Eur Respir J (2017) 50:1700657. doi:10.1183/13993003.00657-2017
365. Honda Y, Takahashi H, Kuroki Y, Akino T, Abe S. Decreased contents of surfactant proteins A and D in BAL fluids of healthy smokers. Chest (1996) 109:1006–9. doi:10.1378/chest.109.4.1006
366. Zou W, Liu S, Hu J, Sheng Q, He F, Li B, et al. Nicotine reduces the levels of surfactant proteins A and D via Wnt/beta-catenin and PKC signaling in human airway epithelial cells. Respir Physiol Neurobiol (2016) 221:1–10. doi:10.1016/j.resp.2015.10.004
367. Haddam N, Samira S, Dumont X, Taleb A, Haufroid V, Lison D, et al. Lung epithelium injury biomarkers in workers exposed to sulphur dioxide in a non-ferrous smelter. Biomarkers (2009) 14:292–8. doi:10.1080/13547500902989088
368. Stockfelt L, Sallsten G, Olin AC, Almerud P, Samuelsson L, Johannesson S, et al. Effects on airways of short-term exposure to two kinds of wood smoke in a chamber study of healthy humans. Inhal Toxicol (2012) 24:47–59. doi:10.3109/08958378.2011.633281
369. Kobayashi H, Kanoh S, Motoyoshi K. Serum surfactant protein-A, but not surfactant protein-D or KL-6, can predict preclinical lung damage induced by smoking. Biomarkers (2008) 13:385–92. doi:10.1080/13547500801903651
370. Ozyurek BA, Ulasli SS, Bozbas SS, Bayraktar N, Akcay S. Value of serum and induced sputum surfactant protein-D in chronic obstructive pulmonary disease. Multidiscip Respir Med (2013) 8:36. doi:10.1186/2049-6958-8-36
371. Ishikawa N, Hattori N, Kohno N, Kobayashi A, Hayamizu T, Johnson M. Airway inflammation in Japanese COPD patients compared with smoking and nonsmoking controls. Int J Chron Obstruct Pulmon Dis (2015) 10:185–92. doi:10.2147/COPD.S74557
372. Duffy S, Weir M, Criner GJ. The complex challenge of chronic obstructive pulmonary disease. Lancet Respir Med (2015) 3:917–9. doi:10.1016/S2213-2600(15)00480-4
373. Um SJ, Lam S, Coxson H, Man SF, Sin DD. Budesonide/formoterol enhances the expression of pro surfactant protein-B in lungs of COPD patients. PLoS One (2013) 8:e83881. doi:10.1371/journal.pone.0083881
374. Liu W, Ju CR, Chen RC, Liu ZG. Role of serum and induced sputum surfactant protein D in predicting the response to treatment in chronic obstructive pulmonary disease. Exp Ther Med (2014) 8:1313–7. doi:10.3892/etm.2014.1865
375. Ju CR, Liu W, Chen RC. Serum surfactant protein D: biomarker of chronic obstructive pulmonary disease. Dis Markers (2012) 32:281–7. doi:10.3233/DMA-2011-0887
376. Sin DD, Leung R, Gan WQ, Man SP. Circulating surfactant protein D as a potential lung-specific biomarker of health outcomes in COPD: a pilot study. BMC Pulm Med (2007) 7:13. doi:10.1186/1471-2466-7-13
377. Tkacova R, McWilliams A, Lam S, Sin DD. Integrating lung and plasma expression of pneumo-proteins in developing biomarkers in COPD: a case study of surfactant protein D. Med Sci Monit (2010) 16:CR540–4.
378. El-Deek SE, Makhlouf HA, Saleem TH, Mandour MA, Mohamed NA. Surfactant protein D, soluble intercellular adhesion molecule-1 and high-sensitivity C-reactive protein as biomarkers of chronic obstructive pulmonary disease. Med Princ Pract (2013) 22:469–74. doi:10.1159/000349934
379. Zaky DSE, Naiem M, Eid HA, Adawy ZR, Abd-Elraheem SE, Mohamed ZAZ. Circulating surfactant protein D as a biomarker of severity in stable chronic pulmonary disease. Egypt J Chest Dis Tuberc (2014) 63:553–9. doi:10.1016/j.ejcdt.2014.03.011
380. Engstrom G, Lindberg C, Gerhardsson de Verdier M, Nihlen U, Anderson M, Svartengren M, et al. Blood biomarkers and measures of pulmonary function – a study from the Swedish twin registry. Respir Med (2012) 106:1250–7. doi:10.1016/j.rmed.2012.05.004
381. Lange P, Celli B, Agusti A, Boje Jensen G, Divo M, Faner R, et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med (2015) 373:111–22. doi:10.1056/NEJMoa1411532
382. Hoegh SV, Sorensen GL, Tornoe I, Lottenburger T, Ytting H, Nielsen HJ, et al. Long-term stability and circadian variation in circulating levels of surfactant protein D. Immunobiology (2010) 215:314–20. doi:10.1016/j.imbio.2009.05.001
383. Sin DD, Man SF, Marciniuk DD, Ford G, FitzGerald M, Wong E, et al. The effects of fluticasone with or without salmeterol on systemic biomarkers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2008) 177:1207–14. doi:10.1164/rccm.200709-1356OC
384. Shakoori TA, Sin DD, Ghafoor F, Bashir S, Bokhari SN. Serum surfactant protein D during acute exacerbations of chronic obstructive pulmonary disease. Dis Markers (2009) 27:287–94. doi:10.3233/DMA-2009-0674
385. Zien Alaabden A, Mohammad Y, Fahoum S. The role of serum surfactant protein D as a biomarker of exacerbation of chronic obstructive pulmonary disease. Qatar Med J (2015) 2015:18. doi:10.5339/qmj.2015.18
386. Johansson SL, Roberts NB, Schlosser A, Andersen CB, Carlsen J, Wulf-Johansson H, et al. Microfibrillar-associated protein 4: a potential biomarker of chronic obstructive pulmonary disease. Respir Med (2014) 108:1336–44. doi:10.1016/j.rmed.2014.06.003
387. Labonte LE, Bourbeau J, Daskalopoulou SS, Zhang M, Coulombe P, Garland K, et al. Club cell-16 and RelB as novel determinants of arterial stiffness in exacerbating COPD patients. PLoS One (2016) 11:e0149974. doi:10.1371/journal.pone.0149974
388. Hurst JR, Vestbo J, Anzueto A, Locantore N, Mullerova H, Tal-Singer R, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med (2010) 363:1128–38. doi:10.1056/NEJMoa0909883
389. Coxson HO, Dirksen A, Edwards LD, Yates JC, Agusti A, Bakke P, et al. The presence and progression of emphysema in COPD as determined by CT scanning and biomarker expression: a prospective analysis from the ECLIPSE study. Lancet Respir Med (2013) 1:129–36. doi:10.1016/S2213-2600(13)70006-7
390. Kokuho N, Ishii T, Kamio K, Hayashi H, Kurahara M, Hattori K, et al. Diagnostic values for club cell secretory protein (CC16) in serum of patients of combined pulmonary fibrosis and emphysema. COPD (2015) 12:347–54. doi:10.3109/15412555.2014.948994
391. Nakano M, Omae K, Uchida K, Michikawa T, Yoshioka N, Hirata M, et al. Five-year cohort study: emphysematous progression of indium-exposed workers. Chest (2014) 146:1166–75. doi:10.1378/chest.13-2484
392. Vestbo J, Agusti A, Wouters EF, Bakke P, Calverley PM, Celli B, et al. Should we view chronic obstructive pulmonary disease differently after ECLIPSE? A clinical perspective from the study team. Am J Respir Crit Care Med (2014) 189:1022–30. doi:10.1164/rccm.201311-2006PP
393. Rennard SI, Locantore N, Delafont B, Tal-Singer R, Silverman EK, Vestbo J, et al. Identification of five chronic obstructive pulmonary disease subgroups with different prognoses in the ECLIPSE cohort using cluster analysis. Ann Am Thorac Soc (2015) 12:303–12. doi:10.1513/AnnalsATS.201403-125OC
394. Zemans RL, Jacobson S, Keene J, Kechris K, Miller BE, Tal-Singer R, et al. Multiple biomarkers predict disease severity, progression and mortality in COPD. Respir Res (2017) 18:117. doi:10.1186/s12931-017-0597-7
395. Groves AM, Gow AJ, Massa CB, Hall L, Laskin JD, Laskin DL. Age-related increases in ozone-induced injury and altered pulmonary mechanics in mice with progressive lung inflammation. Am J Physiol Lung Cell Mol Physiol (2013) 305:L555–68. doi:10.1152/ajplung.00027.2013
396. Ochs M, Knudsen L, Allen L, Stumbaugh A, Levitt S, Nyengaard JR, et al. GM-CSF mediates alveolar epithelial type II cell changes but not emphysema-like pathology in SP-D deficient mice. Am J Physiol Lung Cell Mol Physiol (2004) 287(6):L1333–41. doi:10.1152/ajplung.00137.2004
397. Knudsen L, Atochina-Vasserman EN, Massa CB, Birkelbach B, Guo CJ, Scott P, et al. The role of inducible nitric oxide synthase for interstitial remodeling of alveolar septa in surfactant protein D-deficient mice. Am J Physiol Lung Cell Mol Physiol (2015) 309:L959–69. doi:10.1152/ajplung.00017.2015
398. Schneider JP, Arkenau M, Knudsen L, Wedekind D, Ochs M. Lung remodeling in aging surfactant protein D deficient mice. Ann Anat (2017) 211:158–75. doi:10.1016/j.aanat.2017.01.013
399. Knudsen L, Wucherpfennig K, Mackay RM, Townsend P, Muhlfeld C, Richter J, et al. A recombinant fragment of human surfactant protein D lacking the short collagen-like stalk fails to correct morphological alterations in lungs of SP-D deficient mice. Anat Rec (Hoboken) (2009) 292:183–9. doi:10.1002/ar.20830
400. Pilecki B, Wulf-Johansson H, Støttrup C, Jørgensen PT, Schlosser A, Hansen S, et al. Surfactant protein D deficiency aggravates cigarette smoke-induced inflammation. Abstract. Scand J Immunol (2016) 83:375–6. doi:10.1111/sji.12431
401. Haanes KA, Kruse LS, Wulf-Johansson H, Stottrup CC, Sorensen GL, Edvinsson L. Contractile changes in the vasculature after subchronic smoking: a comparison between wild type and surfactant protein D knock-out mice. Nicotine Tob Res (2016) 18:642–6. doi:10.1093/ntr/ntv243
402. Kierstein S, Poulain FR, Cao Y, Grous M, Mathias R, Kierstein G, et al. Susceptibility to ozone-induced airway inflammation is associated with decreased levels of surfactant protein D. Respir Res (2006) 7:85. doi:10.1186/1465-9921-7-85
403. Mikura S, Wada H, Higaki M, Yasutake T, Ishii H, Kamiya S, et al. Erythromycin prevents the pulmonary inflammation induced by exposure to cigarette smoke. Transl Res (2011) 158:30–7. doi:10.1016/j.trsl.2011.03.001
404. Connor AJ, Laskin JD, Laskin DL. Ozone-induced lung injury and sterile inflammation. Role of toll-like receptor 4. Exp Mol Pathol (2012) 92:229–35. doi:10.1016/j.yexmp.2012.01.004
405. Horie M, Fukui H, Endoh S, Maru J, Miyauchi A, Shichiri M, et al. Comparison of acute oxidative stress on rat lung induced by nano and fine-scale, soluble and insoluble metal oxide particles: NiO and TiO2. Inhal Toxicol (2012) 24:391–400. doi:10.3109/08958378.2012.682321
406. Lee YS, Chen PW, Tsai PJ, Su SH, Liao PC. Proteomics analysis revealed changes in rat bronchoalveolar lavage fluid proteins associated with oil mist exposure. Proteomics (2006) 6:2236–50. doi:10.1002/pmic.200500347
407. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet (2011) 378:1015–26. doi:10.1016/S0140-6736(11)60988-4
408. Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol (2008) 8:183–92. doi:10.1038/nri2254
409. Groves AM, Gow AJ, Massa CB, Laskin JD, Laskin DL. Prolonged injury and altered lung function after ozone inhalation in mice with chronic lung inflammation. Am J Respir Cell Mol Biol (2012) 47:776–83. doi:10.1165/rcmb.2011-0433OC
410. Bridges JP, Davis HW, Damodarasamy M, Kuroki Y, Howles G, Hui DY, et al. Pulmonary surfactant proteins A and D are potent endogenous inhibitors of lipid peroxidation and oxidative cellular injury. J Biol Chem (2000) 275:38848–55. doi:10.1074/jbc.M005322200
411. Atochina EN, Beers MF, Hawgood S, Poulain F, Davis C, Fusaro T, et al. Surfactant protein-D, a mediator of innate lung immunity, alters the products of nitric oxide metabolism. Am J Respir Cell Mol Biol (2004) 30:271–9. doi:10.1165/rcmb.2003-0091OC
412. Atochina-Vasserman EN, Beers MF, Kadire H, Tomer Y, Inch A, Scott P, et al. Selective inhibition of inducible NO synthase activity in vivo reverses inflammatory abnormalities in surfactant protein D-deficient mice. J Immunol (2007) 179:8090–7. doi:10.4049/jimmunol.179.12.8090
413. Knudsen L, Atochina-Vasserman EN, Guo CJ, Scott PA, Haenni B, Beers MF, et al. NOS2 is critical to the development of emphysema in Sftpd deficient mice but does not affect surfactant homeostasis. PLoS One (2014) 9:e85722. doi:10.1371/journal.pone.0085722
414. Korfhagen TR, Sheftelyevich V, Burhans MS, Bruno MD, Ross GF, Wert SE, et al. Surfactant protein-D regulates surfactant phospholipid homeostasis in vivo. J Biol Chem (1998) 273:28438–43. doi:10.1074/jbc.273.43.28438
415. Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol (2002) 169:3978–86. doi:10.4049/jimmunol.169.7.3978
416. Ariestanti DM, Ando H, Hirose S, Nakamura N. Targeted disruption of Ig-Hepta/Gpr116 causes emphysema-like symptoms that are associated with alveolar macrophage activation. J Biol Chem (2015) 290:11032–40. doi:10.1074/jbc.M115.648311
417. Bridges JP, Ludwig MG, Mueller M, Kinzel B, Sato A, Xu Y, et al. Orphan G protein-coupled receptor GPR116 regulates pulmonary surfactant pool size. Am J Respir Cell Mol Biol (2013) 49:348–57. doi:10.1165/rcmb.2012-0439OC
418. Yang MY, Hilton MB, Seaman S, Haines DC, Nagashima K, Burks CM, et al. Essential regulation of lung surfactant homeostasis by the orphan G protein-coupled receptor GPR116. Cell Rep (2013) 3:1457–64. doi:10.1016/j.celrep.2013.04.019
419. Ryu JH, Myers JL, Swensen SJ. Bronchiolar disorders. Am J Respir Crit Care Med (2003) 168:1277–92. doi:10.1164/rccm.200301-053SO
420. Whitsett JA, Wert SE, Weaver TE. Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol (2015) 10:371–93. doi:10.1146/annurev-pathol-012513-104644
421. Hughes DA, Haslam PL. Effect of smoking on the lipid composition of lung lining fluid and relationship between immunostimulatory lipids, inflammatory cells and foamy macrophages in extrinsic allergic alveolitis. Eur Respir J (1990) 3:1128–39.
422. Kitamura T, Tanaka N, Watanabe J, Uchida K, Kanegasaki S, Yamada Y, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med (1999) 190:875–80. doi:10.1084/jem.190.6.875
423. Hoegh SV, Lindegaard HM, Sorensen GL, Hoj A, Bendixen C, Junker P, et al. Circulating surfactant protein D is decreased in early rheumatoid arthritis: a 1-year prospective study. Scand J Immunol (2008) 67:71–6. doi:10.1111/j.1365-3083.2007.02039.x
424. Hoegh SV, Voss A, Sorensen GL, Hoj A, Bendixen C, Junker P, et al. Circulating surfactant protein D is decreased in systemic lupus erythematosus. J Rheumatol (2009) 36:2449–53. doi:10.3899/jrheum.090069
425. Christensen AF, Sorensen GL, Horslev-Petersen K, Holmskov U, Lindegaard HM, Junker K, et al. Circulating surfactant protein-D is low and correlates negatively with systemic inflammation in early, untreated rheumatoid arthritis. Arthritis Res Ther (2010) 12:R39. doi:10.1186/ar2948
426. Christensen AF, Hoegh SV, Lottenburger T, Holmskov U, Tornoe I, Horslev-Petersen K, et al. Circadian rhythm and the influence of physical activity on circulating surfactant protein D in early and long-standing rheumatoid arthritis. Rheumatol Int (2011) 31:1617–23. doi:10.1007/s00296-010-1538-7
427. Fernández-Real JM, Valdes S, Manco M, Chico B, Botas P, Campo A, et al. Surfactant protein D, a marker of lung innate immunity, is positively associated with insulin sensitivity. Diabetes Care (2010) 33:847–53. doi:10.2337/dc09-0542
428. Tanaka M, Arimura Y, Goto A, Hosokawa M, Nagaishi K, Yamashita K, et al. Genetic variants in surfactant, pulmonary-associated protein D (SFTPD) and Japanese susceptibility to ulcerative colitis. Inflamm Bowel Dis (2009) 15:918–25. doi:10.1002/ibd.20936
429. Hill J, Heslop C, Man SF, Frohlich J, Connett JE, Anthonisen NR, et al. Circulating surfactant protein-D and the risk of cardiovascular morbidity and mortality. Eur Heart J (2011) 32:1918–25. doi:10.1093/eurheartj/ehr124
430. Doyle TJ, Patel AS, Hatabu H, Nishino M, Wu G, Osorio JC, et al. Detection of rheumatoid arthritis-interstitial lung disease is enhanced by serum biomarkers. Am J Respir Crit Care Med (2015) 191:1403–12. doi:10.1164/rccm.201411-1950OC
431. Curkendall SM, Lanes S, de Luise C, Stang MR, Jones JK, She D, et al. Chronic obstructive pulmonary disease severity and cardiovascular outcomes. Eur J Epidemiol (2006) 21:803–13. doi:10.1007/s10654-006-9066-1
432. Williams MC, Murchison JT, Edwards LD, Agusti A, Bakke P, Calverley PM, et al. Coronary artery calcification is increased in patients with COPD and associated with increased morbidity and mortality. Thorax (2014) 69:718–23. doi:10.1136/thoraxjnl-2012-203151
433. Gargiulo P, Banfi C, Ghilardi S, Magri D, Giovannardi M, Bonomi A, et al. Surfactant-derived proteins as markers of alveolar membrane damage in heart failure. PLoS One (2014) 9:e115030. doi:10.1371/journal.pone.0115030
434. Kati C, Alacam H, Duran L, Guzel A, Akdemir HU, Sisman B, et al. The effectiveness of the serum surfactant protein D (Sp-D) level to indicate lung injury in pulmonary embolism. Clin Lab (2014) 60:1457–64. doi:10.7754/Clin.Lab.2013.131009
435. Heimer SR, Evans DJ, Mun JJ, Stern ME, Fleiszig SM. Surfactant protein D contributes to ocular defense against Pseudomonas aeruginosa in a murine model of dry eye disease. PLoS One (2013) 8:e65797. doi:10.1371/journal.pone.0065797
436. Liu Z, Shi Q, Liu J, Abdel-Razek O, Xu Y, Cooney RN, et al. Innate immune molecule surfactant protein D attenuates sepsis-induced acute pancreatic injury through modulating apoptosis and NF-kappaB-mediated inflammation. Sci Rep (2015) 5:17798. doi:10.1038/srep17798
437. Liu J, Abdel-Razek O, Liu Z, Hu F, Zhou Q, Cooney RN, et al. Role of surfactant proteins A and D in sepsis-induced acute kidney injury. Shock (2015) 43:31–8. doi:10.1097/SHK.0000000000000270
438. Hu F, Ding G, Zhang Z, Gatto LA, Hawgood S, Poulain FR, et al. Innate immunity of surfactant proteins A and D in urinary tract infection with uropathogenic Escherichia coli. Innate Immun (2016) 22:9–20. doi:10.1177/1753425915609973
439. Murray E, Khamri W, Walker MM, Eggleton P, Moran AP, Ferris JA, et al. Expression of surfactant protein D in the human gastric mucosa and during Helicobacter pylori infection. Infect Immun (2002) 70:1481–7. doi:10.1128/IAI.70.3.1481-1487.2002
440. Khamri W, Worku ML, Anderson AE, Walker MM, Hawgood S, Reid KB, et al. Helicobacter infection in the surfactant protein D-deficient mouse. Helicobacter (2007) 12:112–23. doi:10.1111/j.1523-5378.2007.00480.x
441. Du X, Meng Q, Sharif A, Abdel-Razek OA, Zhang L, Wang G, et al. Surfactant proteins SP-A and SP-D ameliorate pneumonia severity and intestinal injury in a murine model of Staphylococcus aureus pneumonia. Shock (2016) 46(2):164–72. doi:10.1097/SHK.0000000000000587
442. Zhang L, Meng Q, Yepuri N, Wang G, Xi X, Cooney RN. Surfactant proteins-A and -D attenuate LPS-induced apoptosis in primary intestinal epithelial cells (IECs). Shock (2017) 49(1):90–98. doi:10.1371/journal.pone.0174441
443. Madhukaran SP, Koippallil Gopalakrishnan AR, Pandit H, Marri ED, Kouser L, Jamil K, et al. Expression of surfactant proteins SP-A and SP-D in murine decidua and immunomodulatory effects on decidual macrophages. Immunobiology (2016) 221:377–86. doi:10.1016/j.imbio.2015.09.019
444. Wulf-Johansson H, Thinggaard M, Tan Q, Johansson SL, Schlosser A, Christensen K, et al. Circulating surfactant protein D is associated to mortality in elderly women: a twin study. Immunobiology (2013) 218:712–7. doi:10.1016/j.imbio.2012.08.272
445. Nybo M, Andersen K, Sorensen GL, Lolk A, Kragh-Sorensen P, Holmskov U. Serum surfactant protein D is correlated to development of dementia and augmented mortality. Clin Immunol (2007) 123:333–7. doi:10.1016/j.clim.2007.03.001
446. Anthonisen NR, Connett JE, Enright PL, Manfreda J; Lung Health Study Research Group. Hospitalizations and mortality in the lung health study. Am J Respir Crit Care Med (2002) 166:333–9. doi:10.1164/rccm.2110093
447. Hansell AL, Walk JA, Soriano JB. What do chronic obstructive pulmonary disease patients die from? A multiple cause coding analysis. Eur Respir J (2003) 22:809–14. doi:10.1183/09031936.03.00031403
448. Sin DD, Wu L, Man SF. The relationship between reduced lung function and cardiovascular mortality: a population-based study and a systematic review of the literature. Chest (2005) 127:1952–9. doi:10.1378/chest.127.6.1952
449. Curkendall SM, DeLuise C, Jones JK, Lanes S, Stang MR, Goehring E Jr, et al. Cardiovascular disease in patients with chronic obstructive pulmonary disease, Saskatchewan Canada cardiovascular disease in COPD patients. Ann Epidemiol (2006) 16:63–70. doi:10.1016/j.annepidem.2005.04.008
450. Finkelstein J, Cha E, Scharf SM. Chronic obstructive pulmonary disease as an independent risk factor for cardiovascular morbidity. Int J Chron Obstruct Pulmon Dis (2009) 4:337–49. doi:10.2147/COPD.S6400
451. Fisher KA, Stefan MS, Darling C, Lessard D, Goldberg RJ. Impact of COPD on the mortality and treatment of patients hospitalized with acute decompensated heart failure: the Worcester Heart Failure study. Chest (2015) 147:637–45. doi:10.1378/chest.14-0607
452. Hu F, Zhong Q, Gong J, Qin Y, Cui L, Yuan H. Serum surfactant protein D is associated with atherosclerosis of the carotid artery in patients on maintenance hemodialysis. Clin Lab (2016) 62:97–104. doi:10.7754/Clin.Lab.2015.150536
453. Merck E, Gaillard C, Scuiller M, Scapini P, Cassatella MA, Trinchieri G, et al. Ligation of the FcR gamma chain-associated human osteoclast-associated receptor enhances the proinflammatory responses of human monocytes and neutrophils. J Immunol (2006) 176:3149–56. doi:10.4049/jimmunol.176.5.3149
454. Merck E, Gaillard C, Gorman DM, Montero-Julian F, Durand I, Zurawski SM, et al. OSCAR is an FcRgamma-associated receptor that is expressed by myeloid cells and is involved in antigen presentation and activation of human dendritic cells. Blood (2004) 104:1386–95. doi:10.1182/blood-2004-03-0850
455. Merck E, de Saint-Vis B, Scuiller M, Gaillard C, Caux C, Trinchieri G, et al. Fc receptor gamma-chain activation via hOSCAR induces survival and maturation of dendritic cells and modulates toll-like receptor responses. Blood (2005) 105:3623–32. doi:10.1182/blood-2004-07-2809
456. Goettsch C, Rauner M, Sinningen K, Helas S, Al-Fakhri N, Nemeth K, et al. The osteoclast-associated receptor (OSCAR) is a novel receptor regulated by oxidized low-density lipoprotein in human endothelial cells. Endocrinology (2011) 152:4915–26. doi:10.1210/en.2011-1282
457. Hirano Y, Choi A, Tsuruta M, Jaw JE, Oh Y, Ngan D, et al. Surfactant protein-D deficiency suppresses systemic inflammation and reduces atherosclerosis in ApoE knockout mice. Cardiovasc Res (2017) 113:1208–18. doi:10.1093/cvr/cvx067
458. Skaggs BJ, Hahn BH, McMahon M. Accelerated atherosclerosis in patients with SLE – mechanisms and management. Nat Rev Rheumatol (2012) 8:214–23. doi:10.1038/nrrheum.2012.14
459. Lazarus R, Sparrow D, Weiss ST. Baseline ventilatory function predicts the development of higher levels of fasting insulin and fasting insulin resistance index: the Normative Aging study. Eur Respir J (1998) 12:641–5. doi:10.1183/09031936.98.12030641
460. Agusti A, Calverley PM, Celli B, Coxson HO, Edwards LD, Lomas DA, et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res (2010) 11:122. doi:10.1186/1465-9921-11-122
461. Song Y, Klevak A, Manson JE, Buring JE, Liu S. Asthma, chronic obstructive pulmonary disease, and type 2 diabetes in the women’s health study. Diabetes Res Clin Pract (2010) 90:365–71. doi:10.1016/j.diabres.2010.09.010
462. Divo M, Cote C, de Torres JP, Casanova C, Marin JM, Pinto-Plata V, et al. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2012) 186:155–61. doi:10.1164/rccm.201201-0034OC
463. Wannamethee SG, Shaper AG, Rumley A, Sattar N, Whincup PH, Thomas MC, et al. Lung function and risk of type 2 diabetes and fatal and nonfatal major coronary heart disease events: possible associations with inflammation. Diabetes Care (2010) 33:1990–6. doi:10.2337/dc10-0324
464. Kinney GL, Black-Shinn JL, Wan ES, Make B, Regan E, Lutz S, et al. Pulmonary function reduction in diabetes with and without chronic obstructive pulmonary disease. Diabetes Care (2014) 37:389–95. doi:10.2337/dc13-1435
465. Fernández-Real JM, Pickup JC. Innate immunity, insulin resistance and type 2 diabetes. Trends Endocrinol Metab (2008) 19:10–6. doi:10.1016/j.tem.2007.10.004
466. Yeh HC, Punjabi NM, Wang NY, Pankow JS, Duncan BB, Brancati FL. Vital capacity as a predictor of incident type 2 diabetes: the atherosclerosis risk in communities study. Diabetes Care (2005) 28:1472–9. doi:10.2337/diacare.28.6.1472
467. Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature (2006) 444:881–7. doi:10.1038/nature05488
468. Yeh F, Dixon AE, Marion S, Schaefer C, Zhang Y, Best LG, et al. Obesity in adults is associated with reduced lung function in metabolic syndrome and diabetes: the strong heart study. Diabetes Care (2011) 34:2306–13. doi:10.2337/dc11-0682
469. Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol (2016) 12:15–28. doi:10.1038/nrendo.2015.189
470. Bottai M, Pistelli F, Di Pede F, Carrozzi L, Baldacci S, Matteelli G, et al. Longitudinal changes of body mass index, spirometry and diffusion in a general population. Eur Respir J (2002) 20:665–73. doi:10.1183/09031936.02.01282001
471. Lin WY, Yao CA, Wang HC, Huang KC. Impaired lung function is associated with obesity and metabolic syndrome in adults. Obesity (Silver Spring) (2006) 14:1654–61. doi:10.1038/oby.2006.190
472. Leone N, Courbon D, Thomas F, Bean K, Jego B, Leynaert B, et al. Lung function impairment and metabolic syndrome: the critical role of abdominal obesity. Am J Respir Crit Care Med (2009) 179:509–16. doi:10.1164/rccm.200807-1195OC
473. van Huisstede A, Cabezas MC, Birnie E, van de Geijn GJ, Rudolphus A, Mannaerts G, et al. Systemic inflammation and lung function impairment in morbidly obese subjects with the metabolic syndrome. J Obes (2013) 2013:131349. doi:10.1155/2013/131349
474. Naveed B, Weiden MD, Kwon S, Gracely EJ, Comfort AL, Ferrier N, et al. Metabolic syndrome biomarkers predict lung function impairment: a nested case-control study. Am J Respir Crit Care Med (2012) 185:392–9. doi:10.1164/rccm.201109-1672OC
475. López-Cano C, Lecube A, Garcia-Ramirez M, Munoz X, Sanchez E, Seminario A, et al. Serum surfactant protein D as a biomarker for measuring lung involvement in obese patients with type 2 diabetes. J Clin Endocrinol Metab (2017) 102:4109–16. doi:10.1210/jc.2017-00913
476. Sorensen GL, Hjelmborg JV, Leth-Larsen R, Schmidt V, Fenger M, Poulain F, et al. Surfactant protein D of the innate immune defence is inversely associated with human obesity and SP-D deficiency infers increased body weight in mice. Scand J Immunol (2006) 64:633–8. doi:10.1111/j.1365-3083.2006.01853.x
477. Stapleton RD, Dixon AE, Parsons PE, Ware LB, Suratt BT; NHLBI Acute Respiratory Distress Syndrome Network. The association between BMI and plasma cytokine levels in patients with acute lung injury. Chest (2010) 138:568–77. doi:10.1378/chest.10-0014
478. Stidsen JV, Khorooshi R, Rahbek MK, Kirketerp-Moller KL, Hansen PB, Bie P, et al. Surfactant protein D deficiency in mice is associated with hyperphagia, altered fat deposition, insulin resistance, and increased basal endotoxemia. PLoS One (2012) 7:e35066. doi:10.1371/journal.pone.0035066
479. DeSilva NS, Ofek I, Crouch EC. Interactions of surfactant protein D with fatty acids. Am J Respir Cell Mol Biol (2003) 29:757–70. doi:10.1165/rcmb.2003-0186OC
480. Layden BT, Durai V, Newman MV, Marinelarena AM, Ahn CW, Feng G, et al. Regulation of pancreatic islet gene expression in mouse islets by pregnancy. J Endocrinol (2010) 207:265–79. doi:10.1677/JOE-10-0298
481. Aye T, Toschi E, Sharma A, Sgroi D, Bonner-Weir S. Identification of markers for newly formed beta-cells in the perinatal period: a time of recognized beta-cell immaturity. J Histochem Cytochem (2010) 58:369–76. doi:10.1369/jhc.2009.954909
482. Trevino-Alanis M, Ventura-Juarez J, Hernandez-Pinero J, Nevarez-Garza A, Quintanar-Stephano A, Gonzalez-Pina A. Delayed lung maturation of foetus of diabetic mother rats develop with a diminish, but without changes in the proportion of type I and II pneumocytes, and decreased expression of protein D-associated surfactant factor. Anat Histol Embryol (2009) 38:169–76. doi:10.1111/j.1439-0264.2008.00902.x
483. Reading PC, Allison J, Crouch EC, Anders EM. Increased susceptibility of diabetic mice to influenza virus infection: compromise of collectin-mediated host defense of the lung by glucose? J Virol (1998) 72:6884–7.
484. Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol (2004) 4:469–78. doi:10.1038/nri1372
485. Bihlet AR, Karsdal MA, Bay-Jensen AC, Read S, Kristensen JH, Sand JM, et al. Clinical drug development using dynamic biomarkers to enable personalized health care in COPD. Chest (2015) 148:16–23. doi:10.1378/chest.15-0296
486. Jenkins RG, Simpson JK, Saini G, Bentley JH, Russell AM, Braybrooke R, et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir Med (2015) 3:462–72. doi:10.1016/S2213-2600(15)00048-X
487. Nagae H, Takahashi H, Kuroki Y, Honda Y, Nagata A, Ogasawara Y, et al. Enzyme-linked immunosorbent assay using F(ab’)2 fragment for the detection of human pulmonary surfactant protein D in sera. Clin Chim Acta (1997) 266:157–71. doi:10.1016/S0009-8981(97)00124-1
488. Nikolaidis NM, White MR, Allen K, Tripathi S, Qi L, McDonald B, et al. Mutations flanking the carbohydrate binding site of surfactant protein D confer antiviral activity for pandemic influenza A viruses. Am J Physiol Lung Cell Mol Physiol (2014) 306:L1036–44. doi:10.1152/ajplung.00035.2014
489. van Eijk M, Bruinsma L, Hartshorn KL, White MR, Rynkiewicz MJ, Seaton BA, et al. Introduction of N-linked glycans in the lectin domain of surfactant protein D: impact on interactions with influenza A viruses. J Biol Chem (2011) 286:20137–51. doi:10.1074/jbc.M111.224469
490. Reinhardt A, Wehle M, Geissner A, Crouch EC, Kang Y, Yang Y, et al. Structure binding relationship of human surfactant protein D and various lipopolysaccharide inner core structures. J Struct Biol (2016) 195:387–95. doi:10.1016/j.jsb.2016.06.019
491. Gram K, Yang S, Steiner M, Somani A, Hawgood S, Blazar BR, et al. Simultaneous absence of surfactant proteins A and D increases lung inflammation and injury after allogeneic HSCT in mice. Am J Physiol Lung Cell Mol Physiol (2009) 296:L167–75. doi:10.1152/ajplung.90253.2008
492. Thacker S, Moran A, Lionakis M, Mastrangelo MA, Halder T, del Pilar Huby M, et al. Restoration of lung surfactant protein D by IL-6 protects against secondary pneumonia following hemorrhagic shock. J Infect (2014) 68:231–41. doi:10.1016/j.jinf.2013.11.010
493. White J, Ikegami M. Surfactant Protein D for the Treatment Of Disorders Associated with Lung Injury. US20110189104. Cincinatti (OH) US (2011).
494. Clark H, Nadesalingam P, Reid KBM, Strong P. Recombinant Surfactant Protein D Compositions and Methods of Use Thereof. US20120010126. Southhampton GB (2012).
495. Hess G, Horsh A, Zdunek D. Surfactant Proteins B and D for Differential Diagnosis of Dyspnea. US20100261283. Basel (CH) (2010).
496. Panettieri RA, Beers MF, Haczku AF, Sims MW. Surfactant Protein D Is a Biomarker for Steroid Responsiveness in Asthma and Chronic Obstructive Pulmonary Disease. US201001143920. Pennsylvania (2010).
497. Zhang L, Zhang W, Xu J, Yang S. Use of Glycyrrhetinic Acid, Glycyrrhizic Acid and Related Compounds for Prevention and/or Treatment of Pulmonary Fibrosis. US20120053141. Gainesville FL US (2012).
498. Mahajan L, Madan T, Sarma PU, Kishore U. Human Lung Surfactant Protein, SP-D, Modulates Eosinophil Activation and Survival and Enhances Phagocytosis of Apoptotic Basophils. US20080255043. Delhi (In) (2008).
499. Shteyngart B. Intratracheal Administration of Lysozyme with Other Therapeutic Agents in the Prevention and Treatment of Respiratory Disorders. US 20050271645 A1. Brooklyn NY US (2005).
Keywords: surfactant protein D, respiratory distress syndrome, allergic asthma, chronic obstructive lung disease, atherosclerosis
Citation: Sorensen GL (2018) Surfactant Protein D in Respiratory and Non-Respiratory Diseases. Front. Med. 5:18. doi: 10.3389/fmed.2018.00018
Received: 09 October 2017; Accepted: 19 January 2018;
Published: 08 February 2018
Edited by:
Mehdi Mirsaeidi, University of Miami, United StatesReviewed by:
Uday Kishore, Brunel University London, United KingdomTaruna Madan, National Institute for Research in Reproductive Health, India
Copyright: © 2018 Sorensen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grith L. Sorensen, Z2xzb3JlbnNlbkBoZWFsdGguc2R1LmRr