




- 1 Department of Microbiology, Institute of Health Biosciences, The University of Tokushima Graduate School, Tokushima, Japan
- 2 Research Institute for Drug Discovery, School of Pharmacy, Kumamoto University, Kumamoto, Japan
HIV-1 Vpu acts positively on viral infectivity by mediating CD4 degradation in endoplasmic reticulum and enhances virion release by counteracting a virion release restriction factor, tetherin. In order to define the impact of Vpu activity on HIV-1 replication, we have generated a series of site-specific proviral vpu mutants. Of fifteen mutants examined, seven exhibited a replication-defect similar to that of a vpu-deletion mutant in a lymphocyte cell line H9. These mutations clustered in narrow regions within transmembrane domain (TMD) and cytoplasmic domain (CTD). Replication-defective mutants displayed the reduced ability to enhance virion release from a monolayer cell line HEp2 without exception. Upon transfection with Vpu expression vectors, neither TMD mutants nor CTD mutants blocked CD4 expression at the cell surface in another monolayer cell line MAGI. While TMD mutants were unable to down-modulate cell surface tetherin in HEp2 cells, CTD mutants did quite efficiently. Confocal microscopy analysis revealed the difference of intracellular localization between TMD and CTD mutants. In total, replication capability of HIV-1 carrying vpu mutations correlates well with the ability of Vpu to enhance virion release and to impede the cell surface expression of CD4 but not with the ability to down-modulate cell surface tetherin. Our results here suggest that efficient viral replication requires not only down-regulation of cell surface tetherin but also its degradation.
Introduction
Accessory protein Vpu is encoded only by HIV-1 and certain strains of simian immunodeficiency virus (Desrosiers, 2007; Kirchhoff, 2009). HIV-1 Vpu is a phosphorylated type I integral membrane protein and consists of two major domains, that is, an N-terminal transmembrane domain (TMD) and a cytoplasmic domain (CTD) (for review, Nomaguchi et al., 2008). Major functions of HIV-1 Vpu are: (1) to give a positive effect on viral infectivity by mediating CD4 degradation in endoplasmic reticulum (ER) (Bour and Strebel, 2003; Levesque et al., 2003; Tanaka et al., 2003), and (2) to enhance virion release in a cell-type dependent manner by counteracting a host restriction factor, tetherin (Strebel et al., 1989; Terwilliger et al., 1989; Sakai et al., 1995; Neil et al., 2008; Van Damme et al., 2008). For CD4 degradation, Vpu binds to newly synthesized CD4 in ER and recruits β-TrCP, which is a component of E3 ubiquitin ligase complex, via two conserved serine residues (at positions 52 and 56). Subsequently, CD4 is ubiquitinated and degraded by 26S proteasomes (Lenburg and Landau, 1993; Bour et al., 1995; Paul and Jabbar, 1997; Margottin et al., 1998). For virion release enhancement, Vpu counteracts tetherin that is induced by type I interferon (Neil et al., 2007, 2008; Van Damme et al., 2008). Tetherin traps nascent virions at cell surface and restricts virion release of various viruses including retroviruses, arenaviruses, filoviruses, and herpesviruses (for review, Tokarev et al., 2009; Douglas et al., 2010; Sauter et al., 2010). Vpu down-modulates cell surface tetherin and modestly decreases total cellular tetherin (Van Damme et al., 2008; Mitchell et al., 2009). Vpu appears to direct tetherin to trans-Golgi network (TGN) or to early endosomes for β-TrCP dependent proteasomal or lysosomal degradation (Douglas et al., 2009; Goffinet et al., 2009; Iwabu et al., 2009; Mangeat et al., 2009). Since binding of Vpu to tetherin partially relieves the restriction of virion release by tetherin, depletion of total cellular tetherin does not entirely account for counteraction of Vpu (Mangeat et al., 2009; Dubé et al., 2010). It also has been shown that Vpu enhances virion release even in the absence of down-modulation of cell surface and intracellular tetherin (Miyagi et al., 2009). The exact mechanism for antagonism of tetherin by Vpu remains to be elucidated. Early work proposed that two separable biological activities are directed by distinct Vpu domains: TMD is important for virion release enhancement and CTD, especially serine 52 and 56, is critical for CD4 degradation (Schubert and Strebel, 1994; Schubert et al., 1996). Recent studies have suggested that both TMD and CTD are involved in two major functions of Vpu (Van Damme et al., 2008; Douglas et al., 2009; Mangeat et al., 2009; Magadán et al., 2010). Functional domains of Vpu and mechanism by which Vpu mediates CD4 degradation and counteracts tetherin have been intensively studied using mainly a randomized mutants of TMD and serine 52 and/or 56 mutants in CTD. However, functional mapping of Vpu and its effect on HIV-1 replication have not been well studied.
In this study, to better define HIV-1 Vpu functions for viral replication, we performed a comprehensive and systemic analysis using a series of site-specific vpu mutants. Virological analysis of the vpu mutants in a lymphocyte cell line identified two clusters in TMD and CTD important for efficient viral replication. Vpu mutants within these two clusters exhibited the reduced ability to enhance virion release and to disturb CD4 expression at the cell surface. While CTD mutants extensively colocalized with tetherin in intracellular compartments and exhibited the capability to down-modulate cell surface tetherin, TMD mutants failed to do so. In sum, our results show that replication capability of the vpu mutations correlates with ability to augment virion release and to down-modulate CD4 but not to down-regulate cell surface tetherin. This finding suggests that antagonism of tetherin by Vpu for optimal viral replication may require tetherin degradation in addition to down-modulation of cell surface tetherin.
Materials and Methods
Plasmid DNA
Construction of a series of site-specific HIV-1 vpu mutants from pNL4-3 (Adachi et al., 1986) was carried out by the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, USA). Amino acid and nucleotide substitutions are as follows: V9D and V13D (gta to gac), I19D and I24D (ata to gac), RK30AA (agg aaa to gcg gca), Q35A (caa to gca), D43A (gat to gct), E47A (gaa to gca), EDS50AAA (gaa gac agt to gca gca gct), S52A (agt to gct), S56G (agt to ggt), E59K (gaa to aaa), M66V and M70V (atg to gtg), and P75S (cct to tct). A vpu-deletion mutant, pNL-uE65 (Iida et al., 1999), was generated by replacing the initiation codon (ATG) to AGG. For flow cytometry analysis, vectors co-expressing Vpu by cytomegalovirus promoter (CMV P) and GFP via an internal ribosomal entry site (IRES) were constructed as follows. Wild-type (WT) and mutated vpu sequences were amplified by PCR using forward primer (Vpu-5-SacII: TCCCCGCGGATGCAACCTATAATAGTAGC), reverse primer (U down BamHI: CGCGGATCCCTACAGATCATCAATATCCC), and pNL4-3 derived sequences as template. Amplified products were then digested with SacII and BamHI, cloned into pIRES-hrGFP-1a vector (Stratagene), and resultant clones were designated pIRES-hrGFP-Vpu vectors. To analyze intracellular localization of Vpu, vectors expressing C-terminally FLAG-tagged Vpu were generated. Vpu genes were amplified by PCR using forward primer (EcoRI-5-Vpu: CGGAATTCAAGGATGCAACCTATAATAGTAGC), reverse primer (Vpu-3-XhoI: CCGCTCGAGCAGATCATCAATATCCCAAG), and proviral vpu mutant clones as template, and cloned into pSG-cFLAG as described previously (Wang et al., 2005). The resultant clones were designated pSG-VpucFLAG vectors.
Cells
293T (Lebkowski et al., 1985) and HEp2 cells (ATCC CCL-23) were maintained in Eagle’s minimal essential medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS). MAGI cells (Kimpton and Emerman, 1992) were cultured in MEM containing 10% FBS, 200 μg/mL G418 (SIGMA-ALDRICH, St. Louis, USA), and 100 μg/mL hygromycin B (SIGMA-ALDRICH). H9 cells (Mann et al., 1989) were maintained in RPMI1640 medium supplemented with 10% FBS.
Assays for Viral Replication
Input viruses were prepared from 293T cells transfected with proviral clones by the calcium-phosphate co-precipitation method (CPC; Adachi et al., 1986). H9 cells (1 × 106) were infected with equivalent amounts (5 × 106 cpm) of various viruses as determined by reverse transcriptase (RT) assays, and monitored for virus replication by RT assays as previously described (Willey et al., 1988).
Assays for Virion Release from Cells
HEp2 cells were transfected with proviral clones by CPC followed by glycerol treatment (20% glycerol in MEM without FBS). On day 2 post-transfection, culture supernatants were collected and virions released from cells were determined by RT assays. Virion release assays in 293T cells were similarly performed; no glycerol treatment following CPC transfection. For analysis of Vpu expression vectors, subconfluent HEp2 cells in 24 well-dishes were transfected with 300 ng of pNL-uE65, 200 ng of pSG5, and 200 ng of pIRES-hrGFP-Vpu vectors or pSG-VpucFLAG vectors by Lipofectamine 2000 (Invitrogen, Carlsbad, USA). At 6 h post-transfection, culture medium was replaced with fresh 10% FBS-MEM and cells were incubated for 2 days. Viral production in the culture supernatants was determined by RT assays.
Western Blot Analysis
For analysis of Vpu expression by proviral clones, transfected HEp2 cells were lysed with CHAPS buffer (10 mM CHAPS, 150 mM NaCl, 1 mM NaF, 1 mM Na3VO4, and 10 mM Na-phosphate pH 7.4). For analysis of Vpu expression by subgenomic vectors, transfected 293T cells were lysed in TNE buffer (10 mM Tris–HCl pH 8.0, 1% Nonidet P-40, 150 mM NaCl, 1 mM EDTA, and 1% protease inhibitor cocktail (SIGMA-ALDRICH)]. Vpu in cell lysates were monitored as previously described (Yamashita et al., 2008) using anti-FLAG (SIGMA-ALDRICH), anti-β actin (SIGMA-ALDRICH), anti-p24 (NIH AIDS Research and References Reagent Program), or anti-Vpu (NIH AIDS Research and References Reagent Program) antibody.
Flow Cytometry Analysis
For analysis of cell surface CD4, MAGI cells in 60 mm dishes were transfected with 5 μg of pIRES-hrGFP-Vpu vectors by CPC. On day 2 post-transfection, cells were trypsinized, washed with PBS, and resuspended in FACS washing buffer (10% FBS in PBS). Cells were then stained with a PE conjugate mouse anti-human CD4 antibody (BD Biosciences, San Jose, USA). For analysis of cell surface tetherin, HEp2 cells in 60 mm dishes were transfected with pIRES-hrGFP-Vpu vectors and harvested as described above. Cells were then stained with an anti-HM1.24 monoclonal antibody (a generous gift from Chugai Pharmaceutical Co., Tokyo, Japan) and a secondary PE conjugate anti-mouse Ig antibody (BD Biosciences). Stained cells were analyzed by FACSCalibur and CELLQuest software (BD Biosciences).
Confocal Microscopy
HEp2 cells were seeded onto 4-well Lab-Tek chambered coverglasses (Nalge Nunc International, Rochester, USA), incubated overnight, and transfected with a pNL-uE65 (300 ng), pSG-VpucFLAG vectors (200 ng), and pSG5 (200 ng) by Lipofectamine 2000 (Invitrogen). On the next day, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and incubated with a first antibody [anti-TGN46 (Novus Biologicals, Inc., Littleton, USA), anti-FLAG, or anti-HM1.24 antibody]. Cells were then incubated with an appropriate secondary antibody [Alexa Fluor 488 or 555 SFX kit (Invitrogen)] according to manufacturer’s instructions, and cell nuclei were counterstained with DAPI. Cell images were obtained by DIGITAL ECLIPSE C1si-Ready (Nikon Co., Tokyo, Japan) and analyzed by EZ-C1 viewer (Nikon Co.).
Results
Amino Acids in Vpu Important for Efficient HIV-1 Replication are Clustered in Narrow Regions within TMD and CTD
HIV-1 Vpu has been shown to positively modulate viral infectivity by mediating CD4 degradation and also to promote virion release by down-modulating tetherin (Levesque et al., 2003; Neil et al., 2008; Van Damme et al., 2008). The correlation between the viral replication and the two Vpu functions has been examined for a limited number and kind of vpu mutants (Terwilliger et al., 1989; Schubert et al., 1996; Van Damme et al., 2008; Dubé et al., 2010). To study functional role of Vpu in viral replication more clearly and extensively, we generated a series of proviral vpu mutants (Figure 1A). Substitutions were introduced into residues that are well-conserved among viruses of HIV-1 group M. For mutations within TMD, Val9, Val13, Ile19, and Ile24 were replaced with Asp, respectively. By these substitutions, TMD was changed to contain a negatively charged amino acid in place of non-charged amino acids. Some CTD mutants (ArgLys30, Gln35, Asp43, Glu47, GluAspSer50, and Ser52) were generated by Ala-substitution, and the other mutants (Ser56, Glu59, Met66, Met70, and Pro75) contained amino acids with physicochemical property distinct from authentic ones (Gly, Lys, Val, Val, and Ser, respectively). Since Ser52 and Ser56 are key residues to interact with β-TrCP (Schubert and Strebel, 1994; Margottin et al., 1998), it is expected that S52A and S56G mutations disrupt the ability to recruit E3 ubiquitin ligase complex and the following degradation of target proteins.
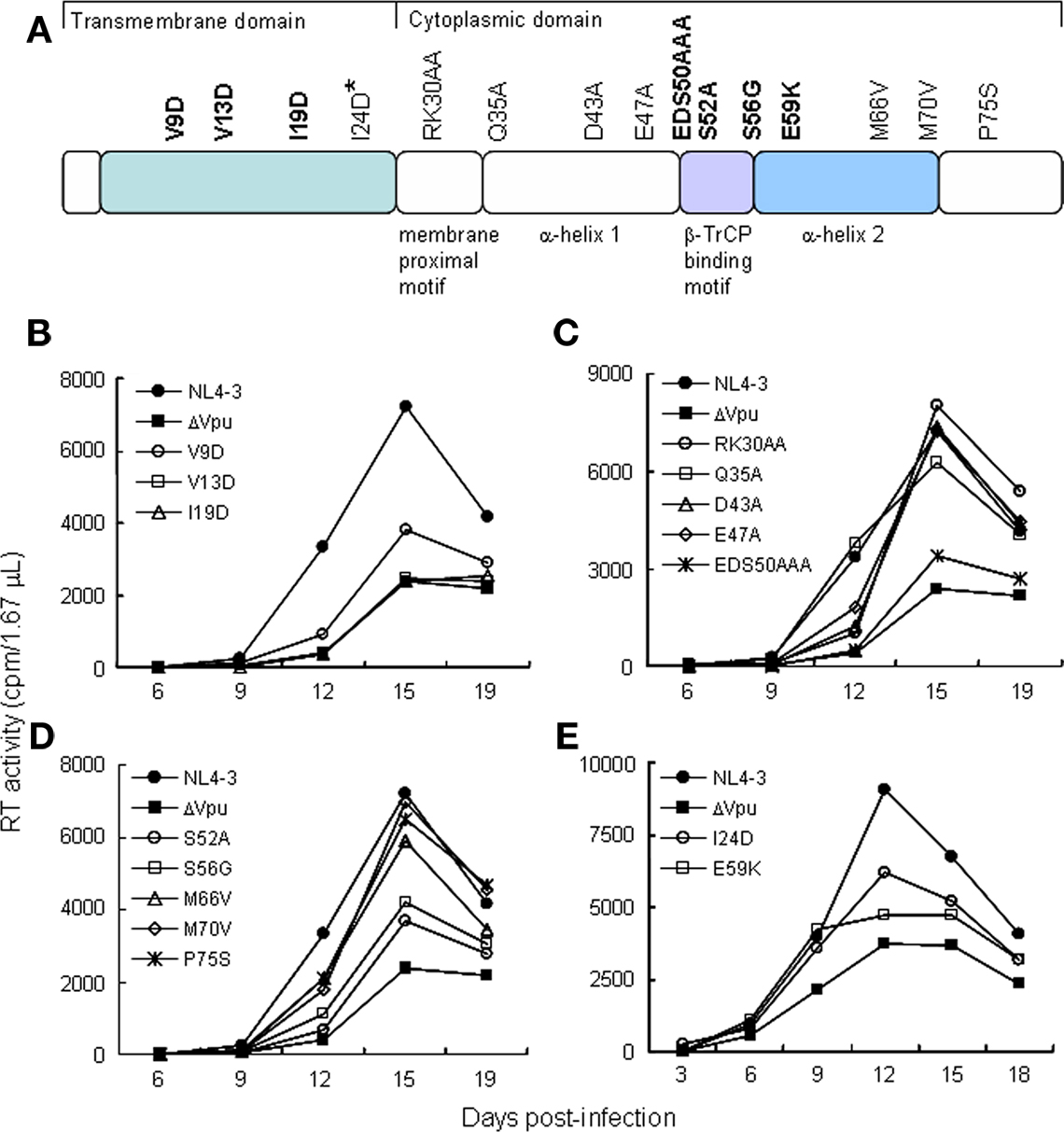
Figure 1. HIV-1 vpu mutants used in this study. (A) Locations of mutations in Vpu (NL4-3 clone) and the mutant designations. Bold letters indicate replication-defective mutants in H9 cells. The I24D mutant displayed a small replication-defect in H9 cells. Colored areas show functional regions of Vpu involved in viral replication. (B–E) Replication kinetics of various vpu mutants in H9 cells. H9 cells (1 × 106) were infected with equivalent RT units (5 × 106) of cell-free viruses prepared from transfected 293T cells. Virus replication was monitored at intervals by RT production in the culture supernatants.
To determine replication kinetics, mutant virus stocks were prepared from transfected 293T cells and inoculated into a Vpu-dependent lymphocyte cell line H9 (Figures 1B–E). Out of fifteen mutants shown in Figure 1A, seven (V9D, V13D, I19D, EDS50AAA, S52A, S56G, and E59K) exhibited a replication-defect similar to that of a vpu-deletion mutant (ΔVpu in Figures 1B–E) in H9 cells. The other mutant viruses were found to have a replication potential similar to that of WT virus. Viral clone carrying I24D mutation had a small replication-defect but appeared not to be significant (Figure 1E). In total, the results described above demonstrate that narrow regions in Vpu TMD and CTD are required for efficient viral replication.
Replication-Defective Vpu Mutants Display Reduced Ability to Enhance Virion Release in HEp2 Cells
It has been shown previously that Vpu is required to promote virion release in tetherin-positive cells (e.g., HeLa, HEp2, T cells, and macrophages) but dispensable in tetherin-negative cells (e.g., 293T, HT1080, and COS-7; Sakai et al., 1995; Neil et al., 2008). To determine if mutations in vpu affect progeny production, proviral mutant clones were transfected into HEp2 and 293T cells, and virion level in the culture supernatants was determined by RT assays. In HEp2 cells, control ΔVpu and seven TMD (V9D, V13D, and I19D) and CTD (EDS50AAA, S52A, S56G, and E59K) mutants exhibited two- to three-fold reduction of virus production relative to WT (Figure 2A). These seven mutants were replication-defective in H9 cells (Figures 1B–E). The other eight mutants behaved similarly with WT with respect to virion production in HEp2 cells. As expected, all mutant clones were indistinguishable from WT in 293T cells (Figure 2B). Vpu expression level in HEp2 cells was different among proviral clones in this particular experiment (Figure 2C), but virion level was not correlated to the amount of Vpu (Figures 2A–C). The results described above indicate that, in the presence of tetherin, efficient viral replication coincidentally occurred with promotion of virion release by Vpu.
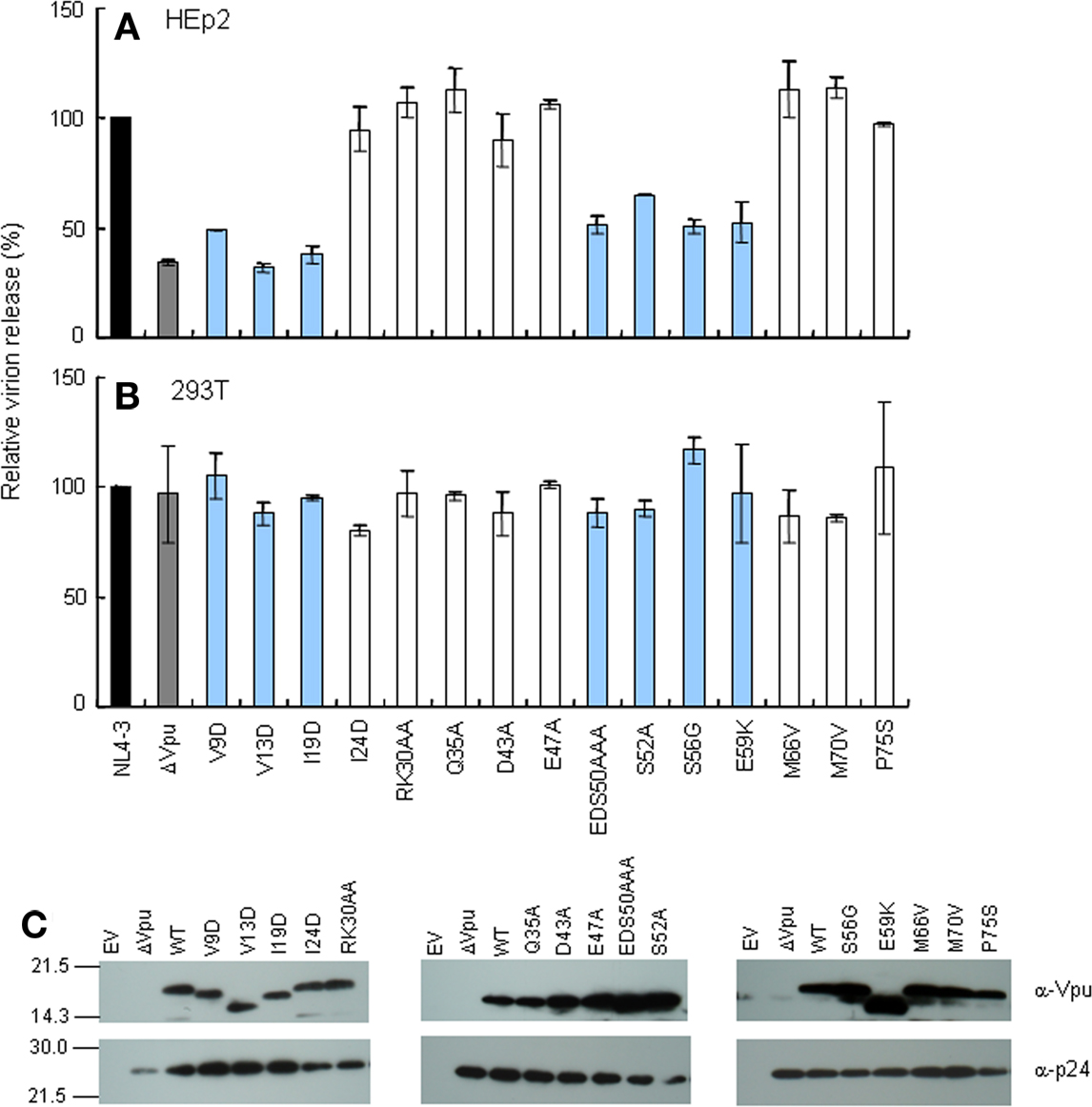
Figure 2. Ability of proviral vpu mutants to enhance virion release from cells. (A,B) Cells indicated were transfected with 10 μg of proviral clones, and on day 2 post-transfection, culture supernatants were collected and virion production was determined by RT assays. Values represent virion production level relative to that by WT clone (NL4-3). Black and gray bars indicate WT (pNL4-3) and ΔVpu (pNL-uE65) proviral constructs, respectively. Replication-defective mutants in H9 cells are shown by blue bars. (C) Western blot analysis of Vpu expression in lysates of transfected HEp2 cells. Protein amounts loaded were 1 ng of Gag-p24. EV, empty vector control.
Replication-Defective TMD (V9D and I19D) and CTD (S56G and E59K) Mutants Lack the Ability to Down-Modulate CD4
Down-modulation of CD4 appears to be important for HIV-1 replication, since numbers of viral proteins (Env, Nef, and Vpu) show this particular activity. For analysis of CD4 and tetherin down-regulation by Vpu, we generated constructs that co-express Vpu (target for study) and GFP (transfection marker). To confirm the activity of these constructs, Vpu expression and virion production level were examined. As shown in Figure 3, these vectors expressed the Vpu proteins and complemented virion production of ΔVpu to a similar extent with that observed in the provirus experiment (Figure 2A).
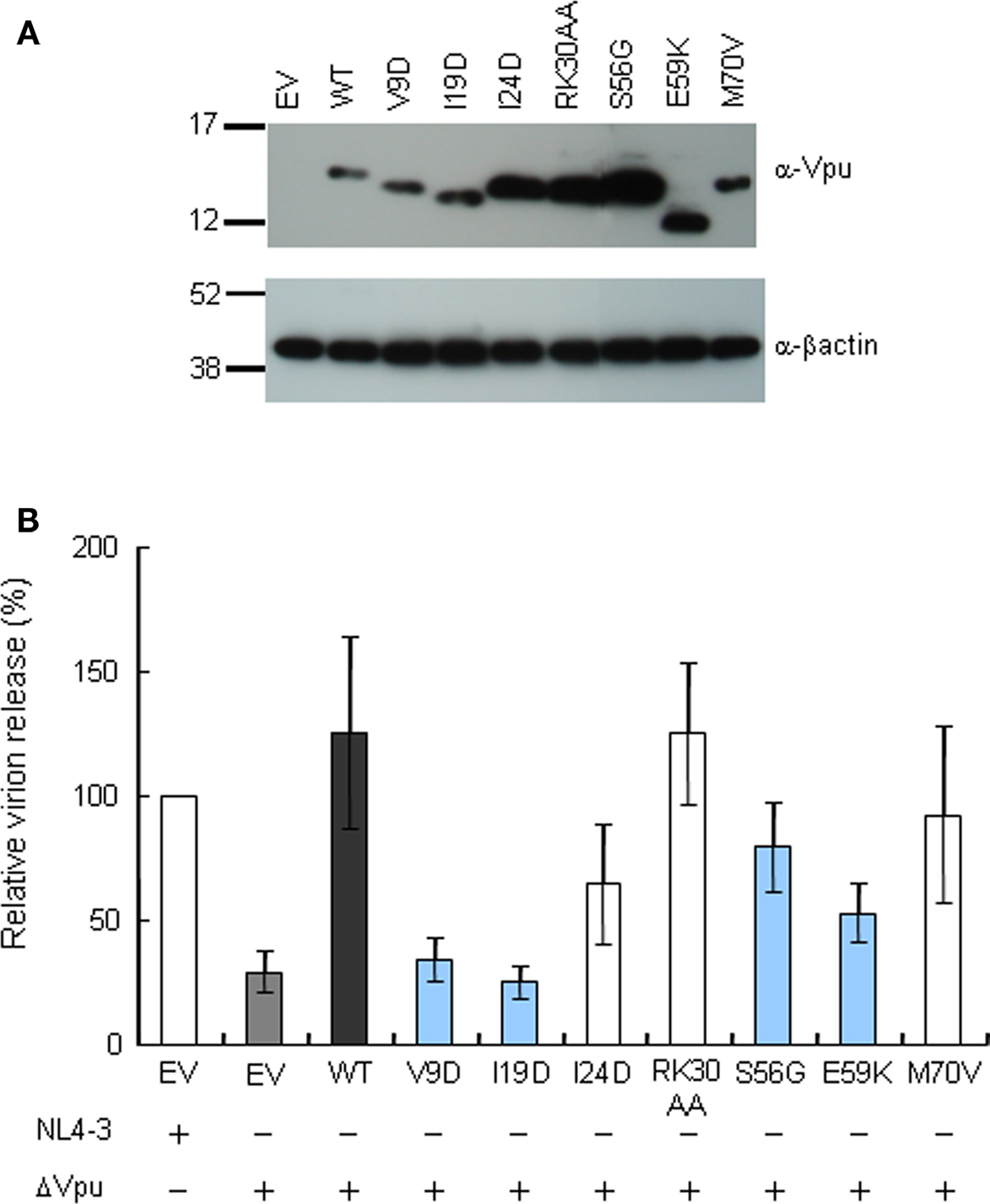
Figure 3. Trans-complementation assay for virion release from HEp2 cells. (A) Western blot analysis of Vpu expression in lysates of 293T cells transfected with various pIRES-hrGFP-Vpu vectors. Equivalent amounts of total protein (15 μg) were loaded and Vpu was detected by anti-Vpu antibody. β-actin was used as a control. EV, empty vector control. (B) Virion release assays. HEp2 cells were transfected with the indicated proviral constructs (300 ng) and pIRES-hrGFP-Vpu vectors (200 ng), and virion production in the culture supernatants on day 2 post-transfection was determined by RT assays. Values represent virion production level relative to that by WT proviral clone (NL4-3). Gray bar shows the results of proviral ΔVpu (NL-uE65). Black bar indicates the results of complementation of ΔVpu (pNL-uE65) by WT Vpu (pIRES-hrGFP-WTVpu). Blue and white bars show the results of expression vectors derived from replication-defective and replication-normal mutants in H9 cells, respectively. EV, empty vector control pSG5 (400 ng).
To determine whether Vpu mutant proteins down-modulate CD4, we evaluated its expression level at the surface of transfected MAGI cells (CD4-positive HeLa cells) (Figure 4). Some mutants (I24D, RK30AA, and M70V) were able to decrease the cell surface level of CD4 equally efficiently with WT Vpu. In contrast, mutants V9D, I19D, S56G, and E59K failed to down-modulate cell surface CD4. These four mutants were all replication-defective in H9 cells (Figure 1). Thus, viral replication potential and ability to down-modulate CD4 by Vpu are correlated.
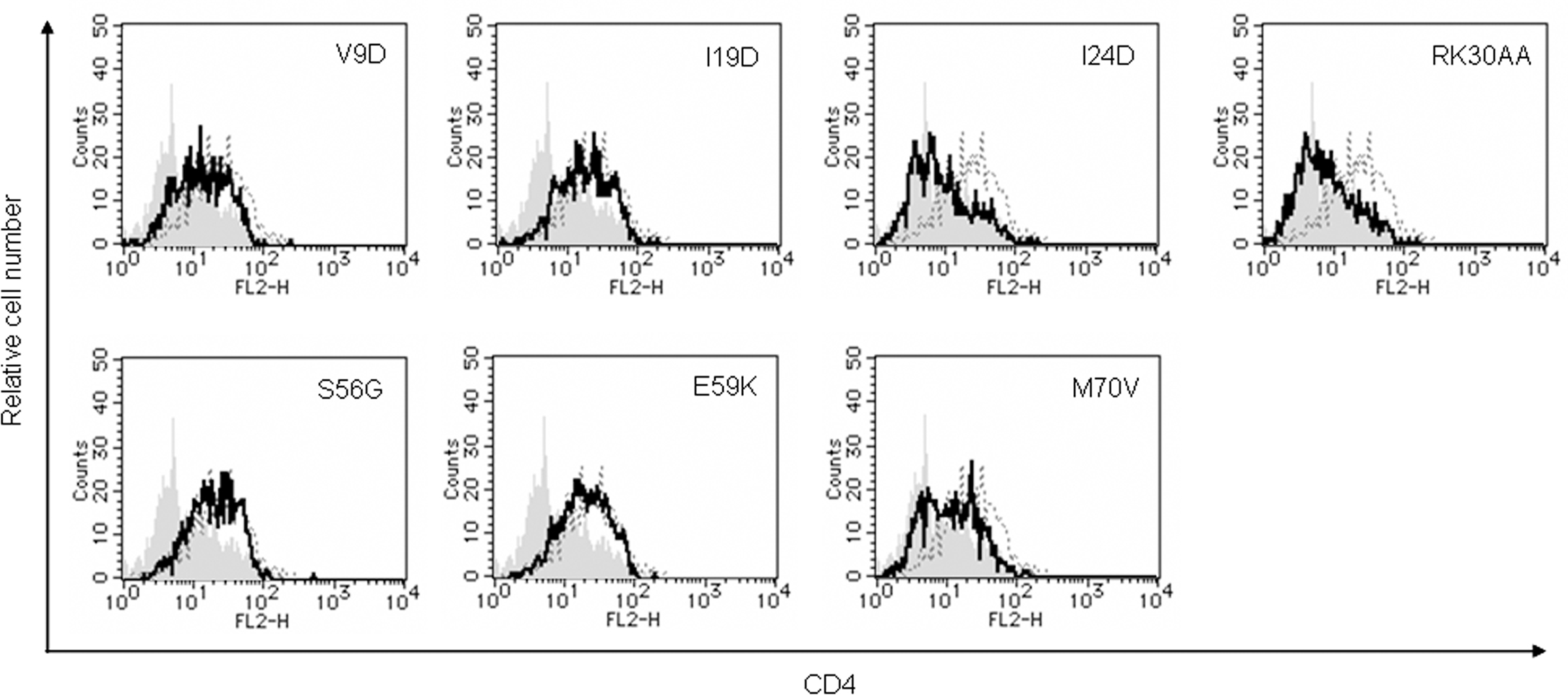
Figure 4. Ability of Vpu mutants to down-modulate cell surface CD4. MAGI cells were transfected with 5 μg of an empty vector, a vector encoding WT Vpu, or vectors encoding the indicated Vpu mutant protein and GFP as in Figure 3. On day 2 post-transfection, cells were strained for cell surface CD4 and analyzed by two-color flow cytometry. Histograms represent relative cell number vs. CD4 fluorescence intensity for GFP-positive cells. Broken lines indicate the results of empty vector control, and gray shaded areas indicate the results of WT Vpu. Solid lines show the results of indicated mutants.
Vpu CTD Mutants (S56G and E59K) are Able to Down-Modulate Tetherin from the Cell Surface of HEp2 Cells but not TMD Mutants (V9D and I19D)
Down-modulation of cell surface tetherin by Vpu has been shown to be important for enhancement of virion release from HeLa cells (Van Damme et al., 2008; Mitchell et al., 2009). It also has been reported that, during infection of T cell lines with viruses carrying S52 and S56 mutations, cell surface level of tetherin was partially reduced (Douglas et al., 2009) or unchanged (Miyagi et al., 2009). To assess the potential of mutant Vpu proteins, we analyzed cell surface tetherin on HEp2 cells expressing Vpu. HEp2 cells were transfected with vectors co-expressing Vpu and GFP as above, and monitored for expression level of cell surface tetherin by flow cytometry (Figure 5). Some mutants (I24D, RK30AA, and M70V), which promote virion release (Figure 3B), down-modulated cell surface tetherin as effective as WT Vpu. In addition, mutants S56G and E59K effectively reduced the tetherin. In contrast, some TMD mutants (V9D and I19D) did not decrease tetherin from cell surface. While these two TMD mutants did not enhance virion release as expected, CTD mutants S56G and E59K did so considerably (Figure 3B). In sum, TMD mutants V9D and I19D did not promote virion release and did not down-modulate cell surface tetherin. CTD mutants S56G and E59K did not enhance virion release but did reduce cell surface tetherin. These results show that, two activities of Vpu, i.e., down-modulation of cell surface tetherin and enhancement of virion release, are not always correlated.
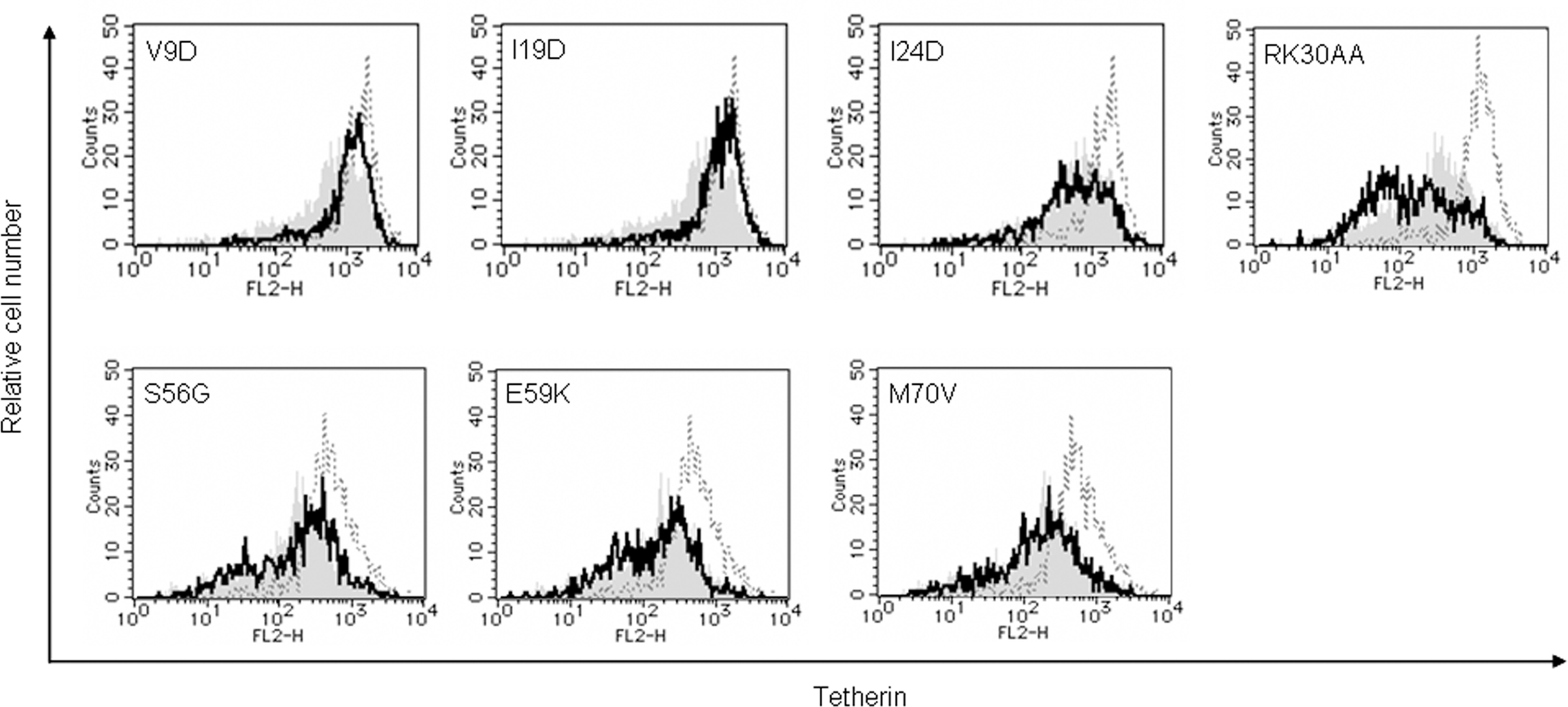
Figure 5. Ability of Vpu mutants to down-modulate cell surface tetherin. HEp2 cells were transfected with 5 μg of an empty vector, a vector encoding WT Vpu, or vectors encoding the indicated Vpu mutant protein and GFP as in Figure 3. On day 2 post-transfection, cells were strained for cell surface tetherin and analyzed by two-color flow cytometry. Histograms represent relative cell number vs. tetherin fluorescence intensity for GFP-positive cells. Broken lines indicate the results of empty vector control, and gray shaded areas indicate the results of WT Vpu. Solid lines show the results of indicated mutants.
The Ability of Vpu to Colocalize with Tetherin is Parallel to its Capability to Down-Modulate Cell Surface Tetherin but not to Enhance Virion Release
Recent studies have suggested that the ability of Vpu to localize to TGN and to colocalize with tetherin is important to elicit anti-tetherin activity, and that this property of Vpu correlates with virion release enhancement (Dubé et al., 2009, 2010). Therefore, we examined mutational effects of Vpu on its intracellular localization. For microscopic analysis, we generated vectors encoding C-terminally FLAG-tagged Vpu, and verified their protein expression and ability to promote virion production from cells. As shown in Figure 6, these vectors expressed the Vpu proteins and complemented virion production of ΔVpu to a similar extent with that observed in the provirus experiment (Figure 2A).
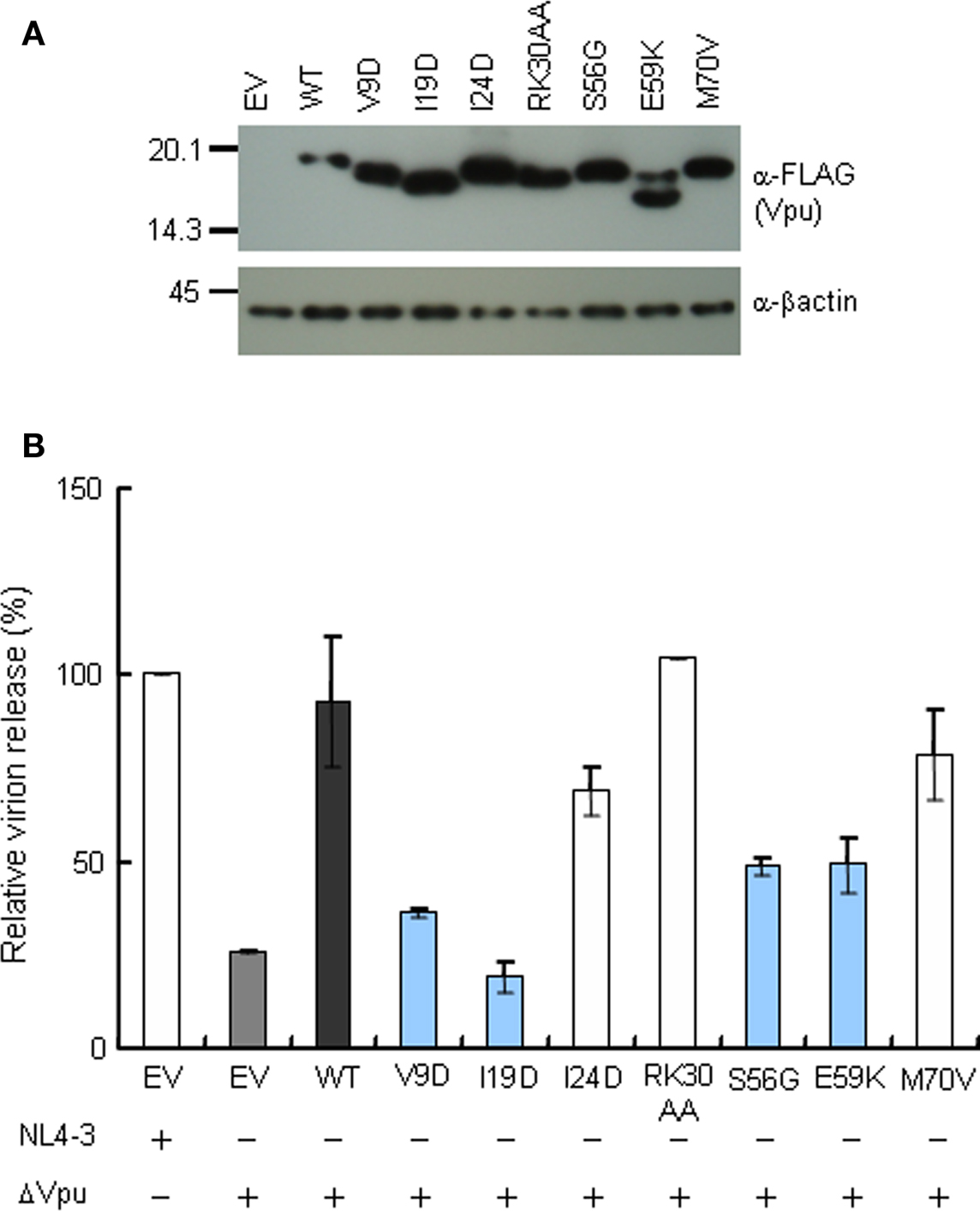
Figure 6. Expression and activity of C-terminally FLAG-tagged Vpu constructs. (A) Western blot analysis of Vpu expression in lysates of 293T cells transfected with pSG-VpucFLAG vectors. Equivalent amounts of total protein (1 μg) were loaded and Vpu was detected by anti-FLAG antibody. β-Actin was used as a control. EV, empty vector control. (B) Virion release assays from HEp2 cells. HEp2 cells were transfected with the indicated proviral construct (300 ng) and pSG-VpucFLAG vectors (200 ng), and virion production in the culture supernatants on day 2 post-transfection was determined by RT assays. Values represent virion production level relative to that by WT proviral clone (NL4-3). Gray bar shows the results of proviral ΔVpu (pNL-uE65). Black bar indicates the results of complementation of ΔVpu (pNL-uE65) by WT Vpu (pSG-WT VpucFLAG). Blue and white bars show the results of expression vectors derived from replication-defective and replication-normal mutants in H9 cells, respectively. EV, empty vector control pSG5 (400 ng).
Using these FLAG-tagged Vpu constructs, we analyzed intracellular localization of Vpu in HEp2 cells. Experiments for microscopic analysis were performed similarly with those for virion release assays to ensure direct functional comparison between the virion release and intracellular localization. As shown in Figure 7, WT Vpu localized in intracellular compartments stained with the TGN marker TGN46, and colocalized with tetherin in good agreement with the results reported previously (Dubé et al., 2009). None of WT and mutant Vpu proteins localized with the ER marker calreticulin, and their colocalization with the late endosome marker CD63 was rarely seen (data not shown). Mutants I24D and RK30AA, which have the ability to promote virion release and to down-modulate cell surface tetherin, were detected in the TGN46-positive compartment and colocalized with tetherin like WT Vpu.
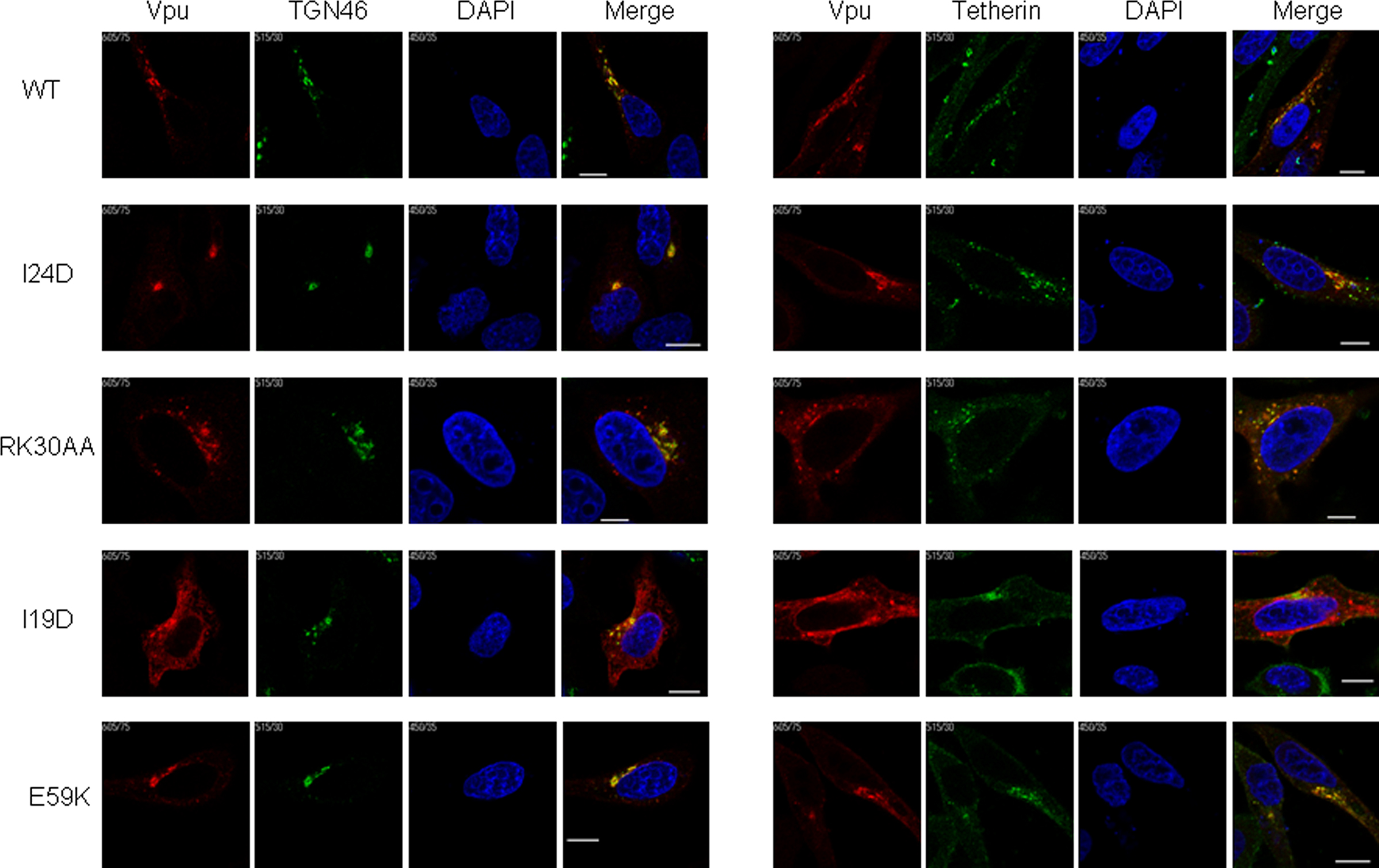
Figure 7. Localization of Vpu to TGN and colocalization with tetherin. HEp2 cells were transfected with ΔVpu (pNL-uE65) and the indicated vectors expressing FLAG-tagged Vpu (Figure 6). On the next day, cells were fixed, permeabilized, and immunostained with an anti-FLAG (red for Vpu), an anti-TGN46 (green), or an anti-HM1.24 (green for tetherin) antibody. Nuclei were counterstained with DAPI (blue). Cells were observed by confocal microscopy. Intracellular localization of each Vpu protein was analyzed for around 15 Vpu-positive cells, and representative results were shown. Except for I19D, the colocalization of WT and mutant Vpu proteins with TGN46 and tetherin was observed in more than 80% cells examined. White bar, 10 μm.
A Vpu TMD mutant I19D, defective for enhancing virion release and for inducing down-modulation of tetherin, showed altered localization and its expression was diffusely observed in the cytoplasm. A portion of the mutant was noted to exist with TGN46. Furthermore, this mutant colocalized less extensively with tetherin. Another Vpu TMD mutant V9D exhibited a similar phenotype with the I19D mutant (data not shown). Such a clear change in subcellular localization of Vpu TMD mutants has not been reported previously (Tiganos et al., 1998; Dubé et al., 2010). A third TMD mutant I24D exhibited a similar intracellular distribution with WT Vpu. Taken together, the evident alteration of intracellular localization by V9D and I19D mutations may have resulted from introducing a negatively charged amino acid into these residues, and show their critical importance for function of TMD.
In contrast to TMD mutants, a CTD mutant E59K was defective for virion release enhancement but efficiently down-modulated cell surface tetherin. As expected, E59K was co-stained with TGN46. Interestingly, this mutant colocalized with tetherin more extensively than WT Vpu. Another CTD mutant S56G also colocalized with tetherin extensively (data not shown). The results for localization of our CTD mutants (S56G and E59K) were consistent with a previous report (Dubé et al., 2010). Extensive colocalization of these CTD mutants could explain efficient down-modulation of cell surface tetherin (Figure 5). The result for E59K also suggests that amino acid residue(s) close to β-TrCP binding motif may play a role in recruiting β-TrCP and subsequent degradation of the target protein. In total, the activity of Vpu to localize optimally in TGN and to colocalize with tetherin is parallel with the capability to down-modulate cell surface tetherin but not to enhance virion release. Colocalization of Vpu and tetherin may be sufficient to induce down-modulation of tetherin from cell surface but subsequent degradation of the restriction factor may also be required for efficient virion release.
On the other hand, it has been reported that Vpu retains newly synthesized CD4 in ER and targets it to proteasomal degradation (Lenburg and Landau, 1993; Bour et al., 1995; Paul and Jabbar, 1997; Margottin et al., 1998; Magadán et al., 2010). Thus, we were interested in analyzing whether Vpu colocalizes with CD4 intracellularly. However, as mentioned earlier, Vpu did not localize the ER marker calreticulin in our assays. Furthermore, colocalization of Vpu and CD4 was not detected in MAGI cells transfected with a WT Vpu construct (data not shown). Evidently, it is necessary to examine more thoroughly the intracellular localization of Vpu and CD4.
Discussion
In this study we analyzed mutational effects on major Vpu activities as summarized in Table 1, and identified two functional clusters within TMD and CTD required for efficient viral replication (Figure 1). Replication-defective vpu mutants were unable to promote virion release and also to down-modulate CD4 expression at the cell surface (V9D, I19D, S56G, and E59K) (Figures 2 and 4). Our results here indicate that these two Vpu functions are necessary for efficient viral replication, and that functional regions for virion release and CD4 down-modulation overlap each other.
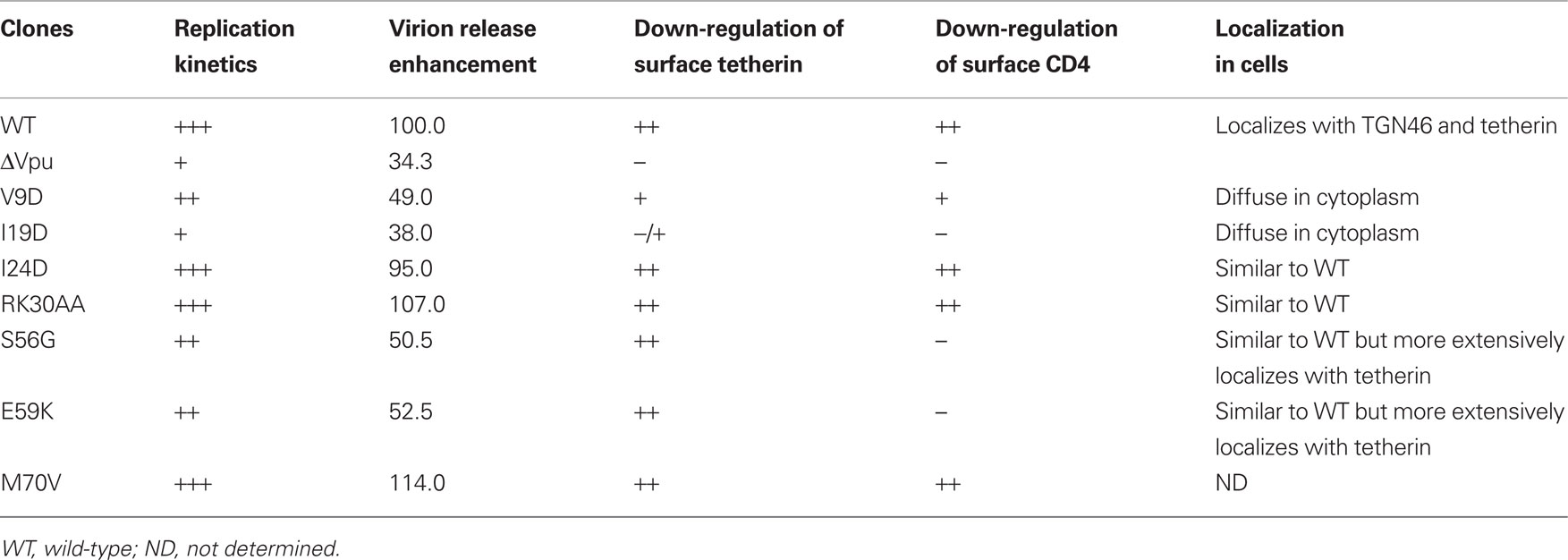
Table 1. Characteristics of HIV-1 vpu mutants.
Transmembrane domain mutants (V9D and I19D) were unable to down-modulate the cell surface tetherin (Figure 5) and to colocalize with tetherin extensively (Figure 7). These results were similar to those obtained using a randomized TMD mutants (Van Damme et al., 2008; Dubé et al., 2010). It is not clear that inability of the TMD mutants to reduce cell surface tetherin is due to an indirect effect by their altered intracellular localization, a direct effect by their lack of binding to tetherin, or both. The I24D mutant had a similar ability with WT Vpu to down-modulate CD4 and tetherin from cell surface, and to localize in TGN (Figures 4, 5, and 7), but showed a small defect in viral replication and trans-complementation assays (Figures 1E, 3B, and 6B). These observations would represent a subtle effect of the I24D mutation within TMD on the Vpu activity. Recently, a mutant (RK30AA) of Vpu membrane proximal region has been shown to reduce virion release (Dubé et al., 2009). Moreover, importance of the membrane proximal region for Vpu localization and virion release has been proposed (Ruiz et al., 2008). In this study, however, we did not observe any functional abnormality for the RK30AA mutant. The reason for this discrepancy is unclear but may be due to difference in cell type, viral strain, and method used in the individual experiment. In contrast to TMD mutants, CTD mutants (S56G and E59K) were able to down-modulate cell surface tetherin without promoting virion release (Figures 2 and 5). This down-modulation of cell surface tetherin could be explained by its sequestration through extensive intracellular colocalization with the CTD mutants (Dubé et al., 2010). In addition, CTD mutants complemented virion release relatively more efficiently than TMD mutants in trans-complementation assay (50–70% for CTD and 20–35% for TMD in Figures 3B and 6B). This could be due to the ability of CTD mutants to down-regulate cell surface tetherin (Figure 5). The TMD mutants did not down-regulate the tetherin from cell surface (Figure 5). The defective nature of the CTD mutants suggests that recruitment of β-TrCP by Vpu and subsequent degradation of tetherin may be required for efficient viral replication. In this regard, it is interesting to determine whether CTD mutants bind to β-TrCP and induce ubiquitination of tetherin and subsequent degradation. In particular, it is important to examine the E59K mutant, which has a mutation outside of, but close to, the β-TrCP binding motif.
There have been arguments regarding levels of tetherin and virion production. It has been shown that tetherin down-modulation is positively correlated with virion release enhancement (Van Damme et al., 2008; Mitchell et al., 2009). It also has been reported that, in T cell lines infected with the S52N/56N mutant, reduction of cell surface tetherin did not occur efficiently relative to WT virus (Douglas et al., 2009). In our experiments, while CTD mutants reduce cell surface tetherin similarly well with WT Vpu, they did not enhance virion production efficiently. It is possible that efficient down-modulation of cell surface tetherin was achieved by overexpressed mutant proteins. Conflicting results with respect to antagonism of Vpu against tetherin have been also reported for requirement of depletion of total cellular tetherin and for tetherin degradation pathway (proteasomal or lysosomal) (for review, Tokarev et al., 2009; Douglas et al., 2010). In addition, a recent report showed that more Vpu is necessary for anti-tetherin activity in macrophages, which express relatively higher level of tetherin, than those in primary blood lymphocytes (Schindler et al., 2010). These observations suggest that Vpu-dependent antagonism of tetherin may be determined by the balance of expression levels between Vpu and tetherin.
Finally, biological relevance of Vpu and tetherin in viral replication and pathogenesis need to be considered well. From this point of view, cell-to-cell transmission of viruses is another important issue to address. Our laboratory has shown previously that ΔVpu virus exhibit more rapid cell-to-cell spread than WT by using genetically engineered marker containing virus (Sakai et al., 1995). Another report proposed that increase of fusogenicity induced by accumulation of virions on the cell surface may lead to potential enhancement of viral replication via cell-to-cell spread (Gummuluru et al., 2000). Quite contrary to the reports described above, it has been shown recently that tetherin restricts productive infection via cell-to-cell spread (Casartelli et al., 2010). In conclusion, impact of Vpu on cell-to-cell spread remains to be elucidated. In order to understand biological significance of Vpu and tetherin in viral replication and pathogenesis, more study using replication-competent viruses and animal models is necessary, and this would provide new strategies against HIV-1.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ms. Kazuko Yoshida of our department for her excellent editorial assistance. We are indebted to Chugai Pharmaceutical Co., Japan and NIH AIDS Research and References Reagent Program, USA for antibodies. This work was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas (21022033) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
Adachi, A., Gendelman, H. E., Koenig, S., Folks, T., Willey, R., Rabson, A., and Martin, M. A. (1986). Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59, 284–291.
Bour, S., Schubert, U., and Strebel, K. (1995). The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: implications for the mechanism of degradation. J. Virol. 69, 1510–1520.
Bour, S., and Strebel K. (2003). The HIV-1 Vpu protein: a multifunctional enhancer of viral particle release. Microbes Infect. 5, 1029–1039.
Casartelli, N., Sourisseau, M., Feldmann, J., Guivel-Benhassine, F., Mallet, A., Marcelin, A. G., Guatelli, J., and Schwartz, O. (2010). Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 6, e1000955. doi: 10.1371/journal.ppat.1000955.
Desrosiers, R. C. (2007). “Nonhuman Lentiviruses,” in Fields Virology, 5th Edn, eds D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (Philadelphia: Lippincott Williams and Wilkins), 2215–2243.
Douglas, J. L., Gustin, J. K., Viswanathan, K., Mansouri, M., Moses, A. V., and Früh, K. (2010). The great escape: viral strategies to counter BST-2/tetherin. PLoS Pathog. 6, e1000913. doi: 10.1371/journal.ppat.1000913.
Douglas, J. L., Viswanathan, K., McCarroll, M. N., Gustin, J. K., Früh, K., and Moses, A. V. (2009). Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a βTrCP-dependent mechanism. J. Virol. 83, 7931–7947.
Dubé, M., Roy, B. B., Guiot-Guillain, P., Binette, J., Mercier, J., Chiasson, A., and Cohen, E. A. (2010). Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 6, e1000856. doi: 10.1371/journal.ppat.1000856.
Dubé, M., Roy, B. B., Guiot-Guillain, P., Mercier, J., Binette, J., Leung, G., and Cohen, E. A. (2009). Suppression of tetherin-restricting activity upon human immunodeficiency virus type 1 particle release correlates with localization of Vpu in the trans-Golgi network. J. Virol. 83, 4574–4590.
Goffinet, C., Allespach, I., Homann, S., Tervo, H. M., Habermann, A., Rupp, D., Oberbremer, L., Kern, C., Tibroni, N., Welsch, S., Krijnse-Locker, J., Banting, G., Kräusslich, H. G., Fackler, O. T., and Keppler, O. T. (2009). HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5, 285–297.
Gummuluru, S., Kinsey, C. M., and Emerman, M. (2000). An in vitro rapid-turnover assay for human immunodeficiency virus type 1 replication selects for cell-to-cell spread of virus. J. Virol. 74, 10882–10891.
Iida, S., Fukumori, T., Oshima, Y., Koyama, A. H., and Adachi, A. (1999). Growth characteristics of T-cell tropic HIV-1 vpu gene mutants in human peripheral blood mononuclear cells. J. Med. Invest. 46, 43–47.
Iwabu, Y., Fujita, H., Kinomoto, M., Kaneko, K., Ishizaka, Y., Tanaka, Y., Sata, T., and Tokunaga, K. (2009). HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 284, 35060–35072.
Kimpton, J., and Emerman, M. (1992). Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J. Virol. 66, 2232–2239.
Kirchhoff, F. (2009). Is the high virulence of HIV-1 an unfortunate coincidence of primate lentiviral evolution? Nat. Rev. Microbiol. 7, 467–476.
Lebkowski, J. S., Clancy, S., and Calos, M. P. (1985). Simian virus 40 replication in adenovirus-transformed human cells antagonizes gene expression. Nature 317, 169–171.
Lenburg, M. E., and Landau, N. R. (1993). Vpu-induced degradation of CD4: requirement for specific amino acid residues in the cytoplasmic domain of CD4. J. Virol. 67, 7238–7245.
Levesque, K., Zhao, Y. S., and Cohen, E. A. (2003). Vpu exerts a positive effect on HIV-1 infectivity by down-modulating CD4 receptor molecules at the surface of HIV-1-producing cells. J. Biol. Chem. 278, 28346–28353.
Magadán, J. G., Pérez-Victoria, F. J., Sougrat, R., Ye, Y., Strebel, K., and Bonifacino, J. S. (2010). Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 6, e1000869. doi: 10.1371/journal.ppat.1000869.
Mangeat, B., Gers-Huber, G., Lehmann, M., Zufferey, M., Luban, J., and Piguet, V. (2009). HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 5, e1000574. doi: 10.1371/journal.ppat.1000574.
Mann, D. L., O’Brien, S. J., Gilbert, D. A., Reid, Y., Popovic, M., Read-Connole, E., Gallo, R. C., and Gazdar, A. F. (1989). Origin of the HIV-susceptible human CD4+ cell line H9. AIDS Res. Hum. Retroviruses 5, 253–255.
Margottin, F., Bour, S. P., Durand, H., Selig, L., Benichou, S., Richard, V., Thomas, D., Strebel, K., and Benarous, R. (1998). A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1, 565–574.
Mitchell, R. S., Katsura, C., Skasko, M. A., Fitzpatrick, K., Lau, D., Ruiz, A., Stephens, E. B., Margottin-Goguet, F., Benarous, R., and Guatelli, J. C. (2009). Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 5, e1000450. doi: 10.1371/journal.ppat.1000450.
Miyagi, E., Andrew, A. J., Kao, S., and Strebel, K. (2009). Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. U.S.A. 106, 2868–2873.
Neil, S. J., Sandrin, V., Sundquist, W. I., and Bieniasz, P. D. (2007). An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe 2, 193–203.
Neil, S. J., Zang, T., and Bieniasz, P. D. (2008). Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430.
Nomaguchi, M., Fujita, M., and Adachi, A. (2008). Role of HIV-1 Vpu protein for virus spread and pathogenesis. Microbes Infect. 10, 960–967.
Paul, M., and Jabbar, M. A. (1997). Phosphorylation of both phosphoacceptor sites in the HIV-1 Vpu cytoplasmic domain is essential for Vpu-mediated ER degradation of CD4. Virology 232, 207–216.
Ruiz, A., Hill, M. S., Schmitt, K., Guatelli, J., and Stephens, E. B. (2008). Requirements of the membrane proximal tyrosine and dileucine-based sorting signals for efficient transport of the subtype C Vpu protein to the plasma membrane and in virus release. Virology 378, 58–68.
Sakai, H., Tokunaga, K., Kawamura, M., and Adachi, A. (1995). Function of human immunodeficiency virus type 1 Vpu protein in various cell types. J. Gen. Virol. 76, 2717–2722.
Sauter, D., Specht, A., and Kirchhoff, F. (2010). Tetherin: holding on and letting go. Cell 141, 392–398.
Schindler, M., Rajan, D., Banning, C., Wimmer, P., Koppensteiner, H., Iwanski, A., Specht, A., Sauter, D., Dobner, T., and Kirchhoff, F. (2010). Vpu serine 52 dependent counteraction of tetherin is required for HIV-1 replication in macrophages, but not in ex vivo human lymphoid tissue. Retrovirology 7, 1.
Schubert, U., Bour, S., Ferrer-Montiel, A. V., Montal, M., Maldarell, F., and Strebel, K. (1996). The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J. Virol. 70, 809–819.
Schubert, U., and Strebel, K. (1994). Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J. Virol. 68, 2260–2271.
Strebel, K., Klimkait, T., Maldarelli, F., and Martin, M. A. (1989). Molecular and biochemical analyses of human immunodeficiency virus type 1 vpu protein. J. Virol. 63, 3784–3791.
Tanaka, M., Ueno, T., Nakahara, T., Sasaki, K., Ishimoto, A., and Sakai, H. (2003). Downregulation of CD4 is required for maintenance of viral infectivity of HIV-1. Virology 311, 316–325.
Terwilliger, E. F., Cohen, E. A., Lu, Y. C., Sodroski, J. G., and Haseltine, W. A. (1989). Functional role of human immunodeficiency virus type 1 vpu. Proc. Natl. Acad. Sci. U.S.A. 86, 5163–5167.
Tiganos, E., Friborg, J., Allain, B., Daniel, N. G., Yao, X. J., and Cohen, E. A. (1998). Structural and functional analysis of the membrane-spanning domain of the human immunodeficiency virus type 1 Vpu protein. Virology 251, 96–107.
Tokarev, A., Skasko, M., Fitzpatrick, K., and Guatelli, J. (2009). Antiviral activity of the interferon-induced cellular protein BST-2/tetherin. AIDS Res. Hum. Retroviruses 25, 1197–1210.
Van Damme, N., Goff, D., Katsura, C., Jorgenson, R. L., Mitchell, R., Johnson, M. C., Stephens, E. B., and Guatelli, J. (2008). The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3, 245–252.
Wang, H., Sakurai, A., Khamsri, B., Uchiyama, T., Gu, H., Adachi, A., and Fujita M. (2005). Unique characteristics of HIV-1 Vif expression. Microbes Infect. 7, 385–390.
Willey, R. L., Smith, D. H., Lasky, L. A., Theodore, T. S., Earl, P. L., Moss, B., Capon, D. J., and Martin, M.A. (1988). In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J. Virol. 62, 139–147.
Keywords: HIV-1, replication, Vpu, CD4, tetherin
Citation: Nomaguchi M, Doi N, Fujiwara S, Fujita M and Adachi A (2010) Site-directed mutagenesis of HIV-1 vpu gene demonstrates two clusters of replication-defective mutants with distinct ability to down-modulate cell surface CD4 and tetherin. Front. Microbio. 1:116. doi: 10.3389/fmicb.2010.00116
Received: 23 July 2010;
Paper pending published: 10 August 2010;
Accepted: 30 September 2010;
Published online: 24 November 2010.
Edited by:
Akihide Ryo, Yokohama City University, JapanReviewed by:
Akifumi Takaori-Kondo, Kyoto University Graduate School of Medicine, JapanYouichi Suzuki, National University of Singapore, Singapore
Copyright: © 2010 Nomaguchi, Doi, Fujiwara, Fujita and Adachi. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Akio Adachi, Department of Microbiology, Institute of Health Biosciences, The University of Tokushima Graduate School, 3-18-15 Kuramoto-cho, Tokushima-shi, Tokushima 770-8503, Japan. e-mail:YWRhY2hpQGJhc2ljLm1lZC50b2t1c2hpbWEtdS5hYy5qcA==