 Jiali Long
Jiali Long Benhua Zeng1,2
Benhua Zeng1,2 Guohong Deng
Guohong Deng- 1Department of Infectious Diseases, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, China
- 2Chongqing Key Laboratory of Viral Infectious Diseases, Chongqing, China
Purpose: This study aimed to comprehensively analyze the global landscape, trends, and research focus of nanopore sequencing technology in the field of pathogenic microorganism diagnosis using bibliometric analysis.
Methods: Literature published between January 2014, and December 2024, was retrieved from the Web of Science Core Collection. A cross-sectional bibliometric analysis was conducted using VOSviewer, CiteSpace, Origin 2024, and R software to extract and evaluate metrics. Publications were categorized by country, institution, author, journal, highly cited papers, and keywords. Variables were compared based on publication output and academic impact, which included citation counts, citation impact, H-index, journal impact factor, total link strength, major pathogens, and research directions.
Results: Initial searches identified 2,098 articles related to nanopore sequencing and pathogenic microorganisms, of which 729 were ultimately included in the analysis. Among the 104 participating countries, the United Kingdom, the United States, and China have led in publication output, citations, and academic influence. The most versatile institution was the University of Oxford, followed by Zhejiang University. The most productive scholars and journals were Crook, Derrick W., and Frontiers in Microbiology, respectively. Keyword analysis revealed that the primary advantages of nanopore sequencing include portability, long-read capabilities, and real-time analysis. Current research hotspots focus on real-time pathogen identification, viral genomic surveillance, and antimicrobial resistance profiling.
Conclusion: Presently, nanopore sequencing is rapidly transitioning from laboratory research to on-site sequencing and public health emergency scenarios. To our knowledge, this study is the first bibliometric analysis to comprehensively delineate the latest developments in nanopore sequencing in pathogenic microorganism diagnosis. It provides researchers with an understanding of the current situation, identifies knowledge gaps, and points out future research directions.
1 Introduction
The rapid and accurate identification of pathogenic microorganisms is critical for clinical diagnosis, outbreak surveillance, and antimicrobial stewardship (1–3). Conventional culture-based methods and polymerase chain reaction (PCR)-dependent assays, while foundational in microbiology, face limitations in turnaround time, resolution, and scalability, particularly when confronting polymicrobial infections, unculturable pathogens, or emerging strains with novel genetic signatures (4). Recently, the advent of third-generation sequencing technologies, such as nanopore sequencing, has revolutionized pathogen detection by enabling real-time, high-throughput, and portable genomic analyses (3, 5).
Nanopore sequencing is characterized by its unique ability to generate ultra-long reads (> 100 kb) and directly detect epigenetic modifications, circumventing the amplification biases inherent in short-read sequencing platforms (6). These features are critical for addressing the complex genomic architectures of pathogens, including antibiotic resistance gene clusters, virulence plasmids, and repetitive elements, which are often fragmented or misassembled by traditional sequencing approaches (7–9). Furthermore, its portability and minimal infrastructure requirements make it a transformative tool for on-site diagnostics in resource-limited settings. This assertion is evidenced by its successful utilization in real-time monitoring of viral transmission during outbreaks of Ebola virus (EBOV) (10), Zika virus (ZIKV) (11), and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (12). Recent studies have demonstrated the utility of nanopore sequencing in a variety of clinical scenarios, including the identification of pathogens in bloodstream infections within 6 h, real-time metagenomic analysis of cerebrospinal fluid, and rapid detection of multidrug-resistant tuberculosis (13–15).
Despite these advances, there is a paucity of comprehensive reviews examining previous publications, academic influence, and research trends in the application of nanopore sequencing technology to pathogenic microorganisms across the globe. Bibliometric analysis has been extensively employed to explore the productivity of countries, institutions, and researchers in specific disciplines, identify key events, and predict emerging trends. The application of bibliometric analysis, which quantifies the distribution patterns and characteristics of scientific data from multiple perspectives, has the potential to reveal the overarching knowledge of architecture and the evolutionary trajectory of this field. Such insights empower experts and newcomers to delineate the scope of their discipline, discover novel topics of interest, and strategically formulate future research plans.
2 Materials and methods
2.1 Data source and search strategy
We retrieved the Web of Science Core Collection (WOSCC) database to identify publications on nanopore sequencing from 2014 to 2024. To comprehensively capture literature relevant to the research topic while minimizing the inclusion of non-relevant records, we used the topical search query: TS = ((“pathogenic microorganism” OR “pathogenic bacteria” OR “pathogen” OR “vir*” OR “bacteri*”) AND (“nanopore sequencing” OR “Oxford Nanopore” OR “MinION” OR “GridION” OR “PromethION”)). Language was limited to English. A literature search and data download was performed on May 25, 2025. The screening process is summarized in Figure 1.
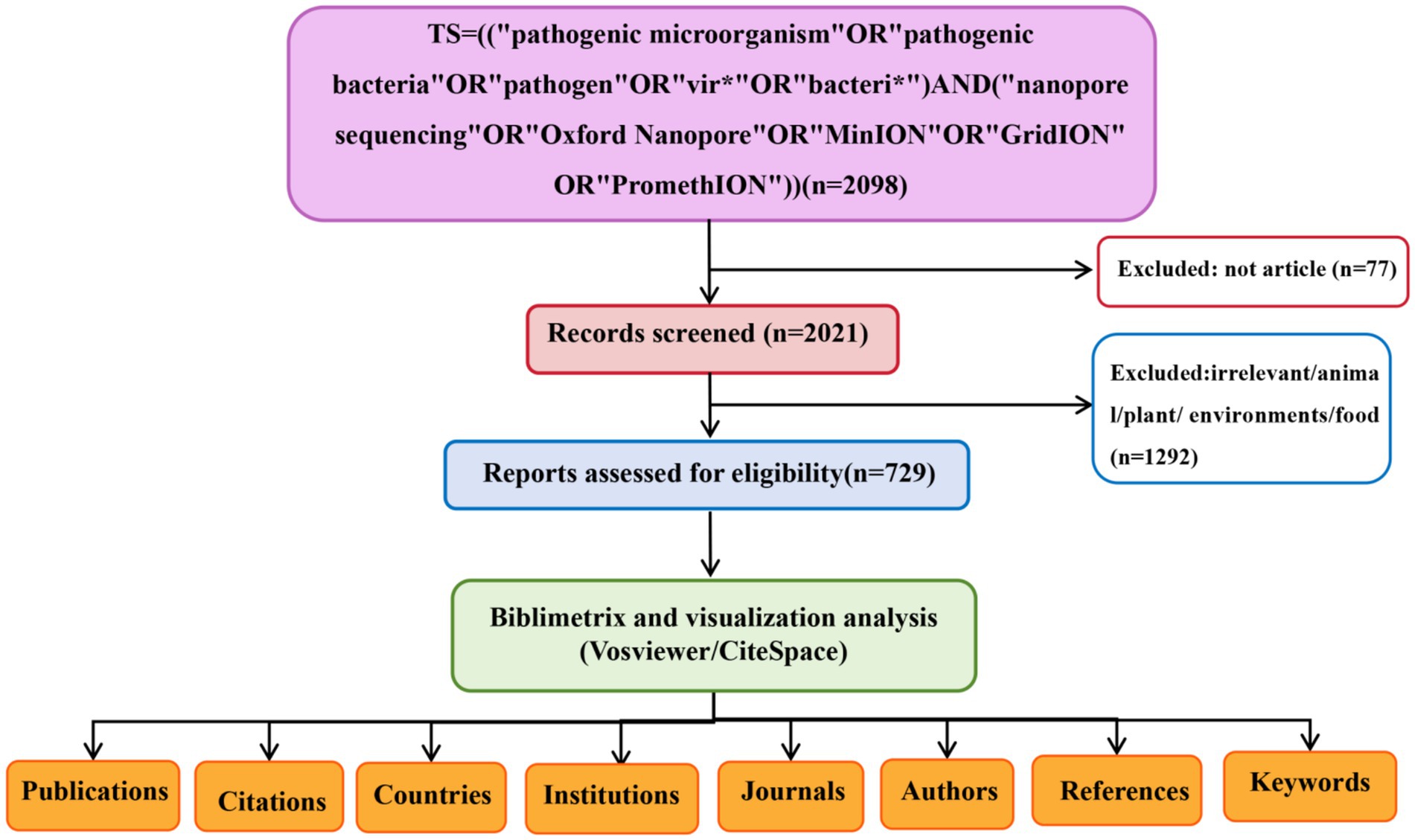
Figure 1. Flowchart of metrology screening.
2.2 Bibliometric analysis
The VOSviewer software (version 1.6.20) was used to cluster countries/regions, institutions, journals, and keywords. The CiteSpace software (version 6.3.1) was used to visualize the institutional centrality cooperation authors’ network, generate keyword timeline maps, and conduct burst word analysis. Origin2024 software was used for statistical analyses. R software (version 4.4.1) was used to perform the geographical distribution of nanopore sequencing in the field of pathogenic microorganisms. Academic influence is a primary criterion for evaluating the academic excellence of the document, journal, institution, researcher, etc., and its measurement is inherent and diverse. We used the composite metrics to measure academic influence, including the following elements: (1) citation count: number of times a country/institution/author/journal is cited; (2) citation impact: average number of citations a document received; (3) Hirsh index (H-index): a researcher has an h-index if they have at least h publications for which they received at least h citations; (4) total link strength: total strength of the co-authorship links of a given country/institution/author with others; (5) impact factor (IF): functional approximation of the mean citation rate per citable item (16).
3 Results
3.1 Baseline characteristics of the eligible documents
The initial search identified 2,098 publications (Figure 1). After excluding non-article types (n = 77), 2,021 articles underwent rigorous title and abstract screenings. Studies unrelated to human pathogens or focusing exclusively on animal, plant, environmental, or food-related contexts (n = 1,292) were excluded, resulting in 729 eligible studies for final inclusion. The annual publication volume and citation counts from 2014 to 2024 are presented in Figure 2A. A consistent growth trend was observed for both metrics, with a notable surge post-2019. Before 2019, fewer than 35 publications annually addressed nanopore sequencing in pathogenic microorganisms. However, annual output has exhibited sustained growth since 2019, peaking at 147 publications in 2024. Citation counts followed a similar trajectory, reaching a maximum of 3,271 in 2024. Compared to 2015, citation impact demonstrated a modest increase, with significant upward shifts observed in 2016 and 2017 (Figure 2B). The 2020 year demonstrated unparalleled scholarly impact, attaining the maximum H-index of 27 (Figure 2B). Regarding the research areas, approximately 1/3 of the papers (n = 287, 31.4%) belonged to the microbiology category, followed by infectious diseases (n = 162, 17.7%), multidisciplinary (n = 88, 9.6%), pharmacology (n = 87, 9.5%), and genetics (n = 82, 9%) (Figure 2C).
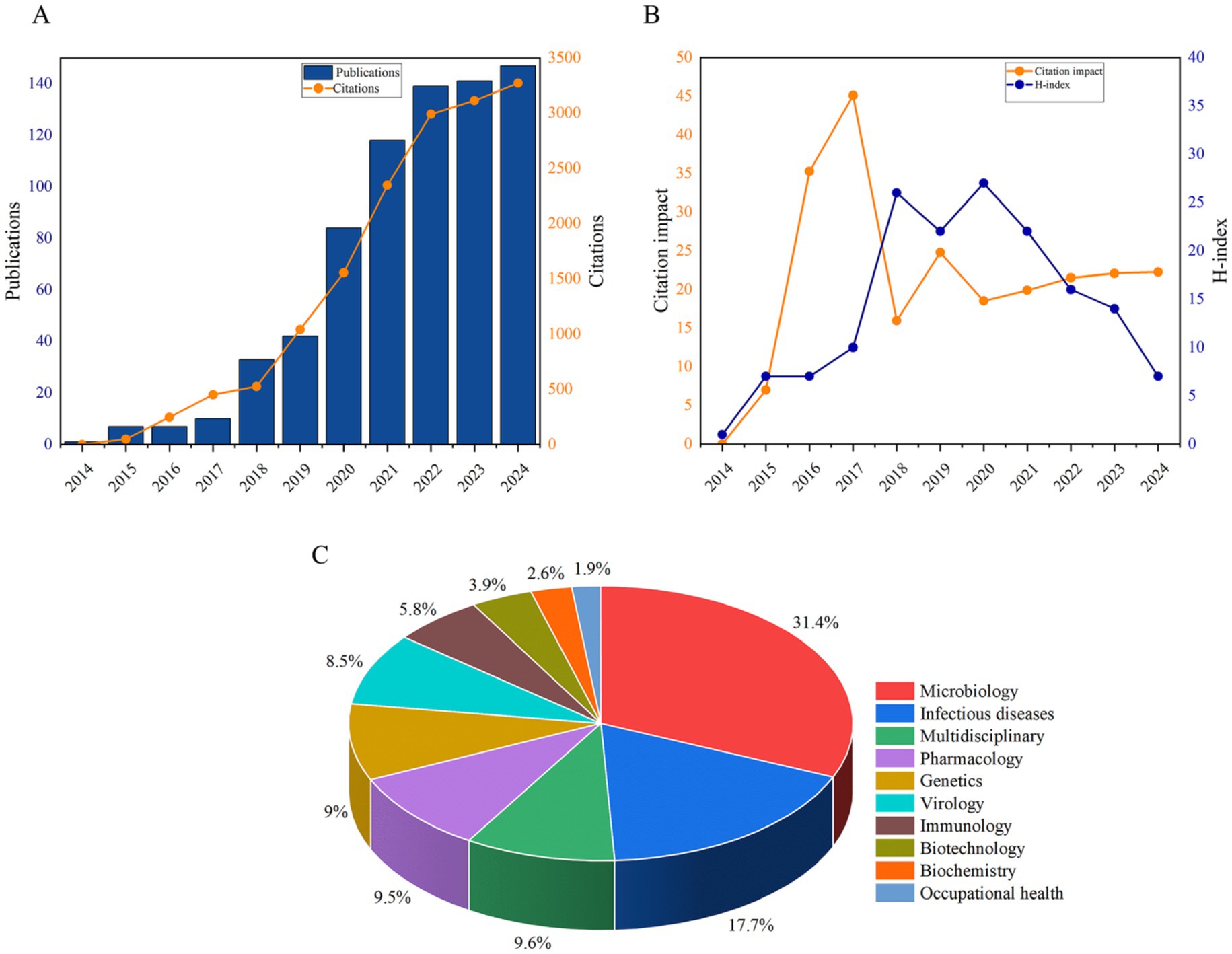
Figure 2. Baseline bibliometric characteristics of all eligible documents. (A) Annual publications and citations. (B) Citation impact per year and H-index. (C) Research areas.
3.2 Analysis of countries and institutions
One hundred and four countries contributed to the global discourse, of which 35.6% were high-income countries, 15.4% were upper-middle-income countries, 12.5% were middle-income countries, 22.1% were lower-middle-income countries, and 14.4% were lower-income countries (Figure 3A). The global productivity map revealed pronounced geographical clustering, with most publications originating from Asia, North America, and Europe (Figure 3B). China, the USA, and England have been identified as the leading contributors to nanopore sequencing research because of their high productivity levels, citations, and H-index (Figure 3C). Subsequently, we analyzed the number of documents, citations, H-index, and main research pathogens of the top 10 most productive countries (Table 1).
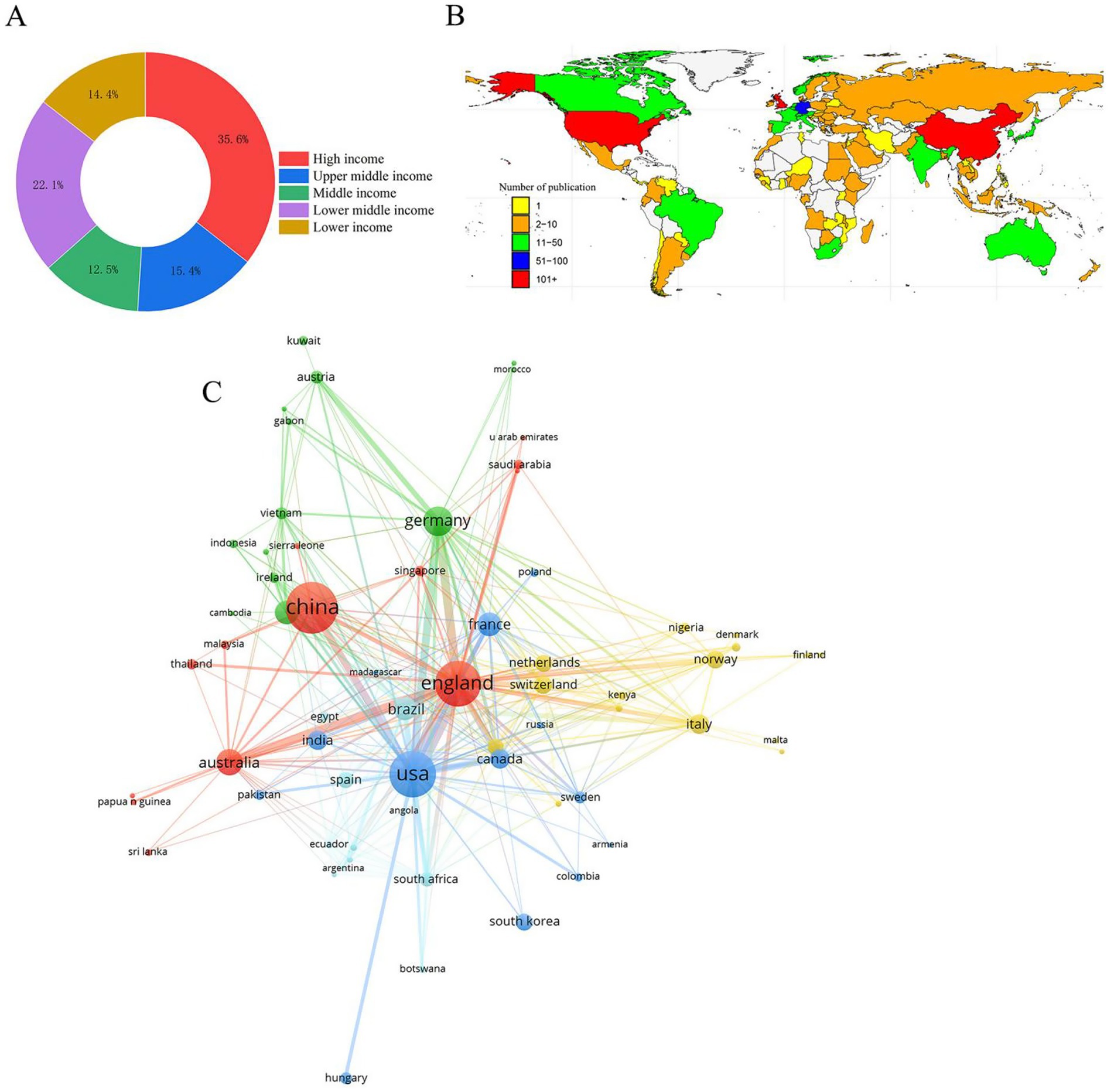
Figure 3. Relationships and clusters of countries. (A) Classification of countries according to income levels. (B) Geographical distribution based on the number of documents. (C) Relationship between international and domestic collaborations.
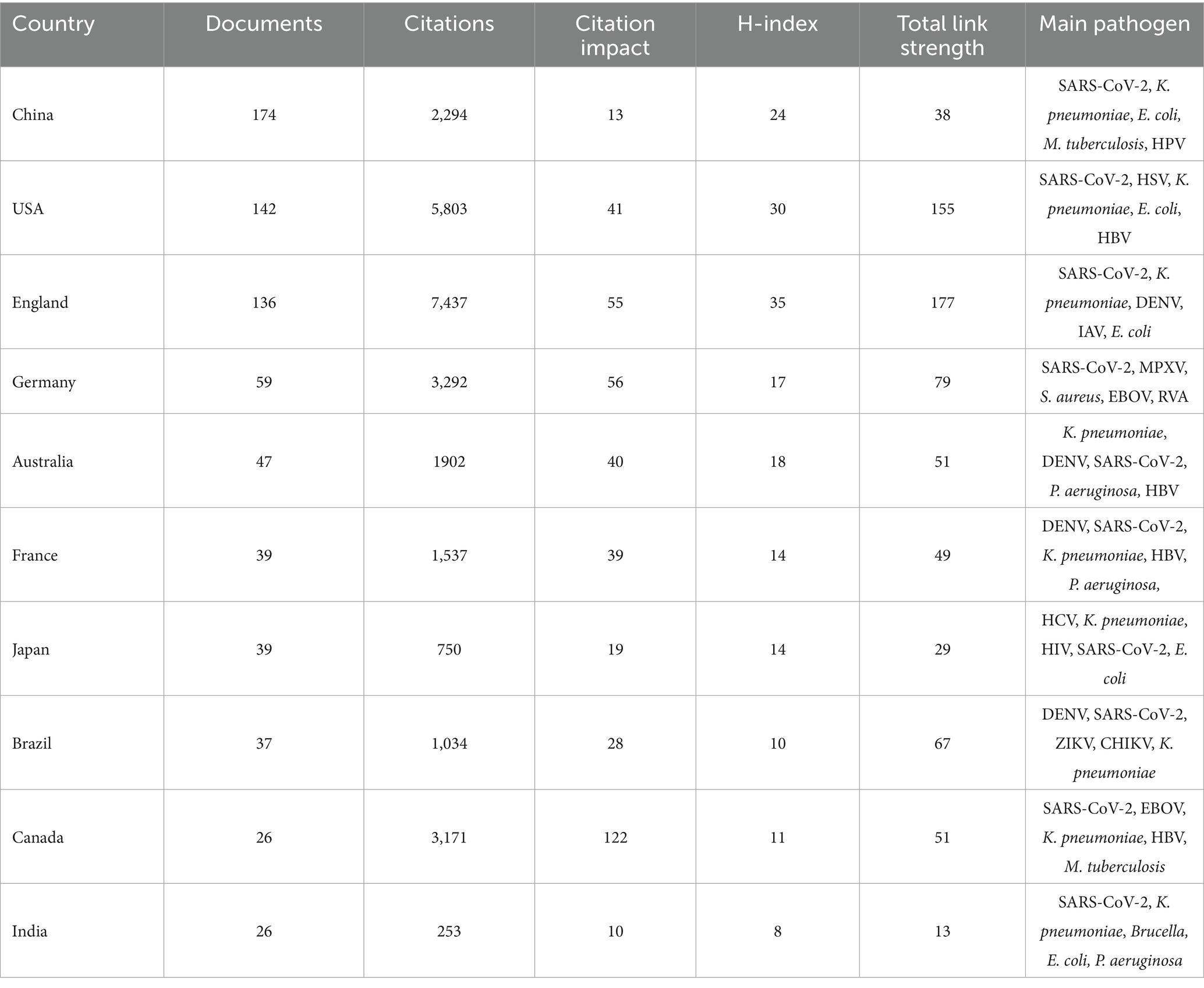
Table 1. Top 10 most productive countries regarding nanopore sequencing and pathogen research from 2014 to 2024.
A total of 281 institutions participated in this collaboration. Figure 4A displays the institutional co-authorship network, comprising 211 institutions and 691 collaborative instances, with the University of Oxford and Zhejiang University serving as representative institutions in their respective countries. Additionally, these two organizations maintain close collaboration with domestic and international academic institutions. Figure 4B and Table 2 present the top 10 institutions with the highest documents in the fields of nanopore sequencing technology and pathogenic microorganisms.
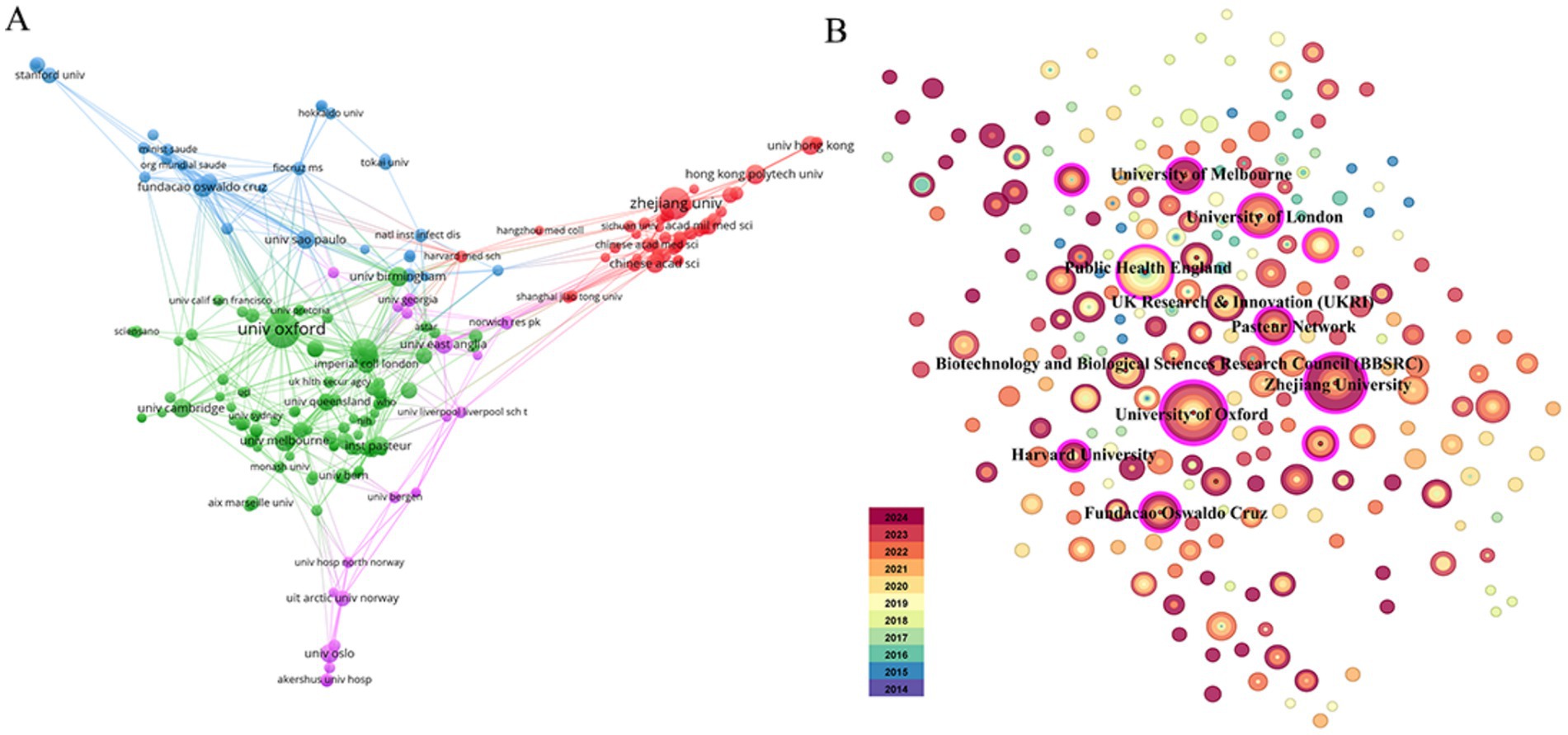
Figure 4. Visualization of institutions. (A) Collaboration network of institutions. (B) Top 10 institutions for related publications.
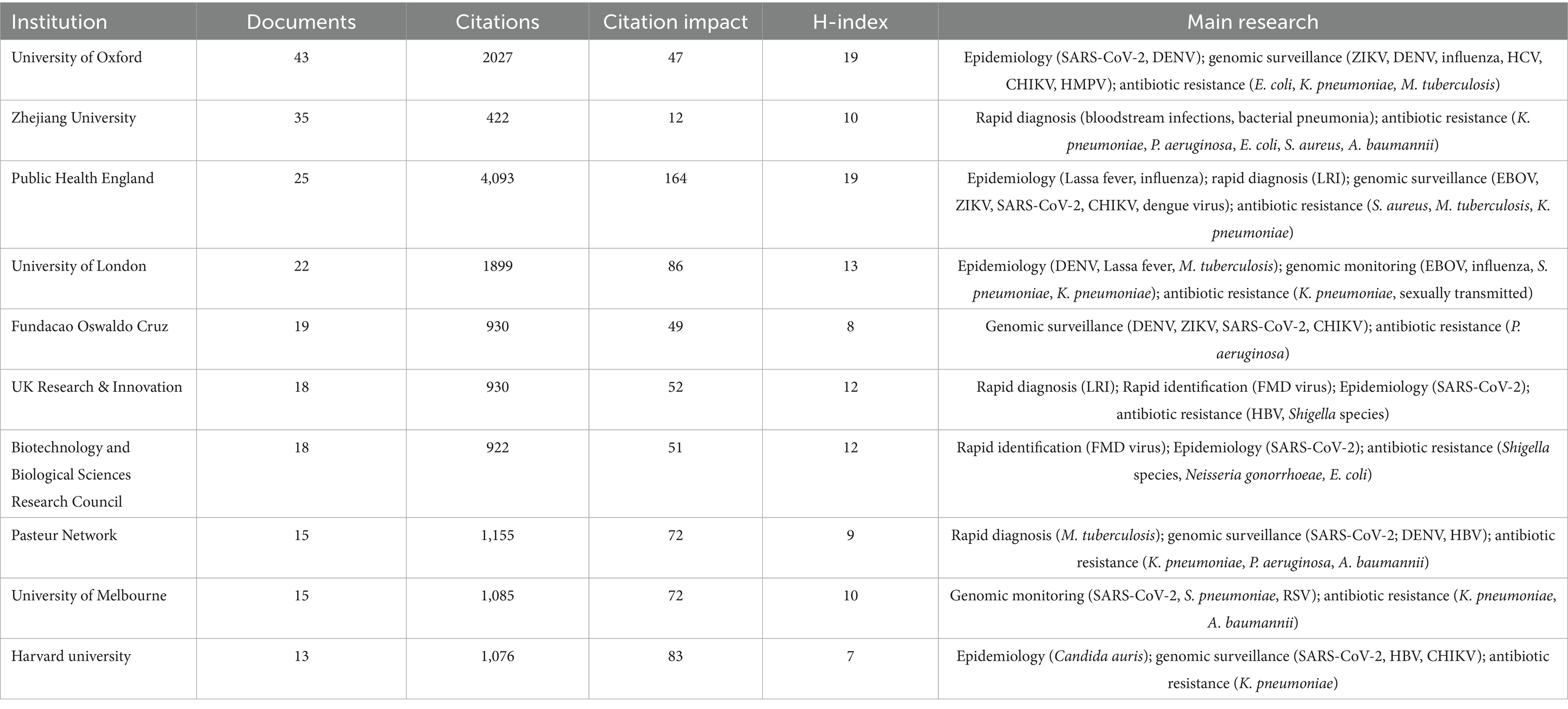
Table 2. Top 10 productive institutions regarding nanopore sequencing and pathogen research from 2014 to 2024.
3.3 Analysis of authors, journals, and highly cited publications
Among the 5,393 authors included, the most prolific authors in the bibliometric analysis were Crook, Derrick W (n = 14), followed by Giovanetti, Marta (n = 12), Alcantara, Luiz Carlos Júnior (n = 12) (Table 3). Giovanetti, Marta and Alcantara, Luiz Carlos Júnior had the highest citation impact; both worked at the Fundacao Oswaldo Cruz. The centralized author clustering revealed a strong cooperative relationship between the authors (Figure 5A). The top three academic journals out of 205 were Frontiers in Microbiology (n = 43; Table 4), Scientific Reports (n = 35), and Viruses-Basel (n = 31). The journal with the highest impact factor was the Frontiers in Cellular and Infection Microbiology (IF = 4.6), followed by the Microorganisms (IF = 4.1). These journals were categorized into four clusters based on their co-citation relationships (Figure 5B).
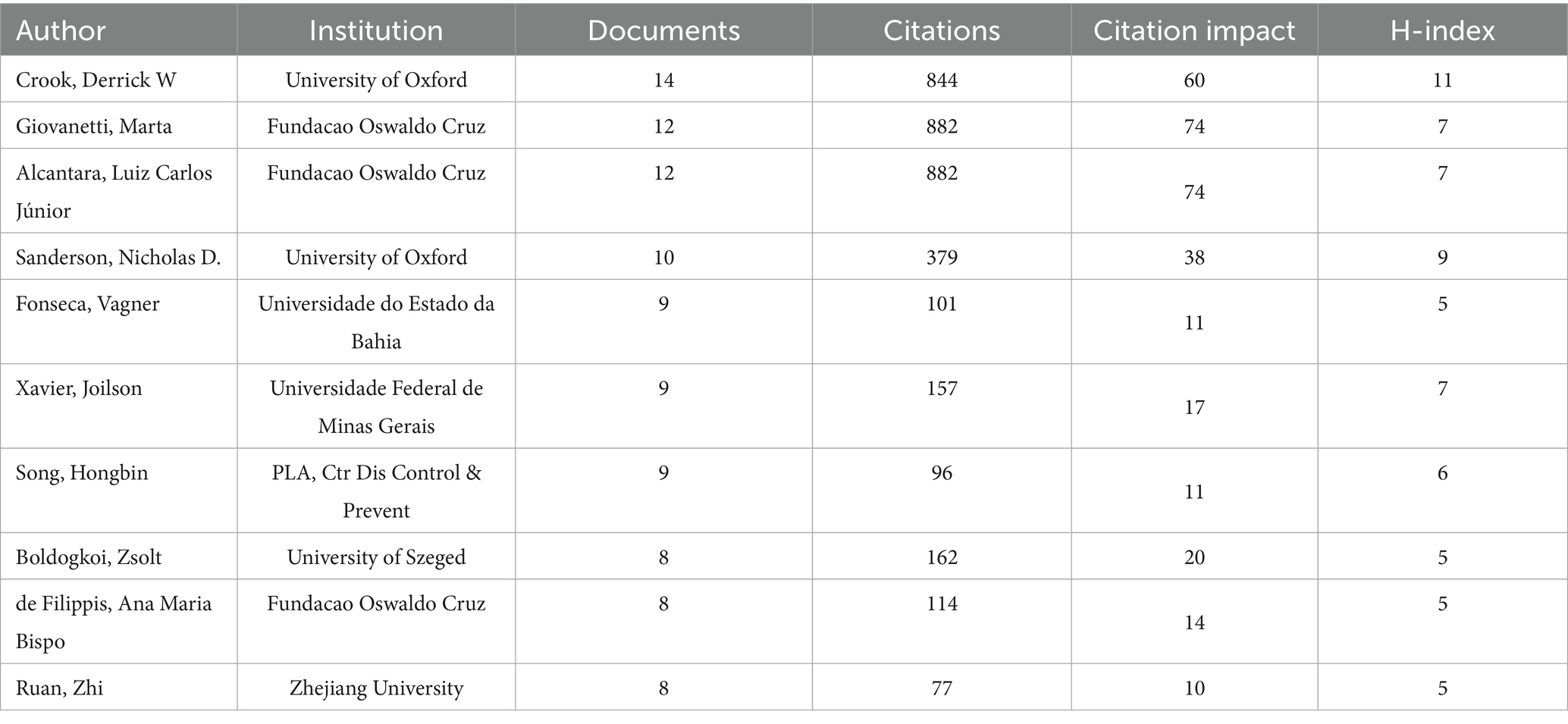
Table 3. Top 10 productive authors of nanopore sequencing and pathogen research from 2014 to 2024.
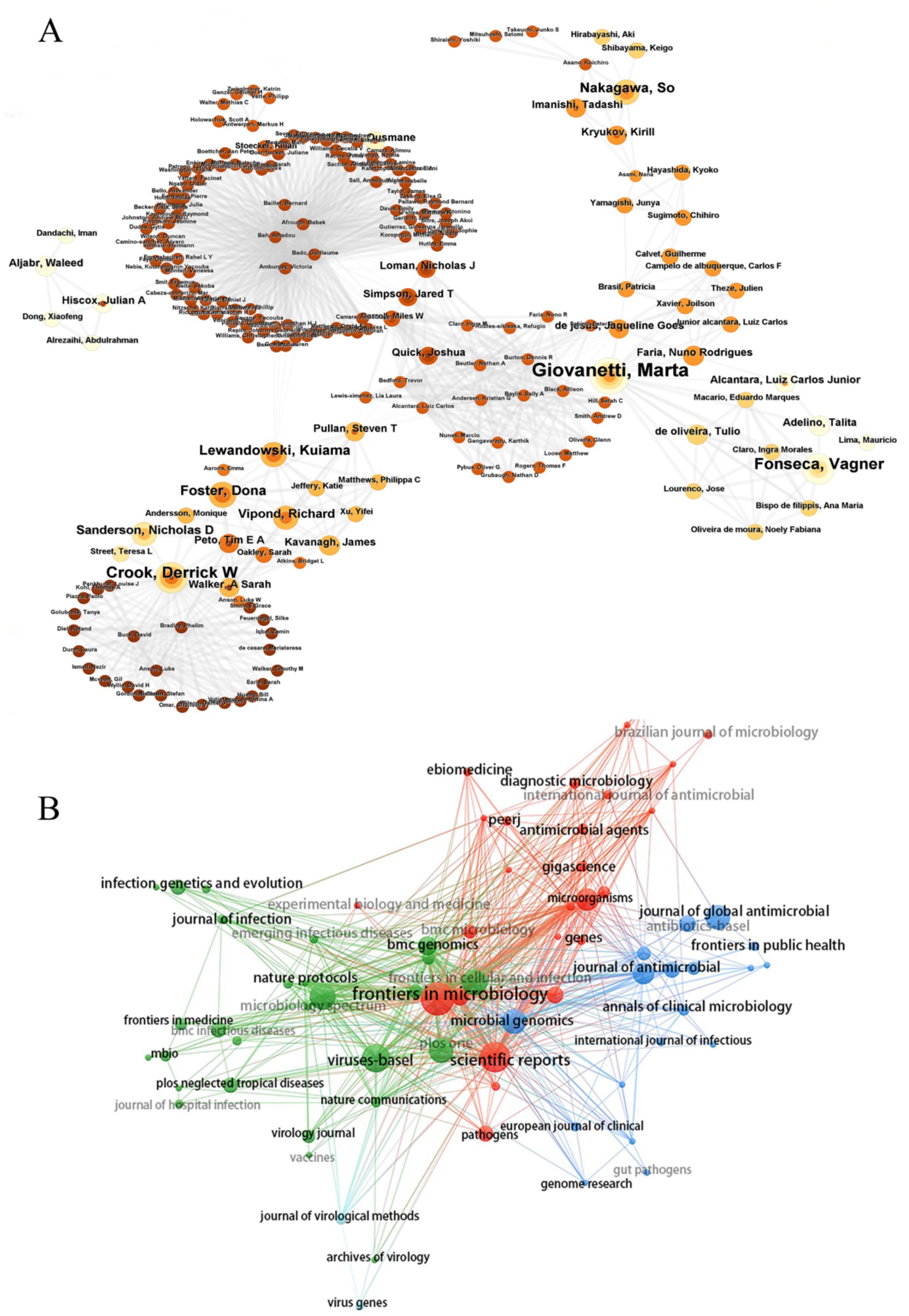
Figure 5. Visualization of authors and journals (A,B).
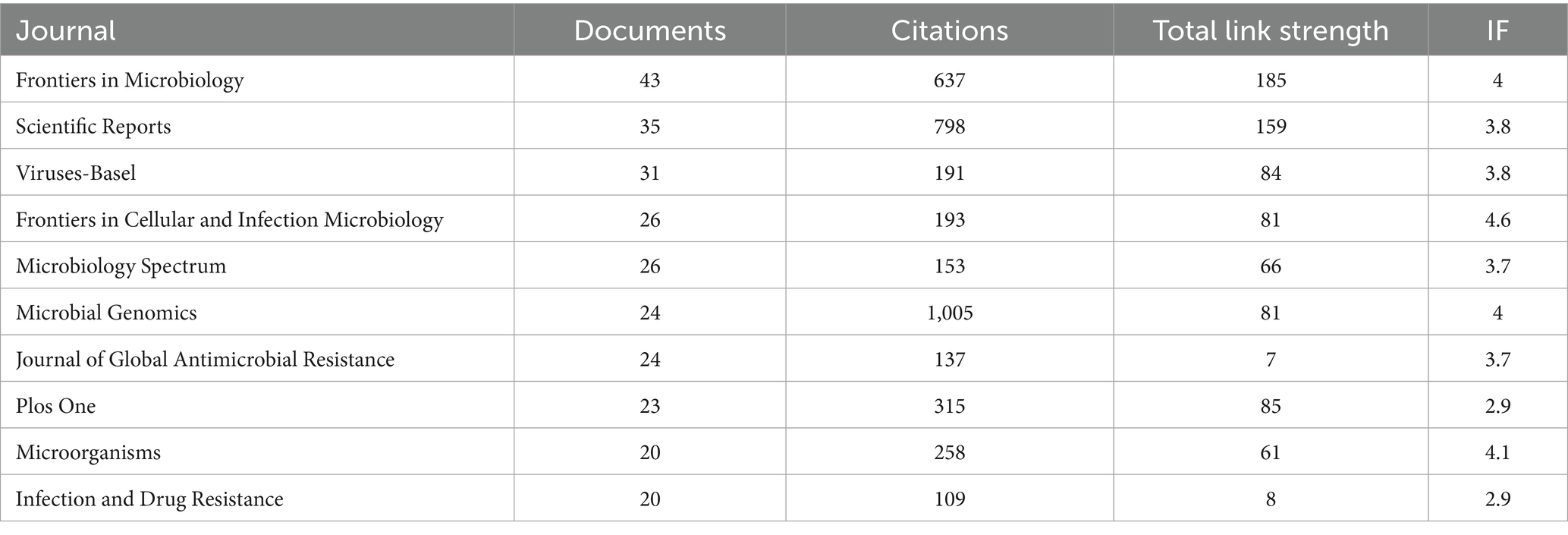
Table 4. Top journals ranked by publication counts.
Table 5 displays the top 10 original articles that received the most attention in the application of nanopore sequencing in the field of pathogenic microorganisms. All HCPs were published before 2021, consistent with the expected trend that older articles inherently accumulate higher citation counts over time compared to recent ones. A study titled “Real-time, portable genome sequencing for Ebola surveillance,” published in “Nature” in 2016, garnered 967 citations, rendering it the most highly cited publication in this domain. In contrast, an analysis of HCPs from the past 3 years indicated that research efforts have increasingly concentrated on integrating targeted amplification with nanopore sequencing. This synergistic approach optimizes sequencing workflows by reducing costs, accelerating turnaround time, and enhancing accuracy (Table 6).
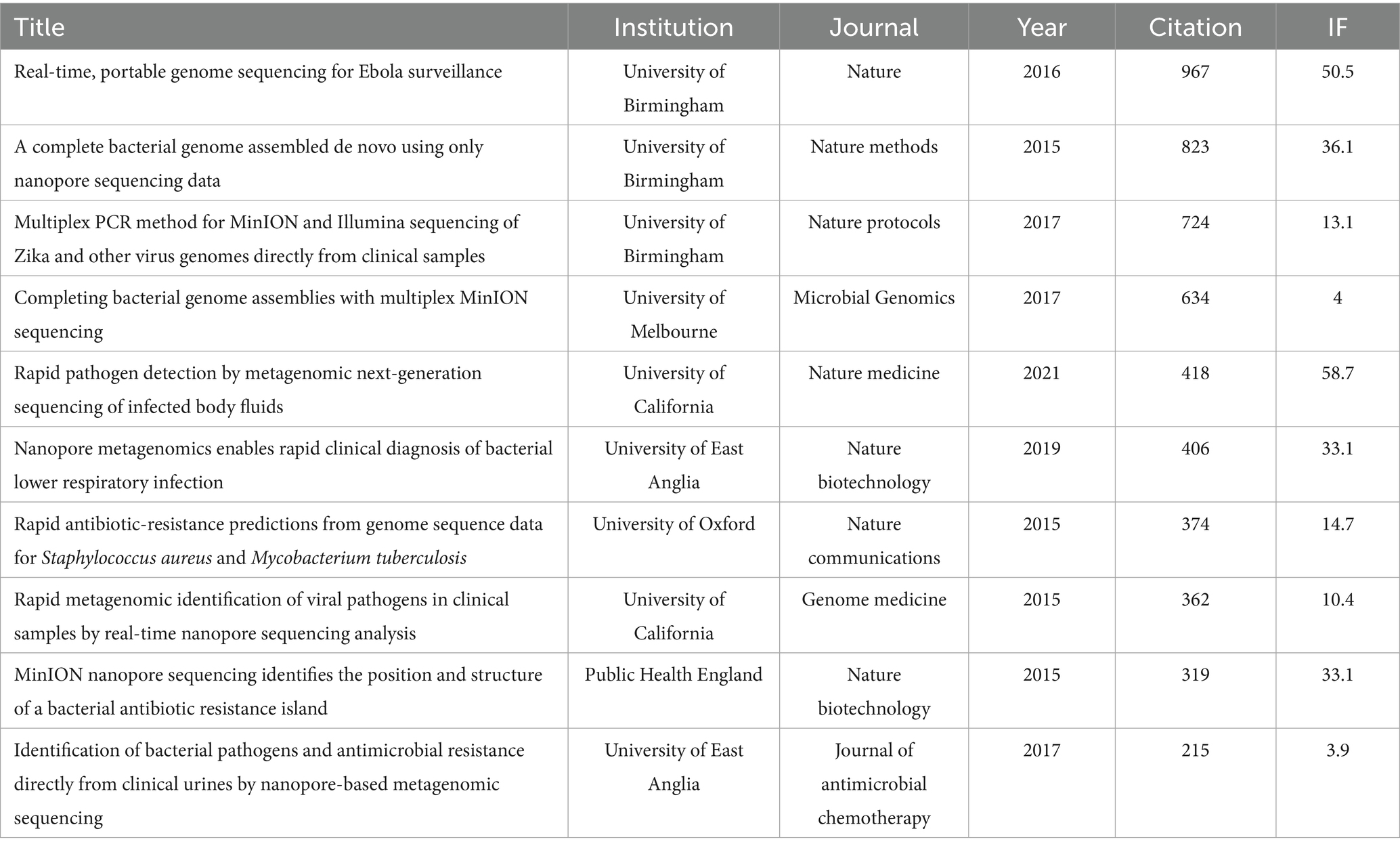
Table 5. Top 10 cited original articles.
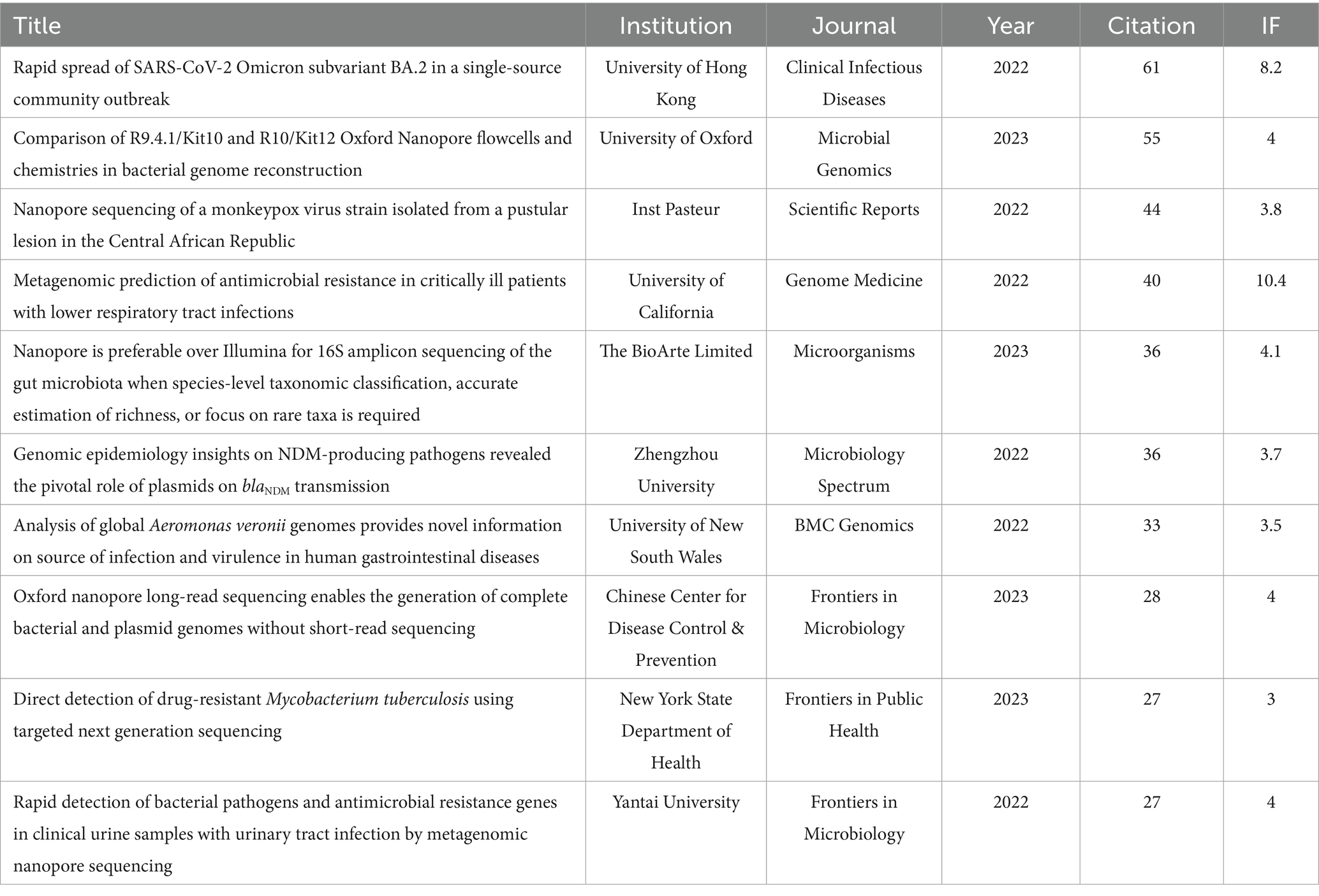
Table 6. Top 10 original articles that have been cited the most times in the last 3 years.
3.4 Keywords of research hotspots
Keywords are of paramount importance in reflecting core themes research interests and future directions in a given discipline. As illustrated in Figure 6A the keywords have been divided into three clusters. The first major cluster displayed in blue focuses on the application of nanopore sequencing in bacterial identification and diagnosis with keywords such as identification diagnosis metagenomics epidemiology and bloodstream infection. The second major cluster is in red and emphasizes the real-time surveillance capabilities of nanopore sequencing with keywords such as real-time SARS-CoV-2 alignment genome and virus. The third major cluster displayed in green highlights the applications of nanopore sequencing in the context of antimicrobial resistance (AMR) with keywords including whole genome sequencing antimicrobial resistance Klebsiella pneumoniae (K. pneumoniae) plasmids and Enterobacteriaceae.
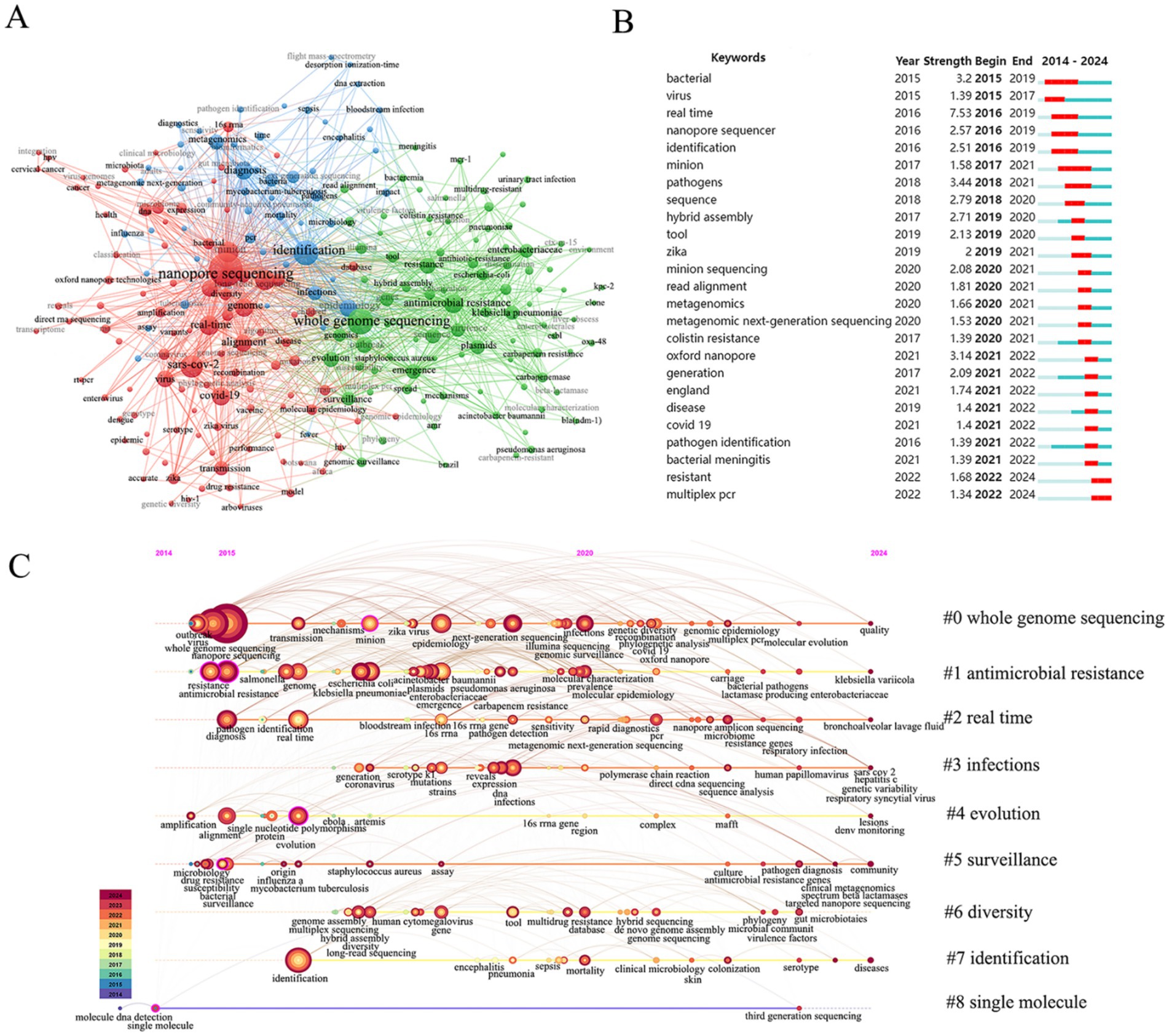
Figure 6. Keyword analysis. (A) Cluster visualization map of keywords analysis. (B) Top 25 keywords with the strongest citation bursts generated. (C) Keyword timeline clustering map.
Most significantly, the terms “resistant” and “multiplex PCR” (2022–2024) exhibited citation burst times that persisted into 2024, indicating sustained recognition and interest in these areas (Figure 6B). Notably, “real-time” (2016–2019, burst strength = 7.53) and “pathogens” (2018–2021, burst strength = 3.44) emerged as the strongest burst terms, reflecting heightened attention to real-time pathogen identification by nanopore sequencing in public health and research during these periods. The keyword timeline revealed a dynamic evolution of nanopore sequencing technology (Figure 6C). Early development and validation (2014–2016) were mainly focused on the functional exploration of nanopore sequencing in the identification and diagnosis of pathogenic microorganisms. The key terms included nanopore sequencing, whole genome sequencing, antibiotic resistance, identification and real-time. Technical optimization and clinical transformation (2017–2021) were widely used in the identification and drug resistance analysis of pathogens such as K. pneumoniae, Pseudomonas aeruginosa (P. aeruginosa), and Enterobacter, and molecular epidemiological analysis of ZIKV and other viruses by whole genome sequencing. Rapid development and diversification (2022–2024) have been deeply applied to real-time screening of drug resistance genes, targeted capture of pathogens, and multi-omics analysis of complex samples.
4 Discussion
4.1 General information
Nanopore sequencing is a revolutionary technology that enables real-time analysis of DNA or RNA by detecting transient changes in the ionic current as nucleic acids traverse engineered protein nanopores (17). Unlike conventional sequencing methods that rely on amplification or fluorescent labeling, this single-molecule approach eliminates PCR-induced biases. It directly identifies epigenetic modifications (5-methylcytosine and N6-methyladenosine), thereby distinguishing itself from short-read sequencing platforms that depend on synthetic probes (18). The defining advantages of this technology, namely ultra-long read lengths, on-site deployable portability, and cost-effectiveness, have propelled its application in identifying pathogenic microorganisms, analysis of antibiotic resistance, and epidemic monitoring (19).
The current bibliometric analysis delineates the status and trends of nanopore sequencing technology in the field of pathogenic microorganisms. The studies were stratified into two distinct phases based on whether the number of publications exceeded 50 per year for two consecutive years. From 2014 to 2019, nanopore sequencing-related research was in a slow growth stage. Since 2019, it has entered a rapid growth stage, with more than 50 publications annually. Notably, a competitive landscape has emerged between China, which contributes the largest number of publications (174 documents), and England, which obtains the most citations (7,437 citations). Among the 10 institutions with the highest productivity, five are in England. These findings highlight the important contributions and leading position of England in nanopore sequencing application research. This leadership may be attributed to the country’s economic situation, high level of investment in healthcare, and the presence of industry leader Oxford Nanopore Technology (ONT) in the country. Since its establishment in 2005, ONT has been committed to the development of DNA sequencing technology based on nanopores and launched the first commercial nanopore sequencer, MinION, in 2014, which has brought revolutionary progress to sequencing technology. Professor Crook, Derrick W, from Oxford University, has published fourteen publications and is the author with the most prolific in the field. Since 2015, Professor Crook, Derrick W has explored the wide application of nanopore sequencing in the field of pathogenic microbiology, including the identification of influenza virus (20), Neisseria gonorrhoeae (21), human metapneumovirus (22), and antibiotic resistance analysis (23, 24). HCPs typically represent fundamental themes in a research field. In this study, the top 10 most-cited publications primarily focused on the rapid diagnosis of pathogens, epidemic surveillance, and antibiotic resistance analysis, aligning with the identified research hotspots. Further exploration of the differential applications of nanopore sequencing in these three key research areas across various pathogens could offer enhanced guidance for clinical practice and scientific investigations.
4.2 Current research status and development trends
4.2.1 Nanopore sequencing in rapid identification of pathogens
The rapid identification of pathogens is vital for shortening diagnostic timelines, guiding targeted antimicrobial therapy, reducing healthcare costs, and minimizing unnecessary hospitalizations, thereby having significant implications for clinical management and public health systems. Compared to conventional diagnostic technologies (for example, culture-based methods and PCR), nanopore sequencing exhibits distinct advantages in the following aspects. Bias-free and ultra-broad detection capability: Without preset targets, it can simultaneously identify bacteria, fungi, viruses, and parasites, particularly advantageous for screening rare or emerging pathogens. For example, identifying pathogens such as R. typhi (25), A. multifidum (26), Tropheryma whipplei (27), Mycobacterium marinum (28), and Streptococcus suis (29) and other pathogens that lack typical symptoms and are easily missed. Real-time detection capability, output results during sequencing, and the quality and status of samples can be understood early in sequencing. Currently, pathogen identification and culture-based antimicrobial susceptibility testing (AST) may take 3 days or longer. Direct sequencing and predictive AST on the positive blood culture broth of 201 patients with suspected sepsis. Results the species-level consistency between AST predicted by nanopore sequencing and conventional species identification methods was 94.2%, which increased to 100% in single microbial infection. The potential turnaround time from labeled positive blood culture to report generation could be achieved within 9–17 h, which was faster than conventional AST methods (up to 48 h or longer). This result indicates that nanopore sequencing can reduce the generation cycle of clinical reports while ensuring diagnostic performance (7). Charalampous et al. (1) developed a nanopore metagenomics protocol for diagnosing bacterial lower respiratory infections (LRI) and conducted experimental tests on respiratory samples from 40 suspected patients with LRI. The results revealed that the sensitivity and specificity were 96.6 and 41.7%, respectively, which were better than those of conventional culture, and the pathogen identification and antibiotic resistance genes could be completed within 6 h. Technological innovation capability, recent analysis of HCPs have revealed a growing body of research focusing on the integration of targeted amplification with nanopore sequencing (Table 6). Nanopore targeted sequencing (NTS) has been developed using amplicon sequencing principles for the rapid detection of hundreds of pathogens and has exhibited significant advantages in pathogen detection in respiratory tract infections (30), bloodstream infections (13), and central nervous system infections (14). In bloodstream infection diagnostics, NTS achieved higher positivity rates (69.5% versus 33.9%) and accuracy (85.3% versus 53.2%) than blood cultures, with 50% of results generated within 223.5 min and 95% within 419 min (13). Moreover, it has a low-biomass pathogen detection capability, which can detect pathogens at ultralow concentrations (< 10 copies/μL) in challenging specimens, such as cerebrospinal fluid. These characteristics are uniquely valuable in diagnosing and treating acute and severe diseases, including sepsis and central nervous system infections (31).
4.2.2 Nanopore sequencing in epidemic surveillance and public health emergency response
Nanopore sequencing technology has emerged as a critical tool for epidemic surveillance and public health emergency response due to its real-time capabilities, portability, and broad-spectrum detection capacity, demonstrating unique value in identifying emerging pathogens and tracking viral mutations. In 2014, the ONT launched the first MinION sequencer, a device whose instrument size is only the size of a U disk and whose weight is less than 100 grams. This device can be connected to desktops or laptops for detection and real-time analysis, making it well-suited for use in settings with limited resources, such as refugee camps and remote clinics, or for on-site testing during outbreaks. As early as 2015, Quick et al. (10) transported the MinION sequencing system to Guinea via standard airline luggage for EBOV genome sequencing. Results were generated within 24 h of receiving Ebola-positive samples, with sequencing taking only 15–60 min. By integrating sequencing data from Guinea and Sierra Leone, this study identified frequent cross-border transmission between the two regions. This pioneering work demonstrates the potential for rapid deployment of the MinION system for outbreak monitoring, thereby shifting epidemic response strategies from “passive detection” to “active genomic surveillance.” In 2016, Brazil’s ZIKV real-time analysis project established a mobile laboratory equipped with MinION systems for amplicon-targeted sequencing, which increased the scale of virus genome monitoring and promoted ZIKV sequencing in various parts of Brazil. Phylogenetic analysis of the virus genome revealed the origins of closely related genotypes, which is vital for studying the evolutionary laws of pathogens, outbreak tracing, and epidemic prevention and control (11). Notwithstanding the aforementioned advantages, nanopore sequencing faces considerable challenges in the context of large-scale genomic initiatives. When ONT data was the only source used, the assemblies contained a high number of indels and frameshifts, resulting in misassemblies and erroneous gene annotations (32). In the prediction of AMR and the phylogenetic analysis of Neisseria gonorrhoeae isolates, ONT data produced phylogenetic tree topologies that were comparable to those produced by Illumina datasets, and all related isolates were clustered similarly. However, the number of isolates was limited and the genomic heterogeneity of the strains was high, which may have exerted a slight bias on the analysis (33). Currently, there are few reports on the on-site nanopore sequencing in sudden outbreak zones, likely due to the unpredictability, dangerousness, and control orientation of such scenarios. Beyond the monitoring of viral genomes through on-site nanopore sequencing, this method has also been employed in a series of critical research studies in the laboratory. The simultaneous sequencing of numerous samples and the real-time surveillance of the viral genome have facilitated the establishment of epidemiological links during periods of sustained viral transmission. In addition to the previously mentioned examples of EBOV and ZIKV, related studies have been conducted on Chikungunya virus (34), dengue virus (35), influenza A virus (36), yellow fever virus (37), Lassa fever virus (38), and SARS-CoV-2 (12). Concurrently, nanopore sequencing has played a pivotal role in bacterial outbreak investigations, including those involving wild-type Klebsiella variicola (39), Clostridioides difficile (40), Salmonella (41), Enterococcus faecium (42), K. pneumoniae (43). These studies integrate genomic and epidemiological data to facilitate public health laboratories in comprehending and monitoring the patterns and diversity of bacterial/viral epidemics, expeditiously tracking and investigating infections in hospital and community settings, and ascertaining opportunities for infection control interventions to further mitigate healthcare-associated infections.
Notably, early sequencing devices exhibited constrained output capabilities, rendering them inadequate for large-scale research initiatives. This limitation was ultimately addressed by innovative advancements in flow cell design, notably with the introduction of the PromethION platform by ONT. The platform can achieve a theoretical throughput of 290 Gb per flow cell, and an impressive 14 Tb for the PromethION 48 (3). However, the cost of nanopore sequencing for DNA methylation studies in Alzheimer’s disease and frontotemporal dementia is $1,000 per sample, representing a 4-fold increase compared to Illumina-based approaches (44). In order to enhance competitiveness, ONT must further reduce costs, thereby improving accessibility and cost-effectiveness across diverse applications.
4.2.3 Nanopore sequencing in AMR detection
AMR has become a major threat to global health. From 1990 to 2021, more than 1 million people died annually from drug-resistant infections. It is estimated that by 2050, AMR will directly cause more than 1.9 million deaths per year and indirectly cause more than 8 million deaths (45). AMR infections are primarily driven by pathogens, including ESKAPE (Enterococcus faecium, Staphylococcus aureus, K. pneumoniae, Acinetobacter baumannii, P. aeruginosa, and Enterobacter) or Mycobacteria, and form antibiotic resistance genes (ARGs) through horizontal gene transfer between microbial communities. Rapid, comprehensive, and accurate detection of ARGs is crucial for guiding early and effective antibacterial treatment (46). Compared to culture-based susceptibility testing (2–5 days), nanopore sequencing, with its long reads and real-time data generation capabilities, can rapidly construct a complete bacterial genome in a matter of hours and align it to a known antibiotic resistance gene database to identify ARGs, thereby guiding clinical targeted drugs in a timely manner. Currently, The nanopore sequencing method has been demonstrated to be effective in the detection of drug resistance in various pathogens, including K. pneumoniae (47), P. aeruginosa (48), Salmonella (49), Mycobacterium tuberculosis (50), Neisseria gonorrhoeae (51, 52). This has resulted in significant advancements and accomplishments in the realm of detecting ARGs, identifying single nucleotide mutations and heterogeneous drug resistance, and analyzing drug resistance mechanisms. For instance, Liu et al. (53) utilized nanopore sequencing to identify three novel K. pneumoniae sequence types (STs) from 1,484 clinical isolates carrying the mobile multidrug-resistant (MDR) efflux pump gene cluster (tmexCD1-toprJ1), including a pan-resistant ST22 strain co-harboring blaKPC-2 and blaNDM-1 (carbapenemase genes), a MDR ST3691 strain, and a MDR ST37 strain. The identification of these novel strains represents a valuable addition to the epidemiological study of K. pneumoniae carrying tmexCD-toprJ, highlighting the ability of nanopore sequencing in detecting AMR caused by horizontal transmission of drug-resistant genes facilitated by mobile elements, such as plasmids. Subsequent to this, the same research team utilized nanopore sequencing to ascertain that the tandem repeats and copy number of the blaSHV-12 gene element of cefiderocol-resistant strains had increased. This finding suggests the potential for a novel mechanism contributing to cefodil resistance in K. pneumoniae, and the hypothesis that was validated through quantitative PCR and in vitro experimentation. In a separate study, Sauerborn et al. (7) demonstrated the great potential of real-time genomics for rapid and accurate analysis of complex bacterial infections in clinical settings through the case of a multidrug-resistant K. pneumoniae infection. In comparison with a clinically established diagnosis, real-time genomics-based drug resistance prediction can identify a novel antibiotic resistance gene variant, blaKPC-14, located on the low-abundance IncN plasmid. This finding is of great significance for clinical decision-making and potential patient prognosis, indicating the potential for change by integrating real-time genomic analysis into clinical practice.
Two significant advancements in nanopore sequencing, namely Q20 + chemistry and adaptive sampling, have had a substantial impact on real-time pathogen identification, viral genome surveillance and AMR analysis. The implementation of the latest Q20 + chemistry and flow cell has been demonstrated to enhance the precision of base detection data and the accuracy of sequencing. In comparison with the old kit LSK109 with R9.4, the average sequence accuracy of SARS-CoV-2 sequencing was increased from 96.25 to 98.34% using the Q20 + kit (LSK112) with flow cell R10.4, and the proportion of sequences with an accuracy of more than 99% was increased from 0.61 to 30.1% (54). Furthermore, three strains of Salmonella carrying MDR plasmids were sequenced using the LSK114 kit with flow cell R10.4.1, and de novo genome assembly was performed with accuracy increased from 92 to 98% and Q20 increased from 13 to 42% (55). Nanopore adaptive sampling (NAS) improves the target reading by real-time alignment, and ejects the uninteresting read by reversing the voltage across the nanopore, which is very important for the targeted enrichment of low-abundance genomes. Herbert J. et al. conducted a study to assess the impact of the nanopore sequencing chemistry version on the output of the novel adaptive sampling approach. In comparison with non-adaptive sequencing, this resulted in a 5-7-fold increase in target enrichment (LSK109: 4.8-fold; LSK112: 6.8-fold) (56). It is noteworthy that no significant differences in adaptive sampling enrichment efficiency were observed between the older and newer ONT sequencing chemistries, suggesting that adaptive sampling performs consistently across different library preparation kits. Hang Cheng et al. developed a metagenomic workflow that is based on the efficient selective “human host depletion” NAS sequencing and species-specific ARGs prediction. The microbial sequence yield was increased by a factor of at least 8-fold in 21 sequenced clinical bronchoalveolar lavage fluid samples, and the process was completed within 4.5 h from sample to result (57). This ultra-sensitive diagnosis of bacterial pathogens and ARGs from clinical samples has the potential to reduce unnecessary prophylactic antibiotic use and the related morbidity and mortality.
4.3 Limitations
To our knowledge, this study is the first bibliometric analysis to comprehensively describe the latest developments in nanopore sequencing in the field of pathogenic microorganisms. It elucidates early collaboration patterns, journal contributions, and research hotspots in this field, aiding scholars in identifying knowledge gaps and future directions. However, this study has some limitations. First, our analysis relied solely on the WOSCC, a constraint inherent to bibliometric methods. We mitigated database bias through rigorous search strategies and manual verification to ensure that the results obtained in this study are a true reflection of the existing literature. Second, while bibliometrics quantifies macro-level trends, it may overlook nuanced insights within the individual studies. Additionally, nanopore sequencing faces challenges, including lower raw read accuracy (96.84% for nanopore versus 99.68% for Illumina), high costs, and underdeveloped bioinformatics pipelines (58).
5 Conclusion
In summary, the rapid growth of nanopore sequencing research from 2014 to 2024 highlights its potential for pathogen diagnostics. While current limitations exist, we believe that they are not insurmountable technical obstacles but catalysts that promote continuous innovation. By integrating bioengineering, artificial intelligence, and public health strategies, this technology may achieve breakthroughs in ultrasensitive detection (detection of pathogens with a load as low as 1 copy/μL within 1 h), fully automated analysis (handheld devices for sample-to-clinical report), and global genomic surveillance networks, ultimately transforming infectious disease management.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material. Further inquiries can be directed to the corresponding authors.
Author contributions
JLo: Methodology, Validation, Writing – review & editing, Visualization, Software, Data curation, Writing – original draft. BZ: Data curation, Validation, Writing – review & editing. JLi: Visualization, Writing – review & editing, Software. JZ: Validation, Project administration, Writing – review & editing. GD: Writing – review & editing, Project administration, Investigation, Funding acquisition.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fmed.2025.1678005.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1610063/full#supplementary-material
References
1. Charalampous, T, Kay, GL, Richardson, H, Aydin, A, Baldan, R, Jeanes, C, et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol. (2019) 37:783–92. doi: 10.1038/s41587-019-0156-5
2. Maimaiti, M, Kong, L, Yu, Q, Wang, Z, Liu, Y, Yang, C, et al. Analytical performance of a novel nanopore sequencing for SARS-CoV-2 genomic surveillance. J Med Virol. (2024) 96:e70108. doi: 10.1002/jmv.70108
3. Zhang, T, Li, H, Jiang, M, Hou, H, Gao, Y, Li, Y, et al. Nanopore sequencing: flourishing in its teenage years. J Genet Genomics. (2024) 51:1361–74. doi: 10.1016/j.jgg.2024.09.007
4. Zhang, X, Ye, J, Wang, L, Zhang, L, Wang, L, and Jin, H. Nanopore sequencing technology: a reliable method for pathogen diagnosis in elderly patients with community-acquired pneumonia. Infect Drug Resist. (2024) 17:3659–67. doi: 10.2147/IDR.S475861
5. Wang, Y, Zhao, Y, Bollas, A, Wang, Y, and Au, KF. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol. (2021) 39:1348–65. doi: 10.1038/s41587-021-01108-x
6. MacKenzie, M, and Argyropoulos, C. An introduction to nanopore sequencing: past, present, and future considerations. Micromachines. (2023) 14:459. doi: 10.3390/mi14020459
7. Sauerborn, E, Corredor, NC, Reska, T, Perlas, A, da Fonseca, V, Atum, S, et al. Detection of hidden antibiotic resistance through real-time genomics. Nat Commun. (2024) 15:5494. doi: 10.1038/s41467-024-49851-4
8. Liu, C, Yi, J, Lu, M, Yang, P, Du, C, Jiang, F, et al. Dynamic within-host cefiderocol heteroresistance caused by Bla SHV-12 amplification in pandrug-resistant and hypervirulent Klebsiella pneumoniae sequence type 11. Drug Resist Updat. (2024) 73:101038. doi: 10.1016/j.drup.2023.101038
9. Battaglia, S, Dong, K, Wu, J, Chen, Z, Najm, FJ, Zhang, Y, et al. Long-range phasing of dynamic, tissue-specific and allele-specific regulatory elements. Nat Genet. (2022) 54:1504–13. doi: 10.1038/s41588-022-01188-8
10. Quick, J, Loman, NJ, Duraffour, S, Simpson, JT, Severi, E, and Cowley, L. Real-time, portable genome sequencing for Ebola surveillance. Nature. (2016) 530:228–32. doi: 10.1038/nature16996
11. Faria, NR, Sabino, EC, Nunes, MRT, Alcantara, LCJ, Loman, NJ, and Pybus, OG. Mobile real-time surveillance of Zika virus in Brazil. Genome Med. (2016) 8:97. doi: 10.1186/s13073-016-0356-2
12. Meredith, LW, Hamilton, WL, Warne, B, Houldcroft, CJ, Hosmillo, M, Jahun, AS, et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect Dis. (2020) 20:1263–71. doi: 10.1016/S1473-3099(20)30562-4
13. Han, D, Yu, F, Zhang, D, Hu, J, Zhang, X, Xiang, D, et al. Molecular rapid diagnostic testing for bloodstream infections: nanopore targeted sequencing with pathogen-specific primers. J Infect. (2024) 88:106166. doi: 10.1016/j.jinf.2024.106166
14. Yang, X, Zhou, S, Chang, Z, Xi, X, Li, J, Miao, M, et al. Nanopore targeted sequencing-based diagnosis of central nervous system infections in HIV-infected patients. Ann Clin Microbiol Antimicrob. (2024) 23:22. doi: 10.1186/s12941-024-00682-7
15. Zhang, Q, Ding, Y, Ren, Q, Zhang, F, Lyu, G, Lu, T, et al. Evaluation of targeted sequencing for pathogen identification in bone and joint infections: a cohort study from China. Ann Clin Microbiol Antimicrob. (2024) 23:77. doi: 10.1186/s12941-024-00733-z
16. Wu, T, Wu, Y, Nie, K, Yan, J, Chen, Y, Wang, S, et al. Bibliometric analysis and global trends in uterus transplantation. Int J Surg. (2024) 110:4932–46. doi: 10.1097/JS9.0000000000001470
17. Liu-Wei, W, van der Toorn, W, Bohn, P, Hölzer, M, Smyth, RP, and Von Kleist, M. Sequencing accuracy and systematic errors of nanopore direct RNA sequencing. BMC Genomics. (2024) 25:528. doi: 10.1186/s12864-024-10440-w
18. Leger, A, Amaral, PP, Pandolfini, L, Capitanchik, C, Capraro, F, Miano, V, et al. RNA modifications detection by comparative nanopore direct RNA sequencing. Nat Commun. (2021) 12:7198. doi: 10.1038/s41467-021-27393-3
19. Kolmogorov, M, Billingsley, KJ, Mastoras, M, Meredith, M, Monlong, J, Lorig-Roach, R, et al. Scalable nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation. Nat Methods. (2023) 20:1483–92. doi: 10.1038/s41592-023-01993-x
20. Lewandowski, K, Xu, Y, Pullan, ST, Lumley, SF, Foster, D, Sanderson, N, et al. Metagenomic nanopore sequencing of influenza virus direct from clinical respiratory samples. J Clin Microbiol. (2019) 58:e00963–19. doi: 10.1128/JCM.00963-19
21. Street, TL, Barker, L, Sanderson, ND, Kavanagh, J, Hoosdally, S, Cole, K, et al. Optimizing DNA extraction methods for nanopore sequencing of Neisseria gonorrhoeae directly from urine samples. J Clin Microbiol. (2020) 58:e01822–19. doi: 10.1128/JCM.01822-19
22. Xu, Y, Lewandowski, K, Jeffery, K, Downs, LO, Foster, D, Sanderson, ND, et al. Nanopore metagenomic sequencing to investigate nosocomial transmission of human metapneumovirus from a unique genetic group among haematology patients in the United Kingdom. J Infect. (2020) 80:571–7. doi: 10.1016/j.jinf.2020.02.003
23. Bradley, P, Gordon, NC, Walker, TM, Dunn, L, Heys, S, Huang, B, et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun. (2015) 6:10063. doi: 10.1038/ncomms10063
24. Street, TL, Sanderson, ND, Kolenda, C, Kavanagh, J, Pickford, H, Hoosdally, S, et al. Clinical metagenomic sequencing for species identification and antimicrobial resistance prediction in orthopedic device infection. J Clin Microbiol. (2022) 60:e0215621–1. doi: 10.1128/jcm.02156-21
25. Qian, P, He, X, Yang, M, Wei, L, Zhang, L, and Xing, X. Detection of severe murine typhus by nanopore targeted sequencing, China. Emerg Infect Dis. (2023) 29:1275–7. doi: 10.3201/eid2906.221929
26. Chen, Q, Yang, Q, Chen, H, Yao, Y, Shen, L, Zhang, R, et al. Zoonotic fungus Arthroderma multifidum causing chronic pulmonary infection. Int J Infect Dis. (2023) 130:17–9. doi: 10.1016/j.ijid.2023.02.010
27. Ruan, W, Xu, J, Yang, F, Wu, X, and Ying, K. Tropheryma whipplei infection in the lung of a patient with long COVID: a case report. BMC Infect Dis. (2024) 24:292. doi: 10.1186/s12879-024-09183-6
28. Huang, YY, Li, QS, Li, ZD, Sun, AH, and Hu, SP. Rapid diagnosis of Mycobacterium marinum infection using targeted nanopore sequencing: a case report. Front Cell Infect Microbiol. (2023) 13:1238872. doi: 10.3389/fcimb.2023.1238872
29. Wang, YY, Huang, Q, and Cheng, Y. Detecting Streptococcus suis by nanopore sequencing in endophthalmitis: a case report. Eur J Inflamm. (2021) 19:1–5. doi: 10.1177/20587392211002657
30. Guo, Y, Li, Z, Li, L, Li, S, Sun, L, Yang, X, et al. A dual-process of targeted and unbiased nanopore sequencing enables accurate and rapid diagnosis of lower respiratory infections. EBioMedicine. (2023) 98:104858. doi: 10.1016/j.ebiom.2023.104858
31. Lopez-Labrador, FX, Huber, M, Sidorov, IA, Brown, JR, Cuypers, L, Laenen, L, et al. Multicenter benchmarking of short and long read wet lab protocols for clinical viral metagenomics. J Clin Virol. (2024) 173:105695. doi: 10.1016/j.jcv.2024.105695
32. Goussarov, G, Mysara, M, Cleenwerck, I, Claesen, J, Leys, N, Vandamme, P, et al. Benchmarking short-, long- and hybrid-read assemblers for metagenome sequencing of complex microbial communities. Microbiology. (2024) 170:001469. doi: 10.1099/mic.0.001469
33. Golparian, D, Donà, V, Sánchez-Busó, L, Foerster, S, Harris, S, Endimiani, A, et al. Antimicrobial resistance prediction and phylogenetic analysis of Neisseria gonorrhoeae isolates using the Oxford Nanopore MinION sequencer. Sci Rep. (2018) 8:17596. doi: 10.1038/s41598-018-35750-4
34. Gräf, T, Vazquez, C, Giovanetti, M, de Bruycker-Nogueira, F, Fonseca, V, Claro, IM, et al. Epidemiologic history and genetic diversity origins of chikungunya and dengue viruses, Paraguay. Emerg Infect Dis. (2021) 27:1393–404. doi: 10.3201/eid2705.204244
35. Adelino, TER, Giovanetti, M, Fonseca, V, Xavier, J, De Abreu, AS, Do Nascimento, VA, et al. Field and classroom initiatives for portable sequence-based monitoring of dengue virus in Brazil. Nat Commun. (2021) 12:2296. doi: 10.1038/s41467-021-22607-0
36. Badar, N, Ikram, A, Salman, M, Saeed, S, Mirza, HA, Ahad, A, et al. Evolutionary analysis of seasonal influenza a viruses in Pakistan 2020–2023. Influenza Other Respir Viruses. (2024) 18:e13262. doi: 10.1111/irv.13262
37. Faria, NR, Kraemer, MUG, Hill, SC, Goes De Jesus, J, Aguiar, RS, Iani, FCM, et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science. (2018) 361:894–9. doi: 10.1126/science.aat7115
38. Kafetzopoulou, LE, Pullan, ST, Lemey, P, Suchard, MA, Ehichioya, DU, Pahlmann, M, et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science. (2019) 363:74–7. doi: 10.1126/science.aau9343
39. White, RT, Balm, M, Burton, M, Hutton, S, Jeram, J, Kelly, M, et al. The rapid detection of a neonatal unit outbreak of a wild-type Klebsiella variicola using decentralized Oxford Nanopore sequencing. Antimicrob Resist Infect Control. (2025) 14:6. doi: 10.1186/s13756-025-01529-2
40. Bloomfield, M, Hutton, S, Burton, M, Tarring, C, Velasco, C, Clissold, C, et al. Early identification of a ward-based outbreak of Clostridioides difficile using prospective multilocus sequence type-based Oxford Nanopore genomic surveillance. Infect Control Hosp Epidemiol. (2024) 45:1–7. doi: 10.1017/ice.2024.77
41. Quick, J, Ashton, P, Calus, S, Chatt, C, Gossain, S, Hawker, J, et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. (2015) 16:114. doi: 10.1186/s13059-015-0677-2
42. Egan, SA, Corcoran, S, McDermott, H, Fitzpatrick, M, Hoyne, A, McCormack, O, et al. Hospital outbreak of linezolid-resistant and vancomycin-resistant ST80 Enterococcus faecium harbouring an optrA-encoding conjugative plasmid investigated by whole-genome sequencing. J Hosp Infect. (2020) 105:726–35. doi: 10.1016/j.jhin.2020.05.013
43. Prussing, C, Southwick, K, Snavely, E, Kidney, A, Randall, L, Sossei, A, et al. Comparative analysis of multiplexed PCR and short- and long-read whole genome sequencing to investigate a large Klebsiella pneumoniae outbreak in New York state. Diagn Microbiol Infect Dis. (2022) 104:115765. doi: 10.1016/j.diagmicrobio.2022.115765
44. Pagano, L, Lagrotteria, D, Facconi, A, Saraceno, C, Longobardi, A, Bellini, S, et al. Evaluation of Illumina and Oxford Nanopore sequencing for the study of DNA methylation in Alzheimer's disease and frontotemporal dementia. Int J Mol Sci. (2025) 26:4198. doi: 10.3390/ijms26094198
45. Bertagnolio, S, Dobreva, Z, Centner, CM, Olaru, ID, Donà, D, Burzo, S, et al. WHO global research priorities for antimicrobial resistance in human health. Lancet Microbe. (2024) 5:100902. doi: 10.1016/S2666-5247(24)00134-4
46. Suzuki, M, Hashimoto, Y, Hirabayashi, A, Yahara, K, Yoshida, M, Fukano, H, et al. Genomic epidemiological analysis of antimicrobial-resistant bacteria with nanopore sequencing. Methods Mol Biol. (2023) 2632:227–46. doi: 10.1007/978-1-0716-2996-3_16
47. Liu, P, Wu, H, Li, Y, Cheng, H, Liou, C, Chen, F, et al. Comprehensive pathogen identification and antimicrobial resistance prediction from positive blood cultures using nanopore sequencing technology. Genome Med. (2024) 16:141. doi: 10.1186/s13073-024-01416-2
48. Liu, B, Gao, J, Liu, XF, Rao, G, Luo, J, Han, P, et al. Direct prediction of carbapenem resistance in Pseudomonas aeruginosa by whole genome sequencing and metagenomic sequencing. J Clin Microbiol. (2023) 61:e0061723–3. doi: 10.1128/jcm.00617-23
49. Xu, F, Ge, C, Luo, H, Li, S, Wiedmann, M, Deng, X, et al. Evaluation of real-time nanopore sequencing for Salmonella serotype prediction. Food Microbiol. (2020) 89:103452. doi: 10.1016/j.fm.2020.103452
50. Hall, MB, Rabodoarivelo, MS, Koch, A, Dippenaar, A, George, S, Grobbelaar, M, et al. Evaluation of nanopore sequencing for Mycobacterium tuberculosis drug susceptibility testing and outbreak investigation: a genomic analysis. Lancet Microbe. (2023) 4:e84–92. doi: 10.1016/S2666-5247(22)00301-9
51. Sanderson, ND, Swann, J, Barker, L, Kavanagh, J, Hoosdally, S, Crook, D, et al. High precision Neisseria gonorrhoeae variant and antimicrobial resistance calling from metagenomic nanopore sequencing. Genome Res. (2020) 30:1354–63. doi: 10.1101/gr.262865.120
52. Zhang, C, Xiu, L, Li, Y, Sun, L, Li, Y, Zeng, Y, et al. Multiplex PCR and nanopore sequencing of genes associated with antimicrobial resistance in Neisseria gonorrhoeae directly from clinical samples. Clin Chem. (2021) 67:610–20. doi: 10.1093/clinchem/hvaa306
53. Liu, C, Guo, J, Lu, M, Shen, N, and Du, P. Dissemination of the mobilised RND efflux pump gene cluster tmexCD-toprJ among Klebsiella pneumoniae. Lancet Microbe. (2023) 4:e135. doi: 10.1016/S2666-5247(22)00325-1
54. Luo, J, Meng, Z, Xu, X, Wang, L, Zhao, K, Zhu, X, et al. Systematic benchmarking of nanopore Q20+ kit in SARS-CoV-2 whole genome sequencing. Front Microbiol. (2022) 13:973367. doi: 10.3389/fmicb.2022.973367
55. Zhao, W, Zeng, W, Pang, B, Luo, M, Peng, Y, Xu, J, et al. Oxford nanopore long-read sequencing enables the generation of complete bacterial and plasmid genomes without short-read sequencing. Front Microbiol. (2023) 14:1179966. doi: 10.3389/fmicb.2023.1179966
56. Herbert, J, Thompson, S, Beckett, AH, and Robson, SC. Impact of microbiological molecular methodologies on adaptive sampling using nanopore sequencing in metagenomic studies. Environ Microbiome. (2025) 20:47. doi: 10.1186/s40793-025-00704-7
57. Cheng, H, Sun, Y, Yang, Q, Deng, M, Yu, Z, Zhu, G, et al. A rapid bacterial pathogen and antimicrobial resistance diagnosis workflow using Oxford nanopore adaptive sequencing method. Brief Bioinform. (2022) 23:bbac453. doi: 10.1093/bib/bbac453
58. Bejaoui, S, Nielsen, SH, Rasmussen, A, Coia, JE, Andersen, DT, Pedersen, B, et al. Comparison of Illumina and Oxford Nanopore sequencing data quality for Clostridioides difficile genome analysis and their application for epidemiological surveillance. BMC Genomics. (2025) 26:92. doi: 10.1186/s12864-025-11267-9
Glossary
EBOV - Ebola virus
ZIKV - Zika virus
SARS-CoV-2 - Severe Acute Respiratory Syndrome Coronavirus 2
WOSCC - Web of Science Core Collection
E. coli - Escherichia coli
HPV - Human papillomavirus
M. tuberculosis - Mycobacterium tuberculosis
HSV - Herpes simplex virus
DENV - Dengue virus
HCV - Hepatitis C virus
IAV - Influenza A virus
MPXV - Monkeypox virus
S. aureus - Staphylococcus aureus
K. pneumoniae - Klebsiella pneumoniae
S. pneumoniae - Streptococcus pneumoniae
P. aeruginosa - Pseudomonas aeruginosa
CHIKV - Chikungunya virus
HAdV - Human adenovirus
AMR - Antimicrobial resistance;
ONT - Oxford Nanopore Technology;
NTS - Nanopore Targeted Sequencing
HCPs - Highly Cited Papers
A. baumannii - Acinetobacter baumannii
LRI - Lower respiratory infection
RSV - Respiratory syncytial virus
NAS - Nanopore adaptive sampling
Keywords: nanopore sequencing, pathogenic microorganisms, bibliometric analysis, real-time, genomic surveillance, antimicrobial resistance
Citation: Long J, Zeng B, Li J, Zhang J and Deng G (2025) Global trends in the application of nanopore sequencing technology in the detection of infectious disease pathogens: a bibliometric analysis from 2014 to 2024. Front. Med. 12:1610063. doi: 10.3389/fmed.2025.1610063
Edited by:
Shisan Bao, The University of Sydney, AustraliaReviewed by:
Alberto Antonelli, University of Florence, ItalyLin Yanfeng, Academy of Military Medical Sciences (AMMS), China
Copyright © 2025 Long, Zeng, Li, Zhang and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Zhang, ZG9jdG9yemhhbmczMzMzQDE2My5jb20=; Guohong Deng, Z2hfZGVuZ0Bob3RtYWlsLmNvbQ==