 Bin Xu1
Bin Xu1 Chi Liu
Chi Liu Xiangrong Song
Xiangrong Song- 1Department of Orthopedics, Deyang Fifth Hospital, Deyang, Sichuan, China
- 2Graduate School of Xinjiang Medical University, Ürümqi, Xinjiang, China
- 3Department of Nephrology and Institute of Nephrology, Sichuan Provincial People’s Hospital, Sichuan Clinical Research Centre for Kidney Diseases, Chengdu, China
- 4Department of Nephrology, Deyang Sixth People’s Hospital, Deyang, Sichuan, China
Chronic kidney disease-mineral and bone disorder (CKD-MBD) is recognized as a systemic syndrome that manifests with a range of complications including mineral dysregulation, skeletal abnormalities, and vascular calcification (VC). Recent research has increasingly pointed toward immune dysregulation as a pivotal factor in the development and progression of this disorder. The current review endeavors to consolidate the latest findings regarding how chronic inflammation, dysfunction of immune cells, and disturbances in the gut-kidney axis contribute to the progression of CKD-MBD. Central to the mechanisms at play are pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin (IL)-6, which are found to facilitate bone resorption through the activation of the receptor activator of NF-kappaB ligand (RANKL)/receptor activator of nuclear factor-kappa B (RANK)/osteoprotegerin (OPG) signaling pathway. Furthermore, macrophage-induced VC is linked to the activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome. Additionally, an imbalance between osteoblasts and osteoclasts, driven by uremic toxins, exacerbates the skeletal manifestations of the disorder. Despite the availability of current therapeutic options, including phosphate binders and vitamin D analogs, these treatments fall short in adequately addressing the immune-mediated aspects of CKD-MBD, indicating an urgent need for innovative strategies that effectively target inflammatory pathways, inhibit sclerostin, or modulate fibroblast growth factor (FGF)-23 levels. Emerging preclinical studies have shown that sodium-glucose cotransporter 2 (SGLT2) inhibitors and anti-sclerostin antibodies hold significant promise in lessening VC and enhancing bone health. However, translating these findings into clinical application encounters hurdles related to the diversity of patient populations and the dependence on surrogate endpoints for efficacy. This review emphasizes the critical need for incorporating immune-centric strategies into the management of CKD-MBD. It advocates for the development of biomarker-driven, personalized therapies and highlights the importance of conducting longitudinal studies to bridge the existing gaps in knowledge and improve patient outcomes.
1 Introduction
Chronic kidney disease (CKD) represents a progressive disorder marked by ongoing kidney damage or a diminished glomerular filtration rate that persists for more than 3 months (1). It is frequently indicated by the presence of albuminuria or structural abnormalities in the kidneys. CKD poses a significant global health concern, impacting more than 10% of the global population, with its prevalence continuing to rise due to aging demographics and increasing risk factors such as diabetes and hypertension (2). In the Asian region, approximately 434.3 million adults are affected by CKD, with China and India together representing 69.1% of the region’s total cases, indicating notable regional disparities (3). The global impact of this disease disproportionately burdens lower socioeconomic groups, who face higher prevalence rates, limited access to medical care, and poorer health outcomes, thus highlighting CKD as a critical equity issue (4). The increasing prevalence of this epidemic, coupled with its links to cardiovascular morbidity and mortality, stresses the urgent requirement for further exploration of its underlying mechanisms, such as chronic kidney disease-mineral and bone disorder (CKD-MBD), to guide prevention and management approaches.
According to the Kidney Disease Improving Global Outcomes (KDIGO) guidelines, CKD-MBD is a systemic syndrome characterized by abnormalities in bone histology and ectopic calcification. This condition fundamentally arises from an imbalance in mineral metabolism parameters, including calcium, phosphorus, parathyroid hormone (PTH), vitamin D, and fibroblast growth factor-23 (FGF-23). Such imbalances impede the bone remodeling process and promote vascular calcification (VC), which significantly impacts patients with stage 5 CKD (5–7). This condition is almost universally observed in patients with advanced CKD, leading to increased risks of fractures, cardiovascular incidents, and higher mortality rates (8). The importance of CKD-MBD stems from its function as a crucial factor in poor health outcomes, with VC–arising from phosphate retention and inflammation–being a major contributor to cardiovascular death among CKD patients (9). Additionally, beyond the susceptibility to bone fractures, CKD-MBD indicates a wider systemic issue, aggravated by new elements such as inflammation and alterations in gut microbiota, which further challenge its treatment. Prompt detection and focused treatment approaches are essential, as CKD-MBD not only deteriorates quality of life but also highlights the pressing need for novel therapeutic methods to alleviate its significant clinical ramifications.
Chronic kidney disease-mineral and bone disorder exhibits a complex interplay with the immune system, driven by chronic inflammation and gut dysbiosis, which significantly influence mineral metabolism and cardiovascular outcomes in CKD patients. Recent evidence underscores that CKD triggers persistent immune activation through uremic toxins, oxidative stress (OS), and microbial imbalances, fostering an inflammatory environment that exacerbates bone resorption and VC, hallmark features of CKD-MBD (5). Pro-inflammatory cytokines, such as Interleukin (IL)-1 and tumor necrosis factor-α (TNF-α), are pivotal in promoting these pathological processes, linking immune dysregulation to skeletal and vascular complications (10). Furthermore, gut dysbiosis compromises intestinal barrier integrity, increasing bacterial product translocation, which amplifies systemic inflammation and perpetuates CKD-MBD progression (5). FGF-23, a critical regulator of mineral metabolism, also interacts with immune cells, modulating inflammatory responses and highlighting a bidirectional relationship between CKD-MBD and immunity (11). This intricate connection suggests that immune-mediated pathways, including inflammation and osteoimmunological mechanisms, are central to CKD-MBD pathogenesis, offering potential therapeutic targets to mitigate its impact. These findings emphasize the need for integrated approaches addressing both immune and mineral dysregulation to improve outcomes in CKD.
This review examines advances in understanding the immune mechanisms of CKD-MBD and explores emerging therapies to mitigate its clinical burden. This review explores the advancements in understanding the immune mechanisms underlying CKD-MBD and investigates emerging therapies aimed at alleviating its clinical burden. It synthesizes the latest evidence from PubMed/Medline, Web of Science, and Cochrane Library databases up to January 2025, focusing on the immune mechanisms, inflammation, and therapeutic strategies related to CKD-MBD.
2 Pathophysiology of CKD-MBD
2.1 Mineral metabolism dysregulation
CKD disrupts systemic mineral homeostasis, primarily through impaired phosphate excretion and reduced renal synthesis of active vitamin D. Hyperphosphatemia, a hallmark of CKD-MBD, triggers FGF-23 elevation to promote urinary phosphate excretion, but progressive renal failure limits compensatory mechanisms, exacerbating phosphate retention (12). CKD also impacts the parathyroid’s internal circadian clock, contributing to the hyperplasia of the parathyroid gland in these patients (13). The calcium-sensing receptor (CaSR), found within the parathyroid gland, acts as the primary regulator of PTH release (14). Notably, recent findings indicate that the CaSR possesses a phosphate-binding site; when phosphate binds to this site, it modifies the receptor’s configuration, driving it into an inactive state and consequently initiating PTH secretion (15). Small fluctuations in plasma ionized calcium are promptly adjusted through the movement of calcium on the surface of bones. In contrast, plasma phosphate levels tend to vary more significantly and respond more slowly to conditions of hyperphosphatemia (16). Concurrently, reduced renal 1α-hydroxylase activity causes vitamin D deficiency, impairing intestinal calcium absorption and contributing to hypocalcemia (17). Secondary hyperparathyroidism (sHPT) develops as hypocalcemia and vitamin D deficiency stimulate PTH secretion, further worsening bone and vascular pathology (5). Emerging evidence also highlights the role of uremic toxins such as indoxyl sulfate (IS) in suppressing klotho expression, amplifying FGF-23 resistance and perpetuating mineral dysregulation (18). These interconnected disturbances create a vicious cycle, driving CKD-MBD progression (19).
2.2 Skeletal abnormalities
Chronic kidney disease-mineral and bone disorder induces heterogeneous bone disorders, ranging from high-turnover osteitis fibrosa to low-turnover adynamic bone disease (ABD). Early CKD stages often exhibit ABD, characterized by suppressed osteoblast activity due to uremic toxin-mediated inhibition of Wnt/β-catenin signaling and elevated sclerostin levels (20). As CKD progresses, sustained sHPT may override these inhibitory signals, leading to excessive bone resorption and osteitis fibrosa (21). Osteomalacia, marked by defective mineralization, is linked to vitamin D deficiency and elevated FGF-23, which impair phosphate availability for bone matrix formation (22). Notably, FGF-23 directly suppresses osteocyte differentiation, exacerbating bone fragility (22). These abnormalities collectively increase fracture risk and correlate with poor clinical outcomes, including cardiovascular mortality (23). In CKD-MBD, the primary clinical features of osteomalacia are associated with defects in bone mineralization. This is particularly evident in tumor-induced osteomalacia (TIO), which arises from hypophosphatemia and FGF-23-related mechanisms, resulting in dysfunction and limited activity (24, 25). High-turnover bone disease is a prevalent form of renal osteodystrophy, characterized by accelerated bone formation and resorption. Elevated PTH levels are indicative of this condition and correlate with bone density and fracture risk (26). ABD represents the most common form of renal osteodystrophy; although asymptomatic, it is closely linked to a poor prognosis, including an increased fracture risk. Fractures can occur independently of VC or premature death, and treatment with anti-resorptive agents may not effectively mitigate this risk. Furthermore, a low bone turnover state can be diagnosed using markers such as the sclerostin/iPTH ratio, which correlates with impaired bone mass and quality (27). The core characteristics of osteoporosis involve the deterioration of bone quality and density, which are directly related to mineral metabolism disorders, such as elevated PTH levels and decreased bone material quality (21, 28, 29).
2.3 Vascular calcification
Vascular calcification, a hallmark of CKD-MBD, arises from the osteogenic transformation of vascular smooth muscle cells (VSMCs) driven by hyperphosphatemia, inflammation, and uremic toxins. Elevated phosphate activates RUNX2 in VSMCs, promoting calcium-phosphate deposition and arterial stiffening (23). Pro-inflammatory cytokines (e.g., TNF-α) and OS further enhance VC by upregulating bone morphogenetic proteins (BMPs) and downregulating calcification inhibitors like matrix Gla protein (17). Uremic toxins, such as IS, exacerbate VSMC apoptosis and extracellular vesicle release, accelerating microcalcification (5). Clinically, VC is strongly associated with cardiovascular mortality, as calcified vessels impair hemodynamics and increase cardiac afterload (30). Emerging biomarkers, including miR-125b-2-3p and sulfatase 1 (SULF1), show promise in predicting VC severity, highlighting opportunities for early intervention (23).
Figure 1 delineates the dysregulated mineral metabolism axis in CKD, wherein FGF-23 induces parathyroid hyperplasia and downregulates calcium-sensing receptors (CaSR), abrogating calcium-mediated suppression of PTH secretion (15); consequent PTH excess stimulates bone resorption but fails to promote phosphaturia due to renal impairment, while FGF-23 exacerbates hyperphosphatemia through dual mechanisms (11, 14): suppressing renal 1α-hydroxylase activity to reduce calcitriol (1,25-dihydroxyvitamin D3) synthesis and directly enhancing renal phosphate excretion, thereby establishing a vicious cycle of calcitriol deficiency that further aggravates secondary hyperparathyroidism through loss of vitamin D receptor (VDR)-mediated transcriptional repression and impairs intestinal calcium absorption, collectively driving hypercalcemia, hyperphosphatemia, progressive VC, and uncoupled bone remodeling characterized by increased resorption and suppressed formation, with therapeutic calcitriol supplementation representing a targeted intervention to partially correct this axis.
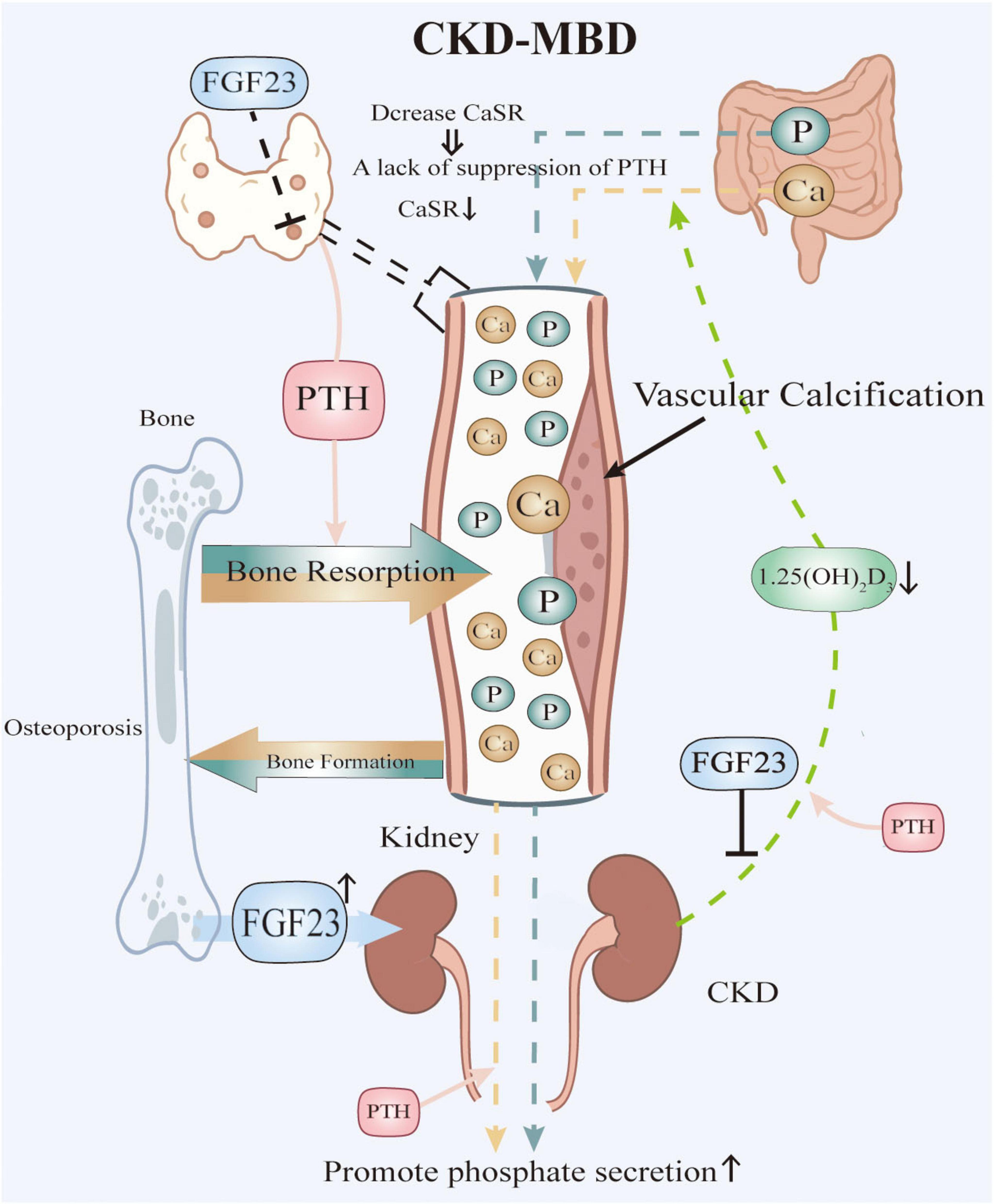
Figure 1. Pathophysiological Interplay in CKD-MBD. Elevated phosphate levels, resulting from renal failure, stimulate the secretion of FGF23. High levels of FGF23 suppress the synthesis of active vitamin D and contribute to the downregulation of CaSR in the parathyroid glands, thereby impairing calcium sensing. The combination of low active vitamin D and impaired calcium sensing leads to increased secretion of PTH, resulting in sHPT. Both PTH and FGF23 promote bone resorption, which releases additional calcium and phosphate into the bloodstream. However, renal impairment hinders adequate phosphate excretion, despite elevated levels of FGF23 and PTH, thereby perpetuating hyperphosphatemia. Additionally, low levels of active vitamin D reduce intestinal calcium absorption. The resultant mineral imbalances, characterized by hyperphosphatemia and fluctuations between hypercalcemia and hypocalcemia, together with inflammation and other contributing factors, drive vascular calcification and disrupt normal bone remodeling.
3 CKD-associated immune dysregulation
3.1 Chronic inflammation and oxidative stress
Chronic kidney disease is marked by a persistent state of low-grade inflammation, a critical factor driving disease progression and its associated complications, such as cardiovascular disease (CVD) (31). This chronic inflammatory state is largely fueled by elevated levels of pro-inflammatory cytokines, notably IL-6 and TNF-α. IL-6, a central mediator of the acute-phase response, is significantly increased in CKD and correlates strongly with disease severity. It amplifies systemic inflammation by triggering the production of acute-phase proteins and is implicated in heightened cardiovascular risk, a leading cause of mortality in CKD patients (32). Similarly, TNF-α exacerbates renal injury by promoting apoptosis of renal tubular cells and stimulating the release of additional inflammatory mediators, thus perpetuating a pro-inflammatory environment (33). These cytokines not only reflect the inflammatory burden but also actively contribute to tissue damage and fibrosis, key hallmarks of CKD progression.
Oxidative stress, another defining feature of CKD, significantly amplifies this inflammatory milieu and impairs immune function. The accumulation of reactive oxygen species (ROS) in CKD results from an imbalance between oxidant production and antioxidant defenses, damaging cellular components such as lipids, proteins, and DNA. This oxidative burden activates inflammatory pathways, including the nuclear factor-kappa B (NF-κB) system, which further upregulate cytokine production, creating a vicious cycle of inflammation and oxidative damage (34). Moreover, OS disrupts immune homeostasis, leading to a dysregulated immune response characterized by both hyperactivation and immunosuppression. For instance, ROS-mediated damage impairs the function of immune cells, reducing their ability to effectively combat infections while simultaneously promoting chronic inflammation (5). This dual impact underscores the complex interplay between OS and inflammation, accelerating renal damage and contributing to the high morbidity observed in CKD. Understanding these mechanisms is pivotal for devising strategies to mitigate inflammation and its deleterious effects in this population.
3.2 Immune cell dysfunction and the gut-kidney axis in CKD
Immune cell dysfunction is a key driver of CKD progression (Figure 2), involving abnormalities in adaptive and innate immune cells, including T cells, B cells, and monocytes/macrophages. T cell populations in CKD exhibit a shift toward pro-inflammatory phenotypes, with reduced regulatory T cell function, which fails to suppress excessive immune responses, thereby sustaining inflammation and tissue injury (5). B cell dysregulation further compounds this immune imbalance, characterized by altered antibody production and increased autoantibody formation, which may exacerbate renal damage through immune complex deposition (35). Monocytes and macrophages, critical components of the innate immune system, show heightened activation and polarization toward pro-inflammatory M1 phenotypes in CKD. This shift promotes the production of fibrogenic cytokines, such as transforming growth factor-beta (TGF-β), driving renal fibrosis and tubular injury (36). These cellular abnormalities collectively contribute to a maladaptive immune environment that accelerates CKD progression.
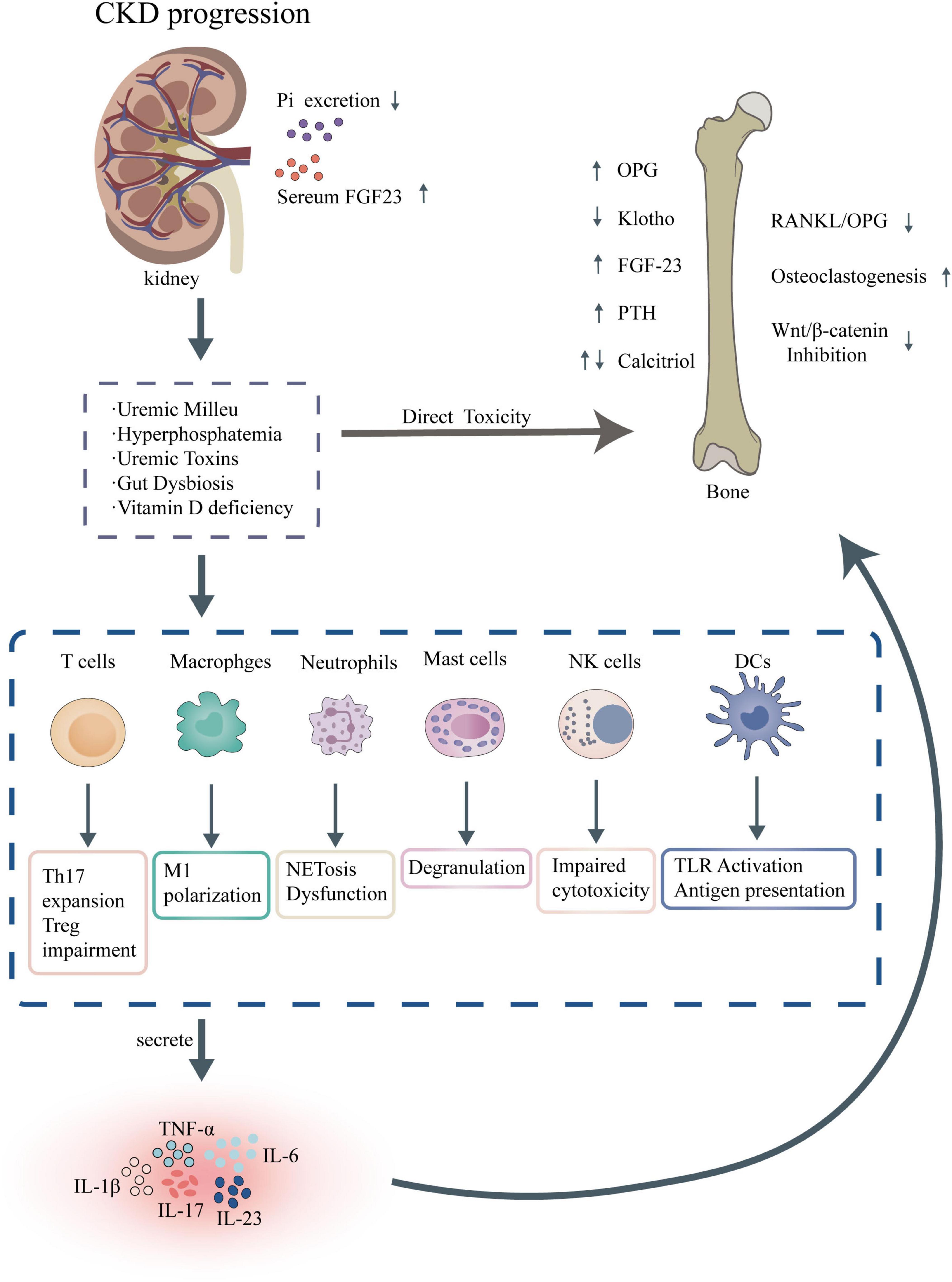
Figure 2. Pathophysiological mechanisms of CKD-MBD and associated immune dysregulation.
Dysbiosis of the gut microbiota leads to the disruption of tryptophan metabolism (resulting in the production of toxins such as IS and the entry of lipopolysaccharides (LPS) into the bloodstream (37). This process activates the AhR/TLR4 complex, which in turn triggers the activation of the NF-κB pathway, resulting in the release of pro-inflammatory factors and the generation of ROS, along with mitochondrial damage (38, 39). Subsequently, the NLRP3 inflammasome is assembled, leading to the activation of caspase-1 (40). This cascade results in the release of IL-1β and IL-18, pyroptosis, VC, and disturbances in bone metabolism, ultimately contributing to the progression of CKD-MBD (41).
4 Immune mechanisms in CKD-MBD
Chronic kidney disease-mineral and bone disorder is a systemic condition marked by disruptions in mineral metabolism, bone pathology, and VC, intricately linked to immune dysregulation. The interplay between immune responses and CKD-MBD pathogenesis has gained significant attention, particularly in how inflammation drives bone resorption and VC. This section delves into the immune mechanisms underpinning CKD-MBD, focusing on inflammatory pathways promoting bone resorption, immune-mediated VC, and the effects of uremic toxins on bone and immune cells, synthesizing recent findings to elucidate their roles in disease progression.
4.1 Inflammatory pathways promoting bone resorption
4.1.1 The RANKL/RANK/OPG axis and inflammatory pathways
The receptor activator of nuclear factor-κB ligand (RANKL)/receptor activator of nuclear factor-Kb (RANK)/osteoprotegerin (OPG) axis is a cornerstone of bone remodeling, and its dysregulation in CKD-MBD significantly contributes to bone resorption. RANKL, expressed by osteoblasts and activated T cells, binds to RANK on osteoclast precursors, stimulating their differentiation into mature osteoclasts and enhancing bone resorption (42). In CKD, systemic inflammation elevates pro-inflammatory cytokines such as IL-6 and TNF-α, which upregulate RANKL expression while suppressing OPG, a decoy receptor that curbs RANKL activity, thereby tipping the balance toward osteoclastogenesis (43). PTH, frequently elevated in CKD, further amplifies this process by boosting RANKL production, exacerbating bone loss. Recent studies in young rats with experimental CKD demonstrate that serum PTH levels correlate with increased RANKL and decreased OPG, alongside altered bone geometry and strength, underscoring the axis’s role in CKD-MBD. Additionally, inflammatory mediators in the bone microenvironment amplify local RANKL/RANK signaling, linking systemic and localized immune responses to bone pathology (44).
4.1.2 Roles of sclerostin and FGF-23
Sclerostin and FGF-23, osteocyte-derived regulators, play pivotal roles in CKD-MBD’s bone abnormalities, modulated by inflammatory cues. Sclerostin, an inhibitor of Wnt/β-catenin signaling, suppresses bone formation and is elevated in CKD, contributing to ABD characterized by low turnover (45). In peritoneal dialysis patients, high sclerostin levels correlate with reduced bone formation rates, a finding supported by bone biopsy data. Conversely, sclerostin may exert a protective effect against VC, as its deficiency in mice exacerbates arterial calcification via enhanced Wnt signaling (46). FGF-23, elevated early in CKD to manage phosphate overload, interacts with inflammatory pathways to influence bone mineralization (44). Its overexpression disrupts osteoblast differentiation and matrix mineralization, potentially through mitogen-activated protein kinase (MAPK) signaling inhibition, as seen in uremic rat models treated with C-type natriuretic peptide (47). The interplay between FGF-23 and inflammation, including IL-6 upregulation, further complicates bone homeostasis in CKD-MBD (44, 48).
4.2 Immune-mediated VC
4.2.1 Roles of macrophages
In CKD-MBD, VC is an immune-driven process in which macrophage-mediated chronic inflammation and cytokines play central roles PMID:39867890. The uremic microenvironment disrupts macrophage function, promoting the release of inflammatory factors, which in turn affects mineral metabolism such as calcium and phosphorus disorders and VC (5, 49). Macrophages participate in the process of VC through miRNA regulation such as the miR-125b-5p/TRAF6 (TNF receptor associated factor 6)/NF-κB axis, which is particularly significant in patients with CKD (50). Hyperphosphatemia in CKD activates macrophages, particularly the pro-inflammatory M1 phenotype, to release cytokines like IL-1β, IL-6, and TNF-α, which promote osteogenic differentiation of VSMCs (51, 52). The inflammatory environment is further aggravated by factors specific to CKD. Increased phosphate levels encourage the polarization of M1 macrophages through miRNA and RNA regulatory mechanisms, while senescent macrophages heighten local inflammation and the transdifferentiation of VSMCs via interferon signaling (50, 53). Senescent macrophages exacerbate local inflammation and the transdifferentiation of VSMCs by interfering with interferon signaling that propagates the senescent phenotype. The decline in Klotho expression leads to phosphate retention and exacerbated calcification, and the levels of sKlotho are negatively correlated with the risk of VC (9, 54). The degree of VC is significantly negatively correlated with bone density, reflecting systemic mineral metabolism disorders such as hyperphosphatemia, elevated FGF-23, and abnormal vitamin D levels (55, 56). This phenomenon stems from the abnormal transfer of calcium and phosphorus from the bones to the blood vessels, resulting in pathological bone formation (57). The NLRP3 inflammasome, a key inflammatory platform in macrophages, is activated by uremic toxins such as IS, leading to IL-1β maturation and enhanced calcification (58). Macrophages also contribute directly by releasing matrix vesicles that nucleate calcium phosphate deposits in vessel walls. Recent evidence highlights that sodium-glucose cotransporter 2 (SGLT2) inhibitors, such as canagliflozin, attenuates VC in CKD rats by suppressing NLRP3-mediated cytokine release, suggesting a therapeutic avenue (59). These findings position macrophages as critical mediators linking inflammation to vascular pathology in CKD-MBD.
M2 macrophages play a complex dual role in renal fibrosis and CKD-MBD (60). On one hand, they exhibit anti-inflammatory and tissue repair functions, such as clearing apoptotic debris and secreting IL-10 (61, 62). On the other hand, especially during the late stages of disease or under specific microenvironmental signals (e.g., high phosphate, uremic toxins), their excessive activation or phenotypic instability (imbalance in phenotypic plasticity) can transform them into pathogenic factors (63). At this stage, M2 macrophages directly promote the activation of myofibroblasts and extracellular matrix deposition by secreting factors like TGF-β, thereby exacerbating renal fibrosis. Some of these cells may even participate directly in the fibrosis process through macrophage-myofibroblast transition (MMT) (63). Furthermore, M2 macrophages promote the osteogenic transformation of VSMCs and VC by secreting inflammatory factors (e.g., TNF-α) and pro-calcifying mediators, which aggravates the instability of atherosclerotic plaques, significantly increasing the risk of cardiovascular complications. The heterogeneity of M2 subtypes (e.g., M2a, M2c) further complicates their functions (61, 64). Therefore, a deeper understanding of the mechanisms regulating M2 polarization direction, stabilizing their reparative phenotype, and overcoming their plasticity is crucial for developing strategies that target M2 macrophages to delay renal fibrosis and mitigate cardiovascular risks associated with CKD-MBD.
4.2.2 Osteogenic transformation of VSMCs
Vascular smooth muscle cells undergo an osteogenic transformation in CKD-MBD, driven by immune signals and uremic conditions, culminating in VC. Inflammatory cytokines and uremic toxins, including trimethylamine-N-oxide (TMAO), activate the NLRP3 inflammasome and NF-κB pathways in VSMCs, upregulating osteogenic markers like Runx2 and BMP2 (65). This phenotypic switch is marked by increased expression of alkaline phosphatase and osteocalcin, mirroring bone-forming cells (66). MicroRNA-34a, upregulated by inflammatory stimuli in CKD, further promotes VSMC senescence and calcification by modulating sirtuin 1 and Notch1 signaling. Phosphate excess, a hallmark of CKD, triggers this transformation via MAPK and NF-κB activation, as demonstrated in rat models where zinc supplementation inhibited calcification by enhancing TNFAIP3-mediated NF-κB suppression (67). These pathways highlight the immune-mediated mechanisms driving VSMC osteogenesis in CKD-MBD (59).
Beyond the intrinsic transformation of VSMCs, the vascular microenvironment in CKD-MBD is profoundly shaped by the involvement of diverse immune cells, which synergistically exacerbate calcification and inflammatory responses.
4.2.3 Roles of other immune cells
In CKD-MBD, mast cells drive renal inflammation and fibrosis processes directly by releasing proteases (such as tryptase and chymase) and inflammatory mediators (such as histamine and heparin) (68). Their activated state may serve as a potential biomarker for assessing CKD progression (69). Neutrophil function is significantly disrupted (with impaired chemotaxis, phagocytosis, and reactive oxygen species generation), which not only leads to decreased anti-infection capabilities and a persistent micro-inflammatory state but also may accelerate atherosclerosis by releasing NETs, thereby indirectly promoting cardiovascular complications (70–72). Natural killer (NK) cells are characterized by a reduced number and weakened cytotoxicity in kidney failure, which undermines immune surveillance and exacerbates systemic inflammation and immune imbalance (73, 74). Mast cells are important contributors to the instability of atherosclerotic plaques, potentially mediated by the release of inflammatory factors such as histamine or tryptase (75, 76). In stone formers, the number of mast cells was found to be significantly correlated with cortical calcification, suggesting that mast cell infiltration may locally drive the calcification process (77). The accumulation of uremic toxins and OS can activate innate immune pathways, such as TLR signaling, promoting inflammatory responses. TLR signaling may promote calcification by activating the osteogenic differentiation pathway of VSMCs, with dendritic cells (DCs) being key responsive cells in the TLR pathway (75, 78). Exosomes released by bone marrow mesenchymal stem cells (BMSCs) carry miRNAs that can inhibit VSMC calcification (79). DCs may participate in microenvironmental regulation through the uptake or secretion of exosomes (80). VC is negatively correlated with bone density reduction, a relationship possibly driven by shared inflammatory pathways such as RANKL/OPG imbalance (81, 82). If DCs activate Th17 cells (which secrete IL-17), they may simultaneously exacerbate bone loss and VC (57). Neutrophils release inflammatory factors such as IL-6 and TNF-α, while mast cell degranulation products and dysfunction of NK cells collectively promote a systemic microinflammatory state. This chronic inflammation may influence CKD-MBD through various pathways, such as accelerating VC and interfering with bone metabolism.
Above all, mast cells are central drivers that release mediators such as IL-6 and CXCL10 in response to estrogen deficiency, which impairs bone repair and promotes vascular inflammation (83, 84). DCs function as regulators, modulating bone immunity and the vascular microenvironment through antigen presentation and cytokine secretion (85). NK cells act more as effector cells, amplifying the pathological processes in bone and vasculature during inflammation (86, 87). The interactions among these three cell types within the bone-vascular axis (e.g., mast cell activation of DCs and DCs supporting NK cells) form an immune network; however, the direct integration mechanisms documented in the literature are limited. These mechanisms hold significant importance in diseases such as osteoporosis, atherosclerosis, and post-traumatic bone repair, and targeting these cells (e.g., inhibiting mast cells) may ameliorate the dysregulation of the bone-vascular axis (88, 89).
4.3 Impact of uremic toxins on bone and immune cells
Uremic toxins accumulating in CKD profoundly affect bone remodeling and immune function, exacerbating CKD-MBD. IS, a protein-bound toxin, impairs osteoblastogenesis by inhibiting Runx2 via the aryl hydrocarbon receptor (AhR) pathway while promoting osteoclastogenesis through NFATc1 upregulation, disrupting bone homeostasis (90). IS also induces OS and inflammation in immune cells, amplifying systemic inflammation that aggravates bone and vascular damage (91). Similarly, p-cresyl sulfate and advanced glycation end products (AGEs) enhance osteoclast activity and inhibit osteoblast function, contributing to bone demineralization (92). In macrophages, IS triggers toxicity by increasing OS and lipid metabolism abnormalities, linking gut microbiota-derived toxins to atherosclerosis and bone loss (58). Strategies reducing uremic toxin levels, such as dialysis optimization or AhR antagonists like resveratrol, may mitigate these effects, offering potential therapeutic benefits.
5 Discussion
The intricate interplay between immune dysregulation and CKD-MBD significantly amplifies clinical risks, notably fracture susceptibility, cardiovascular complications, and mortality. In CKD, chronic inflammation–evidenced by elevated TNF-α–exacerbates bone resorption and compromises bone quality, increasing fracture risk beyond what traditional mineral metabolism markers predict (93). This inflammatory milieu also promotes VC, a key contributor to cardiovascular morbidity, as TNF-α enhances both bone turnover and arterial stiffness (94).
Conventional therapies for CKD-MBD–phosphate binders, vitamin D analogs, and calcimimetics–often fail to address immune-driven pathology (95). For example, denosumab effectively inhibits bone resorption by suppressing RANKL; however, it poses a significant risk of hypocalcemia in patients with CKD-MBD, particularly in those who are dialysis-dependent or have advanced CKD (6, 96–98). This risk arises from the compounded effects of bone resorption inhibition and the inherent calcium-phosphate metabolism disorders associated with CKD, as well as secondary hyperparathyroidism.
Persistent VC despite phosphate control highlights the need for novel strategies targeting inflammation and osteoimmunological pathways (99, 100). Therapeutic strategies targeting immune mechanisms in CKD-MBD are gaining traction, with anti-inflammatory and mineral-regulating approaches showing promise. Cytokine inhibitors and antioxidants, such as those explored in preclinical models, reduce inflammation-driven bone turnover and VC, offering a complementary approach to conventional therapies (100). Cytokine inhibitors such as anti-RANKL and IL-1β targeting pro-inflammatory pathways may improve inflammation and cardiovascular outcomes in CKD (34, 101). Novel strategies involving antioxidants (Nrf2 activators, NOX inhibitors) show greater potential but must balance clinical risks (102–104). The bone protective effects of denosumab are well established; however, its application in advanced CKD requires monitoring of calcium and phosphorus metabolism disorders, including the risk of hypocalcemia (105, 106). Future research should focus on precision anti-inflammatory/antioxidant therapies targeting CKD-MBD, such as combined strategies targeting RANK or the Nrf2 pathway, while strictly evaluating their long-term impacts on mineral metabolism.
Mineral metabolism regulation remains critical, with phosphate binders, vitamin D analogs (e.g., paricalcitol), and calcimimetics effectively lowering PTH and phosphorus levels, yet their impact on inflammation is less consistent (49, 107). Emerging therapies, including anti-sclerostin antibodies and FGF-23 inhibitors, target the osteoimmune axis directly, potentially improving bone integrity while mitigating systemic inflammation (108). For instance, romosozumab, an anti-sclerostin agent, has demonstrated efficacy in hemodialysis patients by enhancing bone mineral density (BMD), though its long-term effects on cardiovascular outcomes remain uncharted (109). Vitamin D receptor activators (VDRAs) directly influence the immune-inflammatory aspects of CKD-MBD by regulating the NF-κB pathway, T cell differentiation, and the expression of pro-inflammatory factors (110–112). This regulation has the potential to improve patients’ immune deficiencies and inflammatory status. SGLT2 inhibitors have led to significant advancements in the treatment of CKD; however, their clinical translation is hindered by cognitive biases, subgroup heterogeneity, and risks associated with bone metabolism. The safety and applicability of anti-sclerostin antibodies in CKD necessitate further research for validation. Future efforts should focus on promoting personalized medication strategies and enhancing clinical safety monitoring (113–115). SGLT2 inhibitors have been confirmed in clinical studies to improve biochemical indicators related to CKD-MBD, and their cardioprotective and nephroprotective effects are clear (114, 116). However, the issue of insufficient evidence in non-diabetic CKD populations needs to be addressed. Preclinical studies on anti-bone sclerosis protein antibodies have shown potential for regulating bone metabolism, but cardiovascular risks have limited clinical translation, necessitating the urgent design of new strategies for safety optimization targeted at the CKD-MBD population (117–119). Existing trials have significant limitations in sample size, follow-up duration, endpoint indicators, and population representativeness. Future research should focus on large-scale, long-term studies targeting heterogeneous populations, particularly emphasizing treatment optimization within the framework of precision medicine. These advances highlight a shift toward personalized interventions, yet their integration into clinical practice awaits robust validation.
Despite these insights, significant limitations hinder the translation of immune-focused CKD-MBD research. The reliance on short-term surrogate endpoints, such as PTH or BMD changes, limits understanding of long-term clinical benefits, particularly regarding fracture prevention and cardiovascular mortality (120). Variability in study designs and patient cohorts–often excluding early-stage CKD or non-dialysis populations–further obscures the generalizability of findings (121). Future research should prioritize longitudinal trials to delineate the causal roles of specific inflammatory pathways (e.g., TNF-α, IL-6) and their therapeutic modulation across all CKD stages. Moreover, developing biomarkers that integrate inflammation, bone turnover, and vascular health could refine risk assessment and treatment monitoring, addressing current gaps in precision medicine for CKD-MBD.
6 Conclusion
This review highlights the pivotal role of immune dysregulation in the pathogenesis of CKD-MBD, demonstrating how chronic inflammation and aberrant immune responses contribute to bone loss, VC, and increased cardiovascular risk in CKD patients. Key findings indicate that pro-inflammatory cytokines, such as TNF-α, drive bone resorption and vascular stiffening, establishing a mechanistic link between immune activation and the skeletal and extra-skeletal manifestations of CKD-MBD (5, 122). Furthermore, immune-mediated pathways, including sclerostin inhibition of Wnt signaling and FGF-23 dysregulation, have emerged as critical regulators of mineral metabolism and potential therapeutic targets beyond conventional phosphate management (123, 124). These insights underscore that addressing immune mechanisms is essential for improving CKD-MBD management, offering a paradigm shift from solely mineral-focused strategies to integrated immune-modulatory approaches.
Looking forward, future research should focus on identifying immune-related biomarkers to enhance risk stratification and personalize treatment in CKD-MBD. Recent studies suggest that gut dysbiosis, a driver of inflammation and mineral imbalance, warrants exploration as a novel therapeutic avenue, potentially through microbiome-targeted interventions (5). Additionally, advanced techniques like single-cell RNA sequencing could elucidate patient-specific immune profiles, paving the way for tailored therapies that address individual variability in CKD-MBD progression (125). The complexity introduced by osteoimmunology and osteomicrobiology highlights both the challenges and opportunities in this field, necessitating longitudinal, collaborative studies to validate these approaches and translate them into clinical practice.
In conclusion, integrating immune mechanisms into CKD-MBD management holds transformative potential for reducing morbidity and mortality in CKD patients. However, significant knowledge gaps remain, particularly regarding the translation of immune-based interventions into effective treatments. We call for further research to bridge these gaps, leveraging emerging technologies and interdisciplinary efforts to improve outcomes in this high-risk population.
Author contributions
BX: Software, Writing – original draft, Writing – review & editing. RM: Writing – original draft, Writing – review & editing. YW: Software, Writing – original draft. CL: Supervision, Writing – review & editing. XS: Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article can solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1678640/full#supplementary-material
Abbreviations
ABD, adynamic bone disease; AhR, aryl hydrocarbon receptor; BMD, bone mineral density; BMPs, bone morphogenetic proteins; BMSCs, bone marrow mesenchymal stem cells; CaSR, calcium-sensing receptor; CVD, cardiovascular disease; CKD-MBD, chronic kidney disease-mineral and bone disorder; CKD, chronic kidney disease; FGF-23, fibroblast growth factor-23; IL, interleukin; IS, indoxyl sulfate; NK cells, natural killer cells; NLRP3, NLR family pyrin domain containing 3; OPG, osteoprotegerin; OS, oxidative stress; PTH, parathyroid hormone; RANK, receptor activator of nuclear factor-kappa B; RANKL, receptor activator of nuclear factor-kappa B ligand; ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α; SGLT2, sodium-glucose cotransporter 2; TGF-β, transforming growth factor-beta; TLRs, toll-like receptors; TIO, tumor-induced osteomalacia; TNF-α, tumor necrosis factor-alpha; TRAF6, TNF receptor associated factor 6; VC, vascular calcification; sHPT, secondary hyperparathyroidism; LPS, lipopolysaccharides; VSMCs, vascular smooth muscle cells; MAPK, mitogen-activated protein kinase; TMAO, trimethylamine-N-oxide; SULF1, sulfatase 1; NF-κ B, nuclear factor-kappa B; NETs, neutrophil extracellular traps; KDIGO, Kidney Disease Improving Global Outcomes; VDR, vitamin D receptor; VDRAs, Vitamin D receptor activators.
References
1. Chen T, Hoenig M, Nitsch D, Grams M. Advances in the management of chronic kidney disease. BMJ. (2023) 383:e074216. doi: 10.1136/bmj-2022-074216
2. Cockwell P, Fisher LA. The global burden of chronic kidney disease. Lancet. (2020) 395:662–4. doi: 10.1016/S0140-6736(19)32977-0
3. Liyanage T, Toyama T, Hockham C, Ninomiya T, Perkovic V, Woodward M, et al. Prevalence of chronic kidney disease in Asia: a systematic review and analysis. BMJ Glob Health. (2022) 7:e007525. doi: 10.1136/bmjgh-2021-007525
4. Shlipak M, Tummalapalli S, Boulware L, Grams M, Ix J, Jha V, et al. The case for early identification and intervention of chronic kidney disease: conclusions from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. (2021) 99:34–47. doi: 10.1016/j.kint.2020.10.012
5. Evenepoel P, Stenvinkel P, Shanahan C, Pacifici R. Inflammation and gut dysbiosis as drivers of CKD-MBD. Nat Rev Nephrol. (2023) 19:646–57. doi: 10.1038/s41581-023-00736-7
6. Bird S, Smith E, Gelperin K, Jung T, Thompson A, Kambhampati R, et al. Severe hypocalcemia with denosumab among older female dialysis-dependent patients. JAMA. (2024) 331:491–9. doi: 10.1001/jama.2023.28239
7. Ketteler M, Evenepoel P, Holden R, Isakova T, Jørgensen H, Komaba H, et al. Chronic kidney disease-mineral and bone disorder: conclusions from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. (2025) 107:405–23. doi: 10.1016/j.kint.2024.11.013
8. Elder G. Current status of mineral and bone disorders in transplant recipients. Transplantation. (2023) 107:2107–19. doi: 10.1097/TP.0000000000004538
9. Kaur R, Singh R. Mechanistic insights into CKD-MBD-related vascular calcification and its clinical implications. Life Sci. (2022) 311:121148. doi: 10.1016/j.lfs.2022.121148
10. Haffner D, Leifheit-Nestler M, Alioli C, Bacchetta J. Muscle and bone impairment in infantile nephropathic cystinosis: new concepts. Cells. (2022) 11:170. doi: 10.3390/cells11010170
11. Figurek A, Rroji M, Spasovski G. FGF23 in chronic kidney disease: bridging the heart and anemia. Cells. (2023) 12:609. doi: 10.3390/cells12040609
12. Covic A, Vervloet M, Massy Z, Torres P, Goldsmith D, Brandenburg V, et al. Bone and mineral disorders in chronic kidney disease: implications for cardiovascular health and ageing in the general population. Lancet Diabetes Endocrinol. (2018) 6:319–31. doi: 10.1016/S2213-8587(17)30310-8
13. Mace ML, Lewin E. Frontiers in bone metabolism and disorder in chronic kidney Disease. Metabolites. (2023) 13:1034. doi: 10.3390/metabo13101034
14. Chandu A, Arana C, Díaz-García J, Cozzolino M, Ciceri P, Torregrosa J. Calcimimetics and Vascular Calcification. Toxins. (2025) 17:297. doi: 10.3390/toxins17060297
15. Ahmad R, Deb-Choudhury S, Subbaraj A, Kim J. Activation of the calcium-sensing receptor by glutathione maillard products: implications for kokumi sensation. Food Chem X. (2025) 29:102616. doi: 10.1016/j.fochx.2025.102616
16. Nakagawa Y, Komaba H. Roles of parathyroid hormone and fibroblast growth factor 23 in advanced chronic kidney disease. Endocrinol Metab. (2024) 39:407–15. doi: 10.3803/EnM.2024.1978
17. Fang Y, Ginsberg C, Sugatani T, Monier-Faugere M, Malluche H, Hruska K. Early chronic kidney disease-mineral bone disorder stimulates vascular calcification. Kidney Int. (2014) 85:142–50. doi: 10.1038/ki.2013.271
18. Komaba H, Kaludjerovic J, Hu D, Nagano K, Amano K, Ide N, et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. (2017) 92:599–611. doi: 10.1016/j.kint.2017.02.014
19. Freedman BI, Register TC. Effect of race and genetics on vitamin D metabolism, bone and vascular health. Nat Rev Nephrol. (2012) 8:459–66. doi: 10.1038/nrneph.2012.112
20. Drueke TB, Massy ZA. Changing bone patterns with progression of chronic kidney disease. Kidney Int. (2016) 89:289–302. doi: 10.1016/j.kint.2015.12.004
21. Haarhaus M, Evenepoel P. Differentiating the causes of adynamic bone in advanced chronic kidney disease informs osteoporosis treatment. Kidney Int. (2021) 100:546–58. doi: 10.1016/j.kint.2021.04.043
22. Shalhoub V, Shatzen E, Ward S, Davis J, Stevens J, Bi V, et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest. (2012) 122:2543–53. doi: 10.1172/JCI61405
23. Chao CT, Yeh HY, Tsai YT, Chiang CK, Chen HW. A combined microRNA and target protein-based panel for predicting the probability and severity of uraemic vascular calcification: a translational study. Cardiovasc Res. (2021) 117:1958–73. doi: 10.1093/cvr/cvaa255
24. Imanishi Y, Ito N, Rhee Y, Takeuchi Y, Shin C, Takahashi Y, et al. Interim analysis of a phase 2 open-label trial assessing burosumab efficacy and safety in patients with tumor-induced osteomalacia. J Bone Miner Res. (2021) 36:262–70. doi: 10.1002/jbmr.4184
25. Hidaka N, Koga M, Kimura S, Hoshino Y, Kato H, Kinoshita Y, et al. Clinical challenges in diagnosis, tumor localization and treatment of tumor-induced osteomalacia: outcome of a retrospective surveillance. J Bone Miner Res. (2022) 37:1479–88. doi: 10.1002/jbmr.4620
26. Chiu H, Lu K, Lin Y, Hou Y, Liao M, Chen Y, et al. Etelcalcetide ameliorates bone loss in chronic kidney disease-mineral and bone disorder by activation of IRF7 and necroptosis pathways. Int J Biol Macromol. (2024) 280:135978. doi: 10.1016/j.ijbiomac.2024.135978
27. Pereira L, Magalhães J, Mendonça L, Neto R, Santos J, Carvalho C, et al. Evaluation of renal osteodystrophy and serum bone-related biomarkers in a peritoneal dialysis population. J Bone Miner Res. (2022) 37:1689–99. doi: 10.1002/jbmr.4636
28. Ott S, Malluche H, Jorgetti V, Elder G. Importance of bone turnover for therapeutic decisions in patients with CKD-MBD. Kidney Int. (2021) 100:502–5. doi: 10.1016/j.kint.2021.05.024
29. Ginsberg C, Ix JH. Diagnosis and management of osteoporosis in advanced kidney disease: a review. Am J Kidney Dis. (2022) 79:427–36. doi: 10.1053/j.ajkd.2021.06.031
30. Xu C, Smith E, Tiong M, Ruderman I, Toussaint N. interventions to attenuate vascular calcification progression in chronic kidney disease: a systematic review of clinical trials. J Am Soc Nephrol. (2022) 33:1011–32. doi: 10.1681/ASN.2021101327
31. Düsing P, Göbel I, Ackerschott A, Reese L, Giavalisco P, Dethloff F, et al. The role of uremic toxin indoxyl sulfate in the pathophysiology of aortic valve stenosis. Cardiovasc Res. (2025): doi: 10.1093/cvr/cvaf106 Online ahead of print
32. Speer T, Dimmeler S, Schunk S, Fliser D, Ridker P. Targeting innate immunity-driven inflammation in CKD and cardiovascular disease. Nat Rev Nephrol. (2022) 18:762–78. doi: 10.1038/s41581-022-00621-9
33. Liu BC, Tang TT, Lv LL, Lan HY. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. (2018) 93:568–79. doi: 10.1016/j.kint.2017.09.033
34. Zoccali C, Mallamaci F. Innate immunity system in patients with cardiovascular and kidney disease. Circ Res. (2023) 132:915–32. doi: 10.1161/CIRCRESAHA.122.321749
35. Cheung C, Alexander S, Reich H, Selvaskandan H, Zhang H, Barratt J. The pathogenesis of IgA nephropathy and implications for treatment. Nat Rev Nephrol. (2025) 21:9–23. doi: 10.1038/s41581-024-00885-3
36. Huang X, Ye L, An N, Wu C, Wu H, Li H, et al. Macrophage autophagy protects against acute kidney injury by inhibiting renal inflammation through the degradation of TARM1. Autophagy. (2025) 21:120–40. doi: 10.1080/15548627.2024.2393926
37. Zhang L, Cheng D, Zhang J, Tang H, Li F, Peng Y, et al. Role of macrophage AHR/TLR4/STAT3 signaling axis in the colitis induced by non-canonical AHR ligand aflatoxin B1. J Hazard Mater. (2023) 452:131262. doi: 10.1016/j.jhazmat.2023.131262
38. Zhang J, Liu H, Shen Y, Cheng D, Tang H, Zhang Q, et al. Macrophage AHR-TLR4 cross-talk drives p-STAT3 (Ser727)-mediated mitochondrial oxidative stress and upregulates IDO/ICAM-1 in the steatohepatitis induced by aflatoxin B1. Sci Total Environ. (2024) 923:171377. doi: 10.1016/j.scitotenv.2024.171377
39. Zhang D, Wu J, Feng H, Tang P, Zhou Y, Zhao C, et al. Gastrodin ameliorates ulcerative colitis via modulating gut microbial tryptophan metabolism and AhR/NLRP3 pathway. Phytomedicine. (2025) 147:157217. doi: 10.1016/j.phymed.2025.157217
40. Zhang C, Huang Y, Ouyang F, Su M, Li W, Chen J, et al. Extracellular vesicles derived from mesenchymal stem cells alleviate neuroinflammation and mechanical allodynia in interstitial cystitis rats by inhibiting NLRP3 inflammasome activation. J Neuroinflammation. (2022) 19:80. doi: 10.1186/s12974-022-02445-7
41. Hao Q, Yan J, Wei J, Zeng Y, Feng L, Que D, et al. Prevotella copri promotes vascular calcification via lipopolysaccharide through activation of NF-κB signaling pathway. Gut Microbes. (2024) 16:2351532. doi: 10.1080/19490976.2024.2351532
42. Sieklucka B, Pawlak D, Domaniewski T, Hermanowicz J, Lipowicz P, Doroszko M, et al. Serum PTH, PTH1R/ATF4 pathway, and the sRANKL/OPG system in bone as a new link between bone growth, cross-sectional geometry, and strength in young rats with experimental chronic kidney disease. Cytokine. (2021) 148:155685. doi: 10.1016/j.cyto.2021.155685
43. Tsirpanlis G. Is inflammation the link between atherosclerosis and vascular calcification in chronic kidney disease? Blood Purif. (2007) 25:179–82. doi: 10.1159/000099011
44. Albrecht LV, Pereira RC, Salusky IB. All the might of the osteocyte: emerging roles in chronic kidney disease. Kidney Int. (2023) 104:910–5. doi: 10.1016/j.kint.2023.08.009
45. de Oliveira R, Barreto F, Mendes M, dos Reis L, Castro J, Britto Z, et al. Peritoneal dialysis per se is a risk factor for sclerostin-associated adynamic bone disease. Kidney Int. (2015) 87:1039–45. doi: 10.1038/ki.2014.372
46. De Mare A, Opdebeeck B, Neven E, D’Haese PC, Verhulst A. Sclerostin protects against vascular calcification development in mice. J Bone Miner Res. (2022) 37:687–99. doi: 10.1002/jbmr.4503
47. Zhang D, Wu Y, Chen W, Xu Y, Liu S, Luo H, et al. C-type natriuretic peptide attenuates renal osteodystrophy through inhibition of FGF-23/MAPK signaling. Exp Mol Med. (2019) 51:1–18. doi: 10.1038/s12276-019-0265-8
48. Delanaye P, Cavalier E, Bouquegneau A, Khwaja A. Sclerostin levels in CKD patients: an important, but not definitive, step on the way to clinical use. Kidney Int. (2015) 88:1221–3. doi: 10.1038/ki.2015.258
49. Sprague SM, Martin KJ, Coyne DW. Phosphate balance and CKD-mineral bone disease. Kidney Int Rep. (2021) 6:2049–58. doi: 10.1016/j.ekir.2021.05.012
50. Li Q, Zhang C, Shi J, Yang Y, Xing X, Wang Y, et al. High-phosphate-stimulated macrophage-derived exosomes promote vascular calcification via let-7b-5p/TGFBR1 axis in chronic kidney disease. Cells. (2022) 12:161. doi: 10.3390/cells12010161
51. Alesutan I, Razazian M, Luong T, Estepa M, Pitigala L, Henze L, et al. Augmentative effects of leukemia inhibitory factor reveal a critical role for TYK2 signaling in vascular calcification. Kidney Int. (2024) 106:611–24. doi: 10.1016/j.kint.2024.07.011
52. Ding N, Lv Y, Su H, Wang Z, Kong X, Zhen J, et al. Vascular calcification in CKD: new insights into its mechanisms. J Cell Physiol. (2023) 238:1160–82. doi: 10.1002/jcp.31021
53. Park H, Kim Y, Kim M, Kim H, Bae S, Bae M. Regulation of vascular calcification by M1-type macrophage-derived Semaphorin 4D. Int J Mol Sci. (2025) 26:5071. doi: 10.3390/ijms26115071
54. Fan Z, Wei X, Zhu X, Yang K, Tian L, Du Y, et al. Correlation between soluble klotho and chronic kidney disease-mineral and bone disorder in chronic kidney disease: a meta-analysis. Sci Rep. (2024) 14:4477. doi: 10.1038/s41598-024-54812-4
55. Uhlinova J, Kuudeberg A, Metsküla K, Lember M, Rosenberg M. Significant associations between bone mineral density and vascular calcification in patients with different stages of chronic kidney disease. BMC Nephrol. (2022) 23:327. doi: 10.1186/s12882-022-02955-9
56. Izzo C, Secondulfo C, Bilancio G, Visco V, Virtuoso N, Migliarino S, et al. Chronic kidney disease with mineral bone disorder and vascular calcification: an overview. Life. (2024) 14:418. doi: 10.3390/life14030418
57. Shen Y, Yu C. The bone-vascular axis: a key player in chronic kidney disease associated vascular calcification. Kidney Dis. (2024) 10:545–57. doi: 10.1159/000541280
58. Wakamatsu T, Yamamoto S, Yoshida S, Narita I. Indoxyl sulfate-induced macrophage toxicity and therapeutic strategies in uremic atherosclerosis. Toxins. (2024) 16:254. doi: 10.3390/toxins16060254
59. Chen A, Lan Z, Li L, Xie L, Liu X, Yang X, et al. Sodium-glucose cotransporter 2 inhibitor canagliflozin alleviates vascular calcification through suppression of nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 inflammasome. Cardiovasc Res. (2023) 119:2368–81. doi: 10.1093/cvr/cvad119
60. Gao X, Liu X, Han Z, Liao H, Li R. Friend or foe? The role of SIRT6 on macrophage polarized to M2 subtype in acute kidney injury to chronic kidney disease. Ren Fail. (2025) 47:2482121. doi: 10.1080/0886022X.2025.2482121
61. Luo L, Wang S, Hu Y, Wang L, Jiang X, Zhang J, et al. Precisely regulating M2 subtype macrophages for renal fibrosis resolution. ACS Nano. (2023) 17:22508–26. doi: 10.1021/acsnano.3c05998
62. Guiteras R, Flaquer M, Cruzado JM. Macrophage in chronic kidney disease. Clin Kidney J. (2016) 9:765–71. doi: 10.1093/ckj/sfw096
63. Meng X, Jin J, Lan HY. Driving role of macrophages in transition from acute kidney injury to chronic kidney disease. Chin Med J. (2022) 135:757–66. doi: 10.1097/CM9.0000000000002100
64. Hu Y, Wang Y, Hong H, Chen Y, Zhou Q, Zhu G, et al. Global trends and prospects related to macrophage in chronic kidney disease: a bibliometric analysis. Ren Fail. (2024) 46:2423846. doi: 10.1080/0886022X.2024.2423846
65. Zhang X, Li Y, Yang P, Liu X, Lu L, Chen Y, et al. Trimethylamine-N-Oxide promotes vascular calcification through activation of NLRP3 (Nucleotide-binding domain, leucine-rich-containing family, Pyrin Domain-Containing-3) Inflammasome and NF-κB (Nuclear Factor κB) signals. Arterioscler Thromb Vasc Biol. (2020) 40:751–65. doi: 10.1161/ATVBAHA.119.313414
66. Raucci A, Macrì F, Castiglione S, Badi I, Vinci M, Zuccolo E. MicroRNA-34a: the bad guy in age-related vascular diseases. Cell Mol Life Sci. (2021) 78:7355–78. doi: 10.1007/s00018-021-03979-4
67. Voelkl J, Tuffaha R, Luong T, Zickler D, Masyout J, Feger M, et al. Zinc inhibits phosphate-induced vascular calcification through TNFAIP3-mediated suppression of NF-κ B. J Am Soc Nephrol. (2018) 29:1636–48. doi: 10.1681/ASN.2017050492
68. Atiakshin D, Morozov S, Dlin V, Kostin A, Volodkin A, Ignatyuk M, et al. Renal mast cell-specific proteases in the pathogenesis of tubulointerstitial fibrosis. J Histochem Cytochem. (2024) 72:495–515. doi: 10.1369/00221554241274878
69. Owens E, Vesey D, Kassianos A, Healy H, Hoy W, Gobe G. Biomarkers and the role of mast cells as facilitators of inflammation and fibrosis in chronic kidney disease. Transl Androl Urol. (2019) 8:S175–83. doi: 10.21037/tau.2018.11.03
70. Lemesch S, Ribitsch W, Schilcher G, Spindelböck W, Hafner-Gießauf H, Marsche G, et al. Mode of renal replacement therapy determines endotoxemia and neutrophil dysfunction in chronic kidney disease. Sci Rep. (2016) 6:34534. doi: 10.1038/srep34534
71. Lauxen J, Vondenhoff S, Junho C, Martin P, Fleig S, Schütt K, et al. Neutrophil function in patients with chronic kidney disease: a systematic review and meta-analysis. Acta Physiol. (2025) 241:e70057. doi: 10.1111/apha.70057
72. Bronze-da-Rocha E, Santos-Silva A. Neutrophil elastase inhibitors and chronic kidney disease. Int J Biol Sci. (2018) 14:1343–60. doi: 10.7150/ijbs.26111
73. Wu I, Wu Y, Yang H, Hsu C, Chang L, Twu Y, et al. Deep immune profiling of patients with renal impairment unveils distinct immunotypes associated with disease severity. Clin Kidney J. (2022) 16:78–89. doi: 10.1093/ckj/sfac196
74. Nagai K. Dysfunction of natural killer cells in end-stage kidney disease on hemodialysis. Ren Replace Ther. (2021) 7:8. doi: 10.1186/s41100-021-00324-0
75. Wang Z, Gui Z, Zhang L, Wang Z. Advances in the mechanisms of vascular calcification in chronic kidney disease. J Cell Physiol. (2025) 240:e31464. doi: 10.5527/wjn.v1.i2.43
76. Skenteris N, Hemme E, Delfos L, Karadimou G, Karlöf E, Lengquist M, et al. Mast cells participate in smooth muscle cell reprogramming and atherosclerotic plaque calcification. Vascul Pharmacol. (2023) 150:107167. doi: 10.1016/j.vph.2023.107167
77. Dejban P, Wilson E, Jayachandran M, Herrera Hernandez L, Haskic Z, Wellik L, et al. Inflammatory cells in nephrectomy tissue from patients without and with a history of urinary stone disease. Clin J Am Soc Nephrol. (2022) 17:414–22. doi: 10.2215/CJN.11730921
78. Voelkl J, Cejka D, Alesutan I. An overview of the mechanisms in vascular calcification during chronic kidney disease. Curr Opin Nephrol Hypertens. (2019) 28:289–96. doi: 10.1097/MNH.0000000000000507
79. Wu Y, Shan S, Lin X, Xu F, Zhong J, Wu F, et al. Cellular crosstalk in the vascular wall microenvironment: the role of exosomes in vascular calcification. Front Cardiovasc Med. (2022) 9:912358. doi: 10.3389/fcvm.2022.912358
80. Guo Y, Bao S, Guo W, Diao Z, Wang L, Han X, et al. Bone marrow mesenchymal stem cell-derived exosomes alleviate high phosphorus-induced vascular smooth muscle cells calcification by modifying microRNA profiles. Funct Integr Genomics. (2019) 19:633–43. doi: 10.1007/s10142-019-00669-0
81. Shen Y. Role of nutritional vitamin D in chronic kidney disease-mineral and bone disorder: a narrative review. Medicine. (2023) 102:e33477. doi: 10.1097/MD.0000000000033477
82. Mace M, Gravesen E, Nordholm A, Egstrand S, Morevati M, Olgaard K, et al. The calcified vasculature in chronic kidney disease secretes factors that inhibit bone mineralization. JBMR Plus. (2022) 6:e10610. doi: 10.1002/jbm4.10610
83. Ragipoglu D, Bülow J, Hauff K, Voss M, Haffner-Luntzer M, Dudeck A, et al. Mast cells drive systemic inflammation and compromised bone repair after trauma. Front Immunol. (2022) 13:883707. doi: 10.3389/fimmu.2022.883707
84. Fischer V, Ragipoglu D, Diedrich J, Steppe L, Dudeck A, Schütze K, et al. Mast cells trigger disturbed bone healing in osteoporotic mice. J Bone Miner Res. (2022) 37:137–51. doi: 10.1002/jbmr.4455
85. Ding X, Yang J, Wei Y, Wang M, Peng Z, He R, et al. The nexus between traditional chinese medicine and immunoporosis: implications in the treatment and management of osteoporosis. Phytother Res. (2025) 39:1826–46. doi: 10.1002/ptr.8397
86. Vallejo J, Cochain C, Zernecke A, Ley K. Heterogeneity of immune cells in human atherosclerosis revealed by scRNA-Seq. Cardiovasc Res. (2021) 117:2537–43. doi: 10.1093/cvr/cvab260
87. Keum B, Kim H, Lee J, Lee M, Hong S, Chang H, et al. Heterogeneous osteoimmune profiles via single-cell transcriptomics in osteoporotic patients who fail bisphosphonate treatment. Proc Natl Acad Sci U S A. (2024) 121:e2316871121. doi: 10.1073/pnas.2316871121
88. Nie H, Yan C, Zhou W, Li T. Analysis of immune and inflammation characteristics of atherosclerosis from different sample sources. Oxid Med Cell Longev. (2022) 2022:5491038. doi: 10.1155/2022/5491038
89. Abe S, Asahi T, Hara T, Cui G, Shimba A, Tani-Ichi S, et al. Hematopoietic cell-derived IL-15 supports NK cell development in scattered and clustered localization within the bone marrow. Cell Rep. (2023) 42:113127. doi: 10.1016/j.celrep.2023.113127
90. Shyu J, Liu W, Zheng C, Fang T, Hou Y, Chang C, et al. Toxic effects of indoxyl sulfate on osteoclastogenesis and osteoblastogenesis. Int J Mol Sci. (2021) 22:11265. doi: 10.3390/ijms222011265
91. Chao CT, Lin SH. Uremic toxins and frailty in patients with chronic kidney disease: a molecular insight. Int J Mol Sci. (2021) 22:6270. doi: 10.3390/ijms22126270
93. Meza K, Biswas S, Zhu Y, Gajjar A, Perelstein E, Kumar J, et al. Tumor necrosis factor-alpha is associated with mineral bone disorder and growth impairment in children with chronic kidney disease. Pediatr Nephrol. (2021) 36:1579–87. doi: 10.1007/s00467-020-04846-3
94. Chen N, Srinivasan S, O’Neill K, Nickolas T, Wallace J, Allen M, et al. Effect of advanced glycation end-products (AGE) lowering drug ALT-711 on biochemical, vascular, and bone parameters in a rat model of CKD-MBD. J Bone Miner Res. (2020) 35:608–17. doi: 10.1002/jbmr.3925
95. Ketteler M, Block G, Evenepoel P, Fukagawa M, Herzog C, McCann L, et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) guideline update: what’s changed and why it matters. Kidney Int. (2017) 92:26–36. doi: 10.1016/j.kint.2017.04.006
96. Lee JH, Heo SJ, Kim KM, Lee JE. Trends in serum phosphate, calcium, and parathyroid hormone levels according to renal function following denosumab. Osteoporos Int. (2025): doi: 10.1007/s00198-025-07601-2 Online ahead of print.
97. Bird S, Gelperin K, Smith E, Jung T, Lyu H, Thompson A, et al. The effect of denosumab on risk for emergently treated hypocalcemia by stage of chronic kidney disease: a target trial emulation. Ann Intern Med. (2025) 178:29–38. doi: 10.7326/M24-0013
98. Fu Q, Bustamante-Gomez N, Reyes-Pardo H, Gubrij I, Escalona-Vargas D, Thostenson J, et al. Reduced osteoprotegerin expression by osteocytes may contribute to rebound resorption after denosumab discontinuation. JCI Insight. (2023) 8:e167790. doi: 10.1172/jci.insight.167790
99. Zhang H, Li G, Yu X, Yang J, Jiang A, Cheng H, et al. Progression of vascular calcification and clinical outcomes in patients receiving maintenance dialysis. JAMA Netw Open. (2023) 6:e2310909. doi: 10.1001/jamanetworkopen.2023.10909
100. Neven E, Vervaet B, Brand K, Gottwald-Hostalek U, Opdebeeck B, De Maré A, et al. Metformin prevents the development of severe chronic kidney disease and its associated mineral and bone disorder. Kidney Int. (2018) 94:102–13. doi: 10.1016/j.kint.2018.01.027
101. Kondegowda N, Filipowska J, Do J, Leon-Rivera N, Li R, Hampton R, et al. RANKL/RANK is required for cytokine-induced beta cell death; osteoprotegerin, a RANKL inhibitor, reverses rodent type 1 diabetes. Sci Adv. (2023) 9:eadf5238. doi: 10.1126/sciadv.adf5238
102. Ram C, Gairola S, Syed A, Verma S, Mugale M, Sahu B. Carvacrol preserves antioxidant status and attenuates kidney fibrosis via modulation of TGF-β1/Smad signaling and inflammation. Food Funct. (2022) 13:10587–600. doi: 10.1039/d2fo01384c
103. Colombijn J, Hooft L, Jun M, Webster A, Bots M, Verhaar M, et al. Antioxidants for adults with chronic kidney disease. Cochrane Database Syst Rev. (2023) 11:CD008176. doi: 10.1002/14651858.CD008176.pub3
104. Barnes P. Oxidative stress in chronic obstructive pulmonary disease. Antioxidants. (2022) 11:965. doi: 10.3390/antiox11050965
105. Seto T, Yukata K, Fujii Z, Shibuya M, Okazaki T, Mihara A, et al. Denosumab associated with accelerated progression of abdominal aortic calcification among patients on dialysis. J Bone Miner Res. (2025): doi: 10.1093/jbmr/zjaf093 Online ahead of print.
106. Ciscar M, Trinidad E, Perez-Chacon G, Alsaleem M, Jimenez M, Jimenez-Santos M, et al. RANK is a poor prognosis marker and a therapeutic target in ER-negative postmenopausal breast cancer. EMBO Mol Med. (2023) 15:e16715. doi: 10.15252/emmm.202216715
107. D’Marco L, Checa-Ros A, Gamero D, Soto C, Salazar J, Nava M, et al. Etelcalcetide and paricalcitol in chronic kidney disease: when the target is inflammation. Healthcare. (2022) 11:72. doi: 10.3390/healthcare11010072
108. Tsuchiya K, Akihisa T. The importance of phosphate control in chronic kidney disease. Nutrients. (2021) 13:1670. doi: 10.3390/nu13051670
109. Saito T, Mizobuchi M, Kato T, Suzuki T, Fujiwara Y, Kanamori N, et al. One-year romosozumab treatment followed by one-year denosumab treatment for osteoporosis in patients on hemodialysis: an observational study. Calcif Tissue Int. (2023) 112:34–44. doi: 10.1007/s00223-022-01031-6
110. Yeung W, Palmer S, Strippoli G, Talbot B, Shah N, Hawley C, et al. Vitamin D therapy in adults with CKD: a systematic review and meta-analysis. Am J Kidney Dis. (2023) 82:543–58. doi: 10.1053/j.ajkd.2023.04.003
111. Rayego-Mateos S, Morgado-Pascual J, Valdivielso J, Sanz A, Bosch-Panadero E, Rodrigues-Díez R, et al. TRAF3 modulation: novel mechanism for the anti-inflammatory effects of the vitamin D receptor agonist paricalcitol in renal disease. J Am Soc Nephrol. (2020) 31:2026–42. doi: 10.1681/ASN.2019111206
112. Capossela L, Ferretti S, D’Alonzo S, Di Sarno L, Pansini V, Curatola A, et al. Bone disorders in pediatric chronic kidney disease: a literature review. Biology. (2023) 12:1395. doi: 10.3390/biology12111395
113. Tsai DH, Chuang AT, Liu KH, Shao SC, Lai EC. Sodium-glucose cotransporter 2 (SGLT2) inhibitors and risk of chronic kidney disease-mineral and bone disorders in patients with type 2 diabetes mellitus and stage 1-3 chronic kidney disease. CMAJ. (2025) 197:E178–89. doi: 10.1503/cmaj.240922
114. Evans M, Morgan A, Whyte M, Hanif W, Bain S, Kalra P, et al. New therapeutic horizons in chronic kidney disease: the role of SGLT2 inhibitors in clinical practice. Drugs. (2022) 82:97–108. doi: 10.1007/s40265-021-01655-2
115. Del Vecchio L, Beretta A, Jovane C, Peiti S, Genovesi SA. Role for SGLT-2 inhibitors in treating non-diabetic chronic kidney disease. Drugs. (2021) 81:1491–511. doi: 10.1007/s40265-021-01573-3
116. Giorgino F, Vora J, Fenici P, Solini A. Renoprotection with SGLT2 inhibitors in type 2 diabetes over a spectrum of cardiovascular and renal risk. Cardiovasc Diabetol. (2020) 19:196. doi: 10.1186/s12933-020-01163-9
117. Zhang B, Swanson W, Durdan M, Livingston H, Dodd M, Vidanapathirana S, et al. Affinity targeting of therapeutic proteins to the bone surface-local delivery of sclerostin-neutralizing antibody enhances efficacy. J Bone Miner Res. (2024) 39:717–28. doi: 10.1093/jbmr/zjae050
118. Yu S, Li D, Zhang N, Ni S, Sun M, Wang L, et al. Drug discovery of sclerostin inhibitors. Acta Pharm Sin B. (2022) 12:2150–70. doi: 10.1016/j.apsb.2022.01.012
119. Xiaohui T, Wang L, Yang X, Jiang H, Zhang N, Zhang H, et al. Sclerostin inhibition in rare bone diseases: molecular understanding and therapeutic perspectives. J Orthop Translat. (2024) 47:39–49. doi: 10.1016/j.jot.2024.05.004
120. Palmer S, Mavridis D, Johnson D, Tonelli M, Ruospo M, Strippoli G. Comparative effectiveness of calcimimetic agents for secondary hyperparathyroidism in adults: a systematic review and network meta-analysis. Am J Kidney Dis. (2020) 76:321–30. doi: 10.1053/j.ajkd.2020.02.439
121. Yuan Q, Wang J, Peng Z, Zhou Q, Xiao X, Xie Y, et al. Neutrophil-to-lymphocyte ratio and incident end-stage renal disease in Chinese patients with chronic kidney disease: results from the Chinese cohort study of chronic kidney disease (C-STRIDE). J Transl Med. (2019) 17:86. doi: 10.1186/s12967-019-1808-4
122. Filipska I, Winiarska A, Knysak M, Stompor T. Contribution of gut microbiota-derived uremic toxins to the cardiovascular system mineralization. Toxins. (2021) 13:274. doi: 10.3390/toxins13040274
123. Pietrzyk B, Smertka M, Chudek J. Sclerostin: intracellular mechanisms of action and its role in the pathogenesis of skeletal and vascular disorders. Adv Clin Exp Med. (2017) 26:1283–91. doi: 10.17219/acem/68739
124. Yoon S, Meyer M, Arevalo C, Tekguc M, Zhang C, Wang J, et al. A parathyroid hormone/salt-inducible kinase signaling axis controls renal vitamin D activation and organismal calcium homeostasis. J Clin Invest. (2023) 133:e163627. doi: 10.1172/JCI163627
Keywords: chronic kidney disease-mineral and bone disorder, immune dysregulation, vascular calcification, osteoimmunology, fibroblast growth factor 23
Citation: Xu B, Ma R, Wu Y, Liu C and Song X (2025) Immune mechanisms in chronic kidney disease-mineral and bone disorder: current insights and therapeutic implications. Front. Med. 12:1678640. doi: 10.3389/fmed.2025.1678640
Received: 03 August 2025; Accepted: 16 September 2025;
Published: 09 October 2025.
Edited by:
Xin Hu, National Center for Child Health and Development (NCCHD), JapanReviewed by:
Shao-Wei Li, Taizhou Hospital Affiliated to Wenzhou Medical University, ChinaLilio Hu, University of Bologna, Italy
Jing Wang, School of Rehabilitation Science, China
Copyright © 2025 Xu, Ma, Wu, Liu and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangrong Song, MTczMjMxMDAzMjlAMTYzLmNvbQ==