 Keli Hočevar1
Keli Hočevar1 Aleš Maver1
Aleš Maver1 Marijana Vidmar Šimic2
Marijana Vidmar Šimic2 Alenka Hodžić1Alexander Haslberger3
Alenka Hodžić1Alexander Haslberger3 Tanja Premru Seršen2,4
Tanja Premru Seršen2,4 Borut Peterlin1*
Borut Peterlin1*- 1Clinical Institute of Medical Genetics, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 2Division of Obstetrics and Gynecology, Department of Perinatology, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 3Department of Nutritional Sciences, University of Vienna, Vienna, Austria
- 4Medical Faculty, University of Ljubljana, Ljubljana, Slovenia
Background: Preterm delivery (PTD) represents an important public health and therapeutic challenge. Despite the reported link between the composition of vaginal microbiome and PTD, previous studies were inconsistent in their conclusions and utilized non-uniform designs. We performed an independent case-control study carried out on the Slovenian population, where we re-evaluated the role of the vaginal microbiome in PTD.
Methods: Vaginal microbiomes of pregnant women who delivered preterm were compared to those delivered at term to examine differences in the microbial richness, diversity, and differential abundance of specific taxa. We obtained vaginal swab samples from 155 Caucasian women who were classified as either term (≥380/7 weeks, n = 107) or preterm (≤366/7 weeks, n = 48) in exclusion of any other medical or obstetric conditions. The vaginal microbiomes of these women were characterized by 16S ribosomal RNA (rRNA) gene sequencing of the V3-V4 region on the MiSeq platform.
Results: Women who experienced PTD had a higher microbial richness (Chao1, P = 0.011) and alpha diversity (Shannon, P = 0.00059) than women with term deliveries. We report that overall vaginal microbial community composition (beta-diversity) was significantly different by delivery gestational age category (PWeightedUnifrac < 0.001). Women who delivered preterm had decreased Lactobacilli spp. abundance as well as increased abundance of Gardnerella and other bacterial vaginosis (BV) and aerobic vaginitis (AV) associated genera including Atopobium, Sneathia, Gemella, Megasphaera, Dorea, Streptococcus, and Escherichia/Shigella.
Conclusions: In the present study, we provide further evidence that vaginal microbiome composition is associated with PTD.
Introduction
Preterm delivery (PTD), defined by the World Health Organization (WHO) as the delivery of an infant before completed 37 weeks of gestation (1), represents a major global public health burden. It is the leading determinant of neonatal morbidity and mortality in developed countries (2) and the second most common cause of death in children under the age of five worldwide (3). Annually, 5–18% of pregnancies end preterm, resulting in 15 million premature babies (3), and prevalence is still escalating. Moreover, survivor premature babies have an increased risk of neonatal complications associated with the immaturity of organ systems, such as respiratory distress syndrome, cerebral palsy, retinopathy, hearing impairment, and bronchopulmonary dysplasia as well as increased long-term risk for a spectrum of cognitive and neurologic disorders (4–6).
Underlying causes of PTD are not completely understood, with more than 65–75% spontaneous PTDs with idiopathic onset (2, 7). Thus, it is still challenging to identify women at higher risk. Epidemiologic data suggest that complex genetic, environmental and sociodemographic factors are involved. Spontaneous PTD is a syndrome of numerous pathological processes (3), including certain maternal or fetal conditions, such as preeclampsia, intrauterine infection, cervical insufficiency, uterine anomalies, preterm premature rupture of membranes (PPROM), polyhydramnios, and fetal malformations (8). Previously observed risk factors also include, prior PTD, multifetal gestation, young or advanced maternal age, assisted reproductive technology (ART), black race, smoking, extremes of body-mass index (BMI), and low socioeconomic status (2).
Multiple lines of evidence support the role of maternal vaginal microbiota as a potential risk factor for PTD. Most notably, as much as 40–50% of premature deliveries are assumed to be due to maternal infection and inflammation (9–11). Prior research has shown that the most frequent cause of bacterial infection represents ascending pathway from the vagina through the cervix and into the uterus (3), where bacteria provoke deciduitis and chorioamnionitis or enter into the amniotic cavity and the fetus. Immediately after passing amniotic membranes, bacteria and their products are detected by pattern recognition receptors, such as toll-like receptors (TLRs), which causes the production of pro-inflammatory cytokines, chemokines, prostaglandins, and proteases leading to myometrial contractions. Additionally, inflammatory cascade leads to remodeling and disruption of fetal membranes (3, 11, 12). Hematogenous dissemination has also been proposed as an alternative route of intrauterine infection, as bacteria involved in periodontal disease have been found in the amniotic fluid (13). Furthermore, in utero microbial colonization of fetus has been suggested to have a role in the early establishment of the infant's microbiome (14, 15).
During pregnancy, a rise in the level of progesterone and estrogens, as well as immunological changes, significantly remodel composition of the human vaginal microbiome toward stable, low richness and diversity community structures dominated by Lactobacillus spp., which provide greater resistance and a protective role against genital tract infections (16, 17). Although dysbiosis (disruption of the vaginal microbiota) is asymptomatic in about 40% of affected women, it may predispose to a higher risk of urogenital conditions including aerobic vaginitis (AV) and bacterial vaginosis (BV) (18). The latest was reported to increase the risk of PTD ~5-fold (19).
The link between bacterial diversity and PTD has previously been demonstrated (20). Additionally, recent advances in the culture independent-techniques, using next-generation sequencing technologies proposed the causative role of overall vaginal microbiota composition in the development of PTD, although findings across studies have been inconsistent and largely ethnically dependent (7, 21–29). Therefore, the role of the vaginal microbiome in the etiology of PTD remains to be delineated. However, studies (7, 17, 25, 30) have unequivocally agreed that racial differences in the composition of vaginal microbiome play an important role in the detection of associations. Additionally, different conclusions about the role of vaginal microbiota in the development of PTD may have been reached due to the following reasons: (1) various definitions of PTD, (2) inclusion criteria varied widely (from low-risk PTDs to medically indicated and high-risk PTDs), (3) small power, (4) diverse designs, including different sequencing technologies and different 16S regions sequenced.
Although most of the previous 16S sequencing studies have identified a degree of association between the vaginal microbiome and PTD, there are inconsistencies in results that cannot currently be attributed to either different methodologies, heterogeneous definitions of PTD, or actual variations between study populations. Therefore, we used high-throughput 16S rRNA sequencing of the V3-V4 region in an independent Caucasian population to characterize whether the composition of maternal vaginal microbiota prior delivery differs between women who subsequently delivered preterm and those who delivered term.
Materials and Methods
Ethics Statement
All the women provided written informed consent prior to recruitment. This study was approved by the Slovenia National Medical Ethics Committee (approval number 90/02/15) and was conducted according to the principles expressed in the Declaration of Helsinki.
Sample Collection and Clinical Assessment
We included 48 pregnant, Caucasian women, with spontaneous onset of premature labor with or without PPROM (≤366/7 weeks) (cases) and 107 pregnant women with a spontaneous onset of labor with or without premature rupture of membranes (PROM) or planned cesarean section at term (≥380/7) (controls). The women gave birth at the Department of Perinatology of the University Medical Center Ljubljana in the period from October 2015 to January 2019. We assessed the gestational age at delivery by last menstrual period and confirmed it through ultrasound examination in the first trimester by the measurement of the fetal crown-rump length (31). Vaginal swabs of the lateral wall of the vagina were collected by experienced obstetricians of the women who exhibited signs of labor based on the presentation of uterine contractions and cervical dilation at the admission to the delivery department.
Women who delivered preterm were subdivided into two groups according to the gestational age of labor; early preterm delivery (EPTD) (≤336/7 weeks) and late preterm delivery (LPTD) (340/7to 366/7 weeks).
The inclusion criteria for both, cases and controls, were as follows: (1) healthy pregnant woman with healthy fetus, (2) singleton pregnancy, (3) spontaneous onset of labor with or without PPROM for cases and a spontaneous onset of labor with or without PROM (380/7 to 406/7 weeks) or a planned cesarean section between 390/7 and 406/7 weeks of gestation for controls (in the absence of exclusion criteria), (4) ≥18 years of age, and (5) ability to provide informed consent. Exclusion criteria for cases and controls were: (1) use of antibiotic within the week prior to inclusion, (2) use of vaginal antimicrobials, antibiotics or lactic acid <1 week prior to inclusion, (3) individuals receiving immunosuppressive therapy, (4) placenta previa, (5) uterine anomaly, (6) chronic disease, including pre-pregnancy hypertension, diabetes mellitus, and morbid obesity (class III WHO), (7) human immunodeficiency virus (HIV) or hepatitis C positive status, (8) bleeding due to abruption or placenta previa, (9) vaginal lavage during or before pregnancy, (10) hypertensive diseases in pregnancy (preeclampsia), (11) fetus anomalies, (12) intrauterine death, and (13) intrauterine growth restriction (IUGR).
National Perinatal Information System of Slovenia (NPIS) was reviewed to collect additional data on the pregnancy, delivery and postpartum. Women participating to this study were also asked to complete a brief in-person questionnaire to collect additional demographic and medical information (age, working status, cigarette smoking, pre-pregnancy BMI, antibiotics use during pregnancy, gestational diabetes, previous PTD, and vaginitis.
Bacterial DNA Isolation, 16S rRNA Gene Amplification and Sequencing
The vaginal swabs for genomic bacterial DNA isolation were transferred to the laboratory in the Amies transport medium and stored at −80°C until isolation. The enzymatic and mechanical cell lysis was performed as described by Ravel et al. (30). The lysate was processed using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany), according to manufacturer's guidelines for DNA purification from buccal swabs (recommendations for cotton or Dacron swabs) with minor modifications, including incubation at 70°C for 10 min instead of 56°C and 5-min incubation step at room temperature after the addition of 100 μl of heated (56°C) elution buffer. The DNA concentrations in the samples were quantified using the Qubit dsDNA high-sensitivity Assay Kit (Thermo Fischer Scientific). After isolation, the selected region (V3-V4) of the species-specific 16S rRNA gene was amplified by a polymerase chain reaction (PCR), using a universal set of Illumina adapter containing primers: 341F (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′) and 805R (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′). V3-V4 region was chosen to also assess differences in Gardernella abundance between groups (32). For the preparation of libraries, we used the standardized 16S Metagenomic Sequencing Library Preparation Protocol (Illumina®), using KAPA HiFi HotStart ReadyMix 2X for amplification. The amplicons of each sample were labeled with Nextera XT Indexes. The PCR products were quantified and quality checked by using Bioanalyzer DNA 1000 chip (Agilent Technologies, Santa Clara, CA, USA). Libraries of pooled samples were sequenced on the Illumina MiSeq sequencer according to the manufacturer's specifications in the 2 × 300 bp pair-end runs (MiSeq Reagent kit v3).
Bioinformatics Analysis
The sequence data were imported into the R statistical environment (version 3.5.1) from demultiplexed FASTQ files, containing sequences with an average Phred score >30. Microbiome analysis was performed using Bioconductor software1 following Callahan et al. (33) workflow for Big Data. Trimming and filtering were carried out with The Divisive Amplicon Denoising Algorithm 2 (DADA2) (34) on paired reads jointly, with forward reads truncated at position 260 nt, and the reverse reads at position 240 nt based on the quality scores. First 17 nucleotides of forward reads and 21 nucleotides of reverse reads were trimmed to remove primers with ambiguous sequences. Filtering parameters were as follows; zero ambiguous base calls; enforcement of a maximum of 2/3 expected errors per forward/reverse reads, respectively. The relationship between quality scores and error rates was assessed separately for each run in order to minimize batch effects resulting from run-to-run variability. DADA2 was used to produce a sequence table by independently inferring amplicon sequence variants (ASVs) from the forward and reverse sequences of each sample using the run-specific error rates and to further merge read pairs. ASV represents a higher-resolution analog of the common operational taxonomic unit (OTU) table (34), such that it clusters the sequence reads into a finer taxonomic resolution without the need for a particular similarity cut-off (34, 35). Sequences identified as chimeric were removed. Taxonomy was assigned to ribosomal sequence variants using the RDP (Ribosomal Database Project) naive Bayesian classifier algorithm in the DADA2 R package and Silva v128 database (36). Only annotations that had more than 0.6 in their bootstrap confidence estimation values were accepted for further analyses. Clinical, taxonomic, AVS and phylogenetic data were combined into a single object using the phyloseq package (37) (version 1.24.2) for R. Due to the limitations of species-level classification with 16S rRNA sequencing, an additional aligning of representative sequences was performed by the Basic Local Alignment Search Tool (BLAST) (38), where necessary.
Statistical Analysis
All statistical analyses were conducted using R programing language (version 3.5.1). Alpha-diversity metrics were calculated at ASV-level, after rarefying at the minimal sequencing depth of 39,998 reads by performing 1,000 iterations of random subsampling. Rarefaction analysis was performed to evaluate whether further sequencing would likely detect additional AVSs, using the vegan2 package for R. The microbial richness (number of unique ASVs) was evaluated by the Chao1 index, and alpha diversity by Shannon diversity index, which takes into account richness as well as the abundance of ASVs for each sample, using the phyloseq package (version 1.24.2) (37). Continuous variables were tested for the normality with the Shapiro-Wilk normality test and visually inspected by histograms. Differences in richness and diversity between the different sample groups were calculated by the nonparametric Wilcoxon rank-sum test and the Kruskal-Wallis test when comparing three groups (39).
To estimate differences in beta-diversity, ASVs occurring in fewer than 3 samples and with a count of <10 across all samples were excluded from raw data. The ASV table normalized to relative abundance values and multidimensional scaling (MDS) was performed on phylogenetic distance matrices; weighted UniFrac (40). Variation in the overall microbial community structure between groups was assessed with permutational multivariate analyses of variance (PERMANOVA), using adonis function implemented in the R package vegan with 999 permutations for significance testing. Differences between the outcome groups were visually presented by generating Principal Coordinates Analysis (PCoA) plots and confidence ellipses in the R package ggplot.
The differential relative abundance of taxa between groups was determined at ASVs level using the Bioconductor package DESeq2 (version 1.20.0) in R (41). After variance-stabilizing transformations, we normalized samples using the nonparametric Wald negative binomial regression. Results were expressed as log2 fold change and taxa were considered significantly differentially abundant between groups if their adjusted P-value was <0.05. ASVs with prevalence >3% and >50 reads study-wide were included in the DESeq2 analysis.
Vaginal community state types (CST) were determined at ASV level, following the script by DiGiulio et al. (24). The Bray-Curtis dissimilarity metric, based on relative abundance was used to calculate the distance between all samples. Medoid clustering approach (pam in R) was applied to PCoA distances. The appropriate number of clusters was determined using the gap statistic (k = 5) (42).
For hypothesis testing of specific-taxa associations, we performed one-sided Wilcox rank-sum tests on the mean relative abundance of a taxon across samples from the same group. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg false-discovery rate (FDR) correction method (43).
Results
Participants Characteristics
Vaginal microbiome sequencing was performed in 158 Caucasian pregnant women who contributed vaginal swabs at the admission to the perinatal delivery department. Three cases were excluded after the medical records were reviewed because the PTD was indicated due to IUGR. The majority of the participants had a vaginal delivery (94.8%), except eight controls, who delivered with a planned C-section (5.2%). Table 1 reports demographic and clinical features of participants. Of the included 155 women, 48 (31.0%) subsequently experienced PTD and 34 (70.8%) of these PTDs were classified as EPTD (<340/7 weeks.). Women who delivered at term did not differ significantly from those who delivered preterm in terms of age, working status, parity, pre-pregnancy BMI, the presence of thyroid disease, gestational diabetes mellitus (GDM), presence of asymptomatic bacteriuria, presence of vaginitis and vaginal treatment during pregnancy, urogenital infection, and smoking status (Table 1). All of the included participants were Caucasians of Slovenian ethnicity.
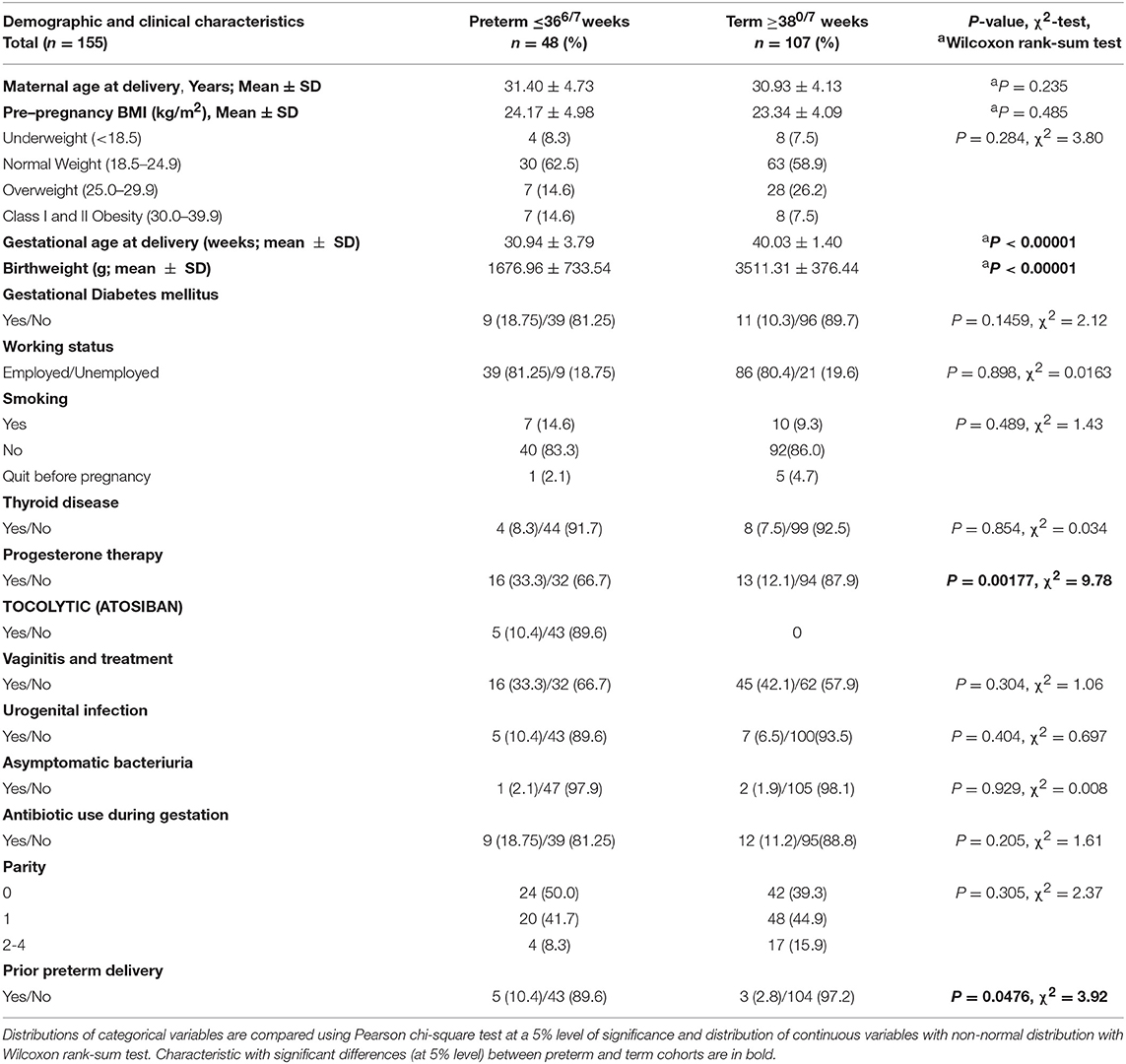
Table 1. Demographic and clinical characteristics of participants.
As reported previously, the women with prior PTD history were more likely to experience subsequent PTD (P = 0.0476). Of the PTD group, 18 women (37.5%) experienced PPROM. As expected, the PTD group had a greater number of women, receiving progesterone therapy (P = 0.00177). None of the women reported vaginal intercourse during the 3 days before sampling.
16S Sequencing Results
A total of 155 swab samples were analyzed, comprising of 48 PTD samples and 107 controls. After initial quality control steps and chimera removal, Illumina MiSeq sequencing of 16S rRNA gene resulted in a total of 12,233,991 processed paired-end V3-V4 reads, with an average read count of 78,928.97 reads per sample (min = 39,998; max = 223,503) (Supplementary Figure S1A).
Next, rarefaction curves were generated by subsampling evenly to the minimal depth of 39,998 reads. Rarefaction analysis showed that curves for each sample reached the plateau and thus no additional ASVs were expected to be found by increasing sequencing depth (Supplementary Figure S1B).
After removal of singletons, doubletons, rare ASVs (defined as prevalence ≥0.018 (in <3 samples), uncharacterized phylum, and taxa with total reads of <10 (8 × 10−7) study-wide, 390 ASVs were identified across 155 samples. 11,984,382 reads remained, presenting 94.2% of the total dataset.
Alpha and Beta Diversity
We demonstrated that the microbiome of participants across EPTD, LPTD, and term gestational delivery groups differed significantly in richness (Figure 1A) (Chao1, Kruskal-Wallis, P = 0.034). Individual comparison using Wilcoxon rank sum test showed that Chao1 index decreased in the term group compared to the all preterm cases (Chao1, P = 0.011) (Supplementary Figure S2) or EPTD group (Chao1, P = 0.012), indicating the reduced richness in women who delivered term babies.
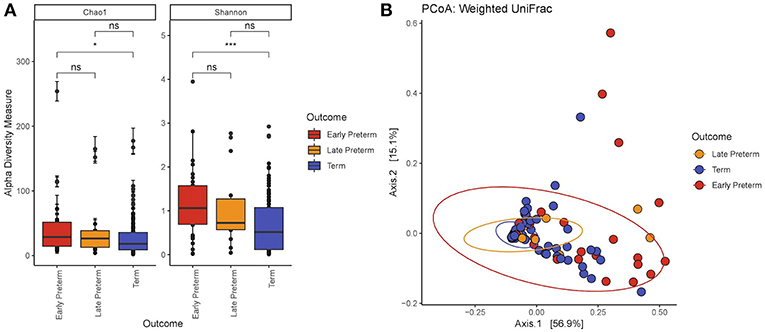
Figure 1. (A) Alpha diversity by the gestational delivery group. Chao1: Kruskal-Wallis χ2 = 6.76, P = 0.0034; Shannon: Kruskal-Wallis χ2 = 12.4, P = 0.0021. The microbiome of EPTD group has increased richness (Chao1) and diversity (Shannon Index) compared to term participants. (B) Principal coordinate analysis (PCoA) plot created at the ASV level generated by the Weighted Unifrac distance. The values in parentheses show the percentages of total community variation explained. The red dots represent EPTD women, the orange dots represent LPTD women and the blue dots represent women who delivered at term. P-values <0.05 and <0.001 are represented as * and ***, respectively.
Comparison of bacterial diversity between EPTD, LPTD, and term, revealed significant differences by Shannon index in the Kruskal-Wallis test (P = 0.0021) (Figure 1A). By Wilcoxon rank sum test we demonstrated a significant decrease in diversity in the term group compared to all preterm cases (Shannon, P = 0.00059) (Supplementary Figure S2A) or EPTD group (Shannon, P = 0.00079).
Next, we independently tested PPROM for alpha diversity, comparing cases who experienced PPROM (n = 18) to cases who delivered spontaneously without PPROM (n = 30). Among mothers with PTD, those who experienced PPROM showed a trend toward greater richness when compared to spontaneous PTD cases without PPROM, although the results were not statistically significant (Wilcoxon test, Chao1, P = 0.097) (Supplementary Figure S3A).
With respect to beta diversity, weighted UniFrac analysis demonstrated that overall microbial community composition of vaginal swabs was significantly different by delivery gestational age category (Figure 1B). This was statistically confirmed with the non-parametric adonis test (PWeightedUniFrac <0.001) (Table 2). Figure 1B shows the two-dimensional principal coordinate analysis (PCoA) plot, which indicates a clustering of EPTD, LPTD, and term groups, away from each other.
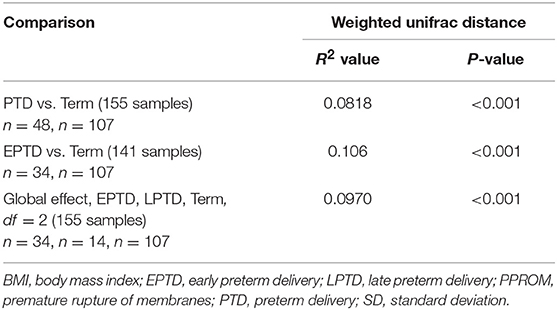
Table 2. Results from adonis (PERMANOVA) analysis (UniFrac distance and similarity measurements).
We did not detect significant differences in beta diversity between preterm cases who experienced PPROM and preterm cases who did deliver spontaneously without PPROM (Supplementary Figure S3B).
Composition of the Vaginal Microbiome
Across 155 participants, the dominant phyla in the vaginal microbiota were Firmicutes and Actinobacteria, which respectively made up 84.4 and 10.8% of total abundance, with lower contributions from Fusobacteria (1.29%), Proteobacteria (0.902%) and Tenericutes (0.874%). At order level, the top five dominant taxa were Lactobacillales (81.9%), Bifidobacteriales (8.07%), Coriobacteriales (2.48%), Fusobacteriales (1.29%), and Enterobacteriales (0.671%). The predominant bacterial families were Lactobacillaceae (77.9%), Bifidobacteriaceae (8.07%), Coriobacteriaceae (2.48%), Streptococcaceae (2.57%), and Leptotrichiaceae (1.23%) (Figure 2A). At the genus level, the dominant members were Lactobacillus (77.9%), Gardnerella (7.73%), Atopobium (2.46%), Streptococcus (2.57%), and Sneathia (1.20%) (Figure 2B). At the species level, L. iners and L. crispatus were most abundant (Figure 2C).
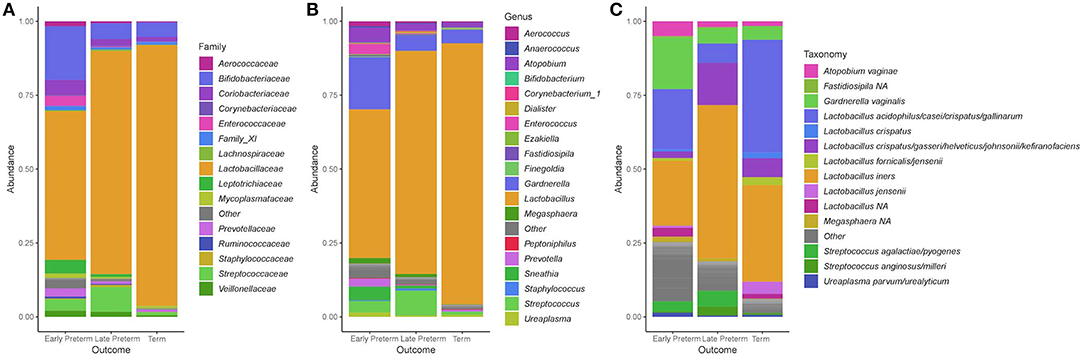
Figure 2. Stacked bar plots of average relative abundance at the (A) family, (B) genus, and (C) species taxonomic levels compared according to the gestational age of delivery. Lactobacillus acidophilus/casei/crispatus/gallinarum: 100% L. crispatus/100% L. gallinarum according to BLAST; Lactobacillus crispatus/gasseri/helveticus/johnsonii/kefiranofaciens: 100% L. gasseri according to BLAST.
In women, who delivered at term, the Lactobacillus genus constituted 88.3% and Gardnerella 4.63% of total bacteria. In that group 91/107 (85%) samples had Lactobacillus abundance above 70%, and 55/107 (51%) samples harbored Lactobacillus genera above 98%. In the EPTD group, Lactobacillus comprised 50.3% and Gardnerella 17.6%, and in LPTD 75.5 and 5.22%, respectively. Atopobium was present at higher percentages in the EPTD (5.05%) and LPTD (2.13%) groups when compared to the term group (1.59%). Relative abundances of genera within each sample according to a week of delivery are presented in Supplementary Figure S4.
Differential Abundance at ASVs Level Between Delivery Groups
Hypothesis-free approach at ASV level, using differential abundance analysis (DESeq2) revealed more than 24-fold higher abundance of ASVs belonging to species Lactobacillus gasseri and more than 6-fold higher abundance of ASVs belonging to Lactobacillus jensenii in women who delivered term compared to women who delivered preterm. 8-fold and 7-fold higher abundance of ASVs belonging to Sneathia sanguinegens and Streptococcus agalactiae/pyogenes were detected in the PTD group when compared to term (Supplementary Figure S5A). When EPTD and term groups were compared, results additionally indicated a higher abundance (3-fold) of Lactobacillus crispatus/gasseri/helveticus/johnsonii/kefiranofaciens (100% L. gasseri according to BLAST) in the term group (Supplementary Figure S5B). The AVSs representing the following taxa were detected differentially abundant in preterm vs. term comparison (Punadj <0.05), bud did not reach the significance after FDR adjustment for multiple testing: Lactobacillus crispatus/gasseri/helveticus/johnsonii/kefiranofaciens (100% L. gasseri according to BLAST), Shewanella algae/indica, Prevotella disiens, Gardnerella vaginalis, Streptococcus anginosus/milleri, Prevotella bivia/denticola, and Enterococcus faecalis. Prevotella timonensis, Anaerococcus hydrogenalis, Lactobacillus acidophilus/casei/crispatus/gallinarum (100% L. crispatus/L. gallinarum, BLAST), Atopobium vaginae, Staphylococcus haemolyticus/petrasii, and Bifidobacterium longum were detected as marginally significant before FDR adjustment (Supplementary Tables S1, S2).
Relative Abundance Comparisons of Individual Taxa
At the genus level, we analyzed and confirmed the reduced mean relative abundance of Lactobacillus in the PTD group when compared to term (Padj = 0.000177), using one sided-Wilcoxon rank-sum test on taxonomically grouped data.
Furthermore, we tested for some of the additional previously reported associations between PTD and genera observed to have increased abundance during BV or AV (24, 25) (Table 3). At the genus level, significant associations for following taxa, previously associated with BV were confirmed in our dataset: Gardnerella, Atopobium, Sneathia, Gemella, Megasphaera, and Dorea. We did not detect significant associations with PTD in mean relative abundances of the following genera: Ureaplasma, Mycoplasma, Mobiluncus, and Peptoniphilus. Among genera associated with AV, we detected an increased abundance of Streptococcus and Escherichia/Shigella (Table 3).
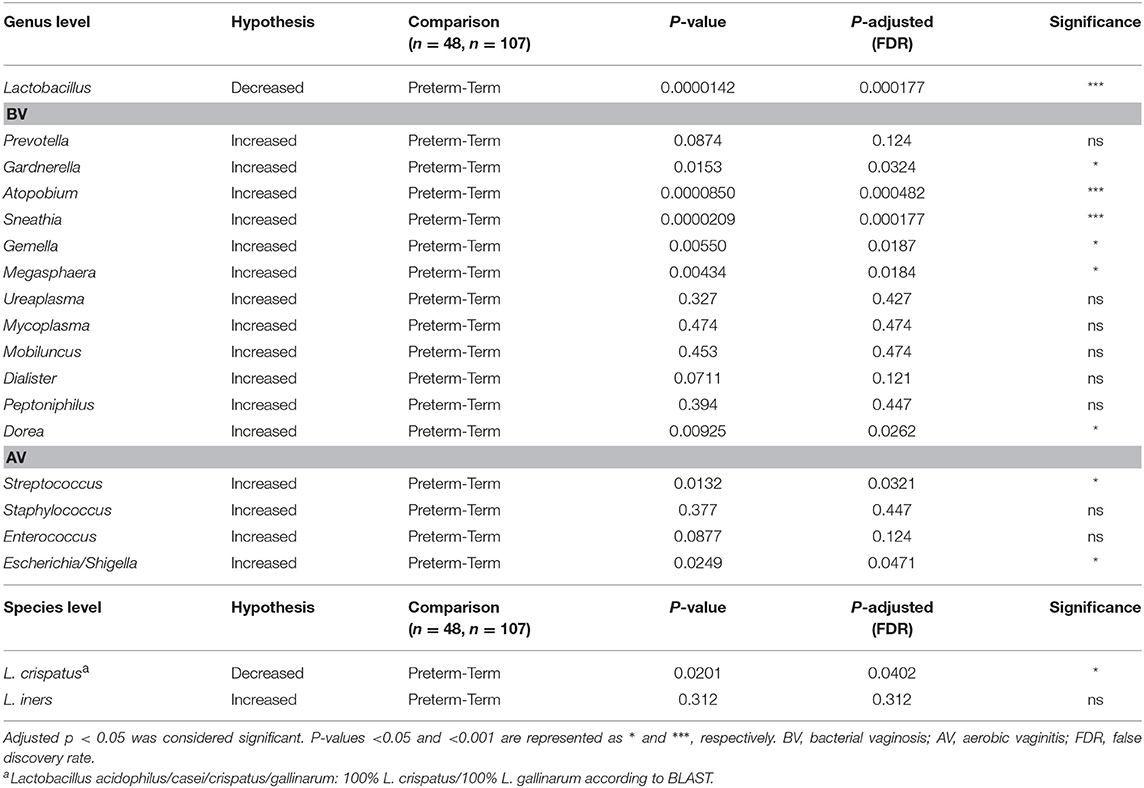
Table 3. Hypothesis testing: The P-values were calculated with the one-sided Wilcoxon rank-sum test and then adjusted for multiple hypotheses by the Benjamini–Hochberg method.
At the species level, we individually tested for L. crispatus and L. iners associations with PTD. The mean relative abundance of ASV belonging to Lactobacillus acidophilus/casei/crispatus/gallinarum (classified as 100% L. crispatus/L. gallinarum according to BLAST) was enriched in the term group compared to PTD (one-sided Wilcox-test, Padj = 0.0201), with 38.1% in the term and 26.9% in the PTD group. Association of L. iners with gestational age delivery group was not significant.
Vaginal Microbiome Community State Types (CSTs)
Hierarchical clustering analysis demonstrated that vaginal microbiota clustered into 5 distinct community profiles, consisting of L. crispatus (CST I), L. gasseri (CST II), L. iners (CST III), and L. jensenii/L. iners-dominated clusters (CST V) and a cluster depleted of Lactobacilli species and enriched for anaerobic bacteria (CST IV) (Figure 3). Frequencies of specific CSTs in the entire sample were as follows: CST I (31.6%), CST II (11.6%), CST III (32.3%), CST IV (16.8%), and CST V (7.74%). In CST IV cluster were 41.2% of EPTD participants, 21.4% of LPTD participants, and only 8.41% of the term participants (Figure 3A). CST I (dominated by L. crispaus) was present in 38.3% of term delivery group, 17.6% in EPTD and 14.3% in the LPTD group (Figure 3A). We observed a significant difference in the overall frequency of five CSTs between three delivery groups (term, LPTD, and EPTD), PFisherexacttest = 0.0025, which was even more profound when only term and the EPTD groups were considered (PFisherexacttest = 0.0005).
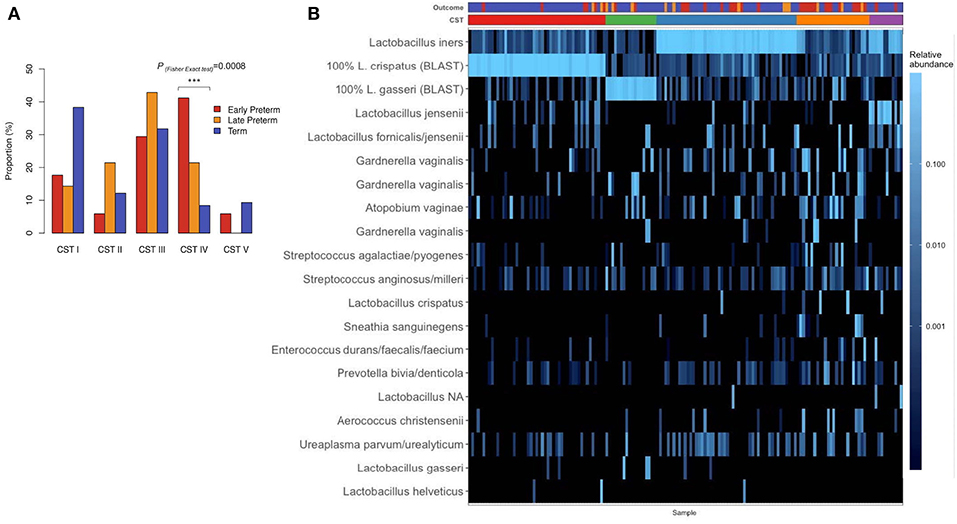
Figure 3. (A) The proportion of delivery group; early preterm (n = 34), late preterm (n = 14), term (n = 107), in specific CST. Significant difference was detected in overall frequency of five CSTs between term, LPTD, and EPTD (PFisher exact test= 0.0025) and between term-EPTD groups (PFisher exact test= 0.0005). (B) The heatmap of the relative abundance of 20 most abundant ASVs in the vaginal microbiome before the delivery of pregnant women by early preterm, late preterm, and term delivery. Frequencies of CSTI, CST II; III; IV; V in the entire sample were: 31.6, 11.6, 32.3, 16.8, 7.74%. P-value <0.001 is represented as ***.
When we tested previously reported association of Lactobacillus-depleted CST IV with PTD, the CST IV was significantly overrepresented in the EPTD group compared to term (PFisher exact test = 0.0008).
Discussion
Our findings showed that pregnant women who delivered preterm relative to term have higher richness and diversity of the vaginal microbiome, along with reduced Lactobacillus species, especially reduced L. crispatus and the increased abundance of the genus Gardnerella. Additionally, we found that overall frequencies of CSTs were significantly different between EPTD, LPTD, and term delivery groups, with CST IV (enriched for BV-associated taxa and higher diversity) observed in the EPTD group more often than in the term group. These observations are in accordance with the majority of the previously published 16S sequencing studies investigating the similar research question in Caucasian populations.
The higher alpha diversity and lower abundances of Lactobacillus spp. (CST IV) in the early stages of pregnancy were previously correlated with gestational age at delivery (24, 25). CSTs were previously shown to differ between term and preterm deliveries also in the study of Stafford et al. (27), where PTD has been associated with CST V (L. jesenii) dominance, while CST I (L. crispatus) and CST II (L. gasseri) were beneficial for term delivery. The association between PTD and vaginal dysbiosis (CST IV) was not confirmed in the study of Kindinger et al. (22). This discrepancy may be explained by the inclusion of only women at high-risk for PTD, possibly making the microbial-cause for PTD less prominent in that cohort. The study of Freitas et al. (44), conducted in Canadian cohort also failed to detect specific vaginal microbiome CSTs early in pregnancy that could predict PTD, however, the findings of increased richness and diversity in PTD are in agreement with our study, despite different methodologies used (pyrosequencing of cpn60).
While the protective role of L. crispatus was repeatedly demonstrated in most previous 16S sequencing studies (21, 22, 25, 27, 29), association of L. iners with PTD yielded conflicting results. The largest 16S sequencing study of white European population investigating the role of vaginal microbiota in PTD (21), concluded that L. iners/Ralstonia solanacearum oligotype was associated with decreased risk of early (<34 weeks), but not late (34–36 weeks) PTD, as well as L. gasseri/L. johnsonii, L. crispatus/L. acidophilus, and Bifidobacterium longum/Bifidobacterium breve oligotypes. In contrast, a recent 16S sequencing study of V1-V3 region by Kindinger et al. (22) showed that dominance of L. iners at 16 weeks of gestation is a potential risk factor for early PTD (<34 weeks). We did not manage to confirm the association with L. iners with either term or preterm delivery, however, our results indicated that its mean relative abundance was the highest in the LPTD group (51.8%), while it was more prevalent in term group (32.6%) compared to EPTD (21. 9%).
Consistent with the previous reports of an association between BV and AV with PTD (24, 25, 29), we detected increased abundance of BV-associated taxa, including Gardnerella, Atopobium, Sneathia (Sneathia sanguinegens), Gemella, Dorea, and AV-associated bacteria, Streptococcus (Streptococcus agalactiae/pyogenes), and Escherichia/Shigella genera in the PTD group, when compared to term. Contrary to the study of Callahan et al. (25), we did not confirm the association of genera Mobiluncus, Peptoniphilus, and Mycoplasma with PTD; however, we did detect the positive association of Megasphaera with PTD, which was not confirmed in a before-mentioned study. The association of Ureaplasma with PTD (24) was not replicated in our cohort, nor was it in the studies of Callahan et al. (25) and Freitas et al. (44). Similarly to our study, Elovitz et al. (7) reported the significant association of Atopobium vaginae with increased rates of PTD in non-Afro-American (AA) women, while Sneathia, Atopobium, and Megasphaera were associated with PTD when all women (AA and non-AA) were considered.
Our results differ from the results of the studies by Stout et al. (26) and Romero et al. (23), where no specific bacterial taxa or community was correlated with pregnancy outcome. Nevertheless, indicated PTDs (preeclampsia, fetal indications) were not excluded from the first study and only taxa that were present in at least 25% of samples were considered in the differential abundance analysis in the study of Romero et al. (23). We suppose that geographical and cultural differences (predominantly AA population), as well as a low number of PTD cases included (n = 23 and n = 18, respectively), might also contribute to negative results of those two studies.
Decreased Lactobacilli abundance manifests in the reduced lactic acid, hydrogen peroxide, and bacteriocins production, which enhances the proliferation of pathogenic bacteria known to cause BV (45, 46). Additionally, increased bacterial diversity has previously been associated with mucus and cytoskeleton alterations, cell death, increased expression of pro-inflammatory cytokines (29) and metalloproteinases and decreased IgG1/2 (47), which potentially leads to the inflammatory activation and stimulation of PTD. Furthermore, in the recent study, women with a non-Lactobacillus spp.-dominant microbiome were more likely to develop premature cervical dilation (48). It was also demonstrated (28, 49) that PPROM, which precedes 30% of all PTDs, is associated with high bacterial diversity and reduced Lactobacillus abundance prior to rupture. In the present study, we detected a tendency toward increased richness when preterm cases with PPROM were compared with preterm cases who delivered spontaneously without PPROM, but the result did not reach statistical significance.
It is already known that 16S sequencing experimental design impacts sensitivity to different taxa, and here we used the V3-V4 primer set, which is also sensitive to Gardnerella. Consequently, in our dataset Gardnerella represented 7.7% of the genus relative abundance, which is similar to study of the Callahan et al. (25) (5% of reads). The higher relative abundance of L. iners in the PTD group in the study of Kindinger et al. (22) may, therefore, be due to undetected Gardernella (V1-V3 primer set), which was shown to often coexist at high frequencies with L. iners (25), rather than a direct association of L. inners with PTD. Another explanation is that increased L. iners abundance during the 16th week of gestation might be associated with increased bacterial instability later in pregnancy, as L. iners is often associated with BV-related vaginal bacteria, due to its possible lack of protection against pathogens (50).
In contrast, L. crispatus has the largest genome of four Lactobacilli spp. (45), including genes encoding for bacteriocin, adhesin (anti-adhesive molecules against Gardnerella vaginalis), lactate, and succinate (51) and was reported to have protection against inflammation induced PTD (51). It has also been demonstrated that D-lactic acid levels were significantly higher when L. crispatus was the dominant vaginal bacterium compared to L. iners or Gardernella dominated vaginal microbiota. It was demonstrated that D-lactic acid enhances activation of anti-microbial innate and acquired immune system and also minimizes matrix metalloproteinase-8 (MMP-8) levels that would otherwise promote proteolytic activity and thus enhance bacterial migration past endocervix into the uterus, leading to infection-related PTD (52).
The strength of the present study is the inclusion of a large number of early PTDs (<34 weeks) compared to previous reports. Furthermore, we utilized strict inclusion criteria, meaning that PTDs indicated due to maternal or fetal complications, such as IUGR, preeclampsia, or fetal anomalies were excluded from the study. With an aim to exclude potential pre-existing risk factors for PTD and factors that could independently influence on microbiota composition, we also excluded individuals with chronic diseases, such as hypertension, diabetes mellitus, or women with the uterine anomalies. Another strength is the homogeneity of the study population, while it is known that microbiota composition is largely influenced by ethnicity (7, 17, 30). All participants were Caucasians and belonged to the same Slovenian ethnicity. Furthermore, compared to the most studies, we utilized bioinformatic approach with improved resolution and comprehensiveness that resolves exact ASVs, instead of imposing the arbitrary dissimilarity thresholds that define molecular OTUs (35).
Our study also has some limitations that need to be addressed. Firstly, sampling was conducted at a single time point; longitudinal sampling through pregnancy could add information about microbiome stability. Although previous studies showed that vaginal microbiome in the pregnancy remains relatively stable (24, 53), the increased Lactobacillus (23) and significant decrease in abnormal vaginal colonization rate were reported as the pregnancy progressed (54). Another limitation of our study is that 16S-sequencing technology does not assess the total abundance load of the taxa in the sample (gene copy measurement) that could be detected using a broad-range 16S rRNA quantitative PCR. Immune factors, that were suggested to lower the risk of PTD associated with vaginal microbiota, such as beta-defensin-2 (7) or proinflammatory cytokines (29), were not measured.
With a more comprehensive understanding of the vaginal microbiota in pregnancy in different populations, and its effect on PTD, detection of mothers at high-risk and development of pharmacological interventions for PTD prevention will become plausible.
In conclusion, our results suggest that maternal vaginal microbiome has an important role in the PTD etiology. In our independent analysis, we confirmed that vaginal microbiome of women who delivered preterm has increased richness and diversity prior delivery, decreased Lactobacillus abundance and the increased abundance of BV-associated taxa. Nevertheless, the role of vaginal microbiota in the etiology of PTD remains to be elucidated in further research considering different ethnicities and ideally with the integration of hosts and fetus genetic data.
Data Availability
Generated Statement: The datasets generated for this study can be found in the Sequence Read Archive (NCBI) repository, BioProject PRJNA544732, http://www.ncbi.nlm.nih.gov/bioproject/544732.
Ethics Statement
The studies involving human participants were reviewed and approved by Slovenia National Medical Ethics Committee, Ministry of Health, Štefanova 5, SI-1000 Ljubljana, Slovenia (approval number 90/02/15). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
KH, MV, TP, and AHo collected the samples. KH performed sequencing experiments and analyzed the data. BP, TP, and KH conceived and designed the study. KH wrote the manuscript. All authors contributed to the manuscript revision and approved the final manuscript.
Funding
This study was funded by the Slovenian Research Agency (ARRS), grant no. P3-0326.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2019.00201/full#supplementary-material
Abbreviations
AA, African American; ART, assisted reproductive technology; ASV, amplicon sequence variant; AV, aerobic vaginitis; BLAST, Basic Local Alignment Search Tool; BMI, body-mass index; BV, bacterial vaginosis; CST, community state type; DADA2, Divisive Amplicon Denoising Algorithm 2; EPTD, early preterm delivery; FDR, false-discovery rate; GDM, gestational diabetes mellitus; GLM, generalized linear model; HIV, human immunodeficiency virus; IUGR, intrauterine growth restriction; LPTD, late preterm delivery; MDS, multidimensional scaling, MMP-8, matrix metalloproteinase-8; OTU, operational taxonomic unit; PBS, phosphate-buffered saline; PCoA, Principal Coordinates Analysis; PCR, polymerase chain reaction; PERIS, Perinatal Information System of Slovenia; PERMANOVA, permutational multivariate analyses of variance; PPROM, premature rupture of membranes; PROM, premature rupture of membranes; PTD, preterm delivery; RDP, Ribosomal Database Project; rRNA, ribosomal RNA; spp, species; TLRs, toll-like receptors; WHO, World Health Organization.
Footnotes
References
1. Howson CP, Kinney MV, Lawn J. Born Too Soon: The Global Action Report on Preterm Birth. World Health Organization, (2013).
2. Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. (2008) 371:75–84. doi: 10.1016/S0140-6736(08)60074-4
3. Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science. (2014) 345:760–5. doi: 10.1126/science.1251816
4. Wood NS, Marlow N, Costeloe K, Chir B, Gibson AT, Wilkinson AE. Neurological and developmental disablitiy after extremely preterm birth. N Engl J Med. (2000) 343:378–84. doi: 10.1056/NEJM200008103430601
5. Ward RM, Beachy JC. Neonatal complications following preterm birth. BJOG An Int J Obstet Gynaecol. (2003) 110:8–16. doi: 10.1046/j.1471-0528.2003.00012.x
6. Mwaniki MK, Atieno M, Lawn JE, Newton CRJC. Long-term neurodevelopmental outcomes after intrauterine and neonatal insults: a systematic review. Lancet. (2012) 379:445–52. doi: 10.1016/S0140-6736(11)61577-8
7. Elovitz MA, Gajer P, Riis V, Brown AG, Humphrys MS, Holm JB, et al. Cervicovaginal microbiota and local immune response modulate the risk of spontaneous preterm delivery. Nat Commun. (2019) 10:1305. doi: 10.1038/s41467-019-09285-9
8. Romero R, Espinoza J, Kusanovic JP, Gotsch F, Hassan S, Erez O, et al. The preterm parturition syndrome. BJOG An Int J Obstet Gynaecol. (2006) 113:17–42. doi: 10.1111/j.1471-0528.2006.01120.x
9. Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N Engl J Med. (2002) 342:1500–7. doi: 10.1056/NEJM200005183422007
10. Oliver RS, Lamont RF. Infection and antibiotics in the aetiology, prediction and prevention of preterm birth. J Obstet Gynaecol. (2013) 33:768–75. doi: 10.3109/01443615.2013.842963
11. Agrawal V, Hirsch E. Intrauterine infection and preterm labor. Semin Fetal Neonatal Med. (2012) 17:12–9. doi: 10.1016/j.siny.2011.09.001
12. Witkin SS. The vaginal microbiome, vaginal anti-microbial defence mechanisms and the clinical challenge of reducing infection-related preterm birth. BJOG An Int J Obstet Gynaecol. (2015) 122:213–8. doi: 10.1111/1471-0528.13115
13. Fardini Y, Chung P, Dumm R, Joshi N, Han YW. Transmission of diverse oral bacteria to murine placenta: evidence for the oral microbiome as a potential source of intrauterine infection. Infect Immun. (2010) 78:1789–96. doi: 10.1128/IAI.01395-09
14. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. (2014) 6:237ra65. doi: 10.1126/scitranslmed.3008599
15. Perez-Muñoz ME, Arrieta M-C, Ramer-Tait AE, Walter J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome. (2017) 5:48. doi: 10.1186/s40168-017-0268-4
16. Aagaard K, Riehle K, Ma J, Segata N, Mistretta TA, Coarfa C, et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE. (2012) 7:0036466. doi: 10.1371/journal.pone.0036466
17. MacIntyre DA, Chandiramani M, Lee YS, Kindinger L, Smith A, Angelopoulos N, et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci Rep. (2015) 5:8988. doi: 10.1038/srep08988
18. Kaambo E, Africa C, Chambuso R, Passmore J-AS. Vaginal microbiomes associated with aerobic vaginitis and bacterial vaginosis. Front Public Heal. (2018) 6:78. doi: 10.3389/fpubh.2018.00078
19. Hay PE, Lamont RF, Taylor-Robinson D, Morgan DJ, Ison C, Pearson J. Abnormal bacterial colonisation of the genital tract and subsequent preterm delivery and late miscarriage. BMJ. (1994) 308:295. doi: 10.1136/bmj.308.6924.295
20. Hyman RW, Fukushima M, Jiang H, Fung E, Rand L, Johnson B, et al. Diversity of the vaginal microbiome correlates with preterm birth. Reprod Sci. (2014) 21:32–40. doi: 10.1177/1933719113488838
21. Tabatabaei N, Eren A, Barreiro L, Yotova V, Dumaine A, Allard C, et al. Vaginal microbiome in early pregnancy and subsequent risk of spontaneous preterm birth: a case-control study. BJOG An Int J Obstet Gynaecol. (2018) 126:349–58. doi: 10.1111/1471-0528.15299
22. Kindinger L, Bennett P, Lee YS, Marchesi JR, Smith A, Cacciatore S, et al. The interaction between vaginal microbiota, cervical length, and vaginal progesterone treatment for preterm birth risk. Microbiome. (2017) 5:1–14. doi: 10.1186/s40168-016-0223-9
23. Romero R, Hassan SS, Gajer P, Tarca AL, Fadrosh DW, Bieda J, et al. The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome. (2014) 2:18. doi: 10.1186/2049-2618-2-18
24. DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci USA. (2015) 112:11060–5. doi: 10.1073/pnas.1502875112
25. Callahan BJ, DiGiulio DB, Goltsman DSA, Sun CL, Costello EK, Jeganathan P, et al. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc Natl Acad Sci USA. (2017) 114:9966–71. doi: 10.1073/pnas.1705899114
26. Stout MJ, Zhou Y, Wylie KM, Tarr PI, Macones GA, Tuuli MG. Early pregnancy vaginal microbiome trends and preterm birth. Am J Obstet Gynecol. (2017) 217:356.e1–e18. doi: 10.1016/j.ajog.2017.05.030
27. Stafford GP, Reynolds S, Anumba DOC, Amabebe E, Stern V, Paley M, et al. Spontaneous preterm birth is associated with differential expression of vaginal metabolites by lactobacilli-dominated microflora. Front Physiol. (2017) 8:615. doi: 10.3389/fphys.2017.00615
28. You Y, Kwon EJ, Choi S, Hwang H, Choi S, Lee SM, et al. Vaginal microbiome profiles of pregnant women in Korea using a 16S metagenomics approach. Am J Reprod Immunol. (2019) 82:e13124. doi: 10.1111/aji.13124
29. Fettweis JM, Serrano MG, Brooks JP, Edwards DJ, Girerd PH, Parikh HI, et al. The vaginal microbiome and preterm birth. Nat Med. (2019) 25:1012–21. doi: 10.1038/s41591-019-0450-2
30. Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA. (2011) 108:4680–7. doi: 10.1073/pnas.1002611107
31. Salomon LJ, Alfirevic Z, Berghella V, Bilardo C, Hernandez-Andrade EJS. ISUOG practice guidelines: performance of first-trimester fetal ultrasound scan. Ultrasound Obstet Gynecol. (2013) 41:102–13. doi: 10.1002/uog.12342
32. Baines JF, Rupp J, Loeper N, Graspeuntner S, Künzel S. Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci Rep. (2018) 8:4–10. doi: 10.1038/s41598-018-27757-8
33. Callahan BJ, Sankaran K, Fukuyama JA, McMurdie PJ, Holmes SP. Bioconductor workflow for microbiome data analysis: from raw reads to community analyses. F1000Res. (2016) 5:1492. doi: 10.12688/f1000research.8986.1
34. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. (2016) 13:581–3. doi: 10.1038/nmeth.3869
35. Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. (2017) 11:2639–43. doi: 10.1038/ismej.2017.119
36. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. (2013) 41:D590–6. doi: 10.1093/nar/gks1219
37. McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. (2013) 8:61217. doi: 10.1371/journal.pone.0061217
38. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. (1990) 215:403–10. doi: 10.1016/S0022-2836(05)80360-2
39. Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. (1952) 47:583–621. doi: 10.1080/01621459.1952.10483441
40. Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. (2011) 5:169–72. doi: 10.1038/ismej.2010.133
41. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:1–21. doi: 10.1186/s13059-014-0550-8
42. Tibshirani R, Walther G, Hastie T. Estimating the number of data clusters via the gap statistic. J R Stat Soc Ser B. (2001) 63:411–23. doi: 10.1111/1467-9868.00293
43. Benjamini YYH. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Source J R Stat Soc Ser C Appl Stat. (1966) 15:216–33.
44. Freitas AC, Bocking A, Hill JE, Money DM. Increased richness and diversity of the vaginal microbiota and spontaneous preterm birth. Microbiome. (2018) 6:1–15. doi: 10.1186/s40168-018-0502-8
45. Witkin SS, Linhares IM. Why do lactobacilli dominate the human vaginal microbiota? BJOG An Int J Obstet Gynaecol. (2017) 124:606–11. doi: 10.1111/1471-0528.14390
46. Amabebe E, Anumba DOC. The vaginal microenvironment: the physiologic role of lactobacilli. Front Med. (2018) 5:181. doi: 10.3389/fmed.2018.00181
47. Borgdorff H, Gautam R, Armstrong SD, Xia D, Ndayisaba GF, Van Teijlingen NH, et al. Cervicovaginal microbiome dysbiosis is associated with proteome changes related to alterations of the cervicovaginal mucosal barrier. Mucosal Immunol. (2016) 9:621–33. doi: 10.1038/mi.2015.86
48. Brown RG, Chan D, Terzidou V, Lee YS, Smith A, Marchesi JR, et al. Prospective observational study of vaginal microbiota pre- and post-rescue cervical cerclage. BJOG. 126:916–25. doi: 10.1111/1471-0528.15600
49. Brown RG, Al-Memar M, Marchesi JR, Lee YS, Smith A, Chan D, et al. Establishment of vaginal microbiota composition in early pregnancy and its association with subsequent preterm prelabor rupture of the fetal membranes. Transl Res. (2019) 207:30–43. doi: 10.1016/j.trsl.2018.12.005
50. Petricevic L, Domig KJ, Nierscher FJ, Sandhofer MJ, Fidesser M, Krondorfer I, et al. Characterisation of the vaginal Lactobacillus microbiota associated with preterm delivery. Sci Rep. (2014) 4:5136. doi: 10.1038/srep05136
51. Ojala T, Kankainen M, Castro J, Cerca N, Edelman S, Westerlund-Wikström B, et al. Comparative genomics of Lactobacillus crispatus suggests novel mechanisms for the competitive exclusion of Gardnerella vaginalis. BMC Genom. (2014) 15:1070. doi: 10.1186/1471-2164-15-1070
52. Witkin SS, Mendes-Soares M. LI, Jayaram Aswathi, J LW, Forney Larry J. Influence of vaginal bacteria and d- and l-lactic acid isomers on vaginal extracellular matrix metal.pdf. MBio. (2013) 4:1–7. doi: 10.1128/mBio.00460-13
53. Romero R, Hassan SS, Gajer P, Tarca AL, Fadrosh DW, Nikita L, et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome. (2014) 2:4. doi: 10.1186/2049-2618-2-10
Keywords: preterm delivery, microbiome, next-generation sequencing, 16S rRNA gene, vaginal microbiome, pregnancy
Citation: Hočevar K, Maver A, Vidmar Šimic M, Hodžić A, Haslberger A, Premru Seršen T and Peterlin B (2019) Vaginal Microbiome Signature Is Associated With Spontaneous Preterm Delivery. Front. Med. 6:201. doi: 10.3389/fmed.2019.00201
Received: 14 June 2019; Accepted: 27 August 2019;
Published: 10 September 2019.
Edited by:
Patrice Mathevet, Lausanne University Hospital (CHUV), SwitzerlandReviewed by:
Milos Stojanov, Lausanne University Hospital (CHUV), SwitzerlandFauziah Binti Jummaat, Management and Science University, Malaysia
Copyright © 2019 Hočevar, Maver, Vidmar Šimic, Hodžić, Haslberger, Premru Seršen and Peterlin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Borut Peterlin, Ym9ydXQucGV0ZXJsaW5AZ3Vlc3QuYXJuZXMuc2k=