 Miguel Marco-Bonilla1
Miguel Marco-Bonilla1 Maria Fresnadillo1†
Maria Fresnadillo1† Macarena de la Riva-Bueno1†
Macarena de la Riva-Bueno1† Gabriel Herrero-Beaumont1
Gabriel Herrero-Beaumont1 Miguel Angel González-Gay1,2*
Miguel Angel González-Gay1,2* Raquel Largo1‡
Raquel Largo1‡ Aránzazu Mediero1‡
Aránzazu Mediero1‡- 1Joint and Bone Research Unit, FIIS Fundación Jiménez Díaz UAM, Madrid, Spain
- 2Medicine and Psychiatry Department, University of Cantabria, Santander, Spain
Joint inflammation is the most prominent feature of rheumatoid arthritis (RA), but this disease can affect practically any organ of the body. The association between RA and comorbidities is multifaceted, involving traditional risk factors, chronic inflammation, and the effects of medications. A large number of animal models have been developed for the study of RA. All of them developed histopathological changes, such as human diseases, and often experienced other comorbidities. The choice of one model or another depends on several factors. It is important to bear in mind, for example, the study of pathophysiological mechanisms, the progression, and the activated autoimmunity, among others. It is also necessary to know what comorbidities are described in each model, as the selection may depend on the possibility of replicating these comorbidities. In this review, we will focus on the study of cardiovascular, musculoskeletal, and hepatic comorbidities in the four most used and induced RA models: collagen-induced arthritis (CIA), adjuvant-induced arthritis (AIA), pristane-induced arthritis (PIA), and serum transfer K/BxN. In this manuscript we offer guidance on how these models replicate RA key comorbidities and how to choose the most suitable RA model.
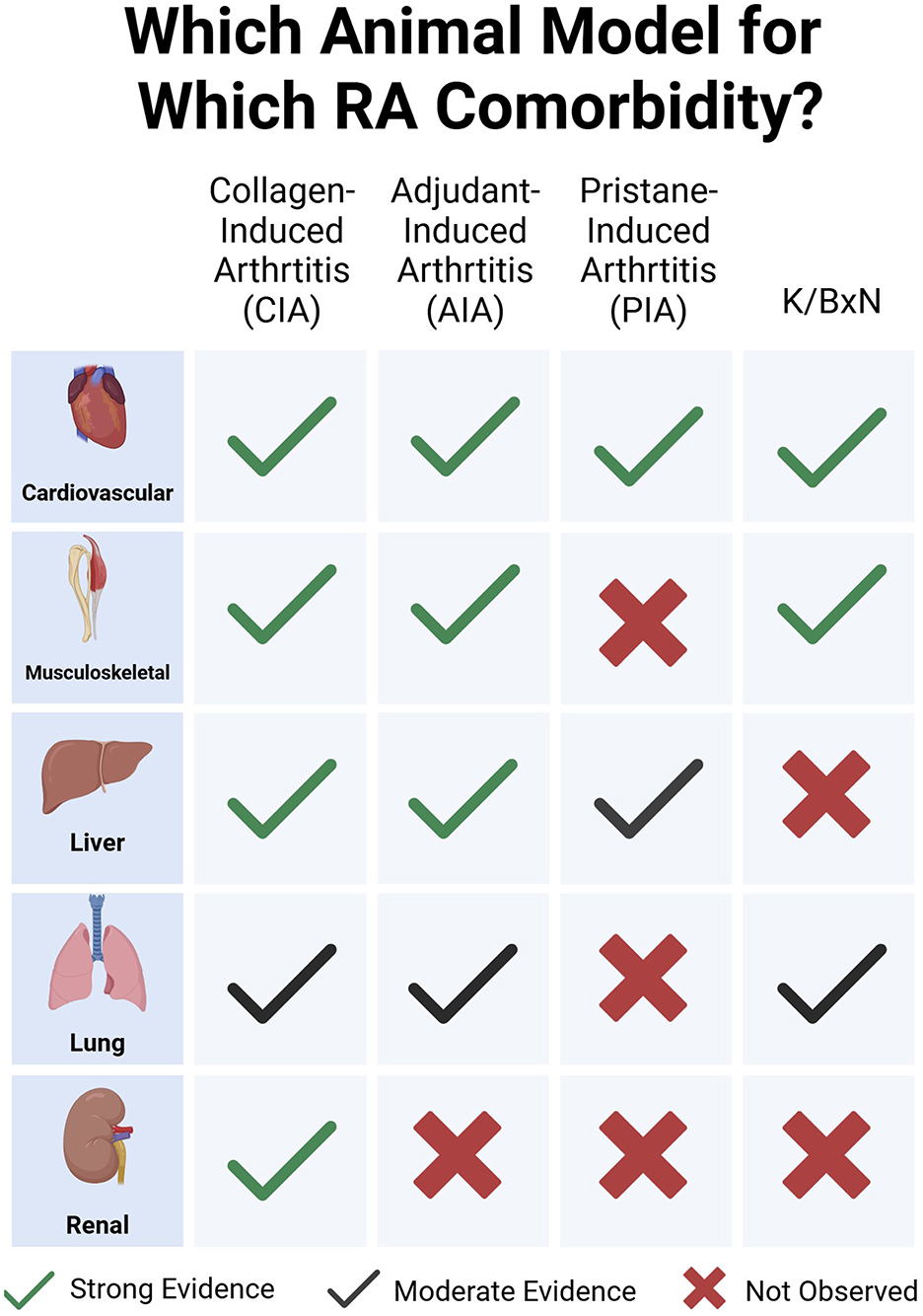
Graphical Abstract.
1 Comorbidities in RA
Rheumatoid arthritis (RA) is a systemic autoimmune disorder marked by chronic synovial inflammation, leading to joint damage and disability. It can occur at any age and affects both sexes, though it is more prevalent in women (1). It is estimated that 31.7 million individuals will be living with RA worldwide by 2050, constituting a major global health burden, as measured in disability-adjusted life years (DALYs) (2). Although joint inflammation is the most prominent feature, RA can affect practically any organ of the body (3). The association between RA and comorbidities is multifaceted, involving the medications used to treat RA, traditional risk factors, and the presence of chronic systemic inflammation (3–5). Based on the cross-sectional COMOrbidities in RA (COMORA) study, the most commonly observed comorbidities (past or current) in patients with RA are depression, asthma, and cardiovascular disease (CVD), the leading cause of death in RA, including myocardial infarction (MI) and stroke, solid-organ malignancies, and chronic obstructive pulmonary disease (4, 5). Moreover, osteoporotic fractures are more commonly observed in patients with RA and they significantly affect the functional decline of the patient (6). Muscle loss is commonly observed in patients with RA (7). According to the updated EWGSOP2 guideline, the prevalence of sarcopenia is 11 times higher in patients with RA than in controls (8). In fact, muscle loss has been recognized as an important contributor to comorbidity and reduced life expectancy in RA (9). Regarding gastrointestinal comorbidity in patients with RA, the most common is liver dysfunction, followed by intrahepatic hemorrhage, hepatosplenomegaly, cirrhosis, and necrotic pancreatitis (4). RA can also affect both the central and peripheral nervous systems, with neurological clinical manifestations undetected or attributed to arthritic pain, causing diagnostic delays (4).
Although our understanding of RA pathogenesis has advanced in recent years, RA remains a global research hotspot due to the lack of preventive or curative treatments, the presence of drug-refractory cases, and the wide range of comorbidities and extra-articular manifestations frequently observed (2, 4, 5).
Recent epidemiological studies in 2024 report that the most prevalent comorbidities in patients with RA are hypertension (36.4%−56% of patients with RA), thyroid disorders (21.5%−34.8%), dyslipidemia (19.5%), and obesity (14.2%−16.9%), followed by osteoporosis (19.1%), osteoarthritis (9.2%), etc. (10, 11). Most patients with RA suffer from these comorbidities. Around 42.8% develop at least one comorbidity, and approximately 28% develop up to three (12). The high prevalence and clinical impact of these comorbidities highlight why it is important to study RA as a systemic disorder. Although studies in humans would be ideal, they have many limitations that lead us to use animal models to study RA and its comorbidities (13). In humans, it is impossible to fully control genetic or environmental variables. However, most of the RA models resemble the pathological characteristics shown in human diseases (14). The same occurs in the case of testing new treatments; studies on patient response have intrinsic limitations, especially in the preclinical phase (15). Furthermore, access to human tissue is scarce and often restricted, limiting the ability to investigate pathological processes in depth. All these features make it essential to use animal models to study RA comorbidities, as they offer insights that would be difficult to obtain from human studies.
2 Animal models of RA
A large number of animal models have been developed for the study of RA (16–19). The choice of one model or another depends on several factors, such as the study of the mechanisms of the disease, its progression, severity, activated autoimmunity, the reliability and simplicity of some models, and the efficiency to predict drug efficacy in humans (18, 20). RA models are relatively easy to perform, have good reproducibility of data, and are generally of short duration. Most RA models exhibit pathological features similar to the human disease, although important differences exist, such as the rapid progression of RA in animal models, primarily due to acute inflammatory responses, and a tendency in rodents to exhibit both marked bone resorption and new bone formation in response to joint inflammation (20). Therefore, when choosing an animal model for RA, we must carefully analyze the specific aspects of the disease and the specific objective of each study.
Animal models of RA can be broadly divided into two main categories: spontaneous models, which include animals developing arthritis via genetic modifications; and induced models in which arthritis arises following chemical or immunological induction. Spontaneous models typically progress naturally and result in chronic, non-resolving disease, whereas induced models often self-resolve over time (17, 19).
Rodents represent the most commonly employed models for investigating the pathogenesis and progression of RA. However, significant genetic divergences between rodents and humans influence the development of RA and complicate the direct translational applicability of experimental findings to clinical settings. Therefore, alternative animal models, including rabbits, guinea pigs, and non-human primates (NHPs), have been used to overcome these limitations and more accurately replicate the human disease (18, 20).
Among the various experimental models of arthritis, the following are among the most widely used:
• The collagen-induced arthritis (CIA) mouse model is one of the most widely used experimental models for studying RA (21). In this mouse model, the most commonly used strains are those that are genetically susceptible to developing autoimmune arthritis in response to type II collagen immunization. In this regard, susceptibility to CIA in mice is closely linked to their MHC haplotype, particularly the H-2q haplotype (22).
• The most widely used strain is DBA/1, particularly the DBA/1J sub-strain. These mice are highly susceptible to CIA when immunized with type II collagen (CII) in combination with adjuvants. As a result, DBA/1 mice are considered the gold standard for CIA studies and are extensively used in both pathogenesis research and the preclinical testing of therapeutic agents (23). In contrast, C57BL/6 mice are naturally resistant to CIA. However, they are frequently used in research due to the availability of numerous transgenic and knockout lines. Therefore, CIA can be induced in C57BL/6 mice under modified conditions, such as with specific adjuvants or genetic alterations, making them valuable for studying the role of individual genes in arthritis (23). Another commonly referenced strain is BALB/c, which is also generally resistant to CIA. The B10.Q strain is another CIA-susceptible model (24). These mice are used to investigate the genetic and immunological basis of arthritis susceptibility.
Mice are injected with a solution of CII, often from bovine or chicken sources, emulsified in an adjuvant, commonly Complete Freund's Adjuvant (CFA), which contains killed mycobacteria and enhances the immune response (21). This injection is usually administered at the base of the tail or in the footpad. In some cases, a second dose of CII, often in Incomplete Freund's Adjuvant (IFA), is given approximately 1 week later to boost the immune response (21). Arthritis typically develops within a few weeks following the sensitization phase, leading to the manifestation of clinical signs such as joint swelling, redness, and limping (25). The model may not fully replicate the episodic nature of RA, including spontaneous remissions and exacerbations (21, 25).
The adjuvant-induced arthritis (AIA) model is a well-established system primarily employed to explore the pathogenesis of RA and evaluate potential therapeutic agents. It is known for its simplicity and reliability (26). A single unilateral subcutaneous injection of CFA is administered, usually in the hind foot or at the base of the tail (26). This injection triggers an inflammatory response, leading to the development of arthritis with joint swelling and stiffness, reduced mobility and pain, and often affects multiple joints, mimicking the symmetrical involvement observed in human RA (26, 27). One of the main limitations of the AIA model is its self-limiting nature, where the disease typically resolves after a few weeks, which may not accurately reflect the chronic progression seen in human RA (18).
The pristane-induced arthritis (PIA) model is primarily utilized to investigate the mechanisms of inflammatory arthritis, particularly those involving T cell activation and the role of autoantibodies in the disease process (28, 29). The model is induced by injecting pristane, a mineral oil derivative, typically administered intradermally or subcutaneously. Arthritis usually develops within a few weeks after pristane injection (28, 29). The onset can be influenced by factors such as mouse strain and the specific injection site. The PIA model exhibits several clinical features reminiscent of human RA, including joint swelling and inflammation, particularly in the hind limbs, pain, and reduced mobility in affected joints (28, 29). The model can present a polyarticular pattern of arthritis, affecting multiple joints symmetrically. The model primarily emphasizes T cell-mediated mechanisms and may not fully represent other aspects of RA pathogenesis, such as antibody-mediated processes (28, 29).
The K/BxN model is designed to investigate the pathogenesis of RA, particularly the role of autoantibodies and T cells in the development of joint inflammation and damage (16). The K/BxN model is derived from a cross between KRN mice, which express a transgenic T cell receptor (TCR) specific for glucose-6-phosphate isomerase (GPI), and Non-Obese Diabetic (NOD) mice, which have a strong autoimmune background. K/BxN mice express a TCR that recognizes GPI, leading to a strong autoimmune response when they are exposed to their specific antigen (30). Unlike other models that require specific antigen immunization, K/BxN mice spontaneously develop arthritis due to the expression of the transgenic TCR and the production of anti-GPI antibodies. While the spontaneous development of arthritis mimics some aspects of human disease, it may not fully capture all features of RA, particularly those that arise from environmental triggers (30). The model focuses heavily on T cell and antibody-mediated mechanisms, potentially overlooking other important pathways involved in RA (16).
All these models developed histopathological changes, such as synovial hyperplasia, infiltration of immune cells (e.g., T cells, B cells, and macrophages), cartilage destruction, and bone erosion (17). In addition to the inflammatory joint pathology, patients frequently suffer from comorbidities, such as muscle loss (rheumatoid sarcopenia) and cardiovascular disease (8, 31). Consequently, the selection of a particular animal model may depend on the possibility of replicating these comorbidities (Figure 1).
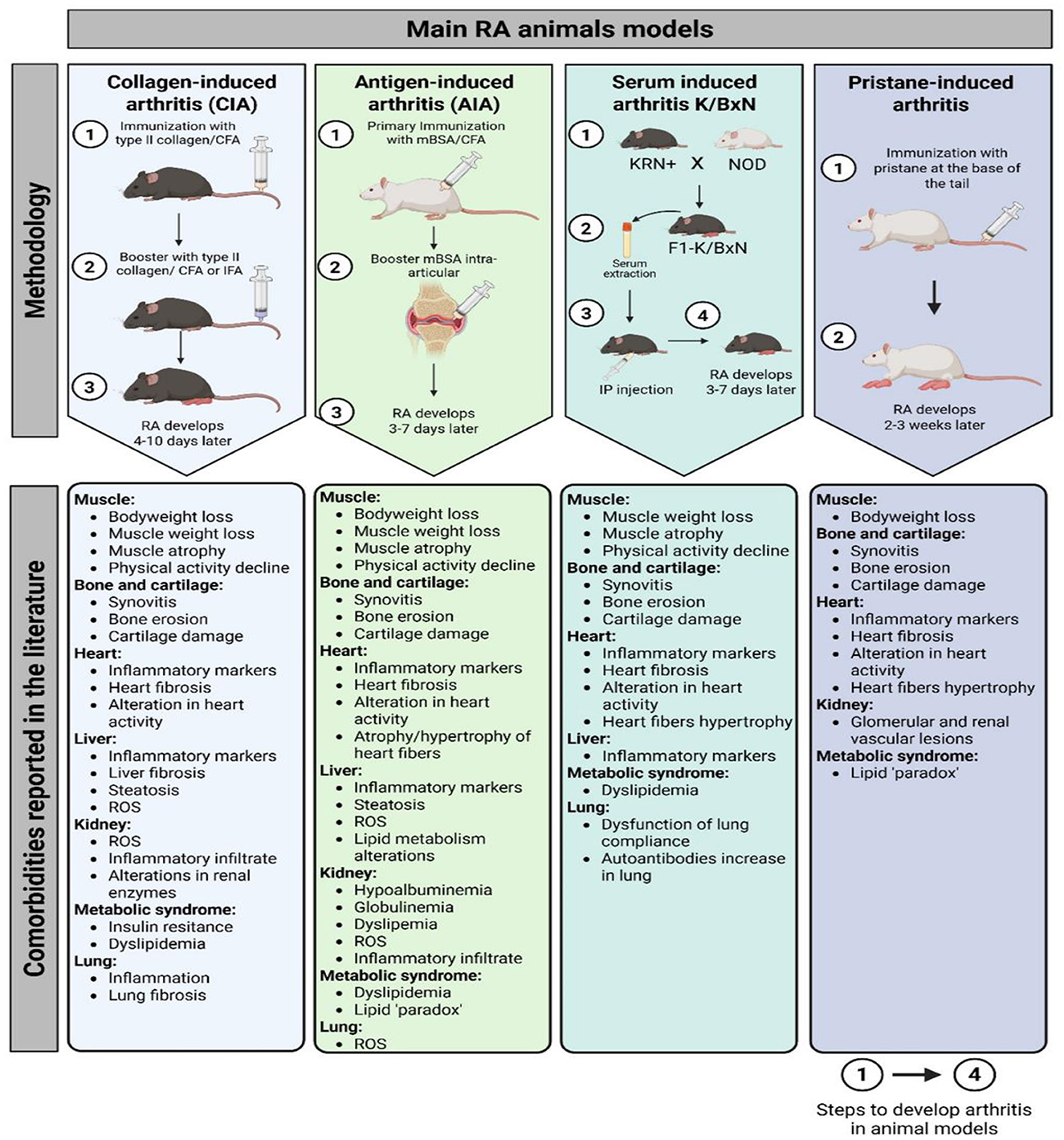
Figure 1. Main RA animal models methodology and related comorbidities reported in the literature. CIA, collagen-induced arthritis; AIA, antigen-induced arthritis; CFA, complete Freund's adjuvant; IFA, incomplete Freund's adjuvant; RA, rheumatoid arthritis; ROS, reactive oxygen species; mBSA, methylated bovine serum albumin; NOD, non-obese diabetic; IP, intraperitoneal.
There is a wide literature already covering the range of arthritis animal models and their characteristics in comparison to human disease pathophysiology and therapeutic responses (32). Being aware of the wide variety of existing arthritis models, in this review, we will focus on four of the induced models described in this section: CIA, AIA, PIA, and K/BxN, and how comorbidities related to the cardiovascular, musculoskeletal, and digestive systems are studied in these models. Although most RA comorbidities are represented in animal models, some are poorly covered. This is the case for neurological affections such as depression or chronic fatigue, for which there is little evidence (33). The same applies to pain (34). Therefore, we will not address them in this review. In addition, some other animal models, apart from the four mentioned, will not be discussed in this review, as they do not adequately mimic RA comorbidities (35).
3 Study of RA comorbidities in different animal models
3.1 Cardiovascular disease (CVD) studies in RA animal models
CVD is the most prevalent comorbidity in patients with RA, resulting in a more severe disease burden, with a 1.5 times higher risk of CVD in patients with RA when compared to the general population (36). Both traditional risks and inflammation contribute to the progression of atherosclerosis and cardiovascular issues in patients with RA (31, 37). Patients with RA have a higher prevalence of coronary artery disease (CAD) compared to the healthy population. In RA, the lipid profile shows reduced levels of total and low-density lipoprotein (LDL) cholesterol during high-grade inflammation (38). CVD in RA is linked to a dyslipidemic pattern and severe systemic inflammation (39). Additionally, traditional CVD risk factors, such as hypertension in RA may be modulated by the inflammatory state (24). Some of the preclinical animal studies discussed above can contribute to interpreting the pathophysiological connection between RA and CVD, as well as to identifying underlying mechanisms and potential therapeutic targets. CVD studies in RA animal models include cardiac morphological and histological abnormalities (such as cardiac hypertrophy, structural impairments, and fibrosis), as well as ultrastructural changes (40). Preclinical studies also explore cardiac functional parameters either using cardiac ultrasound or invasively after catheterization (40). Coronary atherosclerosis, coronary endothelial dysfunction, arterial hypertension, and heart rhythm disorders are also well described in RA animal models, but although myocardial infarction is the leading cause of mortality in RA, there is a lack of RA animal studies (40). Among cardiac arrhythmias, atrial fibrillation (AF) and ventricular arrhythmias are the most studied ones. The study of this AF using animal models holds significant translational relevance, as it enables a more precise understanding of the molecular mechanisms involved in disease development. However, clinical studies face challenges in directly determining the causes of AF. Consequently, experimental investigations are essential to elucidate the relationship between AF and atrial remodeling. In CIA models, findings resemble cardiovascular outcomes observed in humans (41, 42).
The most common studies on cardiac impairments have been performed in CIA, AIA, and PIA animal models, with few studies being carried out in the K/BxN model (Table 1) (40). In this regard, Zihao et al. (43) detected increased infiltration of inflammatory cells and fibrosis in the ventricular tissues of CIA mice. Additionally, elevated expression of pro-inflammatory genes such as TNF-α, IL-6, IL-17, and MMP3 was observed in isolated ventricular cardiomyocytes and cardiac fibroblasts in these mice (43). In a parallel study, the treatment with Liquiritigenin, a triterpene with anti-inflammatory properties, dismissed the expression of inflammatory factors (TNF-α, IL-1β, and IL-6) and pro-fibrotic genes (fibronectin, and collagen I and III) in the heart, and reduced the fibrotic markers, such as TGF-b1 and phos-Smad2/3, in cardiac tissue in mice (44). In male DBA/1J mice, the response of the aorta to norepinephrine and acetylcholine with or without endothelium did not change in comparison with healthy mice, although the CIA-induced RA mice presented an increment of inducible nitric oxide synthase (45). The prevalence of atrial fibrillation (AF) was also studied using the CIA model (41, 46). In atrial tissue of female Wistar rats, both IL-6 and TNF-α levels were incremented with a high AF inducibility and duration in the arthritic animals that correlated with the serum levels of the pro-inflammatory cytokines (41, 42). Furthermore, in this model, increased expression of TGF-β and αSMA in the atrium was observed, indicating a fibrotic process in the cardiac tissue (42). The CIA animal model is also used to study the reduced heart rate variability (HRV) experienced by patients with RA (47). In this regard, reduced HRV can indicate problems with the autonomic nervous system, which is often seen in patients with RA. In rats, the low-frequency/high-frequency ratio was increased in the first weeks of arthritis induction in comparison with healthy animals (48). This alteration correlates with the CIA inflammatory phase (48). Deceleration and acceleration capacity, both measures of heart rate variability, were altered in CIA rats (48).
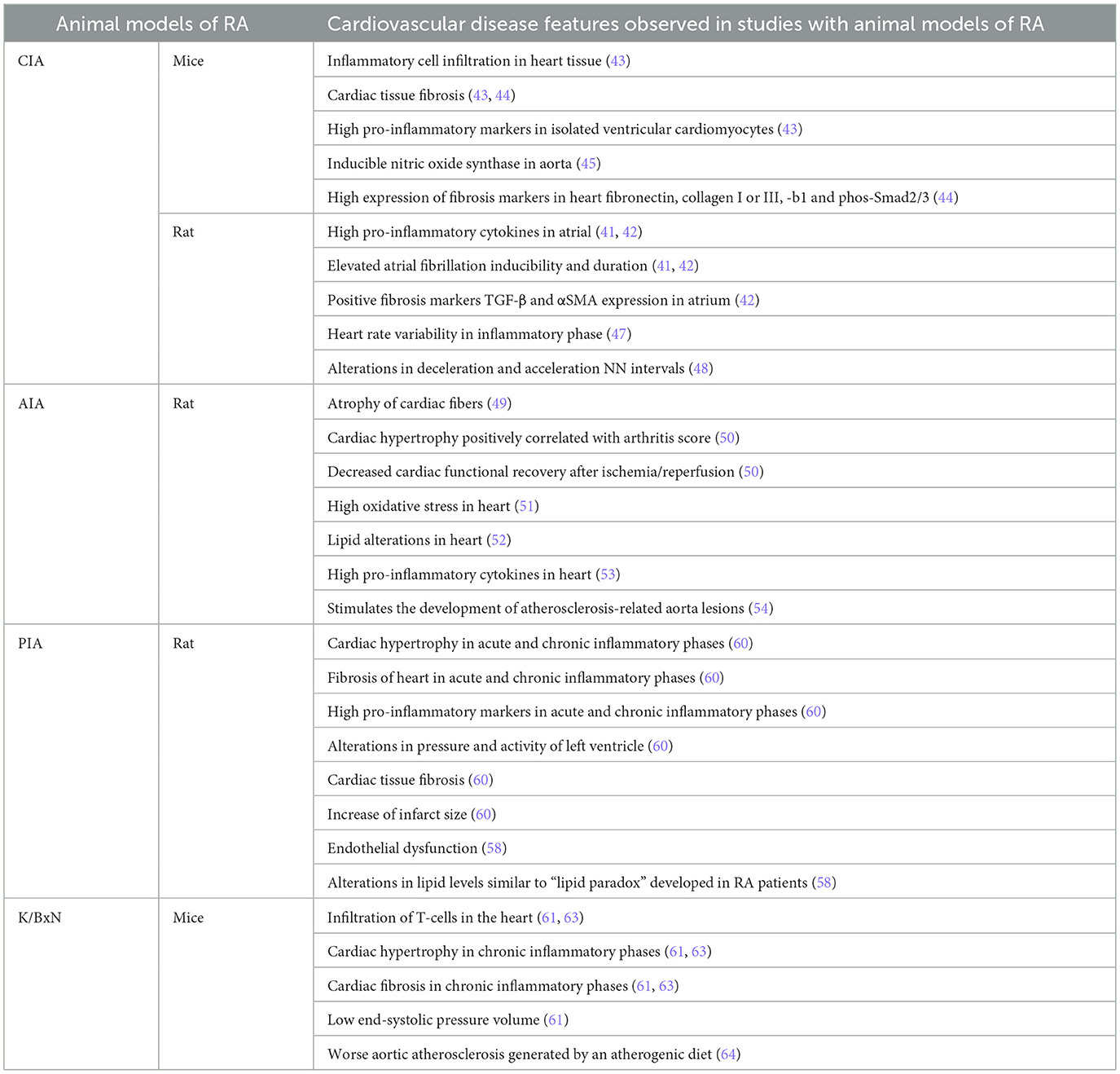
Table 1. Cardiovascular comorbidities in RA animal model studies.
In male AIA rats, the histology of cardiac tissue revealed atrophic fibers at day 40 of arthritis induction (49). In a parallel study in isolated hearts from AIA rats, a reduced coronary acetylcholine-induced relaxation associated with cardiac hypertrophy was developed, whose positivity correlated with plasma levels of endothelin-1, angiotensin-II, and arthritis score (50). The same manuscript reported decreased cardiac functional recovery, and high myeloperoxidase activity and infarct size after ischemia/reperfusion in arthritic animals (50). On the other hand, Shubert et al. (51) reported that AIA rats have several modifications in the oxidative state of the heart, including an increase of oxidative stress, protein damage, and lipid damage in the whole tissue. The use of tofacitinib in AIA rats decreased total cholesterol and low-density lipoprotein (LDL) cholesterol, but did not modulate the alterations in blood pressure or heart rate (52). In another study, PGE synthase, Cyclooxygenase (COX)-2, and IL-1β were upregulated in the heart tissue (53). A few years ago, a study assessed how chronic arthritis causes vascular lesions in rabbits with pre-existing atherosclerosis. For this purpose, the investigators developed an animal model combining chronic inflammatory arthritis with atherosclerosis to study their interaction. Their findings showed that chronic arthritis significantly worsened vascular damage in rabbits, suggesting that sustained inflammation from arthritis can accelerate the progression of atherosclerosis. This model highlighted the link between chronic inflammatory diseases and cardiovascular complications, offering insights into the mechanisms by which arthritis may increase cardiovascular risk (54). This indicates that RA is an independent risk factor for the development of atherosclerotic lesions. In the same animal model, treatment with chondroitin sulfate was able to reduce markers of systemic inflammation as well as PBMCs' pro-inflammatory activation (55). Chondroitin sulfate diminished the size of the femoral neointima lesions, and only 11% of chondroitin sulfate-treated rabbits developed early atherosclerotic lesions in the aorta (55). When these rabbits were treated with glucosamine sulfate, an inhibition in NF-κB activation in PBMCs was observed, indicating that this is probably the mode of action for sulphated glucosamine (56). Finally, when AIA rabbits with induced endothelial injury of the femoral artery were fed with an atherogenic diet and treated with celecoxib, serum levels of CRP and IL-6 were reduced. However, the increased expression of COX-2 and CCL2 remained unchanged. Celecoxib blocked NF-κB activation in PBMCs, but it did not affect the lesions in the femoral artery (57).
The PIA model mimics the features of chronic inflammatory arthritis. It has been found useful for long-term pharmacological studies and to interpret the complexity of CVD in RA (58).
The PIA model is able to reproduce some cardiovascular features of patients with RA, such as the presence of endothelial dysfunction in both macro- and microvascular beds, a link between inflammation and macrovascular endothelial dysfunction. This model also replicates changes in lipid levels mimicking the “lipid paradox” (58). The lipid paradox associated with rheumatoid arthritis is understood as an alteration in the lipid profile of these patients, which occurs due to the high inflammatory burden. An inverse relationship is observed between cholesterol levels and cardiovascular risk in this population (59).
Peyronnel et al. studied the effects of treadmill exercise on cardiac health in rats with PIA. Regular treadmill exercise reduced cardiac fibrosis and inflammation in these rats. Additionally, exercise decreased the heart's vulnerability to ischemia–reperfusion injury, which is the damage caused when blood supply returns to the tissue after a period of ischemia or lack of oxygen. These findings suggest that physical exercise may provide protective cardiovascular benefits in the context of chronic inflammatory arthritis (60).
With regard to the K/BxN animal model, K/BxN F1 mice presented increased infiltration of activated T-cells in the heart at week 8 and cardiac hypertrophy and fibrosis at week 16 in comparison with KRN mice at the same week (61). Cardiomyopathy was also presented in the K/BxN model in comparison with healthy mice (61). In arthritic mice, increased MYH7 expression in the heart and reduced end-systolic pressure-volume relationships were observed, indicating progression toward dilated cardiomyopathy (62). All these pathologies were prevented with 16 weeks of exercise in K/BxN mice (63). Moreover, on an atherogenic diet, K/BxN mice displayed a 22-fold increase in aortic atherosclerosis when compared to control mice (64).
3.2 Musculoskeletal system disorders in RA animal models
3.2.1 Muscle studies in RA animal models
Rheumatoid cachexia (RC) is a syndrome characterized by weight loss, muscle wasting, and overall weakness associated with RA (65). RC affects 11%−26% of patients with RA worldwide, although some studies report a prevalence as high as two-thirds of patients with RA (9, 66). It results from a combination of inflammatory processes, metabolic changes, and the body's response to chronic disease (67). Chronic inflammation in RA leads to the release of pro-inflammatory cytokines (TNFα, IL-1β, and IL-6), which can disrupt normal metabolism and appetite (68). Muscle protein breakdown, driven by proteases activated through the ubiquitin–proteasome pathway involving MuRF1 and Atrogin-1, can exceed muscle protein synthesis, ultimately leading to sarcopenia (69). This is often exacerbated by physical inactivity due to pain and joint damage (70). Patients may experience significant fatigue, contributing to a decreased ability to perform daily activities, further worsening the cycle of inactivity and muscle loss (71).
The CIA model remains one of the most employed animal models for investigating muscle-related comorbidities in RA due to its ability to mimic both joint pathology and systemic manifestations (Table 2). The Filippin group demonstrated progressive weight loss beginning at week 2 following arthritis induction in the CIA model (72). Notably, mice exhibited reduced motor activity and speed within the first week. Histological analysis at 45 days post-induction revealed atrophy of the gastrocnemius (GA) and tibialis anterior (TA) muscles, which correlated with elevated serum IL-6 levels (72). In a long-term CIA model using male DBA/1J mice, a reduction in grip strength, decreased weights of the tibialis anterior (TA), and gastrocnemius (GA) were reported (73). The same CIA model was developed by the Suginohara group. In this study, the arthritic animals presented soleus, plantaris, and GA with less weight than healthy mice, with high systemic IL-6 and TNF-α levels. In this study, treatment with Ninjin'yoeito, a traditional Japanese medicine, reduced inflammatory cytokine levels and prevented muscle loss in arthritic animals at higher doses (74). In the same way, female Wistar rats induced to arthritis presented weight loss and GA atrophy (75). Positive staining for IL-1β and high expression of Murf1 were reported in this study, although myostatin (MSTN) levels did not increase in the GA of CIA rats, as observed in patients with RA (75). In a parallel study with male Sprague–Dawley rats immunized with bovine type II collagen, less muscle CSA was observed, with a decrease in the first protein of muscle differentiation, MyoD in GA, and there was no modulation in the late muscle differentiation protein, myogenin, indicating changes in muscle myogenesis and muscle atrophy (76). In this study, Murf1 was not decreased in GA, suggesting differences in atrogenes expression, which play a critical role in controlling protein turnover in skeletal muscle to maintain muscle function, between genders in rats. MSTN expression was also not modulated, consistent with the previous study in rats (76). In a study of RC pharmacological treatment, the Oliveira group compared the effect of methotrexate and etanercept in male DBA1/J mice. CIA mice treated with methotrexate did not counteract the GA/TA weight loss and the high expression of Murf1 in GA, but etanercept prevented muscle atrophy, and Murf1 decreased the expression in GA, suggesting a different response to muscle loss, dependent on treatment and independent of the inflammatory state (77).
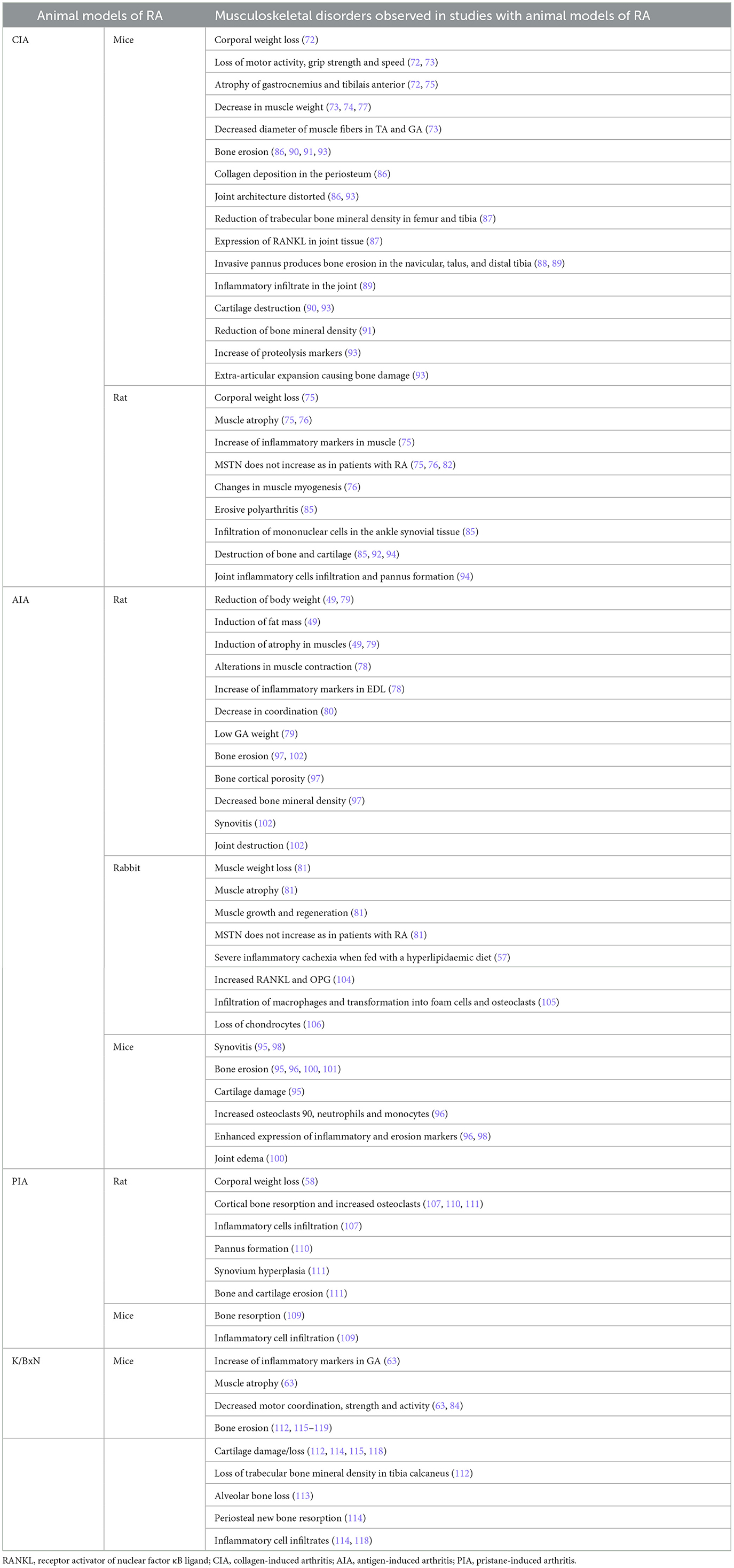
Table 2. Musculoskeletal comorbidities in RA animal model studies.
Muscle loss is not only present in the CIA model. Strong evidence of muscle loss is reported in the AIA model (Table 2). Pita et al. (49) observed weight loss with induction of fat mass and atrophy of the soleus muscle in AIA male Wistar rats at day 15 of induction. The ATPase activity of these myosins was negatively correlated with the duration of muscle contraction, indicating that the ATPase activity of myosin may play a significant role in influencing the speed of muscle contraction (49). In AIA rats, the activity of ATPase and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase expression decreased, suggesting that changes in the transport of Ca2+ cause alterations in the muscle contraction of arthritic AIA mice (78). Also, an increase of TNF-α expression was observed in EDL (78). The decrease in body weight in AIA rats was also reported by the Ghouri group at day 8 of arthritis induction, accompanied by GA atrophy (79). Furthermore, they observed that motor coordination in the rotating rod was dismissed in AIA rats (80). Little et al. showed in rabbit experimental AIA that increased inflammation produces muscle loss, contributes to atrophy, structural derangement, and increased atrogene expression. A myonuclear expansion was also observed, which is an anatomical marker of muscle growth and regeneration (81). Paradoxically, this model presented a decrease in myostatin levels in serum and muscle (81). This does not correlate with levels observed in patients with RA (82). However, these data, together with the downregulation of myostatin signaling, the increase MyoD and myogenin, and decreased pSTAT3 signaling, reflect attempts to repair the catabolic insult of inflammatory arthritis (81, 83). The data suggest the existence of a compensatory anabolic activation in AIA rabbits as these animals displayed signs of simultaneous muscle wasting and repair (81). When AIA rabbits were fed with a hyperlipidemic diet they developed a severe inflammatory cachexia, and the inhibition of COX-2 by celecoxib improved this state, suggesting that COX products may play an important role in cachexia development (57).
Only one reference has been found regarding the use of the PIA model in muscle studies. Chouk et al. (58) reported weight loss in the first days of arthritis induction in rats (Table 2).
Finally, in the K/BxN mice model (Table 2), the Huffman group observed an increase in systemic and GA IL-6 expression, which was negatively correlated with GA weight (63). Also, high levels of atrogin-1 in GA and less motor coordination and strength were evidenced in K/BxN mice. Exercise in K/BxN mice for 12 weeks counteracted the muscle atrophy and atrogenes expression, however, not the high levels of IL6 in GA (63). Doucet et al. (84) used smart cages for locomotor activity measurements for 23-h periods in C57BL/6 mice, finding that arthritic mice had a significant reduction in locomotor activity (speed, number of active movements and rear movements, travel distance) on days 7–8 of arthritis compared to days 0 and 13–14. In the same work, authors indicate that treatment with a fish oil diet induced an impact on both travel distance and rear time, being increased with the diet during the peak of arthritis at day 8. This was accompanied by no changes in clinical index, but a significant attenuation in the ankle when compared to the chow-fed group (84).
In conclusion, based on the evidence reviewed, the CIA model appears to be the most suitable animal model for studying and replicating rheumatoid sarcopenia. It consistently shows muscle mass loss and a decreased CSA of muscle fibers mediated by IL-6, TNF-α, and IL-1β. In addition, the CIA model also reproduces accurately the chronic and progressive course of RA as observed in humans (72, 77). In contrast, the AIA model is the least suitable for this purpose, as it induces an acute and limiting inflammatory response that makes it more difficult to reflect the prolonged muscle wasting characteristic of rheumatoid sarcopenia (78–80). Finally, the K/BxN model replicates muscle atrophy and physical activity decline, but only driven by IL-6 inflammatory signaling (Figure 2).
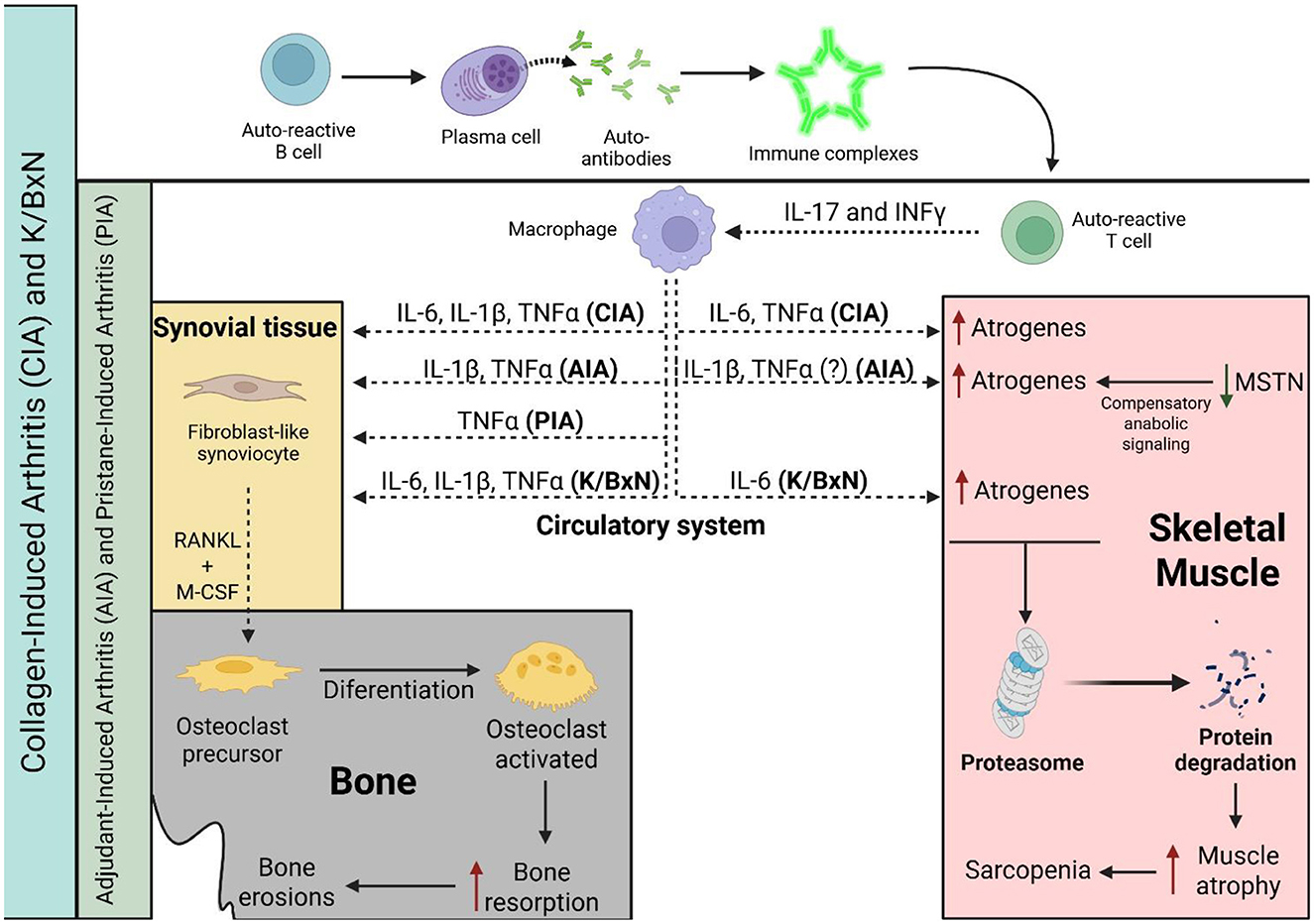
Figure 2. Progression of musculoskeletal and bone comorbidities is associated with specific inflammatory cytokines or autoantibodies production in RA animal models. In the CIA and K/BxN models, cytokine release by macrophages is driven by autoantibody signaling, whereas in the PIA and AIA models, the inflammatory response is primarily T cell-dependent. A solid black line indicates progression or activation, a dashed black line indicates the release of molecules, a red line indicates induction, and a green line indicates a reduction in levels. CIA, collagen-induced arthritis; AIA, antigen-induced arthritis; PIA, pristane-induced arthritis; IL-6, interleukin-6; TNF-α, tumor necrosis factor alpha; IL-1β, interleukin-1 beta; and MSTN, myostatin.
3.2.2 Bone and cartilage studies in RA animal models
Bone remodeling in RA is a frequent comorbidity mediated by local and/or systemic alterations in the levels of proinflammatory cytokines that are known to stimulate bone resorption and can lead to osteoporosis and fractures. Both induced and genetically manipulated arthritis models have been extensively used to investigate bone resorption and formation in RA (Table 2 and Figure 2).
The first model described in 1977 was the CIA model. Trentham et al. (85) showed that immunization of rats developed an erosive polyarthritis that was associated with an autoimmune response against cartilage. The histology of these arthritic rats resembles RA, producing an intense infiltration of mononuclear cells in the ankle synovial tissue and destruction of bone and cartilage (85). Since this first approach, many studies on bone loss in the CIA model have appeared on the scene. The CIA model has been used to analyze the kinetics, histological, and molecular changes in bone and associate them with the clinical disease development (86). In 2015, Denninger et al. (86) demonstrated that the main histopathological changes in inflammation and bone structure happened during the first 2 weeks on the onset of clinical symptoms in the joint, and once inflammation declined, it is the bone remodeling that predominated. This fact makes the CIA model a suitable candidate to study the relationship between inflammation and bone formation in RA. In mice, a reduction of trabecular bone mineral density was observed in the femur and tibia after 45 days of induction, this reduction being enhanced with the administration of adiponectin (87). No differences were observed in cortical bone density analysis, and a positive expression of RANKL was observed in the joint (87). MicroCT imaging of bone volume in the tarsal region in CIA mice showed a decrease at 31–35 days following the initial collagen immunization, with pannus causing bone erosion in the navicular, talus, and distal tibia that was counteracted with prednisone (88). In 2018, Chen et al. (89) showed that treatment with Apremilast, a phosphodiesterase 4 (PDE4) inhibitor, which blocks immune and inflammatory responses, counteracted bone erosion and inflammatory infiltrate in the joint in the murine CIA model. Moreover, when bone erosion was studied at day 42 of immunization after 20 days of treatment with etanercept, abatacept, or zoledronic acid, no changes in bone erosion or cartilage destruction in femorotibial joints were observed, in contrast to dexamethasone treatment (90). A similar effect was observed when mice were treated with a neutralizing anti-RANKL monoclonal antibody (IK22-5) (91). Recently, Lin et al. (92), in order to evaluate the similarity of CIA models with the features of pre-RA (high conversion risk time period before clinical diagnosis), explored changes in antibodies, joint inflammation, erosion, and gut microbiota in rats. These researchers showed that both std-CIA (standard CIA model) and Mono-CIA (single collagen-induced group) could successfully cause RA symptoms, including joint swelling and bone erosion; meanwhile, a much milder model, half-CIA (half-dose collagen-induced group), induced only mild swelling in rats (92). Li et al. (93) reported that CIA mice developed joint space changes and bone damage, with extra-articular expansion being observed. They observed a significant loss of medullary trabecular bone and a higher OARSI score in CIA mice, concomitant with Aggrecan upregulation and metalloproteinase (as MMP3) downregulation (93). Early intra-articular Alpha2-macroglobulin treatment exerted an anti-inflammatory effect and attenuated bone and cartilage damage in this model (93). Yan et al. (94), in a recent manuscript, summarize the clinical signs of RA in the CIA model, including body weight loss, higher arthritis and paw indexes, cartilage degeneration, bone destruction, inflammatory cells infiltration, and pannus formation. All these clinical and molecular signs were ameliorated with Jolkinolide B, an ent-abietane-type diterpenoid found in Euphorbia jolkini, treatment that showed a reduction in arthritis progression and disease severity in a JAK2/STAT3 mechanism (94).
Bone remodeling at the onset of RA has also been studied using the AIA animal model. It has been observed that AIA develops synovitis, bone erosion, and cartilage damage after 14 days of primary immunization (95). Decreased periarticular trabecular bone mineral density and increased presence of osteoclasts, neutrophils, and monocytes have also been found after 14 days of immunization (96). It has been observed that cortical bone deterioration started before AIA onset at day 12 post-immunization (97). Cortical porosity was the earliest structural cortical parameter to be altered, starting at day 8 post-immunization, followed by cortical thickness and mineral density decreased from day 10, and a lower CT area after day 12 (97). One of the characteristics of bone pathology in RA is periarticular bone loss that occurs in early arthritis and happens adjacent to the inflamed joints. Engdahl et al. (96) found that mutated citrullinated vimentin triggered significant periarticular bone loss associated with an increased infiltration of osteoclast precursors and mature osteoclasts in the periarticular bone marrow. In this context, articular injection of murine bovine serum albumin after CFA immunization enhanced the expression of both RANKL and M-CSF, IL-8, IL-1, IL-6, and TNF-α (96). As synovial IL-17 expression is upregulated in RA, it has been observed that neutralization of IL-17 in mice significantly prevented joint swelling at day 1 of flare, suppressing joint inflammation and cartilage proteoglycan depletion at day 4 (98). Blocking IL-17 also clearly reduced bone erosions, Cathepsin K, and synovial RANKL (98). Moreover, using this animal model, it was observed that deficiency of the IL-1ra (a naturally occurring inhibitor of IL-1) gene induced autoimmunity and arthritis, with erosive destruction of the ankle bone among other features, such as infiltration of inflammatory cells, proliferation of the lining cells in the synovial membrane, and neutrophil infiltration, emphasizing the importance of IL-1/IL-1ra balance in maintaining joints physiology and immune system homeostasis (99). Methotrexate (MTX), the reference drug for RA treatment worldwide, decreased joint edema and prevented arthritis-induced alveolar bone loss in mice, probably via a newly described mechanism where oral and gut microbiota are involved (100). Using the AIA animal model, Almeida de Arruda et al. (100) described the impact of MTX on the oral–gut axis microbiota and that the protective role of MTX in RA-induced alveolar bone loss might be mediated via drug-microbiome interaction in the course of RA. Schneider et al. (101) have demonstrated using AIA mice that NETs directly contribute to bone erosions, increasing osteoclast formation. Moreover, in 2017, Vidal et al. (102) studied the impact of tofacitinib on the skeletal bone effects of inflammation. Authors observed that in the AIA rat model, treatment with tofacitinib inhibited synovitis as well as joint destruction, preventing bone erosion. Although tofacitinib was able to reduce RANKL and OPG, reducing bone turnover and bone cortical and trabecular hardness, this drug was not able to reverse the effects of inflammation on mechanical properties or cortical and trabecular bone structure (102). It has also been proved in AIA rats that BCEE and Diclofenac treatments prevent the development of granuloma and destructive lesions in ankle's connective tissue. In addition, both treatments prevented erosions and cystic expansion in the bone (103). In AIA-induced rabbits, an increased expression of both RANKL and OPG was found in the articular cartilage when compared to healthy cartilage, with a higher RANKL/OPG ratio that correlated with a significant bone loss in the subchondral plate (104). The location of this RANKL was also different, with intracellular and extracellular RANKL signals in AIA and no extracellular RANKL signals in healthy cartilage (104). Prieto-Potín et al. (105) have demonstrated, by inducing AIA in rabbits, that hyperlipidemia is capable of enhancing the systemic inflammation produced by RA, inducing damage to joint tissues via massive infiltration of macrophages and their transformation into foam cells and active osteoclasts. In the same AIA rabbit model, it was found that the parathyroid hormone related protein (PTHrP) was detected in diseased cartilage chondrocytes, suggesting that it has a role in this pathological condition, with a decrease in cell and matrix PTHrP in late AIA in parallel with the loss of chondrocytes, as happens in human RA cartilage (106).
PIA-induced arthritis rats also had cortical bone resorption with increased osteoclast number, inflammatory cells infiltrates, and a high range of bone formation on day 130 post-induction (107). Both systemic and local administration of porcine extracellular matrix-bound nanovesicles (MBV) are as effective as MTX in the alleviation of acute and chronic PIA in the rat, including adverse bone remodeling (108). Several antirheumatic drugs have been tested in this animal model in order to assess their therapeutic effects (109). Prednisolone, MTX, Celecoxib, diclofenac, indomethacin, and SB242235 (p38 inhibitor) inhibited bone resorption among other RA features, such as cell infiltration, but etanerceb was not able to alter either clinical or biological manifestations (109). PIA rats expressed heterogeneous nuclear ribonucleoprotein (hnRNP)-A2, maximum being during the acute phase, and its levels correlated with arthritis severity (110). hnRNP-A2 also stimulated lymph node cells to produce inflammatory cytokines in a MyD88-dependent manner, and overexpression of hnRNP-A2 in the PIA rats joints during the acute and chronic phases was found in synoviocytes of the inflammatory pannus tissue, chondrocytes of articular cartilage, and osteoclast-like multinucleated cells, inducing resorption of cortical bone during both phases of PIA (110). Finally, a recent work from Zeng et al. (111) showed that PIA animals had swollen paws with increased arthritis scores, synovium hyperplasia, body weight loss, and bone or cartilage erosion. In this model, treatment with the classical reversible AChE inhibitor pyridostigmine (PYR) abolished PIA-induced inflammation, oxidative stress, bone resorption, and gut microbiota dysbiosis, data that support new pharmacological interventions in animal models of RA (111).
Both male and female K/BxN mice had severe bone erosion and cartilage loss in the ankle, with loss of trabecular bone mineral density in the tibia calcaneus (112). In this model, alveolar bone loss is also found (113), suggesting that K/BxN serum injection is a suitable model to study the bone damage that occurs in arthritis. K/BxN serum transfer mice developed periosteal new bone formation and articular cartilage damage with cartilaginous metaplasia 29 days post-serum transfer (114). Bone resorption, with loss of articular cartilage and inflammatory cells infiltrates, is shown 21–42 days post-serum transfer; meanwhile, it has been observed that 42 days post-serum injection, extra-articular fibroplasia, ulcerated articular cartilage, joint ankylosis with severe bone remodeling, and a few remaining inflammatory cells are the main characteristics of this model (114). K/BxN models have been used to understand the mechanisms underlying the pathogenesis of RA as well as to investigate new treatments. Using this model, it was found that although osteopontin is involved in inflammation, immunity mediated by Th1 cells, and bone remodeling, this molecule did not have any role in either inflammation, bone erosion, or cartilage damage in the K/BxN serum-transfer model (115). It has been described that Budding uninhibited by benzimidazoles 1 (BUB1), which is known as a serine/threonine protein kinase, exerted an inhibitory effect on TNFα or IL-1β-mediated NF-κB signaling in bone marrow-derived macrophages, inhibiting their differentiation to osteoclasts, and attenuating bone loss (116). It has also been reported that blockade of Netrin-1, an axonal guidance molecule that acts as a chemorepulsant and inhibits migration of neutrophils, monocytes, and lymphocytes, and its receptor Unc5b prevented bone destruction and inflammation in K/BxN serum transferred mice (117). It was found that ankle bone erosions were present since week 2 post-serum transfer, and blockade of Netrin-1 or its receptor Unc5b reduces bone lesions as osteoclast differentiation was inhibited (117). García et al. showed that the absence of metalloproteinase MMP8 exacerbated the severity of arthritis but not its time course, onset, and remission, with increased synovial inflammation, bone erosions and overexpression of IL-1β, PROKR2, and PTX3. In this model, authors also observed that the absence of MMP8 did not protect from cartilage damage (118). The K/BxN model has been used to understand the role of nuclear protein heterogeneous nuclear RNP A2/B1 (hnRNP A2/B1), as antibodies against this protein, as found in approximately 30% of patients with RA (119). In both K/BxN and CIA experimental models, the severity of arthritis as well as bone erosions were reduced when hnRNP A2/B1 was silenced (119). Finally, using this model, Brines et al. (120) demonstrated that hemeoxygenase-1 (HO-1) deficiency aggravates arthritis progression with local upregulation of pro-inflammatory IL-6 and MMP-3 cytokines and serum RANKL and osteocalcin levels, suggesting a role for HO-1 in osteoblast function in arthritis.
In conclusion, the AIA model is the most appropriate to reproduce periarticular bone loss characteristic of RA. As shown in studies, this model describes juxta-articular bone loss associated with high osteoclast presence in the periarticular marrow, increased RANKL and proinflammatory cytokines (96–98, 104). In contrast, K/BxN models show more heterogeneous bone remodeling, with early erosions, periosteal bone formation, and, in late stages, even ankylosis, making them less specific for studying localized periarticular bone loss exclusively (114, 117, 118).
3.3 Metabolic syndrome studies in RA animal models
It is well known that RA is associated with several components of metabolic syndrome, such as obesity, insulin resistance, rheumatoid cachexia, dyslipidemia, and altered adipokine profiles, all being linked to an increase of CVD mortality (121). In a healthy population, CVD is usually associated with an increase in LDL levels and a decrease in HDL levels. Paradoxically, systemic inflammation in RA correlates with lower levels of total cholesterol, HDL, and LDL. Congruently, the use of methotrexate and DMARDs (disease modifying anti-rheumatic drugs), which decrease systemic inflammation and disease activity, counteract this lipid profile in patients with RA. Differences in the metabolic state of patients with RA (obesity, adipokine levels, or insulin sensitivity) make the study of the mechanisms associated with this comorbidity difficult (122).
For this reason, preclinical studies have an enormous interest (Table 3) (121).
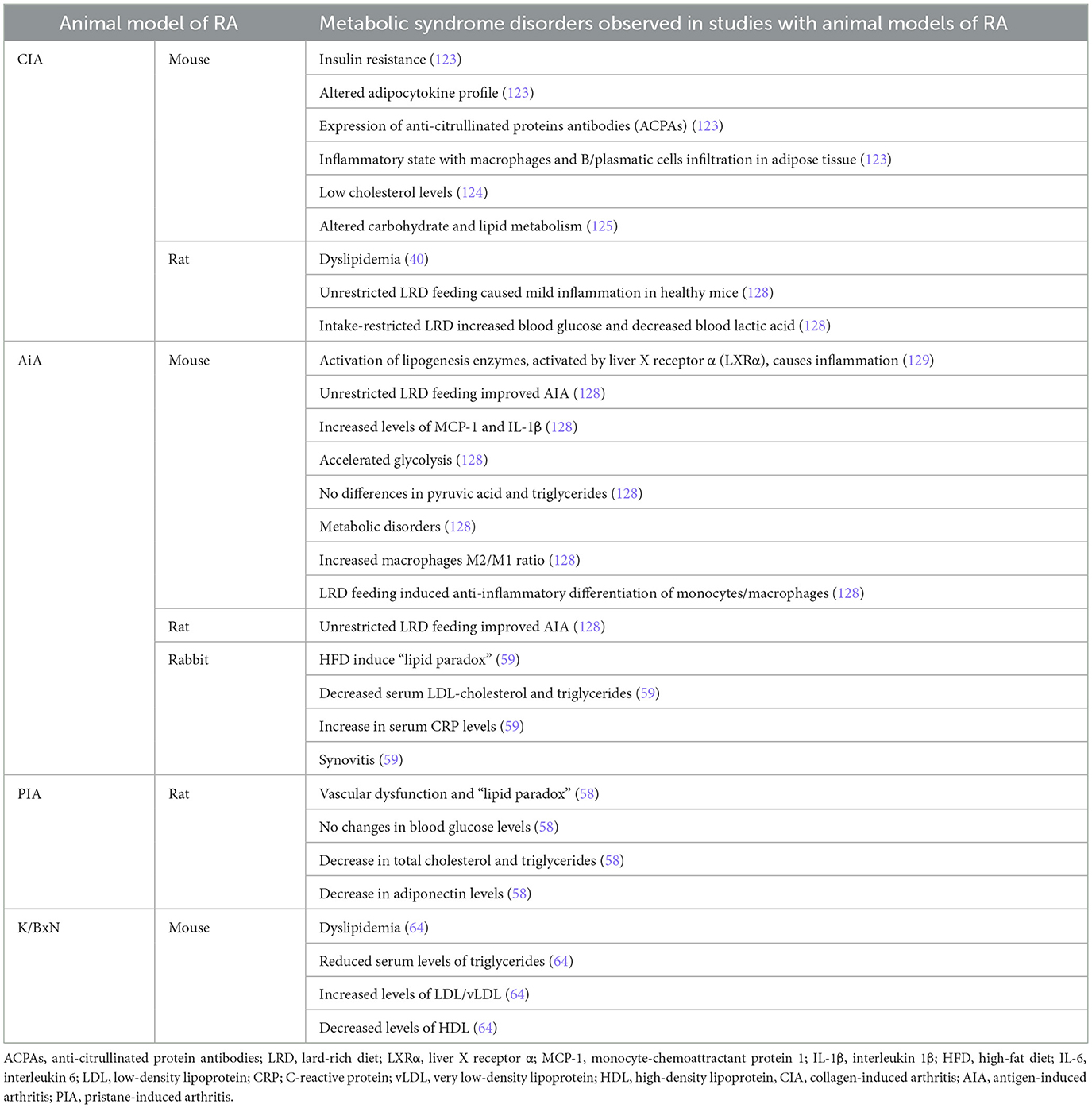
Table 3. Metabolic syndrome comorbidities in RA animal model studies.
In the CIA mouse model, disease activity was associated with insulin resistance and an altered adipocytokine profile together with the presence of anti-citrullinated protein antibodies (ACPAs) (123). In this RA context, adipose tissue is characterized by an inflammatory state with macrophages and B/plasmatic cells infiltration. ACPAs can have a direct effect, inducing inflammation and insulin resistance in macrophages to promote defective adipocyte differentiation, which can be partially restored by biologicals (123). In 2016, Wen et al. (124) described low cholesterol levels in CIA mice. Arias de la Rosa et al. (125) reported that CIA global inflammation (at systemic and tissue levels) was characterized by inadequate carbohydrate and lipid metabolism in different tissues, with the adipose tissue being the most susceptible tissue to the RA-induced metabolic alterations. These authors suggest that the inflammatory state in RA affects the adipose tissue, inducing insulin resistance and lipolysis (reducing lipid accumulation), therefore, the adipose tissue is an early RA target (125). The integration of the whole body glucose test in the CIA mice model is a useful translational model to test compound-induced metabolic derangements in patients with RA (126). This study showed that prednisolone slightly decreased fasted glucose concentrations as well as endogenous glucose production, while increasing insulin secretion (126). Jhun et al. (127) developed an obese CIA model, where obesity accelerated the inflammation and autoimmunity through the upregulation of inflammatory adipokines and cytokines expression. These authors indicate that obesity contributes to inflammation through CII-specific T cell differentiation. Although it may not be pathogenic in triggering arthritis, obesity is crucial in amplifying the inflammatory process via the Th1/Th17 response (127). However, data found in CIA rats are more controversial, and some investigators showed low lipid levels in this model, and other groups found high levels (40). It has been demonstrated in the CIA rat model that unrestricted Lard-rich Diet (LRD) feeding caused mild inflammation in healthy mice, but it can conditionally reduce inflammation in reloading endogenous PPAR-γ agonist fatty acids (128). Intake-restricted LRD increased blood glucose and decreased blood lactic acid in CIA rats, indicating an overall negative effect on inflammation-related glycolysis (128).
CIA models also show pro-atherogenic lipid alterations. These models show decreased levels of total cholesterol (TC) and HDL, and high levels of ox-LDL, mimicking the alterations described in patients with RA (124).
Dyslipidemia has also been studied in AIA mice and rabbits (59). In these animals, inflammation is worse due to the activation of lipogenesis enzymes by Liver X receptor α (LXRα) (129). Unrestricted LRD feeding without intake restriction improved AIA both in mice and rats (128). MCP-1 and IL-1β are increased in AIA mice, but unrestricted LRD feeding attenuated their expression in an opposite effect than in healthy mice (128). These authors did not observe any differences in pyruvic acid and triglycerides among groups, but glycolysis was accelerated in AIA mice (128). SIRT1 expression was not modified, but unrestricted LRD feeding induced an increase in PPAR-γ expression in perirenal fat tissues. This interaction between fat tissue and resident macrophages provides the basis for immune changes associated with metabolic disorders. Increase in ARG-1 expression confirmed that LRD increased the macrophage M2/M1 ratio in AIA mice, indicating that LRD feeding induced anti-inflammatory differentiation of monocytes/macrophages (128). Arthritic rabbits fed with a high-fat diet (HFD) mimicked the “lipid paradox” found in patients with RA. HFD-fed AIA rabbits had decreased serum LDL–cholesterol and triglycerides when compared with control HFD-fed animals. This was accompanied by an increase in serum CRP levels and synovitis. Also in this animal model, inflammation impairs reverse cholesterol transport and promotes lipid accumulation in macrophages. Administration of tofacitinib reestablishes this pathway, normalizes CRP levels, and elevates circulating lipid concentrations, mirroring the effects reported in patients with RA (59).
Limited data are available on PIA and K/BxN animal models. As described above, the PIA dark Agouti rat reproduces vascular dysfunction and the “lipid paradox” observed in RA (58). Chouk et al. (58) found that in arthritic rats, blood glucose was not modified in any phase of the disease, but TC and triglycerides were decreased in PIA at both phases, with adiponectin levels decreased in the acute phase. It has been observed that K/BxN mice under an atherogenic diet developed dyslipidemia, characterized by reduced serum levels of triglycerides, increased LDL/vLDL, and decreased HDL compared with controls (64).
3.4 Liver disease in RA animal models
Liver damage in patients with RA has received considerably less attention than cardiovascular or musculoskeletal comorbidities, and the available evidence remains limited. In patients with RA, liver damage can complicate diagnosis, making it challenging to identify whether it's a hepatic manifestation of RA, an unrelated primary liver disease, or liver toxicity resulting from RA treatment (130). Liver damage in RA often presents as asymptomatic abnormal liver tests, but in some cases, it can progress to cirrhosis (130). Additionally, individuals with RA are at a higher risk of developing associated autoimmune liver diseases (131).
Reports of liver damage in RA models are also limited (Table 4). The CIA model has been most widely used to replicate the liver damage observed in patients with RA. In CIA rats, significant alterations in hepatic lipid metabolism have been reported, such as reductions in fatty acid content during the CIA induction phase.
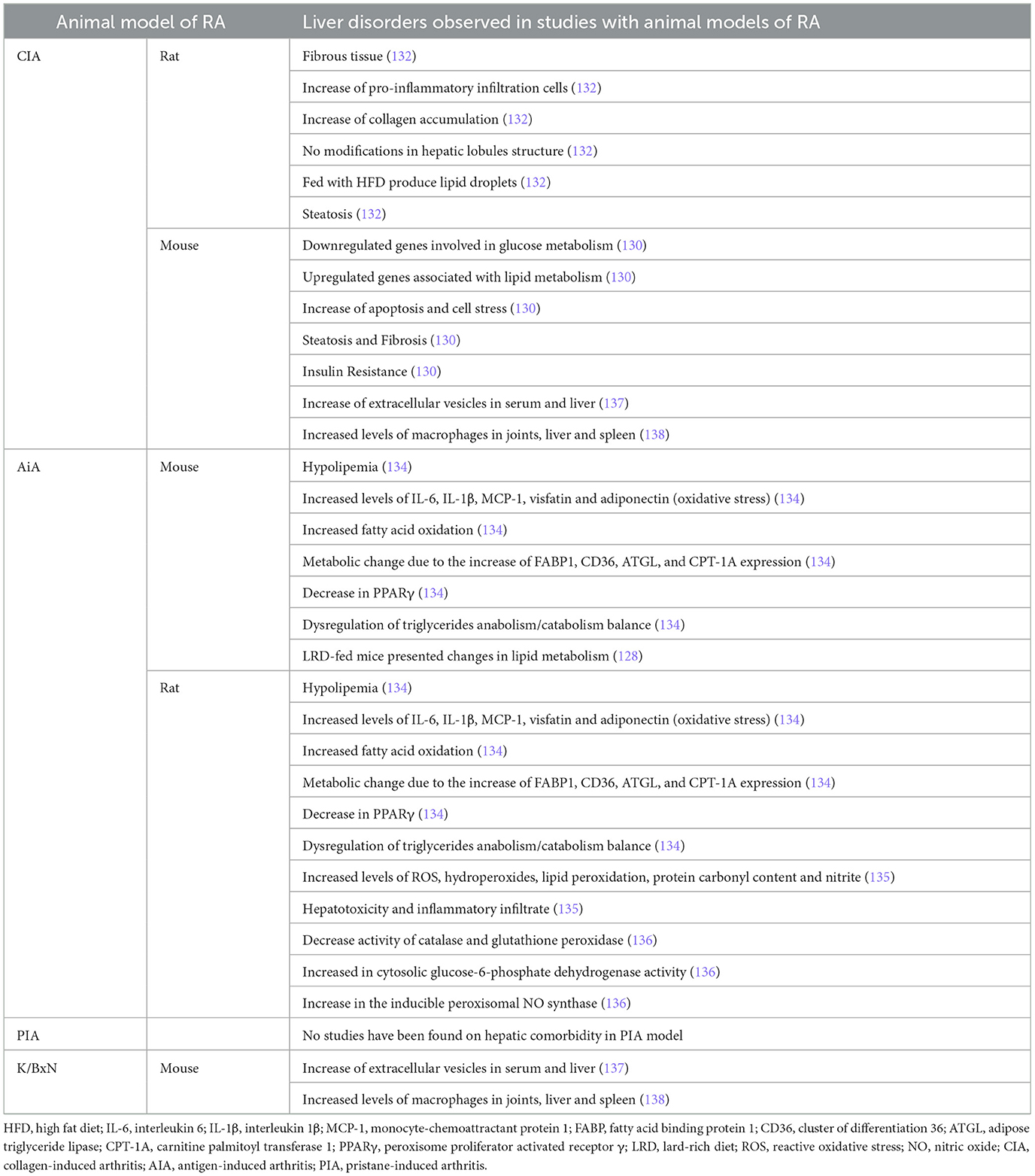
Table 4. Liver Comorbidities in RA animal model studies.
Zhang et al. (132) have recently established an animal model combining CIA with non-alcoholic fatty liver disease (NAFLD) in rats. These authors reported the development of fibrous tissue, an increase of pro-inflammatory infiltration cells, and collagen accumulation in the liver, but the structure of the hepatic lobules remained intact (132). Only the CIA model fed with HFD presented lipid droplets in the cytoplasm and nucleus of liver tissue. Also, steatosis was observed in hepatocytes of the CIA model, and this was higher in the CIA model fed with HFD (132). In a parallel study with CIA DBA1/J male mice, arthritic animals had downregulated genes involved in glucose metabolism and upregulated genes associated with lipid metabolism in the liver (130). Also, the CIA model showed high expression of markers of apoptosis and cell stress in the liver (130). Furthermore, marked hepatocellular fat accumulation and fibrosis were reported in the liver of CIA mice (130). A possibility is that inflammation in RA mice generates insulin resistance, promoting the apoptotic and fibrotic state of the liver (130). The study of how CIA affects the metabolism of tryptophan, kynurenine, and 3-hydroxyanthranilic acid (3-HAA) in the liver was performed and showed that tryptophan was statistically reduced in CIA mice when compared with controls. However, in the pre-arthritic livers, there was a trend toward a decrease in tryptophan concentration as well as kynurenine (133).
Adipokine-caused hepatic changes in RA-related hypolipemia were studied in AIA in both mice and rats. In these models, IL-6, IL-1β, MCP-1, visfatin, and adiponectin were increased in arthritic animals, inducing oxidative stress, liver injuries, and increased fatty acid oxidation (134). This metabolic change was accompanied by an increased FABP1, CD36, ATGL, and CPT-1A expression and a decrease in PPARγ that impaired its inhibition on NFκB in preadipocytes, leading to a mass secretion of inflammatory adipokines (134). These data indicate a disruption of triglyceride anabolism/catabolism balance in the liver, and as hepatocytes use more lipids but synthesize less, hypolipemia is developed in RA animals (134). The same authors indicate changes in the lipid metabolism-related genes (HSL, PPARγ, and SIRT1) expression in LRD-fed mice, indicating an accelerated fat turnover (128). Sundaram et al. (135) reported high levels of ROS, hydroperoxides, lipid peroxidation, protein carbonyl content, and nitrite levels in AIA rats in comparison with healthy animals, together with hepatotoxicity and inflammatory infiltrate in the liver. Comar et al. also revealed an increased oxidative stress in the liver of AIA rats, with higher levels of protein carbonyl groups. They also reported high levels of NO markers, a decrease in catalase and glutathione peroxidase activities, and an increase in cytosolic glucose-6-phosphate dehydrogenase activity. Finally, no changes were observed in superoxide dismutase and glutathione reductase activities (136). These authors indicate that the increased ROS content in the liver seems to be the consequence of both a deficient antioxidant defense and a stimulated pro-oxidant system (136).
However, beyond these animal models, the information about other models like PIA and K/BxN is less clear. This model has been poorly studied with respect to liver pathology, leaving an important gap in our understanding of whether hepatic changes can be representative. Regarding the K/BxN animal model, Liang et al. (137) have recently demonstrated in both CIA and K/BxN animal models the increase of extracellular vesicles in serum and liver of arthritic mice compared to controls. When compared with methotrexate treatment, a decrease in serum ALT activity was observed after decoy extracellular vesicles treatment, together with reduced steatosis and inflammatory infiltration in the liver (137). Comparing both CIA and K/BxN animal models, Paoletti et al. (138) demonstrated that macrophages are increased not only in joints but also in other tissues (such as liver and spleen). Treatment with the antagomiR-155-5p encapsulated in PEG liposomes to deliver small RNA to monocytes and macrophages specifically reduces joint inflammation. Despite the high accumulation in the liver, these liposomes had no or minor effects on liver macrophages.
No studies have been found on hepatic comorbidity in the PIA model.
3.5 Lung comorbidities in RA animal models
Lung complications are the second most common cause of death in patients with RA (139, 140). Among them, patients with RA suffer from interstitial lung disease (ILD), chronic obstructive pulmonary disease (COPD), chronic bronchitis (and other airway-related manifestations), pleural diseases, pulmonary nodules, and inducible bronchus-associated lymphoid tissue (iBALT) (141). The pathogenesis of lung complications in RA has been associated with genetics and environmental exposures, such as smoking. However, further studies are needed to elucidate the underlying pathological mechanisms and identify novel therapeutic options (139). In this regard, studies using different animal models are shedding light on the mechanisms underlying RA comorbidities and potential therapeutic approaches. However, animal modeling of RA-associated lung disease is limited (Table 5).
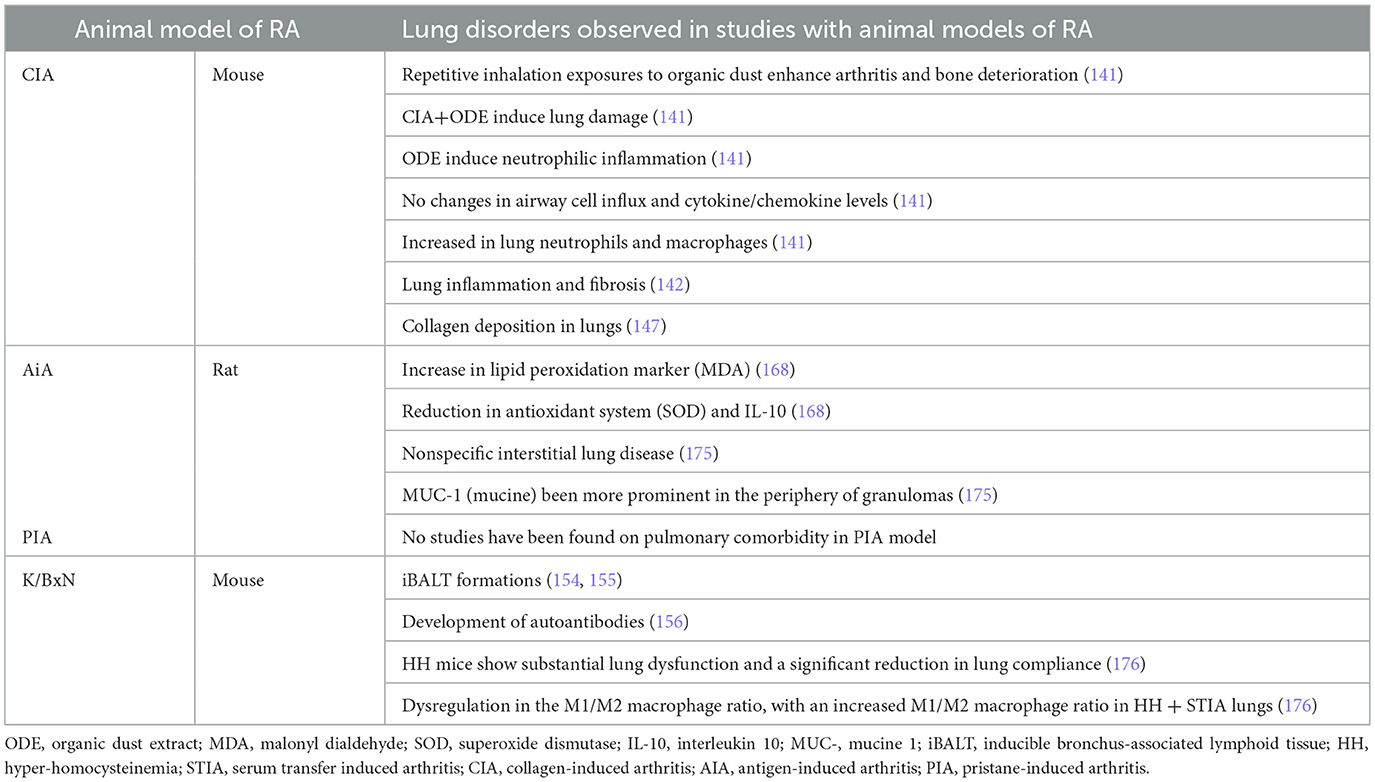
Table 5. Lung comorbidities RA animal model studies.
The CIA and AIA models are the primary animal models used to study RA-associated interstitial lung disease (RA-ILD), as they reproduce key features of the condition in a manner dependent on the response to CFA.
Environmental co-exposures exacerbate both joint and pulmonary pathology in the CIA model (141). Repetitive inhalation of organic dust or LPS enhanced arthritis severity while promoting interstitial inflammation, autoantibody production, and fibrotic remodeling, replicating the clinical features observed in RA-ILD patients (141–143). Combination approaches, particularly CIA plus bleomycin, reproduced lung fibrosis in addition to joint pathology. In this context, multiple therapeutic strategies like melatonin, ethoxyquin, hESC-MSCs, and inhibitors of JAK2/STAT3 or TGF-β/SMAD signaling reduce both articular inflammation and pulmonary fibrosis (144–149). These results suggest that the recruitment of inflammatory macrophages/monocytes and neutrophils might contribute to the pro-fibrotic inflammatory lung responses in the CIA model following airborne biohazard exposures (149). Therefore, although CFA induces the main features of RA-ILD, additional factors such as CII immunization or environmental exposures (e.g., organic dust, LPS, or bleomycin) can further amplify inflammatory infiltration, fibrosis, and autoantibodies deposition in the lungs, making the model more analogous to the comorbidity observed in patients with RA.
Studies have shown that AIA rats develop disrupted lung histology characterized by inflammatory cell infiltration, pleural inflammation, and pleural fibrosis (150, 151). These alterations replicate pathological changes in RA-ILD patients. However, many of these similarities are driven by the CFA component itself: granuloma-like structures and giant cells commonly observed in the model are characteristic of live mycobacterial infection but can also arise from the killed Mycobacterium butyricum present in CFA. This indicates that the adjuvant substantially contributes to the pulmonary phenotype (152). While CFA acts as a potent immune enhancer, CII immunization is essential to amplify the pulmonary manifestations in the CIA model. Indeed, lung cell counts are significantly higher in CIA rats compared with CFA-only rats, indicating that CII is required for robust inflammatory infiltration (153). In addition, ACPA autoantibodies (e.g., anti-citrullinated fibrinogen), which induce the autoimmune response in RA, are deposited in the lung tissue of the CIA model (153).
No studies have been found on pulmonary comorbidity in the PIA model.
Regarding the use of the K/BxN animal model to study lung involvement, few studies have been published. K/BxN have been employed in the study of iBALT pathology. K/BxN lung exhibited multiple areas of lymphocytic infiltration around vessels, airways, and submucosal in the lungs, replicating iBALT comorbidity of patients with RA (154). In the K/BxN model, lung infiltrate was correlated with weight loss, but not with the severity of arthritis (154). However, the K/BxN model does not develop pulmonary fibrosis naturally; it can be induced by bleomycin, similar to the CIA model (154). The use of segmented filamentous bacteria (SFB) colonization also induced iBALT in young K/BxN mice that resembled the iBALT formations in patients with RA (154, 155). In addition, SFB are able to induce autoantibodies in lung during the pre-arthritic phase of K/BxN model (154–156). In a follow-up study, Teng et al. (157) demonstrated that middle-aged K/BxN mice developed more severe arthritis and exhibited more extensive iBALT lesions compared to young mice, regardless of SFB colonization. Furthermore, K/BxN mice presented pulmonary dysfunction, characterized by reduced compliance and an elevated M1/M2 macrophage ratio in lung tissue (146). Pulmonary dysfunction paralleled articular inflammation, as therapeutic interventions produced similar outcomes in both lung and joint compartments (146). Although few studies have directly investigated lung pathology in the K/BxN model, data indicate that K/BxN mice develop multiple lymphocytic infiltration around airways, vessels, and submucosal regions, recapitulating iBALT structures seen in Patients with RA.
3.6 Renal comorbidities in RA animal models
The prevalence of chronic kidney disease (CKD) in Patients with RA is around 25%, proportionally higher than the prevalence in healthy individuals (158). CDK in patients with RA can be divided into two main causes: derived from chronic inflammation (elevated inflammatory markers such as CRP in early stages) and drug-induced kidney diseases (159, 160). Specifically in animal models, renal damage is best reproduced through inflammation, whereas drug-induced toxicity is highly dependent on dose and pharmacokinetics. Patients with RA with CKD have an increased risk of cardiovascular disease regardless of other classical CVD risk factor, and the decrease in kidney function limits RA treatment options (161, 162). This is caused due to the higher disease activity in RA, the greater the influence on kidney function due to nephrotoxic medication use (e.g., NSAIDs), the atherosclerotic renal disease, the secondary amyloidosis, and the direct nephrotoxic effects of chronic inflammation (163). Studying RA-associated renal comorbidities is highly relevant for expanding the range of treatments available to patients. As previously mentioned, many patients have a limited treatment repertoire due to impaired kidney function.
In order to understand the mechanism involved in RA associated kidney disease, several studies in animal models have been performed (Table 6).
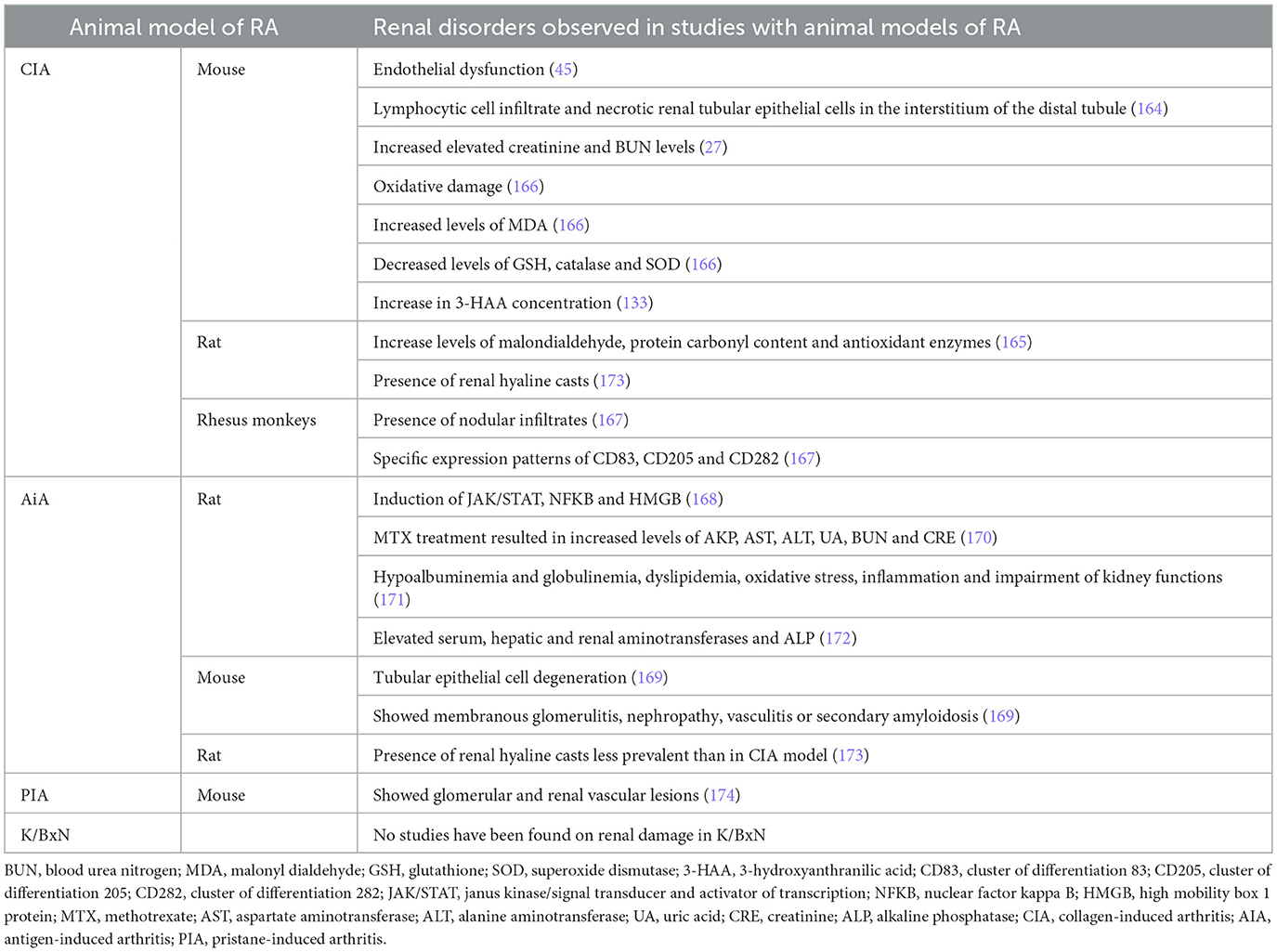
Table 6. Renal comorbidities in RA animal model studies.
The CIA model was used to study endothelial dysfunction, and data showed an increase in iNOS in aorta, heart, and kidney microcirculation, finding iNOS immunostaining in the endothelial layers of microvessels, in the glomeruli, and in the interstitium (45). Kidneys from CIA mice exhibited lymphocytic cell infiltration and necrotic renal tubular epithelial cells in the interstitium of the distal tubule, together with elevated creatinine and BUN levels, which was not prevented with MTX. However, Ganoderma lucidum polysaccharide peptide (GLPP) was able to reduce the systemic immune response and ameliorate kidney injury (164). The CIA model has also been used to study the link between RA and oxidative stress. CIA rats have increased malondialdehyde, protein carbonyl content, and antioxidant enzymes (SOD, catalase, GST, GPx, and GR) in joints, liver, kidney, and spleen, and administration of suramin restored all of them (165). Oxidative damage in CIA mice was also studied by Kim et al. (166), who showed that levels of MDA were increased in kidneys from CIA mice, meanwhile the levels of GSH, catalase, and SOD were reduced. In 2012, Jonker et al. (167) reported in Rhesus monkeys that CIA kidney allografts had nodular infiltrates with increased CD3+ T cells and that CD8+ cells slightly increased in the interstitium when compared with nodular infiltrates. Markers of dendritic cells (CD83), monocyte-derived DC (CD205), and TLR2 (CD282) showed specific expression patterns in the kidney in these animals (167). As urine from patients with RA showed an increase in 3-HAA concentration, this was tested in CIA mice, and data demonstrated that kidneys from both pre-arthritic and animals with established CIA had increased 3-HAA concentration when compared with naive organs (133).
The JAK/STAT, NFKB, and HMGB expressions were substantially increased in AIA kidneys in comparison to the normal control tissue, and were reduced when animals were γ-radiated (168). Histologically, arthritic mice showed tubular epithelial cell degeneration without significant necrosis or apoptosis. Moreover, kidneys from AIA mice showed compression of the renal tubules, disorganization of the glomeruli, and vascularization when compared with control animals, indicating the development of membranous glomerulitis, nephropathy, vasculitis, or secondary amyloidosis (169). These were reverted when AIA mice were treated with nanocapsules of curcumin and vitamin D3 (169). AIA rats were used to analyze liver and kidney injuries of MTX treatment. MTX treatment resulted in obvious toxicity as early as 18 days after induction, with increased levels of alkaline phosphatase (AKP), aspartate aminotransferase (AST), alanine aminotransferase (ALT), uric acid (UA), blood urea nitrogen (BUN), and creatinine (CRE) reinforced on day 35, and probably induced by glycolysis-facilitated intestinal absorption (170). In the same animal model, it an increase of serum TAG, total lipid, LDL cholesterol, total cholesterol, CRP, globulin, urea, creatinine, and NOx levels has been observed, with a decrease in serum total protein, HDL-cholesterol, albumin: globulin ratio and total anti-oxidants in the arthritis animals, all indicating hypoalbuminemia and globulinemia, dyslipidemia, oxidative stress, inflammation, and impairment of kidney functions (171). Serum, liver, and kidney aminotransferases and ALP have been described to be elevated in AIA rats due to inflammation in both liver and kidney impairment in arthritis, as well as leakage of lysosomal enzymes, a consequence of increased endocytic activity (172).
An article has been found where kidney damage is studied in the PIA model, and a systematic comparison between CIA and PIA in Dark Agouti rats was done in 2010. It was observed that kidney hyaline casts were more prevalent in CIA animals than in PIA rats, and the authors indicate that this may be related to the increase of autoimmune circulating antibodies in CIA animals (173). Another PIA study, focused on the lupus pathology, presented glomerular and renal vascular lesions in kidneys after 26 weeks of induction (174).
No studies have been found on renal damage in K/BxN beyond studies to demonstrate the absence of drug toxicity.
4 Synthesis, translational relevance, and recommendations for model selection
While the CIA, AIA, PIA, and K/BxN animal models replicate joint inflammation and bone erosions characteristic of patients with RA, their systemic manifestations significantly differ. The selection of an appropriate animal model must therefore be driven not just by joint pathology, but specifically by the comorbidity of interest and the scientific question being addressed.
The next synthesis provides a comparative guide to leveraging the strengths of each model for RA comorbidity research.
4.1 Recommendations for cardiovascular or metabolic comorbidity studies
CVD is the main cause of mortality in patients with RA, so developing models that reflect the inflammatory–metabolic–vascular pathology is mandatory. This comorbidity is associated with chronic inflammation-accelerated atherosclerosis, insulin resistance, endothelial dysfunction, and cardiac fibrosis.
• All RA models developed circulating lipid alterations observed in patients with RA. However, insulin resistance is only present in the CIA model. This suggests different inflammatory-metabolic associations among different RA models, which could explain the various cardiac and vascular alterations observed.
• CIA and AIA models demonstrate consistent evidence for cardiac pathology, including hypertrophy and reduced functional recovery. These are excellent starting points for general inflammation-driven cardiac research.
• PIA is employed for studies focusing on the vascular components of CVD, replicating systemic endothelial dysfunction and the RA-associated “lipid paradox” (where low cholesterol is a poor prognostic factor). PIA is recommended for long-term pharmacological trials targeting vascular protection.
• The K/BxN mice, supplemented with an atherogenic diet, are used to investigate accelerated atherosclerosis and the progression toward serious heart failure, such as dilated cardiomyopathy, making it ideal for cardiac fibrosis studies.
4.2 Recommendations for musculoskeletal comorbidity
RA-associated muscle wasting (sarcopenia/cachexia) is a highly prevalent systemic complication. The different experimental models show different skeletal muscle responses, affecting atrophy, fat infiltration, and tissue regeneration.
• CIA is the gold standard model for general studies on rheumatoid cachexia. The extensive literature consistently shows progressive weight loss, muscle atrophy, and increased atrogene expression, making it the ideal model for testing treatments targeting chronic muscle wasting and fibrosis.
• AIA is recommended for studying muscle regeneration in RA. Although it exhibits muscle wasting, AIA simultaneously activates anabolic pathways (e.g., upregulation of MSTN) and represents a unique RA model for investigating the interplay between inflammatory atrophy and compensatory regeneration.
• K/BxN seems to be an excellent choice for investigating systemic correlation between inflammatory mediators (particularly high circulating IL-6) and sarcopenia, or for examining treatments focused on systemic myositis and associated muscle fibrosis.
4.3 Recommendations for bone and cartilage pathology
While all RA models develop the periarticular bone loss, their capacity to replicate systemic bone comorbidities (e.g., osteoporosis) varies in both timing and underlying mechanisms.
• AIA is recommended for studies focused on early RA pathogenesis. This model develops cortical bone deterioration starting before the onset of clinical arthritis. This is a useful model for understanding the preclinical or early diagnostic phase of RA-related systemic bone loss.
• K/BxN is employed for antibody-driven bone damage that extends to extra-articular sites. Specifically, this model replicates the alveolar bone damage and potentially other sites associated with antibody deposition.
• CIA is suitable for long-term studies of bone pathology in the context of chronic, resolving, or treatment-modified inflammation.
4.4 Recommendations for liver and kidney comorbidities
Research into these comorbidities is limited across all models, but distinct preferences exist based on available data.
• Liver disease: CIA is the model with more evidence, replicating pathologies like steatosis, fibrosis, and insulin resistance when coupled with dietary challenges (e.g., high-fat diets). It should be prioritized for studies investigating the metabolic syndrome component of RA-associated liver disease. However, further research is needed to evaluate the development of these comorbidities in the CIA model independent of high-fat diets.
• Kidney disease: the CIA model is recommended for studies of kidney involvement, particularly tubular epithelial cell degeneration and hyaline casts. While all RA models (CIA, AIA, PIA, and K/BxN) show systemic inflammatory impact on renal function, CIA is the best choice for specific nephropathy studies.
Selection of CIA, AIA, PIA, and K/BxN models should be guided by the specific RA comorbidity under investigation. By aligning the specific translational question with the model's strengths, researchers can significantly increase the validity and translational relevance of their findings to human RA pathology (Table 7 and Supplementary Table S1).
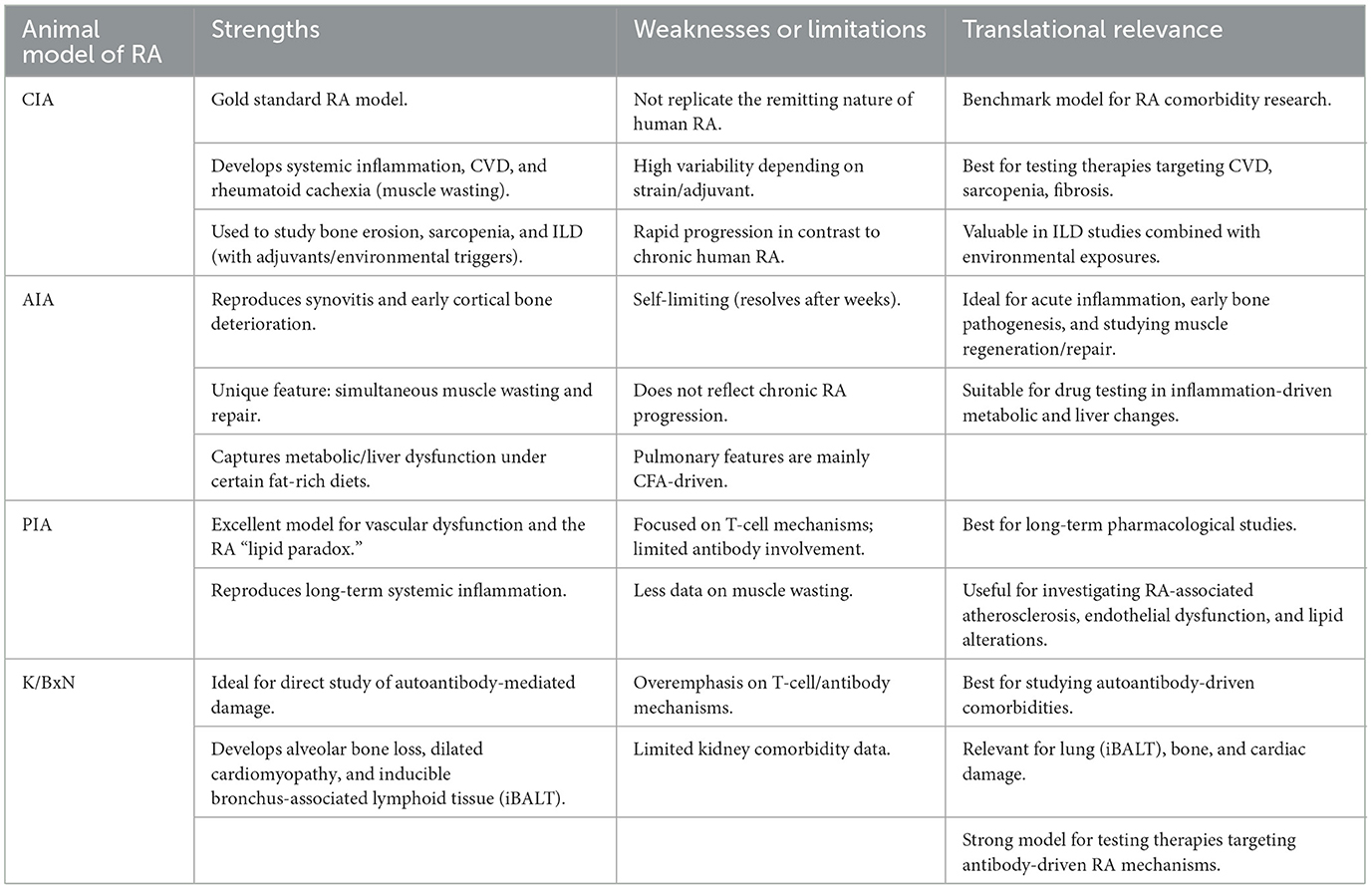
Table 7. Strengths, weaknesses and translational relevance of RA models
5 Conclusion
The selection and design of an animal model for the study of a specific comorbidity are important strategies for developing research translational capacity. In this review article, we have described comorbidities associated with the cardiac, musculoskeletal, liver, lung, and renal systems in four widely used arthritis models with pathologies similar to humans. This summary will help researchers select the best animal model to use for the specific study of the comorbidity in question and thus more effectively evaluate experimental treatments for it. Among the comorbidities studied herein, there is strong evidence of muscle loss and heart disease in CIA and AIA y K/BxN animal models, which reflect the associated comorbidities in patients with RA. The strongest evidence is in the CIA model, due to the greater number of studies performed. In contrast, the study of hepatic comorbidities in these models remains limited and, in many cases, it is more dependent on diet rather than on the inflammatory stage, highlighting an important gap to be addressed. Neurological comorbidities, although mentioned in the introduction, are not discussed in this article, so it would be necessary to focus on this aspect.
Animal models can also contribute to improving precision medicine in patients with RA. By combining and stratifying experimental traditional models depending on genetic susceptibility, immune phenotype or metabolic profile, and systemic biology approaches, it will be possible to reproduce patients' heterogeneity and treatment response more faithfully. This forward-looking perspective emphasizes that animal studies can not only replicate known comorbidities but also serve to develop more effective and individualized therapies.
The technological progress avoids us from generating new RA models, beyond the conventional progress exposed in this work. These new models will solve current limitations and will reduce the differences between human and animal RA pathology. Among these, we found humanized mouse models, multi-omics approaches, and organ-on-chip systems that could complement traditional models by offering mechanisms with higher translational value.
Author contributions
MM-B: Conceptualization, Data curation, Formal analysis, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MF: Data curation, Formal analysis, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. MR-B: Formal analysis, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. GH-B: Conceptualization, Project administration, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MG-G: Conceptualization, Formal analysis, Funding acquisition, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. RL: Conceptualization, Formal analysis, Funding acquisition, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from Instituto de Salud Carlos III (ISCIII) through research grants PI22/00347, PI22/00352, PI24/00554, and co-funded by the European Union; and RD21/0002/0025 and RD24/0007/0031 Health Outcomes-Oriented Cooperative Research Networks, granted by ISCIII and funded by the European Union-NextGenerationEU via Mecanismo de Recuperación, Transformación y Resiliencia.
Conflict of interest
AM, MM-B, MF, RL, and GH-B have filed a patent (#907642) on the use of dipyridamole as a novel therapy for muscular myogenesis disorders and inflammatory arthritis. RL and GH-B have filed a patent for the use of 6-shogaol in osteoarthritis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1693610/full#supplementary-material
References
1. Slobodin G. Rheumatic Disease in Geriatrics: Diagnosis and Management. Cham: Springer Nature (2020). p. 173–83.
2. GBD 2021 Rheumatoid Arthritis Collaborators. Global, regional, and national burden of rheumatoid arthritis, 1990–2020, and projections to 2050: a systematic analysis of the Global Burden of Disease Study 2021. Lancet Rheumatol. (2023) 5:e594–610. doi: 10.1016/S2665-9913(23)00211-4
3. Colquhoun M, Gulati M, Farah Z, Mouyis M. Clinical features of rheumatoid arthritis. Medicine. (2022) 50:138–42. doi: 10.1016/j.mpmed.2021.12.002
4. Figus FA, Piga M, Azzolin I, McConnell R, Iagnocco A. Rheumatoid arthritis: extra-articular manifestations and comorbidities. Autoimmun Rev. (2021) 20:102776. doi: 10.1016/j.autrev.2021.102776
5. Dougados M, Soubrier M, Antunez A, Balint P, Balsa A, Buch MH, et al. Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: results of an international, cross-sectional study (COMORA). Ann Rheum Dis. (2014) 73:62–8. doi: 10.1136/annrheumdis-2013-204223
6. Coulson KA, Reed G, Gilliam BE, Kremer JM, Pepmueller PH. Factors influencing fracture risk, T score, and management of osteoporosis in patients with rheumatoid arthritis in the Consortium of Rheumatology Researchers of North America (CORRONA) registry. J Clin Rheumatol. (2009) 15:155–60. doi: 10.1097/RHU.0b013e3181a5679d
7. Letarouilly J-G, Flipo R-M, Cortet B, Tournadre A, Paccou J. Body composition in patients with rheumatoid arthritis: a narrative literature review. Ther Adv Musculoskelet Dis. (2021) 13:1759720X211015006. doi: 10.1177/1759720X211015006
8. Dietzel R, Wiegmann S, Borucki D, Detzer C, Zeiner KN, Schaumburg D, et al. Prevalence of sarcopenia in patients with rheumatoid arthritis using the revised EWGSOP2 and the FNIH definition. RMD Open. (2022) 8:e002600. doi: 10.1136/rmdopen-2022-002600
9. Walsmith J, Roubenoff R. Cachexia in rheumatoid arthritis. Int J Cardiol. (2002) 85:89–99. doi: 10.1016/S0167-5273(02)00237-1
10. Guła Z, Łosińska K, Kuszmiersz P, Strach M, Nowakowski J, Biedroń G, et al. A comparison of comorbidities and their risk factors prevalence across rheumatoid arthritis, psoriatic arthritis and axial spondyloarthritis with focus on cardiovascular diseases: data from a single center real-world cohort. Rheumatol Int. (2024) 44:2817–28. doi: 10.1007/s00296-024-05740-z
11. Imanuel CA, Sivatheesan S, Koyanagi A, Smith L, Konrad M, Kostev K. Associations between rheumatoid arthritis and various comorbid conditions in Germany-a Retrospective Cohort Study. J Clin Med. (2023) 12:7265. doi: 10.3390/jcm12237265
12. Al-Saleh J, Ali Khan N, Zamani N, AlSaidi H, Rachidi W. Prevalence of comorbidities among patients with rheumatoid arthritis in the UAE: a case-control study. BMJ Open. (2024) 14:e086116. doi: 10.1136/bmjopen-2024-086116
13. Cassotta M, Pistollato F, Battino M. Rheumatoid arthritis research in the 21st century: limitations of traditional models, new technologies, and opportunities for a human biology-based approach. ALTEX. (2020) 37:223–42. doi: 10.14573/altex.1910011
14. Terao C, Raychaudhuri S, Gregersen PK. Recent advances in defining the genetic basis of rheumatoid arthritis. Annu Rev Genomics Hum Genet. (2016) 17:273–301. doi: 10.1146/annurev-genom-090314-045919
15. McNamee K, Williams R, Seed M. Animal models of rheumatoid arthritis: how informative are they? Eur J Pharmacol. (2015) 759:278–86. doi: 10.1016/j.ejphar.2015.03.047
16. Christensen AD, Haase C, Cook AD, Hamilton JA. K/BxN serum-transfer arthritis as a model for human inflammatory arthritis. Front Immunol. (2016) 7:213. doi: 10.3389/fimmu.2016.00213
17. Kong J-S, Jeong GH, Yoo S-A. The use of animal models in rheumatoid arthritis research. J Yeungnam Med Sci. (2022) 40:23–9. doi: 10.12701/jyms.2022.00773
18. Zhao T, Xie Z, Xi Y, Liu L, Li Z, Qin D. How to model rheumatoid arthritis in animals: from rodents to non-human primates. Front Immunol. (2022) 13:887460. doi: 10.3389/fimmu.2022.887460
19. Chenchula S, Najmi A, Atal S, Sadasivam B. In vivo murine models for evaluating anti-arthritic agents: an updated review. Future Health. (2024) 2:138–47. doi: 10.25259/FH_51_2024
20. Asquith DL, Miller AM, McInnes IB, Liew FY. Animal models of rheumatoid arthritis. Eur J Immunol. (2009) 39:2040–4. doi: 10.1002/eji.200939578
21. Holmdahl R, Andersson ME, Goldschmidt TJ, Jansson L, Karlsson M, Malmstrom V, et al. Collagen induced arthritis as an experimental model for rheumatoid arthritis. APMIS. (1989) 97:575–84. doi: 10.1111/j.1699-0463.1989.tb00446.x
22. Luan J, Hu Z, Cheng J, Zhang R, Yang P, Guo H, et al. Applicability and implementation of the collagen-induced arthritis mouse model, including protocols (review). Exp Ther Med. (2021) 22:939. doi: 10.3892/etm.2021.10371
23. Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc. (2007) 2:1269–75. doi: 10.1038/nprot.2007.173
24. Nandakumar KS, Holmdahl R. A genetic contamination in MHC-congenic mouse strains reveals a locus on chromosome 10 that determines autoimmunity and arthritis susceptibility. Eur J Immunol. (2005) 35:1275–82. doi: 10.1002/eji.200425925
25. Iwakura Y. Roles of IL-1 in the development of rheumatoid arthritis: consideration from mouse models. Cytokine Growth Factor Rev. (2002) 13:341–55. doi: 10.1016/S1359-6101(02)00021-7
26. Donaldson LF, Chillingworth NL. Arthritis model, adjuvant-induced arthritis. In:Schmidt RF, Willis WD, , editors. Encyclopedia of Pain. Berlin, Heidelberg: Springer Berlin Heidelberg (2007). p. 111–5. doi: 10.1007/978-3-540-29805-2_282
27. De Castro Costa M, De Sutter P, Gybels J, Van Hees J. Adjuvant-induced arthritis in rats: a possible animal model of chronic pain. Pain. (1981) 10:173–85. doi: 10.1016/0304-3959(81)90193-7
28. Vingsbo C, Sahlstrand P, Brun JG, Jonsson R, Saxne T, Holmdahl R. Pristane-induced arthritis in rats: a new model for rheumatoid arthritis with a chronic disease course influenced by both major histocompatibility complex and non-major histocompatibility complex genes. Am J Pathol. (1996) 149:1675–83.
29. Wooley PH, Seibold JR, Whalen JD, Chapdelaine JM. Pristane-induced arthritis. The immunologic and genetic features of an experimental murine model of autoimmune disease. Arthritis Rheum. (1989) 32:1022–30. doi: 10.1002/anr.1780320812
30. Monach PA, Mathis D, Benoist C. The K/BxN arthritis model. Curr Protoc Immunol. (2008) 81:15.22.1–15.22.12. doi: 10.1002/0471142735.im1522s81
31. Crowson CS, Liao KP, Davis JM, Solomon DH, Matteson EL, Knutson KL, et al. Rheumatoid arthritis and cardiovascular disease. Am Heart J. (2013) 166:622–8.e1. doi: 10.1016/j.ahj.2013.07.010
32. Marco-Bonilla M, Fresnadillo M, De La Riva-Bueno M, Herrero-Beaumont G, Largo R, Mediero A. Animal models in rheumatoid arthritis: is there a correlation between autoantibodies in human pathology and animal models? Biology. (2025) 14:460. doi: 10.3390/biology14050460
33. Meng Q, Guo M-H, Zhang R, Wei J, Chen Q, Zhao X-C, et al. G-protein coupled receptor kinase 2 mediates rheumatoid arthritis-induced depression-like behaviors via the hippocampal CRHR1 signaling pathway. Acta Pharmacol Sin. (2025). doi: 10.1038/s41401-025-01621-8. [Epub ahead of print].
34. Vierboom MPM, Breedveld E, Keehnen M, Klomp R, Bakker J. Pain relief in nonhuman primate models of arthritis. Methods Mol Biol. (2017) 1559:411–7. doi: 10.1007/978-1-4939-6786-5_28
35. Sadasivam B, Jhaj R, Kumar S, Pathan S, Chenchula S. Anti-inflammatory and arthritic activity of zaltoprofen compared to piroxicam in murine models. Bioinformation. (2022) 18:752–6. doi: 10.6026/97320630018752
36. Løgstrup BB, Ellingsen T, Pedersen AB, Darvalics B, Olesen KKW, Bøtker HE, et al. Cardiovascular risk and mortality in rheumatoid arthritis compared with diabetes mellitus and the general population. Rheumatology. (2021) 60:1400–9. doi: 10.1093/rheumatology/keaa374
37. Crowson CS, Rollefstad S, Ikdahl E, Kitas GD, van Riel PLCM, Gabriel SE, et al. Impact of risk factors associated with cardiovascular outcomes in patients with rheumatoid arthritis. Ann Rheum Dis. (2018) 77:48–54. doi: 10.1136/annrheumdis-2017-211735
38. González-Gay MA, González-Juanatey C. Inflammation and lipid profile in rheumatoid arthritis: bridging an apparent paradox. Ann Rheum Dis. (2014) 73:1281–3. doi: 10.1136/annrheumdis-2013-204933
39. Yan J, Yang S, Han L, Ba X, Shen P, Lin W, et al. Dyslipidemia in rheumatoid arthritis: the possible mechanisms. Front Immunol. (2023) 14:1254753. doi: 10.3389/fimmu.2023.1254753
40. Kessler J, Totoson P, Devaux S, Moretto J, Wendling D, Demougeot C. Animal models to study pathogenesis and treatments of cardiac disorders in rheumatoid arthritis: advances and challenges for clinical translation. Pharmacol Res. (2021) 170:105494. doi: 10.1016/j.phrs.2021.105494
41. Dai H, Wang X, Yin S, Zhang Y, Han Y, Yang N, et al. Atrial fibrillation promotion in a rat model of rheumatoid arthritis. JAHA. (2017) 6:e007320. doi: 10.1161/JAHA.117.007320
42. Han X, Gao Y, He M, Luo Y, Wei Y, Duan Y, et al. Evolocumab prevents atrial fibrillation in rheumatoid arthritis rats through restraint of PCSK9 induced atrial remodeling. J Adv Res. (2024) 61:211–21. doi: 10.1016/j.jare.2023.09.007
43. Zihao Z, Zhengyue M, Aishu L, Didi Z, Yan L, Peng L, et al. Identifying a marked inflammation mediated cardiac dysfunction during the development of arthritis in collagen-induced arthritis mice. Clin Exp Rheumatol. (2019) 38:203–11. doi: 10.55563/clinexprheumatol/6kxs1o
44. Ning X, Ni Y, Cao J, Zhang H. Liquiritigenin attenuated collagen-induced arthritis and cardiac complication via inflammation and fibrosis inhibition in mice. Chem Pharm Bull. (2023) 71:269–76. doi: 10.1248/cpb.c22-00684
45. Palma Zochio Tozzato G, Taipeiro EF, Spadella MA, Marabini Filho P, De Assis MR, Carlos CP, et al. Collagen-induced arthritis increases inducible nitric oxide synthase not only in aorta but also in the cardiac and renal microcirculation of mice. Clin Exp Immunol. (2016) 183:341–9. doi: 10.1111/cei.12728
46. Qian Y, Fei Z, Nian F. The association between rheumatoid arthritis and atrial fibrillation: epidemiology, pathophysiology and management. IJGM. (2023) 16:1899–908. doi: 10.2147/IJGM.S406926
47. Evrengül H, Dursunoglu D, Cobankara V, Polat B, Seleci D, Kabukçu S, et al. Heart rate variability in patients with rheumatoid arthritis. Rheumatol Int. (2004) 24:198–202. doi: 10.1007/s00296-003-0357-5
48. Lin T-T, Sung Y-L, Wu C-E, Zhang H, Liu Y-B, Lin S-F. Proarrhythmic risk and determinants of cardiac autonomic dysfunction in collagen-induced arthritis rats. BMC Musculoskelet Disord. (2016) 17:491. doi: 10.1186/s12891-016-1347-6
49. Pita LM, Spadella MA, Montenote MC, Oliveira PB, Chies AB. Repercussions of adjuvant-induced arthritis on body composition, soleus muscle, and heart muscle of rats. Braz J Med Biol Res. (2020) 53:e8969. doi: 10.1590/1414-431x20198969
50. Bordy R, Moretto J, Devaux S, Wendling D, Moretto-Riedweg K, Demougeot C, et al. Adjuvant-induced arthritis is a relevant model to mimic coronary and myocardial impairments in rheumatoid arthritis. Joint Bone Spine. (2021) 88:105069. doi: 10.1016/j.jbspin.2020.09.001
51. Schubert AC, Wendt MMN, de Sá-Nakanishi AB, Amado CAB, Peralta RM, Comar JF, et al. Oxidative state and oxidative metabolism of the heart from rats with adjuvant-induced arthritis. Exp Mol Pathol. (2016) 100:393–401. doi: 10.1016/j.yexmp.2016.03.005
52. Totoson P, Peyronnel C, Quirié A, Pédard M, Cefis M, Bermont L, et al. Tofacitinib improved peripheral endothelial dysfunction and brain-derived neurotrophic factor levels in the rat adjuvant-induced arthritis model. Fundam Clin Pharmacol. (2022) 36:363–74. doi: 10.1111/fcp.12731
53. Mancini JA, Blood K, Guay J, Gordon R, Claveau D, Chan C-C, et al. Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J Biol Chem. (2001) 276:4469–75. doi: 10.1074/jbc.M006865200
54. Largo R, Sánchez-Pernaute O, Marcos ME, Moreno-Rubio J, Aparicio C, Granado R, et al. Chronic arthritis aggravates vascular lesions in rabbits with atherosclerosis: a novel model of atherosclerosis associated with chronic inflammation. Arthritis Rheum. (2008) 58:2723–34. doi: 10.1002/art.23765
55. Herrero-Beaumont G, Marcos ME, Sánchez-Pernaute O, Granados R, Ortega L, Montell E, et al. Effect of chondroitin sulphate in a rabbit model of atherosclerosis aggravated by chronic arthritis. Br J Pharmacol. (2008) 154:843–51. doi: 10.1038/bjp.2008.113
56. Largo R, Martínez-Calatrava MJ, Sánchez-Pernaute O, Marcos ME, Moreno-Rubio J, Aparicio C, et al. Effect of a high dose of glucosamine on systemic and tissue inflammation in an experimental model of atherosclerosis aggravated by chronic arthritis. Am J Physiol Heart Circ Physiol. (2009) 297:H268–76. doi: 10.1152/ajpheart.00142.2009
57. Romero FI, Martínez-Calatrava MJ, Sánchez-Pernaute O, Gualillo O, Largo R, Herrero-Beaumont G. Pharmacological modulation by celecoxib of cachexia associated with experimental arthritis and atherosclerosis in rabbits. Br J Pharmacol. (2010) 161:1012–22. doi: 10.1111/j.1476-5381.2010.00957.x
58. Chouk M, Bordy R, Moretto J, Wendling D, Totoson P, Demougeot C. Pristane-induced arthritis in dark Agouti rat is a relevant model for mimicking vascular dysfunction and lipid paradox in rheumatoid arthritis. Joint Bone Spine. (2019) 86:483–90. doi: 10.1016/j.jbspin.2018.12.001
59. Pérez-Baos S, Barrasa JI, Gratal P, Larrañaga-Vera A, Prieto-Potin I, Herrero-Beaumont G, et al. Tofacitinib restores the inhibition of reverse cholesterol transport induced by inflammation: understanding the lipid paradox associated with rheumatoid arthritis. Br J Pharmacol. (2017) 174:3018–31. doi: 10.1111/bph.13932
60. Peyronnel C, Kessler J, Bobillier-Chaumont Devaux S, Houdayer C, Tournier M, Chouk M, et al. A treadmill exercise reduced cardiac fibrosis, inflammation and vulnerability to ischemia-reperfusion in rat pristane-induced arthritis. Life Sci. (2024) 341:122503. doi: 10.1016/j.lfs.2024.122503
61. Chen J, Norling LV, Garrido Mesa J, Silva MDP, Burton SE, Reutelingsperger C, et al. Annexin A1 attenuates cardiac diastolic dysfunction in mice with inflammatory arthritis. Proc Natl Acad Sci USA. (2021) 118:e2020385118. doi: 10.1073/pnas.2020385118
62. de Frutos F, Ochoa JP, Navarro-Peñalver M, Baas A, Bjerre JV, Zorio E, et al. Natural history of MYH7-related dilated cardiomyopathy. J Am Coll Cardiol. (2022) 80:1447–61. doi: 10.1016/j.jacc.2022.07.023
63. Huffman KM, Andonian BJ, Abraham DM, Bareja A, Lee DE, Katz LH, et al. Exercise protects against cardiac and skeletal muscle dysfunction in a mouse model of inflammatory arthritis. J Appl Physiol. (2021) 130:853–64. doi: 10.1152/japplphysiol.00576.2020
64. Rose S, Eren M, Murphy S, Zhang H, Thaxton CS, Chowaniec J, et al. A novel mouse model that develops spontaneous arthritis and is predisposed towards atherosclerosis. Ann Rheum Dis. (2013) 72:89–95. doi: 10.1136/annrheumdis-2012-201431
65. Ångström L, Hörnberg K, Sundström B, Södergren A. Rheumatoid cachexia in early rheumatoid arthritis: prevalence and associated variables. Scand J Rheumatol. (2023) 52:10–6. doi: 10.1080/03009742.2021.1973678
66. Rall LC, Roubenoff R. Rheumatoid cachexia: metabolic abnormalities, mechanisms and interventions. Rheumatology. (2004) 43:1219–23. doi: 10.1093/rheumatology/keh321
67. Summers GD, Deighton CM, Rennie MJ, Booth AH. Rheumatoid cachexia: a clinical perspective. Rheumatology. (2008) 47:1124–31. doi: 10.1093/rheumatology/ken146
68. Van Den Berg WB, Bresnihan B. Pathogenesis of joint damage in rheumatoid arthritis: evidence of a dominant role for interleukin-1. Best Pract Res Clin Rheumatol. (1999) 13:577–97. doi: 10.1053/berh.1999.0047
69. An HJ, Tizaoui K, Terrazzino S, Cargnin S, Lee KH, Nam SW, et al. Sarcopenia in autoimmune and rheumatic diseases: a comprehensive review. IJMS. (2020) 21:5678. doi: 10.3390/ijms21165678
70. Baker JF, Giles JT, Weber D, George MD, Leonard MB, Zemel BS, et al. Sarcopenic obesity in rheumatoid arthritis: prevalence and impact on physical functioning. Rheumatology. (2022) 61:2285–94. doi: 10.1093/rheumatology/keab710
71. Cooper C, Dere W, Evans W, Kanis JA, Rizzoli R, Sayer AA, et al. Frailty and sarcopenia: definitions and outcome parameters. Osteoporos Int. (2012) 23:1839–48. doi: 10.1007/s00198-012-1913-1
72. Filippin LI, Teixeira VN, Viacava PR, Lora PS, Xavier LL, Xavier RM. Temporal development of muscle atrophy in murine model of arthritis is related to disease severity. J Cachexia Sarcopenia Muscle. (2013) 4:231–8. doi: 10.1007/s13539-013-0102-1
73. Alabarse PVG, Lora PS, Silva JMS, Santo RCE, Freitas EC, De Oliveira MS, et al. Collagen-induced arthritis as an animal model of rheumatoid cachexia. J Cachexia Sarcopenia Muscle. (2018) 9:603–12. doi: 10.1002/jcsm.12280
74. Suginohara T, Kawaguchi M, Michihara S, Fujita N, Han L-K, Takahashi R. Ninjin'yoeito suppressed the onset of arthritis, pain, and muscle atrophy in rheumatoid arthritis model mice. Front Pharmacol. (2022) 13:974380. doi: 10.3389/fphar.2022.974380
75. De Oliveira Nunes Teixeira V, Filippin LI, Viacava PR, De Oliveira PG, Xavier RM. Muscle wasting in collagen-induced arthritis and disuse atrophy. Exp Biol Med. (2013) 238:1421–30. doi: 10.1177/1535370213505961
76. Vial G, Coudy-Gandilhon C, Pinel A, Wauquier F, Chevenet C, Béchet D, et al. Lipid accumulation and mitochondrial abnormalities are associated with fiber atrophy in the skeletal muscle of rats with collagen-induced arthritis. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 1865:158574. doi: 10.1016/j.bbalip.2019.158574
77. Teixeira VON, Bartikoski BJ, Do Espirito Santo RC, Alabarse PVG, Ghannan K, Silva JMS, et al. The role of proteasome in muscle wasting of experimental arthritis. Adv Rheumatol. (2023) 63:14. doi: 10.1186/s42358-023-00292-5
78. Yamada T, Abe M, Lee J, Tatebayashi D, Himori K, Kanzaki K, et al. Muscle dysfunction associated with adjuvant-induced arthritis is prevented by antioxidant treatment. Skelet Muscle. (2015) 5:20. doi: 10.1186/s13395-015-0045-7
79. Gómez-SanMiguel AB, Martín AI, Nieto-Bona MP, Fernández-Galaz C, Villanúa MÁ, López-Calderón A. The melanocortin receptor type 3 agonist d-Trp(8)-γMSH decreases inflammation and muscle wasting in arthritic rats. J Cachexia Sarcopenia Muscle. (2016) 7:79–89. doi: 10.1002/jcsm.12036
80. Ghouri M, Lateef M, Liaquat L, Zulfquar A, Saleem S, Zehra S. Decreased muscle strength in adjuvant-induced rheumatoid arthritis animal model: a relationship to behavioural assessments. Heliyon. (2024) 10:e23264. doi: 10.1016/j.heliyon.2023.e23264
81. Little RD, Prieto-Potin I, Pérez-Baos S, Villalvilla A, Gratal P, Cicuttini F, et al. Compensatory anabolic signaling in the sarcopenia of experimental chronic arthritis. Sci Rep. (2017) 7:6311. doi: 10.1038/s41598-017-06581-6
82. Lin J-Z, Ma J-D, Yang L-J, Zou Y-W, Zhang X-P, Pan J, et al. Myokine myostatin is a novel predictor of one-year radiographic progression in patients with rheumatoid arthritis: a prospective cohort study. Front Immunol. (2022) 13:1005161. doi: 10.3389/fimmu.2022.1005161
83. Bermejo-Álvarez I, Pérez-Baos S, Gratal P, Medina JP, Largo R, Herrero-Beaumont G, et al. Effects of tofacitinib on muscle remodeling in experimental rheumatoid sarcopenia. IJMS. (2023) 24:13181. doi: 10.3390/ijms241713181
84. Doucet MR, Laevski AM, Doiron JA, Boudreau LH, Surette ME. Locomotor activity as an effective measure of the severity of inflammatory arthritis in a mouse model. PLoS ONE. (2024) 19:e0291399. doi: 10.1371/journal.pone.0291399
85. Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen an experimental model of arthritis. J Exp Med. (1977) 146:857–68. doi: 10.1084/jem.146.3.857
86. Denninger KC, Litman T, Marstrand T, Moller K, Svensson L, Labuda T, et al. Kinetics of gene expression and bone remodelling in the clinical phase of collagen-induced arthritis. Arthritis Res Ther. (2015) 17:43. doi: 10.1186/s13075-015-0531-7
87. Sun X, Feng X, Tan W, Lin N, Hua M, Wei Y, et al. Adiponectin exacerbates collagen-induced arthritis via enhancing Th17 response and prompting RANKL expression. Sci Rep. (2015) 5:11296. doi: 10.1038/srep11296
88. Lord AE, Zhang L, Erickson JE, Bryant S, Nelson CM, Gaudette SM, et al. Quantitative in vivo micro-computed tomography for monitoring disease activity and treatment response in a collagen-induced arthritis mouse model. Sci Rep. (2022) 12:2863. doi: 10.1038/s41598-022-06837-w
89. Chen W, Wang J, Xu Z, Huang F, Qian W, Ma J, et al. Apremilast ameliorates experimental arthritis via suppression of Th1 and Th17 cells and enhancement of CD4+Foxp3+ regulatory T cells differentiation. Front Immunol. (2018) 9:1662. doi: 10.3389/fimmu.2018.01662
90. Seeuws S, Jacques P, Praet JV, Drennan M, Coudenys J, Decruy T, et al. A multiparameter approach to monitor disease activity in collagen-induced arthritis. Arthritis Res Ther. (2010) 12:R160. doi: 10.1186/ar3119
91. Kamijo S, Nakajima A, Ikeda K, Aoki K, Ohya K, Akiba H, et al. Amelioration of bone loss in collagen-induced arthritis by neutralizing anti-RANKL monoclonal antibody. Biochem Biophys Res Commun. (2006) 347:124–32. doi: 10.1016/j.bbrc.2006.06.098
92. Lin X, Wang Q, He Z, Huang L, Wen C, Zhou D. Evaluating the similarity of different collagen-induced arthritis models to the pre-clinical phase of RA in female rats. Inflammation. (2022) 45:1559–67. doi: 10.1007/s10753-022-01641-0
93. Li S, Xiang C, Wei X, Sun X, Li R, Li P, et al. Early supplemental α2-macroglobulin attenuates cartilage and bone damage by inhibiting inflammation in collagen II-induced arthritis model. Int J Rheum Dis. (2019) 22:654–65. doi: 10.1111/1756-185X.13457
94. Yan Y, Zhang L-B, Ma R, Wang M-N, He J, Wang P-P, et al. Jolkinolide B ameliorates rheumatoid arthritis by regulating the JAK2/STAT3 signaling pathway. Phytomedicine. (2024) 124:155311. doi: 10.1016/j.phymed.2023.155311
95. Liphardt A-M, Windahl SH, Sehic E, Hannemann N, Gustafsson KL, Bozec A, et al. Changes in mechanical loading affect arthritis-induced bone loss in mice. Bone. (2020) 131:115149. doi: 10.1016/j.bone.2019.115149
96. Engdahl C, Lindholm C, Stubelius A, Ohlsson C, Carlsten H, Lagerquist MK. Periarticular bone loss in antigen-induced arthritis. Arthritis Rheum. (2013) 65:2857–65. doi: 10.1002/art.38114
97. Courbon G, Lamarque R, Gerbaix M, Caire R, Linossier M-T, Laroche N, et al. Early sclerostin expression explains bone formation inhibition before arthritis onset in the rat adjuvant-induced arthritis model. Sci Rep. (2018) 8:3492. doi: 10.1038/s41598-018-21886-w
98. Koenders MI, Lubberts E, Oppers-Walgreen B, Van Den Bersselaar L, Helsen MM, Di Padova FE, et al. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. (2005) 167:141–9. doi: 10.1016/S0002-9440(10)62961-6
99. Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist–deficient mice. J Exp Med. (2000) 191:313–20. doi: 10.1084/jem.191.2.313
100. De Arruda JAA, Corrêa JD, Singh Y, Oliveira SR, Machado CC, Schneider AH, et al. Methotrexate promotes recovery of arthritis-induced alveolar bone loss and modifies the composition of the oral-gut microbiota. Anaerobe. (2022) 75:102577. doi: 10.1016/j.anaerobe.2022.102577
101. Schneider AH, Taira TM, Públio GA, Da Silva Prado D, Donate Yabuta PB, Dos Santos JC, et al. Neutrophil extracellular traps mediate bone erosion in rheumatoid arthritis by enhancing RANKL-induced osteoclastogenesis. Br J Pharmacol. (2024) 181:429–46. doi: 10.1111/bph.16227
102. Vidal B, Cascão R, Finnilä MAJ, Lopes IP, da Glória VG, Saarakkala S, et al. Effects of tofacitinib in early arthritis-induced bone loss in an adjuvant-induced arthritis rat model. Rheumatology. (2018) 57:1461–71. doi: 10.1093/rheumatology/kex258
103. Posinasetty B, Chikatipalli R, Chenchula S, Kuttiappan A, Swapna G, Lakshmi GNA, et al. Anti-arthritic efficacy of Bombax ceiba ethanolic extract in a murine model for rheumatoid arthritis using in vivo, in vitro and radiological analysis. Bioinformation. (2023) 19:833–9. doi: 10.6026/97320630019833
104. Martínez-Calatrava MJ, Prieto-Potín I, Roman-Blas JA, Tardio L, Largo R, Herrero-Beaumont G. RANKL synthesized by articular chondrocytes contributes to juxta-articular bone loss in chronic arthritis. Arthritis Res Ther. (2012) 14:R149. doi: 10.1186/ar3884
105. Prieto-Potín I, Roman-Blas J, Martínez-Calatrava M, Gómez R, Largo R, Herrero-Beaumont G. Hypercholesterolemia boosts joint destruction in chronic arthritis. An experimental model aggravated by foam macrophage infiltration. Arthritis Res Ther. (2013) 15:R81. doi: 10.1186/ar4261
106. Gómez-Barrena E, Sánchez-Pernaute O, Largo R, Calvo E, Esbrit P, Herrero-Beaumont G. Sequential changes of parathyroid hormone related protein (PTHrP) in articular cartilage during progression of inflammatory and degenerative arthritis. Ann Rheum Dis. (2004) 63:917–22. doi: 10.1136/ard.2003.008904
107. Tuncel J, Haag S, Hoffmann MH, Yau ACY, Hultqvist M, Olofsson P, et al. Animal models of rheumatoid arthritis (I): pristane-induced arthritis in the rat. PLoS ONE. (2016) 11:e0155936. doi: 10.1371/journal.pone.0155936
108. Crum RJ, Hall K, Molina CP, Hussey GS, Graham E, Li H, et al. Immunomodulatory matrix-bound nanovesicles mitigate acute and chronic pristane-induced rheumatoid arthritis. npj Regen Med. (2022) 7:1–12. doi: 10.1038/s41536-022-00208-9
109. Patten C, Bush K, Rioja I, Morgan R, Wooley P, Trill J, et al. Characterization of pristane-induced arthritis, a murine model of chronic disease: response to antirheumatic agents, expression of joint cytokines, and immunopathology. Arthritis Rheum. (2004) 50:3334–45. doi: 10.1002/art.20507
110. Hoffmann MH, Tuncel J, Skriner K, Tohidast-Akrad M, Türk B, Pinol-Roma S, et al. The rheumatoid arthritis-associated autoantigen hnRNP-A2 (RA33) is a major stimulator of autoimmunity in rats with pristane-induced arthritis1. J Immunol. (2007) 179:7568–76. doi: 10.4049/jimmunol.179.11.7568
111. Zeng M, Issotina Zibrila A, Li X, Liu X, Wang X, Zeng Z, et al. Pyridostigmine ameliorates pristane-induced arthritis symptoms in dark agouti rats. Scand J Rheumatol. (2023) 52:627–36. doi: 10.1080/03009742.2023.2196783
112. Gonçalves Dos Santos G, Jimenéz-Andrade JM, Woller SA, Muñoz-Islas E, Ramírez-Rosas MB, Ohashi N, et al. The neuropathic phenotype of the K/BxN transgenic mouse with spontaneous arthritis: pain, nerve sprouting and joint remodeling. Sci Rep. (2020) 10:15596. doi: 10.1038/s41598-020-72441-5
113. Montero-Melendez T, Madeira MFM, Norling LV, Alsam A, Curtis MA, Da Silva TA, et al. Association between periodontal disease and inflammatory arthritis reveals modulatory functions by melanocortin receptor type 3. Am J Pathol. (2014) 184:2333–41. doi: 10.1016/j.ajpath.2014.04.009
114. LaBranche TP, Hickman-Brecks CL, Meyer DM, Storer CE, Jesson MI, Shevlin KM, et al. Characterization of the KRN cell transfer model of rheumatoid arthritis (KRN-CTM), a chronic yet synchronized version of the K/BxN mouse. Am J Pathol. (2010) 177:1388–96. doi: 10.2353/ajpath.2010.100195
115. Jacobs JP, Pettit AR, Shinohara ML, Jansson M, Cantor H, Gravallese EM, et al. Lack of requirement of osteopontin for inflammation, bone erosion, and cartilage damage in the K/BxN model of autoantibody-mediated arthritis. Arthritis Rheum. (2004) 50:2685–94. doi: 10.1002/art.20381
116. Yoshida S, Ikedo A, Yanagihara Y, Sakaue T, Saeki N, Imai Y. Bub1 suppresses inflammatory arthritis–associated bone loss in mice through inhibition of TNFα-mediated osteoclastogenesis. J Bone Miner Res. (2024) 39:341–56. doi: 10.1093/jbmr/zjae015
117. Mediero A, Wilder T, Ramkhelawon B, Moore KJ, Cronstein BN. Netrin-1 and its receptor Unc5b are novel targets for the treatment of inflammatory arthritis. FASEB J. (2016) 30:3835–44. doi: 10.1096/fj.201600615R
118. García S, Forteza J, López-Otin C, Gómez-Reino JJ, González A, Conde C. Matrix metalloproteinase-8 deficiency increases joint inflammation and bone erosion in the K/BxN serum-transfer arthritis model. Arthritis Res Ther. (2010) 12:R224. doi: 10.1186/ar3211
119. Herman S, Fischer A, Presumey J, Hoffmann M, Koenders MI, Escriou V, et al. Inhibition of inflammation and bone erosion by RNA interference-mediated silencing of heterogeneous nuclear RNP A2/B1 in two experimental models of rheumatoid arthritis. Arthritis Rheumatol. (2015) 67:2536–46. doi: 10.1002/art.39223
120. Brines R, Maicas N, Ferrándiz ML, Loboda A, Jozkowicz A, Dulak J, et al. Heme oxygenase-1 regulates the progression of K/BxN serum transfer arthritis. PLoS ONE. (2012) 7:e52435. doi: 10.1371/journal.pone.0052435
121. Ferraz-Amaro I, González-Juanatey C, López-Mejias R, Riancho-Zarrabeitia L, González-Gay MA. Metabolic syndrome in rheumatoid arthritis. Mediators Inflamm. (2013) 2013:710928. doi: 10.1155/2013/710928
122. Toms TE, Panoulas VF, John H, Douglas KM, Kitas GD. Methotrexate therapy associates with reduced prevalence of the metabolic syndrome in rheumatoid arthritis patients over the age of 60- more than just an anti-inflammatory effect? A cross sectional study. Arthritis Res Ther. (2009) 11:R110. doi: 10.1186/ar2765
123. Arias-de la Rosa I, Escudero-Contreras A, Ruiz-Ponce M, Cuesta-López L, Román-Rodríguez C, Pérez-Sánchez C, et al. Pathogenic mechanisms involving the interplay between adipose tissue and auto-antibodies in rheumatoid arthritis. iScience. (2022) 25:104893. doi: 10.1016/j.isci.2022.104893
124. Wen W, He M, Liang X, Gao S, Zhou J, Yuan Z. Accelerated transformation of macrophage-derived foam cells in the presence of collagen-induced arthritis mice serum is associated with dyslipidemia. Autoimmunity. (2016) 49:115–23. doi: 10.3109/08916934.2015.1118761
125. Arias de la Rosa I, Escudero-Contreras A, Rodríguez-Cuenca S, Ruiz-Ponce M, Jiménez-Gómez Y, Ruiz-Limón P, et al. Defective glucose and lipid metabolism in rheumatoid arthritis is determined by chronic inflammation in metabolic tissues. J Intern Med. (2018) 284:61–77. doi: 10.1111/joim.12743
126. Toonen EJM, Laskewitz AJ, van Dijk TH, Bleeker A, Grefhorst A, Schouten AE, et al. Glucose kinetics in the collagen-induced arthritis model: an all-in-one model to assess both efficacy and metabolic side effects of glucocorticoids. PLoS ONE. (2014) 9:e98684. doi: 10.1371/journal.pone.0098684
127. Jhun J-Y, Yoon B-Y, Park M-K, Oh H-J, Byun J-K, Lee S-Y, et al. Obesity aggravates the joint inflammation in a collagen-induced arthritis model through deviation to Th17 differentiation. Exp Mol Med. (2012) 44:424–31. doi: 10.3858/emm.2012.44.7.047
128. Wang R, Ji C-L, Feng D-D, Wu Y-J, Li Y, Olatunji OJ, et al. Consumption of saturated fatty acids-rich lard benefits recovery of experimental arthritis by activating PPAR-γ. Mol Nutr Food Res. (2023) 67:2200429. doi: 10.1002/mnfr.202200429
129. Xie Y, Feng S-L, Mai C-T, Zheng Y-F, Wang H, Liu Z-Q, et al. Suppression of up-regulated LXRα by silybin ameliorates experimental rheumatoid arthritis and abnormal lipid metabolism. Phytomedicine. (2021) 80:153339. doi: 10.1016/j.phymed.2020.153339
130. Arias-de La Rosa I, Ruiz-Ponce M, Cuesta-López L, Pérez-Sánchez C, Leiva-Cepas F, Gahete M, et al. Clinical features and immune mechanisms directly linked to the altered liver function in patients with rheumatoid arthritis. Eur J Intern Med. (2023) 118:49–58. doi: 10.1016/j.ejim.2023.08.002
131. Radovanović-Dinić B, Tešić-Rajković S, Zivkovic V, Grgov S. Clinical connection between rheumatoid arthritis and liver damage. Rheumatol Int. (2018) 38:715–24. doi: 10.1007/s00296-018-4021-5
132. Zhang S, Zhu P, Yuan J, Cheng K, Xu Q, Chen W, et al. Non-alcoholic fatty liver disease combined with rheumatoid arthritis exacerbates liver fibrosis by stimulating co-localization of PTRF and TLR4 in rats. Front Pharmacol. (2023) 14:1149665. doi: 10.3389/fphar.2023.1149665
133. Kolodziej L. Systemic metabolism of tryptophan and its catabolites, kynurenine and 3-HAA, in mice with inflammatory arthritis. Gene. (2013) 512:23–7. doi: 10.1016/j.gene.2012.09.122
134. Wang Y, Ruan Y-Q, He L-J, Song M-K, Olatunji OJ, Wang X, et al. PPARγ Functional deficiency expedited fatty acid utilization in the liver: a foundation of inflammatory adipokine-induced hypolipemia in rheumatoid arthritis. ACS Pharmacol Transl Sci. (2024) 7:3969–83. doi: 10.1021/acsptsci.4c00470
135. Sundaram MS, Neog MK, Rasool M, Kumar GS, Hemshekhar M, Kemparaju K, et al. Guggulipid ameliorates adjuvant-induced arthritis and liver oxidative damage by suppressing inflammatory and oxidative stress mediators. Phytomedicine. (2019) 64:152924. doi: 10.1016/j.phymed.2019.152924
136. Comar JF, Babeto de Sá-Nakanishi A, de Oliveira AL, Marques Nogueira Wendt M, Bersani Amado CA, Ishii Iwamoto EL, et al. Oxidative state of the liver of rats with adjuvant-induced arthritis. Free Radic Biol Med. (2013) 58:144–53. doi: 10.1016/j.freeradbiomed.2012.12.003
137. Liang M, Wang K, Wei X, Gong X, Tang H, Xue H, et al. Replenishing decoy extracellular vesicles inhibits phenotype remodeling of tissue-resident cells in inflammation-driven arthritis. Cell Rep Med. (2023) 4:101228. doi: 10.1016/j.xcrm.2023.101228
138. Paoletti A, Ly B, Cailleau C, Gao F, de Ponfilly-Sotier MP, Pascaud J, et al. Liposomal antagomir-155-5p restores anti-inflammatory macrophages and improves arthritis in preclinical models of rheumatoid arthritis. Arthritis Rheumatol. (2024) 76:18–31. doi: 10.1002/art.42665
139. Azam AT, Odeyinka O, Alhashimi R, Thoota S, Ashok T, Palyam V, et al. Rheumatoid arthritis and associated lung diseases: a comprehensive review. Cureus. (2022) 14:e22367. doi: 10.7759/cureus.22367
140. Olson AL, Swigris JJ, Sprunger DB, Fischer A, Fernandez-Perez ER, Solomon J, et al. Rheumatoid arthritis–interstitial lung disease–associated mortality. Am J Respir Crit Care Med. (2011) 183:372–8. doi: 10.1164/rccm.201004-0622OC
141. Poole JA, Thiele GM, Janike K, Nelson AJ, Duryee MJ, Rentfro K, et al. Combined collagen-induced arthritis and organic dust-induced airway inflammation to model inflammatory lung disease in rheumatoid arthritis. J Bone Miner Res. (2019) 34:1733–43. doi: 10.1002/jbmr.3745
142. Huo Y, Gao Y, Li B, Zhang P, Liu H, Wang G, et al. Analysis of how melatonin-upregulated clock genes PER2 and CRY2 alleviate rheumatoid arthritis-associated interstitial lung disease. Eur J Pharmacol. (2025) 986:177136. doi: 10.1016/j.ejphar.2024.177136
143. Mikuls TR, Gaurav R, Thiele GM, England BR, Wolfe MG, Shaw BP, et al. The impact of airborne endotoxin exposure on rheumatoid arthritis-related joint damage, autoantigen expression, autoimmunity, and lung disease. Int Immunopharmacol. (2021) 100:108069. doi: 10.1016/j.intimp.2021.108069
144. Li X, Wang Y, Chen Z, Ruan M, Yang C, Zhou M, et al. Hepatorenal pathologies in TNF-transgenic mouse model of rheumatoid arthritis are alleviated by anti-TNF treatment. Arthritis Res Ther. (2023) 25:188. doi: 10.1186/s13075-023-03178-5
145. Khoroshun K, Bantel C, Hoffmann F, Jobski K. Methotrexate-related drug reactions on kidneys and liver in rheumatoid arthritis: an analysis of spontaneous reports in EudraVigilance. Arthritis Res Ther. (2025) 27:80. doi: 10.1186/s13075-025-03551-6
146. Fu Y, Xiang Y, Wei Q, Ilatovskaya D, Dong Z. Rodent models of AKI and AKI-CKD transition: an update in 2024. Am J Physiol Renal Physiol. (2024) 326:F563–83. doi: 10.1152/ajprenal.00402.2023
147. Huang J-R, Chen L, Li C-Q. Ethoxyquin mediates lung fibrosis and cellular immunity in BLM-CIA mice by inhibiting HSP90. Adv Clin Exp Med. (2024) 34:211–25. doi: 10.17219/acem/186365
148. Ba X, Wang H, Huang Y, Yan J, Han L, Lin W, et al. Simiao pill attenuates collagen-induced arthritis and bleomycin-induced pulmonary fibrosis in mice by suppressing the JAK2/STAT3 and TGF-β/Smad2/3 signalling pathway. J Ethnopharmacol. (2023) 309:116274. doi: 10.1016/j.jep.2023.116274
149. Gaurav R, Mikuls TR, Thiele GM, Nelson AJ, Niu M, Guda C, et al. High-throughput analysis of lung immune cells in a combined murine model of agriculture dust-triggered airway inflammation with rheumatoid arthritis. PLoS ONE. (2021) 16:e0240707. doi: 10.1371/journal.pone.0240707
150. Song L, Kong X, Wang H, Zhan L. Establishment of a rat adjuvant arthritis-interstitial lung disease model. Biomed Res Int. (2016) 2016:2970783. doi: 10.1155/2016/2970783
151. Xu Q, Chen J, Ye W, Zhang C, Wang D, Wei W, et al. Activation of angiotensin II type 2 receptor attenuates lung injury of collagen-induced arthritis by alleviating endothelial cell injury and promoting Ly6Clo monocyte transition. Eur J Pharmacol. (2023) 941:175466. doi: 10.1016/j.ejphar.2022.175466
152. Schurgers E, Mertens F, Vanoirbeek JAJ, Put S, Mitera T, De Langhe E, et al. Pulmonary inflammation in mice with collagen-induced arthritis is conditioned by complete Freund's adjuvant and regulated by endogenous IFN-γ. Eur J Immunol. (2012) 42:3223–34. doi: 10.1002/eji.201242573
153. Sato T, Satooka H, Ichioka S, Maruo Y, Hirata T. Citrullinated fibrinogen is a target of auto-antibodies in interstitial lung disease in mice with collagen-induced arthritis. Int Immunol. (2020) 32:533–45. doi: 10.1093/intimm/dxaa021
154. Shilling RA, Williams JW, Perera J, Berry E, Wu Q, Cummings OW, et al. Autoreactive T and B cells induce the development of bronchus-associated lymphoid tissue in the lung. Am J Respir Cell Mol Biol. (2013) 48:406–14. doi: 10.1165/rcmb.2012-0065OC
155. Naskar D, Teng F, Felix KM, Bradley CP, Joyce Wu H-J. Synthetic retinoid AM80 ameliorates lung and arthritic autoimmune responses by inhibiting Tfh and Th17 cell responses. J Immunol. (2017) 198:1855–64. doi: 10.4049/jimmunol.1601776
156. Bradley CP, Teng F, Felix KM, Sano T, Naskar D, Block KE, et al. Segmented filamentous bacteria provoke lung autoimmunity by inducing gut-lung axis th17 cells expressing dual TCRs. Cell Host Microbe. (2017) 22:697–704.e4. doi: 10.1016/j.chom.2017.10.007
157. Teng F, Felix KM, Bradley CP, Naskar D, Ma H, Raslan WA, et al. The impact of age and gut microbiota on Th17 and Tfh cells in K/BxN autoimmune arthritis. Arthritis Res Ther. (2017) 19:188. doi: 10.1186/s13075-017-1398-6
158. Tokoroyama T, Ando M, Setoguchi K, Tsuchiya K, Nitta K. Prevalence, incidence and prognosis of chronic kidney disease classified according to current guidelines: a large retrospective cohort study of rheumatoid arthritis patients. Nephrol Dial Transplant. (2017) 32:2035–42. doi: 10.1093/ndt/gfw315
159. Hickson LJ, Crowson CS, Gabriel SE, McCarthy JT, Matteson EL. Development of reduced kidney function in rheumatoid arthritis. Am J Kidney Dis. (2014) 63:206–13. doi: 10.1053/j.ajkd.2013.08.010
160. Hanaoka H, Kikuchi J, Hiramoto K, Saito S, Kondo Y, Kaneko Y. Decreased chronic kidney disease in rheumatoid arthritis in the era of biologic disease-modifying anti-rheumatic drugs. Clin Kidney J. (2022) 15:1373–8. doi: 10.1093/ckj/sfac036
161. Sihvonen S, Korpela M, Mustonen J, Laippala P, Pasternack A. Renal disease as a predictor of increased mortality among patients with rheumatoid arthritis. Nephron Clin Pract. (2004) 96:c107–14. doi: 10.1159/000077372
162. Chiu H-Y, Huang H-L, Li C-H, Chen H-A, Yeh C-L, Chiu S-H, et al. Increased risk of chronic kidney disease in rheumatoid arthritis associated with cardiovascular complications - a National Population-Based Cohort Study. PLoS ONE. (2015) 10:e0136508. doi: 10.1371/journal.pone.0136508
163. Kapoor T, Bathon J. Renal manifestations of rheumatoid arthritis. Rheum Dis Clin North Am. (2018) 44:571–84. doi: 10.1016/j.rdc.2018.06.008
164. Meng M, Wang L, Yao Y, Lin D, Wang C, Yao J, et al. Ganoderma lucidum polysaccharide peptide (GLPP) attenuates rheumatic arthritis in rats through inactivating NF-κB and MAPK signaling pathways. Phytomedicine. (2023) 119:155010. doi: 10.1016/j.phymed.2023.155010
165. Sahu D, Sharma S, Singla RK, Panda AK. Antioxidant activity and protective effect of suramin against oxidative stress in collagen induced arthritis. Eur J Pharm Sci. (2017) 101:125–39. doi: 10.1016/j.ejps.2017.02.013
166. Kim KR, Park K-K, Chun K-S, Chung W-Y. Honokiol inhibits the progression of collagen-induced arthritis by reducing levels of pro-inflammatory cytokines and matrix metalloproteinases and blocking oxidative tissue damage. J Pharmacol Sci. (2010) 114:69–78. doi: 10.1254/jphs.10070fp
167. Jonker M, Wubben J, Haanstra K, Vierboom M, ‘t Hart B. Comparative analysis of inflammatory infiltrates in collagen-induced arthritis, kidney graft rejection and delayed-type hypersensitivity in non-human primates. Inflamm Res. (2013) 62:181–94. doi: 10.1007/s00011-012-0564-1
168. Maarouf RE, Abdel-Rafei MK, Thabet NM, Azab KS, Rashed L, El Bakary NM. Ondansetron or beta-sitosterol antagonizes inflammatory responses in liver, kidney, lung and heart tissues of irradiated arthritic rats model. Int J Immunopathol Pharmacol. (2024) 38:03946320241260635. doi: 10.1177/03946320241260635
169. da Silva JLG, Passos DF, Bernardes VM, Cabral FL, Schimites PG, Manzoni AG, et al. Co-nanoencapsulation of vitamin D3 and curcumin regulates inflammation and purine metabolism in a model of arthritis. Inflammation. (2019) 42:1595–610. doi: 10.1007/s10753-019-01021-1
170. Wang Q-H, Pan S, Yang K, Wu Y-J, Cheng X-P, Olatunji OJ, et al. Glycolysis aggravates methotrexate toxicity by fueling RFC1-controlled intestinal absorption in rheumatic rats. Biomed Pharmacother. (2022) 150:113067. doi: 10.1016/j.biopha.2022.113067
171. Ramadan G, El-Menshawy O. Protective effects of ginger-turmeric rhizomes mixture on joint inflammation, atherogenesis, kidney dysfunction and other complications in a rat model of human rheumatoid arthritis. Int J Rheum Dis. (2013) 16:219–29. doi: 10.1111/1756-185X.12054
172. Mythilypriya R, Shanthi P, Sachdanandam P. Therapeutic effect of Kalpaamruthaa, a herbal preparation on adjuvant induced arthritis in wistar rats. Inflammopharmacology. (2008) 16:21–35. doi: 10.1007/s10787-007-1602-4
173. Hou W, Meng L, Tian L, Zhu W, Jiang C, Lu S. A systematic comparison between collagen-induced arthritis and pristane-induced arthritis in Dark Agouti rats. Clin Exp Rheumatol. (2010) 28:532–8.
174. Liao P, He Y, Yang F, Luo G, Zhuang J, Zhai Z, et al. Polydatin effectively attenuates disease activity in lupus-prone mouse models by blocking ROS-mediated NET formation. Arthritis Res Ther. (2018) 20:254. doi: 10.1186/s13075-018-1749-y
175. Dalix E, Paret C, Maalouf M, Peyroche S, Vanden-Bossche A, Marotte H. AB0051 description of pulmonary involvement in the rat adjuvant-induced arthritis model. Ann Rheum Dis. (2023) 82:1204. doi: 10.1136/annrheumdis-2023-eular.3716
Keywords: rheumatoid arthritis, animal models, comorbidities, inflammation, innate immunity
Citation: Marco-Bonilla M, Fresnadillo M, Riva-Bueno Mdl, Herrero-Beaumont G, González-Gay MA, Largo R and Mediero A (2025) Beyond joints: the importance of animal models in exploring rheumatoid arthritis comorbidities. Front. Med. 12:1693610. doi: 10.3389/fmed.2025.1693610
Received: 27 August 2025; Accepted: 17 October 2025;
Published: 05 November 2025.
Edited by:
Cong-Qiu Chu, Oregon Health and Science University, United StatesReviewed by:
Ahed J. Alkhatib, Jordan University of Science and Technology, JordanSantenna Chenchula, All India Institute of Medical Sciences, Bhopal, India
Copyright © 2025 Marco-Bonilla, Fresnadillo, Riva-Bueno, Herrero-Beaumont, González-Gay, Largo and Mediero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Angel González-Gay, bWlndWVsYWdnYXlAaG90bWFpbC5jb20=
†These authors have contributed equally to this work and share second authorship
‡These authors have contributed equally to this work and share last authorship