 Yahson Fernando Varela-Quitián
Yahson Fernando Varela-Quitián Fabio Enrique Mendez-Rivera
Fabio Enrique Mendez-Rivera David Andres Bernal-Estévez
David Andres Bernal-Estévez- Immunology and Clinical Oncology Research Group (GIIOC), Fundación Salud de Los Andes, Bogotá, Colombia
Cancer is a major global health concern and one of the leading causes of death worldwide. According to the World Health Organization (WHO), there is an urgent need for novel therapeutic agents to treat this disease. Some antimicrobial peptides (AMPs) have demonstrated activity against both microbial pathogens and cancer cells. Among these, cationic AMPs (CAMPs) have garnered significant attention because of their ability to selectively interact with the negatively charged surfaces of cancer cell membranes. CAMPs present several advantages such as high specificity for targeting cancer cells, minimal toxicity to normal cells, reduced probability of inducing resistance, stability under physiological conditions, ease of chemical modification, and low production costs. This review focuses on CAMPs with anticancer properties such as KLA, bovine lactoferricin derivatives, and LTX-315, and briefly explores common bioinformatics tools for Anticancer Peptides (ACPs) selection pipeline from AMPs.
1 Introduction
Cancer is one of the leading causes of mortality worldwide, according to the World Health Organization (WHO). In 2022, global statistics reported nearly 20 million new cancer cases and 9.7 million cancer-related deaths. Over the past 40 years, the number of deaths due to cancer has doubled in women and tripled in men (1, 2). This public health problem is closely related to the increase and aging of the global population (3).
Cancer treatment approaches that attempt to kill cancer cells directly, such as surgery, radiotherapy, chemotherapy, or combinations thereof, are frequently used (4). Surgery is only effective for localized tumors; however, incomplete removal of cancerous tissues increases the risk of recurrence (5). Radiotherapy specifically targets tumors, but has adverse effects on the surrounding healthy tissues, and its efficacy varies with the type and location of the tumor (6). Chemotherapy is one of the most widely used mode of treatment, but it is toxic to both cancer and healthy cells, and cancer cells may become resistant over time, making the treatment progressively less effective (7). In recent years, immunotherapy approaches that try to help the immune system fight cancer have been developed. However, tumor complexity, high resistance to solid tumors, and the ability of certain cancer cells to evade anticancer drugs result in a low percentage of treatment-surviving patients (8). These challenges highlight the urgent need for novel therapeutic agents (9–11).
Anticancer peptides (ACPs) are currently being explored as new alternatives to conventional cancer therapies, with the potential to selectively kill cancer cells. ACPs do not depend on unique receptors or a specific signal transduction pathway for their action, making it more difficult for tumors to develop resistance (12).
Many ACPs are derived from antimicrobial peptides (AMPs), particularly cationic AMPs (CAMPs), which bind selectively to the negative charged membrane of cancer cells. The membrane integrity and the mitochondrial function are compromised by CAMPs resulting in apoptosis or necrosis, thus CAMPs are promising candidates for anticancer therapy (13–17).
This review explores the potential applications of CAMPs derived from natural or artificial sources and examines how their unique characteristics can serve as inspiration for the development of novel anticancer agents. Notable examples of the use of CAMPs as templates for producing anticancer agents include derivatives that have improved the design of KLA, bovine lactoferricins, and LTX-315 peptides. Additionally, it briefly presents computational tools employed in the pipeline for selecting ACPs from AMP.
2 AMPs classified by charge
Antimicrobial peptides can be classified into two main groups based on their charge, cationic and non-cationic peptides (Figure 1). Each group can include peptides from diverse sources with varying activities, structures, and amino acid compositions (18–20).
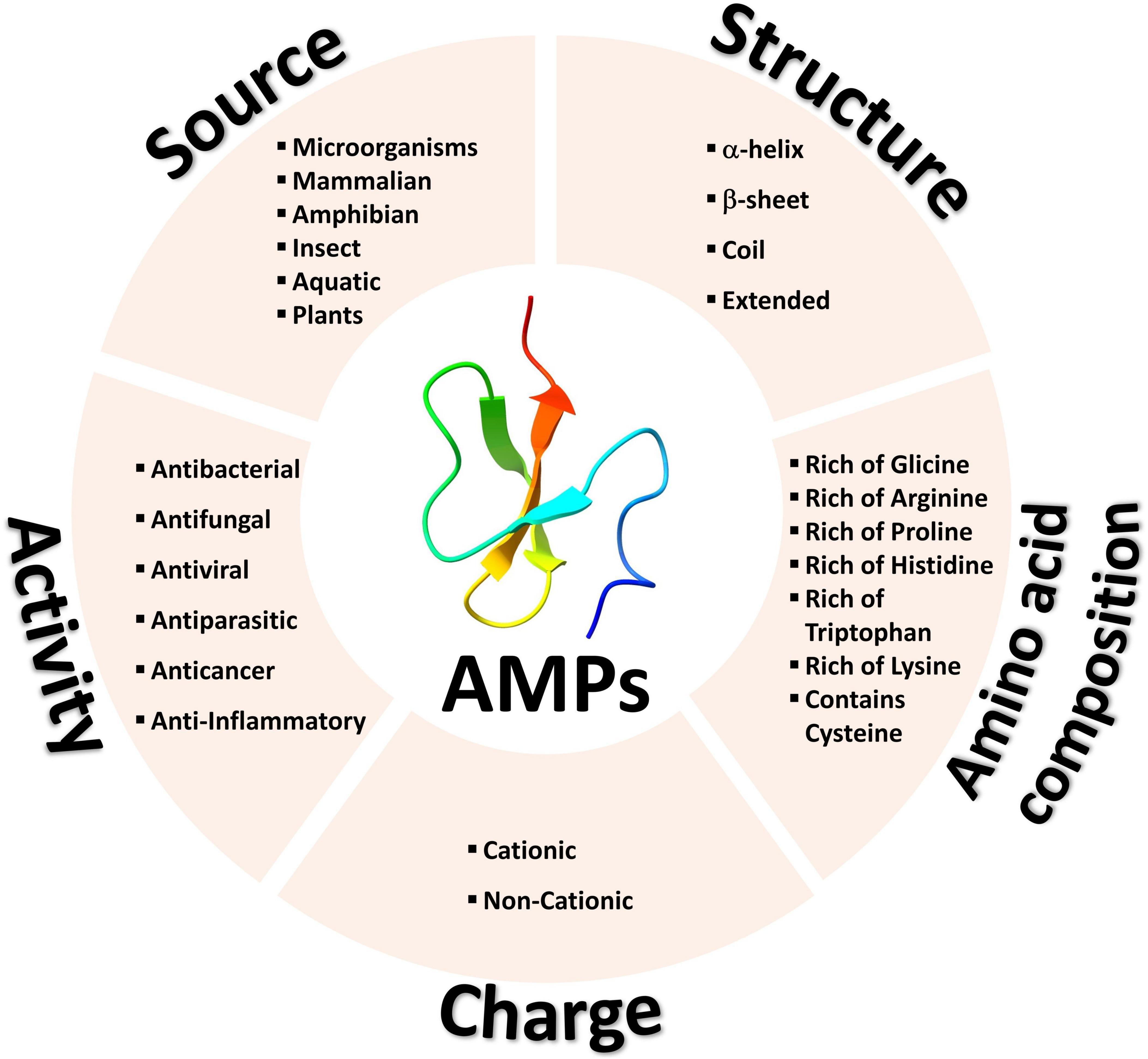
Figure 1. Common methods for classifying antimicrobial peptides.
CAMPs constitute nearly 90% of all AMPs and carry a positive net charge, enabling an initial interaction with the negatively charged microbial cell membrane (21). Notably, some CAMPs also exhibit anticancer activity, selectively targeting tumor cells while sparing normal cells. Similar to their electrostatic interaction with microbial cell membranes, CAMPs engage with tumor cells owing to their more negative charge compared to normal cells (22, 23). Due to this fact, CAMPs are considered as potential tool for cancer therapy and have been used in pharmaceutical sciences, drug design and discovery (24, 25), and cancer clinical trials (26–28).
3 Mode of action of CAMPs with anticancer activity
The membrane composition of tumor cells is considerably different and more anionic than the membrane of normal cells, which enables CAMPs to interact with the tumor membrane, similar to the microbial membrane (21). Two mechanisms of action have been identified for CAMPs against cancer cells: membrane- and non-membrane-targeting mechanisms (29).
The first mechanism involves positively charged amphipathic CAMPs that interact with negatively charged cell membranes. Then, they are adsorbed into the membrane, which results in a conformational change and membrane disruption according to different models, including carpet detergent-like, barrel-stave (formation of transmembrane pores), and toroidal (lipid rearrangement for pore formation) (Figure 2) (30, 31).
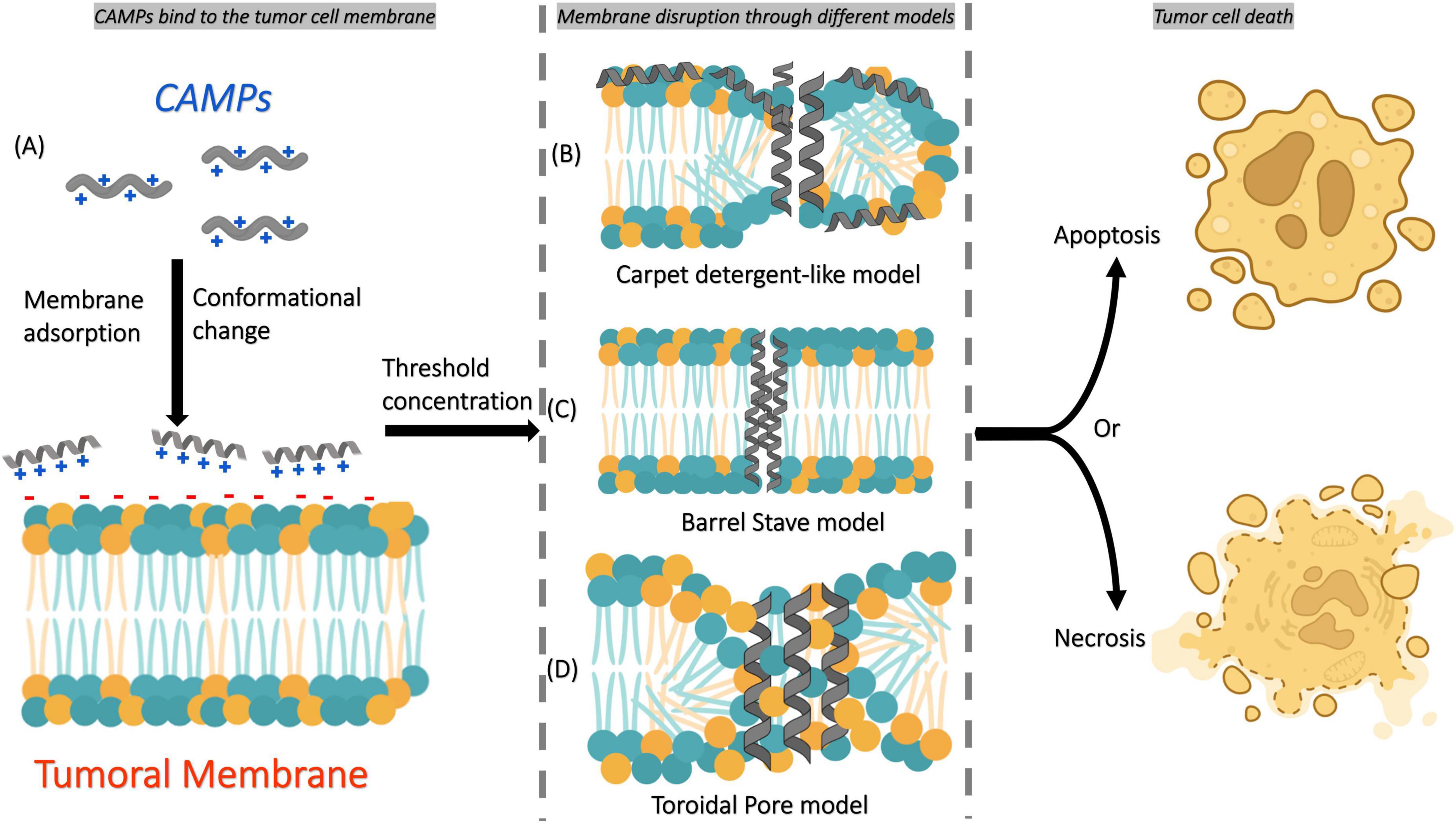
Figure 2. Simple scheme of the main mechanism of action of CAMPs on tumor cell membranes. (A) CAMPs first bind to the tumor cell membrane through electrostatic interactions and become adsorbed onto its surface, inducing conformational changes in their structure. Once a threshold concentration is reached, membrane disruption occurs through different mechanisms: (B) carpet detergent-like model, (C) barrel-stave model, or (D) toroidal pore model. These disruptions ultimately lead to tumor cell death by apoptosis or necrosis, depending on the mechanism involved.
The second mechanism involves CAMPs that penetrate the cells directly or by endocytosis. Inside the cytoplasm, they target important cellular structures and processes by inhibiting protein biosynthesis, nucleic acid biosynthesis, protease activity, and cell division (15).
These interactions, which are described by the two mechanisms of action, ultimately leading to membrane lysis or pore formation, which triggers apoptosis or necrosis in tumor cells with minimal impact on normal cells (25).
4 Physicochemical factors influencing the anticancer activity of CAMPs
4.1 Attributes of CAMPs
Several physicochemical factors, such as amino acid composition, net charge, hydrophobicity, amphipathicity, structural folding, peptide concentration, oligomerization, and membrane composition, affect the anticancer activity of CAMPs (32, 33). Understanding the relationship between the peptide sequence and function is crucial for the rational design of novel CAMP-based therapeutics with improved anticancer efficacy.
4.1.1 Amino acid composition
A systematic analysis of amino acid-rich AMPs from the Antimicrobial Peptide Database (APD) as well as their distribution across different life kingdoms and animal classes, reveals a low abundance of Asp, Glu and Met; a moderate abundance of Phe, Ser, Thr, Trp and Tyr; and a high abundance of Ala, Cys, Gly, His, Ile, Lys, Leu, Pro, Arg and Val (32, 33).
In general, AMPs are characterized by a high prevalence of amino acid residues classified as hydrophobic, cationic (basic), and aromatic (32–38). ACPs derived from AMPs were expected to show a similar trend in amino acid abundance (Figure 3). In silico models and exploratory data analysis confirmed a preference for certain amino acids (Ala, Cys, Gly, Lys, and Leu) in both AMPs and ACPs global datasets (39, 40). Additionally, Arg and Trp amino acids were also found in many AMPs (20, 41–43) and ACPs (44, 45).
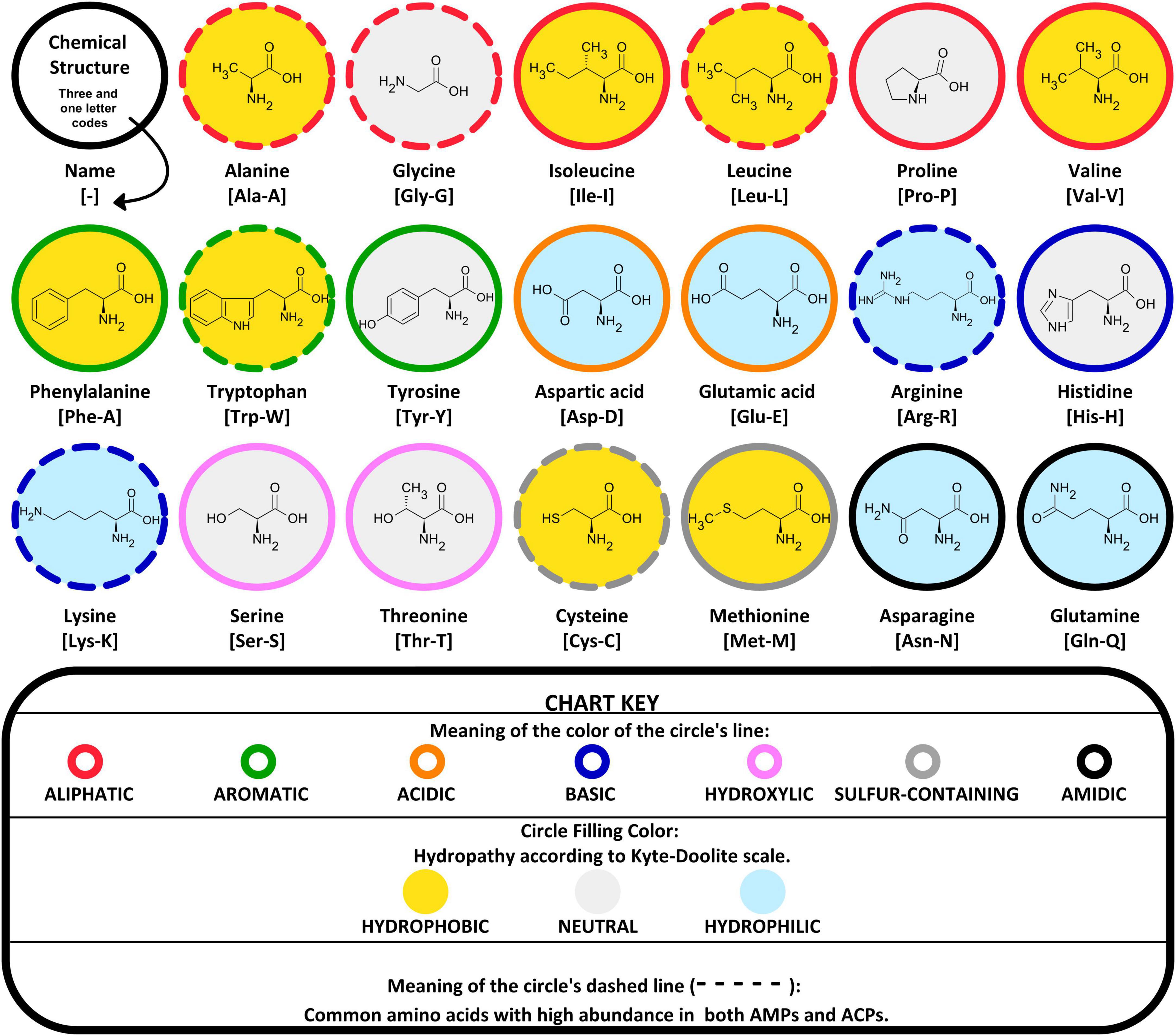
Figure 3. Grouping of 20 common amino acids. Based on their chemical characteristics and hydropathy, they are derived from the physicochemical properties of the amino acid side chains. Additionally, the circle dashed line corresponds to amino acids with high preference for both AMPs and ACPs.
4.1.2 Net positive charge
AMPs are generally cationics (CAMPs), with their charge varying from +2 to +11, owing to the overrepresentation of Arg and/or Lys residues. It is widely accepted that cationicity conferred by Arg and/or Lys is primarily responsible for the initial interaction of CAMPs with the negatively charged membrane surface of cancer cells. Studies have demonstrated a direct correlation between the peptide charge and cytotoxicity against tumor cells (46–48).
In addition, these basic amino acids (charged Arg or Lys) can deform the lipid bilayer by pulling water molecules and lipid head groups into the hydrocarbon core of the lipid membrane, as demonstrated by atomistic molecular dynamics. However, Arg causes greater membrane perturbations by attracting more lipid phosphate groups because Arg forms more hydrogen bonds with lipid phosphates than Lys (49).
In general, peptides enriched in Lys within their hydrophilic regions exhibits more selective anticancer activity, whereas those containing Arg tend to display higher toxicity in normal cells (50), as observed with AMP/ACP tritrpticin (RRFPWWWPFLRR) when arginine was substituted with lysine, which improved the selectivity of peptides toward Jurkat cancer cells compared with normal peripheral blood mononuclear cells (PBMCs) (51).
4.1.3 Amphipathicity
Amphipathicity refers to the spatial distribution of hydrophilic and hydrophobic peptide residues, which allows AMPs to align themselves at the membrane interface, with hydrophobic residues facing the lipid core and hydrophilic residues interacting with the aqueous environment (52, 53).
The initial interaction between CAMPs and microbial or tumor membranes is driven by electrostatic forces through the interaction of the positively charged hydrophilic region of CAMP and negatively charged components of these membranes; subsequently, the hydrophobic region of CAMPs becomes embedded in the membrane through van der Waals interactions. This leads to compromised membrane functionality and increased permeability (54–57).
4.1.4 Hydrophobicity
Hydrophobicity, an important physicochemical characteristic of AMPs, is the percentage of hydrophobic amino acids in the peptide. Typically, AMPs have hydrophobicity values close to 50%, and are expected to contain both hydrophilic and hydrophobic amino acid residues (58). AMPs can interact with membranes, and certain ratios of hydrophilic charged residues affect peptide-membrane interactions (59) as they control the partitioning of the peptide into the membrane hydrophobic core (60).
Peptides can adopt different structures based on hydrophobicity and environmental factors, such as folding into an α-helical conformation in the presence of certain micelles (61). Manipulation of peptide hydrophobicity can enhance anticancer activity by altering their amphipathicity and secondary structure conformation. Studies indicate that increasing the hydrophobicity of peptides may increase their self-association and cytotoxicity against cancer cells (62), but sometimes also increase hemolytic activity (61, 63).
For example, the N-terminus of AMP CM4 (RWKIFKKIEKVGQNIRDGIVKAGPAVAVVGQAATI) was modified by conjugating fatty acids of varying lengths (4–16 carbons) to enhance hydrophobicity, helical content, and anticancer activity. The best anticancer effects were observed in breast cancer cells when AMPs were conjugated with fatty acids of 12–16 carbons (64). Hence, hydrophobicity is a critical parameter that must be optimized for the development of CAMP/ACP-based drugs.
4.1.5 Structural folding
The conformation of a peptide structure can be affected by the environment in which it is immersed. In aqueous solutions, linear AMPs are mostly unstructured; however, upon interaction with the hydrophobic environment of the lipid bilayers, they undergo significant conformational changes. By adopting specific secondary structures, such as α-helices, β-sheets, or extended polyproline-like helices, which increase amphipathicity, this structural flexibility allows AMPs to adapt to various targets (53).
ACPs may adopt α-helical, β-sheet, or linear conformations, with α-helical ACPs representing one of the largest groups recognized (65, 66). α-helical AMP/ACP showed higher activity than linear peptides, which has been attributed to the fact that the α-helical conformation can project a clear hydrophobic surface and another hydrophilic surface that allows effective interaction with microbial and tumor membranes.
4.1.6 Peptide concentration
Antimicrobial peptides with anticancer activity display concentration-dependent cytotoxicity toward cancer cells and solid tumors in various studies conducted (57). Another important feature is the behavior of AMP monomers in solution and their proximity to the cancer cell membrane, which facilitates membrane aggregation and subsequent pore formation, leading to cell death (65). For example, to induce lytic activity, the cancer cell membrane needs to be exposed to a minimum threshold concentration of monomers, termed the critical concentration, and this concentration-dependent process is critical for the therapeutic efficacy of AMPs (23).
4.1.7 Oligomerization
The capacity for peptide self-association, also known as oligomerization, is an important characteristic of AMPs. This oligomerization process is dependent on the amino acid composition, conformation, and ability of the peptide to align hydrophobic and hydrophilic regions toward adjacent peptides and the membrane (67).
4.2 Membrane composition of cancer cells
Cancer cells differ from normal cells, which is important for understanding the anticancer activity of CAMPs and their interaction with tumor cells. The initial recognition between CAMPs and cancer cells is mainly due to electrostatic interactions. Cancer cells are more negatively charged than normal cells due to increased expression of anionic molecules. In addition, cancer cells have different cholesterol content, microvilli structure, and extracellular pH, which also affect CAMP interactions and selectivity (25, 68).
4.2.1 Negative charge
Cancer cell surfaces are more negatively charged due to the increased expression of anionic molecules such as phosphatidylserine (PS), O-glycosylated mucins, and heparan sulfates (HS), and their exposure to cancer cells, which improves their interaction with CAMPs (68).
In normal cells, PS is found on the surface of the interior membranes, whereas in cancer cells, it is externalized, producing an immune suppressor environment that can promote tumor growth (69–71), the externalization of PS is a universal feature of cancer cells, as observed in tumor endothelial cells (ECs) (72). In addition, PS overexpression has been observed in some cancer cells, including glioblastoma (Gli), astrocytoma (U373), and breast cancer (MDA-MB-231-D3H2LN) (70).
In cancer cells, anionic molecules derived from O-glycosylated mucins are primarily obtained by sialylation and sulfation, which involve the addition of sialic acid and/or sulfate groups, respectively. O-glycosylated mucins are crucial for cancer progression, metastasis, and immune evasion (73).
HS is a type of glycosaminoglycan; it is an unbranched chain of disaccharide repeats that is heavily sulfated at various positions on their sugar residues. HS can modulate the effects of various growth factors and is involved in angiogenesis and metastasis of cancer cells, and its overexpression has been reported in several tumors (74–76). HS serves as an initial anchor for cell-penetrating peptides (CPPs) via electrostatic interactions between the sulfates or carboxylic groups of HS and the basic amino acids (Arg and/or Lys) of CPPs (77).
4.2.2 Cholesterol content
Cholesterol is a key part in the formation of cell membranes and relevant component in maintaining the integrity and organization of the of phospholipid bilayers (78). Cholesterol content affects the regulation of membrane fluidity and modulates the chemotherapy resistance and metastatic properties of cancer cells. When cancer cells prepare for metastasis, they tend to decrease their membrane cholesterol levels to maximize membrane fluidity and plasticity, allowing neoplastic cells to modulate their shape easily (79).
Notably, leukemia and lung cancer cells are more fluid than healthy cells, owing to their lower cholesterol levels. In contrast, the opposite trend was observed in breast and prostate cancer cells (65). Malignant cells with reduced cholesterol content are more susceptible to lysis, which can facilitate AMP-induced apoptosis (80, 81).
4.2.3 Microvilli
A notable difference between cancer and normal cells is the significantly higher number of microvilli in cancer cell membranes, which increases their surface area (68). This structural change is believed to affect the action of AMPs, as cancer cells increase their membrane fluidity and microvilli density. The increased number of microvilli augments surface contact and peptide attraction, making cancer cells more sensitive to AMP interactions. Consequently, this positive feedback mechanism enhances the efficacy of AMPs against cancer cells while reducing the risk of resistance development compared with traditional chemotherapy approaches (65, 80).
4.2.4 Extracellular pH acidification
Cancer tissues have a hypoxic environment caused by limited blood supply; under these conditions, cancer cells elevate their production of carbonic anhydrase IX, an enzyme that catalyzes the reversible conversion of carbon dioxide to bicarbonate and a proton, referring to its contribution to an acidic microenvironment, so the extracellular pH (pHe) of cancer cells is lower (pH 6.2–6.9) than that of normal cells (pH 7.3–7.4) (68).
This phenomenon has been well documented, as shown by the Guanyu Hao group, who used data collected from several different sources to compare the average extracellular pHe values of different normal tissues with those of cancerous tissues (malignant melanoma, vulvar tumor, urine tumor, lung tumor, breast tumor, glioblastoma, astrocytoma, and sarcomas). Their findings make evident the tendency of the higher acidity of the extracellular tumor environment over normal cells, showing 0.3–0.7 pH units lower of pHe of cancer cells than normal cells (82). This lower extracellular pHe has been exploited to induce selective toxicity in cancer cells using peptide conjugates (83–85).
5 Cationic antimicrobial peptides with antitumor properties
Chemotherapy remains the predominant treatment for controlling tumor cells, many anticancer drugs lack specificity and affect cancer and normal cells, causing toxic side effects. Furthermore, tumor cells are prone to developing resistance, which further diminishes the effectiveness of treatment (86).
Given these challenges, the development of new therapeutic approaches which specifically attack cancer cells without damaging normal tissues represents an essential requirement (24). In this context, the exploration of novel CAMPs as prospective anticancer agents has emerged as a significant field.
5.1 KLA
KLA peptide (KLAKLAK)2 is a de novo-designed CAMP developed by Javadpour et al. in 1996, and it was designed to adopt an amphipathic helical conformation. Initially, its antibacterial activity was evaluated against Gram-negative Bacteria Escherichia coli and Pseudomonas aeruginosa, and Gram-positive bacteria Staphylococcus aureus, with minimal inhibitory concentrations (MICs) of 6, 3, and 6 μM, respectively. In contrast, KLA exhibited low cytotoxicity against 3T3 mouse fibroblasts (>517 μM) and human erythrocytes, suggesting a favorable safety profile (>750 μM) (87).
Subsequently, KLA was evaluated in vitro against various human cancer cell lines (breast, prostate, and bladder cancer cells) and in vivo in a breast cancer xenograft model in mice. The results demonstrated its potent anticancer effects, including of cell death in most of the cancer cell lines tested. Among them, breast cancer cell line MCF-7 exhibited the highest sensitivity to KLA (Table 1), while the peptide had minimal effects on healthy control cells (peripheral blood lymphocytes and embryonic kidney epithelial cells). Furthermore, KLA significantly inhibited tumor growth and prolonged the overall survival of tumor-bearing mice compared with that of the control group (88).
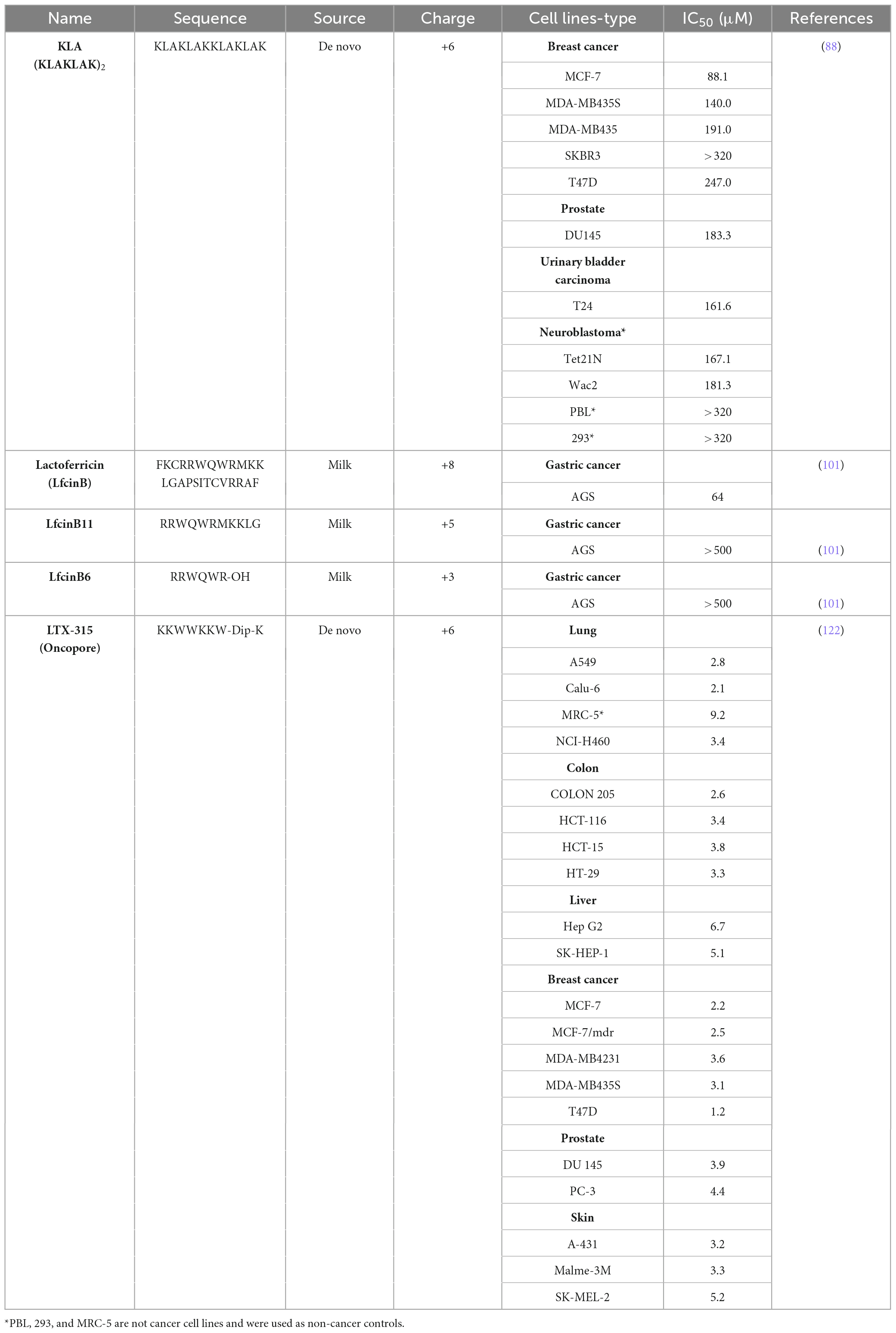
Table 1. Cytotoxic effects of some representative CAMPs with anticancer activity evaluated in vitro in different cancer cell lines.
Jeffrey et al. designed DP1 (RRQRRTSKLMKRGGKLAKLAKKLAKLAK), a conjugated tumor targeting peptide. DP1 consists of an N-terminal fragment derived from Protein Transduction Domain-5 (PTD-5), which facilitates the delivering protein complexes into solid tumors, and C-terminal KLA sequence. When DP1 was directly added at low concentrations to the culture medium of the mouse fibrosarcoma cell line MCA205, *the results showed a significant reduction of cell viability (LC50 < 50 μM) and triggered significant apoptosis (programmed cell death) in vivo; in contrast, no significantly effect of KLA or PTD-5 alone were observed on MCA205 viability (89).
Similarly, a targeted cancer-killing peptide named TP-tox (LTVSPWYGGKLAKLAKKLAKLAK) was developed to mimic antibody-drug conjugates (ADCs) without the size restrictions of conventional ADCs. TP-Tox exhibited selective toxicity in breast, prostate, and neuroblastoma cancer cell lines. It was more effective at killing cancer cells than the individual targeting or killing peptides components. Additionally, weekly injections of TP-ox significantly slowed tumor growth and improved survival in mice with breast cancer tumors (MDA-MB-435S) (90).
A set of pH-dependent targeting KLA-conjugated peptides was developed to prevent off-target effects. It was conducted using a pH-low insertion peptide (pHLIP) and three KLA analogs. pHLIP peptides undergo conformational changes in acidic microenvironments and promote peptide translocation across the cytoplasmic membrane, such as in tumor microenvironments (29). Therefore, these conjugated peptides can translocate KLA to the cytoplasm of breast cancer cells. Each KLA was synthesized individually with a cysteine residue at the N-terminus, while pHLIP, containing a cysteine at the C-terminus (GGEQNPIYWARYADWLFTTPLLLLDLALLVDADEGTCG), was conjugated to each KLA analog by disulfide bonds and purified. The resulting chimeric peptides [pHLIP-(KLAKLAK)2, pHLIP-KLAKLAK, or pHLIP-KLAK] were evaluated in MDA-MB-231 cancer cells and showed concentration- and pH-dependent cell growth inhibition, with little or no significant decrease in cell viability at pH 7.4, and 90% inhibition at pH 5.0. pHLIP-KLAKLAK was identified as a lead conjugate with a IC50 of 0.5 μM to MDA-MB-231 cancer cells (84).
Several other conjugated peptides incorporating the KLA fragment have been tested as potential cancer therapeutics, showing promising results (11, 91–93). Additionally, KLA-conjugated peptides with replacement of all L-to D-amino acids in the KLA fragment (D-KLA) were evaluated. These D-KLA peptides retain membrane-disrupting properties similar to those of their L-counterparts but exhibit enhanced stability in biological systems (94–96).
5.2 Bovine lactoferricin
Bovine lactoferricin (LfcinB) is 25-amino acids peptide (17FKCRRWQWRMKKLGAPSITCVRRAF41) generated from enzymatic digestion of the N-terminal region of bovine lactoferrin. Its sequence contains eight hydrophilic residues (five arginine and three lysine), conferring a net charge of +8, along with four aromatic and hydrophobic residues (two phenylalanine and two tryptophan), and two cysteines forming disulfide bond. In aqueous solution, LfcinB adopts a β-sheet secondary structure, where hydrophobic residues are positioned on one side and hydrophilic residues on the opposite side (97).
Due to its cationic and amphipathic nature, LfcinB interacts with negatively charged molecules on cell surfaces and exhibits broad-spectrum antimicrobial activity against Gram-positive and Gram-negative bacteria, fungi, and viruses, primarily by interrupting the microbial cell membranes. LfcinB also exhibits anticancer properties by selectively targeting negatively charged cancer cell surfaces, leading to apoptosis or necrosis (57, 98).
LfcinB has demonstrated cytotoxic activity in vitro in various human and murine cancer cell lines, including colon carcinoma, lung cancer, liver cancer, melanoma, fibrosarcoma, leukemia, and breast cancer. Notably, LfcinB treatment does not significantly affect the viability of normal human lymphocytes, erythrocytes, endothelial cells, or fibroblasts (99). Additionally, many synthetic peptides derived from LfcinB exhibit anticancer properties, highlighting their potential as novel therapeutic agents for cancer treatment (100).
A study by Pan et al. evaluated bovine lactoferricin peptide fragments (Table 1) in AGS gastric cancer cells. The assays revealed that full-length LfcinB peptide selectively inhibited AGS cell proliferation in a dose-dependent manner, with a half-maximal inhibitory concentration (IC50) of 64 μM. Furthermore, treatment with LfcinB results in an increased sub-G1 population within the cell cycle, indicating the induction of apoptosis (101).
5.2.1 Synthetic derivates inspired by LfcinB
The hexapeptide LfcinB6 (RRWQWR-NH2), which contains a C-terminal carboxamide functional group, retains the antimicrobial activity of LfcinB (102–105). However, unlike native LfcinB, LfcinB6 lacks inherent cytotoxic activity against T-leukemia or breast cancer cells. This difference is attributed to weak binding to isolated mitochondria, which prevents membrane permeabilization or causes cytochrome C release. Interestingly, when LfcinB6 was delivered using fusogenic liposome formulations, it was efficiently transported into the cytosol of cancer cells, where it exhibited strong cytotoxic activity. The mechanism of cytotoxicity was found to be caspase-and cathepsin B-dependent but not reliant on reactive oxygen species (ROS), suggesting a mode of action distinct from that of native LfcinB (106).
To further enhance the anticancer activity of LfcinB6, it was conjugated to CPP peptide containing seven arginine residues, resulting in MPLfcinB6 (RRRRRRRGGRRWQWR). This modified peptide demonstrated selective cytotoxicity against T-leukemia and B-lymphoma cells, while sparing normal T cells. MPLfcinB6 rapidly induced extensive cancer cell membranes damage, triggered ROS production, and disrupted mitochondrial integrity. Its high selectivity was attributed to strong electrostatic interaction between its highly cationic seven-arginine motif and the negatively charged membranes of cancer cells (107).
Inspired by the core sequence of LfcinB6, which contains cationic terminal residues, a lipophilic central core, and a well-defined amphipathic structure upon interaction with negatively charged micelles, Torfoss et al. synthesized eight heptapeptides (H-KKWβ 2,2WKK-NH2), each with different central lipophilic β2,2-amino acid building blocks. Among these, one peptide demonstrated notable anticancer activity, with IC50 values 23 μM against Ramos cancer cells and 22 μM against A20 carcer cells. Additionally, this peptide exhibited a high selectivity index, low toxicity, and improved protein stability (108). Notably, the promising peptide contains two p-trifluoromethyl benzylic substituents within its β2,2 amino acid side chain. These modifications are known to enhance pharmacokinetic properties, alter the electronic distribution of aromatic side chains, and increase lipophilicity, thereby optimizing peptide interactions with the cell membrane components (109–111).
5.3 LTX-315
Rekdal et al. designed, synthesized, and screened a series of peptide inspired by LfcinB derivatives, which demonstrated preferential cytotoxicity against cancer cells over normal cells (112–115). The culmination of their efforts led to the development of a novel and optimized CAMP/ACP named LTX-315 (KKWWKKW-Dip-K) (116).
LTX-315 was obtained by screening a set of lytic nonapeptides using the sequence template KKWWKKWWK, in which tryptophan (Trp) residues were systematically substituted with non-coding amino acids: Ath: 9-anthracenylalanine, Bip: Biphenylalanine, Dip: 3,3-diphenylalanine, 1-Nal: 1-naphthylalanine, and 2-Nal: 2-naphthylalanine. The KKWWKKWWK peptide has a net charge of +6 at physiological pH and is modeled as an α-helical amphipathic structure, with aromatic residues on one side of the helix and cationic residues on the other. Notably, the incorporation of non-coding amino acids has allowed the production of CAMPs (117) and CAPs, enhancing their structural diversity and functional properties (118–121).
Initially, 18 peptides, template peptide and 17 modified peptides, were tested against human A20 lymphoma and murine AT84 squamous cell carcinoma cell lines. A lead series of five peptides, including LTX-315, were identified. These lead candidates were subsequently evaluated against a broad range of cancer cell lines, including drug-resistant strains, were LTX-315 emerged as a highly effective candidate with low activity toward healthy cells (Table 1). Consequently, LTX-315 peptide was selected as the lead candidate for preclinical and clinical studies (122).
LTX-315 is not harmful to human red blood cells (hRBCs), with EC50 > 695 μM. In contrast, it has been observed to rapidly induce cancer cell death in vitro, triggering the release of several danger signals patterns (DAMPs) associated with immunogenic cell death (ICD) and enhanced adaptive immunity. Additionally, LTX-315 induces inflammation and activation of immune cells, such as cytotoxic CD8+ T cells, resulting in tumor shrinkage and systemic immune reactions, as shown in preclinical studies (119, 122, 123).
Two notables preclinical studies explored the combination of LTX-315 with doxorubicin or anti-CTLA-4 antibody. First, administration of LTX-315 with CAELYX® (bran name of the chemotherapy drug doxorubicin) in a murine model triple-negative breast cancer (TNBC) yielded promising results. 4T1 breast cancer cells were implanted, which subsequently mice received LTX-315, CAELYX, or a combination of both. The combination therapy significantly reduced the tumor size, leading to complete regression in 50% of the cases. It also causes extensive tumor necrosis and increases the infiltration of CD8+ T cells into tumors (124). The second study involved the combination of LTX-315 and anti-CTLA-4 antibody, which block the interaction of CTLA-4 with its ligands B7.1 and B7.2 to enhance immune responses, including antitumor immunity. In murine models of sarcoma and melanoma, LTX-315 and anti-CTLA-4 were injected directly into tumors and responses in both treated and untreated tumors, as well as changes in immune cells, were monitored. The combination therapy proved more effective in reducing and eliminating tumors than individual treatments; where LTX-315 decreases the population of suppressor cells and increases active cancer-fighting cells, altering the tumor environment, whereas anti-CTLA-4 enhances the immune response (125). These two promising preclinical studies were conducted in animal models, and further research is required to evaluate the safety and efficacy of these combination therapies in clinical trials.
In a phase I clinical trial (NTCNCT01986426), LTX-315 was evaluated in a dose-escalation study involving intratumoral administration, to assess its safety, tolerability, and efficacy. A total of 39 patients with various advanced solid tumors, including melanoma, breast, head and neck, sarcoma, gastrointestinal, desmoid, pancreas, primary vaginal cancer, and carcinoma of unknown primary, were enrolled. These results indicate that LTX-315 has an acceptable safety profile, is clinically active, induces alterations in the tumor microenvironment, and promotes immune-mediated anticancer activity (126).
Currently, a phase II clinical trial (NCT04796194) is being conducted to investigate the use of LTX-315 in combination with pembrolizumab, focusing on patients with advanced melanoma who have access to percutaneous injections (127). Pembrolizumab is a human antibody approved by the FDA for use in cancer immunotherapy to bind to and block the PD-1 receptor on lymphocytes. It is an immune checkpoint inhibitor (ICI) that avoids the mechanisms used by many cancer cells expressing PD-L1 on their surfaces that could interact with PD-1 in T cells and send a signal to deactivate T cells, effectively preventing them from attacking the tumor (128). To date, LTX-315 has undergone six clinical trials (NCT01986426, NCT01223209, NCT03725605, NCT05188729, NCT01058616, NCT04796194), demonstrating its great potential and versatility as a short cationic peptide with ACP properties.
5.3.1 NTP-217: a synthetic derivate inspired by LTX-315
A derivative of LTX-315 conjugated to rhodamine B (NTP-217) exhibited remarkable enhancement in anticancer activity, achieving a 2.4 to 37.5-fold increase in potency across a diverse panel of adherent cancer cell lines (129). Notably, the efficacy of the hybrid peptide NTP-217 (H-rhodamine B-GABA-KKWWKKWDipK-NH2, where GABA refers to gamma-aminobutyric acid) was rigorously evaluated against liver cancer cells in both in vitro and in vivo models.
Comprehensive assessments, including cell proliferation, cell migration assays, as well as in vivo tumor growth experiments, confirmed NTP-217’s potent anticancer effects. The study concluded that NTP-217 significantly outperformed the parent peptide LTX-315, which proved to be more potent in inhibiting proliferation and migration of liver cancer cells (130).
6 Chemical modifications to enhance peptide resistance to proteolytic degradation
Peptides, particularly AMPs, offer remarkable versatility in cancer treatment and possess substantial potential for overcoming the shortcomings of alternative therapeutic methods (11, 131). However, a key challenge in developing peptide-based medications is that AMPs, which are sometimes effective in vitro, often lose their activity in vivo because of proteolytic degradation in serum (132).
To prevent or mitigate proteolytic degradation, several chemical modification strategies have been employed in the development of synthetic antimicrobial peptides, such as terminal protection, backbone modification, glycosylation, PEGylation, and cyclization (17, 63, 133, 134).
6.1 Terminal protection
Terminal protection strategies help to protect peptides against proteolytic degradation by peptidases, which can cleave both the N-terminal and C-terminal regions of the peptide. This chemical modification can also enhance the half-life and therapeutic efficacy. N-terminal acetylation or C-terminal amidation can fulfill this purpose (135) and similar objectives can be achieved by modifying the termini with unnatural amino acid analogs (136).
6.2 Backbone modification
Backbone modifications include substitution reactions, such as replacing the carbonyl oxygen with sulfur or substitution hydrogen atoms in nitrogen or α-carbon. D-amino acids can be considered a form of backbone modification, and their incorporation into the peptide backbone can increase the resistance to proteolytic degradation. These modifications can also produce profound changes in molecular chirality and conformation, thereby improving peptides stability and bioavailability (17, 137, 138).
6.3 Glycosylation
Glycosylation is the peptide-carbohydrate bond formation which is utilized to improve the cancer treatment outcomes. It improves the tissue targeting, enhances the delivery to the tumor and extends the serum half-life through the inhibition of enzyme degradation. Glycosylated peptides generally show greater permeability across the membrane, which leads to increased cellular uptake and reduced off-target toxicity. Furthermore, glycosylation can affect the immunogenicity of the peptide, which enhances the biocompatibility and the in vivo circulation time. The performance of glycosylated peptide conjugates is a function of the arrangement, type, and number of sugar units which affect the stability, cellular interaction, and therapeutic potential of these compounds (134).
6.4 PEGylation
PEGylation is the attachment of one or more chains of polyethylene glycol (PEG) to a peptide. The application of this modification leads to an increase in peptide solubility and an inhibition of renal clearance together with protection against enzymatic degradation (139), resulting in an extension of the half-life and increased systemic circulation (140). The use of PEGylated peptides has been shown to produce decreased immunogenicity along with enhanced tumor penetration properties, resulting in sustained drug release and improved bioavailability (141).
6.5 Cyclization
Cyclization enhances peptide stability and bioactivity by reducing conformational flexibility and providing better protection against degradation. The formation of a covalent bond, either at the termini or side chains, increases structural rigidity that results in a longer half-life. The anticancer potency of AMPs has been improved through several cyclization methods including head-to-tail cyclization, side-chain cyclization, and peptide stapling (142, 143).
7 Computational pipeline for the selection, optimization, and translation of anticancer peptides from antimicrobial peptides
Early exploration experiments require enormous budgets because the identification and improvement of novel ACPs are labor-intensive, expensive, and time-consuming. In this context, bioinformatics tools present compelling methodologies for ACP discovery (144), that could be applied to enhance CAMPs, which are naturally anticancer, by making them more specific and potent with less toxicity to normal cells (145).
The selection pipeline for ACPs from AMPs is organized systematically, encompassing phases of candidate selection, computational optimization, experimental validation, and clinical translation (Figure 4).
Figure 4. Anticancer peptides selection pipeline. The selection of ACPs from AMPs follows a structured method that begins with identifying potential candidates, then moves on to computational refinement and experimental validation, before advancing to clinical use.
7.1 Selection of candidate peptides
The candidate selection process requires the evaluation of AMPs with anticancer activity retrieved from specialized databases such as APD3, CAMP, CAMPR4, DRAMP, DBAASP, and others. The evaluation includes assessment of key physicochemical properties including charge, hydrophilicity, hydrophobicity, amphipathicity, isoelectric point (pI) and propensity to in vitro aggregate (146–149). While removing toxic or hemolytic peptides using bioinformatic tools such as ToxinPred (150) and HemoPI (Table 2) (151–153), respectively.
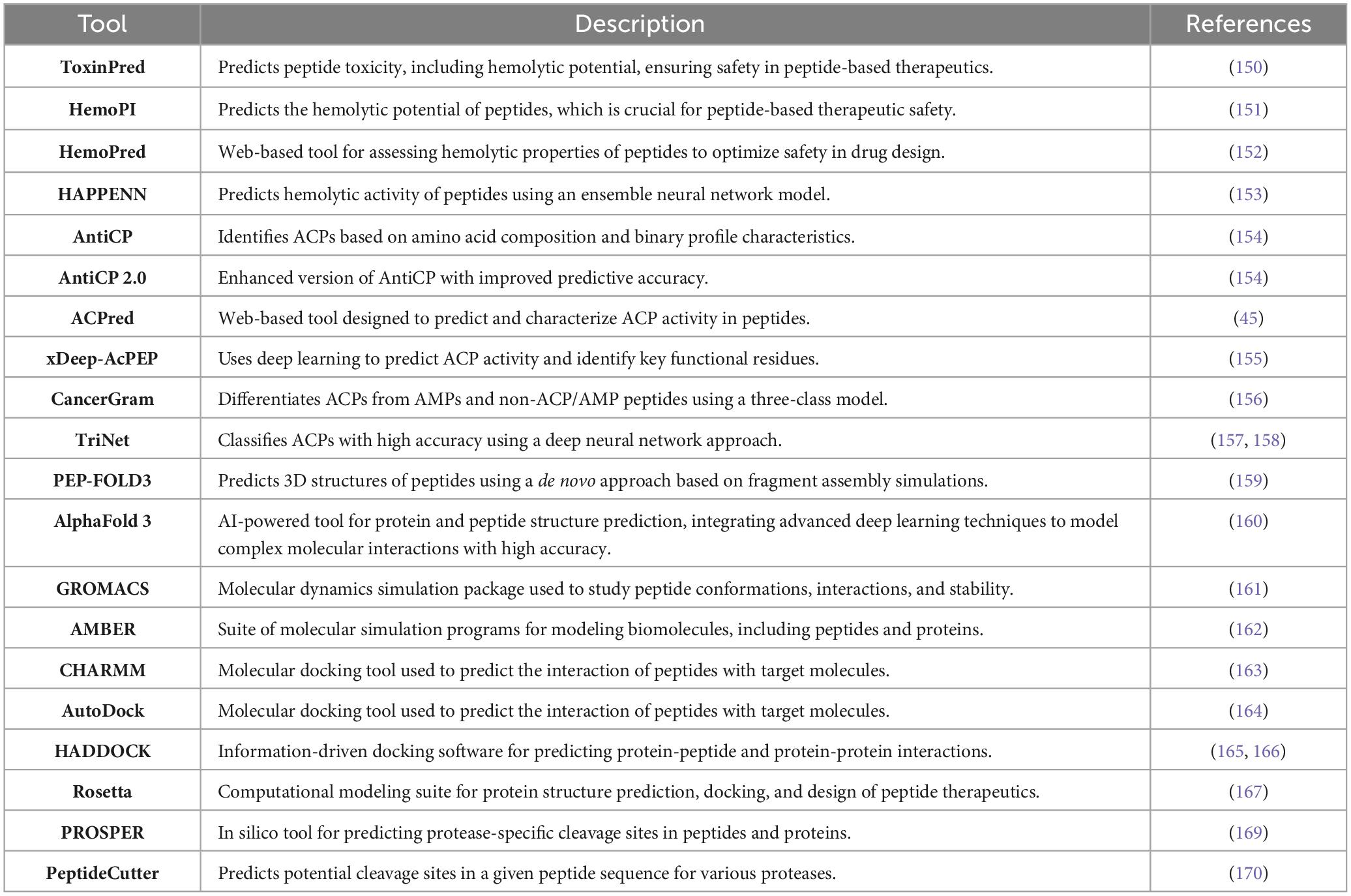
Table 2. Computational tools for predicting ACPs.
7.2 Computational optimization
The candidate peptides receive refinement through computational predictions and optimization which depends on bioinformatics tools such as AntiCP, ACPred and xDeep-AcPEP to evaluate anticancer activity (45, 154, 155). The classification tools such as CancerGram and TriNet server to distinguish antimicrobial from anticancer peptide (156–158).
Molecular modeling techniques predict peptide structures using tools such as PEP-FOLD3 and AlphaFold3 (159, 160), peptide-membrane interaction simulations are conducted with tools like GROMACS, AMBER, or CHARMM (161–163), and binding affinity assessment through docking studies using tools like AutoDock, HADDOCK or Rosetta (164–167).
Furthermore, Quantitative Structure–Activity Relationship (QSAR) models and AI-driven tools optimize sequences based on physicochemical correlations, machine learning algorithms (168), and stability predictions (PROSPER or PeptideCutter) (169, 170).
7.3 Experimental validation
Experimental validation involves conducting in vitro cytotoxicity assays, such as MTT colorimetric assay, assessing selectivity through hemolysis assays on erythrocytes, and evaluating toxicity on normal cells (171, 172). Additionally, membrane disruption assays, including flow cytometry, confocal microscopy, or calcein leakage assays, are examined (173).
Subsequently, in vivo validation is performed using tumor models, where applicable, encompassing efficacy evaluation through xenograft mouse models and toxicity assessment via hematological and histopathological analyses (174, 175).
7.4 Lead optimization and clinical translation
The phases of lead optimization and clinical translation necessitate modifications to improve stability, while formulation strategies are devised to ensure effective delivery. Preclinical evaluations concentrate on pharmacokinetics, immunogenicity, and scalability to facilitate the advancement of therapeutic development (176, 177).
8 Conclusion
Cationic antimicrobial peptides (CAMPs) are considered as potential anticancer agents because they can bind to the negative charged membrane of tumor cells without affecting normal cells. The anticancer activity of CAMPs is dependent on several physicochemical characteristics such as amino acid composition, net charge, hydrophobicity, amphipathicity and secondary structure. Examples such as KLA, bovine lactoferricin derivatives and LTX-315 have been shown to have potent anticancer activity, thus highlighting the potential of CAMPs as anticancer agents.
In order to improve the stability and bioavailability of CAMPs, several chemical modifications like terminal protection, backbone modification, glycosylation, PEGylation and cyclization have been used to enhance the resistance to proteolytic degradation. Moreover, to design next generation anticancer peptides, machine learning driven computational tools have been developed to improve the prediction. However, due to the existing problems such as high production costs, short half-life and possible off-target toxicity, CAMPs still stand as a valuable scaffold for the development of peptide-based anticancer therapeutics. Further structural optimization, chemical modifications and computational advancements will be crucial to fully exploit the potential of CAMPs for clinical uses.
Author contributions
YV-Q: Conceptualization, Data curation, Investigation, Methodology, Visualization, Writing – original draft, Writing – review and editing. FM-R: Funding acquisition, Project administration, Writing – review and editing. DB-E: Conceptualization, Methodology, Project administration, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Fundación Salud de Los Andes, which provided financial support for both the research and the publication of this article.
Acknowledgments
We express their gratitude to Fundación Salud de Los Andes for their generous support. We would like to express our sincere gratitude to Dr. Carolina López and Dr. Willian Agudelo for their valuable feedback and critical reading of the manuscript. Their insightful comments greatly contributed to improving the clarity and quality of this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that Generative AI was used in the creation of this manuscript. In preparing this manuscript, the authors used Paperpal, an AI tool to improve the text. It was applied to review grammar and style, optimize coherence and fluency, and refine the manuscript for publication. This support was especially helpful for ensuring the manuscript met language and editorial standards. Additionally, as the authors are not native English speakers, the tool helped improve the language and ensure the manuscript was ready for publication.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lewandowska A, Rudzki G, Lewandowski T, Rudzki S. The problems and needs of patients diagnosed with cancer and their caregivers. Int J Environ Res Public Health. (2021) 18:87. doi: 10.3390/ijerph18010087
2. Bray F, Laversanne M, Sung H, Ferlay J, Siegel R, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
3. Mandal R, Basu P. Cancer screening and early diagnosis in low and middle income countries : Current situation and future perspectives. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. (2018) 61:1505–12. doi: 10.1007/s00103-018-2833-9
4. Debela D, Muzazu S, Heraro K, Ndalama M, Mesele B, Haile D, et al. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. (2021) 9:20503121211034366. doi: 10.1177/20503121211034366
5. Tohme S, Simmons R, Tsung A. Surgery for cancer: A trigger for metastases. Cancer Res. (2017) 77:1548–52. doi: 10.1158/0008-5472.CAN-16-1536
6. Wang K, Tepper J. Radiation therapy-associated toxicity: Etiology, management, and prevention. CA Cancer J Clin. (2021) 71:437–54. doi: 10.3322/caac.21689
7. Anand U, Dey A, Chandel A, Sanyal R, Mishra A, Pandey D, et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. (2022) 10:1367–401. doi: 10.1016/j.gendis.2022.02.007
8. Nussinov R, Tsai C, Jang H. Anticancer drug resistance: An update and perspective. Drug Resist Updat. (2021) 59:100796. doi: 10.1016/j.drup.2021.100796
9. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: A brief review. Adv Pharm Bull. (2017) 7:339–48. doi: 10.15171/apb.2017.041
10. Emran T, Shahriar A, Mahmud A, Rahman T, Abir M, Siddiquee M, et al. Multidrug resistance in cancer: Understanding molecular mechanisms, immunoprevention and therapeutic approaches. Front Oncol. (2022) 12:891652. doi: 10.3389/fonc.2022.891652
11. Vadevoo S, Gurung S, Lee H, Gunassekaran G, Lee S, Yoon J, et al. Peptides as multifunctional players in cancer therapy. Exp Mol Med. (2023) 55:1099–109. doi: 10.1038/s12276-023-01016-x
12. Hilchie A, Hoskin D, Power Coombs M. In: K Matsuzaki editor. Antimicrobial Peptides: Basics for Clinical Application. Singapore: Springer Singapore (2019). p. 131–47. doi: 10.1007/978-981-13-3588-4_9
13. Mader J, Hoskin D. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin Investig Drugs. (2006) 15:933–46. doi: 10.1517/13543784.15.8.933
14. Gaspar D, Veiga A, Castanho M. From antimicrobial to anticancer peptides. A review. Front Microbiol. (2013) 4:294. doi: 10.3389/fmicb.2013.00294
15. Mahlapuu M, Björn C, Ekblom J. Antimicrobial peptides as therapeutic agents: Opportunities and challenges. Crit Rev Biotechnol. (2020) 40:978–92. doi: 10.1080/07388551.2020.1796576
16. Mabrouk D. Antimicrobial peptides: Features, applications and the potential use against covid-19. Mol Biol Rep. (2022) 49:10039–50. doi: 10.1007/s11033-022-07572-1
17. Barman P, Joshi S, Sharma S, Preet S, Sharma S, Saini A. Strategic approaches to improvise peptide drugs as next generation therapeutics. Int J Pept Res Ther. (2023) 29:61. doi: 10.1007/s10989-023-10524-3
18. Harris F, Dennison S, Phoenix D. Anionic antimicrobial peptides from eukaryotic organisms. Curr Protein Pept Sci. (2009) 10:585–606. doi: 10.2174/138920309789630589
19. Hassan F, Al-aamery R, Mahdi S. A systematic review of Antimicrobial peptides and their current applications. J Popul Ther Clin Pharmacol. (2023) 30:337–43. doi: 10.47750/jptcp.2023.30.06.038
20. Huan Y, Kong Q, Mou H, Yi H. Antimicrobial peptides: Classification, design, application and research progress in multiple fields. Front Microbiol. (2020) 11:582779. doi: 10.3389/fmicb.2020.582779
21. Singh T, Choudhary P, Singh S. Antimicrobial peptides: Mechanism of action. IntechOpen. (2022) 11:1–20. doi: 10.5772/intechopen.99190
22. Le W, Chen B, Cui Z, Liu Z, Shi D. Detection of cancer cells based on glycolytic-regulated surface electrical charges. Biophys Rep. (2019) 5:10–8. doi: 10.1007/s41048-018-0080-0
23. Tornesello A, Borrelli A, Buonaguro L, Buonaguro F, Tornesello M. Antimicrobial peptides as anticancer agents: Functional properties and biological activities. Molecules. (2020) 25:2850. doi: 10.3390/molecules25122850
24. Deslouches B, Di Y. Antimicrobial peptides with selective antitumor mechanisms: Prospect for anticancer applications. Oncotarget. (2017) 8:46635–51. doi: 10.18632/oncotarget.16743
25. Jafari A, Babajani A, Sarrami Forooshani R, Yazdani M, Rezaei-Tavirani M. Clinical applications and anticancer effects of antimicrobial peptides: From bench to bedside. Front Oncol. (2022) 12:819563. doi: 10.3389/fonc.2022.819563
26. Hancock R. Cationic antimicrobial peptides: Towards clinical applications. Expert Opin Investig Drugs. (2000) 9:1723–9. doi: 10.1517/13543784.9.8.1723
27. Felício M, Silva O, Gonçalves S, Santos N, Franco O. Peptides with dual antimicrobial and anticancer activities. Front Chem. (2017) 5:5. doi: 10.3389/fchem.2017.00005
28. Divyashree M, Mani M, Reddy D, Kumavath R, Ghosh P, Azevedo V, et al. Clinical applications of antimicrobial peptides (AMPs): Where do we stand now? Protein Pept Lett. (2020) 27:120–34. doi: 10.2174/0929866526666190925152957
29. Aghamiri S, Zandsalimi F, Raee P, Abdollahifar M, Tan S, Low T, et al. Antimicrobial peptides as potential therapeutics for breast cancer. Pharmacol Res. (2021) 171:105777. doi: 10.1016/j.phrs.2021.105777
30. Mahlapuu M, Håkansson J, Ringstad L, Björn C. Antimicrobial peptides: An emerging category of therapeutic agents. Front Cell Infect Microbiol. (2016) 6:194. doi: 10.3389/fcimb.2016.00194
31. Gan B, Gaynord J, Rowe S, Deingruber T, Spring D. The multifaceted nature of antimicrobial peptides: Current synthetic chemistry approaches and future directions. Chem Soc Rev. (2021) 50:7820–80. doi: 10.1039/d0cs00729c
32. Pirtskhalava M, Vishnepolsky B, Grigolava M, Managadze G. Physicochemical features and peculiarities of interaction of AMP with the membrane. Pharmaceuticals. (2021) 14:471. doi: 10.3390/ph14050471
33. Decker A, Mechesso A, Wang G. Expanding the landscape of amino acid-rich antimicrobial peptides: Definition, deployment in nature, implications for peptide design and therapeutic potential. Int J Mol Sci. (2022) 23:12874. doi: 10.3390/ijms232112874
34. Katchalski-Katzir E, Kasher R, Fridkin M. Amino Acids: Physicochemical Properties BT - Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine. Berlin: Springer (2006). p. 55–68. doi: 10.1007/3-540-29623-9_2260
35. Wimley W. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem Biol. (2010) 5:905–17. doi: 10.1021/cb1001558
36. Kyte J, Doolittle R. A simple method for displaying the hydropathic character of a protein. J Mol Biol. (1982) 157:105–32. doi: 10.1016/0022-2836(82)90515-0
37. Pommié C, Levadoux S, Sabatier R, Lefranc G, Lefranc M. IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J Mol Recognit. (2004) 17:17–32. doi: 10.1002/jmr.647
38. Clark S, Jowitt T, Harris L, Knight C, Dobson C. The lexicon of antimicrobial peptides: A complete set of arginine and tryptophan sequences. Commun Biol. (2021) 4:605. doi: 10.1038/s42003-021-02137-7
39. Tyagi A, Kapoor P, Kumar R, Chaudhary K, Gautam A, Raghava G. In silico models for designing and discovering novel anticancer peptides. Sci Rep. (2013) 3:2984. doi: 10.1038/srep02984
40. Saini S, Rathore A, Sharma S, Saini A. Exploratory data analysis of physicochemical parameters of natural antimicrobial and anticancer peptides: Unraveling the patterns and trends for the rational design of novel peptides. Bioimpacts. (2024) 14:26438. doi: 10.34172/bi.2023.26438
41. Chan D, Prenner E, Vogel H. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim Biophys Acta. (2006) 1758:1184–202. doi: 10.1016/j.bbamem.2006.04.006
42. Mishra A, Choi J, Moon E, Baek K. Tryptophan-rich and proline-rich antimicrobial peptides. Molecules. (2018) 23:815. doi: 10.3390/molecules23040815
43. Straus S. Tryptophan- and arginine-rich antimicrobial peptides: Anti-infectives with great potential. Biochim Biophys Acta Biomembr. (2024) 1866:184260. doi: 10.1016/j.bbamem.2023.184260
44. Chiangjong W, Chutipongtanate S, Hongeng S. Anticancer peptide: Physicochemical property, functional aspect and trend in clinical application (Review). Int J Oncol. (2020) 57:678–96. doi: 10.3892/ijo.2020.5099
45. Schaduangrat N, Nantasenamat C, Prachayasittikul V, Shoombuatong W. ACPred: A computational tool for the prediction and analysis of anticancer peptides. Molecules. (2019) 24:1973. doi: 10.3390/molecules24101973
46. Li J, Koh J, Liu S, Lakshminarayanan R, Verma C, Beuerman R. Membrane active antimicrobial peptides: Translating mechanistic insights to design. Front Neurosci. (2017) 11:73. doi: 10.3389/fnins.2017.00073
47. Pasupuleti M, Schmidtchen A, Malmsten M. Antimicrobial peptides: Key components of the innate immune system. Crit Rev Biotechnol. (2012) 32:143–71. doi: 10.3109/07388551.2011.594423
48. Hoskin D, Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta. (2008) 1778:357–75. doi: 10.1016/j.bbamem.2007.11.008
49. Li L, Vorobyov I, Allen T. The different interactions of lysine and arginine side chains with lipid membranes. J Phys Chem B. (2013) 117:11906–20. doi: 10.1021/jp405418y
50. Hadianamrei R, Tomeh M, Brown S, Wang J, Zhao X. Rationally designed short cationic α-helical peptides with selective anticancer activity. J Colloid Interface Sci. (2022) 607(Pt 1):488–501. doi: 10.1016/j.jcis.2021.08.200
51. Arias M, Haney E, Hilchie A, Corcoran J, Hyndman M, Hancock R, et al. Selective anticancer activity of synthetic peptides derived from the host defence peptide tritrpticin. Biochim Biophys Acta Biomembr. (2020) 1862:183228. doi: 10.1016/j.bbamem.2020.183228
52. Jiang Z, Vasil A, Hale J, Hancock R, Vasil M, Hodges R. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers. (2008) 90:369–83. doi: 10.1002/bip.20911
53. Bello-Madruga R, Torrent Burgas M. The limits of prediction: Why intrinsically disordered regions challenge our understanding of antimicrobial peptides. Comput Struct Biotechnol J. (2024) 23:972–81. doi: 10.1016/j.csbj.2024.02.008
54. Eisenberg D. Three-dimensional structure of membrane and surface proteins. Annu Rev Biochem. (1984) 53:595–623. doi: 10.1146/annurev.bi.53.070184.003115
55. Hancock R, Chapple D. Peptide antibiotics. Antimicrob Agents Chemother. (1999) 43:1317–23. doi: 10.1128/AAC.43.6.1317
56. Huang Y, Huang J, Chen Y. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell. (2010) 1:143–52. doi: 10.1007/s13238-010-0004-3
57. Dong Z, Zhang X, Zhang Q, Tangthianchaichana J, Guo M, Du S, et al. Anticancer mechanisms and potential anticancer applications of antimicrobial peptides and their nano agents. Int J Nanomedicine. (2024) 19:1017–39. doi: 10.2147/IJN.S445333
58. Gagat P, Ostrówka M, Duda-Madej A, Mackiewicz P. Enhancing antimicrobial peptide activity through modifications of charge, hydrophobicity, and structure. Int J Mol Sci. (2024) 25:10821. doi: 10.3390/ijms251910821
59. Perumal P, Pandey V. Antimicrobial peptides: The role of hydrophobicity in the alpha helical structure. J Pharm. (2013) 1:39–53. doi: 10.56499/jppres13.005_1.2.39
60. Yount N, Bayer A, Xiong Y, Yeaman M. Advances in antimicrobial peptide immunobiology. Biopolymers. (2006) 84:435–58. doi: 10.1002/bip.20543
61. Chen Y, Guarnieri M, Vasil A, Vasil M, Mant C, Hodges R. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob Agents Chemother. (2007) 51:1398–406. doi: 10.1128/AAC.00925-06
62. Huang Y, Wang X, Wang H, Liu Y, Chen Y. Studies on mechanism of action of anticancer peptides by modulation of hydrophobicity within a defined structural framework. Mol Cancer Ther. (2011) 10:416–26. doi: 10.1158/1535-7163.MCT-10-0811
63. Xie M, Liu D, Yang Y. Anti-cancer peptides: Classification, mechanism of action, reconstruction and modification. Open Biol. (2020) 10:200004. doi: 10.1098/rsob.200004
64. Yang Y, Zhang H, Wanyan Y, Liu K, Lv T, Li M, et al. Effect of hydrophobicity on the anticancer activity of fatty-acyl-conjugated CM4 in breast cancer cells. ACS Omega. (2020) 5:21513–23. doi: 10.1021/acsomega.0c02093
65. Trinidad-Calderón P, Varela-Chinchilla C, García-Lara S. Natural peptides inducing cancer cell death: Mechanisms and properties of specific candidates for cancer therapeutics. Molecules. (2021) 26:7453. doi: 10.3390/molecules26247453
66. Quemé-Peña M, Juhász T, Kohut G, Ricci M, Singh P, Szigyártó I, et al. Membrane association modes of natural anticancer peptides: Mechanistic details on helicity, orientation, and surface coverage. Int J Mol Sci. (2021) 22:8613. doi: 10.3390/ijms22168613
67. Lee T, Hall K, Aguilar M. Antimicrobial peptide structure and mechanism of action: A focus on the role of membrane structure. Curr Top Med Chem. (2016) 16:25–39. doi: 10.2174/1568026615666150703121700
68. Hwang J, Kim S, Shin T, Jang Y, Kwon D, Lee G. Development of anticancer peptides using artificial intelligence and combinational therapy for cancer therapeutics. Pharmaceutics. (2022) 14:997. doi: 10.3390/pharmaceutics14050997
69. Kaynak A, Davis H, Kogan A, Lee J, Narmoneva D, Qi X. Phosphatidylserine: The unique dual-role biomarker for cancer imaging and therapy. Cancers. (2022) 14:2536. doi: 10.3390/cancers14102536
70. Sharma B, Kanwar S. Phosphatidylserine: A cancer cell targeting biomarker. Semin Cancer Biol. (2018) 52(Pt 1):17–25. doi: 10.1016/j.semcancer.2017.08.012
71. Wang W, Wu S, Cen Z, Zhang Y, Chen Y, Huang Y, et al. Mobilizing phospholipids on tumor plasma membrane implicates phosphatidylserine externalization blockade for cancer immunotherapy. Cell Rep. (2022) 41:111582. doi: 10.1016/j.celrep.2022.111582
72. De M, Ghosh S, Sen T, Shadab M, Banerjee I, Basu S, et al. A novel therapeutic strategy for cancer using phosphatidylserine targeting stearylamine-bearing cationic liposomes. Mol Ther Nucleic Acids. (2018) 10:9–27. doi: 10.1016/j.omtn.2017.10.019
73. Pinho S, Reis C. Glycosylation in cancer: Mechanisms and clinical implications. Nat Rev Cancer. (2015) 15:540–55. doi: 10.1038/nrc3982
74. Liu D, Shriver Z, Venkataraman G, El Shabrawi Y, Sasisekharan R. Tumor cell surface heparan sulfate as cryptic promoters or inhibitors of tumor growth and metastasis. Proc Natl Acad Sci U S A. (2002) 99:568–73. doi: 10.1073/pnas.012578299
75. Kleeff J, Ishiwata T, Kumbasar A, Friess H, Büchler M, Lander A, et al. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J Clin Invest. (1998) 102:1662–73. doi: 10.1172/JCI4105
76. Nadanaka S, Kitagawa H. Heparan sulfate signaling in cancer. Seikagaku. (2017) 89:681–8. doi: 10.14952/SEIKAGAKU.2017.890681
77. Chen C, Tsai K, Kuo P, Chang P, Wang W, Chuang Y, et al. A heparan sulfate-binding cell penetrating peptide for tumor targeting and migration inhibition. Biomed Res Int. (2015) 2015:237969. doi: 10.1155/2015/237969
78. Xi Y, Yani Z, Jing M, Yinhang W, Xiaohui H, Jing Z, et al. Mechanisms of induction of tumors by cholesterol and potential therapeutic prospects. Biomed Pharmacother. (2021) 144:112277. doi: 10.1016/j.biopha.2021.112277
79. Szlasa W, Zendran I, Zalesińska A, Tarek M, Kulbacka J. Lipid composition of the cancer cell membrane. J Bioenerg Biomembr. (2020) 52:321–42. doi: 10.1007/s10863-020-09846-4
80. Liu X, Li Y, Li Z, Lan X, Leung P, Li J, et al. Mechanism of anticancer effects of antimicrobial peptides. J Fiber Bioeng Informatics. (2015) 8:25–36. doi: 10.3993/jfbi03201503
81. Manrique-Moreno M, Santa-González G, Gallego V. Bioactive cationic peptides as potential agents for breast cancer treatment. Biosci Rep. (2021) 41:BSR20211218C. doi: 10.1042/BSR20211218C
82. Hao G, Xu Z, Li L. Manipulating extracellular tumour pH: An effective target for cancer therapy. RSC Adv. (2018) 8:22182–92. doi: 10.1039/c8ra02095g
83. Zoonens M, Reshetnyak Y, Engelman D. Bilayer interactions of pHLIP, a peptide that can deliver drugs and target tumors. Biophys J. (2008) 95:225–35. doi: 10.1529/biophysj.107.124156
84. Burns K, McCleerey T, Thévenin D. pH-selective cytotoxicity of pHLIP-antimicrobial peptide conjugates. Sci Rep. (2016) 6:28465. doi: 10.1038/srep28465
85. Song Q, Chuan X, Chen B, He B, Zhang H, Dai W, et al. A smart tumor targeting peptide-drug conjugate, pHLIP-SS-DOX: Synthesis and cellular uptake on MCF-7 and MCF-7/Adr cells. Drug Deliv. (2016) 23:1734–46. doi: 10.3109/10717544.2015.1028601
86. Gao Y, Shang Q, Li W, Guo W, Stojadinovic A, Mannion C, et al. Antibiotics for cancer treatment: A double-edged sword. J Cancer. (2020) 11:5135–49. doi: 10.7150/jca.47470
87. Javadpour M, Juban M, Lo W, Bishop S, Alberty J, Cowell S, et al. De novo antimicrobial peptides with low mammalian cell toxicity. J Med Chem. (1996) 39:3107–13. doi: 10.1021/jm9509410
88. Jä,kel C, Meschenmoser K, Kim Y, Weiher H, Schmidt-Wolf I. Efficacy of a proapoptotic peptide towards cancer cells. In Vivo. (2012) 26: 419–26.
89. Mai J, Mi Z, Kim S, Ng B, Robbins P. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. (2001) 61:7709–12.
90. Meschenmoser K, Kim Y, Franken S, Nowak M, Feldmann G, Bendas G, et al. Targeting cancer with a bi-functional peptide: In vitro and in vivo results. In Vivo. (2013) 27:431–42.
91. Boohaker R, Lee M, Vishnubhotla P, Perez J, Khaled A. The use of therapeutic peptides to target and to kill cancer cells. Curr Med Chem. (2012) 19:3794–804. doi: 10.2174/092986712801661004
92. Yavari B, Mahjub R, Saidijam M, Raigani M, Soleimani M. The potential use of peptides in cancer treatment. Curr Protein Pept Sci. (2018) 19:759–70. doi: 10.2174/1389203719666180111150008
93. Borrelli A, Tornesello A, Tornesello M, Buonaguro F. Cell penetrating peptides as molecular carriers for anti-cancer agents. Molecules. (2018) 23:295. doi: 10.3390/molecules23020295
94. Ellerby H, Arap W, Ellerby L, Kain R, Andrusiak R, Rio G, et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. (1999) 5:1032–8. doi: 10.1038/12469
95. Ma X, Xi L, Luo D, Liu R, Li S, Liu Y, et al. Anti-tumor effects of the peptide TMTP1-GG-D(KLAKLAK)(2) on highly metastatic cancers. PLoS One. (2012) 7:e42685. doi: 10.1371/journal.pone.0042685
96. Lim C, Won W, Moon J, Sim T, Shin Y, Kim J, et al. Co-delivery of d-(KLAKLAK)2 peptide and doxorubicin using a pH-sensitive nanocarrier for synergistic anticancer treatment. J Mater Chem B. (2019) 7:4299–308. doi: 10.1039/c9tb00741e
97. Shagaghi N, Palombo E, Clayton A, Bhave M. Archetypal tryptophan-rich antimicrobial peptides: Properties and applications. World J Microbiol Biotechnol. (2016) 32:31. doi: 10.1007/s11274-015-1986-z
98. Cutone A, Rosa L, Ianiro G, Lepanto M, Bonaccorsi di Patti M, Valenti P, et al. Lactoferrin’s anti-cancer properties: Safety, selectivity, and wide range of action. Biomolecules. (2020) 10:456. doi: 10.3390/biom10030456
99. Sadiq I, Babagana K, Danlami D, Abdullahi L, Khan A. Molecular therapeutic cancer peptides: A closer look at bovine lactoferricin. Asian J Biochem Genet Mol Biol. (2018) 1:1–9. doi: 10.9734/ajbgmb/2018/v1i2471
100. de la Rosa Arbeláez M, Aleixo D, Barragán Cárdenas A, Pittella F, Tavares G. The role of synthetic peptides derived from bovine lactoferricin against breast cancer cell lines: A mini-review. Oncologie. (2023) 25:629–33. doi: 10.1515/oncologie-2023-0297
101. Pan W, Chen P, Chen Y, Hsu H, Lin C, Chen W. Bovine lactoferricin B induces apoptosis of human gastric cancer cell line AGS by inhibition of autophagy at a late stage. J Dairy Sci. (2013) 96:7511–20. doi: 10.3168/jds.2013-7285
102. Tomita M, Takase M, Bellamy W, Shimamura S. A review: The active peptide of lactoferrin. Acta Paediatr Jpn. (1994) 36:585–91. doi: 10.1111/j.1442-200x.1994.tb03250.x
103. Schibli D, Hwang P, Vogel H. The structure of the antimicrobial active center of lactoferricin B bound to sodium dodecyl sulfate micelles. FEBS Lett. (1999) 446:213–7. doi: 10.1016/s0014-5793(99)00214-8
104. Farnaud S, Spiller C, Moriarty L, Patel A, Gant V, Odell E, et al. Interactions of lactoferricin-derived peptides with LPS and antimicrobial activity. FEMS Microbiol Lett. (2004) 233:193–9. doi: 10.1016/j.femsle.2004.01.039
105. Vega S, Martínez D, Chalá M, Vargas H, Rosas J. Design, synthesis and evaluation of branched RRWQWR-based peptides as antibacterial agents against clinically relevant gram-positive and gram-negative pathogens. Front Microbiol. (2018) 9:329. doi: 10.3389/fmicb.2018.00329
106. Richardson A, de Antueno R, Duncan R, Hoskin D. Intracellular delivery of bovine lactoferricin’s antimicrobial core (RRWQWR) kills T-leukemia cells. Biochem Biophys Res Commun. (2009) 388:736–41. doi: 10.1016/j.bbrc.2009.08.083
107. Hilchie A, Vale R, Zemlak T, Hoskin D. Generation of a hematologic malignancy-selective membranolytic peptide from the antimicrobial core (RRWQWR) of bovine lactoferricin. Exp Mol Pathol. (2013) 95:192–8. doi: 10.1016/j.yexmp.2013.07.006
108. Tørfoss V, Ausbacher D, Cavalcanti-Jacobsen Cde A, Hansen T, Brandsdal B, Havelkova M, et al. Synthesis of anticancer heptapeptides containing a unique lipophilic β(2,2) -amino acid building block. J Pept Sci. (2012) 18:170–6. doi: 10.1002/psc.1434
109. Giménez D, Andreu C, del Olmo M, Varea T, Diaz D, Asensio G. The introduction of fluorine atoms or trifluoromethyl groups in short cationic peptides enhances their antimicrobial activity. Bioorg Med Chem. (2006) 14:6971–8. doi: 10.1016/j.bmc.2006.06.027
110. Hagmann W. The many roles for fluorine in medicinal chemistry. J Med Chem. (2008) 51:4359–69. doi: 10.1021/jm800219f
111. Gillis E, Eastman K, Hill M, Donnelly D, Meanwell N. Applications of fluorine in medicinal chemistry. J Med Chem. (2015) 58:8315–59. doi: 10.1021/acs.jmedchem.5b00258
112. Rekdal Ø, Andersen J, Vorland L, Svendsen J. Construction and synthesis of lactoferricin derivatives with enhanced antibacterial activity. J Pept Sci. (1999) 5:32–45. doi: 10.1002/(SICI)1099-1387(199901)5:13.0.CO;2-9
113. Yang N, Lejon T, Rekdal O. Antitumour activity and specificity as a function of substitutions in the lipophilic sector of helical lactoferrin-derived peptide. J Pept Sci. (2003) 9:300–11. doi: 10.1002/psc.457
114. Berge G, Eliassen L, Camilio K, Bartnes K, Sveinbjørnsson B, Rekdal O. Therapeutic vaccination against a murine lymphoma by intratumoral injection of a cationic anticancer peptide. Cancer Immunol Immunother. (2010) 59:1285–94. doi: 10.1007/s00262-010-0857-6
115. Fadnes B, Uhlin-Hansen L, Lindin I, Rekdal Ø. Small lytic peptides escape the inhibitory effect of heparan sulfate on the surface of cancer cells. BMC Cancer. (2011) 11:116. doi: 10.1186/1471-2407-11-116
116. Camilio K, Rekdal O, Sveinbjörnsson B. LTX-315 (Oncopore™): A short synthetic anticancer peptide and novel immunotherapeutic agent. Oncoimmunology. (2014) 3:e29181. doi: 10.4161/onci.29181
117. Khemaissa S, Walrant A, Sagan S. Tryptophan, more than just an interfacial amino acid in the membrane activity of cationic cell-penetrating and antimicrobial peptides. Q Rev Biophys. (2022) 55:e10. doi: 10.1017/S0033583522000105
118. Eliassen L, Haug B, Berge G, Rekdal O. Enhanced antitumour activity of 15-residue bovine lactoferricin derivatives containing bulky aromatic amino acids and lipophilic N-terminal modifications. J Pept Sci. (2003) 9:510–7. doi: 10.1002/psc.472
119. Camilio K, Berge G, Ravuri C, Rekdal O, Sveinbjørnsson B. Complete regression and systemic protective immune responses obtained in B16 melanomas after treatment with LTX-315. Cancer Immunol Immunother. (2014) 63:601–13. doi: 10.1007/s00262-014-1540-0
120. Midura-Nowaczek K, Markowska A. Antimicrobial peptides and their analogs: Searching for new potential therapeutics. Perspect Medicin Chem. (2014) 6:73–80. doi: 10.4137/PMC.S13215
121. Cheah Y, Liu C, Yip B, Wu C, Peng K, Cheng J. Strategy to enhance anticancer activity and induced immunogenic cell death of antimicrobial peptides by using non-nature amino acid substitutions. Biomedicines. (2022) 10:1097. doi: 10.3390/biomedicines10051097
122. Haug B, Camilio K, Eliassen L, Stensen W, Svendsen J, Berg K, et al. Discovery of a 9-mer cationic peptide (LTX-315) as a potential first in class oncolytic peptide. J Med Chem. (2016) 59:2918–27. doi: 10.1021/acs.jmedchem.5b02025
123. Sveinbjørnsson B, Camilio K, Haug B, Rekdal Ø. LTX-315: A first-in-class oncolytic peptide that reprograms the tumor microenvironment. Future Med Chem. (2017) 9:1339–44. doi: 10.4155/fmc-2017-0088
124. Camilio K, Wang M, Mauseth B, Waagene S, Kvalheim G, Rekdal Ø, et al. Combining the oncolytic peptide LTX-315 with doxorubicin demonstrates therapeutic potential in a triple-negative breast cancer model. Breast Cancer Res. (2019) 21:9. doi: 10.1186/s13058-018-1092-x
125. Yamazaki T, Pitt J, Vétizou M, Marabelle A, Flores C, Rekdal Ø, et al. The oncolytic peptide LTX-315 overcomes resistance of cancers to immunotherapy with CTLA4 checkpoint blockade. Cell Death Differ. (2016) 23:1004–15. doi: 10.1038/cdd.2016.35
126. Spicer J, Marabelle A, Baurain J, Jebsen N, Jøssang D, Awada A, et al. Safety, antitumor activity, and T-cell responses in a dose-ranging phase I trial of the oncolytic peptide LTX-315 in patients with solid tumors. Clin Cancer Res. (2021) 27:2755–63. doi: 10.1158/1078-0432.CCR-20-3435
127. Dalle S, Marron T, Robert C, Aljabri B, Nyakas M, Bruins Slot K, et al. Sveinbjornsson B, Currie G. 1051P Intratumoral injection of LTX-315 in combination with pembrolizumab in patients with advanced melanoma refractory to prior PD-1/PD-L1 therapy: Interim results from the ATLAS-IT-05 trial. Ann Oncol. (2023) 34:S636. doi: 10.1016/j.annonc.2023.09.2190
128. Kwok G, Yau T, Chiu J, Tse E, Kwong Y. Pembrolizumab (Keytruda). Hum Vaccin Immunother. (2016) 12:2777–89. doi: 10.1080/21645515.2016.1199310
129. Yin H, Chen X, Chi Q, Ma Y, Fu X, Du S, et al. The hybrid oncolytic peptide NTP-385 potently inhibits adherent cancer cells by targeting the nucleus. Acta Pharmacol Sin. (2023) 44:201–10. doi: 10.1038/s41401-022-00939-x
130. Yin H, Fu X, Gao H, Gao H, Ma Y, Chen X, et al. Hybrid peptide NTP-217 triggers ROS-mediated rapid necrosis in liver cancer cells by induction of mitochondrial leakage. Front Oncol. (2023) 12:1028600. doi: 10.3389/fonc.2022.1028600
131. Rossino G, Marchese E, Galli G, Verde F, Finizio M, Serra M, et al. Peptides as therapeutic agents: Challenges and opportunities in the green transition era. Molecules. (2023) 28:7165. doi: 10.3390/molecules28207165
132. Ciulla M, Civera M, Sattin S, Kumar K. Nature-inspired and medicinally relevant short peptides. Explor Drug Sci. (2023) 1:140–71. doi: 10.37349/eds.2023.00011
133. Lucana M, Arruga Y, Petrachi E, Roig A, Lucchi R, Oller-Salvia B. Protease-resistant peptides for targeting and intracellular delivery of therapeutics. Pharmaceutics. (2021) 13:2065. doi: 10.3390/pharmaceutics13122065
134. Bellavita R, Braccia S, Galdiero S, Falanga A. Glycosylation and lipidation strategies: approaches for improving antimicrobial peptide efficacy. Pharmaceuticals. (2023) 16:439. doi: 10.3390/ph16030439
135. D’Accolti M, Bellotti D, Dzień E, Leonetti C, Leveraro S, Albanese V, et al. Impact of C- and N-terminal protection on the stability, metal chelation and antimicrobial properties of calcitermin. Sci Rep. (2023) 13:18228. doi: 10.1038/s41598-023-45437-0
136. Brango-Vanegas J, Leite M, Macedo M, Cardoso M, Franco O. Capping motifs in antimicrobial peptides and their relevance for improved biological activities. Front Chem. (2024) 12:1382954. doi: 10.3389/fchem.2024.1382954
137. Lu J, Xu H, Xia J, Ma J, Xu J, Li Y, et al. D- and unnatural amino acid substituted antimicrobial peptides with improved proteolytic resistance and their proteolytic degradation characteristics. Front Microbiol. (2020) 11:563030. doi: 10.3389/fmicb.2020.563030
138. Pei J, Gao X, Pan D, Hua Y, He J, Liu Z, et al. Advances in the stability challenges of bioactive peptides and improvement strategies. Curr Res Food Sci. (2022) 5:2162–70. doi: 10.1016/j.crfs.2022.10.031
139. Veronese F, Mero A. The impact of PEGylation on biological therapies. BioDrugs. (2008) 22:315–29. doi: 10.2165/00063030-200822050-00004
140. Sahsuvar S, Kocagoz T, Gok O, Can O. In vitro efficacy of different PEGylation designs on cathelicidin-like peptide with high antibacterial and antifungal activity. Sci Rep. (2023) 13:11213. doi: 10.1038/s41598-023-38449-3
141. Manteghi R, Pallagi E, Olajos G, Csóka I. Pegylation and formulation strategy of Anti-Microbial Peptide (AMP) according to the quality by design approach. Eur J Pharm Sci. (2020) 144:105197. doi: 10.1016/j.ejps.2019.105197
142. Ji X, Nielsen A, Heinis C. Cyclic peptides for drug development. Angew Chem Int Ed Engl. (2024) 63:e202308251. doi: 10.1002/anie.202308251
143. Ngambenjawong C, Gustafson H, Sylvestre M, Pun S. A facile cyclization method improves peptide serum stability and confers intrinsic fluorescence. Chembiochem. (2017) 18:2395–8. doi: 10.1002/cbic.201700446
144. Padhy I, Biswas A, Nayak C, Sharma T. Rational design of peptides and protein molecules in drug discovery. In: M Rudrapal editor. Computational Methods for Rational Drug Design. Hoboken, NJ: John Wiley & Sons, Ltd (2025). p. 327–62. doi: 10.1002/9781394249190.ch15
145. Abbas N, Tan H, Goh B, Yap W, Tang Y. In silico study of anticancer and antimicrobial peptides derived from cycloviolacin O2 (CyO2). Biointerface Res Appl Chem. (2023) 13:1–16. doi: 10.33263/BRIAC135.437
146. Wang G, Li X, Wang Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. (2016) 44:D1087–93. doi: 10.1093/nar/gkv1278
147. Waghu F, Gopi L, Barai R, Ramteke P, Nizami B, Idicula-Thomas S. CAMP: Collection of sequences and structures of antimicrobial peptides. Nucleic Acids Res. (2014) 42(Database issue):D1154–8. doi: 10.1093/nar/gkt1157
148. Ramazi S, Mohammadi N, Allahverdi A, Khalili E, Abdolmaleki P. A review on antimicrobial peptides databases and the computational tools. Database. (2022) 2022:baac011. doi: 10.1093/database/baac011
149. Gawde U, Chakraborty S, Waghu F, Barai R, Khanderkar A, Indraguru R, et al. CAMPR4: A database of natural and synthetic antimicrobial peptides. Nucleic Acids Res. (2023) 51:D377–83. doi: 10.1093/nar/gkac933
150. Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Consortium Open Source Drug Discovery, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS One. (2013) 8:e73957. doi: 10.1371/journal.pone.0073957
151. Chaudhary K, Kumar R, Singh S, Tuknait A, Gautam A, Mathur D, et al. A web server and mobile app for computing hemolytic potency of peptides. Sci Rep. (2016) 6:22843. doi: 10.1038/srep22843
152. Win T, Malik A, Prachayasittikul V, Wikberg J, Nantasenamat C, Shoombuatong W. HemoPred: A web server for predicting the hemolytic activity of peptides. Future Med Chem. (2017) 9:275–91. doi: 10.4155/fmc-2016-0188
153. Timmons P, Hewage C. HAPPENN is a novel tool for hemolytic activity prediction for therapeutic peptides which employs neural networks. Sci Rep. (2020) 10:10869. doi: 10.1038/s41598-020-67701-3
154. Agrawal P, Bhagat D, Mahalwal M, Sharma N, Raghava G. AntiCP 2.0: An updated model for predicting anticancer peptides. Brief Bioinform. (2021) 22:bbaa153. doi: 10.1093/bib/bbaa153
155. Chen J, Cheong H, Siu S. xDeep-AcPEP: Deep learning method for anticancer peptide activity prediction based on convolutional neural network and multitask learning. J Chem Inf Model. (2021) 61:3789–803. doi: 10.1021/acs.jcim.1c00181
156. Burdukiewicz M, Sidorczuk K, Rafacz D, Pietluch F, Ba̧kała M, Słowik J, et al. CancerGram: An effective classifier for differentiating anticancer from antimicrobial peptides. Pharmaceutics. (2020) 12:1045. doi: 10.3390/pharmaceutics12111045
157. Zhou W, Liu Y, Li Y, Kong S, Wang W, Ding B, et al. TriNet: A tri-fusion neural network for the prediction of anticancer and antimicrobial peptides. Patterns. (2023) 4:100702. doi: 10.1016/j.patter.2023.100702
158. Han J, Zhang S, Liu J. Protocol for predicting peptides with anticancer and antimicrobial properties by a tri-fusion neural network. STAR Protoc. (2023) 4:102541. doi: 10.1016/j.xpro.2023.102541
159. Lamiable A, Thévenet P, Rey J, Vavrusa M, Derreumaux P, Tufféry P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. (2016) 44:W449–54. doi: 10.1093/nar/gkw329
160. Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. (2024) 630:493–500. doi: 10.1038/s41586-024-07487-w
161. Lemkul J. Introductory tutorials for simulating protein dynamics with GROMACS. J Phys Chem B. (2024) 128:9418–35. doi: 10.1021/acs.jpcb.4c04901
162. Case D, Aktulga H, Belfon K, Cerutti D, Cisneros G, Cruzeiro V, et al. AmberTools. J Chem Inf Model. (2023) 63:6183–91. doi: 10.1021/acs.jcim.3c01153
163. Brooks B, Brooks C, Mackerell A, Nilsson L, Petrella R, Roux B, et al. CHARMM: the biomolecular simulation program. J Comput Chem. (2009) 30:1545–614. doi: 10.1002/jcc.21287
164. Forli S, Huey R, Pique M, Sanner M, Goodsell D, Olson A. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. (2016) 11:905–19. doi: 10.1038/nprot.2016.051
165. van Zundert G, Rodrigues J, Trellet M, Schmitz C, Kastritis P, Karaca E, et al. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J Mol Biol. (2016) 428:720–5. doi: 10.1016/j.jmb.2015.09.014
166. Geng C, Narasimhan S, Rodrigues J, Bonvin A. In: O Schueler-Furman, N London editors. Information-Driven, Ensemble Flexible Peptide Docking Using HADDOCK BT - Modeling Peptide-Protein Interactions: Methods and Protocols. New York, NY: Springer (2017). p. 109–38. doi: 10.1007/978-1-4939-6798-8_8
167. Leman J, Weitzner B, Lewis S, Adolf-Bryfogle J, Alam N, Alford R, et al. Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nat Methods. (2020) 17:665–80. doi: 10.1038/s41592-020-0848-2
168. Mao J, Akhtar J, Zhang X, Sun L, Guan S, Li X, et al. Comprehensive strategies of machine-learning-based quantitative structure-activity relationship models. iScience. (2021) 24:103052. doi: 10.1016/j.isci.2021.103052
169. Song J, Tan H, Perry A, Akutsu T, Webb G, Whisstock J, et al. PROSPER: An integrated feature-based tool for predicting protease substrate cleavage sites. PLoS One. (2012) 7:e50300. doi: 10.1371/journal.pone.0050300
170. Li F, Wang Y, Li C, Marquez-Lago T, Leier A, Rawlings N, et al. Twenty years of bioinformatics research for protease-specific substrate and cleavage site prediction: A comprehensive revisit and benchmarking of existing methods. Brief Bioinform. (2018) 20:2150–66. doi: 10.1093/bib/bby077
171. Kao H, Weng T, Chen C, Chen Y, Chi Y, Huang K, et al. Integrating in silico and in vitro approaches to identify natural peptides with selective cytotoxicity against cancer cells. Int J Mol Sci. (2024) 25:6848. doi: 10.3390/ijms25136848
172. Sæbø I, Bjørås M, Franzyk H, Helgesen E, Booth J. Optimization of the hemolysis assay for the assessment of cytotoxicity. Int J Mol Sci. (2023) 24:2914. doi: 10.3390/ijms24032914
173. Li R, Chen C, Zhu S, Wang X, Yang Y, Shi W, et al. CGA-N9, an antimicrobial peptide derived from chromogranin A: Direct cell penetration of and endocytosis by Candida tropicalis. Biochem J. (2019) 476:483–97. doi: 10.1042/BCJ20180801
174. Liu Y, Wu W, Cai C, Zhang H, Shen H, Han Y. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduct Target Ther. (2023) 8:160. doi: 10.1038/s41392-023-01419-2
175. Sifontes-Rodríguez S, Hernández-Aceves J, Salas-Garrido C, Rocha D, Pérez-Osorio I, Villalobos N, et al. In silico, in vitro and in vivo toxicity assessment of the antitumoral peptide GK-1. Toxicol Rep. (2025) 14:101962. doi: 10.1016/j.toxrep.2025.101962
176. Al Musaimi O, Lombardi L, Williams D, Albericio F. Strategies for improving peptide stability and delivery. Pharmaceuticals. (2022) 15:1283. doi: 10.3390/ph15101283
Keywords: cationic antimicrobial peptides (CAMPs), anticancer peptides (ACPs), peptide-based cancer therapeutics, synthetic peptide design, proteolytic stability enhancement, computational peptide engineering, clinical translation
Citation: Varela-Quitián YF, Mendez-Rivera FE and Bernal-Estévez DA (2025) Cationic antimicrobial peptides: potential templates for anticancer agents. Front. Med. 12:1548603. doi: 10.3389/fmed.2025.1548603
Received: 19 December 2024; Accepted: 07 April 2025;
Published: 24 April 2025.
Edited by:
Anna Lucia Tornesello, G. Pascale National Cancer Institute Foundation (IRCCS), ItalyReviewed by:
Annarita Falanga, University of Naples Federico II, ItalyMadhukiran Parvathaneni, Harrisburg University of Science and Technology, United States
Pratima Gurung, Boston Children’s Hospital and Harvard Medical School, United States
Copyright © 2025 Varela-Quitián, Mendez-Rivera and Bernal-Estévez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yahson Fernando Varela-Quitián, eXZhcmVsYUBzYWx1ZGRlbG9zYW5kZXMuY29t