 Qianying Kong1,2,3,4
Qianying Kong1,2,3,4 Huifang Peng
Huifang Peng Qian Zhao
Qian Zhao Hongwei Jiang
Hongwei Jiang- 1Henan Key Laboratory of Rare Diseases, Endocrinology and Metabolism Center, The First Affiliated Hospital, and College of Clinical Medicine of Henan University of Science and Technology, Luoyang, China
- 2State Key Laboratory of Female Fertility Promotion, Department of Human Anatomy, Histology and Embryology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China
- 3Department of Pediatrics, Children's Medical Center, Peking University First Hospital, Beijing, China
- 4Neuroscience Research Institute, Department of Neurobiology, School of Basic Medical Sciences, Key Laboratory for Neuroscience, Ministry of Education/National Health Commission of China, Peking University, Beijing, China
Normal craniofacial development depends on the precise specification, migration, and differentiation of cranial neural crest cells (CNCCs). Perturbations in these processes result in a wide spectrum of congenital craniofacial anomalies, which represent a major cause of birth defects worldwide. Xenopus has emerged as a particularly powerful model for investigating craniofacial morphogenesis, owing to its external fertilization, large and experimentally accessible embryos, and evolutionarily conserved developmental pathways. These advantages allow direct in vivo visualization and manipulation of CNCCs behaviors at single-cell resolution, providing opportunities not readily achievable in mammalian models. With the integration of advanced techniques such as high-resolution imaging, lineage tracing, microsurgical manipulation, and genome editing, the utility of Xenopus in craniofacial biology has been greatly expanded. In this review, we outline the key stages of craniofacial development, summarize representative craniofacial developmental disorders studied using Xenopus as a model, and highlight how this system has provided critical mechanistic insights. Importantly, the amenability of Xenopus embryos to small-molecule screening underscores their translational potential as a rapid preclinical platform, linking human genetic variants to disease pathogenesis and accelerating therapeutic discovery for craniofacial disorders, as well as its translational potential as a rapid preclinical platform, linking human genetic variants to disease pathogenesis and accelerating therapeutic discovery for craniofacial disorders.
1 Introduction
Craniofacial morphogenesis is a tightly regulated and dynamic developmental process that governs the formation of head and facial structures through precise spatial and temporal patterning during embryogenesis. This process depends on a transient, pluripotent stem cell-like population known as neural crest cells (NCCs). Among these, CNCCs serve as key contributors to the regulation and execution of craniofacial morphogenesis (1). Any genetic or environmental perturbations affecting the induction, migration, proliferation, or fate determination of these cells may result in pronounced craniofacial developmental disorders (2).
It is estimated that congenital craniofacial developmental disorders represent about one-third of all congenital anomalies globally (3). Among these, cleft lip and/or palate (CL/P) is one of the most common, with a global incidence of around 1.7 per 1,000 live births (4). Another prominent condition, craniosynostosis, occurs at a rate of approximately 5.2 per 10,000 live births, with non-syndromic cases being the most frequent (5, 6). Craniofacial developmental disorders not only impact essential functions like feeding, breathing, and speech, but also impose substantial psychological stress on patients. The lifelong medical care, surgical interventions, and rehabilitative support required place a considerable burden on affected families and healthcare systems, underscoring the urgent need for deeper mechanistic understanding and preventive strategies.
Congenital craniofacial developmental disorders often arise from disruptions during early embryogenesis. To investigate the underlying mechanisms, a variety of model organisms have been employed, including the mouse (Mus musculus), chicken (Gallus gallus), zebrafish (Danio rerio), and the African clawed frog (Xenopus laevis). Among these, Xenopus stand out as a classical and highly tractable model system, offering several key advantages: external fertilization and development, high fecundity, a stable genetic background, and large, easily manipulable embryos. These features are particularly well-suited for live imaging, microsurgical manipulations, and microinjection-based gene perturbation, enabling precise analysis of early embryonic events and organogenesis (7). Importantly, Xenopus exhibits a high degree of genetic conservation with humans, sharing over 80% of known human disease-associated gene orthologs, including most genes implicated in craniofacial development, and its branchial arch (BA) structures are homologous to the pharyngeal arches (PA) that shape human facial development (8, 9). This evolutionary conservation, combined with its experimental accessibility, makes Xenopus a powerful and efficient model for dissecting the molecular and cellular mechanisms underlying congenital craniofacial disorders.
This review summarizes the key stages of craniofacial development and highlights recent advances using Xenopus models to study congenital craniofacial disorders. By integrating developmental genetics with functional modeling, Xenopus continues to offer critical insights into the etiology and potential treatment strategies for these complex conditions.
2 Key stages of craniofacial development
Craniofacial development is a temporally and spatially coordinated process that progresses through a series of well-defined stages, beginning at gastrulation and culminating in postnatal tissue remodeling. The Xenopus model provides a robust platform to investigate these stages in vivo due to its external development, accessibility for molecular manipulation, and conserved developmental pathways. Below, we summarize the key stages of craniofacial development, highlighting critical cellular events, regulatory signaling pathways, and clinically relevant disease associations (Table 1).

Table 1. Key stages of craniofacial development: developmental events, molecular regulators, and disease associations.
2.1 Gastrulation and germ layer formation
During gastrulation, occurring around stages 10–12 in Xenopus, the three primary germ layers—ectoderm, mesoderm, and endoderm—are formed through highly coordinated cellular movements such as invagination, involution, and epiboly (10). These germ layers serve as the embryonic source for all craniofacial tissues: the ectoderm gives rise to the neural crest and surface epithelium, the mesoderm contributes to vasculature and musculature, and the endoderm lines the pharyngeal foregut. Key signaling pathways involved in this process include Nodal, BMP, and Wnt, which establish the body axes and control cell fate decisions (11, 12). Disruptions in these early patterning signals can lead to severe craniofacial malformations. For example, defects in midline specification and forebrain patterning may result in holoprosencephaly, a condition often associated with facial dysmorphisms such as cyclopia, cleft lip, and nasal anomalies (13, 14).
2.2 Neural plate border formation and neural crest induction
NCCs possess unique multipotency, giving rise to a diverse range of derivatives, including craniofacial bone and cartilage, peripheral neurons, glial cells, melanocytes, and various connective tissue types (Figure 1). NCCs are induced at the neural plate border, a region between the prospective neural and non-neural ectoderm. This induction is a result of a precise balance between BMP, Wnt, and FGF signaling gradients (15). Transcription factors such as Pax3/7, ZIC1, and Msx1/2 function as early neural crest specifiers, integrating these signals to define the neural crest territory (16). The induced neural crest progenitors then undergo further maturation and are primed for delamination and migration. Abnormal regulation of this process can impair neural crest formation, contributing to a spectrum of craniofacial syndromes. For instance, Treacher Collins syndrome arises from mutations affecting ribosome biogenesis in neural crest progenitors, whereas CHARGE syndrome involves mutations in CHD7, a chromatin remodeler essential for NCCs specification (17).
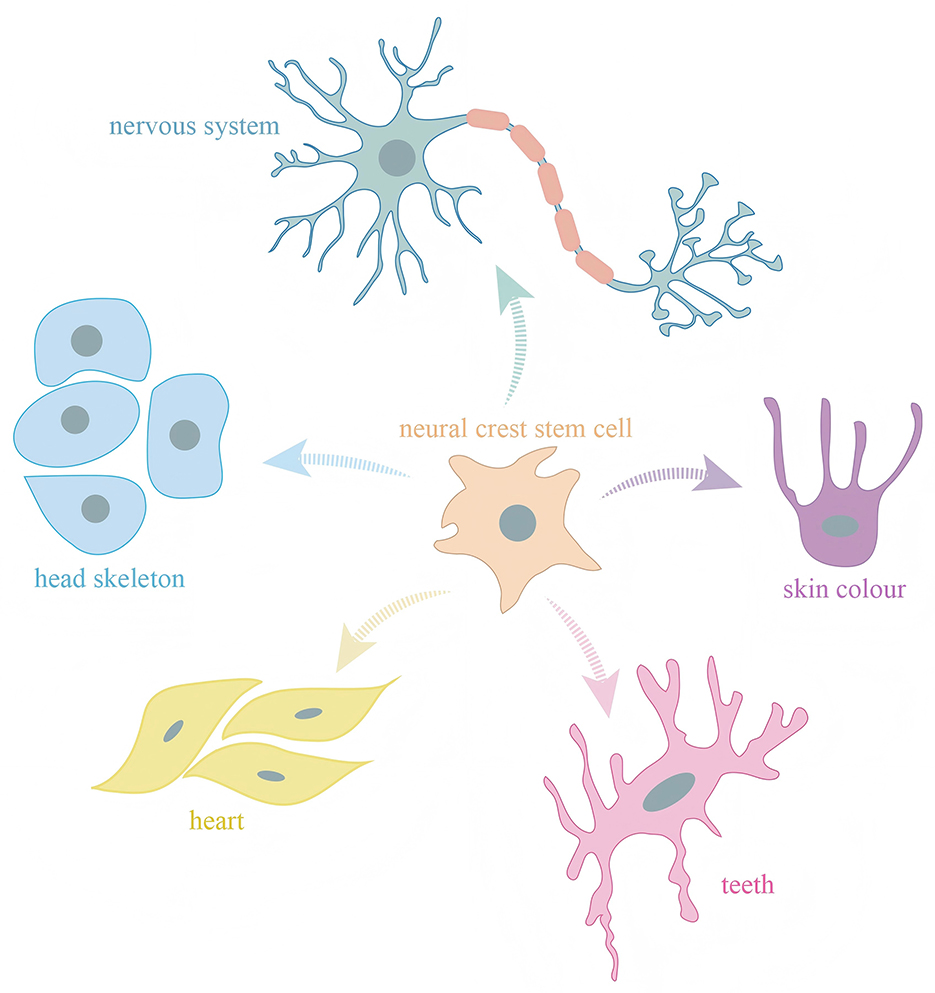
Figure 1. Neural crest cells possess multipotent differentiation potential and give rise to diverse cell types that contribute to a wide range of tissues and structures in vertebrates.
2.3 Neural crest cell migration
Following their specification, CNCCs undergo epithelial-to-mesenchymal transition (EMT), delaminate from the neural tube, and migrate along stereotypical pathways to populate the facial prominences and PA (Figure 2). This migration is tightly regulated by intrinsic transcriptional programs and extracellular cues. Key EMT regulators include SNAIL, SLUG, TWIST, and SOX10, which suppress epithelial traits and promote motility (18–20). In addition, SOX2 modulates EMT during neural crest development, acting as a rheostat that fine-tunes the epithelial-to-mesenchymal transition and thereby influences cranial neural crest cell migration (21). Guidance signals, such as semaphorins, ephrins, and chemokines, help direct CNCCs to their appropriate destinations (22, 23). Defective migration or guidance can result in craniofacial malformations due to failed colonization of target tissues. DiGeorge syndrome, commonly caused by 22q11.2 deletions affecting TBX1, exemplifies how disrupted CNCCs migration can impair pharyngeal arch development, leading to mandibular hypoplasia, cleft palate, and cardiovascular defects (24).
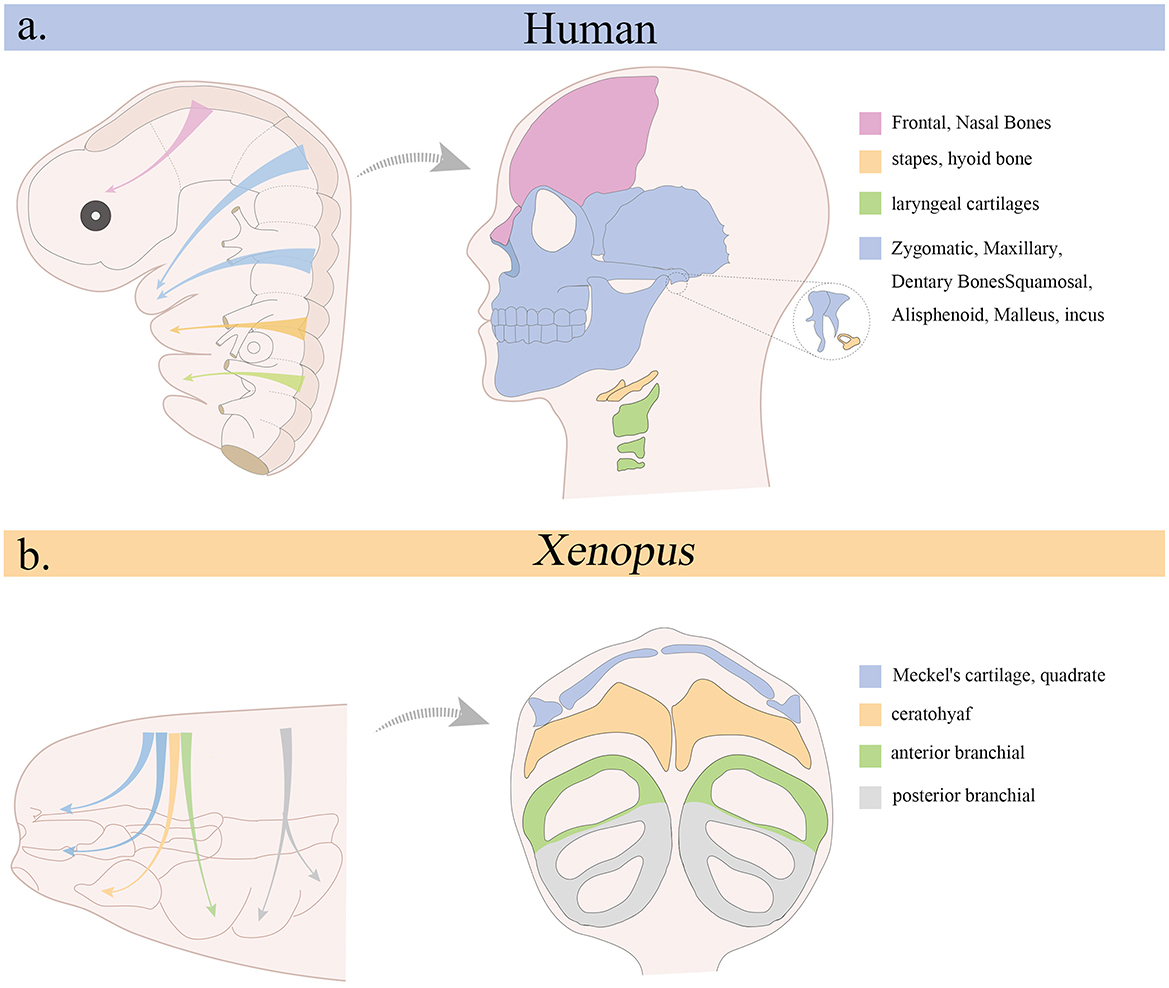
Figure 2. Migration of cranial neural crest and its contribution to cranial skeletal structures. (a) Origin of the cranial neural crest in the human embryo (left) and its derivatives in the adult head (right). (b) Migration pattern of cranial neural crest in Xenopus (left) and its contribution to cranial skeleton during the tadpole stage (right).
2.4 Pharyngeal arch formation and patterning
The pharyngeal arches are transient, segmented structures composed of ectoderm, mesoderm, endoderm, and migrating CNCCs, which differentiate into diverse derivatives including bone, cartilage, nerves, and muscle. In Xenopus, the pharyngeal arches begin to form at stage 23 and are sequentially patterned along the anterior-posterior axis (25). Patterning is controlled by combinatorial expression of transcription factors such as HOX, TBX1, and the DLX family, along with signaling cues from RA, endothelin-1, and FGF (26–28). The first arch gives rise to the maxilla, mandible, and associated muscles and nerves, while the second arch contributes to the hyoid bone and facial musculature (29). Aberrant arch patterning leads to structural and functional anomalies in the craniofacial region. Mutations in HOXA2 (30), for example, can cause duplication or transformation of arch derivatives, while defects in TBX1 underlie many of the craniofacial and cardiovascular features in DiGeorge syndrome (31).
2.5 Craniofacial skeletal morphogenesis
Craniofacial skeletal development involves the differentiation of CNCCs-derived mesenchyme into cartilage and bone, guided by both genetic and environmental cues. Two primary modes of ossification are employed: intramembranous ossification, which forms flat bones such as the frontal and parietal bones of the skull, and endochondral ossification, which produces bones at the cranial base and parts of the pharyngeal skeleton (32, 33). Key transcription factors regulating this process include RUNX2 for osteoblast differentiation, SOX9 for chondrogenesis, and COL2A1 as a cartilage matrix component (34–36). In Xenopus, these genes are expressed in a temporally and spatially conserved manner relative to mammals, making it an effective system for studying craniofacial skeletogenesis. Dysregulation in these pathways can lead to skeletal disorders such as craniosynostosis, characterized by the premature fusion of cranial sutures, often associated with mutations in FGFR2, TWIST1, or EFNB1 (37).
2.6 Facial prominence fusion and palatogenesis
The vertebrate face forms through the coordinated growth and fusion of several facial prominences: the frontonasal, maxillary, and mandibular processes. Failure of these prominences to merge appropriately leads to cleft lip and/or palate. Palatogenesis involves the elevation, growth, and midline fusion of the palatal shelves, derived from the maxillary prominences (38). This process is intricately regulated by signaling molecules such as Shh (epithelial patterning), TGF-β3 (palatal shelf fusion), and IRF6 (epithelial adhesion and remodeling) (39, 40). In Xenopus, palatal analog structures allow for the study of early patterning and epithelial-mesenchymal interactions relevant to human pathology. Mutations in TGF-β3 or IRF6 result in cleft palate or syndromic forms of orofacial clefting such as Van der Woude syndrome and Pierre Robin sequence, highlighting the clinical relevance of this developmental stage (40, 41).
2.7 Postnatal growth and remodeling
Although Xenopus does not undergo mammalian-like postnatal growth, early developmental processes related to bone remodeling and jaw patterning provide insights into later craniofacial maturation (42). Craniofacial bones continue to grow and reshape through the coordinated actions of osteoblasts and osteoclasts, influenced by hormonal, nutritional, and mechanical stimuli. Remodeling is essential for accommodating dental eruption, reshaping the jaw for mastication, and aligning cranial sutures. Disruptions in these processes can lead to postnatal conditions such as malocclusion, temporomandibular joint disorders, and facial asymmetry (43). Studying early regulators of osteogenic and chondrogenic remodeling in Xenopus can inform our understanding of these pathologies in humans (44).
3 Model advantages of Xenopus in craniofacial research
Currently, commonly used model organisms for studying craniofacial development include the mouse, chick, zebrafish, and Xenopus. As a mammalian model, the mouse remains the gold standard for investigating the genetic underpinnings of craniofacial disorders due to its extensive genetic toolbox, including conditional knockouts and CRISPR/Cas9-based genome editing (45, 46). However, the in utero development of mouse embryos presents several limitations: a relatively long gestational period (~21 days), high maintenance and experimental costs, and technical challenges associated with real-time imaging. These factors reduce the efficiency of using mice for high-throughput screening or live imaging–based studies of early craniofacial development (47, 48) (Table 2).
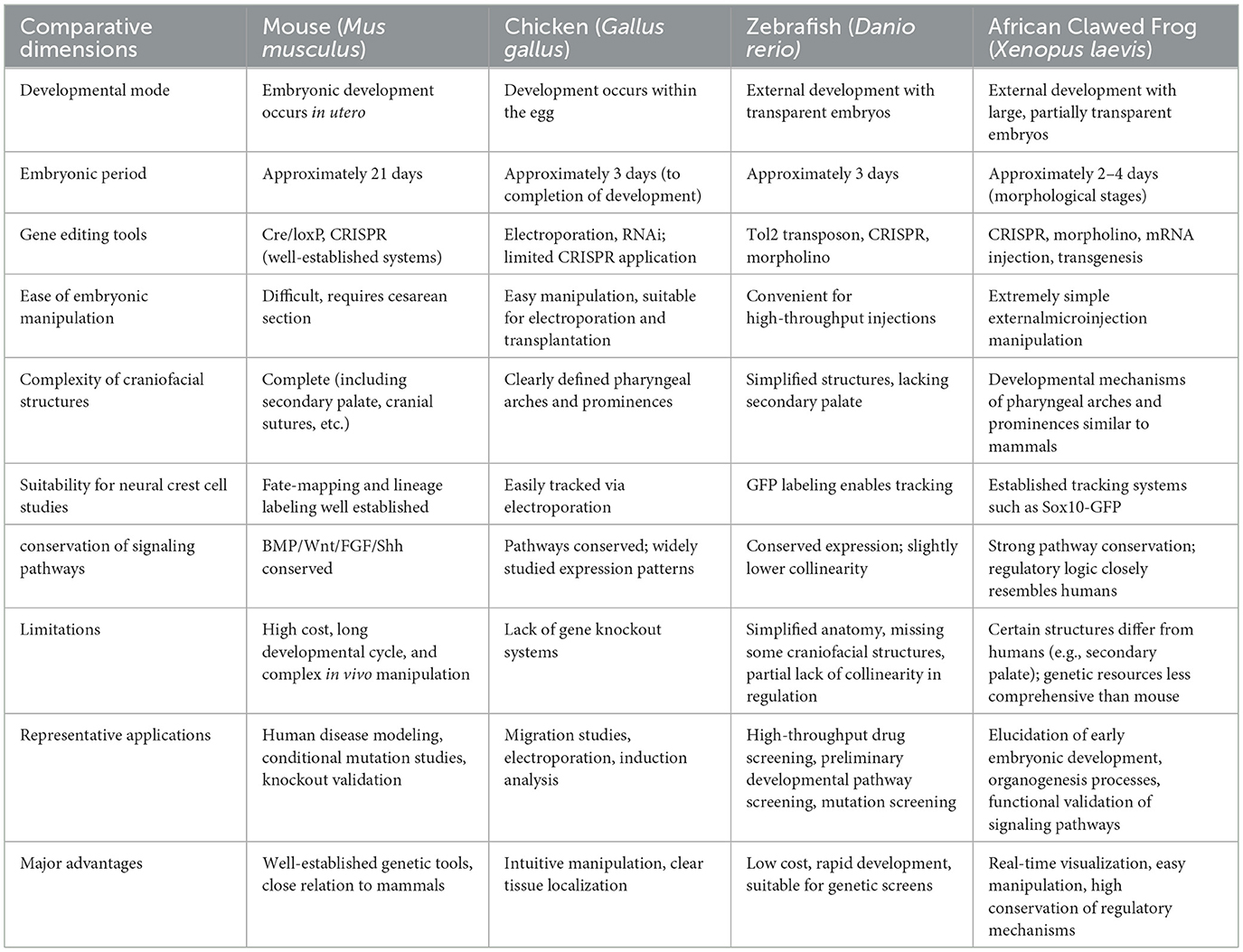
Table 2. Comparative overview of vertebrate model organisms used in craniofacial developmental studies.
Chick embryos provide ex utero development, micromanipulation feasibility, and established developmental atlases (49–51). However, systematic genetic tools (e.g., conditional KO) remain limited (52, 53).
Zebrafish embryos are transparent, develop ex utero, and possess a well-characterized genetic background, making them highly suitable for high-throughput compound screening and live imaging–based analyses (54). With a mature and versatile neural crest cell labeling system, zebrafish offer unique advantages for investigating the early migratory phases of neural crest development (55). However, as a basal teleost species, zebrafish display a relatively simplified craniofacial anatomy and lack several mammalian-specific structures, such as the secondary palate, thereby limiting their capacity to fully recapitulate complex human craniofacial disease phenotypes (56). Moreover, the zebrafish genome has undergone extensive chromosomal rearrangements and transposon-driven expansions (57), resulting in a marked loss of synteny with many human genomic loci (58, 59). These structural alterations disrupt the spatial architecture between cis-regulatory elements (e.g., enhancers) and their target genes, complicating the conservation of transcriptional regulation. Consequently, gene regulatory elements in zebrafish often exhibit a more dispersed genomic organization and context-dependent activity, which can hinder efforts to model higher-order regulatory dynamics underlying human craniofacial disorders (60, 61).
Compared to zebrafish, Xenopus, as an amphibian model organism, offers an optimal balance between evolutionary conservation and experimental accessibility (62, 63). Xenopus exhibits a higher degree of conservation with mammals in terms of gene sequences, chromosomal synteny, cis-regulatory architecture, and transcriptional regulatory networks (64, 65). These features make Xenopus a powerful and versatile model for dissecting the genetic and molecular mechanisms underlying craniofacial development and associated disorders (64, 66). Xenopus is increasingly recognized as a critical phylogenetic bridge between lower vertebrate models and mammals, particularly with respect to genome architecture and the organization of impose substantial psychological stress on patients transcriptional regulatory networks (44, 65).
Additionally, invertebrate models such as Drosophila melanogaster and Caenorhabditis elegans have played significant roles in elucidating conserved developmental pathways, including Notch, Hedgehog, and Wnt signaling (67, 68). However, due to the absence of craniofacial structures, neural crest cells, and vertebrate-specific tissues such as bone and cartilage, these organisms cannot serve as direct models for studying craniofacial development and its associated disorders.
4 Advances in modeling craniofacial disorders using the Xenopus system
Compared with other vertebrate models, Xenopus uniquely combines close evolutionary proximity to mammals with high experimental accessibility, enabling direct in vivo visualization and manipulation of CNCCs development. Its external development and large embryos facilitate single-cell–level imaging of CNCCs specification, migration, and differentiation—advantages that murine models lack. While zebrafish excel in real-time neural crest tracking, their craniofacial complexity and regulatory architecture are less representative of mammals. Amphibians thus strike an optimal balance, retaining key mammalian craniofacial features and gene networks while allowing high-throughput genetic perturbation and microsurgical studies, making CNCCs biology particularly tractable in Xenopus.
Clinically, craniofacial disorders are broadly classified into two categories based on the presence or absence of associated systemic anomalies: non-syndromic craniofacial malformations, which occur in isolation, and syndromic craniofacial anomalies, which are accompanied by defects in other organ systems.
4.1 Non-syndromic craniofacial malformations
Non-syndromic craniofacial malformations are defined by isolated structural defects confined to the craniofacial region, without involvement of other organ systems. Representative conditions in this category include non-syndromic orofacial clefts (NSOC), non-syndromic craniosynostosis (NCS), and isolated microtia. NSOC can be further subdivided into non-syndromic cleft lip (NSCL), cleft lip with or without cleft palate (NSCLP), and cleft palate alone (NSCP). NSOC are among the most common congenital malformations, which can lead to feeding difficulties, speech and language delays, and other developmental challenges. Common clinical features include cleft lip and/or palate and associated functional impairments. The condition can be caused by pathogenic variants in IRF6, RYK, TBX22, FGFR1, NAT2, or GSTT1.
Studies utilizing the Xenopus model have significantly advanced our understanding of NSOC pathogenesis. Inhibition of the RA biosynthetic enzyme RALDH2 or its receptor RARγ induces clefting of the upper lip, suggesting that RA signaling modulates the expression of homeobox genes such as LHX8 and MSX2, which are critical for boundary specification and tissue patterning during craniofacial development (69). Disrupting folate metabolism (via dhfr knockdown) combined with RA antagonism induces NSOC-like phenotypes. These include cleft lip, maxillofacial hypoplasia, and midfacial narrowing. Mechanistically, dhfr deficiency impairs cell proliferation, promotes DNA damage and apoptosis, and results in downregulation of FGF8, RARγ, and Wnt8. Notably, folate supplementation partially rescues these defects, underscoring a functional interaction between folate metabolism and RA signaling (70).
Additional studies have identified ISM1 as a crucial regulator of craniofacial morphogenesis. Loss of ISM1 function leads to cleft lip/palate, abnormal development of the pharyngeal arches, reduced lhx8 expression, and disruption of FGF8–Sprouty signaling, along with impaired expression of cell adhesion molecules such as integrin αvβ5. The identification of copy number variations and missense mutations in ISM1 among human patients further supports its conserved role in orofacial development (71).
Together, these findings highlight the utility of the Xenopus model in uncovering the genetic and signaling mechanisms underlying non-syndromic orofacial clefts, and support its relevance for studying multifactorial and environmentally modulated craniofacial anomalies.
4.2 Syndromic craniofacial anomalies
This group of disorders features craniofacial malformations as a central component of complex syndromes, often accompanied by abnormalities in other organ systems. In Xenopus models, studies of such syndromic conditions can be broadly categorized based on the affected systems:
4.2.1 Multiple craniofacial malformation syndromes
4.2.1.1 Treacher collins syndrome
Treacher Collins Syndrome (TCS) is an autosomal dominant disorder. Features of TCS include microtia with conductive hearing loss, slanting palpebral fissures with possible coloboma of the lateral part of the lower eyelids, midface hypoplasia, micrognathia, as well as sporadically cleft palate and choanal atresia or stenosis. TCS is caused by pathogenic variants in the TCOF1, POLR1D, POLR1C, and POLR1B genes, with mutations in TCOF1 accounting for more than 90% of cases (72, 73). The TCOF1 gene codes for the nucleolar phosphoprotein Treacle (known as xtreacle in Xenopus), which is essential for ribosomal DNA transcription. Mutations in TCOF1 lead to impaired rRNA synthesis, nucleolar stress, and apoptosis of neural crest cells (74, 75). In Xenopus, knockdown of xtreacle results in rDNA transcriptional defects consistent with the ribosome biogenesis abnormalities observed in human TCS patients. The resulting reduction in ribosome production mimics the nucleolar stress caused by TCOF1 haploinsufficiency in TCS (76).
4.2.1.2 Auriculocondylar syndrome
Auriculocondylar syndrome (ARCND) can be inherited in either an autosomal dominant or autosomal recessive manner. Features of ARCND include congenital ear clefts, mandibular condyle hypoplasia, temporomandibular joint abnormalities, micrognathia, small mouth, round facial appearance, and prominent cheeks. ARCND is caused by pathogenic variants in GNAI3, PLCB4, or EDN1 (77–79). Xenopus models have demonstrated that both wild-type and ACS-mutant forms of gαi3 can result in embryonic developmental defects. The mutant gαi3, which is unable to bind GTP, interferes with downstream signaling and disrupts neural crest differentiation by antagonizing Gαq activity. These findings highlight a conserved cross-species mechanism underlying ACS pathogenesis (80).
4.2.2 Multisystem syndromes
4.2.2.1 Floating-Harbor syndrome
Floating-Harbor Syndrome (FHS) is an autosomal dominant disorder. Features of FHS include short stature, facial dysmorphism, delayed bone mineralization, speech impairment, and intellectual disability. FHS is caused by heterozygous truncating variants in the SRCAP gene (81, 82). A Xenopus model of FHS was generated using morpholino oligonucleotides targeting srcap to mimic human C-terminal truncating mutations, inducing typical craniofacial malformations. SRCAP mutations impaired CNCCs migration and downregulated key genes, such as twist1 and sox9, suggesting neural crest dysfunction underlies the phenotype. The defects were rescued by wild-type SRCAP but not by the FHS-associated mutant. Mechanistically, SRCAP mutations disrupted nuclear localization, H2A.Z.2 deposition, and AT-rich enhancer activity, highlighting the role of H2A.Z.2 in neural crest development (8).
4.2.2.2 CHARGE syndrome
CHARGE syndrome is an autosomal dominant disorder that presents with a broad range of congenital malformations affecting multiple organ systems. Common clinical features include external ear malformations, cranial nerve dysfunction (often with facial palsy), semicircular canal dysplasia or aplasia, choanal atresia, and craniofacial abnormalities such as cleft lip and/or palate. In addition, anosmia, genital hypoplasia, congenital heart defects, and tracheoesophageal anomalies are frequently observed. The syndrome is caused by heterozygous loss-of-function mutations in the CHD7 gene (83). Patients often exhibit severe feeding difficulties, motor developmental delays, intellectual disabilities, and growth retardation. The majority of CHARGE syndrome cases result from haploinsufficiency of CHD7, a chromatin remodeling protein (84). In Xenopus embryos, knockdown of chd7 leads to impaired expression of NCCs effector genes (e.g., twist1, sox9), disruption of its interaction with the PBAF complex, and aberrant pharyngeal arch development, recapitulating phenotypes such as choanal atresia. These effects are associated with dysregulation of guidance molecules such as Sema3a (85, 86).
4.2.2.3 Axenfeld-Rieger syndrome
Axenfeld-Rieger Syndrome (ARS) is an autosomal dominant disorder. Features of ARS include anterior segment dysgenesis, glaucoma, dental anomalies, craniofacial dysmorphism, growth retardation, cardiovascular defects, redundant periumbilical skin, and pituitary defects leading to secondary endocrine disorders. ARS can be caused by pathogenic variants in the PITX2, FOXC1, and FOXO1A genes (87, 88). Studies using Xenopus models have demonstrated that foxc1 expression is regulated by VegT via the Nodal signaling pathway. Loss of foxc1 results in downregulation of adhesion molecules such as E-cadherin and the Ephrin/Eph system, causing mesodermal cell dissociation, axial shortening, and neural tube malformations. Injection of mutant FOXC1 mRNA can partially rescue these phenotypes (89).
4.2.2.4 Mowat-Wilson syndrome
Mowat-Wilson Syndrome (MWS) is an autosomal dominant disorder that presents with neural crest defects and distinctive craniofacial features, accompanied by a broad spectrum of multisystem abnormalities. Common clinical features include mild to severe intellectual disability, epilepsy, and congenital Hirschsprung disease. In addition, congenital malformations such as genital anomalies, congenital heart defects, corpus callosum agenesis, and ocular abnormalities are frequently observed. The syndrome results from heterozygous deletions or truncating mutations of the ZFHX1B gene (90). Patients typically exhibit a characteristic facial phenotype accompanied by a spectrum of multisystem abnormalities including mild to severe intellectual disability (ID), epilepsy, congenital Hirschsprung disease (HSCR), and frequently associated congenital malformations such as genital anomalies (most commonly hypospadias), congenital heart defects, corpus callosum agenesis, and ocular abnormalities (91). Knockdown of mi-2β in Xenopus reduces the expression of neural marker genes induced by zfhx1b and attenuates repression of BMP signaling pathway genes. Mutant ZFHX1B fails to effectively suppress BMP signaling, indicating that defective recruitment of the NuRD complex is a key pathogenic mechanism underlying craniofacial developmental defects (92).
4.2.2.5 Cardiofaciocutaneous syndrome
Cardiofaciocutaneous syndrome (CFC) is an autosomal dominant disorder that presents with multisystem congenital anomalies and moderate to severe intellectual disability. Common clinical features include postnatal growth retardation with relative macrocephaly, characteristic craniofacial features such as prominent forehead, bitemporal narrowing, absent eyebrows, downslanting palpebral fissures with epicanthal folds, depressed nasal bridge, bulbous nasal tip, and distal limb anomalies. In addition, skin abnormalities such as dry, hyperkeratotic, scaly skin, sparse curly hair, and capillary malformations are frequently observed, along with congenital heart defects, most commonly pulmonary valve stenosis and hypertrophic cardiomyopathy. The syndrome is the result of heterozygous pathogenic variants in BRAF, MAP2K1/MEK1, MAP2K2/MEK2, KRAS, or YWHAZ (93). In Xenopus models, injection of the S230W variant of YWHAZ mRNA induces severe craniofacial malformations. Co-expression with Craf enhances GFP-Erk2 phosphorylation and rescues mesodermal defects, implicating sustained activation of the RAF–ERK signaling pathway in the pathogenesis (94).
4.2.2.6 DDX3 syndrome
DDX3 syndrome is an X-linked dominant disorder that presents with neurodevelopmental delay, and intellectual disability, accompanied by a spectrum of multisystem abnormalities. Common clinical features include motor and language delay, autism spectrum disorder, epilepsy, and congenital brain and cardiac malformations (95), craniofacial anomalies are observed in the majority of affected individuals (96). The syndrome is the result of pathogenic variants in the DDX3 gene. In Xenopus embryos, ddx3 is specifically expressed at the neural plate border, pharyngeal arches (regions of neural crest cell migration), and head tissues. Knockdown of ddx3 results in craniofacial cartilage hypoplasia, such as reduced size of the ceratohyal cartilage, along with downregulation of neural crest cell markers, indicating impaired neural crest induction (97).
4.2.2.7 Fetal alcohol spectrum disorders
Fetal Alcohol Spectrum Disorders (FASD) affect 1%−5% of newborns in high-risk populations, characterized by smooth philtrum, thin vermilion border, and micrognathia. These craniofacial dysmorphisms correlate with neurocognitive deficits and require lifelong supportive care. Prenatal alcohol exposure is a critical factor causing FASD, which are characterized primarily by neurodevelopmental abnormalities and may be accompanied by craniofacial malformations, congenital organ defects, and growth retardation among other multisystem impairments (98). Exposure of Xenopus embryos to ethanol disrupts the expression of genes associated with later-stage NCCs migration. Supplementation with 5-methyltetrahydrofolate can partially rescue the observed phenotypes (99).
4.2.2.8 Kabuki syndrome
Kabuki Syndrome (KS) is a rare neuro-developmental disorder caused by variants in genes of histone modification, including KMT2D and KDM6A (100) it is considered a Mendelian disorder of epigenetic regulation, affecting multiple organ systems including the nervous, sensory, immune, cardiac, renal, and skeletal systems, significantly impacting patients' quality of life (101). Knockdown of the kmt2d gene in Xenopus models recapitulates phenotypes of mandibular, hyoid, and pharyngeal arch cartilage hypoplasia. The underlying mechanism involves defects in CNCCs formation and migration, evidenced by downregulation of marker genes such as twist, abnormal cell dispersal capacity, and decreased levels of histone modifications H3K4me1 and H3K27ac. Further studies revealed that kmt2d regulates CNCCs migration by modulating the secreted factor Sema3F, and overexpression of Sema3F can partially rescue the migration defects (102).
4.2.2.9 Wolf-Hirschhorn syndrome
Wolf-Hirschhorn syndrome (WHS) is araredisorderwithan estimated prevalence being around 1 in 50,000 births. The syndrome is caused by the deletion of a critical region (Wolf-Hirschhorn Syndrome Critical region—WHSCR) on chromosome 4p16.3. WHS is clinically characterized by pre-and postnatal growth restriction, hypotonia, intellectual disability, craniofacial dysmorphismand congenital fusion anomalies (103, 104), which are associated with aberrant development of CNCCs. Modeling this in Xenopus revealed that whsc1/tacc3 knockdown disrupts neural crest migration, directly linking genetic loss to craniofacial pathogenesis (105).
4.2.2.10 Musculocontractural Ehlers–Danlos syndrome
Musculocontractural Ehlers–Danlos syndrome (MC-EDS) is an autosomal recessive disease characterized primarily by increased connective tissue fragility, craniofacial structural abnormalities, congenital contractures, and impaired growth and development (106). It is caused by mutations in DSE or CHST14, which result in defects in craniofacial cartilage development. In Xenopus models, dse knockdown results in the downregulation of key EMT regulators in neural crest cells, including snail2 and twist1, and impairs cell migration on fibronectin substrates, providing a cellular dynamic basis for the craniofacial anomalies associated with the disease (107).
4.2.2.11 Branchio-oto-renal syndrome
Branchio-oto-renal (BOR) syndrome is an autosomal dominant disorder typically characterized by branchial cleft anomalies, ear developmental defects, and renal malformations. Additional clinical features may include abnormalities of the face, maxilla, ureters, and bladder, lacrimal system dysfunction, otitis media, and shoulder anomalies (108). About 40% of patients with BOS carry aberrations of EYA1 gene which is the most important cause of BOS. A total of 240 kinds of pathogenic variations of EYA1 have been reported in different populations so far, including frameshift, nonsense, missense, aberrant splicing, deletion and complex rearrangements (109). Knockdown of the eya1 gene in Xenopus leads to defects in otic vesicle development (inner ear structural abnormalities) and hypoplasia of branchial arch cartilages, such as Meckel's cartilage, thereby recapitulating the craniofacial skeletal malformations observed in patients. Mechanistically, eya1 deficiency downregulates neural crest cell marker genes, including sox10 and snail2, suggesting that eya1 regulates craniofacial development through modulation of neural crest cell migration and differentiation. Furthermore, wild-type human EYA1 mRNA can partially rescue the phenotype (110, 111).
4.2.2.12 Campomelic dysplasia
Campomelic Dysplasia (CD) is an autosomal dominant, perinatal lethal skeletal dysplasia characterized by a small chest and short long bones with bowing of the lower extremities. CD is the result of heterozygosity for mutations in the gene encoding the chondrogenesis master regulator, SOX9 (112). It is caused by SOX9 haploinsufficiency, manifesting as mandibular hypoplasia and craniosynostosis. Knockdown of sox9 in Xenopus suppresses the expression of neural crest effector genes such as snail2 and foxd3, resulting in complete loss of pharyngeal arch cartilages, including Meckel's cartilage, and malformation of the otic capsule, thereby recapitulating sensorineural hearing loss phenotypes (35).
4.2.2.13 Oral-facial-digital syndrome
Oral-facial-digital (OFD) represent a heterogeneous group of rare developmental disorders affecting the mouth, the face and the digits. Additional signs may involve brain, kidneys and other organs thus better defining the different clinical subtypes. With the exception of OFD types I and VIII, which are X-linked, the majority of OFDS is transmitted as an autosomal recessive syndrome. A number of genes have already found to be mutated in OFDS and most of the encoded proteins are predicted or proven to be involved in primary cilia/basal body function (113). Injection of morpholino oligonucleotides targeting ints13 into Xenopus embryos to knock down ints13 expression results in developmental defects including microcephaly, shortened body axis, and tail curvature. Additionally, differentiation of multiciliated cells (MCCs) is impaired, accompanied by reduced cilia number and functional abnormalities, thereby recapitulating craniofacial hypoplasia phenotypes (114)
4.2.2.14 Hamamy syndrome
Hamamy syndrome is a rare genetic disorder characterized by intellectual disability, sensorineural hearing loss, congenital cardiac anomalies with intraventricular conduction delay, hypopigmented microcytic anemia, and skeletal abnormalities of the long bones with recurrent fractures. Craniofacial features include severe hypertelorism and craniofacial skeletal dysmorphisms (115). Homozygous mutations in IRX5 are associated with this syndrome. Knockdown of irx5 in Xenopus disrupts the expression of the key chemokine cxcl12 involved in cranial neural crest cell migration, impeding pharyngeal arch mesenchymal condensation. Moreover, knockdown of cxcl12 partially rescues the phenotype, suggesting that IRX5 regulates neural crest migration through negative modulation of chemotactic signaling (1).
4.2.2.15 Nager syndrome
Nager syndrome is a rare human developmental disorder characterized by craniofacial defects including the downward slanting of the palpebral fissures, cleft palate, limb deformities, mandibular hypoplasia, hypoplasia or absence of thumbs, microretrognathia, and ankylosis of the temporomandibular joint. There is evidence of autosomal dominant and autosomal recessive inheritance for Nager syndrome, suggesting genetic heterogeneity. The majority of the described causes of Nager syndrome include pathogenic variants in the SF3B4 gene, which encodes a component of the spliceosome; therefore, the syndrome belongs to the spliceosomopathy group of diseases (116). Knockdown of sf3b4 in Xenopus results in downregulation of neural crest marker genes (sox10, twist1) and induces ectodermal apoptosis, suggesting that aberrant RNA splicing leads to craniofacial defects by impairing the survival of neural crest progenitor cells (117).
4.2.2.16 Coronal craniosynostosis
Craniosynostosis is one of the most common congenital cranial malformations affecting approximately 6/10,000 live births (118), with increasing incidence trends reported over the recent decades (119). The condition, which is characterized by premature fusion of one or more cranial sutures, is classified according to the type (e.g., sagittal, coronal, lambdoid) and/or number (single/multiple) of affected sutures. Involvement of the coronal suture is relatively rare and accounts for only 11%−13% of all cases of single-suture craniosynostosis. Injection of mRNA encoding human ZIC1 mutations (such as p. Ser388* and p. Glu402) into Xenopus embryos enhances the expression of the downstream target gene engrailed-2 (en-2), leading to aberrant neural crest signaling. Truncating variants, such as p. Ala437, retain the zinc finger domain and aberrantly activate En-2, thereby recapitulating craniosynostosis-like phenotypes in the model organism (120).
4.2.2.17 Smith-Magenis syndrome
Smith-Magenis syndrome (SMS) is a complex genetic disorder characterized by distinctive physical features, developmental delay, cognitive impairment, and a typical behavioral phenotype. SMS is caused by interstitial 17p11.2 deletions (90%), encompassing multiple genes and including the retinoic acid-induced 1 gene (RAI1), or by pathogenic variants in RAI1 itself (10%). RAI1 is a dosage-sensitive gene expressed in many tissues and acting as transcriptional regulator. The majority of individuals exhibit a mild-to-moderate range of intellectual disability (121). Knockdown of ra11 has been shown to reduce the expression of the neural crest cell marker tfap2a, leading to decreased craniofacial cartilage formation and increased apoptosis in the forebrain (122).
The Xenopus model has been employed to faithfully recapitulate a wide spectrum of craniofacial disease phenotypes—including cartilage hypoplasia, craniosynostosis, and hypertelorism—thus enabling precise in vivo and in vitro functional analyses such as gene knockdown/overexpression, phenotype rescue assays, pathway interrogation, and cellular behavior studies. These investigations have collectively revealed a shared pathological basis underlying craniofacial malformations: disrupted developmental programs of CNCCs, and the central roles of key signaling pathways including FGF, Wnt, BMP/TGFβ, RA, and Shh. Furthermore, critical cellular processes such as EMT, directed migration, differentiation, and apoptosis are shown to be perturbed. The Xenopus model therefore provides robust in vivo evidence and a unique experimental platform for elucidating disease mechanisms, identifying pathogenic genes, and developing therapeutic strategies.
5 Translational applications and challenges in clinical implementation
The clinical translational potential of Xenopus laevis in the study of craniofacial developmental disorders is becoming increasingly evident, offering multiple avenues for application. Its remarkable self-regeneration capacity has elucidated cartilage-dependent repair mechanisms involving matrix metalloproteinases such as Mmp1 and Mmp13, highlighting the possibility of developing therapeutic strategies that do not rely on exogenous interventions (123). Through gene editing and embryological approaches, the Xenopus model has also clarified the central role of the RSPO2–RNF43/ZNRF3–WNT signaling axis in limb regeneration, offering key molecular targets and theoretical frameworks for regenerative medicine (124, 125). Additionally, serotonergic signaling has been shown to mediate structural repair, suggesting that neurotransmitter pathways may serve as novel therapeutic avenues (126). Xenopus efficiently screens teratogens (e.g., e-cigarette-derived vanillin), which disrupt retinoic acid signaling to induce malformations (127, 128). Some recent studies have used Xenopus oocytes to test new targets and identify new active drug candidates. Romero et al. developed the novel peptide RgIA4 through high-throughput screening of >200 synthetic analogs on α9α10 nAChR-expressing Xenopus oocytes, identifying a candidate with 100-fold higher potency than conventional analgesics for human and rodent targets (129).
Despite the unique advantages of Xenopus in the study of craniofacial developmental disorders, several limitations continue to hinder its translational applications. First, Xenopus laevis is an allotetraploid species (130), and its genomic complexity renders gene knockout and editing more technically challenging than in diploid models, thereby limiting precise genetic manipulation (131). Second, notable physiological and immunological differences exist between Xenopus and mammals, particularly the absence of placental and lactational systems, which constrains its ability to faithfully model certain human syndromes. Moreover, Xenopus is not suitable for investigating complex behavioral phenotypes or modeling chronic adult-onset diseases over extended periods, areas where murine models remain indispensable. Although Xenopus excels in elucidating developmental mechanisms, its findings must often be validated in human organoids, cell lines, or mammalian systems to ensure clinical relevance and translatability (132). Finally, the experimental toolkit for Xenopus remains underdeveloped; tissue-specific Cre-lox systems, comprehensive reporter gene libraries, and high-resolution imaging technologies are still lacking, which limits its utility for high-throughput screening and fine-scale mechanistic dissection.
6 Conclusion
In summary, Xenopus serves as a powerful model to investigate craniofacial development and associated disorders, offering insights into fundamental mechanisms and translational relevance. Future work should focus on integrating high-resolution single-cell and multi-omics approaches with in vivo functional assays in Xenopus, to systematically link candidate human variants to developmental mechanisms and accelerate the discovery of diagnostic and therapeutic targets for craniofacial anomalies.
Author contributions
QK: Supervision, Writing – review & editing, Resources, Formal analysis, Writing – original draft, Methodology, Visualization, Conceptualization, Software, Data curation, Validation, Investigation. HP: Funding acquisition, Visualization, Project administration, Validation, Resources, Supervision, Data curation, Methodology, Investigation, Writing – original draft, Conceptualization. QZ: Validation, Methodology, Visualization, Data curation, Investigation, Funding acquisition, Resources, Supervision, Project administration, Conceptualization, Writing – original draft. HJ: Resources, Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Project administration, Visualization, Validation. XZ: Funding acquisition, Resources, Visualization, Project administration, Conceptualization, Validation, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Beijing Natural Science Foundation [Z240020]; the Peking University Medicine Plus X Pilot Program-Platform Construction Project [2024YXXLHPT012] and the Clinical Medicine Plus X - Young Scholars Project of Peking University, the Fundamental Research Funds for the Central Universities [PKU2024LCXQ005 and PKU2025PKULCXQ036].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bonnard C, Strobl AC, Shboul M, Lee H, Merriman B, Nelson SF, et al. Mutations in IRX5 impair craniofacial development and germ cell migration via SDF1. Nat Genet. (2012) 44:709–13. doi: 10.1038/ng.2259
2. Laberthonnière C, Delourme M, Chevalier R, Dion C, Ganne B, Hirst D, et al. In skeletal muscle and neural crest cells, SMCHD1 regulates biological pathways relevant for Bosma syndrome and facioscapulohumeral dystrophy phenotype. Nucleic Acids Res. (2023) 51:7269–87. doi: 10.1093/nar/gkad523
3. Trainor PA, Krumlauf R. Patterning the cranial neural crest: hindbrain segmentation and Hox gene plasticity. Nat Rev Neurosci. (2000) 1:116–24. doi: 10.1038/35039056
4. Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. (2009) 374:1773–85. doi: 10.1016/S0140-6736(09)60695-4
5. Shlobin NA, Baticulon RE, Ortega CA, Du L, Bonfield CM, Wray A, et al. Global epidemiology of craniosynostosis: a systematic review and meta-analysis. World Neurosurg. (2022) 164:413–23.e3. doi: 10.1016/j.wneu.2022.05.093
6. Alperovich, M., Tonello C, Mayes LC, Kahle KT. Non-syndromic craniosynostosis. Nat Rev Dis Primers. (2025) 11:24. doi: 10.1038/s41572-025-00607-4
7. Bowes JB, Snyder KA, Segerdell E, Gibb R, Jarabek C, Noumen E, et al. Xenbase: a Xenopus biology and genomics resource. Nucleic Acids Res. (2008) 36(Database issue):D761–7. doi: 10.1093/nar/gkm826
8. Medina-Cuadra L, Monsoro-Burq AH. Xenopus, an emerging model for studying pathologies of the neural crest. Curr Top Dev Biol. (2021) 145:313–48. doi: 10.1016/bs.ctdb.2021.03.002
9. Greenberg RS, Long HK, Swigut T, Wysocka J. Single amino acid change underlies distinct roles of H2A.Z subtypes in human syndrome. Cell. (2019) 178:1421–36.e24. doi: 10.1016/j.cell.2019.08.002
10. Winklbauer R. Mesoderm and endoderm internalization in the Xenopus gastrula. Curr Top Dev Biol. (2020) 136:243–70. doi: 10.1016/bs.ctdb.2019.09.002
11. Yoon J., Kumar V, Goutam RS, Kim SC, Park S, Lee U, et al. Bmp signal gradient modulates convergent cell movement via Xarhgef3.2 during gastrulation of Xenopus embryos. Cells. (2021) 11:44. doi: 10.3390/cells11010044
12. Samuel LJ, Latinkić BV. Early activation of FGF and nodal pathways mediates cardiac specification independently of Wnt/beta-catenin signaling. PLoS ONE. (2009) 4:e7650. doi: 10.1371/journal.pone.0007650
13. Houston DW, Wylie C. Maternal Xenopus Zic2 negatively regulates nodal-related gene expression during anteroposterior patterning. Development. (2005) 132:4845–55. doi: 10.1242/dev.02066
14. Lake BB, Kao KR. Early head specification in Xenopus laevis. ScientificWorldJournal. (2003) 3:655–76. doi: 10.1100/tsw.2003.54
15. Monsoro-Burq AH, Wang E, Harland R. Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev Cell. (2005) 8:167–78. doi: 10.1016/j.devcel.2004.12.017
16. Bronner-Fraser M. Gene Regulatory Interactions Mediating Neural Crest Formation. Wiley Online Library. (2008). doi: 10.1096/fasebj.22.1_supplement.85.1
17. Pegoraro C, Monsoro-Burq AH. Signaling and transcriptional regulation in neural crest specification and migration: lessons from Xenopus embryos. Wiley Interdiscip Rev Dev Biol. (2013) 2:247–59. doi: 10.1002/wdev.76
18. Sakai D, Suzuki T, Osumi N, Wakamatsu Y. Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development. (2006) 133:1323–33. doi: 10.1242/dev.02297
19. Honoré SM, Aybar MJ, Mayor R. Sox10 is required for the early development of the prospective neural crest in Xenopus embryos. Dev Biol. (2003) 260:79–96. doi: 10.1016/S0012-1606(03)00247-1
20. Aybar MJ, Nieto MA, Mayor R. Snail precedes slug in the genetic cascade required for the specification and migration of the Xenopus neural crest. Development. (2003) 130:483–94. doi: 10.1242/dev.00238
21. Mandalos N, Rhinn M, Granchi Z, Karampelas I, Mitsiadis T, Economides AN, et al. Sox2 acts as a rheostat of epithelial to mesenchymal transition during neural crest development. Front Physiol. (2014) 5:345. doi: 10.3389/fphys.2014.00345
22. Theveneau E, Mayor R. Neural crest migration: interplay between chemorepellents, chemoattractants, contact inhibition, epithelial-mesenchymal transition, and collective cell migration. Wiley Interdiscip Rev Dev Biol. (2012) 1:435–45. doi: 10.1002/wdev.28
23. Olesnicky Killian EC, Birkholz DA, Artinger KB. A role for chemokine signaling in neural crest cell migration and craniofacial development. Dev Biol. (2009) 333:161–72. doi: 10.1016/j.ydbio.2009.06.031
24. Duband J, Escot S, Fournier-Thibault C. SDF1-CXCR4 signaling: a new player involved in DiGeorge/22q11-deletion syndrome. Rare Dis. (2016) 4:1195050. doi: 10.1080/21675511.2016.1195050
25. Sater AK, Jacobson AG. The restriction of the heart morphogenetic field in Xenopus laevis. Dev Biol. (1990) 140:328–36. doi: 10.1016/0012-1606(90)90083-U
26. Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. (2002) 129:4605–11. doi: 10.1242/dev.129.19.4605
27. Miller CT, Yelon D, Stainier DY, Kimmel CB. Two endothelin 1 effectors, hand2 and bapx1, pattern ventral pharyngeal cartilage and the jaw joint. Development. (2003) 130:1353–65. doi: 10.1242/dev.00339
28. Abe M, Maeda T, Wakisaka S. Retinoic acid affects craniofacial patterning by changing Fgf8 expression in the pharyngeal ectoderm. Dev Growth Differ. (2008) 50:717–29. doi: 10.1111/j.1440-169X.2008.01069.x
29. Baltzinger M, Ori M, Pasqualetti M, Nardi I, Rijli FM. Hoxa2 knockdown in Xenopus results in hyoid to mandibular homeosis. Dev Dyn. (2005) 234:858–67. doi: 10.1002/dvdy.20567
30. Kitazawa T, Minoux M, Ducret S, Rijli FM. Different ectopic Hoxa2 expression levels in mouse cranial neural crest cells result in distinct craniofacial anomalies and homeotic phenotypes. J Dev Biol. (2022) 10:9. doi: 10.3390/jdb10010009
31. Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. (2001) 410:97–101. doi: 10.1038/35065105
32. Gross JB, Hanken J. Cranial neural crest contributes to the bony skull vault in adult Xenopus laevis: insights from cell labeling studies. J Exp Zool B Mol Dev Evol. (2005) 304:169–76. doi: 10.1002/jez.b.21028
33. Allas L, Boumédiene K, Baugé C. Epigenetic dynamic during endochondral ossification and articular cartilage development. Bone. (2019) 120:523–32. doi: 10.1016/j.bone.2018.10.004
34. Miura S, Hanaoka K, Togashi S. Skeletogenesis in Xenopus tropicalis: characteristic bone development in an anuran amphibian. Bone. (2008) 43:901–9. doi: 10.1016/j.bone.2008.07.005
35. Spokony RF, Aoki Y, Saint-Germain N, Magner-Fink E, Saint-Jeannet JP. The transcription factor Sox9 is required for cranial neural crest development in Xenopus. Development. (2002) 129:421–32. doi: 10.1242/dev.129.2.421
36. Kerney R, Hall BK, Hanken J. Regulatory elements of Xenopus col2a1 drive cartilaginous gene expression in transgenic frogs. Int J Dev Biol. (2010) 54:141–50. doi: 10.1387/ijdb.092848rk
37. Peskett E, Kumar S, Baird W, Jaiswal J, Li M, Patel P, et al. Analysis of the Fgfr2(C342Y) mouse model shows condensation defects due to misregulation of Sox9 expression in prechondrocytic mesenchyme. Biol Open. (2017) 6:223–31. doi: 10.1242/bio.022178
38. Llabata M, Faure L, Chacon B, Adameyko I, Slleri L. Disentangling the heterogeneity of the midfacial epithelium to elucidate the pathogenesis of cleft lip/palate. FASEB J. (2020) 34:1–1. doi: 10.1096/fasebj.2020.34.s1.05728
39. Lan Y, Xu J, Jiang R. Cellular and molecular mechanisms of palatogenesis. Curr Top Dev Biol. (2015) 115:59–84. doi: 10.1016/bs.ctdb.2015.07.002
40. Ke CY, Xiao WL, Chen CM, Lo LJ, Wong FH. IRF6 is the mediator of TGFβ3 during regulation of the epithelial mesenchymal transition and palatal fusion. Sci Rep. (2015) 5:12791. doi: 10.1038/srep12791
41. Won HJ, Kim JW, Won HS, Shin JO. Gene regulatory networks and signaling pathways in palatogenesis and cleft palate: a comprehensive review. Cells. (2023) 12:1954. doi: 10.3390/cells12151954
42. Slater BJ, Liu KJ, Kwan MD, Quarto N, Longaker MT. Cranial osteogenesis and suture morphology in Xenopus laevis: a unique model system for studying craniofacial development. PLoS ONE. (2009) 4:e3914. doi: 10.1371/journal.pone.0003914
43. Tunheim EG, Skallevold HE, Rokaya D. Role of hormones in bone remodeling in the craniofacial complex: a review. J Oral Biol Craniofac Res. (2023) 13:210–7. doi: 10.1016/j.jobcr.2023.01.009
44. Castillo H, Godoy F, Gilbert C, Aguilera F, Spicuglia S. Architecture and evolutionary conservation of Xenopus tropicalis osteoblast-specific regulatory regions shed light on bone diseases and early skeletal evolution. bioRxiv. (2023). doi: 10.1101/2023.08.30.555543
45. Rosen B, Schick J, Wurst W. Beyond knockouts: the International Knockout Mouse Consortium delivers modular and evolving tools for investigating mammalian genes. Mamm Genome. (2015) 26:456–66. doi: 10.1007/s00335-015-9598-3
46. Xiao D, Zhang W, Wang Q, Li X, Zhang Y, Rasouli J, et al. CRISPR-mediated rapid generation of neural cell-specific knockout mice facilitates research in neurophysiology and pathology. Mol Ther Methods Clin Dev. (2021) 20:755–64. doi: 10.1016/j.omtm.2021.02.012
47. Pritchard CEJ, Kroese LJ, Huijbers IJ. Direct generation of conditional alleles using CRISPR/Cas9 in mouse zygotes. Methods Mol Biol. (2017) 1642:21–35. doi: 10.1007/978-1-4939-7169-5_2
48. Shang R, Zhang H. Generation PBi of mouse conditional knockout alleles in one step using the i-GONAD method. Genome Res. (2021) 31:121–30. doi: 10.1101/gr.265439.120
49. Vergara MN, Canto-Soler MV. Rediscovering the chick embryo as a model to study retinal development. Neural Dev. (2012) 7:22. doi: 10.1186/1749-8104-7-22
50. Ribatti D, Annese T. Chick embryo in experimental embryology and more. Pathol Res Pract. (2023) 245:154478. doi: 10.1016/j.prp.2023.154478
51. Santagati F, Rijli FM. Cranial neural crest and the building of the vertebrate head. Nat Rev Neurosci. (2003) 4:806–18. doi: 10.1038/nrn1221
52. Antin PB, Fallon JF, Schoenwolf GC. The chick embryo rules (still)! Dev Dyn. (2004) 229:413. doi: 10.1002/dvdy.20014
53. Nakamoto M, Nakamoto C. Transposon-mediated stable suppression of gene expression in the developing chick retina. Methods Mol Biol. (2020) 2092:91–108. doi: 10.1007/978-1-0716-0175-4_8
54. Williams AL, Bohnsack BL. Multi-photon time lapse imaging to visualize development in real-time: visualization of migrating neural crest cells in zebrafish embryos. J Vis Exp. (2017) 126:56214. doi: 10.3791/56214-v
55. Xia Z, Tong X, Liang F, Zhang Y, Kuok C, Zhang Y, et al. Eif3ba regulates cranial neural crest development by modulating p53 in zebrafish. Dev Biol. (2013) 381:83–96. doi: 10.1016/j.ydbio.2013.06.009
56. Shull LC, Sen R, Menzel J, Goyama S, Kurokawa M, Artinger KB. The conserved and divergent roles of Prdm3 and Prdm16 in zebrafish and mouse craniofacial development. Dev Biol. (2020) 461:132–44. doi: 10.1016/j.ydbio.2020.02.006
57. Kawakami K. Transposon tools and methods in zebrafish. Dev Dyn. (2005) 234:244–54. doi: 10.1002/dvdy.20516
58. Fueyo R, Judd J, Feschotte C, Wysocka J. Roles of transposable elements in the regulation of mammalian transcription. Nat Rev Mol Cell Biol. (2022) 23:481–97. doi: 10.1038/s41580-022-00457-y
59. Sundaram V, Wysocka J. Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philos Trans R Soc Lond B Biol Sci. (2020) 375:20190347. doi: 10.1098/rstb.2019.0347
60. Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. (2013) 496:498–503. doi: 10.1038/nature12111
61. Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E, et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. (2004) 431:946–57. doi: 10.1038/nature03025
62. Harapas CR, Robinson KS, Lay K, Wong J, Moreno Traspas R, Nabavizadeh N, et al. DPP9 deficiency: an inflammasomopathy that can be rescued by lowering NLRP1/IL-1 signaling. Sci Immunol. (2022) 7:eabi4611. doi: 10.1126/sciimmunol.abi4611
63. Reversade B, Kuroda H, Lee H, Mays A, De Robertis EM. Depletion of Bmp2, Bmp4, Bmp7 and Spemann organizer signals induces massive brain formation in Xenopus embryos. Development. (2005) 132:3381–92. doi: 10.1242/dev.01901
64. Khokha MK, Loots GG. Strategies for characterising cis-regulatory elements in Xenopus. Brief Funct Genomic Proteomic. (2005) 4:58–68. doi: 10.1093/bfgp/4.1.58
65. Kaaij LJT, van der Weide RH, Ketting RF, de Wit E. Systemic loss and gain of chromatin architecture throughout zebrafish development. Cell Rep. (2018) 24:1–10.e4. doi: 10.1016/j.celrep.2018.06.003
66. Allende ML, Manzanares M, Tena JJ, Feijóo CG, Gómez-Skarmeta JL. Cracking the genome's second code: enhancer detection by combined phylogenetic footprinting and transgenic fish and frog embryos. Methods. (2006) 39:212–9. doi: 10.1016/j.ymeth.2005.12.005
67. Elouej S, Harhouri K, Le Mao M, Baujat G, Nampoothiri S, Kayserili H, et al. Loss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology. Nat Commun. (2020) 11:4589. doi: 10.1038/s41467-020-18146-9
68. Weinmaster G, Roberts VJ, Lemke G. A homolog of Drosophila Notch expressed during mammalian development. Development. (1991) 113:199–205. doi: 10.1242/dev.113.1.199
69. Kennedy AE, Dickinson AJ. Median facial clefts in Xenopus laevis: roles of retinoic acid signaling and homeobox genes. Dev Biol. (2012) 365:229–40. doi: 10.1016/j.ydbio.2012.02.033
70. Wahl SE, Kennedy AE, Wyatt BH, Moore AD, Pridgen DE, Cherry AM, et al. The role of folate metabolism in orofacial development and clefting. Dev Biol. (2015) 405:108–22. doi: 10.1016/j.ydbio.2015.07.001
71. Lansdon LA, Darbro BW, Petrin AL, Hulstrand AM, Standley JM, Brouillette RB, et al. Identification of Isthmin 1 as a novel clefting and craniofacial patterning gene in humans. Genetics. (2018) 208:283–96. doi: 10.1534/genetics.117.300535
72. de Peralta MS, Mouguelar VS, Sdrigotti MA, Ishiy FA, Fanganiello RD, Passos-Bueno MR, et al. CNBP ameliorates Treacher collins syndrome craniofacial anomalies through a pathway that involves redox-responsive genes. Cell Death Dis. (2016) 7:e2397. doi: 10.1038/cddis.2016.299
73. Marszałek-Kruk BA, Wójcicki P, Dowgierd K, Smigiel R. Treacher collins syndrome: genetics, clinical features and management. Genes. (2021) 12:1392. doi: 10.3390/genes12091392
74. Gonzales B, Yang H, Henning D, Valdez BC. Cloning and functional characterization of the Xenopus orthologue of the Treacher Collins syndrome (TCOF1) gene product. Gene. (2005) 359:73–80. doi: 10.1016/j.gene.2005.04.042
75. Calo E, Flynn RA, Martin L, Spitale RC, Chang HY, Wysocka J. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature. (2015) 518:249–53. doi: 10.1038/nature13923
76. Calo E, Gu B, Bowen ME, Aryan F, Zalc A, Liang J, et al. Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature. (2018) 554:112–7. doi: 10.1038/nature25449
77. Rieder MJ, Green GE, Park SS, Stamper BD, Gordon CT, Johnson JM, et al. A human homeotic transformation resulting from mutations in PLCB4 and GNAI3 causes auriculocondylar syndrome. Am J Hum Genet. (2012) 90:907–14. doi: 10.1016/j.ajhg.2012.04.002
78. Masotti C, Oliveira KG, Poerner F, Splendore A, Souza J, Freitas RS, et al. Auriculo-condylar syndrome: mapping of a first locus and evidence for genetic heterogeneity. Eur J Hum Genet. (2008) 16:145–52. doi: 10.1038/sj.ejhg.5201955
79. Li Q, Jiang Z, Zhang L, Cai S, Cai Z. Auriculocondylar syndrome: Pathogenesis, clinical manifestations and surgical therapies. J Formos Med Assoc. (2023) 122:822–42. doi: 10.1016/j.jfma.2023.04.024
80. Marivin A, Leyme A, Parag-Sharma K, DiGiacomo V, Cheung AY, Nguyen LT, et al. Dominant-negative Gα subunits are a mechanism of dysregulated heterotrimeric G protein signaling in human disease. Sci Signal. (2016) 9:ra37. doi: 10.1126/scisignal.aad2429
81. Dong S, Han J, Chen H, Liu T, Huen MSY, Yang Y, et al. The human SRCAP chromatin remodeling complex promotes DNA-end resection. Curr Biol. (2014) 24:2097–110. doi: 10.1016/j.cub.2014.07.081
82. Hood RL, Schenkel LC, Nikkel SM, Ainsworth PJ, Pare G, Boycott KM, et al. The defining DNA methylation signature of Floating-Harbor Syndrome. Sci Rep. (2016) 6:38803. doi: 10.1038/srep38803
83. Basson MA, van Ravenswaaij-Arts C. Functional insights into chromatin remodelling from studies on CHARGE syndrome. Trends Genet. (2015) 31:600–11. doi: 10.1016/j.tig.2015.05.009
84. Stathopoulou A, Wang P, Thellier C, Kelly RG, Zheng D, Scambler PJ. CHARGE syndrome-associated CHD7 acts at ISL1-regulated enhancers to modulate second heart field gene expression. Cardiovasc Res. (2023) 119:2089–105. doi: 10.1093/cvr/cvad059
85. Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. (2010) 463:958–62. doi: 10.1038/nature08733
86. Ufartes R, Schwenty-Lara J, Freese L, Neuhofer C, Möller J, Wehner P, et al. Sema3a plays a role in the pathogenesis of CHARGE syndrome. Hum Mol Genet. (2018) 27:1343–52. doi: 10.1093/hmg/ddy045
87. Ali Z, Charan P, Said JM, Stark Z. Axenfeld-Rieger syndrome as rare cause of umbilical abnormality. Ultrasound Obstet Gynecol. (2019) 54:276–7. doi: 10.1002/uog.20129
88. French CR. Mechanistic insights into Axenfeld-Rieger syndrome from Zebrafish foxc1 and pitx2 mutants. Int J Mol Sci. (2021) 22:10001. doi: 10.3390/ijms221810001
89. Cha JY, Birsoy B, Kofron M, Mahoney E, Lang S, Wylie C, et al. The role of FoxC1 in early Xenopus development. Dev Dyn. (2007) 236:2731–41. doi: 10.1002/dvdy.21240
90. Charney RM, Prasad MS, Juan-Sing C, Patel LJ, Hernandez JC, Wu J, et al. Mowat-Wilson syndrome factor ZEB2 controls early formation of human neural crest through BMP signaling modulation. Stem Cell Rep. (2023) 18:2254–67. doi: 10.1016/j.stemcr.2023.10.002
91. Ivanovski I, Djuric O, Caraffi SG, Santodirocco D, Pollazzon M, Rosato S, et al. Phenotype and genotype of 87 patients with Mowat-Wilson syndrome and recommendations for care. Genet Med. (2018) 20:965–75. doi: 10.1038/gim.2017.221
92. Verstappen G, van Grunsven LA, Michiels C, Van de Putte T, Souopgui J, Van Damme J, et al. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum Mol Genet. (2008) 17:1175–83. doi: 10.1093/hmg/ddn007
93. Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, et al. The cardiofaciocutaneous syndrome. J Med Genet. (2006) 43:833–42. doi: 10.1136/jmg.2006.042796
94. Popov IK, Hiatt SM, Whalen S, Keren B, Ruivenkamp C, van Haeringen A, et al. A YWHAZ variant associated with cardiofaciocutaneous syndrome activates the RAF-ERK pathway. Front Physiol. (2019) 10:388. doi: 10.3389/fphys.2019.00388
95. von Mueffling A, Garcia-Forn M, De Rubeis S. DDX3X syndrome: from clinical phenotypes to biological insights. J Neurochem. (2024) 168:2147–54. doi: 10.1111/jnc.16174
96. Dikow N, Granzow M, Graul-Neumann LM, Karch S, Hinderhofer K, Paramasivam N, et al. DDX3X mutations in two girls with a phenotype overlapping Toriello-Carey syndrome. Am J Med Genet A. (2017) 173:1369–73. doi: 10.1002/ajmg.a.38164
97. Perfetto M, Xu X, Lu C, Shi Y, Yousaf N, Li J, et al. The RNA helicase DDX3 induces neural crest by promoting AKT activity. Development. (2021) 148:dev184341. doi: 10.1242/dev.184341
98. Popova S, Charness ME, Burd L, Crawford A, Hoyme HE, Mukherjee RAS, et al. Fetal alcohol spectrum disorders. Nat Rev Dis Primers. (2023) 9:11. doi: 10.1038/s41572-023-00420-x
99. Shi Y, Li J, Chen C, Gong M, Chen Y, Liu Y, et al. 5-Mehtyltetrahydrofolate rescues alcohol-induced neural crest cell migration abnormalities. Mol Brain. (2014) 7:67. doi: 10.1186/s13041-014-0067-9
100. Shpargel KB, Starmer J, Wang C, Ge K, Magnuson T. UTX-guided neural crest function underlies craniofacial features of Kabuki syndrome. Proc Natl Acad Sci U S A. (2017) 114:E9046–E9055. doi: 10.1073/pnas.1705011114
101. Mertens M, Khalife L, Ma X, Bodamer O. Animal models of Kabuki syndrome and their applicability to novel drug discovery. Expert Opin Drug Discov. (2025) 20:253–65. doi: 10.1080/17460441.2025.2457624
102. Schwenty-Lara J, Nehl D, Borchers A. The histone methyltransferase KMT2D, mutated in Kabuki syndrome patients, is required for neural crest cell formation and migration. Hum Mol Genet. (2020) 29:305–19. doi: 10.1093/hmg/ddz284
103. Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. (2005) 21:188–95. doi: 10.1016/j.tig.2005.01.008
104. Wiel LC, Bruno I, Barbi E, Sirchia F. From Wolf-Hirschhorn syndrome to NSD2 haploinsufficiency: a shifting paradigm through the description of a new case and a review of the literature. Ital J Pediatr. (2022) 48:72. doi: 10.1186/s13052-022-01267-w
105. Mills A, Bearce E, Cella R, Kim SW, Selig M, Lee S, et al. Wolf-Hirschhorn syndrome-associated genes are enriched in motile neural crest cells and affect craniofacial development in Xenopus laevis. Front Physiol. (2019) 10:431. doi: 10.3389/fphys.2019.00431
106. Syx D, Van Damme T, Symoens S, Maiburg MC, van de Laar I, Morton J, et al. Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum Mutat. (2015) 36:535–47. doi: 10.1002/humu.22774
107. Gouignard N, Maccarana M, Strate I, von Stedingk K, Malmström A, Pera EM. Musculocontractural Ehlers-Danlos syndrome and neurocristopathies: dermatan sulfate is required for Xenopus neural crest cells to migrate and adhere to fibronectin. Dis Model Mech. (2016) 9:607–20. doi: 10.1242/dmm.024661
108. Kochhar A, Fischer SM, Kimberling WJ, Smith RJ. Branchio-oto-renal syndrome. Am J Med Genet A. (2007) 143a:1671–8. doi: 10.1002/ajmg.a.31561
109. Pao J, D'Arco F, Clement E, Picariello S, Moonis G, Robson CD, et al. Re-examining the cochlea in Branchio-Oto-Renal syndrome: genotype-phenotype correlation. AJNR Am J Neuroradiol. (2022) 43:309–14. doi: 10.3174/ajnr.A7396
110. Li Y, Manaligod JM, Weeks DL. EYA1 mutations associated with the branchio-oto-renal syndrome result in defective otic development in Xenopus laevis. Biol Cell. (2010) 102:277–92. doi: 10.1042/BC20090098
111. Moody SA, Neilson KM, Kenyon KL, Alfandari D, Pignoni F. Using Xenopus to discover new genes involved in branchiootorenal spectrum disorders. Comp Biochem Physiol C Toxicol Pharmacol. (2015) 178:16–24. doi: 10.1016/j.cbpc.2015.06.007
112. Ming Z, Vining B, Bagheri-Fam S, Harley V. SOX9 in organogenesis: shared and unique transcriptional functions. Cell Mol Life Sci. (2022) 79:522. doi: 10.1007/s00018-022-04543-4
113. Thauvin-Robinet C, Lee JS, Lopez E, Herranz-Pérez V, Shida T, Franco B, et al. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat Genet. (2014) 46:905–11. doi: 10.1038/ng.3031
114. Mascibroda LG, Shboul M, Elrod ND, Colleaux L, Hamamy H, Huang KL, et al. INTS13 variants causing a recessive developmental ciliopathy disrupt assembly of the integrator complex. Nat Commun. (2022) 13:6054. doi: 10.1038/s41467-022-33547-8
115. Duman SB, Dedeoglu N, Arikan B, Altun O. Sphenoid sinus agenesis and sella turcica hypoplasia: very rare cases of two brothers with Hamamy syndrome. Surg Radiol Anat. (2020) 42:1377–80. doi: 10.1007/s00276-020-02558-9
116. Ulhaq ZS, You MS, Yabe T, Takada S, Chen JK, Ogino Y, et al. Fgf8 contributes to the pathogenesis of Nager syndrome. Int J Biol Macromol. (2024) 280(Pt 3):135692. doi: 10.1016/j.ijbiomac.2024.135692
117. Devotta A, Juraver-Geslin H, Gonzalez JA, Hong CS, Saint-Jeannet JP. Sf3b4-depleted Xenopus embryos: a model to study the pathogenesis of craniofacial defects in Nager syndrome. Dev Biol. (2016) 415:371–82. doi: 10.1016/j.ydbio.2016.02.010
118. Sacks D, Baxter B, Campbell BCV, Carpenter JS, Cognard C, Dippel D, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke. (2018) 13:612–32. doi: 10.1016/j.jvir.2017.11.026
119. Tønne E, Due-Tønnessen BJ, Wiig U, Stadheim BF, Meling TR, Helseth E, et al. Epidemiology of craniosynostosis in Norway. J Neurosurg Pediatr. (2020) 26:68–75. doi: 10.3171/2020.1.PEDS2051
120. Twigg SR, Forecki J, Goos JA, Richardson IC, Hoogeboom AJ, van den Ouweland AM, et al. Gain-of-function mutations in ZIC1 are associated with coronal craniosynostosis and learning disability. Am J Hum Genet. (2015) 97:378–88. doi: 10.1016/j.ajhg.2015.07.007
121. Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet. (2003) 33:466–8. doi: 10.1038/ng1126
122. Tahir R, Kennedy A, Elsea SH, Dickinson AJ. Retinoic acid induced-1 (Rai1) regulates craniofacial and brain development in Xenopus. Mech Dev. (2014) 133:91–104. doi: 10.1016/j.mod.2014.05.004
123. Pinet K, Deolankar M, Leung B, McLaughlin KA. Adaptive correction of craniofacial defects in pre-metamorphic Xenopus laevis tadpoles involves thyroid hormone-independent tissue remodeling. Development. (2019) 146:dev175893. doi: 10.1242/dev.175893
124. Szenker-Ravi E, Altunoglu U, Leushacke M, Bosso-Lefèvre C, Khatoo M, Thi Tran H, et al. RSPO2 inhibition of RNF43 and ZNRF3 governs limb development independently of LGR4/5/6. Nature. (2018) 557:564–9. doi: 10.1038/s41586-018-0118-y
125. McKeown CR, Sharma P, Sharipov HE, Shen W, Cline HT. Neurogenesis is required for behavioral recovery after injury in the visual system of Xenopus laevis. J Comp Neurol. (2013) 521:2262–78. doi: 10.1002/cne.23283
126. Familia JS, Pinet K, McLaughlin KAJTFJ. Role of serotonin signaling in the remodeling of thioridazine-induced craniofacial deformities in Xenopus laevis pre-metamorphic tadpoles. FASEB J. (2017) 31:lb143. doi: 10.1096/fasebj.31.1_supplement.lb143
127. Dickinson AJG, Turner SD, Wahl S, Kennedy AE, Wyatt BH, Howton DA. E-liquids and vanillin flavoring disrupts retinoic acid signaling and causes craniofacial defects in Xenopus embryos. Dev Biol. (2022) 481:14–29. doi: 10.1016/j.ydbio.2021.09.004
128. Gao J, Shen W. Xenopus in revealing developmental toxicity and modeling human diseases. Environ Pollut. (2021) 268(Pt B):115809. doi: 10.1016/j.envpol.2020.115809
129. Romero HK, Christensen SB, Di Cesare Mannelli L, Gajewiak J, Ramachandra R, Elmslie KS, et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc Natl Acad Sci U S A. (2017) 114:E1825–32. doi: 10.1073/pnas.1621433114
130. Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S, et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. (2016) 538:336–43. doi: 10.1038/nature19840
131. DeLay BD, Corkins ME, Hanania HL, Salanga M, Deng JM, Sudou N, et al. Tissue-specific gene inactivation in Xenopus laevis: knockout of lhx1 in the kidney with CRISPR/Cas9. Genetics. (2018) 208:673–86. doi: 10.1534/genetics.117.300468
Keywords: Xenopus, craniofacial developmental disorders, CNCCs, disease model, gene editing, signaling pathway
Citation: Kong Q, Peng H, Zhao Q, Jiang H and Zhu X (2025) The Xenopus model as a tool for investigating craniofacial developmental disorders. Front. Med. 12:1671687. doi: 10.3389/fmed.2025.1671687
Received: 23 July 2025; Accepted: 25 August 2025;
Published: 08 September 2025.
Edited by:
Ian James Martins, University of Western Australia, AustraliaReviewed by:
Wanhua Shen, Hangzhou Normal University, ChinaNikolaos Mandalos, Medical School of University of Athens, Greece
Copyright © 2025 Kong, Peng, Zhao, Jiang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuechen Zhu, emh1eGNAYmptdS5lZHUuY24=; Hongwei Jiang, amlhbmdod0BoYXVzdC5lZHUuY24=