 Cong Duan1,2,3,4†
Cong Duan1,2,3,4† Yuan Zong2,3,4†Qian Chen2,3,4Wei Liu2,3,4Qing Chang2,3,4Cheng Xiong1Zhu-Lin Hu1*
Yuan Zong2,3,4†Qian Chen2,3,4Wei Liu2,3,4Qing Chang2,3,4Cheng Xiong1Zhu-Lin Hu1* Ge-Zhi Xu2,3,4*
Ge-Zhi Xu2,3,4* Feng-Juan Gao2,3,4*
Feng-Juan Gao2,3,4*- 1Yunnan Eye Institute & Key Laboratory of Yunnan Province, Yunnan Eye Disease Clinical Medical Center, Affiliated Hospital of Yunnan University, Yunnan University, Yunnan, China
- 2Eye Institute and Department of Ophthalmology, Eye & ENT Hospital, Fudan University, Shanghai, China
- 3Shanghai Key Laboratory of Visual Impairment and Restoration, Shanghai, China
- 4NHC Key Laboratory of Myopia and Related Eye Diseases; Key Laboratory of Myopia and Related Eye Diseases, Chinese Academy of Medical Sciences, Shanghai, China
Cystoid macular edema (CME) is a common complication in various retinal disorders, often leading to significant central vision impairment. However, the underlying genetic causes and detailed clinical features in patients with fluctuating CME remain unclear. This retrospective, observational case series analyzed two patients from a single family with fluctuating CME, focusing on both clinical and genetic aspects. Data were collected and analyzed from September 2022 to January 2023 at a single center. Comprehensive ocular examinations, including best-corrected visual acuity tests, color fundus photography, fundus fluorescein angiography (FFA), optical coherence tomography (OCT), visual field tests, flash electroretinography, multifocal electroretinography, and electrooculography, were performed. Genetic analysis was conducted using whole exome sequencing, with confirmation through Sanger sequencing and co-segregation analysis. The results identified two compound heterozygous variants in the MYO7A gene: c.562C>G p.Q188E and c.5929C>T p.R1977W in both patients. Fundus fluorescein angiography revealed cystoid hyperfluorescence in a petaloid pattern in the foveal area and a honeycomb pattern parafoveally. OCT showed that macular cystoid changes were primarily located in the outer nuclear layer (ONL), and full-field electroretinography indicated rod-cone dysfunction. Over a 108-day follow-up period, CME in both patients exhibited fluctuating changes without any treatment. This case series suggests that the identified MYO7A variants are likely associated with fluctuating CME, expanding the phenotypic spectrum of MYO7A and providing new insights into the mechanisms underlying CME. Identifying these MYO7A variants bridges genetic research with clinical diagnostics, potentially offering more precise and personalized treatment strategies for retinal disorders.
Introduction
Macular edema is a major cause of visual impairment in the course of several retinal diseases, such as diabetic retinopathy, retina vein occlusions, uveitis, and inherited retinal diseases (1). Cystoid macular edema (CME) can be defined as an abnormal accumulation of fluid in the neural retina layers and presents as cystoid spaces on optical coherence tomography (OCT) B-scans (2). Mechanisms associated with CME include breakdown of the inner and outer blood-retinal barrier, dysfunction of Müller or retinal pigment epithelium cells (3), and vitreous traction (4). Although the pathogenesis of CME is not clearly understood, it is a well-described feature of several inherited retinal diseases, such as retinitis pigmentosa (RP) [CME is observed in 10%−50% of RP patients (5)], juvenile X-linked retinoschisis, gyrate atrophy, and chloridemia (6).
MYO7A (OMIM: 276903) is located at 11q13.5 and has 49 exons. It encodes an unconventional myosin of 2,215 amino acids, myosin VIIA (7), which is a member of the large myosin family. Myosin VIIA plays an important role in retinal photoreceptor cells, the pigment epithelium and inner ear; therefore, mutations in MYO7A result in a spectrum of phenotypes ranging from Usher syndrome type 1B (8) to recessive non-syndromic hearing loss (DFNB2) and autosomal dominant hearing loss (DFNA11) (9). To date, 759 MYO7A variants classified as likely pathogenic or pathogenic have been reported in humans (https://www.ncbi.nlm.nih.gov/clinvar; accessed 21 Dec 2023), most of which cause Usher syndrome type 1B. DFNB2 and DFNA11 manifest hearing loss and vestibular dysfunction, but not usually retinal pathology. Patients with Usher syndrome type 1B usually show congenital deafness, vestibular dysfunction, and prepubertal onset RP, which leads to blindness. Although rare, a MYO7A variant also correlated with Usher syndrome type 2, which presents less severe features than type 1B (10). CME is not a rare phenotype in MYO7A-associated USH, and it is often significantly correlated with alterations in photoreceptor segments (11). In the present study, we report an unusual MYO7A mutation-associated phenotype in two patients from one Chinese family, who both present fluctuating CME and rod-cone dysfunction, without other RP-related symptoms or hearing impairment.
Case description
Two 23-year-old twin sisters presented with progressive vision deterioration persisting for over 6 months. Clinical data of the two patients II1 and II2 are summarized in Table 1. The family history was negative for ocular diseases, and both patients reported no history of systemic or other ocular diseases since childhood. No abnormalities were observed in the external and anterior segment. Fundus examination revealed no abnormalities, with only an absence of the foveal reflex (Figure 1A). OCT showed macular cystoid changes in both eyes; large and elongated cavities were mainly in the outer nuclear layer (ONL), while smaller, round cystoid cavities were observed at the level of the inner nuclear layer (INL; Figure 1B). Early arteriovenous phase fundus fluorescein angiography (FFA; 4 min) showing cystoid hyperfluorescence in a petaloid pattern in the foveal area with a honeycomb pattern parafoveally. No leakage or abnormal fluorescence change was present in the periphery of the retina (Figure 1C). FfERG showed notable attenuation of rod responses, while cone responses were prolonged in both eyes corresponding to a rod-cone dystrophy dysfunction pattern (Figure 1D). The Arden ratios of electrooculography were mildly reduced (right/left 1.5/1.7) in both eyes, while mfERG showed that the amplitudes of rings 2–5 of both eyes were mild reduced (Figure 1E). Furthermore, all routine laboratory tests related to vasculitis, including erythrocyte sedimentation rate, C-reactive protein (CRP), blood count, serum creatinine, urinalysis, specific autoantibodies, complement, immunoglobulin, cryoglobulin, and hepatitis B and C serology, were negative. The younger sister (II2) showed similar clinical findings (Table 1 and Supplementary Figure S1).
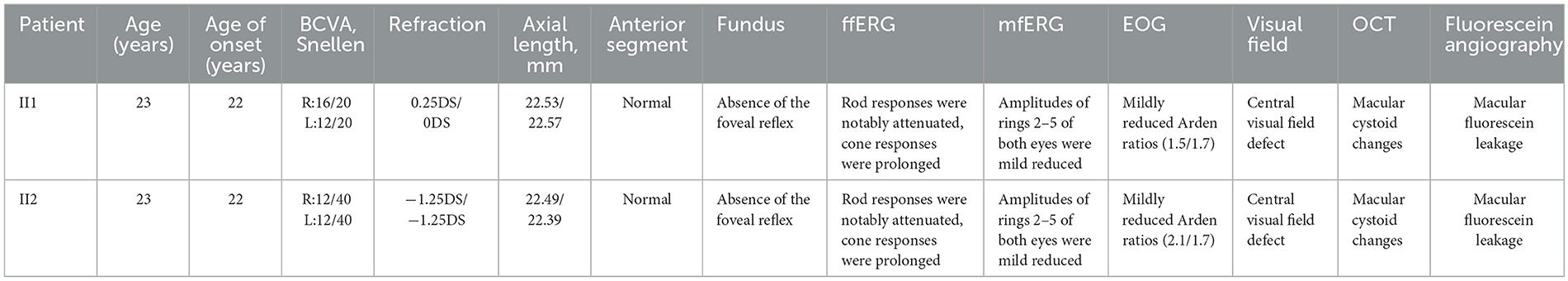
Table 1. Clinical characteristics of patients.
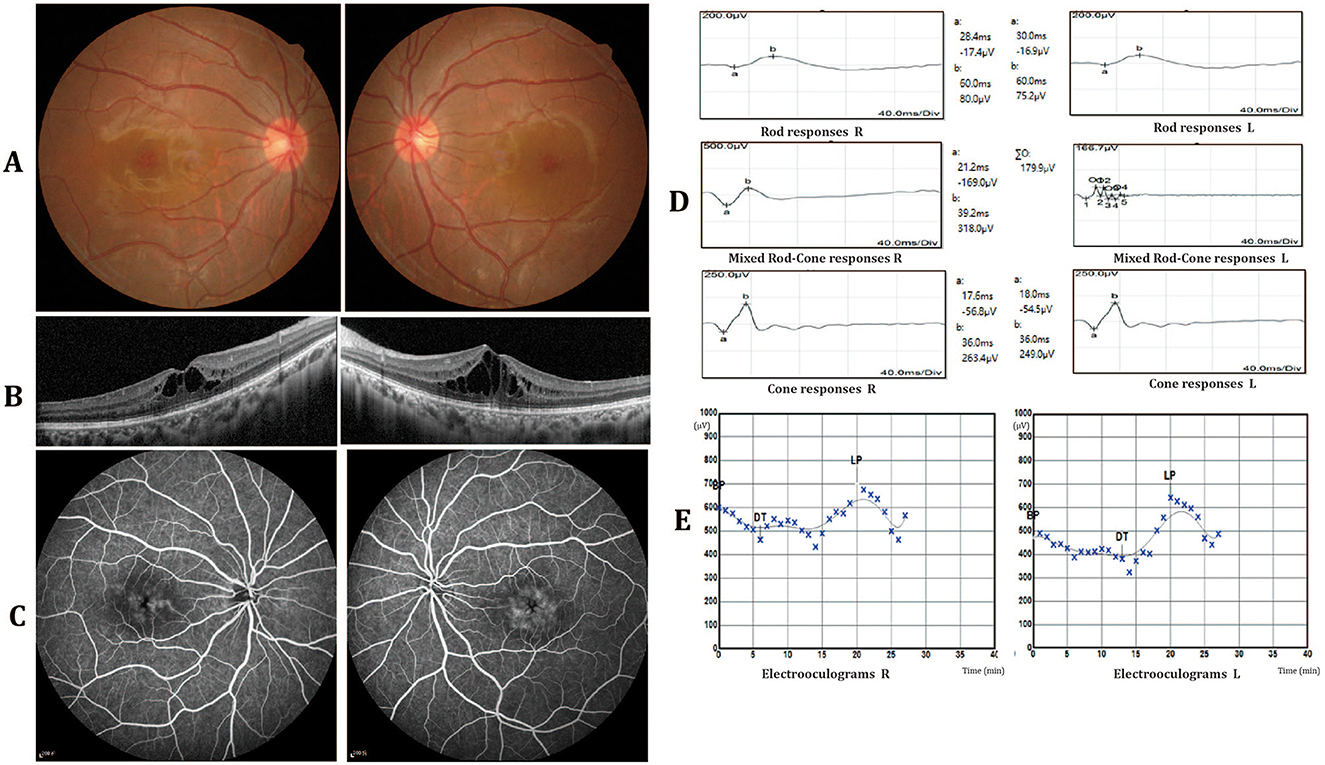
Figure 1. Clinical characteristics of patient II1. (A) Fundus photography revealed no abnormalities, with only an absence of the foveal reflex. (B) Optical coherence tomography (OCT) showed macular cystoid changes in both eyes. The cystoid cavities are mainly involved in the outer nuclear layer (ONL). (C) Fundus fluorescein angiography (FFA) showing cystoid hyperfluorescence in a petaloid pattern in the foveal area with a honeycomb pattern parafoveally. No leakage or abnormal fluorescence change was present in the periphery of the retina. (D) Full-field electroretinography (ffERG) showed rod responses to be notably attenuated and cone responses to be prolonged in both eyes. (E) The electrooculography (EOG) arden ratios were mildly reduced in both eyes (right/left 1.5/1.7).
Follow-up data were available for both patients, with a mean follow-up time of 108 days. Fluctuation of cystoid macular edema (CME) was observed in both eyes without any treatment during this period. The degree of CME was decreased on the second day. The intraretinal cystoid spaces in the fovea were almost absent in the right eye on the third visit (seventh day) but were enlarged in the left eye (Figure 2). However, CME recurred and increased in the right eye on the fourth visit (16th day), but was almost absent from both eyes at 108 days. Final best-corrected visual acuity after 108 days was 10/10 in both eyes, although OCT revealed disorganization of the outer retina at the fovea region. The younger sister (II2) also manifested fluctuation of CME (Supplementary Figures S2, S3).
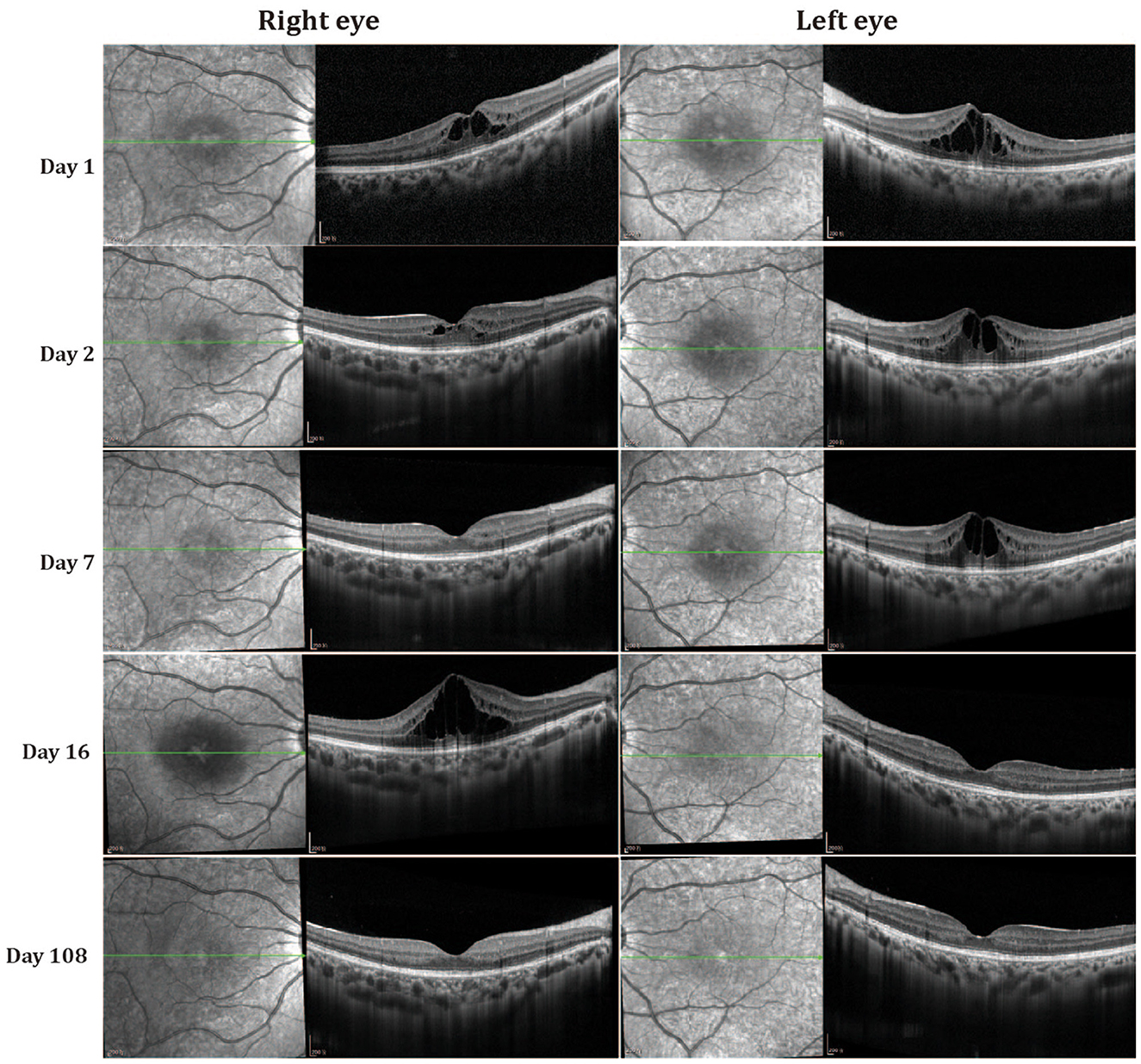
Figure 2. Optical coherence tomography (OCT) changes in patient II1 at different follow-up times.
Whole-exome sequencing was performed on the family, and two previously reported mutations, MYO7A c.562C>G p.Q188E (maternally inherited) and MYO7A c.5929C>T p.R1977W (paternally inherited) (12, 13), were found in the two patients (Figures 3a, b). These mutations co-segregated with the phenotype. Of the two mutations, p.R1977W is located in the FERM domain, and p.Q188E is located in the motor domain of the MYO7A protein (Supplementary Figure S4A). Multiple orthologous sequence alignment revealed that MYO7A codon 188 and subsequent sequences encode highly conserved amino acids across different species, indicating that mutation at any of those codons may lead to a deleterious effect (Supplementary Figure S4B). Three-dimensional predictions of the two mutations are shown in Supplementary Figure S4C. Both mutations were extremely rare in the control population. Further analysis using SIFT, MutationTaster, FATHMM, and PolyPhen-2 predicted both mutations to be pathogenic. Details of the weighted allele scores are given in Supplementary Table S1. No additional pathogenic or likely pathogenic variants known to be associated with inherited eye diseases were found in either patient.
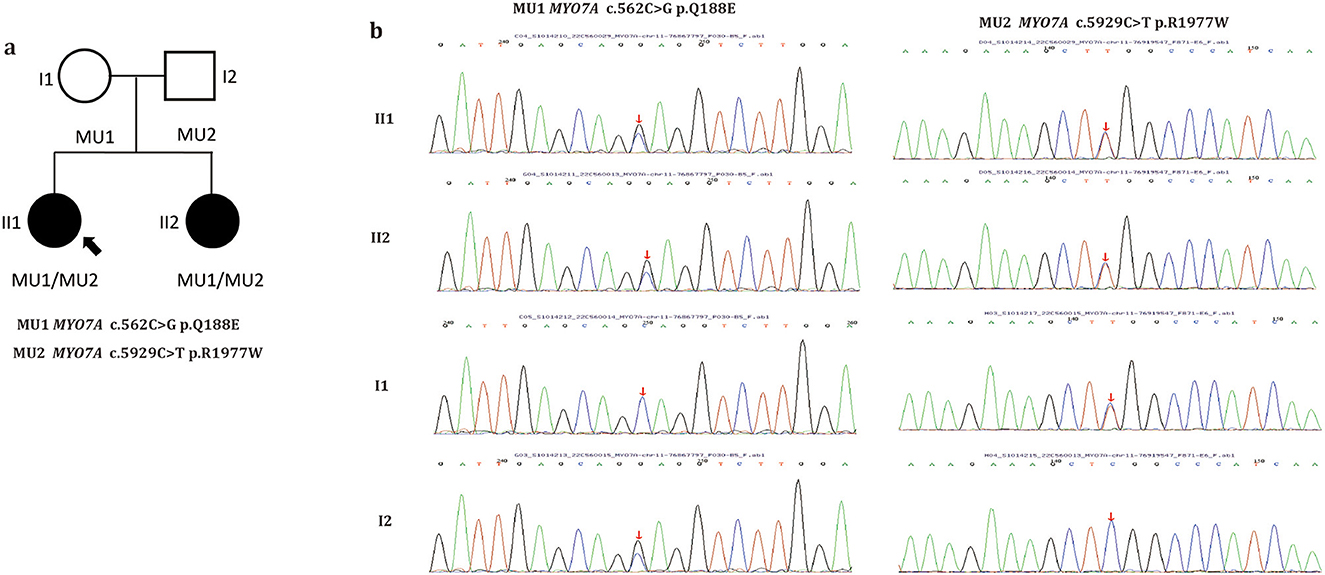
Figure 3. Pedigree of the family (a). Filled symbols indicate patients. Unfilled symbols indicate unaffected family members. Arrow indicates the proband. Squares indicate males and circles females. (b) Sanger sequencing of the two MYO7A variants identified in this study. Arrows indicate the positions of the mutated nucleotides.
Discussion
We describe a family with an unusual CME phenotype associated with two previously reported mutations in MYO7A. The two affected family members were sisters with no ocular history and a negative family history. The appearance of CME in both eyes of both patients was the first and most prominent abnormality, which fluctuated spontaneously during the follow-up period. The retinal pigment epithelium and external limiting membrane bands were intact while the ellipsoid zone was slightly disrupted and disorganized in the central foveal region on OCT images. These cystoid abnormalities corresponded to FFA examinations. Dye diffusion could be detected in the macular area which effectively ruled out a diagnosis of retinoschisis. No vascular leakage was present in the periphery of the retina on FFA images, indicting no uveitis-related CME. Furthermore, all routine laboratory tests related to vasculitis (14), such as EST, CRP, blood count, serum creatinine, urinalysis, specific autoantibodies, complement, immunoglobulin, cryoglobulin, and hepatitis B and C serology, were negative. ERG showed a rod-cone dystrophy dysfunction pattern, and rod function was more impaired than cone function. Furthermore, mfERG showed mild retinal dysfunction and the EOG Arden ratios were mildly reduced. These results, thus indicating rod, cone and retinal pigment epithelial cells were all affected. These changes are different from RP, where the electrophysiological damage is more severe and begins in the peripheral area. Given all this evidence, it is reasonable to speculate that the CME was caused by a breakdown of the barrier function of the RPE. Long-term follow-up is needed to more precisely characterize this family.
The two identified MYO7A variants (c.562C>G and c.5929C>T) were considered associated with fluctuation CME for the following reasons. (1) The variants co-segregated with CME in this family. (2) Although the two variants were classified as variants of uncertain significance, they had previously been reported in individuals with hearing loss or Usher syndrome (12, 13), and (3) both mutations were single nucleotide substitutions, c.562C>G leading to p.Gln188Glu and c.5929C>T leading to p.Arg1977Trp. Generation of three-dimensional structures using PyMOL, enabled us to classify the two variants as variants as potentially pathogenic. The challenge in our study was to evaluate the genotype-phenotype correlation of the affected family members. In humans, MYO7A is widely localized, such as in cochlea neuroepithelia, vestibular neuroepithelia, retinal photoreceptor cells, and retinal pigment epithelium cells. Mutations in MYO7A lead to USH1 in most cases, including a relatively high incidence of CME (15) which was consistent with our affected family members. However, nearly all pathogenic mutations in MYO7A are linked to hearing loss but both the proband and their sister presented normal hearing. The molecular and physiological functions of MYO7A have been well-characterized; however, clinical heterogeneity and tissue-specific differences of MYO7A variants are well-known (7). Patients with an atypical phenotype associated with MYO7A are not infrequent, and mutations in MYO7A have been associated with diverse clinical phenotypes. Previous studies have indicated that mutations in MYO7A might have different effects in the eye but similar effects in the inner ear; however, our patients show that the effects of MYOYA variants on the inner ear may also be different. Moreover, MYO7A can cause autosomal recessive rod-cone dystrophy in the retina, which may explain why the two patients with fluctuation CME exhibited abnormalities and phenotypic characteristics of rod-cone dysfunction.
There are many reasons why different mutations in the same gene can cause diverse phenotypes. Firstly, MYO7A spans ~87 kb of genomic sequence and encodes myosin VIIA, which has a predicted molecular mass of 254 kDa. This protein contains 2,215 amino acids and has three typical domains: the N terminal head or motor (amino acids 1–729); the neck or regulatory domain consisting of five isoleucine glutamine motifs (IQ; IQ1–IQ5: amino acids 745–857); and the tail which begins with a short, coiled-coil domain (amino acids 858–935). These are followed by two large MyTH4-FERM repeat elements, separated by an SH3 domain (amino acids 1,603–1,672) (16). Clinical heterogeneity associated with MYO7A variants was recently shown to rely on affected domains. Missense variants in the motor domain present as adult-onset and slowly progressive, leading to a flat configuration, while patients carrying MyTH4 domain variants showed adult-onset, rapid progression, and a down sloping tendency (7). The two compound heterozygous variants found in our pedigree were in different domains of myosin VIIA, one variant was located in the motor domain, and the other in FERM2. Pathogenic variants are most frequently located in motor and MyTH4 domains (17), while variants in FERM2 are rare. Schwander et al. (18) demonstrated that the FERM2 domain of MYO7A is associated with the transport function of retinal pigment epithelium cells. They also found that a MYO7A point mutation can differentially affect gene expression in the inner ear and retina. These findings might explain why the proband and their sister presented as fluctuating CME, but without hearing impairment. Also consistent with our findings is that compound heterozygous mutations in different domains may cause a mild phenotype (19–21). In addition, a growing number of modifiers have been identified that can affect the phenotype of MYO7A variants. Street et al. (22) described a single nucleotide polymorphism as a modifier that contributes to the severe hearing loss phenotype by reducing expression of the wild-type MYO7A allele. Morgan et al. (23) showed that PDZD7, which was originally identified as a modifier gene for Usher syndrome type 2, also interacted with MYO7A. It is also possible that modifiers may affect the phenotype of our patients.
CME was the first clinically visible abnormality in the two patients, indicating that CME is the direct result of the primary genetic defect. Large and elongated cavities are mainly present in the outer nuclear layer, indicating a primary pathological process affecting retinal photoreceptor cells and retinal pigment epithelium cells. This is supported by ffERG and EOG results, and can be explained by MYO7A mutations. As the disease progressed, Müller cell function was disrupted and cystoid fluid accumulation appeared in the inner nuclear layer. It is possible that cell function was not completely destroyed in the early stage leading to CME presenting in a fluctuating form.
This study has some limitations. First, it had a small sample size with only one Chinese family enrolled. Second, we did not explore the effects of the identified variants by molecular investigations. Our future studies will further investigate genotype-phenotype associations of MYO7A variants.
In conclusion, we used whole-exome sequencing to identify two compound heterozygous variants in MYO7A (both previously reported) that are likely to be associated with fluctuating CME. Our findings expand the phenotypic spectrum of MYO7A variants and may enhance understanding of genotype-phenotype associations for compound heterozygous MYO7A variants.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
This study was approved by the ethics boards of the EENT Hospital of Fudan University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
CD: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Writing – review & editing. YZ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. QC: Conceptualization, Methodology, Validation, Writing – review & editing. WL: Investigation, Supervision, Validation, Writing – review & editing. QC: Formal analysis, Supervision, Validation, Visualization, Writing – review & editing. CX: Methodology, Validation, Visualization, Writing – review & editing. Z-LH: Conceptualization, Data curation, Investigation, Methodology, Supervision, Visualization, Writing – review & editing. G-ZX: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Visualization, Writing – review & editing. F-JG: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The report was supported by Shanghai Municipal Science and Technology Commission (25ZR1402058), NationalNatural Science Foundation of China (Grant NSFC82201204), and Shanghai Municipal Science and Technology Commission (25J32800203). This study adhered to the tenets of the Declaration of Helsinki, and was approved by the Ethics Committee of the Eye and ENT Hospital of Fudan University.
Acknowledgments
We are grateful to the technical staff at Mygenostics for technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1582930/full#supplementary-material
Supplementary Figure 1 | Fundus photography (A and B), fundus fluorescein angiography (FFA, C and D) and optical coherence tomography (OCT, E and F) findings of patient II2. FFA showing cystoid hyperfluorescence in a petaloid pattern in the foveal area with a honeycomb pattern parafoveally. OCT showed macular cystoid changes in both eyes, and cystoid cavities mainly in the outer nuclear layer (ONL).
Supplementary Figure 2 | Optical coherence tomography (OCT) changes in patient II1 at different follow-up times.
Supplementary Figure 3 | Fundus fluorescein angiography (FFA) changes in patient II2 at different follow-up times.
Supplementary Figure 4 | Location of the variants in the MYO7A protein domains (A). Alignment of human MYO7A protein sequence (B) with orthologs. Three-dimensional prediction of the two mutations (variants and wild-type, C). II2 at different follow-up times.
References
1. Daruich A, Matet A, Moulin A, Kowalczuk L, Nicolas M, Sellam A, et al. Mechanisms of macular edema: beyond the surface. Prog Retin Eye Res. (2018) 63:20–68. doi: 10.1016/j.preteyeres.2017.10.006
2. Govetto A, Su D, Farajzadeh M, Megerdichian A, Platner E, Ducournau Y, et al. Microcystoid macular changes in association with idiopathic epiretinal membranes in eyes with and without glaucoma: clinical insights. Am J Ophthalmol. (2017) 181:156–65. doi: 10.1016/j.ajo.2017.06.023
3. Bringmann A, Reichenbach A, Wiedemann P. Pathomechanisms of cystoid macular edema. Ophthalmic Res. (2004) 36:241–9. doi: 10.1159/000081203
4. Johnson MW. Tractional cystoid macular edema: a subtle variant of the vitreomacular traction syndrome. Am J Ophthalmol. (2005) 140:184–92. doi: 10.1016/j.ajo.2005.01.033
5. Strong S, Liew G, Michaelides M. Retinitis pigmentosa-associated cystoid macular oedema: pathogenesis and avenues of intervention. Br J Ophthalmol. (2017) 101:31–7. doi: 10.1136/bjophthalmol-2016-309376
6. Salvatore S, Fishman GA, Genead MA. Treatment of cystic macular lesions in hereditary retinal dystrophies. Surv Ophthalmol. (2013) 58:560–84. doi: 10.1016/j.survophthal.2012.11.006
7. Joo SY, Na G, Kim JA, Yoo JE, Kim DH, Kim SJ, et al. Clinical heterogeneity associated with MYO7a variants relies on affected domains. Biomedicines. (2022) 10:798. doi: 10.3390/biomedicines10040798
8. Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, et al. Defective myosin viia gene responsible for usher syndrome type 1b. Nature. (1995) 374:60–1. doi: 10.1038/374060a0
9. Li L, Yuan H, Wang H, Guan J, Lan L, Wang D, et al. Identification of a MYO7a mutation in a large Chinese Dfna11 family and genotype-phenotype review for Dfna11. Acta Otolaryngol. (2018) 138:463–70. doi: 10.1080/00016489.2017.1397743
10. Zong L, Chen K, Wu X, Liu M, Jiang H. Compound heterozygous Myo7a mutations segregating usher syndrome type 2 in a Han family. Int J Pediatr Otorhinolaryngol. (2016) 90:150–5. doi: 10.1016/j.ijporl.2016.09.010
11. Subira O, Catala-Mora J, Diaz-Cascajosa J, Padron-Perez N, Claveria MA, Coll-Alsina N, et al. Retinal findings in pediatric patients with usher syndrome type 1 due to mutations in Myo7a gene. Eye. (2020) 34:499–506. doi: 10.1038/s41433-019-0536-6
12. Jespersgaard C, Fang M, Bertelsen M, Dang X, Jensen H, Chen Y, et al. Molecular genetic analysis using targeted Ngs analysis of 677 individuals with retinal dystrophy. Sci Rep. (2019) 9:1219. doi: 10.1038/s41598-018-38007-2
13. Sun T, Xu K, Ren Y, Xie Y, Zhang X, Tian L, et al. Comprehensive molecular screening in Chinese usher syndrome patients. Invest Ophthalmol Vis Sci. (2018) 59:1229–37. doi: 10.1167/iovs.17-23312
14. Csernok E, Bossuyt X. Investigations in systemic vasculitis. The role of the laboratory. Best Pract Res Clin Rheumatol. (2018) 32:52–62. doi: 10.1016/j.berh.2018.07.005
15. Testa F, Melillo P, Rossi S, Marcelli V, de Benedictis A, Colucci R, et al. Prevalence of macular abnormalities assessed by optical coherence tomography in patients with usher syndrome. Ophthalmic Genet. (2018) 39:17–21. doi: 10.1080/13816810.2017.1329445
16. Jaijo T, Aller E, Oltra S, Beneyto M, Najera C, Ayuso C, et al. Mutation profile of the Myo7a gene in Spanish patients with usher syndrome type I. Hum Mutat. (2006) 27:290–1. doi: 10.1002/humu.9404
17. Galbis-Martinez L, Blanco-Kelly F, Garcia-Garcia G, Avila-Fernandez A, Jaijo T, Fuster-Garcia C, et al. Genotype-phenotype correlation in patients with usher syndrome and pathogenic variants in Myo7a: implications for future clinical trials. Acta Ophthalmol. (2021) 99:922–30. doi: 10.1111/aos.14795
18. Schwander M, Lopes V, Sczaniecka A, Gibbs D, Lillo C, Delano D, et al. A novel allele of myosin viia reveals a critical function for the C-terminal ferm domain for melanosome transport in retinal pigment epithelial cells. J Neurosci. (2009) 29:15810–8. doi: 10.1523/JNEUROSCI.4876-09.2009
19. Rafalska A, Tracewska AM, Turno-Krecicka A, Szafraniec MJ, Misiuk-Hojlo M. A mild phenotype caused by two novel compound heterozygous mutations in CEP290. Genes. (2020) 11:1240. doi: 10.3390/genes11111240
20. Steenhof M, Kibaek M, Larsen MJ, Christensen M, Lund AM, Brusgaard K, et al. Compound heterozygous mutations in two different domains of Aldh18a1 do not affect the amino acid levels in a patient with hereditary spastic paraplegia. Neurogenetics. (2018) 19:145–9. doi: 10.1007/s10048-018-0547-7
21. Al-Jezawi NK, Al-Shamsi AM, Suleiman J, Ben-Salem S, John A, Vijayan R, et al. Compound heterozygous variants in the multiple Pdz Domain Protein (Mpdz) cause a case of mild non-progressive communicating hydrocephalus. BMC Med Genet. (2018) 19:34. doi: 10.1186/s12881-018-0540-x
22. Street VA Li J, Robbins CA, Kallman JC. A DNA variant within the Myo7a promoter regulates Yy1 transcription factor binding and gene expression serving as a potential dominant Dfna11 auditory genetic modifier. J Biol Chem. (2011) 286:15278–86. doi: 10.1074/jbc.M111.228304
Keywords: cystoid macular edema, MYO7A gene, phenotype, genetic heterogeneity, optical coherence tomography
Citation: Duan C, Zong Y, Chen Q, Liu W, Chang Q, Xiong C, Hu Z-L, Xu G-Z and Gao F-J (2025) Case Report: A family of fluctuating cystoid macular edema caused by MYO7A gene mutations. Front. Med. 12:1582930. doi: 10.3389/fmed.2025.1582930
Received: 25 February 2025; Accepted: 22 July 2025;
Published: 07 August 2025.
Edited by:
Pei-Kang Liu, Kaohsiung Medical University, TaiwanReviewed by:
Hemakumar M. Reddy, Nabsys Inc, United StatesDyah Ayu Windy, Hasanuddin University, Indonesia
Copyright © 2025 Duan, Zong, Chen, Liu, Chang, Xiong, Hu, Xu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng-Juan Gao, Z2FvZmVuZ2p1YW4wODE1QHNpbmEuY29t; Ge-Zhi Xu, ZHJ4dWdlemhpQDE2My5jb20=; Zhu-Lin Hu, aHpsNzdAMjYzLm5ldA==
†These authors have contributed equally to this work