 Xiaoming Lian
Xiaoming Lian Claudia Matthaeus
Claudia Matthaeus Mario Kaßmann
Mario Kaßmann Oliver Daumke2
Oliver Daumke2 Maik Gollasch
Maik Gollasch- 1Experimental and Clinical Research Center—A Joint Cooperation Between the Charité–University Medicine Berlin and the Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany
- 2Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany
- 3Medical Clinic for Nephrology and Internal Intensive Care, Berlin, Germany
Caveolae, flask-shaped cholesterol-, and glycosphingolipid-rich membrane microdomains, contain caveolin 1, 2, 3 and several structural proteins, in particular Cavin 1–4, EHD2, pacsin2, and dynamin 2. Caveolae participate in several physiological processes like lipid uptake, mechanosensitivity, or signaling events and are involved in pathophysiological changes in the cardiovascular system. They serve as a specific membrane platform for a diverse set of signaling molecules like endothelial nitric oxide synthase (eNOS), and further maintain vascular homeostasis. Lack of caveolins causes the complete loss of caveolae; induces vascular disorders, endothelial dysfunction, and impaired myogenic tone; and alters numerous cellular processes, which all contribute to an increased risk for hypertension. This brief review describes our current knowledge on caveolae in vasculature, with special focus on their pathophysiological role in hypertension.
Introduction
In the 1950s, 60- to 100-nm caves in the plasma membrane of the cell were first described using an electron microscope and named caveolae (1). It was later identified that most tissues and cell types contain caveolae, but the quantity varied (2). Because of the lack of experimental approaches and technologies, caveolae functions were mostly unclear until the 1990s. With the development of molecular techniques, the major membrane proteins of caveolae, caveolins, were dissected, and the secrets of function of these bulb-like caves were subsequently revealed. There are three caveolins, which are named caveolin 1 (Cav1), caveolin 2 (Cav2), and caveolin 3 (Cav3) (3). These proteins are encoded by different genes, CAV1, CAV2, and CAV3. Cav1 is expressed in most of the cell types and is essential for caveolae biogenesis; Cav3 is predominantly expressed in muscle cells (i.e., cardiac, striated skeletal, and smooth muscle cells) and is required for caveolar morphogenesis; Cav2 is generally expressed together with Cav1 in adipocytes, endothelial cells, pneumocytes, and fibroblasts, but appears to be dispensable for caveolar formation (4). Loss of either Cav1 or Cav3 results in a complete lack of caveolae (5, 6). A few years later, a series of additional proteins were identified, which play important roles in the formation of caveolae. Cavin 1–4 are essential for caveolae formation (7, 8) and function (8, 9). Together with caveolins, cavins preserve the stable coat around the bulb of caveolae (10, 11). In addition, Eps15 homology domain containing protein 2 (EHD2) is involved in mediating caveolar stabilization at the plasma membrane (Figure 1) (13); pacsin2 is a protein that is involved in the membrane bending to form caveolae as well as in mediating caveolar stabilization and scission (14, 15). Dynamin 2 is a caveolae neck-forming protein and plays a role in caveolar internalization and scission (16). Cav1 is transported from the Golgi complex to bulb from the plasma membrane and associates with cavin complex and pacsin2 to form Cav1-rich domains (11). Dynamin 2 is able to restrict the caveolar neck and EHD2 is located in this neck to stabilize the caveolae.
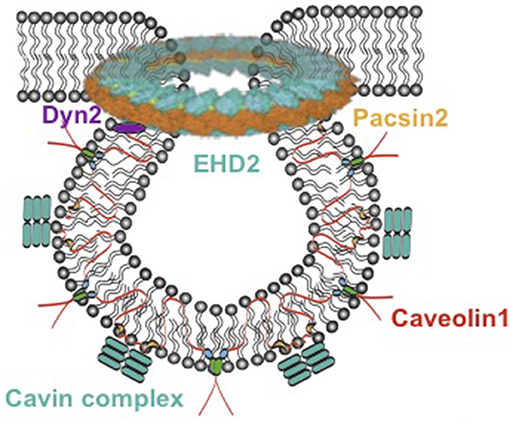
Figure 1. Model of caveolae. Caveolae model modified from Matthäus et al. (12). EHD2, Eps15 homology domain containing protein 2; Dyn2, Dynamin 2.
In accordance with major knowledge and understanding of caveolae structure, increasing interest has been focused on caveolae functions in physiology and pathophysiology. The physiological roles of caveolae vary depending on the organ systems and cell types examined (17). In the cardiovascular system, they contribute to maintaining a normal vascular tone and act as signal platforms (18–20). Signal transduction is critically important in the regulation of vascular homeostasis. Caveolae is reported to act as signaling platforms to a set of signaling molecules and receptors such as angiotensin II type 1 receptors (AT1R) (21), endothelial nitric oxide synthase (eNOS), several ion channels, and tyrosine kinase receptors (RTK) in vasculature (Figure 1) (19, 22, 23). The modification of caveolae structure affects its physiological function; e.g., alterations of lipid and proteins in caveolae induced by eicosapentaenoic acid (EPA) changed eNOS activation (24); lowering of cholesterol content in caveolae by simvastatin inhibited Akt1 serine–threonine kinase/protein kinase B (Akt/PKB) signaling pathway (25).
Loss of caveolae caused numerous vascular disorders, e.g., vascular smooth muscle hypertrophy (26), endothelial dysfunction (27), and impaired myogenic tone (28), which are all risk factors for the development of hypertension (29). A study pointed that renal hypertensive rats expressed a lower number of caveolae in aortic smooth muscle cells (SMCs) and endothelial cells, which induced an impaired effect of acetylcholine (30). Therapeutic targeting of caveolae in vascular diseases is also under study. For instance, a mutant cell-permeable scaffolding domain peptide called Cavnoxin, which can increase basal NO release in eNOS-expressing cells, has been recently identified to reduce vascular tone ex vivo and lower blood pressure in mice (31).
Here, we provide an overview of caveolae expression and function in the vasculature and discuss their putative role in pathophysiology of hypertension.
Vascular Endothelial Caveolae
Electron microscopic, biochemical, and immunochemical analyses demonstrated that caveolae are highly expressed in endothelial cells (32–34). Importantly, various signaling molecules and receptors of endothelial cells enriched in caveolae, in particular eNOS (31), G-proteins (35), protein kinase A (PKA) (36), protein kinase C (PKC) (37), and various receptors (38). They have been suggested to bind and be inhibited by Cav1 through its caveolin scaffolding domain (CSD), a conserved amphipathic region for caveolae formation as well as for regulating signal transduction (35, 39, 40).
Among these caveolae-localized signaling molecules, eNOS has attracted great attention for its critical effects on vascular homeostasis and blood pressure regulation (Figure 2) (41–43). Both biochemical analysis and immunogold labeling showed that a majority of eNOS resides in caveolae of endothelial cells (44, 45). Caveolae represent a predominant location of eNOS in endothelial cells (45, 46). The studies emphasize a critical role of endothelial caveolae in regulating activation of eNOS (Figure 2). In inactive endothelial cells, eNOS is shown to associate with Cav1, which inhibits calmodulin complex (CaM) binding to eNOS (47). This combination interrupts the electrons from NADPH to eNOS. M2-muscarinic acetylcholine receptor activation or other stimulation (e.g., increasing vascular flow and pressure) initiates an influx of Ca2+ that binds to calmodulin. In succession, eNOS dissociates from Cav1 and then combines to CaM. The flow of electrons from NADPH is therefore restored and consequently NO is produced (Figure 2) (48, 49). This NO generation results in the association of Cav1 and eNOS as previously shown, thus terminating the signal transduction (32). Furthermore, due to the increased cytosolic Ca2+ concentration, eNOS translocated from the cell membrane to the Golgi complex and is fully activated. After all, eNOS localizes back to the plasma membrane (35, 50, 51). Moreover, it has been demonstrated in vivo and in vitro that caveolin 1 is able to bind eNOS and therefore to inhibit the synthesis of NO (47, 52, 53). Increased expression of Cav1 is known to appear in patients with insulin resistance and type 2 diabetes (54), associated with impaired acetylcholine-induced NO production and vasodilation (55). Cav1 knockout mice show chronic and dramatic elevation in systemic NO levels and enhanced acetylcholine-induced arterial relaxation (56, 57). Conversely, the Bendhack group (58) reported that caveolae disassembled by methyl-β-cyclodextrin (mβcd) treatment cause an impaired acetylcholine-induced relaxation in the rat isolated aorta. This is in agreement with another study showing that caveolar disruption results in a decreased release of endothelial-derived NO in femoral arteries (59). This difference is supposed to be due to chronic Cav1 deficiency vs. acute caveolar disruption, as upon chronic lack of Cav1, an adjustment of an attenuated myogenic tone can be observed over a longer time period, i.e., active constriction induced by pressure, as compensation at the level of the vascular wall (60, 61).
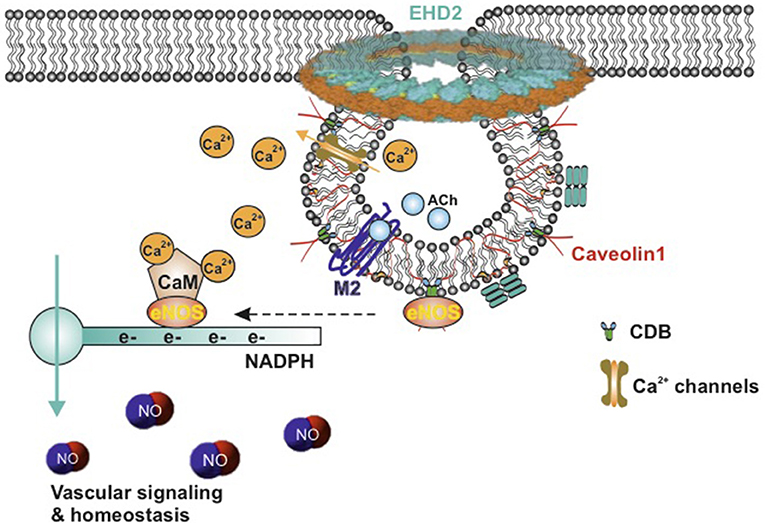
Figure 2. Schematic model of active eNOS in endothelial caveolae. Caveolae model modified from Matthäus et al. (12). M2-muscarinic acetylcholine receptor activation initiates an influx of Ca2+ that bind to calmodulin, eNOS dissociates from CBD of caveolin-1, CaM binds to eNOS, and the flow of electrons from NADPH to eNOS is restored, and then NO is produced. eNOS, endothelial nitric oxide synthase; NO, nitric oxide; CaM, calcium–calmodulin complex; CBD, caveolin scaffolding domain; ACh, acetylcholine; EHD2, Eps15 homology domain containing protein 2.
The elevation of systemic NO levels and impaired myogenic tone in Cav1 knockout mice would be expected to show a lower systemic blood pressure (6, 28). However, there are several studies reporting no difference in systolic and diastolic blood pressure between Cav1 knockout and wild-type mice (62–64). Of note, Wunderlich et al. (65) reported reduced systemic blood pressure whereas Pojoga et al. (66) reported an elevated systolic blood pressure in Cav1 knockout mice. Another study showed that Cav1 knockout mice have a slightly increased heart rate (60), suggesting possible compensation via increased baroreceptor reflex activation leading to increased sympathetic activity and neurogenic tone. Moreover, what should also be mentioned is that the deletion of Cav1 impaired the Mg2+ absorption and increases K+ excretion in renal distal convoluted tubule (67). The involvements of baroreceptor reflex and electrolytes disturbance in blood pressure regulation of Cav1 knockout mice make this scenario even more complicated.
Last but not least, endothelial transient receptor potential vanilloid receptor 4 (TRPV4) channels, where the NO-dependent vasodilation in arteries is triggered by Ca2+ entry (68), colocalize with Cav1 in the caveolae-enriched membrane fractions (69). These channels are potent Ca2+ influx channels (Figure 2). Cav1 knockout results in total absence of TRPV4-induced relaxation, suggesting that caveolae are essential for TRPV4 function and Ca2+ signaling in endothelial cells (68).
Vascular Smooth Muscle Caveolae
Caveolae are also abundant in SMCs, which are known to express Cav1–3 (70). Similar to the endothelial cells, caveolae are also important for SMC function, providing a platform for signal transductions through G-protein-coupled receptors and ion channels, therefore helping to maintain vascular homeostasis (18).
Angiotensin II (Ang II) working through AT1R is a well-known signaling pathway in vascular SMCs, which plays a great role in renal hypertension. Dysfunction of this pathway shows a predominant role in the pathophysiology of renal hypertension and several renal diseases (71, 72). In vascular SMCs, Ang II induces rapid translocation of a subset of AT1Rs to caveolae, where AT1Rs bind to Cav1 (73, 74) (Figure 3), which, in succession, activate the downstream signaling events, such as NADPH oxidase activation (cell migration and growth) (75), Ca2+ mobilization (arterial contractile responses) (6), epidermal growth factor receptor (EGFR) transactivation (tyrosine phosphorylation) (73, 76), and vascular SMC hypertrophy (77). Cav1 showed a beneficial effect in hypertensive mice. Cav1 protected against the development of systemic high blood pressure and enhanced resistance artery constriction through its binding to AT1R, which delays AT1R reactivation after Ang II stimulation (78). Ang II-induced hypertensive vascular remodeling is attenuated in Cav1 knockout mice (79).
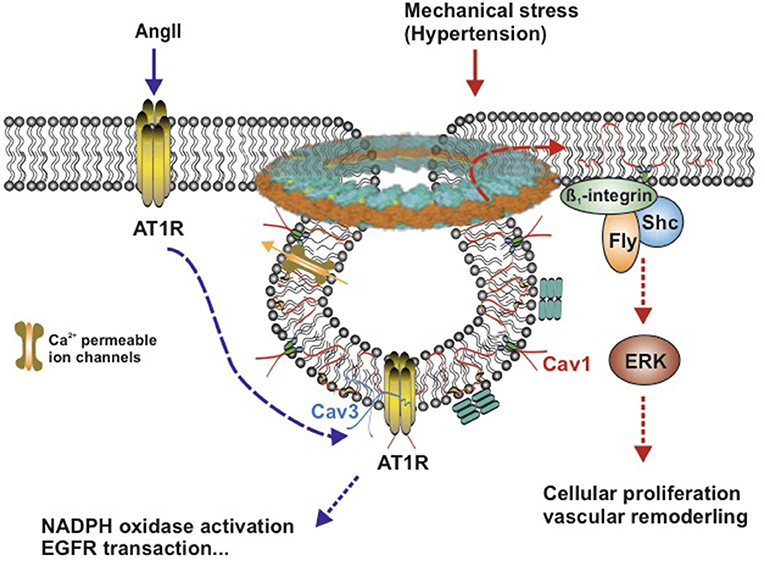
Figure 3. Schematic model of AngII-induced and mechanical stress-induced signaling pathway in vascular smooth muscle caveolae. Caveolae model modified from Matthäus et al. (12). Left: Ang II induces rapid translocation of AT1R to caveolae, AT1R and Caveolin 1 associate with each other. Caveolin 3 accompanies with AT1R. Right: Mechanical stress induces translocation of Caveolin 1 to non-caveolar sites and is associated with β1-integrins/Fyn/Shc to activate ERK signaling pathway. Cav1, Caveolin 1; Cav3, Caveolin 3; AngII, Angiotensin II; AT1R, AngII type1 receptor; ERK, extracellular signal-regulated kinase; Ca2+-permeable ion channels, such as TRPC, transient receptor potential channels, and EGFR, epidermal growth factor receptor.
Besides endogenous hormones, hypertension-induced mechanical stress contributes to the genesis of vascular hypertrophy and vascular remodeling, which can induce translocation of Cav1 in vascular SMCs (80). In response to chronic shear stress, Cav1 is translocated to non-caveolar sites and then combined to β1-integrins/Fyn/Shc, which mediates stretch-induced extracellular signal-regulated kinase (ERK) activation (80–82) (Figure 3). When exposed to acute mechanical stresses, caveolae disassemble completely and rapidly lead to the translocation of caveolins in the plasma membrane (<30 s), which flattens out caveolae in the plasma membrane to provide additional membrane and extra buffer tension (83). This reversible and rapid disassembly of caveolae provides a basic vascular response to an acute shear stress (Figure 3).
In vascular smooth muscle, most of the physiological processes are known to require Ca2+ (84). Ca2+ flux and intracellular Ca2+ level take part in numerous physiological processes of smooth muscle (85). Vascular caveolae are known to provide functional organization of ion channels, in particular calcium channels. Although multiple Ca2+ handling molecules [the plasma membrane Ca2+ pump (PMCA) (86), Na+-Ca2+ exchanger (NCX1) (87), T-type Cav3.2 channels (88), and transient receptor potential canonical channels (TRPCs) (89) are shown to localize or associate with Cav1 or Cav3 within caveolae (Figure 3); the exact function of most Ca2+ handling molecules in vascular SMCs regarding intracellular Ca2+ signaling remains elusive (90).
Myogenic tone, which serves to regulate blood flow and protect downstream vessels from pressure-induced damage, is largely dependent on an influx of extracellular Ca2+ via voltage-operated calcium channels (91). In large cerebral vessels in vitro, the myogenic tone is mainly regulated by L-type Cav1.2 channels (92). However, as the vessel size decreases (<40 μm), L-type Cav1.2 channels have been reported to disappear (93). Recent studies showed that T-type Cav3.2 channels functionally located in caveolae activate BKCa channels to limit vasoconstriction (88, 94–96). This spatial functional organization between T-type Cav3.2 channels, ryanodine receptors, and BKCa channels contrasts the role of L-type Cav1.2 channels in non-caveolar membrane sites to produce primary Ca2+ influx into vascular SMCs and release of Ca2+ sparks via indirect ryanodine type 2 receptor (RyR) activation through sarcoplasmic reticulum Ca2+ content (88, 97–99) (Figure 4). In both cerebral and mesenteric arteries, T-type calcium currents show increased amplitudes as vessel size decreases (93, 100). Genetic deletion of Cav1 or mβcd treatment of vascular SMCs impairs caveolae formation and impacts either the activity or localization of T-type Cav3.2 channels (88, 95, 101). Together, the data support the idea that T-type Cav3.2 channels within caveolae play an important role in the regulation of myogenic tone in small peripheral resistance vessels, which may represent an attractive explanation for attenuated myogenic arterial tone observed in Cav1 knockout models.
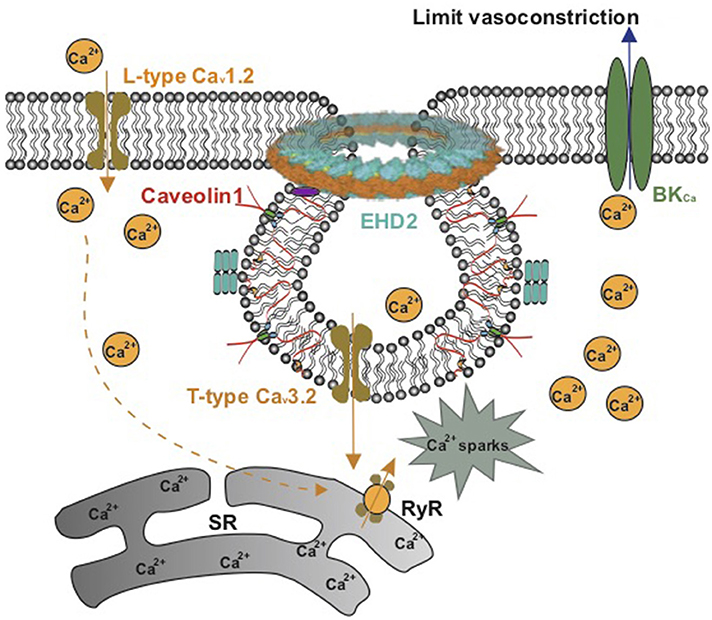
Figure 4. Schematic model on the role of caveolae in Ca2+ signaling in vascular smooth muscle. L-type Cav1.2 channels in noncaveolar membrane sites and T-type Cav3.2 channels in caveolae produce Ca2+ influx into vascular SMCs to release Ca2+ sparks via ryanodine type 2 receptors (RyR) in the sarcoplasmic reticulum (SR). The Ca2+ sparks produce a negative-feedback effect on vasoconstriction by activating maxi Ca2+-activated K+ (BKCa) channels.
Another remarkable ion channel group located in vascular SMC caveolae are TRPC channels, which are suggested to work as store-operated Ca2+ entry (SOCE) channels and are essential for the restoration of internal Ca2+ (102). As SOCE channels, TRPC channel proteins need to associate with other regulatory signaling molecules within caveolae, where they provide a platform for the assembly of TRPC signalplex, including Cav1, G protein, and G-protein-coupled receptor (103). Cav1 deficiency reduced agonist-stimulated Ca2+ secretion and disrupted TRPC signalplex assembly (104). TRPC/TRPC signalplex shows an important role not only in the pathogenesis of pulmonary hypertension (105, 106) but also in essential hypertension and renal hypertension (107–109), which indicated that TRPC or TRPC signalplex may act as important new targets for treatment of hypertension.
Perspective and Conclusions
Caveolae, cholesterol- and glycosphingolipid-rich membrane microdomains, serve as a platform for signal transduction in endothelial cells and vascular SMCs. Within the caveolae membrane domain, in particular Cav1, is a critical molecule, allowing for the rapid activation by posttranslational protein modification. Deletion of caveolin genes is not lethal but caveolin knockout mice show several vascular disorders and dysfunction (110–113) (Table 1).
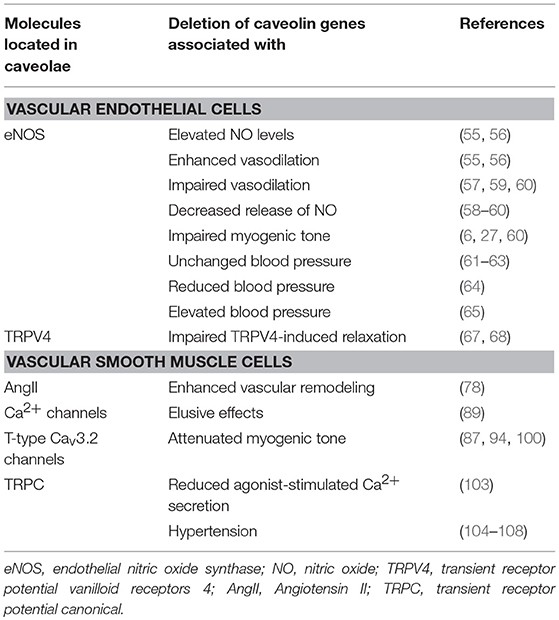
Table 1. Molecules located in caveolae and their effects in Cav 1 deficient mice in vasculature.
In this review, we discussed pathophysiological roles of caveolae in endothelial and vascular SMCs in hypertension, although caveolae most likely also have an impact on vascular function in different tissues like adipose tissue. In the vasculature, except cerebral arteries, blood vessels are directly surrounded by perivascular adipose tissue (PVAT), which directly expresses and secretes Cav1 (18, 113, 114). Numerous studies point out a putative role of caveolae in adipocytes in regulating lipid trafficking, storage, and modulating insulin signal transduction and metabolism (115–118). Moreover, EHD2, which is located in the neck of caveolae, seems to act as a negative regulator of caveolae-dependent lipid uptake (12). These data suggest that adipocyte-secreted Cav1 and EHD2 could contribute to vascular metabolic diseases; however, direct evidence is missing and remains to be determined.
An increasing number of caveolae-associated diseases are explored in order to achieve a more precise assessment of caveolae function in various pathophysiological conditions in vasculature. With the convinced and diversified roles of caveolae in the vasculature, studies on future therapeutic targeting of caveolae in hypertension are necessary.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Deutsche Forschungsgemeinschaft (DFG) (MG), Deutsche Akademische Austauschdienst (DAAD) (MG), and Shanghai Tongji Hospital (XL) support our studies. We also acknowledge support from the Open Access Publication Fund of Charité–Universitätsmedizin Berlin.
References
1. Anderson RG. The caveolae membrane system. Annu Rev Biochem. (1998) 67:199–225. doi: 10.1146/annurev.biochem.67.1.199
2. Yamada E. The fine structure of the gall bladder epithelium of the mouse. J Biophys Biochem Cytol. (1955) 1:445–58. doi: 10.1083/jcb.1.5.445
3. Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. (1992) 68:673–82. doi: 10.1016/0092-8674(92)90143-Z
4. Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci USA. (1996) 93:131–5. doi: 10.1073/pnas.93.1.131
5. Razani B, Lisanti MP. Caveolin-deficient mice: insights into caveolar function human disease. J Clin Invest. (2001) 108:1553–61. doi: 10.1172/JCI14611
6. Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. (2001) 293:2449–52. doi: 10.1126/science.1062688
7. Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, et al. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. (2008) 8:310–7. doi: 10.1016/j.cmet.2008.07.008
8. Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. (2008) 132:113–24. doi: 10.1016/j.cell.2007.11.042
9. Senju Y, Itoh Y, Takano K, Hamada S, Suetsugu S. Essential role of PACSIN2/syndapin-II in caveolae membrane sculpting. J Cell Sci. (2011) 124:2032–40. doi: 10.1242/jcs.086264
10. Hansen CG, Bright NA, Howard G, Nichols BJ. SDPR induces membrane curvature and functions in the formation of caveolae. Nat Cell Biol. (2009) 11:807–14. doi: 10.1038/ncb1887
11. Stoeber M, Schellenberger P, Siebert CA, Leyrat C, Helenius A, Grunewald K. Model for the architecture of caveolae based on a flexible, net-like assembly of Cavin1 and caveolin discs. Proc Natl Acad Sci USA. (2016) 113:E8069–78. doi: 10.1073/pnas.1616838113
12. Matthäus C, Lahmann I, Kunz S, Jonas W, Melo AA, Lehmann M, et al. EHD2-mediated restriction of caveolar dynamics regulates cellular lipid uptake. bioRxiv. (2019) 511709. doi: 10.1101/511709
13. Stoeber M, Stoeck IK, Hanni C, Bleck CK, Balistreri G, Helenius A. Oligomers of the ATPase EHD2 confine caveolae to the plasma membrane through association with actin. EMBO J. (2012) 31:2350–64. doi: 10.1038/emboj.2012.98
14. Senju Y, Suetsugu S. Possible regulation of caveolar endocytosis and flattening by phosphorylation of F-BAR domain protein PACSIN2/Syndapin II. Bioarchitecture. (2015) 5:70–7. doi: 10.1080/19490992.2015.1128604
15. Seemann E, Sun M, Krueger S, Troger J, Hou W, Haag N, et al. Deciphering caveolar functions by syndapin III KO-mediated impairment of caveolar invagination. Elife. (2017) 6:29854. doi: 10.7554/eLife.29854
16. Yao Q, Chen J, Cao H, Orth JD, McCaffery JM, Stan RV, et al. Caveolin-1 interacts directly with dynamin-2. J Mol Biol. (2005) 348:491–501. doi: 10.1016/j.jmb.2005.02.003
17. Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. (2002) 54:431–67. doi: 10.1124/pr.54.3.431
18. Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. (2013) 14:98–112. doi: 10.1038/nrm3512
19. Nassoy P, Lamaze C. Stressing caveolae new role in cell mechanics. Trends Cell Biol. (2012) 22:381–9. doi: 10.1016/j.tcb.2012.04.007
20. Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res. (2016) 118:994–1007. doi: 10.1161/CIRCRESAHA.115.305376
21. Pojoga LH, Yao TM, Opsasnick LA, Siddiqui WT, Reslan OM, Adler GK, et al. Cooperative role of mineralocorticoid receptor and caveolin-1 in regulating the vascular response to low nitric oxide-high angiotensin II-induced cardiovascular injury. J Pharmacol Exp Ther. (2015) 355:32–47. doi: 10.1124/jpet.115.226043
22. Repetto S, Salani B, Maggi D, Cordera R. Insulin and IGF-I phosphorylate eNOS in HUVECs by a caveolin-1 dependent mechanism. Biochem Biophys Res Commun. (2005) 337:849–52. doi: 10.1016/j.bbrc.2005.09.125
23. Li XC, Gu V, Miguel-Qin E, Zhuo JL. Role of caveolin 1 in AT1a receptor-mediated uptake of angiotensin II in the proximal tubule of the kidney. Am J Physiol Renal Physiol. (2014) 307:F949–61. doi: 10.1152/ajprenal.00199.2014
24. Li Q, Zhang Q, Wang M, Zhao S, Ma J, Luo N, et al. Eicosapentaenoic acid modifies lipid composition in caveolae and induces translocation of endothelial nitric oxide synthase. Biochimie. (2007) 89:169–77. doi: 10.1016/j.biochi.2006.10.009
25. Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. (2005) 115:959–68. doi: 10.1172/JCI200519935
26. Garcia-Nino WR, Correa F, Rodriguez-Barrena JI, Leon-Contreras JC, Buelna-Chontal M, Soria-Castro E, et al. Cardioprotective kinase signaling to subsarcolemmal and interfibrillar mitochondria is mediated by caveolar structures. Basic Res Cardiol. (2017) 112:15. doi: 10.1007/s00395-017-0607-4
27. Liu Y, Yoo E, Mahler GJ, Doiron AL. Endothelial barrier dysfunction induced by nanoparticle exposure through actin remodeling via caveolae/raft-regulated calcium signalling. NanoImpact. (2018) 11:82–91. doi: 10.1016/j.impact.2018.02.007
28. Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. (2001) 276:38121–38.
29. Menne J, Wachter R. Invasive treatment of hypertension: update 2016. Internist. (2016) 57:871–8. doi: 10.1007/s00108-016-0116-9
30. Rodrigues GJ, Restini CB, Lunardi CN, Moreira JE, Lima RG, da Silva RS, et al. Caveolae dysfunction contributes to impaired relaxation induced by nitric oxide donor in aorta from renal hypertensive rats. J Pharmacol Exp Ther. (2007) 323:831–7. doi: 10.1124/jpet.107.127241
31. Bernatchez P, Sharma A, Bauer PM, Marin E, Sessa WC. A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. J Clin Invest. (2011) 121:3747–55. doi: 10.1172/JCI44778
32. Oliveira SDS, Minshall RD. Caveolin and endothelial NO signaling. Curr Top Membr. (2018) 82:257–79. doi: 10.1016/bs.ctm.2018.09.004
33. Wang KY, Lee MF, Ho HC, Liang KW, Liu CC, Tsai WJ, et al. Serum caveolin-1 as a novel biomarker in idiopathic pulmonary artery hypertension. Biomed Res Int. (2015) 2015:173970. doi: 10.1155/2015/173970
34. Oh P, Horner T, Witkiewicz H, Schnitzer JE. Endothelin induces rapid, dynamin-mediated budding of endothelial caveolae rich in ET-B. J Biol Chem. (2012) 287:17353–62. doi: 10.1074/jbc.M111.338897
35. Li S, Okamoto T, Chun M, Sargiacomo M, Casanova JE, Hansen SH, et al. Evidence for a regulated interaction between heterotrimeric G proteins and caveolin. J Biol Chem. (1995) 270:15693–701. doi: 10.1074/jbc.270.26.15693
36. Bryant S, Kimura TE, Kong CH, Watson JJ, Chase A, Suleiman MS, et al. Stimulation of ICa by basal PKA activity is facilitated by caveolin-3 in cardiac ventricular myocytes. J Mol Cell Cardiol. (2014) 68:47–55. doi: 10.1016/j.yjmcc.2013.12.026
37. Shakirova Y, Bonnevier J, Albinsson S, Adner M, Rippe B, Broman J, et al. Increased Rho activation and PKC-mediated smooth muscle contractility in the absence of caveolin-1. Am J Physiol Cell Physiol. (2006) 291:C1326–35. doi: 10.1152/ajpcell.00046.2006
38. Wang L, Miller SE, Yuan F. Ultrastructural analysis of vesicular transport in electrotransfection. Microsc Microanal. (2018) 24:553–63. doi: 10.1017/S143192761801509X
39. Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J Biol Chem. (1996) 271:9690–7. doi: 10.1074/jbc.271.16.9690
40. Patel HH, Murray F, Insel PA. G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handb Exp Pharmacol. (2008) 186:167–84. doi: 10.1007/978-3-540-72843-6_7
41. Mineo C, Shaul PW. Circulating cardiovascular disease risk factors and signaling in endothelial cell caveolae. Cardiovasc Res. (2006) 70:31–41. doi: 10.1016/j.cardiores.2006.01.025
42. Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. (2007) 75:247–60. doi: 10.1016/j.cardiores.2007.03.023
43. Van Vliet BN, Chafe LL, Montani JP. Characteristics of 24 h telemetered blood pressure in eNOS-knockout and C57Bl/6J control mice. J Physiol. (2003) 549:313–25. doi: 10.1113/jphysiol.2003.041897
44. Reiner M, Bloch W, Addicks K. Functional interaction of caveolin-1 and eNOS in myocardial capillary endothelium revealed by immunoelectron microscopy. J Histochem Cytochem. (2001) 49:1605–10. doi: 10.1177/002215540104901214
45. Sowa G, Liu J, Papapetropoulos A, Rex-Haffner M, Hughes TE, Sessa WC. Trafficking of endothelial nitric-oxide synthase in living cells. Quantitative evidence supporting the role of palmitoylation as a kinetic trapping mechanism limiting membrane diffusion. J Biol Chem. (1999) 274:22524–31. doi: 10.1074/jbc.274.32.22524
46. Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci USA. (1996) 93:6448–53. doi: 10.1073/pnas.93.13.6448
47. Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem. (1997) 272:18522–5. doi: 10.1074/jbc.272.30.18522
48. Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, et al. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. (2000) 6:1362–7. doi: 10.1038/82176
49. Sowa G, Pypaert M, Sessa WC. Distinction between signaling mechanisms in lipid rafts vs. caveolae. Proc Natl Acad Sci USA. (2001) 98:14072–7. doi: 10.1073/pnas.241409998
50. Cao S, Yao J, Shah V. The proline-rich domain of dynamin-2 is responsible for dynamin-dependent in vitro potentiation of endothelial nitric-oxide synthase activity via selective effects on reductase domain function. J Biol Chem. (2003) 278:5894–901. doi: 10.1074/jbc.M212546200
51. Brouet A, Sonveaux P, Dessy C, Balligand JL, Feron O. Hsp90 ensures the transition from the early Ca2+-dependent to the late phosphorylation-dependent activation of the endothelial nitric-oxide synthase in vascular endothelial growth factor-exposed endothelial cells. J Biol Chem. (2001) 276:32663–9. doi: 10.1074/jbc.M101371200
52. Gu X, Fliesler SJ, Zhao YY, Stallcup WB, Cohen AW, Elliott MH. Loss of caveolin-1 causes blood–retinal barrier breakdown, venous enlargement, and mural cell alteration. Am J. Pathol. (2014) 184:541–55. doi: 10.1016/j.ajpath.2013.10.022
53. Seto SW, Krishna SM, Yu H, Liu D, Khosla S, Golledge J. Impaired acetylcholine-induced endothelium-dependent aortic relaxation by caveolin-1 in angiotensin II-infused apolipoprotein-E (ApoE–/–) knockout mice. PLoS ONE. (2013) 8:e58481. doi: 10.1371/journal.pone.0058481
54. Catalan V, Gomez-Ambrosi J, Rodriguez A, Silva C, Rotellar F, Gil MJ, et al. Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin Endocrinol. (2008) 68:213–9. doi: 10.1111/j.1365-2265.2007.03021.x
55. Yue L, Bian JT, Grizelj I, Cavka A, Phillips SA, Makino A, et al. Apolipoprotein E enhances endothelial-NO production by modulating caveolin 1 interaction with endothelial NO synthase. Hypertension. (2012) 60:1040–6. doi: 10.1161/HYPERTENSIONAHA.112.196667
56. Cao X, Ye Z, Jin M, Yan S, Song X, Huang R. Downregulated caveolin-1 expression serves a potential role in coronary artery spasm by inducing nitric oxide production in vitro. Exp Ther Med. (2018) 16:3567–573. doi: 10.3892/etm.2018.6646
57. Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA. (2002) 99:11375–80. doi: 10.1073/pnas.172360799
58. Rodrigues GJ, Restini CB, Lunardi CN, Neto Mdos A, Moreira JE, Bendhack LM. Decreased number of caveolae in endothelial cells impairs the relaxation induced by acetylcholine in hypertensive rat aortas. Eur J Pharmacol. (2010) 627:251–7. doi: 10.1016/j.ejphar.2009.11.010
59. Al-Brakati AY, Kamishima T, Dart C, Quayle JM. Caveolar disruption causes contraction of rat femoral arteries via reduced basal NO release and subsequent closure of BKCa channels. PeerJ. (2015) 3:e966. doi: 10.7717/peerj.966
60. Albinsson S, Shakirova Y, Rippe A, Baumgarten M, Rosengren BI, Rippe C, et al. Arterial remodeling and plasma volume expansion in caveolin-1-deficient mice. Am J Physiol Regul Integr Comp Physiol. (2007) 293:R1222–31. doi: 10.1152/ajpregu.00092.2007
61. Dubroca C, Loyer X, Retailleau K, Loirand G, Pacaud P, Feron O, et al. RhoA activation and interaction with Caveolin-1 are critical for pressure-induced myogenic tone in rat mesenteric resistance arteries. Cardiovasc Res. (2007) 73:190–7. doi: 10.1016/j.cardiores.2006.10.020
62. Desjardins F, Lobysheva I, Pelat M, Gallez B, Feron O, Dessy C, et al. Control of blood pressure variability in caveolin-1-deficient mice: role of nitric oxide identified in vivo through spectral analysis. Cardiovasc Res. (2008) 79:527–36. doi: 10.1093/cvr/cvn080
63. Sward K, Albinsson S, Rippe C. Arterial dysfunction but maintained systemic blood pressure in cavin-1-deficient mice. PLoS ONE. (2014) 9:e92428. doi: 10.1371/journal.pone.0092428
64. Rosengren BI, Rippe A, Rippe C, Venturoli D, Sward K, Rippe B. Transvascular protein transport in mice lacking endothelial caveolae. Am J Physiol Heart Circ Physiol. (2006) 291:H1371–7. doi: 10.1152/ajpheart.01364.2005
65. Wunderlich C, Schober K, Lange SA, Drab M, Braun-Dullaeus RC, Kasper M, et al. Disruption of caveolin-1 leads to enhanced nitrosative stress and severe systolic and diastolic heart failure. Biochem Biophys Res Commun. (2006) 340:702–8. doi: 10.1016/j.bbrc.2005.12.058
66. Pojoga LH, Yao TM, Opsasnick LA, Garza AE, Reslan OM, Adler GK, et al. Dissociation of hyperglycemia from altered vascular contraction and relaxation mechanisms in caveolin-1 null mice. J Pharmacol Exp Ther. (2014) 348:260–70. doi: 10.1124/jpet.113.209189
67. Wang L, Zhang C, Su X, Lin DH, Wang W. Caveolin-1 deficiency inhibits the basolateral K+ channels in the distal convoluted tubule and impairs renal K+ and Mg2+ transport. J Am Soc Nephrol. (2015) 26:2678–90. doi: 10.1681/ASN.2014070658
68. Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, et al. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol. (2006) 26:1495–502. doi: 10.1161/01.ATV.0000225698.36212.6a
69. Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, et al. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation. (2008) 117:1065–74. doi: 10.1161/CIRCULATIONAHA.107.731679
70. Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, et al. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. (1996) 271:15160–5. doi: 10.1074/jbc.271.25.15160
71. Musial DC, Miranda-Ferreira R, Padin JF, Arranz-Tagarro JA, Parra-Vitela AJ, Jurkiewicz A, et al. Function of AT1 and AT2 receptors in atrial contractions from spontaneous hypertensive and diabetic-induced streptozotocin rats. Clin Exp Pharmacol Physiol. (2018) 45:1274–85. doi: 10.1111/1440-1681.13019
72. Touyz RM, Berry C. Recent advances in angiotensin II signaling. Braz J Med Biol Res. (2002) 35:1001–15. doi: 10.1590/S0100-879X2002000900001
73. Zuo L, Ushio-Fukai M, Ikeda S, Hilenski L, Patrushev N, Alexander RW. Caveolin-1 is essential for activation of Rac1 and NAD(P)H oxidase after angiotensin II type 1 receptor stimulation in vascular smooth muscle cells: role in redox signaling and vascular hypertrophy. Arterioscler Thromb Vasc Biol. (2005) 25:1824–30. doi: 10.1161/01.ATV.0000175295.09607.18
74. Ishizaka N, Griendling KK, Lassegue B, Alexander RW. Angiotensin II type 1 receptor: relationship with caveolae and caveolin after initial agonist stimulation. Hypertension. (1998) 32:459–66. doi: 10.1161/01.HYP.32.3.459
75. Zhang AY, Yi F, Zhang G, Gulbins E, Li PL. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension. (2006) 47:74–80. doi: 10.1161/01.HYP.0000196727.53300.62
76. Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. (2000) 52:639–72.
77. Ushio-Fukai M, Zuo L, Ikeda S, Tojo T, Patrushev NA, Alexander RW. cAbl tyrosine kinase mediates reactive oxygen species- and caveolin-dependent AT1 receptor signaling in vascular smooth muscle: role in vascular hypertrophy. Circ Res. (2005) 97:829–36. doi: 10.1161/01.RES.0000185322.46009.F5
78. Czikora I, Feher A, Lucas R, Fulton DJ, Bagi Z. Caveolin-1 prevents sustained angiotensin II-induced resistance artery constriction and obesity-induced high blood pressure. Am J Physiol Heart Circ Physiol. (2015) 308:H376–85. doi: 10.1152/ajpheart.00649.2014
79. Forrester SJ, Elliott KJ, Kawai T, Obama T, Boyer MJ, Preston KJ, et al. Caveolin-1 deletion prevents hypertensive vascular remodeling induced by angiotensin II. Hypertension. (2017) 69:79–86. doi: 10.1161/HYPERTENSIONAHA.116.08278
80. Kawabe J, Okumura S, Lee MC, Sadoshima J, Ishikawa Y. Translocation of caveolin regulates stretch-induced ERK activity in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. (2004) 286:H1845–52. doi: 10.1152/ajpheart.00593.2003
81. Lodyga M, Bai XH, Mourgeon E, Han B, Keshavjee S, Liu M. Molecular cloning of actin filament-associated protein: a putative adaptor in stretch-induced Src activation. Am J Physiol Lung Cell Mol Physiol. (2002) 283:L265–74. doi: 10.1152/ajplung.00492.2001
82. Oka N, Yamamoto M, Schwencke C, Kawabe J, Ebina T, Ohno S, et al. Caveolin interaction with protein kinase C. Isoenzyme-dependent regulation of kinase activity by the caveolin scaffolding domain peptide. J Biol Chem. (1997) 272:33416–21. doi: 10.1074/jbc.272.52.33416
83. Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. (2011) 144:402–13. doi: 10.1016/j.cell.2010.12.031
84. Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, Morgan KG. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol Rev. (2016) 68:476–532. doi: 10.1124/pr.115.010652
85. Zhuge R, Bao R, Fogarty KE, Lifshitz LM. Ca2+ sparks act as potent regulators of excitation–contraction coupling in airway smooth muscle. J Biol Chem. (2010) 285:2203–10. doi: 10.1074/jbc.M109.067546
86. Cho WJ, Daniel EE. Proteins of interstitial cells of Cajal and intestinal smooth muscle, colocalized with caveolin-1. Am J Physiol Gastrointest Liver Physiol. (2005) 288:G571–85. doi: 10.1152/ajpgi.00222.2004
87. Daniel EE, Jury J, Wang YF. nNOS in canine lower esophageal sphincter: colocalized with Cav-1 and Ca2+-handling proteins? Am J Physiol Gastrointest Liver Physiol. (2001) 281:G1101–14. doi: 10.1152/ajpgi.2001.281.4.G1101
88. Fan G, Kassmann M, Hashad AM, Welsh DG, Gollasch M. Differential targeting and signalling of voltage-gated T-type Cav 3.2 and L-type Cav 1.2 channels to ryanodine receptors in mesenteric arteries. J Physiol. (2018) 596:4863–77. doi: 10.1113/JP276923
89. Ambudkar IS, Brazer SC, Liu X, Lockwich T, Singh B. Plasma membrane localization of TRPC channels: role of caveolar lipid rafts. Novartis Found Symp. (2004) 258:63–70. doi: 10.1002/0470862580.ch5
90. Daniel EE, El-Yazbi A, Cho WJ. Caveolae and calcium handling, a review and a hypothesis. J Cell Mol Med. (2006) 10:529–44. doi: 10.1111/j.1582-4934.2006.tb00418.x
91. Wesselman JP, VanBavel E, Pfaffendorf M, Spaan JA. Voltage-operated calcium channels are essential for the myogenic responsiveness of cannulated rat mesenteric small arteries. J Vasc Res. (1996) 33:32–41. doi: 10.1159/000159129
92. Kuo IY, Ellis A, Seymour VA, Sandow SL, Hill CE. Dihydropyridine-insensitive calcium currents contribute to function of small cerebral arteries. J Cereb Blood Flow Metab. (2010) 30:1226–39. doi: 10.1038/jcbfm.2010.11
93. Jensen LJ, Salomonsson M, Jensen BL, Holstein-Rathlou NH. Depolarization-induced calcium influx in rat mesenteric small arterioles is mediated exclusively via mibefradil-sensitive calcium channels. Br J Pharmacol. (2004) 142:709–18. doi: 10.1038/sj.bjp.0705841
94. Harraz OF, Brett SE, Zechariah A, Romero M, Puglisi JL, Wilson SM, et al. Genetic ablation of CaV3.2 channels enhances the arterial myogenic response by modulating the RyR-BKCa axis. Arterioscler Thromb Vasc Biol. (2015) 35:1843–51. doi: 10.1161/ATVBAHA.115.305736
95. Hashad AM, Harraz OF, Brett SE, Romero M, Kassmann M, Puglisi JL, et al. Caveolae link CaV3.2 Channels to BKCa-mediated feedback in vascular smooth muscle. Arterioscler Thromb Vasc Biol. (2018) 38:2371–81. doi: 10.1161/ATVBAHA.118.311394
96. Harraz OF, Abd El-Rahman RR, Bigdely-Shamloo K, Wilson SM, Brett SE, Romero M, et al. Ca(V)3.2 channels and the induction of negative feedback in cerebral arteries. Circ Res. (2014) 115:650–61. doi: 10.1161/CIRCRESAHA.114.304056
97. Essin K, Welling A, Hofmann F, Luft FC, Gollasch M, Moosmang S. Indirect coupling between Cav1.2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. J Physiol. (2007) 584:205–19. doi: 10.1113/jphysiol.2007.138982
98. Lohn M, Furstenau M, Sagach V, Elger M, Schulze W, Luft FC, et al. Ignition of calcium sparks in arterial and cardiac muscle through caveolae. Circ Res. (2000) 87:1034–9. doi: 10.1161/01.RES.87.11.1034
99. Kassmann M, Szijarto IA, Garcia-Prieto CF, Fan G, Schleifenbaum J, Anistan YM, et al. Role of ryanodine type 2 receptors in elementary Ca(2+) signaling in arteries and vascular adaptive responses. J Am Heart Assoc. (2019) 8:e010090. doi: 10.1161/JAHA.118.010090
100. Abd El-Rahman RR, Harraz OF, Brett SE, Anfinogenova Y, Mufti RE, Goldman D, et al. Identification of L- and T-type Ca2+ channels in rat cerebral arteries: role in myogenic tone development. Am J Physiol Heart Circ Physiol. (2013) 304:H58–71. doi: 10.1152/ajpheart.00476.2012
101. Cheng X, Jaggar JH. Genetic ablation of caveolin-1 modifies Ca2+ spark coupling in murine arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. (2006) 290:H2309–19. doi: 10.1152/ajpheart.01226.2005
102. Parekh AB, Putney JW Jr. Store-operated calcium channels. Physiol Rev. (2005) 85:757–810. doi: 10.1152/physrev.00057.2003
103. Pani B, Singh BB. Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium. (2009) 45:625–33. doi: 10.1016/j.ceca.2009.02.009
104. Pani B, Liu X, Bollimuntha S, Cheng KT, Niesman IR, Zheng C, et al. Impairment of TRPC1–STIM1 channel assembly and AQP5 translocation compromise agonist-stimulated fluid secretion in mice lacking caveolin1. J Cell Sci. (2013) 126:667–75. doi: 10.1242/jcs.118943
105. Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, et al. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA. (2004) 101:13861–6. doi: 10.1073/pnas.0405908101
106. Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, et al. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J. (2007) 21:2970–9. doi: 10.1096/fj.07-8424com
107. Liu D, Scholze A, Zhu Z, Kreutz R, Wehland-von-Trebra M, Zidek W, et al. Increased transient receptor potential channel TRPC3 expression in spontaneously hypertensive rats. Am. J. Hypertens. (2005) 18:1503–7. doi: 10.1016/j.amjhyper.2005.05.033
108. Liu D, Scholze A, Zhu Z, Krueger K, Thilo F, Burkert A, et al. Transient receptor potential channels in essential hypertension. J Hypertens. (2006) 24:1105–14. doi: 10.1097/01.hjh.0000226201.73065.14
109. Mene P, Punzo G, Pirozzi N. TRP channels as therapeutic targets in kidney disease and hypertension. Curr Top Med Chem. (2013) 13:386–97. doi: 10.2174/1568026611313030013
110. Pojoga LH, Adamova Z, Kumar A, Stennett AK, Romero JR, Adler GK, et al. Sensitivity of NOS-dependent vascular relaxation pathway to mineralocorticoid receptor blockade in caveolin-1-deficient mice. Am J Physiol Heart Circ Physiol. (2010) 298:H1776–88. doi: 10.1152/ajpheart.00661.2009
111. Saito H, Godo S, Sato S, Ito A, Ikumi Y, Tanaka S, et al. Important role of endothelial caveolin-1 in the protective role of EDH against NO-mediated nitrative stress in microcirculation in mice. J Cardiovasc Pharmacol. (2018) 71:113–126. doi: 10.1097/FJC.0000000000000552
112. Kuo A, Lee MY, Yang K, Gross RW, Sessa WC. Caveolin-1 regulates lipid droplet metabolism in endothelial cells via autocrine prostacyclin-stimulated, cAMP-mediated lipolysis. J Biol Chem. (2018) 293:973–83. doi: 10.1074/jbc.RA117.000980
113. Chang CC, Chen CY, Wen HC, Huang CY, Hung MS, Lu HC, et al. Caveolin-1 secreted from adipose tissues and adipocytes functions as an adipogenesis enhancer. Obesity. (2017) 25:1932–40. doi: 10.1002/oby.21970
114. Muller G, Schneider M, Biemer-Daub G, Wied S. Microvesicles released from rat adipocytes and harboring glycosylphosphatidylinositol-anchored proteins transfer RNA stimulating lipid synthesis. Cell Signal. (2011) 23:1207–23. doi: 10.1016/j.cellsig.2011.03.013
115. Chaudhary N, Gomez GA, Howes MT, Lo HP, McMahon KA, Rae JA, et al. Endocytic crosstalk: cavins, caveolins, and caveolae regulate clathrin-independent endocytosis. PLoS Biol. (2014) 12:e1001832. doi: 10.1371/journal.pbio.1001832
116. Breen MR, Camps M, Carvalho-Simoes F, Zorzano A, Pilch PF. Cholesterol depletion in adipocytes causes caveolae collapse concomitant with proteosomal degradation of cavin-2 in a switch-like fashion. PLoS ONE. (2012) 7:e34516. doi: 10.1371/journal.pone.0034516
117. Travez A, Rabanal-Ruiz Y, Lopez-Alcala J, Molero-Murillo L, Diaz-Ruiz A, Guzman-Ruiz R, et al. The caveolae-associated coiled-coil protein, NECC2, regulates insulin signalling in adipocytes. J Cell Mol Med. (2018) 22:5648–61. doi: 10.1111/jcmm.13840
Keywords: caveolae, caveolin 1, endothelial nitric oxide synthase, Ca2+ channels, hypertension
Citation: Lian X, Matthaeus C, Kaßmann M, Daumke O and Gollasch M (2019) Pathophysiological Role of Caveolae in Hypertension. Front. Med. 6:153. doi: 10.3389/fmed.2019.00153
Received: 29 January 2019; Accepted: 20 June 2019;
Published: 10 July 2019.
Edited by:
Steven Gerard Achinger, Watson Clinic, United StatesReviewed by:
Rajeev Rohatgi, Northport VA Medical Center, United StatesSwapnil Hiremath, University of Ottawa, Canada
Copyright © 2019 Lian, Matthaeus, Kaßmann, Daumke and Gollasch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoming Lian, eGlhb21pbmcubGlhbkBjaGFyaXRlLmRl