 Michael D. Donnan
Michael D. Donnan Rizaldy P. Scott
Rizaldy P. Scott Tuncer Onay
Tuncer Onay Antoine Tarjus
Antoine Tarjus Ummiye Venus Onay
Ummiye Venus Onay Susan E. Quaggin
Susan E. Quaggin- Division of Nephrology and Hypertension, Feinberg Cardiovascular and Renal Research Institute, Northwestern University Feinberg School of Medicine, Chicago, IL, United States
Nephrotic syndrome is one of the most common glomerular diseases in children and can be classified on the basis of steroid responsiveness. While multiple genetic causes have been discovered for steroid resistant nephrotic syndrome, the genetics of steroid sensitive nephrotic syndrome remains elusive. Mutations in Epithelial Membrane Protein 2 (EMP2), a member of the GAS3/PMP22 tetraspan family of proteins, were recently implicated as putative monogenic cause of steroid sensitive nephrotic syndrome. We investigated this hypothesis by developing Emp2 reporter and knockout mouse models. In lacZ reporter mice (engineered to drive expression of the enzyme β-galactosidase under the control of the endogenous murine Emp2 promoter), Emp2 promoter activity was not observed in podocytes but was particularly prominent in medium- and large-caliber arterial vessels in the kidney and other tissues where it localizes specifically in vascular smooth muscle cells (vSMCs) but not in the endothelium. Strong Emp2 expression was also found in non-vascular smooth muscle cells found in other organs like the stomach, bladder, and uterus. Global and podocyte-specific Emp2 knockout mice were viable and did not develop nephrotic syndrome showing no evidence of abnormal glomerular histology or ultrastructure. Altogether, our results do not support that loss of function of EMP2 represent a monogenic cause of proteinuric kidney disease. However, the expression pattern of Emp2 indicates that it may be relevant in smooth muscle function in various organs and tissues including the vasculature.
Introduction
Nephrotic syndrome (NS) encompasses a triad of clinical features including proteinuria (≥3 g/24 h), low serum albumin (hypoalbuminemia), and edema, and is often accompanied by complications such as secondary hyperlipidemia, thrombotic events, and infections caused by encapsulated organisms (1). Approximately 90% of nephrotic patients respond to corticosteroid treatment and are classified as having idiopathic steroid-sensitive NS (SSNS). The remaining 10% of cases which are refractory to corticosteroids are categorized as steroid-resistant NS (SRNS). Mutations in genes highly expressed in the podocytes have been identified in about two thirds of SRNS patients (2). Many of these SRNS-linked genes encode for proteins that are integral components of the slit diaphragms, regulators of actin cytoskeleton dynamics and cell-matrix interactions, essential for mitochondrial function, and podocyte integrity (3). In contrast, the etiology and pathological mechanism of SSNS remain largely unknown. In ~3% of SSNS patients affected siblings were identified hinting at a possible heritable cause for SSNS (4, 5), although earlier mutation and genome-wide linkage analysis in SSNS-afflicted families fail to uncover a definitive causative gene (6–8). Now, a recent study involving three families suggested that mutations in Epithelial Membrane Protein 2 (EMP2) as potential monogenic autosomal recessive causes of childhood onset SSNS (9).
EMP2 is a member of the growth arrest-specific gene 3/peripheral myelin protein 22 (GAS3/PMP22) family of tetraspan proteins (10–12). Mapped to chromosome 16p13.2, EMP2 was initially identified by its close homology with PMP22 (~40% amino acid sequence similarity) (13, 14). EMP2 translates to an 18 kD protein with four transmembrane domains and multiple consensus sites for N-linked glycosylation in the first hydrophilic domain (13, 15). Tissue distribution and cellular localization of EMP2 has not been fully characterized before although EMP2 is known to be prominently expressed in the lungs (10, 14, 16–18). Several cellular functions have been ascribed to EMP2 including: (1) regulation of cell adhesion involving FAK/Src and β1-integrin; (2) facilitation of trafficking of glycophosphatidylinositol (GPI)-anchored proteins into lipid rafts and regulation of caveolar organization; and, (4) regulation of angiogenesis through induction of VEGFA expression (10, 12, 17, 19–21). EMP2 expression is upregulated in a number of human cancers including endometrial cancers, ovarian cancers, glioblastomas, urinary bladder urothelial carcinoma, and nasopharyngeal carcinomas (10, 14, 22) where it likely plays a role in tumorigenesis through modulation of integrin-dependent cell adhesion (23, 24) and/or promotion of neoplastic angiogenesis (21, 23). EMP2 has been shown to be important for blastocyst implantation and has been identified as a susceptibility factor for chlamydial infectivity, both likely via its role in regulation of integrin-dependent cell signaling (25–27).
The exact function of EMP2 in the kidneys remains poorly understood. EMP2 deficiency has been shown to exacerbate drug-induced injury of zebrafish and cultured human podocytes (28). In this study, we established Emp2 reporter and conditional mutant alleles in the mouse allowing us to: (1) assess comprehensively the tissue distribution of Emp2 expression; and (2) address whether loss of function of Emp2 is a monogenic cause for nephrosis. We conclude that Emp2 is not highly expressed in glomeruli and that global genetic inactivation of Emp2 does not affect viability or cause proteinuric kidney disease in mice.
Results
Renal Expression of Emp2
Using quantitative real-time PCR (qRT-PCR) analysis, we surveyed the relative expression of Emp2 transcript across multiple organs and tissues in mice (Figure 1A). Emp2 is most strongly expressed in the lungs. High levels of expression were also found in the brain, eyes and the uterus. Modest expression levels were found in the heart, spleen, pancreas, liver, and kidneys. Within the kidney, Emp2 expression is >2-fold higher in the renal medulla compared to whole kidneys (Figure 1B). Emp2 expression is not particularly enriched in the renal cortex or isolated glomeruli relative to whole kidneys. In contrast, the transcript level of Nphs1 (a podocyte-specific gene encoding for nephrin) is ~12-fold higher in isolated glomeruli compared to whole kidneys (Figure 1C).
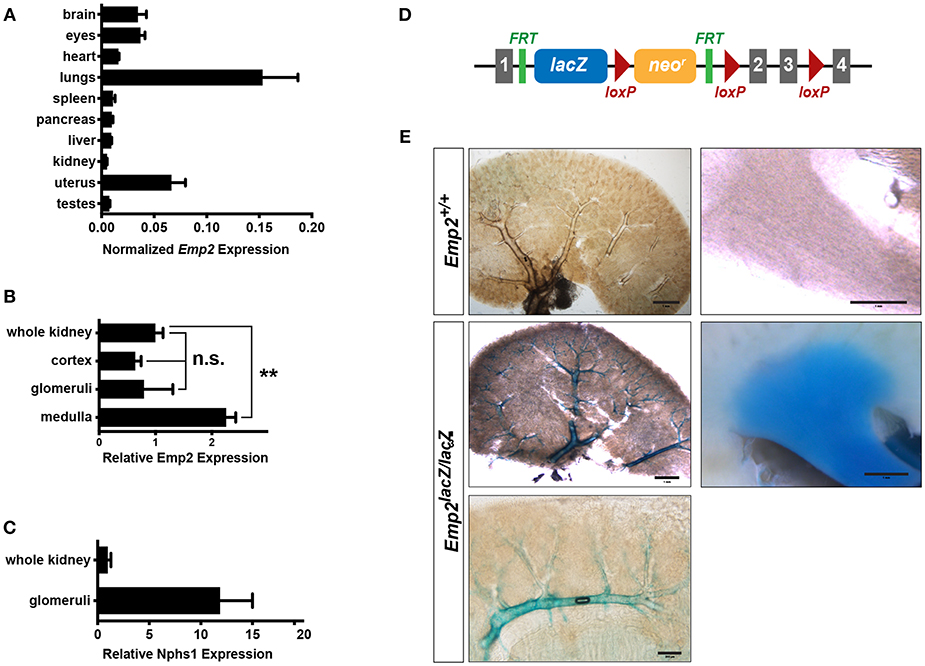
Figure 1. Renal expression of Emp2. (A) Survey of tissue expression of Emp2 by qRT-PCR analysis. Emp2 transcript levels were normalized to that of Gapdh. n ≥ 3 animals per group. (B) Emp2 expression is not particularly enriched in glomeruli. Comparison of Emp2 transcript level normalized to Gapdh relative to whole kidney expression in renal cortex, purified glomeruli, and renal medulla. n = 3 animal per group. **P < 0.005 (C) Nphs1 expression is enriched in glomeruli. Comparison of normalized Nphs1 level between purified glomeruli and whole kidneys. n = 3 animal per group. (D) Schematic diagram of the in vivo Emp2 expression reporter construct used in this study. A dual lacZ-neomycin resistance cassette was inserted between exons 1 and 2 in the Emp2 locus to create the Emp2lacZ reporter allele, where β-galactosidase (β-gal) expression is driven by the endogenous mouse Emp2 promoter. The conditional floxed allele of Emp2 (Emp2flx) was subsequently derived from this reporter construct. Gray boxes, Emp2 exons; green ticks, FRT or Flp recombinase recognition sites; and red triangles, loxP or Cre recombinase recognition sites. (E) Emp2 expression pattern in the kidney. Comparison of Emp2 promoter activity (based on β-gal histochemical staining) in vibratome slices of kidneys from control (Emp2+/+) and Emp2lacZ reporter mice revealing strong Emp2 expression localized to medium and high-caliber renal vessels and also the renal medulla. Scale bars: 2 mm, middle left panel; 1 mm, upper left/right panel, middle right panel; 200 μm lower panel.
Tissue localization of Emp2 expression was analyzed by whole-mount β-galactosidase (β-gal) histochemistry in reporter mice homozygous for the Emp2lacZ gene trapped allele where prokaryotic β-gal expression is driven by the endogenous Emp2 promoter (Figure 1D). Control wild-type mice without the Emp2lacZ allele lack or have negligibly low-levels of background β-gal activity. In 400 μm-thick vibratome sections of the kidney, prominent β-gal activity localizes within large caliber blood vessels (including cortical radial, arcuate, and interlobar vessels), and the renal papilla (Figure 1E). Within the cortex β-gal activity is limited to arcuate vessels and their immediate radial branches but is notably absent in glomeruli and their associated efferent and afferent arterioles. Altogether, these results indicate that in the kidney Emp2 has minimal expression within podocytes and has a predominantly vascular pattern of expression.
Vascular Expression of Emp2
A distinctive vascular pattern of expression of β-gal is found in multiple organs of the Emp2lacZ reporter mice including the brain, eye sclera, cornea, heart, dorsal aorta, thigh musculature, and gut mesentery (Figure 2A). We cryosectioned thick vibratome slices of kidneys and dorsal aorta stained by X-gal histochemistry so we can determine specific tissue localization of β-gal activity in Emp2lacZ mice (Figure 2B). In the kidneys, β-gal activity is restricted to high caliber renal arterial vessels where it is particularly located in Tagln+ (SM22α+) vascular smooth muscle cells (vSMCs) and is absent in Podxl+ arterial endothelial cells and renal podocytes. In contrast, Emcn+ renal veins, and glomerular endothelial cells completely lack staining for β-gal activity. Likewise, in the dorsal aorta, β-gal activity is confined to the Tagln+ vSMCs and is not seen in the Podxl+ endothelial compartment. To validate specific cellular localization of Emp2 expression in the vasculature, we generated tamoxifen-inducible endothelial-specific conditional Emp2 knock out mice (Cdh5-CreERT2Tg/+:Emp2flx/flx). Emp2 expression within the kidney is unchanged upon endothelial specific knockout of the Emp2 gene (Figure 2C) consistent with the lack of β-gal activity in the endothelial compartment of Emp2lacZ reporter mice. We conclude from these results that vSMCs but not their associated endothelial cells account for the vascular pattern of expression of Emp2.
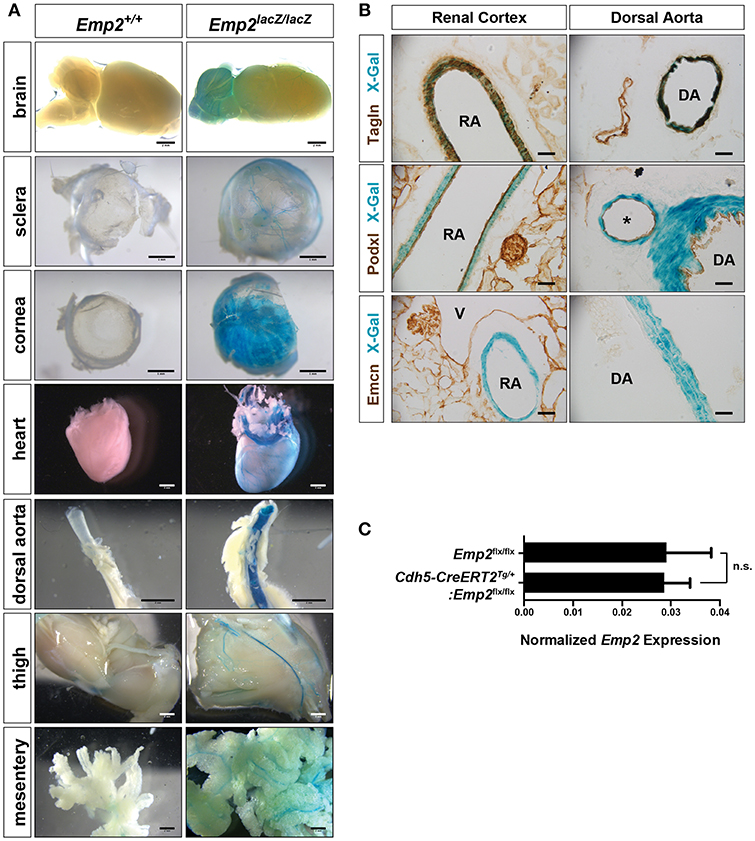
Figure 2. Vascular expression of Emp2 in multiple tissues. (A) β-gal histochemical staining of whole-mount organs and tissues (brain, eye sclera, cornea, heart, dorsal aorta, hind limbs, and mesentery) from control and Emp2lacZ reporter mice showing vascular pattern of expression. Staining was absent in control mice. Scale bars: 2 mm, (A) brain, heart, dorsal aorta, thigh, mesentery; 1 mm, sclera, cornea. (B) Vascular expression of Emp2 is restricted to arterial smooth muscles. In the renal cortex, Emp2 promoter activity (blue X-gal stain) localizes to Tagln+ vascular smooth muscles but not Podxl+ arterial endothelium in the renal cortex and dorsal aorta. Emcn+ veins are unstained. RA, renal artery; DA, dorsal aorta; V, vein; and *, branching artery. Scale bars: 60 μm. (C) Emp2 expression in the kidney is unchanged upon endothelial-specific deletion of the Emp2 gene. Normalized Emp2 expression in kidneys, measured by qRT-PCR analysis, from homozygous Emp2flx/flx animals and tamoxifen-induced endothelial-specific conditional Emp2 knockout mice (Cdh5-CreERT2Tg/+:Emp2flx/flx). n = 5 animals per group.
Non-vascular Expression of Emp2
In adult mice, pronounced β-gal activity was additionally seen in several smooth muscle tissues including the esophagus, stomach, uterus, and bladder (Figure 3A) but was absent from skeletal muscle tissue such as the thigh (Figure 2A). In cryosections of these tissues, β-gal activity was found primarily in Tagln+ or Des+ smooth muscle cells (SMCs) and absent in the CD31+ or endothelia (Figure 3B). Emp2 is therefore broadly expressed in different subsets of SMCs both vascular and non-vascular but is absent in skeletal muscle.
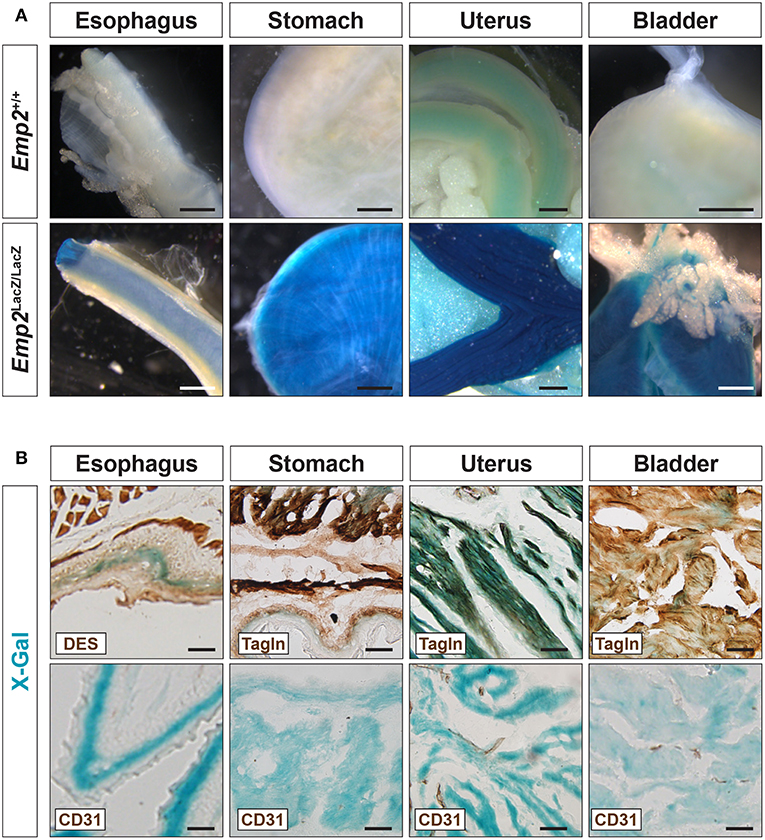
Figure 3. Emp2 is expressed in smooth muscles. (A) Emp2 is expressed in non-vascular smooth muscles. Strong β-gal histochemical staining of the esophagus, stomach, uterus, and bladder of Emp2lacZ mice. Scale bars: 1 mm. (B) Immunohistochemical staining reveals that the Emp2 expression is overlaps with Des+ (esophagus) and Tagln+ (stomach, uterus, and bladder) smooth muscles but not to CD31+ endothelia. Scale bars: 200 μm.
Emp2 expression was additionally observed in non-smooth muscle tissues in several organs. Pronounced β-gal reporter activity was seen in the lungs, eye lens, ear, testes (Figure 4A) and renal papilla (Figure 1E). β-gal activity in the lungs are confined to alveolar pneumocytes that co-stain with antibodies against pan-cytokeratins (PCK) (Figure 4B). β-gal activity is additionally seen in the Tagln+ vSMCs of the high caliber arterial vessels within the lung however appears absent from the Tagln+ airway smooth muscles. There is no co-localization of Emcn immunoreactivity with β-gal activity in the lungs indicating that Emp2 expression is negligible or absent in alveolar endothelium.
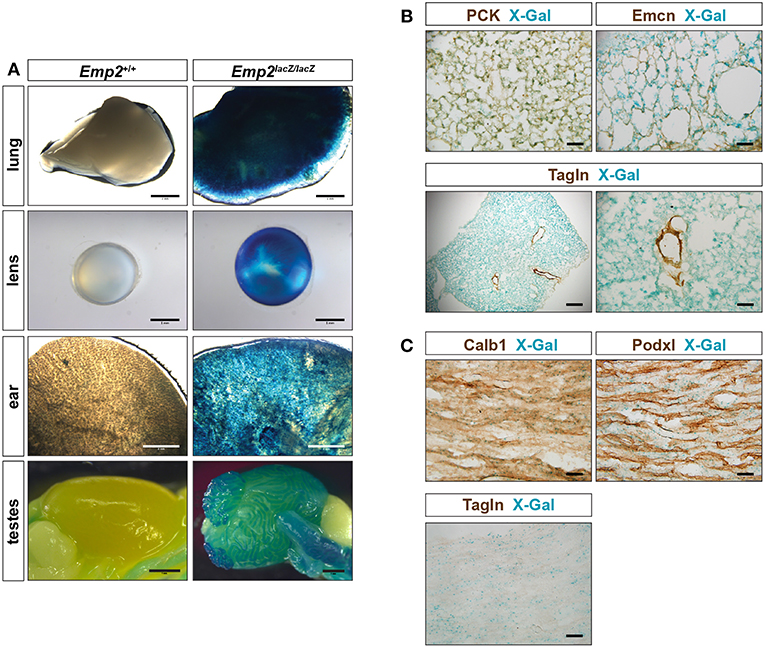
Figure 4. Emp2 is expressed beyond smooth muscles. (A) Emp2 also has non-smooth muscle expression in other organs. Strong Emp2 promoter activity in Emp2lacZ reporter mice in the lungs, eye lens, ear, and testes. Scale bars: 2 mm, lung and ear; 1 mm lens and testes. (B) Pulmonary expression of Emp2 is primarily in pneumocytes. Emp2 promoter activity (X-gal staining) in the lungs co-localizes with pan-cytokeratin (PCK) immunoreactivity in alveolar epithelial cells (pneumocytes) but not with Emcn (endothelial cells) or smooth muscles (Tagln). Scale bars: 200 μm, lower left; 50 μm remaining. (C) Non-vascular expression of Emp2 in the renal papilla. Prominent Emp2 promoter activity in the renal papilla co-localizes with Calb1 (principal collecting duct epithelia) and not Podxl (endothelial cells), or Tagln (smooth muscle) immunoreactivities. Scale bars: 50 μm.
In the kidney, non-vascular expression of Emp2 was found primarily in the renal papilla. Emp2 promoter activity within the renal papilla co-localizes with Calb1 (principal collecting duct epithelia) and not with Podxl (endothelial cells), or Tagln (smooth muscle) immunoreactivities (Figure 4C).
Genetic Inactivation of Emp2 Does Not Cause Nephrosis
An Emp2 null mutant allele (Emp2Δ) was generated by crossing conditional floxed Emp2 (Emp2flx) mice with EIIa-Cre transgenic mice (Supplementary Figure 1). Homozygous Emp2 null mutants (Emp2Δ/Δ) were viable, live to adulthood, and are fertile. Emp2Δ/Δ mutant animals have complete absence of Emp2 expression in their lungs and kidneys by qRT-PCR analysis (Figure 5A). Emp2Δ/Δ mutants had comparable body weights relative to their littermates control cohorts and lack any overt abnormality (Figure 5B).
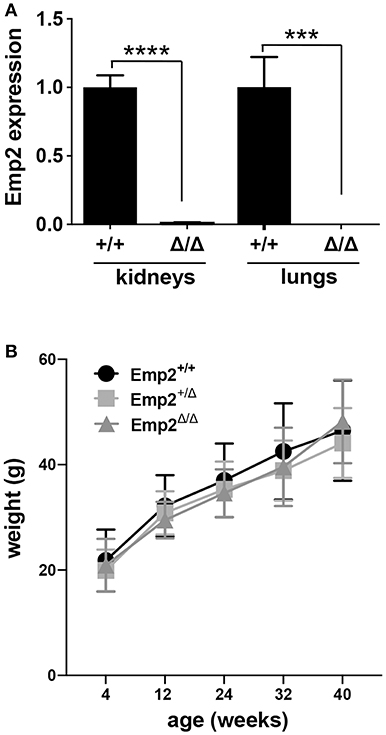
Figure 5. Emp2 null mutant mice are viable. (A) Complete loss of Emp2 expression in global Emp2 null mutant mice. Gapdh-normalized Emp2 transcript levels measured by qRT-PCR analysis showing total ablation of Emp2 expression in kidneys and lungs of homozygous Emp2 null mutants (Emp2Δ/Δ). Mice > 4 weeks of age. n ≥ 3 animals per group. ***P < 0.0005, ****P < 0.0001. (B) Body weight is unaffected by the Emp2 null allele. Comparable body weights between wild-type, heterozygous and homozygous Emp2 null mutant mice (Emp2+/+, Emp2Δ/+, and Emp2Δ/Δ). n ≥ 3 animals per group.
Since mutations in the human EMP2 gene have been previously linked to hereditary congenital nephrosis in a small subset of patients, we analyzed Emp2 null mutant mice for signs of proteinuric kidney disease and glomerular defects. Mice at 10 months of age show no sign of proteinuria based on protein gel urinalysis (Figure 6A) and mice at 1 and 10 months of age show no sign of proteinuria base on urinary protein-creatinine ratios (Figures 6B,C). Gross renal and glomerular histology at 10 months of age also reveals normal overall kidney and glomerular morphology without any evidence of renal tubular protein casts or fibrosis in Emp2 null mutant mice (Figure 6D). Gross pulmonary and renal arterial histology were unremarkable and comparable between control wild-type and Emp2 null mutants even though Emp2 is most prominently expressed in the lungs and vSMCs (Supplementary Figure 2). We also genetically inactivated Emp2 exclusively in podocytes by crossing Emp2flx mice with the podocyte-specific Nphs1-Cre deletor strain. Similarly, mice with targeted loss of Emp2 in podocytes have normal overall renal histology and glomerular morphology (Figure 6E) and were non-proteinuric (not shown). Kidneys from Emp2 null mutants have normal and regular expression of slit diaphragm proteins nephrin (Nphs1) and podocin (Nphs2) as seen by immunofluorescence (Figure 7A). Ultrastructure analysis by transmission electron microscopy shows that the structural integrity of the glomerular filtration barrier appears intact in Emp2 null mutants with normal presence of interdigitating foot processes conjoined by slit diaphragms, unimpressive glomerular basement membrane (GBM), and fenestrated endothelial cells (Figure 7B).
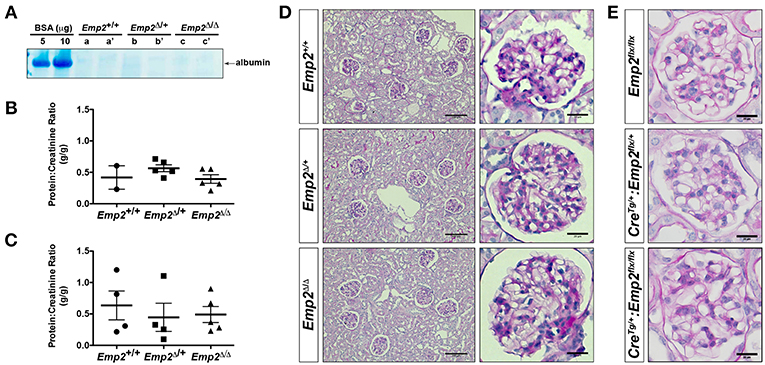
Figure 6. Emp2 null mutant mice do not develop nephrosis. (A) Protein gel urinalysis do not show evidence of albuminuria in homozygous Emp2 null mutant mice (Emp2Δ/Δ). Urine from 10-month-old animals run on SDS-PAGE alongside bovine serum albumin (BSA) standards and stained with Coomasie Brilliant Blue. (B) Urinary protein-creatinine ratios reveal lack of proteinuria in Emp2 null mutants at 1-month of age. (C) No evidence of proteinuria at 10-months of age based on urinary protein-creatinine ratios. (D) Emp2 null mutants have unremarkable overall renal and glomerular histology. (E) Emp2 deletion in podocytes do not disrupt normal glomerular histology. Podocyte-specific Emp2 conditional knockout (Nphs1-CreTg+/:Emp2flx/flx) mice have unremarkable and comparable glomerular histology relative to their wild-type control littermates (Emp2flx/flx and Nphs1-CreTg/+:Emp2flx/+). Scale bars: 100 μm, D column 1; 20 μm D column 2 and E.
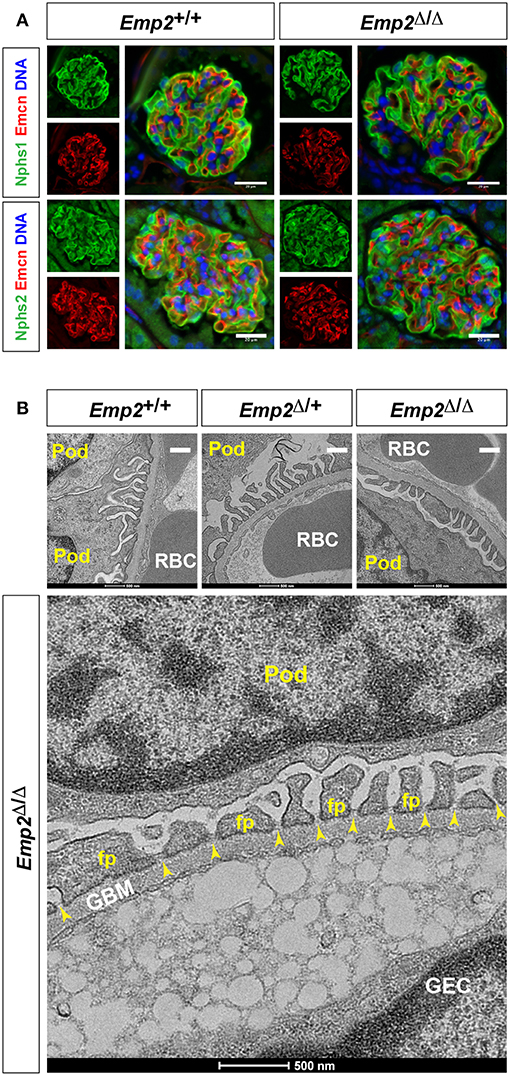
Figure 7. Slit diaphragms are intact in Emp2 null mutants. (A) Comparable and regular expression of Nphs1 (nephrin) and Nphs2 (podocin) in wild-type control and homozygous Emp2 null mutant (Emp2Δ/Δ) glomeruli. Immunofluorescence images of glomeruli stained for Emcn for the glomerular endothelium together with either Nphs1 or Nphs2. Scale bars: 20 μm. (B) Emp2 null mutants have normal glomerular ultrastructure. Transmission electron micrographs of glomeruli showing normal organization, interdigitating podocyte foot processes and intact slit diaphragms. Pod, podocyte; GBM, glomerular basement membrane; GEC, glomerular endothelial cell; RBC, red blood cell; fp, podocyte foot process; and, yellow arrowheads, slit diaphragms. Scale bars: 500 nm.
Altogether, mutant mice aged up to 10 months were not visibly different from their littermate control cohorts and were non-proteinuric. At 10 months, renal histology and glomerular cytoarchitecture remain normal. These findings therefore indicate that Emp2 is non-essential for normal glomerular development and function, and is consistent with lack of glomerular expression of Emp2 in mice.
Discussion
Steroid responsiveness is the most important prognostic factor in patients with nephrotic syndrome (29). As such, determining the underlying and potentially differing pathogenesis of SSNS in contrast to the known genetic variants of SRNS is of significant interest. Given the beneficial response of SSNS to immunosuppression such as steroids, potential immune mediated mechanisms of SSNS pathogenesis have been hypothesized with several risk loci being identified within the HLA-DQ and HLA-DR regions of the human leukocyte antigen (HLA) gene (4). Now a new Mendelian form of SSNS has been proposed with the discovery of biallelic mutations of EMP2 in four individuals from three families with SSNS and SRNS (9). Homozygosity mapping and whole-exome sequencing initially identified a homozygous truncating variant in exon 4 of EMP2 (Q62*) in a Turkish family with two siblings with SSNS. Further evaluation of a cohort of 1,600 individuals with NS revealed two additional individuals with biallelic mutations in EMP2 demonstrating previously unreported missense mutations in exon 2 of EMP2 (F7L and A10T). However, since this discovery of mutations in EMP2 in these 4 individuals with NS, now tentatively classified as nephrotic syndrome type 10, evaluation for EMP2 mutations in larger cohorts of nephrotic patients have not identified additional mutations or cases. In evaluation of a further 33 families with autosomal recessive SSNS, no evidence of pathogenic mutations of EMP2 were found by Sanger sequencing (30). Additionally, in a whole-exome sequencing of 3,315 patients with chronic kidney disease, only two individuals were found to have heterozygous variants of in EMP2 of unclear significance, and these mutations were deemed unlikely related to their underlying kidney disease (31). Whole exome sequencing of 363 unrelated family units with FSGS and 363 ancestry-matched controls also only detected suspicious variants of EMP2 in control individuals (32). Altogether, these data suggest that pathogenic mutations in EMP2 likely do not represent a significant portion of incident SSNS and may not be a monogenic etiology of proteinuric kidney disease.
Our data demonstrate that genetic deletion of Emp2 does not cause proteinuric kidney disease in mice. Using quantitative real-time PCR analysis and histochemical characterization of Emp2lacZ reporter mice, expression of Emp2 is not enriched in the glomerulus or the podocyte as previously suggested (9, 28). Whole-mount β-gal histochemistry was used in lieu of antibodies against Emp2 as commercially available antibodies gave only non-specific background signals by tissue immunostainings. The Emp2lacZ reporter expression pattern we observed qualitatively correlates with relative mRNA expression in tissues based on qRT-PCR analysis, with most prominent expressions seen in lungs followed by the uterus by both methods. Furthermore, mice with global deletion of the Emp2 gene were healthy, viable, and fertile, consistent with what has been reported independently by another group (26). Additionally, these mice did not manifest significant proteinuria by 10 months of age. Consistent with this, glomerular histology and cytoarchitecture are normal in mice with podocyte and global deletion of Emp2. Emp2 null mutants show no evidence of podocyte effacement or perturbation of slit diaphragms. Together these data suggest that Emp2 is non-essential for normal glomerular development and maintenance of integrity of the glomerular filtration barrier in mice. Our data, in addition to published reports of suspicious variants of EMP2 in individuals without proteinuric kidney disease, suggest that mutations in EMP2 may not be solely responsible for the development of early-onset childhood NS as previously reported (9). It is possible other modifying genes or environmental factors may account for the clinical heterogeneity seen in these cases.
Mouse Emp2 protein has a strong homology with its human counterpart and shares a significant amino acid sequence identity (78%) (Supplementary Figures 3, 4), thereby justifying the use of a murine model to validate the hypothesis that loss of function of EMP2 is causative for certain cases of SNSS. Additionally, two of the three reported missense mutations in EMP2 (F7L and Q62*) occur in conserved amino acid residues between mice and humans further supporting the use of mice to model this condition (Supplementary Figure 3). However, the possibility remains that there are interspecies differences in expression and function of EMP2. And animal models of kidney disease do not always match the expected phenotype of human disease (33). Our study did not address these potential differences in EMP2 expression and function in human tissues. Given that we did not observe glomerular expression of Emp2, we decided against examining the effect of Emp2 deficiency in the context of a podocyte injury model. We suspect that a genetic cause of childhood onset NS would be less dependent on environmental injury for the development of the expected phenotype. Additionally, RNA-seq analysis of gene expression in mouse kidneys following unilateral ureteral obstruction injury reveals that Emp2 expression was not particularly enriched in podocytes but rather expressed more significantly in proliferating proximal tubule and principal collecting duct epithelial cells (34).
Interestingly, we found that expression of Emp2 was predominantly in arterial vascular structures across multiple tissues including the kidney. This finding is concordant to multiple recent studies that link EMP2 to vascular function. EMP2 has been previously linked to angiogenesis through both VEGF-dependent and VEGF-independent pathways in multiple tumor models (21, 23). In addition to tumorigenesis, alterations in EMP2 expression has now been linked to failure of appropriate placental angiogenesis and pathogenic corneal neovascularization (26, 35). EMP2 is currently being investigated as a novel target for cancer treatment and to reduce pathologic neovascularization (23, 24, 35–37). As such, it is essential to further define the role of EMP2 in the vasculature as well as other tissues. In this study we found that expression of Emp2 within the vasculature localized primarily to surrounding vSMCs. This is supported by a recent study implicating Emp2 in a maladaptive pathway of vSMC proliferation in diabetic rats and raises the possibility of targeting EMP2 for the treatment of vasculo-proliferative diseases in diabetes (38).
We also found expression of Emp2 in multiple other smooth muscle tissues, including the esophagus, stomach, uterus and bladder, however, not in skeletal muscle tissues such as the thigh. Expression of Emp2 was found in certain non-smooth muscle cells most prominently in the lung as well as the renal papilla. In the renal papilla, Emp2 promoter activity co-localizes with calbindin (Calb1, a marker expressed in principal collecting duct epithelia), which is consistent with human and mouse RNA-seq data demonstrating prominent Emp2 expression within the principal cells of the renal collecting duct (39, 40). While mice with loss of Emp2 were viable and without overt pathology in tissues with high Emp2 expression, such as the lung, it will be important to further define the role of Emp2 in these tissues if future therapeutics targeting this pathway are to be developed.
Our data conclude that genetic deletion of Emp2 does not cause proteinuric kidney disease in mice. Rather our findings further support a potential role of Emp2 within the vasculature of multiple tissues including the kidney. Additional studies are warranted to further explore the specific role of Emp2 in angiogenesis and vascular disease.
Materials and Methods
Mouse Strains and Husbandry
A floxed conditional allele of Emp2 was created using a targeting vector obtained from the Knockout Mouse Project Repository (KOMP, https://www.komp.org). This vector incorporates a tandem bacterial lacZ (β-galactosidase) and neomycin-resistance selection (neor) cassettes in between exons 1 and 2 of the mouse Emp2 locus that are excisable with Flp recombinase. This vector also contains loxP (Cre recombinase recognition) sites flanking exons 2 and 3. DNA electroporation and clonal drug-selection of R1 (129X1 X 129S1) mouse embryonic stem (ES) cells were as described previously (41). Correctly targeted ES clones screened by Southern blot hybridization were used for blastocyst injection and generation of mouse chimera. Mouse lines were subsequently outbred onto a mixed genetic background. Mouse offspring harboring the targeted allele (Emp2lacZ) were used as reporter animals to assess Emp2 promoter activity and tissue expression. A conditional floxed Emp2 allele (Emp2flx) was generated by breeding Emp2lacZ mice with Flp recombinase transgenic mice resulting in the removal of the lacZ and neor expression cassettes (42). Crossing of Empflx mice with the transgenic EIIa-Cre [Tg(EIIa-cre)C5379Lmgd] mouse line (43) results in germline inactivation of the Emp2 locus and the establishment of first-generation heterozygous Emp2 null mice (Emp2Δ/+). Mice with the genotype Emp2Δ/Δ are classified as null mutants while those with genotypes Emp2+/+or Emp2Δ/+ are collectively classified as wild-type controls. Endothelial- and podocyte-specific knockout mice were derived from crosses between Empflx mice with the driver strains Cdh5-CreERT2 [Tg(Cdh5-cre/ERT2)1Rha] (44) and Nphs1-Cre [Tg(Nphs1-cre)#Seq] (45), respectively. Animals were genotyped by genomic PCR analysis using primers indicated in Supplementary Table 1. Mice were reared, bred, and characterized according to strict ethical guidelines approved by the Institutional Animal Care and Use Committee of Northwestern University.
Gene Expression Analysis
Trizol reagent (Thermo Fisher Scientific, Waltham, MA) was used for extraction and purification of total RNA from isolated cells, tissues and organs. For isolation of glomeruli, anesthetized mice were systemically perfused with 0.3% (w/v) Fe2O3 particles in Hank's Balanced Buffer Solution (HBSS). Fe2O3-perfused kidneys were minced finely and digested with collagenase (10 mg/mL) at 37°C for 30 min. Dissociated kidney tissues were sieved through a 100-μm cell strainer, and the glomeruli captured using a magnet. Reverse transcription was completed using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) and real-time PCR was performed on an ABI 7500 thermocycler (Applied Biosystems, Foster City, CA) using iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA). DNA amplification threshold cycles values (Ct) were used to evaluate gene transcript expression levels between genes of interests normalized to the housekeeping gene Gapdh. PCR primers used are listed on Supplementary Table 1.
Histology and Histochemistry
Tissues and organs were routinely fixed in 4% formaldehyde in phosphate buffered saline (PBS, pH7.5) overnight at 4°C. Fixed tissues were embedded in paraffin blocks to produce 5-μm thick sections for routine histology (hematoxylin-eosin and periodic acid-aldehyde Schiff staining), and immunostainings. Standard methods for immunofluorescence and immunohistochemistry were carried out using antibodies listed in Supplementary Table 2. For lacZ reporter expression analysis, anesthetized mice with the Emp2lacZ gene-trapped allele were perfusion-fixed with 300 μM chloroquine in PBS followed by 0.4% formaldehyde in PBS through the right cardiac ventricle. Subsequently, mice were perfused with lacZ rinse buffer (2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% IGEPAL CA-630, and 300 μM chloroquine in PBS). Perfused and dissected organs were stained using an X-gal solution [1 g/L X-gal, 5 mM K4Fe(CN)6, and 5 mM K3Fe(CN)6 in lacZ rinse buffer] and incubated overnight at room temperature. Staining was quenched by incubation of tissues in phosphate buffered formalin solution.
Urine Analysis
Urinary protein was measured using Bio-Rad Protein Assay (Bio-Rad, Hercules, CA) and urinary creatinine was analyzed through a microtiter-format colorimetric Jaffe reaction assay using alkaline picrate. Qualitative albuminuria was assessed on colloidal Coomasie dye-stained protein gels of urine samples alongside bovine serum albumin standards.
Ultrastructure Analysis
Kidney specimens were fixed in 0.1 M cacodylate buffer (pH 7.5) containing 4% formaldehyde and 2% glutaraldehyde and post-fixed in 1% OsO4 prior to dehydration and embedding in epoxy resin for sectioning. Ultrathin resin sections were stained with uranyl acetate and lead acetate solution, and viewed in a FEI Tecnai G2 transmission electron microscope (ThermoFisher Scientific, Waltham, MA).
Statistical Analyses
Quantitative data are shown as mean ± standard error of the mean (SEM). Statistical significance of quantitative results was evaluated using Student's t-test and ANOVA using GraphPad Prism version 8.00 (www.graphpad.com). P values of <0.05 were considered statistically significant.
Data Availability
All datasets generated for this study are included in the manuscript/Supplementary Files.
Author Contributions
MD, RS, TO, and SQ contributed to the design of the experiments. Experiments were performed by MD, RS, TO, AT, and UO. Emp2 mouse lines were developed by TO. MD, RS, TO, and SQ contributed to analysis of the data. The manuscript was written by MD, RS, and SQ. RS and SQ supervised the study. All authors contributed to the review and approval of the manuscript.
Funding
This work was funded with a fellowship grant from the Martha Hofmann Levin and Anna C. Hofmann Research Endowment (MD) and research grants from the National Institutes of Health: T32DK108738 (MD), 5R01HL124120-04 (SQ), and P30DK114857 (SQ).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The genetically engineered mice were generated with the assistance of Northwestern University Transgenic and Targeted Mutagenesis Laboratory (NIH grant CA060553). We are indebted to the staff of the Center for Comparative Medicine of Northwestern University Feinberg School of Medicine for assistance in animal care. We thank AnnaWoo and Megan Kelly for the technical support provided for the study. Microscopy work was performed at the Northwestern University Center for Advanced Microscopy (CAM) with funding from the National Cancer Institute Cancer Center support grant P30CA060533. We are grateful to Lennell Reynolds, Jr. of CAM for help with tissue processing for electron microscopy. We would also like to thank the generous support of the Northwestern University George M. O'Brien Kidney Core Center (NU-GoKIDNEY). Histology services were in part provided by the Northwestern University Research Histology and Phenotyping Laboratory which was supported by NCI P30- CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2019.00189/full#supplementary-material
References
1. Orth SR, Ritz E. The nephrotic syndrome. N Engl J Med. (1998) 338:1202–11. doi: 10.1056/NEJM199804233381707
2. Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol. (2010) 25:1621–32. doi: 10.1007/s00467-010-1495-0
3. Yu SM, Nissaisorakarn P, Husain I, Jim B. Proteinuric kidney diseases: a podocyte's slit diaphragm and cytoskeleton approach. Front Med. (2018) 5:221. doi: 10.3389/fmed.2018.00221
4. Lane BM, Cason R, Esezobor CI, Gbadegesin RA. Genetics of childhood steroid sensitive nephrotic syndrome: an update. Front Pediatr. (2019) 7:8. doi: 10.3389/fped.2019.00008
5. Xia Y, Mao J, Jin X, Wang W, Du L, Liu A. Familial steroid-sensitive idiopathic nephrotic syndrome: seven cases from three families in China. Clinics (Sao Paulo). (2013) 68:628–31. doi: 10.6061/clinics/2013(05)08
6. Gbadegesin RA, Adeyemo A, Webb NJ, Greenbaum LA, Abeyagunawardena A, Thalgahagoda S, et al. HLA-DQA1 and PLCG2 are candidate risk loci for childhood-onset steroid-sensitive nephrotic syndrome. J Am Soc Nephrol. (2015) 26:1701–10. doi: 10.1681/ASN.2014030247
7. Parker L, Bahat H, Appel MY, Baum DV, Forer R, Pillar N, et al. Phospholipase C-gamma 2 activity in familial steroid-sensitive nephrotic syndrome. Pediatr Res. (2019) 85:719–23. doi: 10.1038/s41390-018-0259-6
8. Fuchshuber A, Gribouval O, Ronner V, Kroiss S, Karle S, Brandis M, et al. Clinical and genetic evaluation of familial steroid-responsive nephrotic syndrome in childhood. J Am Soc Nephrol. (2001) 12:374–8.
9. Gee HY, Ashraf S, Wan X, Vega-Warner V, Esteve-Rudd J, Lovric S, et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet. (2014) 94:884–90. doi: 10.1016/j.ajhg.2014.04.010
10. Chung LK, Bhatt NS, Lagman C, Pelargos PE, Qin Y, Gordon LK, et al. Epithelial membrane protein 2: Molecular interactions and clinical implications. J Clin Neurosci. (2017) 44:84–8. doi: 10.1016/j.jocn.2017.06.044
11. Berditchevski F, Odintsova E. Characterization of integrin-tetraspanin adhesion complexes: role of tetraspanins in integrin signaling. J Cell Biol. (1999) 146:477–92. doi: 10.1083/jcb.146.2.477
12. Wadehra M, Iyer R, Goodglick L, Braun J. The tetraspan protein epithelial membrane protein-2 interacts with beta1 integrins and regulates adhesion. J Biol Chem. (2002) 277:41094–100. doi: 10.1074/jbc.M206868200
13. Taylor V, Suter U. Epithelial membrane protein-2 and epithelial membrane protein-3: two novel members of the peripheral myelin protein 22 gene family. Gene. (1996) 175:115–20. doi: 10.1016/0378-1119(96)00134-5
14. Ashki N, Gordon L, Wadehra M. Review of the GAS3 family of proteins and their relevance to cancer. Crit Rev Oncog. (2015) 20:435–47. doi: 10.1615/CritRevOncog.v20.i5-6.140
15. Chen Q, Yao L, Burner D, Minev B, Lu L, Wang M, et al. Epithelial membrane protein 2: a novel biomarker for circulating tumor cell recovery in breast cancer. Clin Transl Oncol. (2019) 21:433–42. doi: 10.1007/s12094-018-1941-1
16. Wang CX, Wadehra M, Fisk BC, Goodglick L, Braun J. Epithelial membrane protein 2. a 4-transmembrane protein that suppresses B-cell lymphoma tumorigenicity. Blood. (2001) 97:3890–5. doi: 10.1182/blood.V97.12.3890
17. Lee EJ, Park MK, Kim HJ, Kim EJ, Kang GJ, Byun HJ, et al. Epithelial membrane protein 2 regulates sphingosylphosphorylcholine-induced keratin 8 phosphorylation and reorganization: Changes of PP2A expression by interaction with alpha4 and caveolin-1 in lung cancer cells. Biochim Biophys Acta. (2016) 1863(6 Pt A):1157–69. doi: 10.1016/j.bbamcr.2016.02.007
18. Wadehra M, Sulur GG, Braun J, Gordon LK, Goodglick L. Epithelial membrane protein-2 is expressed in discrete anatomical regions of the eye. Exp Molecul Pathol. (2003) 74:106–12. doi: 10.1016/S0014-4800(03)00009-1
19. Fu M, Rao R, Sudhakar D, Hogue CP, Rutta Z, Morales S, et al. Epithelial membrane protein-2 promotes endometrial tumor formation through activation of FAK and Src. PLoS ONE. (2011) 6:e1. doi: 10.1371/journal.pone.0019945
20. Forbes A, Wadehra M, Mareninov S, Morales S, Shimazaki K, Gordon LK, et al. The tetraspan protein EMP2 regulates expression of caveolin-1. J Biol Chem. (2007) 282:26542–51. doi: 10.1074/jbc.M702117200
21. Gordon LK, Kiyohara M, Fu M, Braun J, Dhawan P, Chan A, et al. EMP2 regulates angiogenesis in endometrial cancer cells through induction of VEG. Oncogene. (2013) 32:5369–76. doi: 10.1038/onc.2012.622
22. Fu M, Brewer S, Olafsen T, Wu AM, Gordon LK, Said J, et al. Positron emission tomography imaging of endometrial cancer using engineered anti-EMP2 antibody fragments. Mol Imaging Biol. (2013) 15:68–78. doi: 10.1007/s11307-012-0558-y
23. Qin Y, Takahashi M, Sheets K, Soto H, Tsui J, Pelargos P, et al. Epithelial membrane protein-2 (EMP2) promotes angiogenesis in glioblastoma multiforme. J Neurooncol. (2017) 134:29–40. doi: 10.1007/s11060-017-2507-8
24. Qin Y, Fu M, Takahashi M, Iwanami A, Kuga D, Rao RG, et al. Epithelial membrane protein-2 (EMP2) activates Src protein and is a novel therapeutic target for glioblastoma. J Biol Chem. (2014) 289:13974–85. doi: 10.1074/jbc.M113.543728
25. Wadehra M, Dayal M, Mainigi M, Ord T, Iyer R, Braun J, et al. Knockdown of the tetraspan protein epithelial membrane protein-2 inhibits implantation in the mouse. Dev Biol. (2006) 292:430–41. doi: 10.1016/j.ydbio.2006.01.015
26. Williams CJ, Chu A, Jefferson WN, Casero D, Sudhakar D, Khurana N, et al. Epithelial membrane protein 2 (EMP2) deficiency alters placental angiogenesis, mimicking features of human placental insufficiency. J Pathol. (2017) 242:246–59. doi: 10.1002/path.4893
27. Shimazaki K, Wadehra M, Forbes A, Chan AM, Goodglick L, Kelly KA, et al. Epithelial membrane protein 2 modulates infectivity of Chlamydia muridarum (MoPn). Microbes Infect. (2007) 9:1003–10. doi: 10.1016/j.micinf.2007.04.004
28. Wan X, Chen Z, Choi WI, Gee HY, Hildebrandt F, Zhou W. Loss of Epithelial Membrane Protein 2 Aggravates Podocyte Injury via Upregulation of Caveolin-1. J Am Soc Nephrol. (2016) 27:1066–75. doi: 10.1681/ASN.2014121197
29. Özlü SG, Demircin G, Tökmeci N, Çaltik Yilmaz A, Aydog Ö, Bülbül M, et al. Long-term prognosis of idiopathic nephrotic syndrome in children. Ren Fail. (2015) 37:672–7. doi: 10.3109/0886022X.2015.1010940
30. Dorval G, Gribouval O, Martinez-Barquero V, Machuca E, Tête MJ, Baudouin V, et al. Clinical and genetic heterogeneity in familial steroid-sensitive nephrotic syndrome. Pediatr Nephrol. (2018) 33:473–83. doi: 10.1007/s00467-017-3819-9
31. Groopman E, Goldstein D, Gharavi A. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. (2019) 380:142–51. doi: 10.1056/NEJMc1903250
32. Wang M, Chun J, Genovese G, Knob AU, Benjamin A, Wilkins MS, et al. Contributions of rare gene variants to familial and sporadic FSGS. J Am Soc Nephrol. (2019). doi: 10.1681/ASN.2019020152. [Epub ahead of print].
33. Becker GJ, Hewitson TD. Animal models of chronic kidney disease: useful but not perfect. Nephrol Dial Transplant. (2013) 28:2432–8. doi: 10.1093/ndt/gft071
34. Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol. (2019) 30:23–32. doi: 10.1681/ASN.2018090912
35. Sun MM, Chan AM, Law SM, Duarte S, Diaz-Aguilar D, Wadehra M, et al. Epithelial membrane protein-2 (EMP2) antibody blockade reduces corneal neovascularization in an in vivo model. Invest Ophthalmol Vis Sci. (2019) 60:245–54. doi: 10.1167/iovs.18-24345
36. Fu M, Maresh EL, Helguera GF, Kiyohara M, Qin Y, Ashki N, et al. Rationale and preclinical efficacy of a novel anti-EMP2 antibody for the treatment of invasive breast cancer. Mol Cancer Ther. (2014) 13:902–15. doi: 10.1158/1535-7163.MCT-13-0199
37. Telander DG, Yu AK, Forward KI, Morales SA, Morse LS, Park SS, et al. Epithelial membrane protein-2 in human proliferative vitreoretinopathy and epiretinal membranes. Invest Ophthalmol Vis Sci. (2016) 57:3112–7. doi: 10.1167/iovs.15-17791
38. Torella D, Iaconetti C, Tarallo R, Marino F, Giurato G, Veneziano C, et al. miRNA regulation of the hyperproliferative phenotype of vascular smooth muscle cells in diabetes. Diabetes. (2018) 67:2554–68. doi: 10.2337/db17-1434
39. Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, et al. Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci USA. (2017) 114:E9989–98. doi: 10.1073/pnas.1710964114
40. Wu H, Malone AF, Donnelly EL, Kirita Y, Uchimura K, Ramakrishnan SM, et al. Single-cell transcriptomics of a human kidney allograft biopsy specimen defines a diverse inflammatory response. J Am Soc Nephrol. (2018) 29:2069–80. doi: 10.1681/ASN.2018020125
41. Cinà DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J, et al. Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol. (2012) 23:412–20. doi: 10.1681/ASN.2011070690
42. Dymecki SM. Flp recombinase promotes site-specific DNA recombination in embryonic stem cells and transgenic mice. Proc Natl Acad Sci USA. (1996) 93:6191–6. doi: 10.1073/pnas.93.12.6191
43. Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA. (1996) 93:5860–5. doi: 10.1073/pnas.93.12.5860
44. Sörensen I, Adams RH, Gossler A. DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood. (2009) 113:5680–8. doi: 10.1182/blood-2008-08-174508
Keywords: Emp2, nephrotic syndrome, proteinuria, smooth muscle, vascular
Citation: Donnan MD, Scott RP, Onay T, Tarjus A, Onay UV and Quaggin SE (2019) Genetic Deletion of Emp2 Does Not Cause Proteinuric Kidney Disease in Mice. Front. Med. 6:189. doi: 10.3389/fmed.2019.00189
Received: 24 May 2019; Accepted: 09 August 2019;
Published: 27 August 2019.
Edited by:
Heon Yung Gee, Yonsei University, South KoreaReviewed by:
Sandra Merscher, University of Miami, United StatesShuta Ishibe, Yale University, United States
Copyright © 2019 Donnan, Scott, Onay, Tarjus, Onay and Quaggin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan E. Quaggin, cXVhZ2dpbkBub3J0aHdlc3Rlcm4uZWR1