 Silvia Guil-Luna1,2*†
Silvia Guil-Luna1,2*† Rafael Mena1†Carmen Navarrete-Sirvent1Laura María López-Sánchez1,2Karima Khouadri1Marta Toledano-Fonseca1,2Ana Mantrana1Ipek Guler1Carlos Villar3Cesar Díaz4Francisco Javier Medina-Fernández4Juan Rafael De la Haba-Rodríguez1,2,5Enrique Aranda1,2,5,6‡
Rafael Mena1†Carmen Navarrete-Sirvent1Laura María López-Sánchez1,2Karima Khouadri1Marta Toledano-Fonseca1,2Ana Mantrana1Ipek Guler1Carlos Villar3Cesar Díaz4Francisco Javier Medina-Fernández4Juan Rafael De la Haba-Rodríguez1,2,5Enrique Aranda1,2,5,6‡ Antonio Rodríguez-Ariza1,2,5‡
Antonio Rodríguez-Ariza1,2,5‡- 1Instituto Maimónides de Investigación Biomédica de Córdoba, Córdoba, Spain
- 2Centro de Investigación Biomédica en Red de Cáncer (CIBERONC), Madrid, Spain
- 3Unidad de Gestión Clínica de Anatomía Patológica, Hospital Universitario Reina Sofía, Córdoba, Spain
- 4Unidad de Gestión Clínica de Cirugía General y del Aparato Digestivo, Hospital Universitario Reina Sofía, Córdoba, Spain
- 5Unidad de Gestión Clínica de Oncología Médica, Hospital Universitario Reina Sofía, Córdoba, Spain
- 6Departamento de Medicina, Facultad de Medicina de Córdoba, Universidad de Córdoba, Córdoba, Spain
Tumor budding has been found to be of prognostic significance for several cancers, including colorectal cancer (CRC). Additionally, the molecular classification of CRC has led to the identification of different immune microenvironments linked to distinct prognosis and therapeutic response. However, the association between tumor budding and the different molecular subtypes of CRC and distinct immune profiles have not been fully elucidated. This study focused, firstly, on the validation of derived xenograft models (PDXs) for the evaluation of tumor budding and their human counterparts and, secondly, on the association between tumor budding and the immune tumor microenvironment by the analysis of gene expression signatures of immune checkpoints, Toll-like receptors (TLRs), and chemokine families. Clinical CRC samples with different grades of tumor budding and their corresponding PDXs were included in this study. Tumor budding grade was reliably reproduced in early passages of PDXs, and high-grade tumor budding was intimately related with a poor-prognosis CMS4 mesenchymal subtype. In addition, an upregulation of negative regulatory immune checkpoints (PDL1, TIM-3, NOX2, and IDO1), TLRs (TLR1, TLR3, TLR4, and TLR6), and chemokine receptors and ligands (CXCR2, CXCR4, CXCL1, CXCL2, CXCL6, and CXCL9) was detected in high-grade tumor budding in both human samples and their corresponding xenografts. Our data support a close link between high-grade tumor budding in CRC and a distinctive immune-suppressive microenvironment promoting tumor invasion, which may have a determinant role in the poor prognosis of the CMS4 mesenchymal subtype. In addition, our study demonstrates that PDX models may constitute a robust preclinical platform for the development of novel therapies directed against tumor budding in CRC.
Introduction
Tumor budding has recently received much attention in the setting of progression and invasion in several malignancies including colorectal cancer (CRC). Tumor budding is defined as a single tumor cell or cluster of up to 4 cells at the invasive front (1, 2). High-grade tumor budding is now established as an independent prognostic factor since it has been associated with shorter disease-free survival (DFS) and overall survival (OS) in several types of cancer (2–4). Currently, it is widely believed that tumor buds provide the histological basis for invasion and metastasis, but it is still a matter of controversy if it is directly related with the epithelial–mesenchymal transition (EMT) (5, 6).
High-grade tumor budding has been inversely correlated with the presence of immune infiltrate at the invasive front. In addition, an overexpression of stem-cell related genes as ZEB1, ZEB2, DES, and VIM, and the activation of both WNT and TGF-β signaling, has been demonstrated to be expressed in tumor buds (7–9).
In this context, tumor budding has been recently associated with the poor-prognosis CMS4 subtype of CRC (10). This mesenchymal-like subtype is characterized by overexpression of stem cell markers, neoangiogenesis, and activation of TGF-β and WNT/β-catenin pathways which modulate immune evasion and the metastasis process (11–13). CMS4 tumors display low content of immune cells and exhibited the worst DFS and OS, demonstrating an urgent need to develop therapies for this subtype (11, 12). These findings are in line with the described profiles of tumor buds. However, the potential relationship between the immunosuppressive microenvironment of this poor-prognosis subtype and tumor buds still remains unknown (7).
Recognizing that tumor budding is an important contributor of the tumor invasion prognosis and the close relation with the CMS4 subtype, the translation of tumor budding to preclinical models has major challenges. Patient-derived xenografts (PDXs) generated by direct engraftment of human tumor tissue into immunodeficient mice have emerged as powerful preclinical platforms for analysis of predictive biomarkers, therapeutic targets, and drug discovery in cancer (14).
In this study, firstly, we examined early passages of CRC PDXs as potential models to analyze tumor budding and, secondly, we elucidated a link between high-grade budding and CMS4 subtype and specific signatures of immune evasion. PDXs may greatly help in the understanding of tumor budding and the involved mechanisms in the tumor microenvironment, which will provide new strategies and prospects for more effective treatments. In addition, treatments which simultaneously tackle the interactions between tumor buds and surrounding stroma could more effectively kill tumor cells or at least limit tumor progression and metastatic dissemination.
Materials and Methods
Patient and Inclusion Criteria
A consecutive, population-based series of forty-five patients over 18 years of age with resectable colon cancer submitted to Reina Sofía Hospital (Córdoba, Spain) was prospectively included. To avoid the bias of neoadjuvant treated patients, all rectal cancer patients were excluded. The study was approved by the Reina Sofía Hospital ethical committee (Protocol number PI-0150-2017) in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki). Informed consent was obtained from each patient, and clinical and pathological information was prospectively collected. The clinicopathological characteristics of patients are summarized in Table 1.
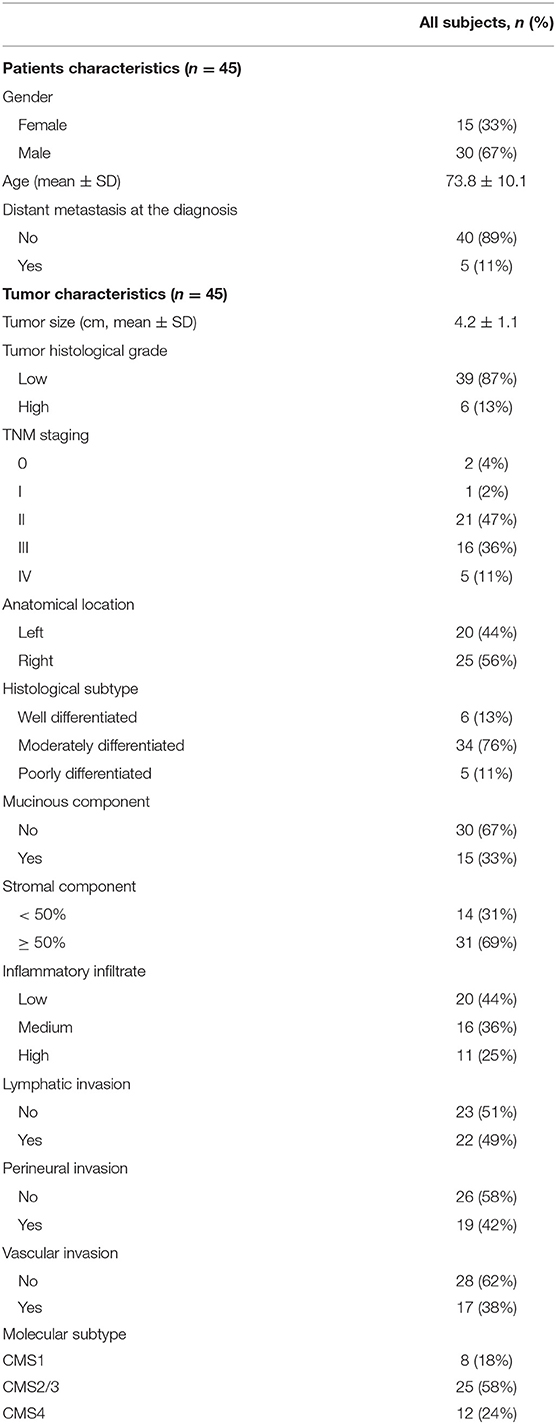
Table 1. Clinicopathological data of the patients included in the study.
Processing of Tumor Samples and Establishment of PDX Models
A total of 45 tumor samples were obtained just after surgical resection. Three adjacent tumor pieces were immediately collected in sterile conditions. One tumor piece was snap frozen and stored in liquid nitrogen for gene expression profiling, another one was fixed in 4% buffered formalin and then embedded in paraffin (FFPE) for hematoxylin and eosin staining and IHC studies, and the third fresh tumor piece was included in sterile PBS and used for establishment of PDXs.
The PDX engraftment was performed according to Puig and coauthors in NOD-SCID mice (NOD.CB17-Prkdcscid/Rj) (Janvier Laboratory, Paris, France) of 4–6 weeks of age (15). The animals were fed with a standard diet (D03-SAFE, Augy, France) and provided with drinking water ad libitum. Mice were daily monitored, and tumor growth was weekly measured until tumor volume was 1 cm3. Mice were ethically sacrificed under isoflurane anesthesia followed by cervical dislocation when tumor reached that size, if they appeared to be suffering, or after 6 months without tumor growth. Samples were immediately collected, fresh-frozen, formalin-fixed, and reimplanted (P1) as described above. This process was repeated to produce subsequent passages (until P3). Animal care and experimental procedures were approved by the University of Córdoba Bioethics Committee and followed the regulations of the European Union normative (26/04/2016/066).
Histological and Immunohistochemical Analysis of Patient Tumor Samples and PDX Models
Hematoxylin and eosin-stained tissue sections were evaluated by 2 trained pathologists (CVP and SGL) for the following criteria: histological subtype, invasion (lymphatic, vascular, or perineural), and stromal and inflammatory component. The degree of inflammatory cell infiltration was assessed in the center of the tumor and invasive margin of the tumor as reported previously (16). For the analysis of stroma, a representative 10 × magnification area of the invasive margin was selected and the percentage of the stroma for each sample was calculated as described by Gujam et al. (17).
On the other hand, IHC staining was performed on 4-μm FFPE sections using the antibodies detailed in Supplementary Table 1 for molecular classification of patient tumors and PDXs. Tissue sections were incubated in 10 mM citrate buffer (pH 6.0) for 5 min at 120°C for antigen retrieval. Endogenous peroxidase was neutralized by using the EnVision FLEX peroxidase-blocking reagent (Dako, Glostrup, Denmark) for 10 min. After blocking with 3% bovine serum albumin or following mouse-on-mouse staining protocol (Abcam, Cambridge, UK) in the case of PDXs, sections were incubated with the primary antibodies overnight at 4°C. Then, after incubation with the corresponding EnVision FLEX+ mouse or rabbit linker (Dako, Glostrup, Denmark) (30 min at room temperature), sections were incubated for 1 h with the secondary antibody EnVision FLEX/HRP (Dako, Glostrup, Denmark). The staining was visualized using 3,3-diaminobenzidine chromogen (Dako, Glostrup, Denmark) and counterstained with Harris hematoxylin. Negative controls without incubation with primary antibodies were also performed.
Immunohistochemistry-Based Molecular CMS Classification
Molecular classification by IHC was performed as described elsewhere (18). Individual cores were scored by trained pathologists (CVP and SGL) for FRMD6, ZEB1, HTR2B, AE1AE3, and CDX2 intensity and content. For MSI status, an analysis was performed with specific antibodies against hMLH1, hMSH2, hMSH6, and hPMS2, as described above. Immunohistochemical scores for each antibody were entered in the online classification tool (crclassifier.shinyapps.io/appTesting/) as described elsewhere (18). Using this classification, tumors were classified as CMS1, CMS2/3, or CMS4 subtypes.
Tumor Budding Determination
Tumor budding was defined as single tumor cells or tumor cell clusters of up to four cells in the stroma of the invasive front as previously reported (1). Tumor buds were assessed on pan-cytokeratin (clone AE1/AE3) immunostaining in a single hot spot measuring 0.785 mm2 for more accurate identification in cases of obscuring factors like inflammation or reactive stroma. Cutoffs as defined by International Tumor Budding Consensus Conference (ITBCC) were used: low (BD1, 0–4 buds; intermediate (BD2), 5–9 buds; and high (BD3) ≥10 buds (18).
Immune Gene Expression Profiling of Patient Tumors and PDX Models
The expression of genes encoding molecules involved in immune checkpoints, Toll-like receptors (TLRs), and chemokine receptors and their ligands was analyzed using the nCounter PanCancer immune-profiling panel from NanoString (Seattle, WA, USA) both in patient tumors and their corresponding PDXs. In order to minimize the variability between patients and xenografts, P0 passage was used for immune gene expression profiling. For this purpose, total RNA extraction was performed using RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's recommendations. Quantification and determination of the RNA purity were performed using a NanoDrop™ 1000 spectrophotometer (NanoDrop® ND-1000 UV-Vis Spectrophotometer, NanoDrop Technology), and RNA integrity Number (RIN) was measured using an Agilent 2200 TapeStation equipment. Data analysis was performed using nSolver software (NanoString Technologies, Seattle, WA, USA) to manage the raw data generated from the expression of each gene (19). The positive or negative expression of one particular gene indicates that the number of RNA molecules is higher or lower than the mean, respectively.
Statistical Analysis
Data were analyzed using GraphPad Prism 7 and R Software (version 3.5.0). Previously, in order to assess normality of the data, D'Agostino and Pearson Normality test was performed. The clinicopathological data were compared using Fisher's exact test or Mann–Whitney's test for qualitative and quantitative variables, respectively. Multivariate regression analysis was carried out with multinomial regression model for budding grades, including the variables selected by using the Akaike information criterion (AIC) with step-wise model selection. Differences in disease-free survival (DFS) were expressed as hazard ratios (HR) with 95% confidence intervals, and survival curves were constructed using the Kaplan–Meier method. All p values ≤ 0.05 were considered statistically significant.
Results
Tumor Budding Is Robustly Recapitulated in PDX Models and Is Closely Associated With the CMS4 Molecular Subtype of CRC
Overall, 82% (37/45) tumors were successfully engrafted with a mean latency period (time from day of inoculation to palpable tumor) of 30.7 ± 26.9 days for P0, which was shortened in subsequent passages (15.1 ± 9.8 for P1, 10.7 ± 5.0 for P2, and 8.1 ± 3.0 for P3).
Histopathological analysis of clinical tumors and their corresponding PDXs showed the preservation of the general tumor architecture and the histological subtype over several passages (Supplementary Figure 1). Remarkably, the determination of tumor budding status revealed a strong correlation between patient tumors and xenograft models (r = 0.72, p < 0.001) (Figure 1A).
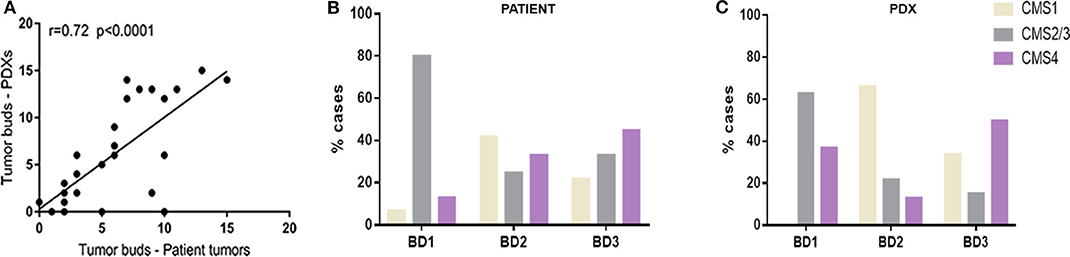
Figure 1. (A) Correlation between number of tumor buds in clinical tumors and in their corresponding PDX models. (B) Distribution of CMS molecular subtypes according to tumor budding grade in patient tumors. (C) Distribution of CMS molecular subtypes according to tumor budding grade in xenograft models (PDX).
In order to analyze the relationship between tumor budding and molecular subtypes of CRC, a molecular classification of patient tumors and xenografts was performed following the IHC-based method implemented by Trinh et al. (18). A strong concordance in the IHC expression patterns and consequently with the molecular CMS subtypes was observed between patient tumors and their corresponding PDXs with a Cohen's kappa coefficient of 0.96 (Figure 2A). Just in one case did the molecular subtype in the patient tumor (CMS4) shift to a different subtype (CMS2/3) in its PDX model (Figure 2B).
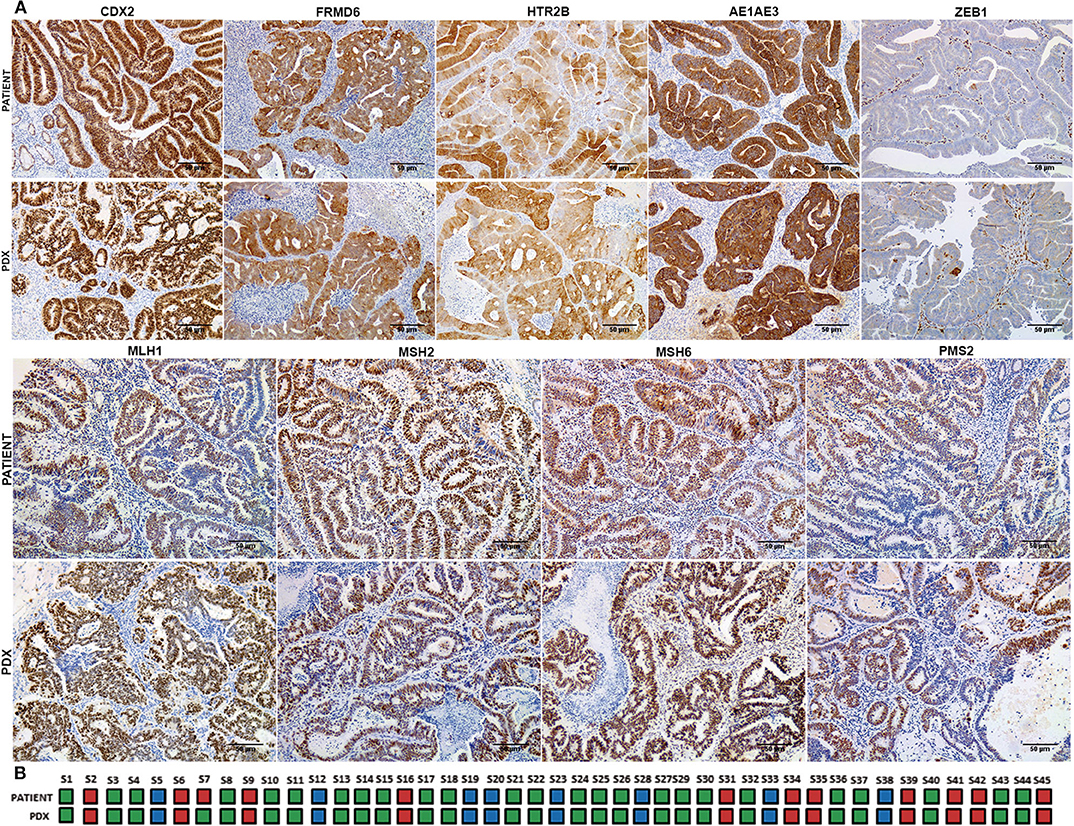
Figure 2. Immunohistochemical classification into CMS subtypes of patient tumor samples and their corresponding PDX models. (A) Representative immunohistochemical staining for CDX2, FRMD6, HTR2B, AE1AE3, ZEB1, MLH1, MSH2, MSH6, and PMS2 of a clinical tumor and its corresponding PDX model. (B) CMS classification concordance between patient tumors and their corresponding PDX models. Blue color corresponds to CMS1 subtype, green color corresponds to CMS2/3 subtype, and red color corresponds to CMS4 subtype. Scale bars: 100 μm.
In particular, while most of the BD1 tumors (80% in tumor patients and 63% in PDXs) were classified as CMS2/3 subtype, BD3 tumors were more abundantly present in the poor-prognosis CMS4 subtype in both patient tumors and xenografts (Figures 1B,C). In addition, only 13% of human CMS4 subtypes were classified with low grade of tumor budding (BD1).
High-Grade Budding (BD3) Is Associated With Adverse Clinicopathological Factors
Table 1 summarizes clinicopathological characteristics of patients included in this study. A high-grade tumor budding (BD3) was identified in 18 (40%) patients, followed by 12 (27%) patients with BD2 tumor budding and 15 (33%) patients with low-grade budding (BD1). The relationship between tumor budding and clinicopathological characteristics of patients is presented in Table 2. On univariate analysis, high-grade tumor budding was associated with poorly differentiated carcinomas (p = 0.02), higher stromal component (p = 0.02), tumor vascular invasion (p = 0.005), and presence of distant metastasis (p = 0.02). The histological subtype, tumor size, and stromal component were entered as covariates into the final multivariate model, based on the variable selection with the Akaike information criterion (AIC) using stepwise selection (Table 3). Regarding survival analysis, no event data (disease progression) were observed in low-grade budding. The intermediate- and high-grade tumor budding (BD2 and BD3) was significantly associated with poor DFS (p = 0.03) when compared with low-grade budding (Figure 3). Additionally, survival probability of intermediate- and high-grade tumor budding was compared but no significant difference was found [HR: 95% CI De-long BD3 vs. BD2: 1.38 (0.31–6.21)] (Figure 3).
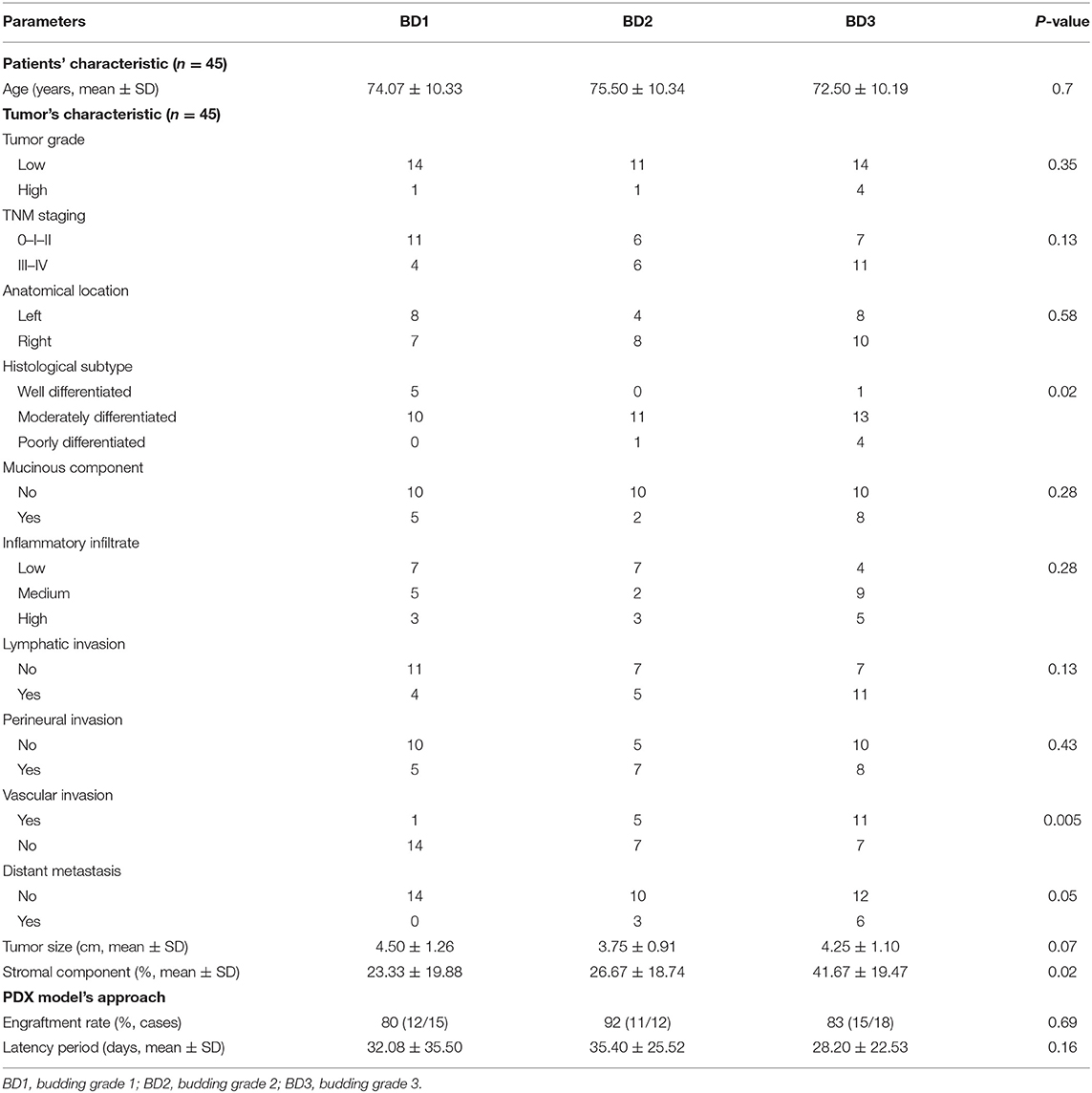
Table 2. Association between clinicopathological data of tumors and budding grade on univariate analysis.
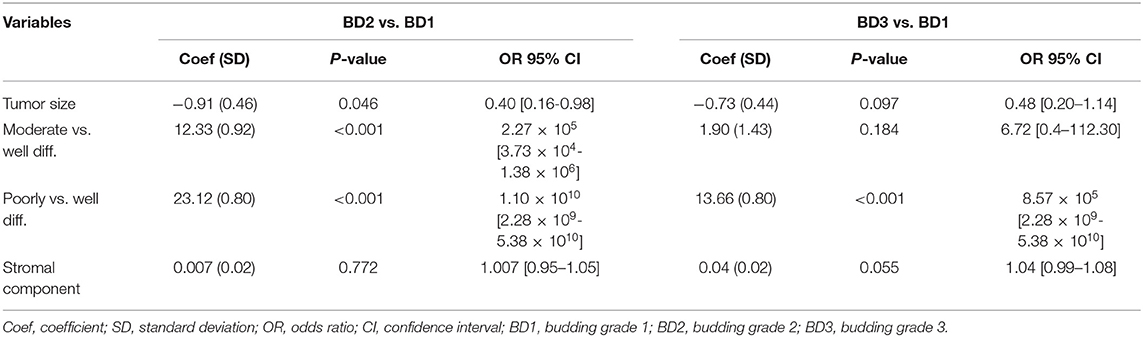
Table 3. Multivariate analysis of clinicopathological data of the tumors.
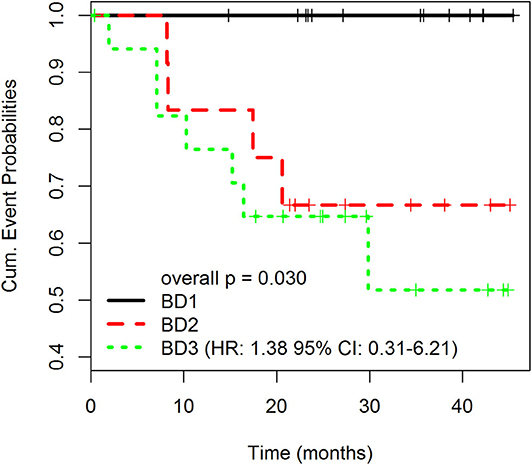
Figure 3. Disease-free survival (DFS) rates according to different grades of tumor budding (BD1, BD2, BD3). Overall p is 0.030. The p-values for pairwise comparisons are BD1 vs. BD2 = 0.034; BD1 vs. BD3 = 0.022; BD2 vs. BD3 = 0.595. HR: 1.38 95% CI: 0.31–6.21).
Gene Expression of Immune Checkpoint, TLRs, and Chemokine Profiles Reveals Similar Distinct Patterns According to Tumor Budding Grade in Patients and Xenografts
By using a PanCancer immune-profiling panel from the NanoString platform, we identified those immune-related genes overexpressed in high-grade tumor budding compared with low-grade budding. In addition, the immune gene expression profiles of patient tumors were compared with the gene expression profiles of their corresponding xenograft models (P0).
The expression of inhibitory immune checkpoints according to tumor budding grade is displayed in Figure 4. The comparative analysis revealed a general upregulation of immune checkpoint-related genes in tumors with BD3 tumors in comparison with BD1 tumors (Figure 4). Interestingly, these immune signatures were remarkably preserved in their corresponding PDX models. Particularly, a higher expression of PDL1, TIM-3, NOX2, and IDO1 genes was observed in BD3 tumors. However, PD1 and CTLA4 genes were less expressed in the higher tumor budding grades in both patients and xenografts (Figure 4).
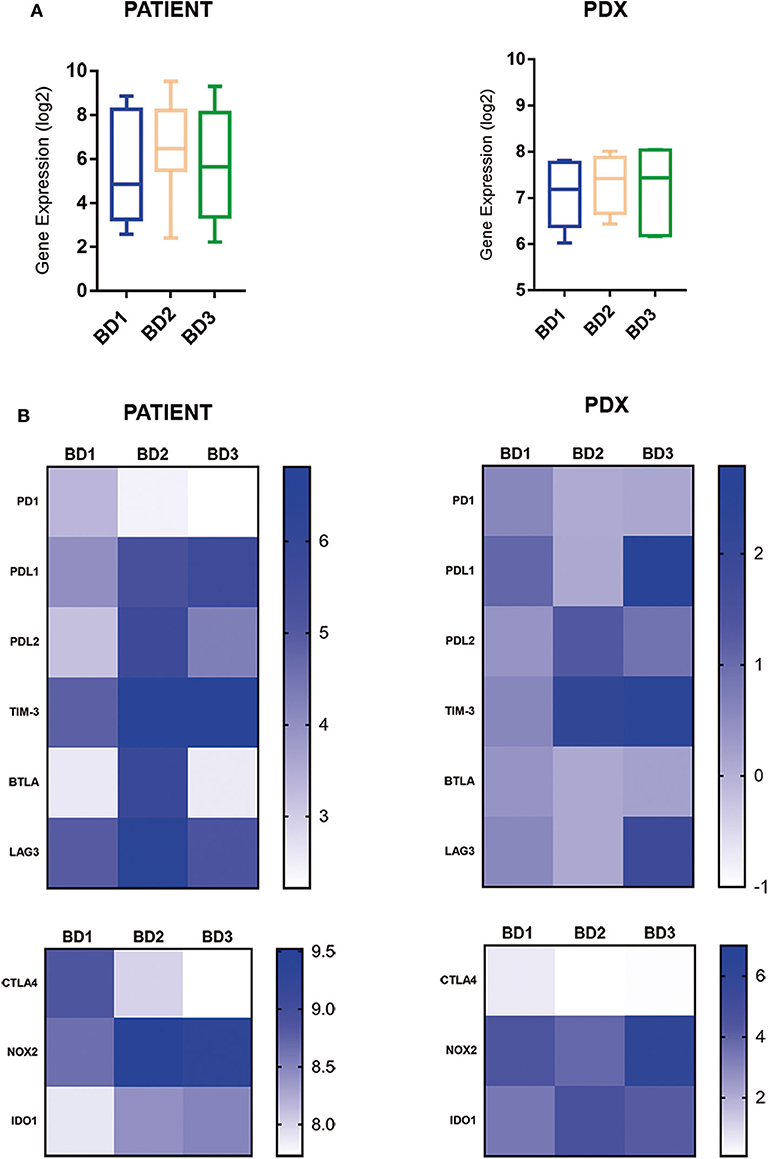
Figure 4. Global (A) and single (B) gene expression levels of immune inhibitor checkpoints in patient tumors and their corresponding PDX models, according to different grades of tumor budding (BD1, BD2, BD3).
The expression of the TLR superfamily also displayed a high correspondence between patients and PDXs with the highest values for high-grade tumor budding (Figure 5). Of note, TLR1, TLR3, TLR4, and TLR6 were overexpressed in tumors with BD3 compared to BD1 tumors (Figure 5).
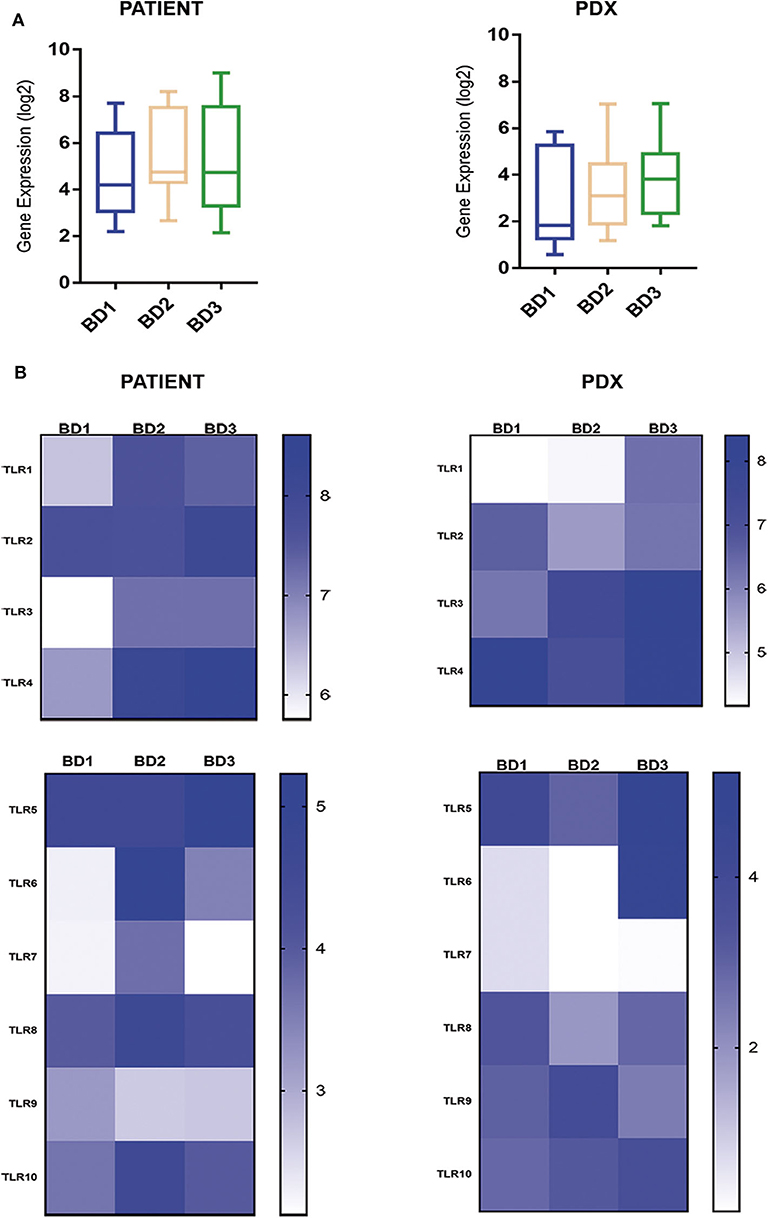
Figure 5. Global (A) and single (B) gene expression levels of the TLR gene family in patient tumors and their corresponding PDX models, according to different grades of tumor budding (BD1, BD2, BD3).
Regarding the CX chemokine receptor family, the results showed that BD3 tumors were associated with a higher expression of CXCR2 and CXCR4 (Figure 6) than BD1 tumors were. Among the chemokine ligands, CXCL1, CXCL2, CXCL6, and CXCL9 genes also displayed a higher expression in tumors with BD3 compared to low-grade tumor budding (Figure 7). Notably, these distinct gene expression profiles of chemokine receptors and ligands depending on different budding statuses were in general, with some exceptions, preserved in the PDX models.
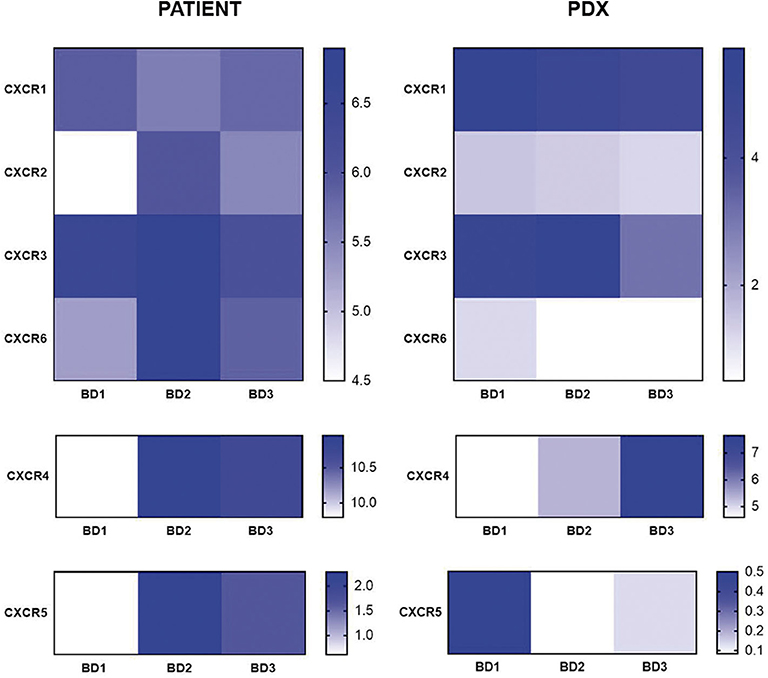
Figure 6. Gene expression of CX chemokine receptors in patient tumors and their corresponding PDX models, according to different grades of tumor budding (BD1, BD2, BD3).
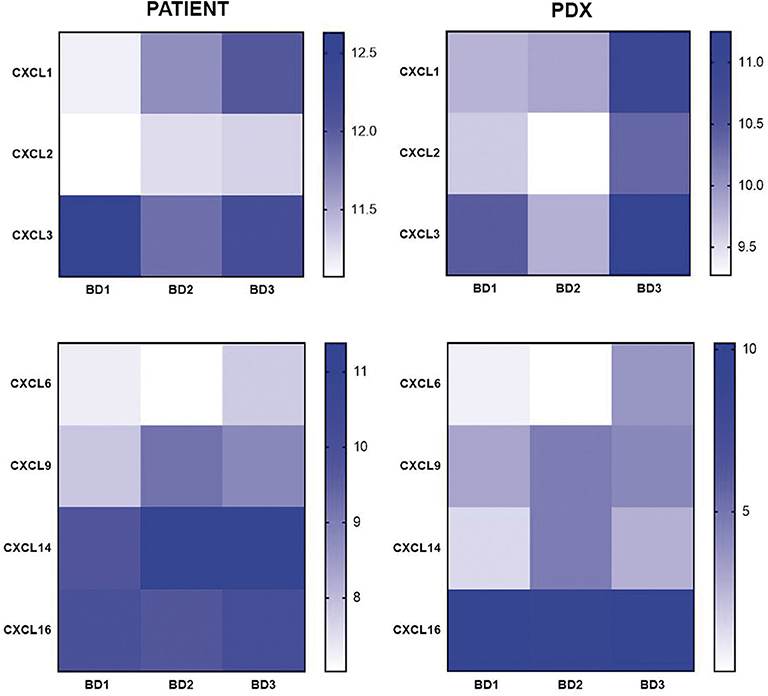
Figure 7. Gene expression of CX chemokine ligands in patient tumors and their corresponding PDX models, according to different grades of tumor budding (BD1, BD2, BD3).
Despite the similar immune-related gene expression patterns found between primary tumors and PDXs with BD1 and BD3 grades, this correspondence was not as evident in PDXs with BD2 grade.
Discussion
Taking into account that tumor heterogeneity is one of the major obstacles in the success of the new personalized therapies for CRC, preclinical platforms which faithfully represent the complex tumor biology are urgently needed. Furthermore, the significance of tumor budding as an independent prognostic factor has now been well established, reinforcing the notion that may constitute a promising target for cancer therapy. However, the interaction between tumor budding and the immune tumor microenvironment still remains unclear. The present study demonstrates that tumor budding is reliably reproduced in early passages of PDXs of CRC. Moreover, our data support that high-grade tumor budding is intimately connected with poor-prognosis CMS4 subtype and with specific gene signatures related to tumor immune evasion.
Our data confirmed that tumor budding is associated with adverse clinicopathological characteristics, such as tumor size, poor histological differentiation, vascular invasion, and poor outcome, as previously reported in several type of cancers (2, 7, 10, 20). An interesting finding of the present study was the high level of correspondence between the budding score in clinical tumors and their corresponding PDX models. Intriguingly, the immune deficiency in host mice did not promote an increase in tumor budding. Pu et al. (21) demonstrated that patient-derived immune cells coexist in the first and second passages with a survival rate of 290 days in the mouse. Recently, tumor budding has also been demonstrated in both center and the invasion front in CRC cell-line xenografts (22). These findings strongly suggest that early passages of PDXs preserve the distinctive cross talk between cancer cells and the immune microenvironment and determine the suitability of this preclinical platform as a model of tumor budding in CRC.
Recently, Trihn et al. (10) reported the potential association of high-grade budding with the CMS4 subtype of CRC in a series of CMS2/3 and CMS4 patient tumors. In the present study, we found similar results with the CMS4 subtype enriched by high-grade tumor budding compared with CMS1 and CMS2/3 subtypes. The fact that tumor buds are well-established independent adverse prognostic factors in CRC (1, 2) as well as the correspondence of CMS subtypes and tumor budding grade between PDXs and their human counterparts observed in our study supports the use of PDX models as powerful tools for the development of targeted therapies against mechanisms involved in tumor budding.
We found that high-grade budding was also significantly associated with stroma-rich tumors. Earlier reports in CRC and breast cancer have suggested an association between tumor budding and the presence of a high density of stromal myofibroblasts (23, 24). Tumor-associated stroma has been shown to facilitate EMT by inducing growth factors, which has been linked with higher capacity of migration and invasion of bud cells (24, 25). Thus, these findings highlight the potential role of the stroma in establishing a microenvironment supportive of the formation of tumor buds. Taken together, the budding phenotype seems to be associated with the high stromal component, which is also accentuated in the mesenchymal CMS4 subtype of CRC.
It is important to note the remarkable overexpression of inhibitory immune checkpoint-related genes (PDL1, TIM-3, NOX2, and IDO1) in BD3 tumors in comparison to BD1 observed in this study. All these upregulated genes have been previously related with tumor invasion and metastasis. However, limited studies have analyzed the expression of immune checkpoint genes in relation with tumor budding (26, 27). In agreement with our data, an upregulation of PDL1 expression has been reported in high-grade tumor budding of CRC suggesting that PDL1 might be specifically overexpressed during EMT to allow invasion and immune escape (27–29). On the other hand, TIM-3, which has been shown to inhibit antitumor immunity by mediating CD8 T-cell exhaustion and pathways involved in metastasis, is an emerging immune checkpoint in several cancers including CRC (30–32). IDO1 and NOX2 are known to exert a potent immunosuppressive effect in a variety of human solid tumors by reducing both tumor-infiltrating T cells as well as B cells (33, 34). Recent studies suggest that NOX2 knockdown reduces metastasis via mechanisms involving amelioration of immune-mediated clearance of metastatic tumor cells (33, 35). The overexpression of these inhibitory immune checkpoints in BD3 tumors observed in our study could explain the immune-permissive microenvironment that facilitates tumor bud formation, invasion, and progression even in early passages of PDXs. Nevertheless, PD1 and CTLA4 genes were more expressed in low tumor budding grade in both patients and xenografts. The distinct expression of PD1 and CTLA4 in immune cells and PDL1 in tumor cells, respectively, would explain these apparent contradictory findings. Hence, the high expression of PDL1 in BD3 tumors would be associated with the immune evasion mechanisms deployed by cancer cells at the invasive front in these tumors, while the overexpression of PD1 and CTLA4 genes in BD1 tumors would reflect their comparative higher immunogenicity. In this regard, the overexpression of these immune checkpoints has been observed in tumors with high immunogenicity and with good clinical response to anti-PD1 and anti-CTLA4 therapy (36, 37). Moreover, high PD1 expression has been recently reported to be associated with a favorable outcome in CRC patients while high-level PDL1 expression, either alone or in combination with PD1, was associated with a worse recurrence-free survival (38). The prognostic value of PD1 expression in lymphocytes and tumor cells and its interaction with PDL1 expression for the prognosis impact in CRC remain to be more deeply investigated. In this context, it may be plausible that CRC patients with low-grade budding will most likely benefit from anti-PD1 and anti-CTLA4 therapies.
TLRs are a diverse family of receptors that regulate gut inflammation but also found to be aberrantly expressed and associated with poor survival and with invasive and metastatic phenotypes in tumors (39, 40). In our study, TLR family expression, specifically TLR1, TLR3, TLR4, and TLR6, was upregulated in BD3 tumors in comparison with low-grade budding tumors (both in patient and PDX tumors) suggesting the presence of TLR-mediated alterations in the tumor invasive front. Overexpression of these TLRs has been previously detected in CRC (39–44). Although the specific mechanisms of TLR-mediated immune escape are still unknown, the current evidences indicated that the high expression of TLRs in tumors can contribute to tumor-cell resistance to apoptosis, malignant transformation of epithelial cells, and tumor progression (40). Results from our study support that TLR upregulation is closely related to BD3 of CRC, which marks them as promising targets for tumor therapy. In addition, it has been previously reported that the activation of TLRs is also accompanied by the expression of PDL1 in tumor cells and other inhibitory molecules as we have observed in this study (41).
Many cancer types show altered chemokine secretion profiles, favoring the recruitment of pro-tumorigenic immune cells such as myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TAN), tumor-associated macrophages (TAM), and regulatory T cells. Particularly, CXCR2 and CXCR4 are chemokine receptors for T-cells implicated in cancer invasion and metastasis (45, 46). Interestingly, these chemokines were overexpressed in BD3 tumors in patients and xenografts in our study. These two chemokine receptors play a crucial role in establishing the “pre-metastatic niche” for tumor cells and are now emerging as key players in the regulation of antitumor immunity (41, 47–50). In addition to these chemokine receptors, chemokine ligands such as CXCL1, CXCL2, CXCL5, CXCL6, CXCL8, and CXCL9 have been also significantly correlated with poor survival and metastasis in several cancers by recruiting MDSCs and suppressing the antitumoral activity of CD8+ T effectors cells. In agreement with these reports, our study reinforces the notion that many different chemokines contribute to antitumoral T cell recruitment and likely some of them may be related to the establishment of a pro-metastatic niche for the tumor buds.
Taken together, our data support a close association between TLRs, chemokines, and tumor budding, raising the exciting hypothesis that the activation of these immune targets may have a determinant role in tumor budding, especially in the case of the CMS4 subtype.
In summary, our findings support that tumor budding in CRC is strongly associated with the mesenchymal poor-prognosis subtype and the presence of a combination of immunosuppressive mechanisms to evade antitumor immunity. Besides, our study suggests that PDXs constitute robust preclinical platforms for reproducing CMS subtypes and tumor budding, hence allowing the development of novel challenging therapies directed against tumor budding in CRC, with special focus in the most aggressive CMS4 subtype.
Data Availability Statement
The datasets have been uploaded to the repository, European Nucleotide Archive (ENA). The accession number is: PRJEB38274.
Ethics Statement
The studies involving human participants were reviewed and approved by Reina Sofía Hospital ethical committee (Protocol number PI-0150-2017) in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki). The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by University of Córdoba Bioethics Committee and followed the regulations of the European Union normative (26/04/2016/066).
Author Contributions
SG-L and RM performed most of the experiments, analyzed the data, and wrote the manuscript. CN-S, AM, KK, and LL-S helped to perform the patient-derived xenograft experiments. KK and MT-H helped to perform the CRC molecular classification. IG analyzed the statistical data of the manuscript. CV, FM-F, CD, and JD contributed to the collection and clinical characterization of human samples. AR-A and EA conceived the project, designed the experiments, and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from Instituto de Salud Carlos III through the project PI16/01508 (Plan Estatal de I+D+I, ISCIII-Subdirección General de Evaluación y Fomento de la Investigación, Spanish Government, and European fund for regional development) and Consejería de Salud de la Junta de Andalucía through the project PI-0150-2017. SG-L was funded with a researcher contract through the program Juan de la Cierva-Incorporación from the Spanish Ministry of Science, Innovation and Universities (IJCI-2016-29786). AM was funded through a predoctoral fellowship from the Asociación Española contra el cáncer (AECC). AR-A was funded with a researcher contract through the program Nicolás Monardes from Junta de Andalucía.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the patients and the technical help of Alvaro Jiménez from the Genomics Units at the IMIBIC.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2020.00264/full#supplementary-material
References
1. Lugli A, Kirsch R, Ajioka Y, Bosman F, Cathomas G, Dawson H, et al. Recommendations for reporting tumor budding in colorectal cancer based on the international tumor budding consensus conference (ITBCC) 2016. Mod Pathol. (2017) 30:1299–311. doi: 10.1038/modpathol.2017.46
2. Dawson H, Galuppini F, Träger P, Berger MD, Studer P, Brügger L, et al. Validation of the international tumor budding consensus conference 2016 recommendations on tumor budding in stage i-IV colorectal cancer. Hum Pathol. (2019) 85:145–51. doi: 10.1016/j.humpath.2018.10.023
3. Mitrovic B, Schaeffer DF, Riddell RH, Kirsch R. Tumor budding in colorectal carcinoma: time to take notice. Mod Pathol. (2012) 25:1315–25. doi: 10.1038/modpathol.2012.94
4. Graham RP, Vierkant RA, Tillmans LS, Wang AH, Laird PW, Weisenberger DJ, et al. Tumor budding in colorectal carcinoma: confirmation of prognostic significance and histologic cutoff in a population-based cohort. Am J Surg Pathol. (2015) 39:1340–6. doi: 10.1097/PAS.0000000000000504
5. Grigore A, Jolly M, Jia D, Farach-Carson M, Levine H. Tumor budding: the name is eMT. Partial EMT. J Clin Med. (2016) 5:51. doi: 10.3390/jcm5050051
6. Li H, Xu F, Li S, Zhong A, Meng X, Lai M. The tumor microenvironment: an irreplaceable element of tumor budding and epithelial-mesenchymal transition-mediated cancer metastasis. Cell Adhes Migr. (2016) 10:434–446. doi: 10.1080/19336918.2015.1129481
7. De Smedt L, Palmans S, Andel D, Govaere O, Boeckx B, Smeets D, et al. Expression profiling of budding cells in colorectal cancer reveals an eMT-like phenotype and molecular subtype switching. Br J Cancer. (2017) 116:58–65. doi: 10.1038/bjc.2016.382
8. Galván JA, Zlobec I, Wartenberg M, Lugli A, Gloor B, Perren A, et al. Expression of e-cadherin repressors sNAIL, zEB1 and zEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br J Cancer. (2015) 112:1944–50. doi: 10.1038/bjc.2015.177
9. Jensen DH, Dabelsteen E, Specht L, Fiehn AMK, Therkildsen MH, Jønson L, et al. Molecular profiling of tumour budding implicates tGFβ-mediated epithelial-mesenchymal transition as a therapeutic target in oral squamous cell carcinoma. J Pathol. (2015) 236:505–16. doi: 10.1002/path.4550
10. Trinh A, Lädrach C, Dawson HE, ten Hoorn S, Kuppen PJK, Reimers MS, et al. Tumour budding is associated with the mesenchymal colon cancer subtype and rAS/RAF mutations: a study of 1320 colorectal cancers with consensus molecular subgroup (CMS) data. Br J Cancer. (2018) 119:1244–51. doi: 10.1038/s41416-018-0230-7
11. Guinney J, Dienstmann R, Wang X, De Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. (2015) 21:1350–6. doi: 10.1038/nm.3967
12. Becht E, De Reyniès A, Giraldo NA, Pilati C, Buttard B, Lacroix L, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. (2016) 22:4057–66. doi: 10.1158/1078-0432.CCR-15-2879
13. López-Sánchez LM, Jiménez-Izquierdo R, Peñarando J, Mena R, Guil-Luna S, Toledano M, et al. SWATH-based proteomics reveals processes associated with immune evasion and metastasis in poor prognosis colorectal tumours. J Cell Mol Med. (2019) 23:8219–32 doi: 10.1111/jcmm.14693
14. Bhimani J, Ball K, Stebbing J. Patient-derived xenograft models—the future of personalised cancer treatment. Br J Cancer. (2020) 122:601–2. doi: 10.1038/s41416-019-0678-0
15. Puig I, Chicote I, Tenbaum SP, Arqu es O, Ra ul Herance J, Gispert JD, et al. A personalized preclinical model to evaluate the metastatic potential of patient-Derived colon cancer initiating cells. Clin Cancer Res. (2013) 19:6787–801. doi: 10.1158/1078-0432.CCR-12-1740
16. Klintrup K, Mäkinen JM, Kauppila S, Väre PO, Melkko J, Tuominen H, et al. Inflammation and prognosis in colorectal cancer. Eur J Cancer. (2005) 41:2645–54. doi: 10.1016/j.ejca.2005.07.017
17. Gujam FJA, Edwards J, Mohammed ZMA, Going JJ, McMillan DC. The relationship between the tumour stroma percentage, clinicopathological characteristics and outcome in patients with operable ductal breast cancer. Br J Cancer. (2014) 111:157–65. doi: 10.1038/bjc.2014.279
18. Trinh A, Trumpi K, De Sousa E, Melo F, Wang X, De Jong JH, Fessler E, et al. Practical and robust identification of molecular subtypes in colorectal cancer by immunohistochemistry. Clin Cancer Res. (2017) 23:387–98. doi: 10.1158/1078-0432.CCR-16-0680
19. Peñarando J, López-Sánchez LM, Mena R, Guil-Luna S, Conde F, Hernández V, et al. A role for endothelial nitric oxide synthase in intestinal stem cell proliferation and mesenchymal colorectal cancer. BMC Biol. (2018) 16:3. doi: 10.1186/s12915-017-0472-5
20. Giger OT, Comtesse SCM, Lugli A, Zlobec I, Kurrer MO. Intra-tumoral budding in preoperative biopsy specimens predicts lymph node and distant metastasis in patients with colorectal cancer. Mod Pathol. (2012) 25:1048–53. doi: 10.1038/modpathol.2012.56
21. Pu X, Zhang R, Wang L, Chen Y, Xu Y, Pataer A, et al. Patient-derived tumor immune microenvironments in patient-derived xenografts of lung cancer. J Transl Med. (2018) 16:1–2. doi: 10.1186/s12967-018-1704-3
22. Georges LMC, De Wever O, Galván JA, Dawson H, Lugli A, Demetter P, et al. Cell line derived xenograft mouse models are a suitable in vivo model for studying tumor budding in colorectal cancer. Front Med. (2019) 6:139. doi: 10.3389/fmed.2019.00139
23. Gujam FJA, McMillan DC, Mohammed ZMA, Edwards J, Going JJ. The relationship between tumour budding, the tumour microenvironment and survival in patients with invasive ductal breast cancer. Br J Cancer. (2015) 113:1066–74. doi: 10.1038/bjc.2015.287
24. Van Wyk HC, Park JH, Edwards J, Horgan PG, McMillan DC, Going JJ. The relationship between tumour budding, the tumour microenvironment and survival in patients with primary operable colorectal cancer. Br J Cancer. (2016) 115:156–63. doi: 10.1038/bjc.2016.173
25. Masugi Y, Yamazaki K, Hibi T, Aiura K, Kitagawa Y, Sakamoto M. Solitary cell infiltration is a novel indicator of poor prognosis and epithelial-mesenchymal transition in pancreatic cancer. Hum Pathol. (2010) 41:1061–8. doi: 10.1016/j.humpath.2010.01.016
26. Prall F, Maletzki C, Hühns M, Krohn M, Linnebacher M. Colorectal carcinoma tumour budding and podia formation in the xenograft microenvironment. PLoS ONE. (2017) 12:1–2. doi: 10.1371/journal.pone.0186271
27. Martinez-Ciarpaglini C, Oltra S, Roselló S, Roda D, Mongort C, Carrasco F, et al. Low miR200c expression in tumor budding of invasive front predicts worse survival in patients with localized colon cancer and is related to pD-L1 overexpression. Mod Pathol. (2019) 32:306–13. doi: 10.1038/s41379-018-0124-5
28. Prall F, Hühns M. PD-L1 expression in tumour buds of colorectal carcinoma. Histopathology. (2016) 69:158–60. doi: 10.1111/his.12915
29. Korehisa S, Oki E, Iimori M, Nakaji Y, Shimokawa M, Saeki H, et al. Clinical significance of programmed cell death-ligand 1 expression and the immune microenvironment at the invasive front of colorectal cancers with high microsatellite instability. Int J Cancer. (2018) 142:822–32. doi: 10.1002/ijc.31107
30. Zhou E, Huang Q, Wang J, Fang C, Yang L, Zhu M, et al. Up-regulation of tim-3 is associated with poor prognosis of patients with colon cancer. Int J Clin Exp Pathol. (2015) 8:8018–27.
31. Nair VS, Toor SM, Taha RZ, Ahmed AA, Kurer MA, Murshed K, et al. Transcriptomic profiling of tumor-infiltrating cD4+ tIM-3+ t cells reveals their suppressive, exhausted, and metastatic characteristics in colorectal cancer patients. Vaccines. (2020) 8:71. doi: 10.3390/vaccines8010071
32. Yu H, Borsotti C, Schickel JN, Zhu S, Strowig T, Eynon EE, et al. A novel humanized mouse model with significant improvement of class-switched, antigen-specific antibody production. Blood. (2017) 129:959–69. doi: 10.1182/blood-2016-04-709584
33. van der Weyden L, Speak AO, Swiatkowska A, Clare S, Schejtman A, Santilli G, et al. Pulmonary metastatic colonisation and granulomas in nOX2-deficient mice. J Pathol. (2018) 246:300–10. doi: 10.1002/path.5140
34. Zhang W, Zhang J, Zhang Z, Guo Y, Wu Y, Wang R, et al. Overexpression of indoleamine 2,3-Dioxygenase 1 promotes epithelial-Mesenchymal transition by activation of the iL-6/STAT3/PD-L1 pathway in bladder cancer. Transl Oncol. (2019) 12:485–92. doi: 10.1016/j.tranon.2018.11.012
35. Aydin E, Johansson J, Nazir FH, Hellstrand K, Martner A. Role of nOX2-derived reactive oxygen species in nK cell–mediated control of murine melanoma metastasis. Cancer Immunol Res. (2017) 5:804–11. doi: 10.1158/2326-6066.CIR-16-0382
36. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. (2018) 8:86. doi: 10.3389/fonc.2018.00086
37. Rotte A. Combination of cTLA-4 and pD-1 blockers for treatment of cancer. J Exp Clin Cancer Res. (2019) 38:1–12. doi: 10.1186/s13046-019-1259-z
38. Lee LH, Cavalcanti MS, Segal NH, Hechtman JF, Weiser MR, Smith JJ, et al. Patterns and prognostic relevance of pD-1 and pD-L1 expression in colorectal carcinoma. Mod Pathol. (2016) 29:1433–42. doi: 10.1038/modpathol.2016.139
39. Ridnour LA, Cheng RYS, Switzer CH, Heinecke JL, Ambs S, Glynn S, et al. Molecular pathways: toll-like receptors in the tumor microenvironment-poor prognosis or new therapeutic opportunity. Clin Cancer Res. (2013) 19:1340–6. doi: 10.1158/1078-0432.CCR-12-0408
40. Sato Y, Goto Y, Narita N, Hoon DSB. Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenviron. (2009) 2:205–14. doi: 10.1007/s12307-009-0022-y
41. Wang K, Wang J, Wei F, Zhao N, Yang F, Ren X. Expression of tLR4 in non-small cell lung cancer is associated with pD-L1 and poor prognosis in patients receiving pulmonectomy. Front Immunol. (2017) 8:456. doi: 10.3389/fimmu.2017.00456
42. Lu C-C, Kuo H-C, Wang F-S, Jou M-H, Lee K-C, Chuang J-H. Upregulation of tLRs and iL-6 as a marker in human colorectal cancer. Int J Mol Sci. (2015) 16:159–77. doi: 10.3390/ijms16010159
43. Li Y, Chen S. Toll-like receptors expressed in tumor cells: targets for therapy. Cancer Immunol Immunother. (2008) 57:1271–8. doi: 10.1007/s00262-008-0459-8
44. Semlali A, Almutairi M, Pathan AAK, Azzi A, Parine NR, AlAmri A, et al. Toll-like receptor 6 expression, sequence variants, and their association with colorectal cancer risk. J Cancer. (2019) 10:2969–81. doi: 10.7150/jca.31011
45. Verbeke H, Struyf S, Laureys G, Van Damme J. The expression and role of cXC chemokines in colorectal cancer. Cytokine Growth Factor Rev. (2011) 22:345–58. doi: 10.1016/j.cytogfr.2011.09.002
46. Susek KH, Karvouni M, Alici E, Lundqvist A. The role of CXC chemokine receptors 1-4 on immune cells in the tumor microenvironment. Front Immunol. (2018) 9:2159. doi: 10.3389/fimmu.2018.02159
47. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-Associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell. (2017) 32:654–68.e5. doi: 10.1016/j.ccell.2017.10.005
48. Yang J, Kumar A, Vilgelm AE, Chen SC, Ayers GD, Novitskiy S V., et al. Loss of cXCR4 in myeloid cells enhances antitumor immunity and reduces melanoma growth through nK cell and fASL mechanisms. Cancer Immunol Res. (2018) 6:1186–98. doi: 10.1158/2326-6066.CIR-18-0045
49. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of tGFβ signaling in mammary carcinomas recruits gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. (2008) 13:23–35. doi: 10.1016/j.ccr.2007.12.004
Keywords: patient-derived xenografts, tumor budding, colorectal cancer, immune evasion, toll-like receptors, chemokines
Citation: Guil-Luna S, Mena R, Navarrete-Sirvent C, López-Sánchez LM, Khouadri K, Toledano-Fonseca M, Mantrana A, Guler I, Villar C, Díaz C, Medina-Fernández FJ, De la Haba-Rodríguez JR, Aranda E and Rodríguez-Ariza A (2020) Association of Tumor Budding With Immune Evasion Pathways in Primary Colorectal Cancer and Patient-Derived Xenografts. Front. Med. 7:264. doi: 10.3389/fmed.2020.00264
Received: 01 April 2020; Accepted: 14 May 2020;
Published: 03 July 2020.
Edited by:
Luigi Tornillo, University of Basel, SwitzerlandReviewed by:
Lucia Bongiovanni, San Raffaele Hospital (IRCCS), ItalyThomas Menter, University Hospital of Basel, Switzerland
Copyright © 2020 Guil-Luna, Mena, Navarrete-Sirvent, López-Sánchez, Khouadri, Toledano-Fonseca, Mantrana, Guler, Villar, Díaz, Medina-Fernández, De la Haba-Rodríguez, Aranda and Rodríguez-Ariza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Guil-Luna, djIyZ3VsdXNAdWNvLmVz
†These authors have contributed equally to this work
‡These authors share senior authorship