 Ilse Sofia Daehn
Ilse Sofia Daehn- Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, The Charles Bronfman Institute for Personalized Medicine, New York City, NY, United States
Diabetic kidney disease (DKD) is one of the major causes of morbidity and mortality in diabetic patients and also the leading single cause of end-stage renal disease in the United States. A large proportion of diabetic patients develop DKD and others don’t, even with comparable blood glucose levels, indicating a significant genetic component of disease susceptibility. The glomerulus is the primary site of diabetic injury in the kidney, glomerular hypertrophy and podocyte depletion are glomerular hallmarks of progressive DKD, and the degree of podocyte loss correlates with severity of the disease. We know that chronic hyperglycemia contributes to both microvascular and macrovascular complications, as well as podocyte injury. We are beginning to understand the role of glomerular endothelial injury, as well as the involvement of reactive oxygen species and mitochondrial stress, which play a direct role in DKD and in other diabetic complications. There is, however, a gap in our knowledge that links genetic susceptibility to early molecular mechanisms and proteinuria in DKD. Emerging research that explores glomerular cell’s specific responses to diabetes and cell cross-talk will provide mechanistic clues that underlie DKD and provide novel avenues for therapeutic intervention.
Introduction
Diabetic kidney disease (DKD) is one of the major causes of morbidity and mortality in diabetic patients and also the leading single cause of end-stage renal disease (ESRD) in the United States (1) and now a global epidemic (2–4). There has been little progress in treating DKD since angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) were shown to delay progression of nephropathy over 20 years ago (5, 6). The current treatments for DKD are limited to hyperglycemic control, blood pressure control, ACE inhibitors, or renin–angiotensin system (RAS) blockade, which can delay the progression to ESRD; however, the absolute risk of renal and cardiovascular morbidity and mortality remains overwhelmingly high (7). Recent renal outcome trails with agents such as thiazolidinediones, DPP-4 inhibitors, bardoxolone, and sulodexide have been unsuccessful; however, exciting new data from trials with GLP1R agonists and SGLT2 inhibitors show beneficial renal effects in patients with type 2 diabetes mellitus (T2DM) (8, 9). Although these new therapies seem promising, they may not reduce occurrence of ESRD (9). Hence, there is urgency from the community for new effective therapies to halt or reverse disease progression.
The prevalence of ESRD is up to 10 times higher in people with diabetes and with much higher health expenditures compared with those without diabetes (7, 10). The complex pathogenesis of DKD is influenced by genetics and by the environment (11–14). The majority of patients with diabetes do not develop DKD despite similar levels of hyperglycemia (15), and this is also the case with experimental mouse models (16, 17). Initially, patients present with microalbuminuria [urinary albumin excretion rate (UAER) 30–300 mg/24 h], which can progress to macroalbuminuria (UAER ≥ 300 mg/24 h) and eventually ESRD (18). However, there is a large proportion of diabetic patients that have decreased renal function in absence of substantial proteinuria (19), suggesting that other mechanisms contribute to kidney damage adding to the complexity of DKD, as well as highlighting the need for better biomarker predictors of progressive kidney failure in this population. It is thought that this will come from a better understanding of the pathogenesis of DKD.
This review aims to highlight the emerging evidence of intra-glomerular cell cross-talk in DKD, with particular focus on endothelial–podocyte communications, redox, cellular targets, and potential opportunities for discovery of new targets for the prevention and treatment of progressive DKD.
Hyperglycemia and Glomerular Injury—First Hit
Hyperglycemia is the driving force for diabetic complications, and the glomerulus is the primary site of diabetic injury in the kidney. The early stage of DKD is characterized by glomerular hyperfiltration, hypertrophy, mesangial expansion with altered matrix composition, and thickening of the glomerular basement membrane (BM). Podocyte injury resulting in podocyte foot process effacement (20–22) and depletion is a hallmark of progressive DKD (23–25). Podocytes contribute to glomerular permeability with their exceptionally complex morphology that is the key to their physiological function. The development of proteinuria is associated with marked morphological changes in these cells, and this relationship has been described in patients with diabetes both with microalbuminuria and macroalbuminuria (26, 27). There is a correlation between diabetes effects on podocyte morphology and their pathophysiology, all principal features that clinically manifests as proteinuria. Podocytes are terminally differentiated visceral epithelial cells with a limited capacity to regenerate (21), thus the “podocyte depletion paradigm” correlates closely with the development of proteinuria and progressive glomerulosclerosis, and therefore podocytes have been studied extensively as key targets in the evolution of segmental sclerosis lesions.
Many research groups are actively investigating the pathomechanisms of podocyte injury and depletion in DKD and other glomerular diseases. In DKD, the glomeruli are exposed to various noxious stimuli such as high glucose, uric acid, fatty acids, growth factors, cytokines, and hormones, and all these have been associated with podocyte loss in experimental models of DKD. A handful of potential mechanisms for podocyte depletion in DKD have been suggested and discussed, including the induction epithelial-to-mesenchymal transition (EMT). During EMT, cell–cell and cell–extracellular matrix interactions are altered to release epithelial cells from the host tissue (28). This process involves reorganization of the cytoskeleton to enable migration, and an altered transcriptional program is induced to maintain these cells in a mesenchymal phenotype while losing their hallmark epithelial characteristics (28–30). Diabetes-induced podocyte EMT may be mediated through several molecular mechanisms such as TGF-β/Smad classic pathway, Wnt/β-catenin signaling pathway (31), integrins/integrin-linked kinase signaling pathway (32), mitogen-activated protein kinases (MAPKs) signaling pathway, Jagged/Notch signaling pathway (33), and NF-κB signaling pathway (34). Although it is still debatable, podocyte depletion may result from decreased podocyte adhesion in DKD, which is a potential consequence of EMT. Podocyte transition to a more mesenchymal nature could result in their detachment facilitated by mechanical forces and shear stress derived from filtrate flow through the filtration slits acting on the foot-processes challenging the attachment of podocytes to the GBM (35–37) and consequent impairment of renal filtration.
In DKD podocyte loss through cell death was first demonstrated by Susztak et al. (38), podocytes exposed to high glucose had increased reactive oxygen species (ROS) via NADPH oxidase resulting in apoptosis by activation of MAPK and the caspase-3 cascade in vitro. An increase in transferase-mediated dUTP nick-end labeling positive podocyte nuclei coincided with onset of hyperglycemia and with onset of albuminuria in both T1DM and T2DM models. Although apoptosis has been described as the major mechanism by which podocytes die in diabetes and progressive glomerular disease by our group and others (39–42), the topic of podocyte cell death, detachment, and loss in glomerular diseases has been discussed extensively elsewhere (43) and remains controversial. Whether the diabetic milieu effects on podocytes could promote podocyte detachment, i.e., preceded by any kind of podocyte cell death, such as apoptosis, necrosis, necroptosis, ferroptosis, podoptosis, and mitotic catastrophe (44–48), still remains under debate and not the focus of this review. The maintenance of the glomerular ultrafiltration barrier requires the interaction of all three glomerular cell types (49, 50) including a multidirectional cross-talk between podocytes, mesangial cells and endothelial cells (51–53), as well as parietal epithelial cells (54, 55). Understanding the key interactions between all the cells in the glomerulus during the development of DKD may provide us with an opportunity for identifying novel interventions that can either stop or reverse its progression.
Endothelial Dysfunction in DKD
Chronic hyperglycemia contributes to both diabetic microvascular and macrovascular complications of diabetes resulting in thrombotic microaneurysms, which are typical manifestations of endothelial dysfunction in the glomerulus. Glomerular endothelial cells are highly specialized cells with fenestrae and a charged luminal glycocalyx layer (56, 57), which contribute to the filtration barrier (58). Studies have shown that the severity of DKD is correlated with endothelial dysfunction in T1DM and T2DM (59, 60). DKD progression may be tied to increased plasma levels of endothelial cells derived von Willebrand factor, soluble vascular cell adhesion molecule-1, and soluble intercellular cell adhesion molecule-1 in patients (61, 62) and reduction of the endothelial glycocalyx (63, 64). However, the role of endothelial injury in the pathogenesis of DKD remains relatively unexplored.
In DKD-susceptible mouse strains, we have reported changes in the morphology and potential dysfunction of glomerular endocapillaries upon early onset of hyperglycemia by examining the overall glomerular 3D structure with scanning electron microscopy (42). Compared to control non-diabetic glomerular capillary vessels, 3-week diabetic mice showed distinct morphological changes including a significant decrease in number of fenestrae, there were cytoplasmic protrusions, and increased in number of non-fenestrated ridges. Interestingly, podocyte foot-processes widening or effacement were not evident at this time point, suggesting to us that endothelial cell injury precedes podocyte foot-processes effacement and this was an important feature of early DKD with potential consequences in the progression of disease in genetically susceptible mice and potentially in humans.
Endothelial dysfunction could result from hyperglycemia increased ROS, uric acid, and endothelial nitric oxide synthase (eNOS) inactivation, which reduces nitric oxide (NO) levels (65). Endothelial NO plays a critical role in maintaining renal blood flow, regulating glomerular filtration rate, as well as salt and fluid homeostasis; hence, the inactivation of NO particularly through increased oxidative stress has been linked to altered kidney and vascular function [for review, see Ref. (66)]. A vital role for eNOS-derived NO in the pathogenesis of DKD was illustrated in studies comparing diabetic control wild-type and diabetic eNOS knockout (KO) on the C57BL6 DKD resistant background (67). eNOS KO developed overt albuminuria, hypertension, and glomerular mesangiolysis, injured endothelial morphology, thickened glomerular BM, and focal foot process effacement, whereas control mice did not. By correcting changes in eNOS dimerization and phosphorylation, Cheng et al. were able to reduced albuminuria, GBM thickness, and urinary excretion of oxidative stress markers; moreover, it did not affect hyperfiltration or mesangial expansion (68). Along with this observation, diabetic mice with endothelial dysfunction induced by genetic deficiency of eNOS get podocyte injury with heavy albuminuria (69) and maintenance of endothelial levels of the essential eNOS cofactor tetrahydrobiopterin was shown to ameliorate DKD in these mice (70). Finally, the levels of asymmetric dimethylarginin, an endogenous inhibitor of endothelial eNOS, contribute to increased risk of progressive DKD in patients with T1DM (71–73) and eNOS polymorphisms that result in reduced enzyme function have been associated with more advanced diabetic nephropathy (74, 75). Altogether, these reports support the role of endothelial dysfunction in the pathogenesis and progression of DKD.
The ROS Paradox in DKD
Oxidative stress represents overproduction of ROS relative to antioxidant defenses (76). An imbalance in the oxidation–reduction state has been identified as a major culprit for diabetic complications (77, 78), including DKD (79–81). Increased ROS production triggers renal fibrosis and inflammation and causes significant tissue damage by promoting lipid peroxidation, protein and DNA damage, and mitochondrial dysfunction. In the kidney, ROS are generated by xanthine oxidase, cytochrome P450 systems, uncoupled NO synthase, mitochondrial respiratory chain, and NOXs. Among NOX isoforms, NOX1 has been shown to modulate the p38/p27(Kip1) signaling pathway via PKC activation and promotes premature senescence in early stage DKD (82). NOX4- and NOX5-derived ROS are important in glomerular and podocyte injury; however, their regulation and function relative to DKD remains unclear (83). Studies by Babelova et al. (84) demonstrated an increase in albuminuria in diabetic mice with genetic deletion of NOX4, although they reported prominent expression of NOX4 to be in tubular cells. Nox4 was also shown to have anti-apoptotic properties protecting kidneys after ischemia reperfusion injury (85). On the other hand, a study using podocyte-specific inducible NOX4 transgenic mice showed glomerular injury characteristic of DKD (86); hence, the context of NOX4 may be important. You et al. and others (87–89) have shown that NOX1/NOX4 inhibition has renoprotective effects in experimental DKD. More research is needed to understand the roles of NOXs in DKD.
The mitochondria are the main cellular source of energy and of ROS production within cells in response to metabolic demands and/or cellular stress signals (90) and are (among other roles) the central regulators of the intrinsic apoptosis pathway. In fact, abnormalities of glomerular podocyte function linked to mitochondrial disorders are involved in the etiology of glomerular pathology with nephrotic syndrome (91–93). An excessive production of mtROS can damage macromolecules within mitochondria, including lipids, proteins, and mitochondrial DNA (mtDNA) (94), this in turn impairs the synthesis of components of the ETC, as well as reduce the capacity to generate ATP, and potentiate further ROS production (vicious cycle: oxidative stress) resulting in mitochondrial dysfunction (95, 96). mtDNA is a vulnerable oxidation target with a higher mutation rate than nuclear DNA due to vicinity to ETC, a higher rate of replication, lack of protection from histones, and DNA repair mechanisms (96–98). An assessment of metabolites from stage 3 to 4 CKD patients versus controls demonstrated that urinary excretion of citric acid cycle metabolites and of genes regulating these metabolites were reduced in patients with CKD, supporting the emerging view of CKD as a state of mitochondrial dysfunction (99).
Increased mtROS and mitochondrial dysfunction could play a critical role in the pathogenesis of DKD (78). Mitochondrial stress in DKD has been demonstrated to be linked to induction Rho-associated coiled-coil-containing protein kinase1 expression and thus resulting in mitochondria fission by promoting phosphorylation and translocation of dynamin-related protein-1 into the mitochondria (100). This study suggests a critical role for hyperglycemia induced mtROS production, mitochondrial fission, and consequent mitochondrial dysfunction in podocytes and endothelial cells (100). By contrast, superoxide production has been shown to be reduced in the kidneys of a STZ-induced T1DM, and using multiple in vivo and ex vivo approaches the authors did not observe evidence for enhanced mitochondrial superoxide production in diabetic kidneys. The authors showed evidence of mitochondrial dysfunction, reduced overall mitochondrial content, decreased biogenesis, and increased total urinary ROS (101, 102). This contrasting view to the widely held unifying theory that suggests high glucose drives overproduction of superoxide from mitochondria in diabetic complications, encompasses “mitochondrial hormesis,” supported by the reduction of AMPK, sirtuins, and PGC1α pathways and increased mTOR, reduced mitochondrial ROS, reduced biogenesis, and disease progression (103). This brings up an important consideration, as ROS moieties are essential for specific intracellular signaling pathways that regulate physiological processes and represent normal mitochondrial function. Increased ROS act as second messengers essential in vascular homeostasis (104) and significantly contribute to the immune response (105, 106). Therefore, scavenging all ROS by antioxidants may be dangerous, as we have learned from failures of several large clinical trials using antioxidant therapies on DKD patients (107, 108). With these considerations, Galvan and colleagues developed a two-photon imaging method using a genetically encoded redox biosensor to monitor the dynamic mitochondrial redox state of mice kidneys in real-time. Using this approach, the authors confirmed an increased production of mtROS in the kidneys of diabetic mice (109). As we evolve our understanding of ROS chemistry and cell biology, it becomes clearer that different ROS and their location can have distinct and important roles in the kidney.
Our recently published data is in accordance with mitochondrial-dependent glomerular pathomechanisms in DKD susceptibility (42). We were guided initially by unbiased transcriptomic profiling of glomeruli after onset of diabetes in DKD-susceptible DBA/2J and -resistant C57BL/6J inbred mouse strains. Among differentially expressed transcripts in diabetic DBA/2J mice, genes with well-established functional roles in “oxidative phosphorylation” and “mitochondrial function” were most significantly enriched and antioxidants were downregulated in diabetic DBA/2J susceptible mice, suggesting a redox imbalance in glomeruli. The increase in mitochondria oxidative stress resulted in mtDNA instability and damage, and we discovered that the damage accumulated exclusively in the glomerular endothelial cells, resulting in decreased NO and overall endothelial dysfunction (42), this occurred despite the fact that ROS and metabolic changes may be increased in podocytes (22) as well as other glomerular cells upon chronic exposure to a diabetic milieu. Moreover, prevention of mtROS with a mitochondrial-specific scavenger prevented podocyte loss, albuminuria, and glomerulosclerosis (42). Another study evaluated the mRNA profile of glomeruli and isolated podocytes from diabetic mice kidneys (110). The authors demonstrated distinct upregulated pathways involving mitochondrial function and oxidative stress in the endothelium compartment, in isolated glomeruli. By contrast, isolated podocytes showed changes in the regulation of actin cytoskeleton-related genes as major pathways affected in diabetic mice (110). We showed that increased oxidative damage in glomerular endothelial cells was also detected in human subjects diagnosed with DKD, and oxidized DNA lesion excretion in the urine (8-OHdG) was significantly increased only in patients with progressive DKD (42). In accordance with our data, higher plasma concentrations of 8-OHdG were found to be independently associated with increased risk of progression of kidney disease in T1DM (111). Altogether, there are convincing data showing that oxidative damage is context dependent in DKD and that the glomerular endothelium could be the weakest link under chronic stress, and suggests that podocyte depletion in DKD-susceptible mice could be contingent on endothelial cell mitochondrial dysfunction.
Second Hit Hypothesis Leading to Proteinuria is Context Dependent
In studying the highly complex nature of diabetes-mediated glomerular cell injury, conflicting results from in vivo and/or in vitro studies driven hypothesis have led some researchers to propose a “second hit hypothesis” to explain podocyte loss in experimental models of DKD and other glomerular diseases (47, 112, 113). Glomerular cells are tightly intertwined interdependently for proper function, with signal processings that must interpret the environment under normal and stress conditions. Podocytes control endothelial cell growth and survival via cross-talk of essential paracrine vascular endothelial growth factor alpha (VEGFA and VEGF-R) (114, 115). Cross-talk also exists between endothelial and mesangial cells (PDGF-B and PDGFR-β) and between podocytes and mesangial cells (CCL21 and CCR7) (116, 117). This bidirectional signal cross-talk enables cells to function effectively. However, in response to changes in the microenvironment (e.g., diabetes), a communication network could provide feedback that may enable cells to tune their signaling activity to organize cytoskeletal dynamics, metabolic output, etc. The molecular mechanisms for glomerular cell cross-talk and feedback regulation in proteinuric glomerular diseases remain poorly understood.
In most diseases causing glomerulosclerosis, transforming growth factor β (Tgfb) expression in podocytes is a stress response signal associated with segmental sclerosis and podocyte loss (118–120). In a model of FSGS, podocyte injury initiated by activating Tgfb signaling specifically in podocytes, we showed an increase in the release of endothelin-1 by podocytes, which acted on increased endothelin receptor type A (Ednra) on adjacent glomerular endothelial cells resulted in endothelial cell mitochondrial oxidative stress and endothelial cell dysfunction (121). Surprisingly, glomerular endothelial mitochondrial oxidative stress and dysfunction was absolutely essential for subsequent podocyte loss, illustrating a podocyte-to-endothelial-to-podocyte cross-talk (121). A similar stressed endothelial-to-podocyte cross-talk via mitochondrial oxidative stress in endothelial cells downstream from Edn-1/Ednra could also underlie segmental lesions in DKD and highlight a potential mechanism for the proven renoprotective activities of EDNRA inhibitors (42). Work from our laboratory and others have shown that Tgfb signaling in podocytes increases mitochondrial activity (respiration rate) (122, 123). Although Abe et al. (122) reported a concomitant increase in ROS production and mitochondrial membrane potential via and activation of mTOR, we did not detect ROS damage in podocytes (123). Podocytes are quite resilient with effective high level of constitutive autophagy (124–126) that could efficiently remove damaged organelles. Perhaps Tgfb signaling activation increases podocytes susceptibility to cross-talk messaging factors from neighboring endothelial cells with sustained injury that might represent a “second hit” for podocytes, leading to depletion and proteinuria in vivo. Recently, a study showed that secreted exosomes derived from high glucose-treated endothelial cells could mediate EMT and dysfunction of podocytes in a paracrine manner and activating canonical Wnt/β-catenin signaling (127). Interestingly, podocyte injury in a DKD resistant mice strain was detected when KLF2 expression was decreased specifically in endothelial cells of these mice (128). Figure 1 illustrates hypothetical cross-talk between glomerular endothelial cells and podocytes, ROS, ROS damage, mitochondrial dysfunction in DKD.
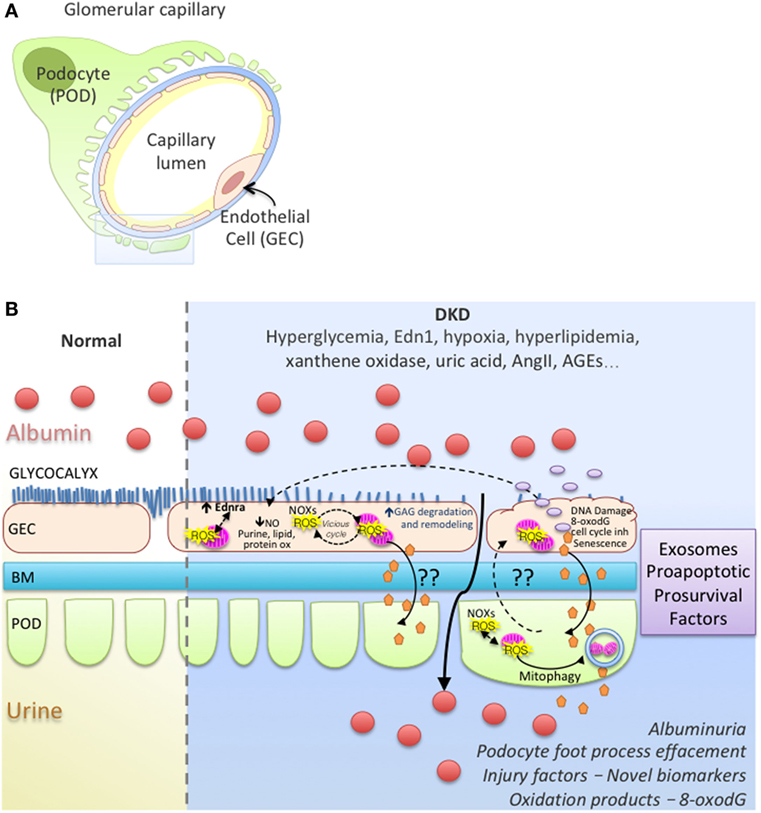
Figure 1. Working model of endothelial cell–podocyte cross-talk in diabetic kidney disease (DKD). (A) Glomerular capillary showing a podocyte (POD; Green), basement membrane (BM; blue), endothelial cell (GEC; pink). (B) Magnified insert from (A) illustrates normal and DKD environment; GEC injury with ROS vicious cycle, mitochondrial dysfunction, glycocalyx loss, endothelial remodeling and cross-talk with podocytes, albuminuria, podocyte foot process effacement in DKD.
A Need for Disruption in Our Approach to Treating DKD
The need for renal replacement, the mortality risk, and the financial burden are tremendously high among diabetics with kidney disease. Hence, halting or reversing disease progression will significantly impact patients living with this debilitating disease.
New treatments that aim to restore endothelial function could be an effective strategy for treating DKD as shown in a study that targeted cGMP; a key messenger for NO signaling. The study showed reduced progression of renal damage in the ZSF1 rat with diabetic nephropathy in the absence of significant hemodynamic effects (129). This approach is currently been examined clinically. Renoprotection in T2DM has been reported by preservation of mitochondrial function with CoQ10, which acts as both an acceptor of electrons and a ROS scavenger (130). Also, the mitochondria-targeted analog mitoubiquinone (131) has been shown to protect kidneys from diabetic injury by prevention of nuclear accumulation of pro- phospho- Smad2/3 and β-catenin or by Nrf2/PINK1-mediated mitophagy in tubules (132, 133). Preclinical studies with mitochondrial-targeted scavanger Szeto-Schiller peptide (Bendavia™) have demonstrated benefits in rodent models of DKD by improving mitochondrial bioenergetics (134), alleviating proteinuria, urinary 8-OHdG levels, glomerular hypertrophy, and accumulation of renal fibronectin and collagen IV (135). Clinical trials are underway to evaluate the cardiac and renal benefits using this targeted antioxidant mechanism in patients. Although mitochondria are a promising therapeutic target, the question of whether mitochondrial stabilization and mtROS inhibition can improve patient endpoints in large, randomized DKD clinical trials still remains.
We are at an exciting time, where as a community we understand that this multi-dimensional, multi-cellular condition requires interdisciplinary efforts and new technologies, that can potentially encompass and integrate the different dimensions of the glomerulus and the complexity of the diabetic milieu. These efforts will lead us to new insights into the pathogenesis of DKD and the discovery of novel therapeutic targets.
Author Contributions
ID drafted, prepared, and edited the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by National Institutes of Health grant R01DK097253 (to ID).
References
1. Saran R, Li Y, Robinson B, Abbott KC, Agodoa LY, Ayanian J, et al. US renal data system 2015 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis (2016) 67(3 Suppl 1):S1–305. doi:10.1053/j.ajkd.2015.12.014
2. Collins AJ, Kasiske B, Herzog C, Chavers B, Foley R, Gilbertson D, et al. Excerpts from the United States renal data system 2004 annual data report: atlas of end-stage renal disease in the United States. Am J Kidney Dis (2005) 45(1 Suppl 1):A5–7. doi:10.1053/j.ajkd.2004.10.009
3. Chan JC, Lau ES, Luk AO, Cheung KK, Kong AP, Yu LW, et al. Premature mortality and comorbidities in young-onset diabetes: a 7-year prospective analysis. Am J Med (2014) 127(7):616–24. doi:10.1016/j.amjmed.2014.03.018
4. Thomas MC, Cooper ME, Zimmet P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat Rev Nephrol (2016) 12(2):73–81. doi:10.1038/nrneph.2015.173
5. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The collaborative study group. N Engl J Med (1993) 329(20):1456–62.
6. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med (2001) 345(12):861–9. doi:10.1056/NEJMoa011161
7. Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al. IDF diabetes atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract (2017) 128:40–50. doi:10.1016/j.diabres.2017.03.024
8. Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med (2016) 375(4):323–34. doi:10.1056/NEJMoa1515920
9. Mann JFE, Orsted DD, Brown-Frandsen K, Marso SP, Poulter NR, Rasmussen S, et al. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med (2017) 377(9):839–48. doi:10.1056/NEJMoa1616011
10. Honeycutt AA, Segel JE, Zhuo X, Hoerger TJ, Imai K, Williams D. Medical costs of CKD in the Medicare population. J Am Soc Nephrol (2013) 24(9):1478–83. doi:10.1681/ASN.2012040392
11. Seaquist ER, Goetz FC, Rich S, Barbosa J. Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med (1989) 320(18):1161–5. doi:10.1056/NEJM198905043201801
12. Krolewski M, Eggers PW, Warram JH. Magnitude of end-stage renal disease in IDDM: a 35 year follow-up study. Kidney Int (1996) 50(6):2041–6. doi:10.1038/ki.1996.527
13. Knowler WC, Coresh J, Elston RC, Freedman BI, Iyengar SK, Kimmel PL, et al. The family investigation of nephropathy and diabetes (FIND): design and methods. J Diabetes Complications (2005) 19(1):1–9. doi:10.1016/j.jdiacomp.2003.12.007
14. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science (2005) 308(5729):1801–4. doi:10.1126/science.1106215
15. The Diabetes Control and Complications (DCCT) Research Group. Effect of intensive therapy on the development and progression of diabetic nephropathy in the diabetes control and complications trial. The diabetes control and complications (DCCT) research group. Kidney Int (1995) 47(6):1703–20.
16. Breyer MD, Bottinger E, Brosius FC III, Coffman TM, Harris RC, Heilig CW, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol (2005) 16(1):27–45. doi:10.1681/ASN.2004080648
17. Qi Z, Fujita H, Jin J, Davis LS, Wang Y, Fogo AB, et al. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes (2005) 54(9):2628–37. doi:10.2337/diabetes.54.9.2628
18. de Boer IH, Rue TC, Cleary PA, Lachin JM, Molitch ME, Steffes MW, et al. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: an analysis of the diabetes control and complications trial/epidemiology of diabetes interventions and complications cohort. Arch Intern Med (2011) 171(5):412–20. doi:10.1001/archinternmed.2011.16
19. Retnakaran R, Cull CA, Thorne KI, Adler AI, Holman RR, Group US. Risk factors for renal dysfunction in type 2 diabetes: U.K. prospective diabetes study 74. Diabetes (2006) 55(6):1832–9. doi:10.2337/db05-1620
20. Foster RR, Saleem MA, Mathieson PW, Bates DO, Harper SJ. Vascular endothelial growth factor and nephrin interact and reduce apoptosis in human podocytes. Am J Physiol Renal Physiol (2005) 288(1):F48–57. doi:10.1152/ajprenal.00146.2004
21. Petermann A, Floege J. Podocyte damage resulting in podocyturia: a potential diagnostic marker to assess glomerular disease activity. Nephron Clin Pract (2007) 106(2):c61–6. doi:10.1159/000101799
22. Stieger N, Worthmann K, Teng B, Engeli S, Das AM, Haller H, et al. Impact of high glucose and transforming growth factor-beta on bioenergetic profiles in podocytes. Metabolism (2012) 61(8):1073–86. doi:10.1016/j.metabol.2011.12.003
23. Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest (1997) 99(2):342–8. doi:10.1172/JCI119163
24. Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia (1999) 42(11):1341–4. doi:10.1007/s001250051447
25. Steffes MW, Schmidt D, McCrery R, Basgen JM, International Diabetic Nephropathy Study Group. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int (2001) 59(6):2104–13. doi:10.1046/j.1523-1755.2001.00725.x
26. Ellis EN, Steffes MW, Chavers B, Mauer SM. Observations of glomerular epithelial cell structure in patients with type I diabetes mellitus. Kidney Int (1987) 32(5):736–41. doi:10.1038/ki.1987.268
27. Bjorn SF, Bangstad HJ, Hanssen KF, Nyberg G, Walker JD, Viberti GC, et al. Glomerular epithelial foot processes and filtration slits in IDDM patients. Diabetologia (1995) 38(10):1197–204. doi:10.1007/BF00422369
28. Radisky DC. Epithelial-mesenchymal transition. J Cell Sci (2005) 118(Pt 19):4325–6. doi:10.1242/jcs.02552
29. Yamaguchi Y, Iwano M, Suzuki D, Nakatani K, Kimura K, Harada K, et al. Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis (2009) 54(4):653–64. doi:10.1053/j.ajkd.2009.05.009
30. Kumar Pasupulati A, Chitra PS, Reddy GB. Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy. Biomol Concepts (2016) 7(5–6):293–309. doi:10.1515/bmc-2016-0021
31. Xiao L, Wang M, Yang S, Liu F, Sun L. A glimpse of the pathogenetic mechanisms of Wnt/beta-catenin signaling in diabetic nephropathy. Biomed Res Int (2013) 2013:987064. doi:10.1155/2013/987064
32. Chen T, Zheng LY, Xiao W, Gui D, Wang X, Wang N. Emodin ameliorates high glucose induced-podocyte epithelial-mesenchymal transition in-vitro and in-vivo. Cell Physiol Biochem (2015) 35(4):1425–36. doi:10.1159/000373963
33. Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/smad and jagged1/notch signalling in epithelial-to-mesenchymal transition. EMBO J (2004) 23(5):1155–65. doi:10.1038/sj.emboj.7600069
34. Ying Q, Wu G. Molecular mechanisms involved in podocyte EMT and concomitant diabetic kidney diseases: an update. Ren Fail (2017) 39(1):474–83. doi:10.1080/0886022X.2017.1313164
35. Kriz W, Hahnel B, Hosser H, Rosener S, Waldherr R. Structural analysis of how podocytes detach from the glomerular basement membrane under hypertrophic stress. Front Endocrinol (2014) 5:207. doi:10.3389/fendo.2014.00207
36. Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol (2015) 26(2):258–69. doi:10.1681/ASN.2014030278
37. Kriz W, Lemley KV. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int (2017) 91(6):1283–6. doi:10.1016/j.kint.2017.02.032
38. Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy 2. Diabetes (2006) 55(1):225–33. doi:10.2337/diabetes.55.01.06.db05-0894
39. Bottinger EP. TGF-beta in renal injury and disease 4. Semin Nephrol (2007) 27(3):309–20. doi:10.1016/j.semnephrol.2007.02.009
40. Ziyadeh FN, Wolf G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev (2008) 4(1):39–45. doi:10.2174/157339908783502370
41. Jain S, De Petris L, Hoshi M, Akilesh S, Chatterjee R, Liapis H. Expression profiles of podocytes exposed to high glucose reveal new insights into early diabetic glomerulopathy. Lab Invest (2011) 91(4):488–98. doi:10.1038/labinvest.2010.188
42. Qi H, Casalena G, Shi S, Yu L, Ebefors K, Sun Y, et al. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes (2017) 66(3):763–78. doi:10.2337/db16-0695
43. Tharaux PL, Huber TB. How many ways can a podocyte die? Semin Nephrol (2012) 32(4):394–404. doi:10.1016/j.semnephrol.2012.06.011
44. Liapis H, Romagnani P, Anders HJ. New insights into the pathology of podocyte loss: mitotic catastrophe. Am J Pathol (2013) 183(5):1364–74. doi:10.1016/j.ajpath.2013.06.033
45. Thomasova D, Bruns HA, Kretschmer V, Ebrahim M, Romoli S, Liapis H, et al. Murine double minute-2 prevents p53-overactivation-related cell death (podoptosis) of podocytes. J Am Soc Nephrol (2015) 26(7):1513–23. doi:10.1681/ASN.2014040345
46. Kers J, Leemans JC, Linkermann A. An overview of pathways of regulated necrosis in acute kidney injury. Semin Nephrol (2016) 36(3):139–52. doi:10.1016/j.semnephrol.2016.03.002
47. Pedigo CE, Ducasa GM, Leclercq F, Sloan A, Mitrofanova A, Hashmi T, et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J Clin Invest (2016) 126(9):3336–50. doi:10.1172/JCI85939
48. Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Nino MD, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol (2017) 28(1):218–29. doi:10.1681/ASN.2015121376
49. Schlondorff D. Putting the glomerulus back together: per aspera ad astra (“a rough road leads to the stars”). Kidney Int (2014) 85(5):991–8. doi:10.1038/ki.2014.51
50. Dimke H, Maezawa Y, Quaggin SE. Crosstalk in glomerular injury and repair. Curr Opin Nephrol Hypertens (2015) 24(3):231–8. doi:10.1097/MNH.0000000000000117
51. Siddiqi FS, Advani A. Endothelial-podocyte crosstalk: the missing link between endothelial dysfunction and albuminuria in diabetes. Diabetes (2013) 62(11):3647–55. doi:10.2337/db13-0795
52. Daehn I. Shift in focus-to explore the role of the endothelium in kidney disease. HSOA J Nephrol Ren Ther (2016) 2(1):4. doi:10.24966/NRT-7313/100004
53. Ebefors K, Nystrom J. New insights into crosstalk in the kidney. Curr Opin Nephrol Hypertens (2017) 26(3):143–7. doi:10.1097/MNH.0000000000000310
54. Ohse T, Vaughan MR, Kopp JB, Krofft RD, Marshall CB, Chang AM, et al. De novo expression of podocyte proteins in parietal epithelial cells during experimental glomerular disease. Am J Physiol Renal Physiol (2010) 298(3):F702–11. doi:10.1152/ajprenal.00428.2009
55. Luque Y, Lenoir O, Bonnin P, Hardy L, Chipont A, Placier S, et al. Endothelial Epas1 deficiency is sufficient to promote parietal epithelial cell activation and FSGS in experimental hypertension. J Am Soc Nephrol (2017) 28(12):3563–78. doi:10.1681/ASN.2016090960
56. Ballermann BJ. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol (2007) 106(2):19–25. doi:10.1159/000101796
57. Fogo AB, Kon V. The glomerulus – a view from the inside – the endothelial cell. Int J Biochem Cell Biol (2010) 42(9):1388–97. doi:10.1016/j.biocel.2010.05.015
58. Haraldsson B, Nystrom J. The glomerular endothelium: new insights on function and structure. Curr Opin Nephrol Hypertens (2012) 21(3):258–63. doi:10.1097/MNH.0b013e3283522e7a
59. Persson F, Rossing P, Hovind P, Stehouwer CD, Schalkwijk C, Tarnow L, et al. Irbesartan treatment reduces biomarkers of inflammatory activity in patients with type 2 diabetes and microalbuminuria: an IRMA 2 substudy. Diabetes (2006) 55(12):3550–5. doi:10.2337/db06-0827
60. Toyoda M, Najafian B, Kim Y, Caramori ML, Mauer M. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes (2007) 56(8):2155–60. doi:10.2337/db07-0019
61. Lamberton RP, Goodman AD, Kassoff A, Rubin CL, Treble DH, Saba TM, et al. Von Willebrand factor (VIII R:Ag), fibronectin, and insulin-like growth factors I and II in diabetic retinopathy and nephropathy. Diabetes (1984) 33(2):125–9. doi:10.2337/diabetes.33.2.125
62. Persson F, Rossing P, Hovind P, Stehouwer CD, Schalkwijk CG, Tarnow L, et al. Endothelial dysfunction and inflammation predict development of diabetic nephropathy in the irbesartan in patients with type 2 diabetes and microalbuminuria (IRMA 2) study. Scand J Clin Lab Invest (2008) 68(8):731–8. doi:10.1080/00365510802187226
63. Singh A, Friden V, Dasgupta I, Foster RR, Welsh GI, Tooke JE, et al. High glucose causes dysfunction of the human glomerular endothelial glycocalyx. Am J Physiol Renal Physiol (2011) 300(1):F40–8. doi:10.1152/ajprenal.00103.2010
64. Boels MG, Avramut MC, Koudijs A, Dane MJ, Lee DH, van der Vlag J, et al. Atrasentan reduces albuminuria by restoring the glomerular endothelial glycocalyx barrier in diabetic nephropathy. Diabetes (2016) 65(8):2429–39. doi:10.2337/db15-1413
65. Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest (2001) 108(9):1341–8. doi:10.1172/JCI11235
66. Modlinger PS, Wilcox CS, Aslam S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin Nephrol (2004) 24(4):354–65. doi:10.1016/j.semnephrol.2004.04.007
67. Kanetsuna Y, Takahashi K, Nagata M, Gannon MA, Breyer MD, Harris RC, et al. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol (2007) 170(5):1473–84. doi:10.2353/ajpath.2007.060481
68. Cheng H, Wang H, Fan X, Paueksakon P, Harris RC. Improvement of endothelial nitric oxide synthase activity retards the progression of diabetic nephropathy in db/db mice. Kidney Int (2012) 82(11):1176–83. doi:10.1038/ki.2012.248
69. Yuen DA, Stead BE, Zhang Y, White KE, Kabir MG, Thai K, et al. eNOS deficiency predisposes podocytes to injury in diabetes. J Am Soc Nephrol (2012) 23(11):1810–23. doi:10.1681/ASN.2011121170
70. Kidokoro K, Satoh M, Channon KM, Yada T, Sasaki T, Kashihara N. Maintenance of endothelial guanosine triphosphate cyclohydrolase I ameliorates diabetic nephropathy. J Am Soc Nephrol (2013) 24(7):1139–50. doi:10.1681/ASN.2012080783
71. Lajer M, Tarnow L, Jorsal A, Teerlink T, Parving HH, Rossing P. Plasma concentration of asymmetric dimethylarginine (ADMA) predicts cardiovascular morbidity and mortality in type 1 diabetic patients with diabetic nephropathy. Diabetes Care (2008) 31(4):747–52. doi:10.2337/dc07-1762
72. Hanai K, Babazono T, Nyumura I, Toya K, Tanaka N, Tanaka M, et al. Asymmetric dimethylarginine is closely associated with the development and progression of nephropathy in patients with type 2 diabetes. Nephrol Dial Transplant (2009) 24(6):1884–8. doi:10.1093/ndt/gfn716
73. Shibata R, Ueda S, Yamagishi S, Kaida Y, Matsumoto Y, Fukami K, et al. Involvement of asymmetric dimethylarginine (ADMA) in tubulointerstitial ischaemia in the early phase of diabetic nephropathy. Nephrol Dial Transplant (2009) 24(4):1162–9. doi:10.1093/ndt/gfn630
74. Zanchi A, Moczulski DK, Hanna LS, Wantman M, Warram JH, Krolewski AS. Risk of advanced diabetic nephropathy in type 1 diabetes is associated with endothelial nitric oxide synthase gene polymorphism. Kidney Int (2000) 57(2):405–13. doi:10.1046/j.1523-1755.2000.00860.x
75. Dellamea BS, Pinto LC, Leitao CB, Santos KG, Canani LH. Endothelial nitric oxide synthase gene polymorphisms and risk of diabetic nephropathy: a systematic review and meta-analysis. BMC Med Genet (2014) 15:9. doi:10.1186/1471-2350-15-9
76. Oberley LW. Free radicals and diabetes. Free Radic Biol Med (1988) 5(2):113–24. doi:10.1016/0891-5849(88)90036-6
77. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature (2000) 404(6779):787–90. doi:10.1038/35008121
78. Brownlee M. Biochemistry and molecular cell biology of diabetic complications 26. Nature (2001) 414(6865):813–20. doi:10.1038/414813a
79. Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes (1991) 40(4):405–12. doi:10.2337/diab.40.4.405
80. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell (2005) 120(4):483–95. doi:10.1016/j.cell.2005.02.001
81. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res (2010) 107(9):1058–70. doi:10.1161/CIRCRESAHA.110.223545
82. Zhu K, Kakehi T, Matsumoto M, Iwata K, Ibi M, Ohshima Y, et al. NADPH oxidase NOX1 is involved in activation of protein kinase C and premature senescence in early stage diabetic kidney. Free Radic Biol Med (2015) 83:21–30. doi:10.1016/j.freeradbiomed.2015.02.009
83. Jha JC, Banal C, Chow BS, Cooper ME, Jandeleit-Dahm K. Diabetes and kidney disease: role of oxidative stress. Antioxid Redox Signal (2016) 25(12):657–84. doi:10.1089/ars.2016.6664
84. Babelova A, Avaniadi D, Jung O, Fork C, Beckmann J, Kosowski J, et al. Role of Nox4 in murine models of kidney disease. Free Radic Biol Med (2012) 53(4):842–53. doi:10.1016/j.freeradbiomed.2012.06.027
85. Nlandu-Khodo S, Dissard R, Hasler U, Schafer M, Pircher H, Jansen-Durr P, et al. NADPH oxidase 4 deficiency increases tubular cell death during acute ischemic reperfusion injury. Sci Rep (2016) 6:38598. doi:10.1038/srep38598
86. You YH, Quach T, Saito R, Pham J, Sharma K. Metabolomics reveals a key role for fumarate in mediating the effects of NADPH oxidase 4 in diabetic kidney disease. J Am Soc Nephrol (2016) 27(2):466–81. doi:10.1681/ASN.2015030302
87. Sedeek M, Gutsol A, Montezano AC, Burger D, Nguyen Dinh Cat A, Kennedy CR, et al. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of type 2 diabetes. Clin Sci (Lond) (2013) 124(3):191–202. doi:10.1042/CS20120330
88. Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol (2014) 25(6):1237–54. doi:10.1681/ASN.2013070810
89. Gorin Y, Cavaglieri RC, Khazim K, Lee DY, Bruno F, Thakur S, et al. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am J Physiol Renal Physiol (2015) 308(11):F1276–87. doi:10.1152/ajprenal.00396.2014
90. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J (2009) 417(1):1–13. doi:10.1042/BJ20081386
91. Guery B, Choukroun G, Noel LH, Clavel P, Rotig A, Lebon S, et al. The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA(Leu) gene mutation. J Am Soc Nephrol (2003) 14(8):2099–108. doi:10.1097/01.ASN.0000080180.51098.02
92. Lowik MM, Hol FA, Steenbergen EJ, Wetzels JF, van den Heuvel LP. Mitochondrial tRNALeu(UUR) mutation in a patient with steroid-resistant nephrotic syndrome and focal segmental glomerulosclerosis. Nephrol Dial Transplant (2005) 20(2):336–41. doi:10.1093/ndt/gfh546
93. Casalena G, Krick S, Daehn I, Yu L, Ju W, Shi S, et al. Mpv17 in mitochondria protects podocytes against mitochondrial dysfunction and apoptosis in vivo and in vitro. Am J Physiol Renal Physiol (2014) 306(11):F1372–80. doi:10.1152/ajprenal.00608.2013
94. Palmeira CM, Rolo AP, Berthiaume J, Bjork JA, Wallace KB. Hyperglycemia decreases mitochondrial function: the regulatory role of mitochondrial biogenesis. Toxicol Appl Pharmacol (2007) 225(2):214–20. doi:10.1016/j.taap.2007.07.015
95. Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) (2006) 5(2):145–52. doi:10.1016/j.dnarep.2005.03.002
96. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis (2007) 12(5):913–22. doi:10.1007/s10495-007-0756-2
97. Mandavilli BS, Santos JH, Van Houten B. Mitochondrial DNA repair and aging. Mutat Res (2002) 509(1–2):127–51. doi:10.1016/S0027-5107(02)00220-8
98. Daehn I, Brem R, Barkauskaite E, Karran P. 6-Thioguanine damages mitochondrial DNA and causes mitochondrial dysfunction in human cells. FEBS Lett (2011) 585(24):3941–6. doi:10.1016/j.febslet.2011.10.040
99. Hallan S, Afkarian M, Zelnick LR, Kestenbaum B, Sharma S, Saito R, et al. Metabolomics and gene expression analysis reveal down-regulation of the citric acid (TCA) cycle in non-diabetic CKD patients. EBioMedicine (2017) 26:68–77. doi:10.1016/j.ebiom.2017.10.027
100. Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab (2012) 15(2):186–200. doi:10.1016/j.cmet.2012.01.009
101. Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest (2013) 123(11):4888–99. doi:10.1172/JCI66218
102. Sharma K, Karl B, Mathew AV, Gangoiti JA, Wassel CL, Saito R, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol (2013) 24(11):1901–12. doi:10.1681/ASN.2013020126
103. Sharma K. Mitochondrial hormesis and diabetic complications. Diabetes (2015) 64(3):663–72. doi:10.2337/db14-0874
104. Vara D, Pula G. Reactive oxygen species: physiological roles in the regulation of vascular cells. Curr Mol Med (2014) 14(9):1103–25. doi:10.2174/1566524014666140603114010
105. Valacchi G, Cervellati C, Evelson P, Rahman I. Redox regulation of inflammatory processes. Int J Biochem Cell Biol (2016) 81(Pt B):234–5. doi:10.1016/j.biocel.2016.11.002
106. Lorenzen I, Mullen L, Bekeschus S, Hanschmann EM. Redox regulation of inflammatory processes is enzymatically controlled. Oxid Med Cell Longev (2017) 2017:8459402. doi:10.1155/2017/8459402
107. Lonn E, Yusuf S, Hoogwerf B, Pogue J, Yi Q, Zinman B, et al. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: results of the HOPE study and MICRO-HOPE substudy. Diabetes Care (2002) 25(11):1919–27. doi:10.2337/diacare.25.11.1919
108. de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med (2013) 369(26):2492–503. doi:10.1056/NEJMoa1306033
109. Galvan DL, Badal SS, Long J, Chang BH, Schumacker PT, Overbeek PA, et al. Real-time in vivo mitochondrial redox assessment confirms enhanced mitochondrial reactive oxygen species in diabetic nephropathy. Kidney Int (2017) 92(5):1282–7. doi:10.1016/j.kint.2017.05.015
110. Fu J, Wei C, Lee K, Zhang W, He W, Chuang P, et al. Comparison of glomerular and podocyte mRNA Profiles in streptozotocin-induced diabetes. J Am Soc Nephrol (2016) 27(4):1006–14. doi:10.1681/ASN.2015040421
111. Sanchez M, Roussel R, Hadjadj S, Moutairou A, Marre M, Velho G, et al. Plasma concentrations of 8-hydroxy-2'-deoxyguanosine and risk of kidney disease and death in individuals with type 1 diabetes. Diabetologia (2017) 61(4):977–84. doi:10.1007/s00125-017-4510-1
112. Wang L, Jirka G, Rosenberg PB, Buckley AF, Gomez JA, Fields TA, et al. Gq signaling causes glomerular injury by activating TRPC6. J Clin Invest (2015) 125(5):1913–26. doi:10.1172/JCI76767
113. Raij L, Tian R, Wong JS, He JC, Campbell KN. Podocyte injury: the role of proteinuria, urinary plasminogen, and oxidative stress. Am J Physiol Renal Physiol (2016) 311(6):F1308–17. doi:10.1152/ajprenal.00162.2016
114. Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol (2010) 21(10):1691–701. doi:10.1681/ASN.2010030295
115. Jeansson M, Gawlik A, Anderson G, Li C, Kerjaschki D, Henkelman M, et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest (2011) 121(6):2278–89. doi:10.1172/JCI46322
116. Vaughan MR, Quaggin SE. How do mesangial and endothelial cells form the glomerular tuft? J Am Soc Nephrol (2008) 19(1):24–33. doi:10.1681/ASN.2007040471
117. Schlondorff D, Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol (2009) 20(6):1179–87. doi:10.1681/ASN.2008050549
118. Schiffer M, von Gersdorff G, Bitzer M, Susztak K, Bottinger EP. Smad proteins and transforming growth factor-beta signaling. Kidney Int (2000) 58(Suppl 77):S45–52. doi:10.1046/j.1523-1755.2000.07708.x
119. Schiffer M, Bitzer M, Roberts ISD, Kopp JB, tenDijke P, Mundel P, et al. Apoptosis in podocytes induced by transforming growth factor-beta and Smad7. J Clin Invest (2001) 108(6):807–16. doi:10.1172/JCI200112367
120. Woroniecki RP, Schiffer M, Shaw AS, Kaskel FJ, Bottinger EP. Glomerular expression of transforming growth factor-beta (TGF-beta) isoforms in mice lacking CD2-associated protein 3. Pediatr Nephrol (2006) 21(3):333–8. doi:10.1007/s00467-005-2102-7
121. Daehn I, Casalena G, Zhang T, Shi S, Fenninger F, Barasch N, et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest (2014) 124(4):1608–21. doi:10.1172/JCI71195
122. Abe Y, Sakairi T, Beeson C, Kopp JB. TGF-beta1 stimulates mitochondrial oxidative phosphorylation and generation of reactive oxygen species in cultured mouse podocytes, mediated in part by the mTOR pathway. Am J Physiol Renal Physiol (2013) 305(10):F1477–90. doi:10.1152/ajprenal.00182.2013
123. Casalena G, Bottinger E, Daehn I. TGFbeta-induced actin cytoskeleton rearrangement in podocytes is associated with compensatory adaptation of mitochondrial energy metabolism. Nephron (2015) 131(4):278–84. doi:10.1159/000442051
124. Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest (2010) 120(4):1084–96. doi:10.1172/JCI39492
125. Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, et al. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol (2015) 26(5):1040–52. doi:10.1681/ASN.2013111202
126. Lenoir O, Jasiek M, Henique C, Guyonnet L, Hartleben B, Bork T, et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy (2015) 11(7):1130–45. doi:10.1080/15548627.2015.1049799
127. Wu X, Gao Y, Xu L, Dang W, Yan H, Zou D, et al. Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci Rep (2017) 7(1):9371. doi:10.1038/s41598-017-09907-6
128. Zhong F, Chen H, Wei C, Zhang W, Li Z, Jain MK, et al. Reduced Kruppel-like factor 2 expression may aggravate the endothelial injury of diabetic nephropathy. Kidney Int (2014) 87(2):382–95. doi:10.1038/ki.2014.286
129. Boustany-Kari CM, Harrison PC, Chen H, Lincoln KA, Qian HS, Clifford H, et al. A soluble guanylate cyclase activator inhibits the progression of diabetic nephropathy in the ZSF1 rat. J Pharmacol Exp Ther (2016) 356(3):712–9. doi:10.1124/jpet.115.230706
130. Sourris KC, Harcourt BE, Tang PH, Morley AL, Huynh K, Penfold SA, et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic Biol Med (2012) 52(3):716–23. doi:10.1016/j.freeradbiomed.2011.11.017
131. James AM, Sharpley MS, Manas AR, Frerman FE, Hirst J, Smith RA, et al. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem (2007) 282(20):14708–18. doi:10.1074/jbc.M611463200
132. Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, et al. Prevention of diabetic nephropathy in Ins2(+/)(-)(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem J (2010) 432(1):9–19. doi:10.1042/BJ20100308
133. Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol (2017) 11:297–311. doi:10.1016/j.redox.2016.12.022
134. Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol (2013) 24(8):1250–61. doi:10.1681/ASN.2012121216
Keywords: diabetic kidney disease, glomerulus, cross-talk, podocyte, endothelial cell, reactive oxygen species
Citation: Daehn IS (2018) Glomerular Endothelial Cell Stress and Cross-Talk With Podocytes in Early Diabetic Kidney Disease. Front. Med. 5:76. doi: 10.3389/fmed.2018.00076
Received: 12 December 2017; Accepted: 07 March 2018;
Published: 23 March 2018
Edited by:
Sandra Merscher, University of Miami, United StatesReviewed by:
Andrea Huwiler, Universität Bern, SwitzerlandTetsuhiro Tanaka, The University of Tokyo, Japan
Copyright: © 2018 Daehn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilse Sofia Daehn, aWxzZS5kYWVobkBtc3NtLmVkdQ==