 Eva Haenssler1,2 Ralph R. Isberg1,2*
Eva Haenssler1,2 Ralph R. Isberg1,2*
- 1 Department of Molecular Biology and Microbiology, Tufts University School of Medicine Boston, MA, USA
- 2 Howard Hughes Medical Institute, Tufts University School of Medicine Boston, MA, USA
Phosphorylation is one of the most frequent modifications in intracellular signaling and is implicated in many processes ranging from transcriptional control to signal transduction in innate immunity. Many pathogens modulate host cell phosphorylation pathways to promote growth and establish an infectious disease. The intracellular pathogen Legionella pneumophila targets and exploits the host phosphorylation system throughout the infection cycle as part of its strategy to establish an environment beneficial for replication. Key to this manipulation is the L. pneumophila Icm/Dot type IV secretion system, which translocates bacterial proteins into the host cytosol that can act directly on phosphorylation cascades. This review will focus on the different stages of L. pneumophila infection, in which host kinases and phosphatases contribute to infection of the host cell and promote intracellular survival of the pathogen. This includes the involvement of phosphatidylinositol 3-kinases during phagocytosis as well as the role of phosphoinositide metabolism during the establishment of the replication vacuole. Furthermore, L. pneumophila infection modulates the NF-κB and mitogen-activated protein kinase pathways, two signaling pathways that are central to the host innate immune response and involved in regulation of host cell survival. Therefore, L. pneumophila infection manipulates host cell signal transduction by phosphorylation at multiple levels.
Introduction
In the eukaryotic genome, protein kinases comprise one of the largest families of proteins (Manning et al., 2002a) and together with their counteracting protein phosphatases, they regulate a common post-translational modification observed in intracellular signaling. Activation of protein kinases and phosphatases typically occurs in response to extracellular stimuli as well as to intracellular stresses, and the resulting changes in the phosphorylation state of proteins lead to specific cellular responses (Cohen, 2000; Moorhead et al., 2009; Pidoux and Tasken, 2009). Eukaryotic kinases themselves often require activation by phosphorylation (Nolen et al., 2004) and are distinguished by their target residue specificities, which for the purposes of this review are either serine/threonine or tyrosine residues (Olsen et al., 2006; Moorhead et al., 2009; Pidoux and Tasken, 2009). Changes in the phosphorylation state have a broad impact on the cell, altering many processes such as the subcellular localization of proteins, the activity, or substrate specificity of enzymes, as well as specific protein–protein interactions. Furthermore, signals can be amplified within the cell by a cascade of substrate phosphorylation events, while fine-tuning and temporal control can be modulated by opposing phosphatases. This interplay allows spatial and temporal separation of intracellular signaling (Cohen, 2000; Moorhead et al., 2009; Pidoux and Tasken, 2009; Scott and Pawson, 2009).
Cellular processes that rely on phosphorylation have been extensively described in prokaryotes and eukaryotes and include transcription, translation, transport and energy flux, cell cycle, phagocytosis, and the innate immune response to pathogens (Manning et al., 2002a,b; Ryan and Shapiro, 2003; Lindmo and Stenmark, 2006; Back et al., 2009). In order to ensure a proper balance between the unphosphorylated and the phosphorylated state of proteins, tight regulation of protein kinases, and phosphatases is of crucial importance for the cell and imbalance often results in aberrant signaling leading to disease (Manning et al., 2002b).
Interference with the host phosphorylation machinery is a common strategy used by pathogens to promote growth and survival in host tissues. For instance, the gastric pathogen Helicobacter pylori directs host cell cytoskeletal rearrangements by delivery of the CagA protein into host cells (Covacci et al., 1993; Segal et al., 1996, 1999; Backert et al., 2000; Stein et al., 2002). Following translocation into the cell, CagA is phosphorylated by Src host tyrosine kinases and subsequently induces changes in cell morphology due to cytoskeleton rearrangement (Segal et al., 1996; Stein et al., 2002). Src kinases along with the host focal adhesion kinase are activated as a result of the interaction of the bacterial CagL protein with the integrin α5β1 receptor (Kwok et al., 2007). Therefore, H. pylori proteins can both activate and serve as substrates for host kinases.
Many intracellular pathogens exploit the host phosphorylation machinery by interfering with phosphoinositide (PI) metabolism and thereby target a major signaling pathway controlling membrane trafficking, actin rearrangement, and cell survival (Toker and Cantley, 1997; De Matteis and Godi, 2004; Krauss and Haucke, 2007; Duronio, 2008; Weber et al., 2009a). Phosphatidylinositol 3-kinases (PI3Ks) play a major role in signal transduction during phagocytosis and therefore in the uptake of many pathogens (Ireton et al., 1996; Toker and Cantley, 1997; Lindmo and Stenmark, 2006; Weber et al., 2009a). Listeria monocytogenes is one such pathogen that is internalized in a PI3K-dependent fashion, as 3-phosphorylated phosphatidylinositol phosphates are required for cytoskeletal rearrangements involved in this process (Ireton et al., 1996; Mostowy and Cossart, 2009). Mycobacterium tuberculosis controls PI metabolism in a different fashion, forcing entry into a replication compartment that has a low phosphatidylinositol-3-phosphate [PI(3)P] content. This association appears to be linked with evasion of the lysosomal network, as PI(3)P is a key component of early endosomes that mature into endolysosomes (Lindmo and Stenmark, 2006; Philips, 2008). One of the strategies utilized by Mycobacteria to keep the PI(3)P content low in the membrane surrounding the replication compartment involves the secretion of PI, protein, and lipid phosphatases (Vergne et al., 2005; Beresford et al., 2007). M. tuberculosis proteins, therefore directly target phosphorylation events associated with host PI metabolism.
Similar to M. tuberculosis, the intracellular pathogen Legionella pneumophila resides and replicates within a specialized vacuole in the host cytosol (Horwitz, 1983a,b; Horwitz and Maxfield, 1984). The proper formation of this replication vacuole relies on the Icm/Dot type IV secretion system (Marra et al., 1992; Berger et al., 1994; Segal et al., 1998; Vogel et al., 1998). Up to 200 bacterial proteins are translocated into the cytosol, targeting a variety of host pathways contributing to efficient intracellular growth of L. pneumophila (Burstein et al., 2009; Huang et al., 2010). Characterized translocated proteins are known to target ER → Golgi membrane trafficking (Murata et al., 2006; Ingmundson et al., 2007; Machner and Isberg, 2007), modulate host cell survival (Laguna et al., 2006; Banga et al., 2007), or inhibit the eukaryotic translation elongation complex (de Felipe et al., 2005; Belyi et al., 2006, 2008).
In this review we discuss selected host phosphorylation pathways that are targeted by L. pneumophila during interactions with host cells. The strategies used by the microorganism include translocation of kinases that directly manipulate host cell phosphorylation, but also include indirect effects that result in alteration of host cell signaling in response to formation of the L. pneumophila replication vacuole.
L. pneumophila Protein Kinases and Phosphatases
While M. tuberculosis has both kinases and phosphatases that directly impact host signal transduction controlled by phosphorylation (Walburger et al., 2004; Vergne et al., 2005; Beresford et al., 2007), of the close to 200 known and putative Icm/Dot translocated substrates (Burstein et al., 2009; Huang et al., 2010) none show sequence similarity to known phosphatases or tyrosine kinases. Four translocated proteins, LegK1, LegK2, LegK3, and LegK4 contain domains that show homology to eukaryotic Ser/Thr kinases (de Felipe et al., 2005, 2008; Bruggemann et al., 2006a; Shin et al., 2008; Hervet et al., 2011). Of the LegK homologs, LegK1 and LegK2 are the best characterized. As is the case with the majority of translocated substrates, LegK1 is dispensible for intracellular growth in bone marrow-derived macrophages isolated from A/J mice and in the environmental host Acanthamoeba castellanii (de Felipe et al., 2005; Losick et al., 2010). LegK1 exhibits kinase activity in vitro and it has been proposed that it interferes with the host innate immune system by directly activating the NF-κB pathway, because ectopic expression of the protein in mammalian cells results in activation of an NF-κB-dependent promoter (Ge et al., 2009; Losick et al., 2010). The kinase activity is necessary for this activation, as a point mutation in the ATP binding domain or a catalytic residue abolishes NF-κB activity (Ge et al., 2009; Losick et al., 2010). In vitro, NF-κB activation by LegK1 occurs through direct phosphorylation of a component in the signaling cascade, the inhibitor IκB, resulting in degradation of the inhibitor and release of NF-κB into the host cell nucleus (Ge et al., 2009). Whether or not LegK1-mediated phosphorylation of IκB plays a role in NF-κB activation during macrophage challenge by L. pneumophila is unclear, as a legK1 deletion mutant is able to efficiently activate an NF-κB regulated promoter (Losick et al., 2010). The role of LegK1 during growth within the natural host ameba is similarly unclear, as there are no known NF-κB orthologs in any sequenced amebal species.
As is true with LegK1 (Ge et al., 2009), LegK2 exhibits protein kinase activity in vitro, however its specific host target is not known (Hervet et al., 2011). In the amebal host A. castellanii, LegK2 activity plays some role in the recruitment of the ER marker calnexin and is required during early time points of intracellular replication, as a legK2 deletion mutant displays a delayed onset of growth (Hervet et al., 2011). Less is known about the functions of LegK3 and LegK4. LegK3 has been studied in the context of NF-κB activation and the mitogen-activated protein kinase (MAPK) pathway, but failed to show an impact on either (Shin et al., 2008; Ge et al., 2009). Regarding their expression during bacterial growth in broth as well as during infection of A. castellanii, the LegK homologs share a similar pattern. In post-exponential phase the expression of LegK1-4 is slightly reduced in the L. pneumophila Lens isolate (Hervet et al., 2011) and during host cell infection, expression levels of all four genes do not significantly change (Bruggemann et al., 2006b).
Compared to other pathogens, such as pathogenic Yersinia species, which have well characterized kinases and phosphatases that have impact on the disease process (Viboud and Bliska, 2005; Ribet and Cossart, 2010), less is known about L. pneumophila proteins that directly change the phosphorylation state of host targets during infection. As will be illustrated below, L. pneumophila appears to modulate host cell phosphorylation pathways indirectly, via processes associated with the uptake and replication of L. pneumophila.
Host Phosphatases and kinases Targeted During L. pneumophila Infection
To obtain a comprehensive understanding of which host cell pathways are necessary for intracellular growth of L. pneumophila, the global transcriptional host cell response has been investigated by several groups, using mouse bone marrow-derived macrophages, human macrophage-like cell lines and amebae (Farbrother et al., 2006; Losick and Isberg, 2006; Abu-Zant et al., 2007; Shin et al., 2008; Li et al., 2009; Fontana et al., 2011). These data were complemented by chemical genetics to identify host factors that are essential for the early steps of infection and for Icm/Dot-dependent protein translocation into a macrophage cell line (Charpentier et al., 2009). The latter approach provided substantial knowledge of the host factors necessary for phagocytosis of L. pneumophila and Icm/Dot substrate translocation including PI3Ks (see below). In the natural host amebae, besides inducing a stress response, major transcriptional changes occurred at various time points of L. pneumophila challenge, including increased transcription of aminoacyl-tRNA synthetases and decreased expression of ribosomal protein genes (Farbrother et al., 2006; Li et al., 2009). In human macrophage-like cells, encounter with L. pneumophila also resulted in increased transcription of stress response genes, however, the most striking transcriptional response in mammalian cells was demonstrated to be the upregulated expression of genes encoding components of the innate immune system (Losick and Isberg, 2006; Shin et al., 2008). In human macrophage-like cells these included genes regulated by NF-κB, genes encoding anti-apoptotic proteins as well as dual specificity phosphatases (DUSPs) known to be negative regulators of the MAPK pathway (Losick and Isberg, 2006). The transcriptional profile of murine bone marrow-derived macrophages also showed enhanced transcription of dusp genes (Shin et al., 2008). These transcriptional responses were specific to virulent L. pneumophila, since they were dependent on the presence of a functional Icm/Dot system.
The transcriptional analyses as well as the application of chemical genetics indicate that the host cell response to L. pneumophila involves differential regulation of a variety of signaling cascades that are controlled by phosphorylation and dephosphorylation. The following sections will focus on different stages of the infection cycle and the impact of host kinases and phosphatases on these pathways (Figure 1).
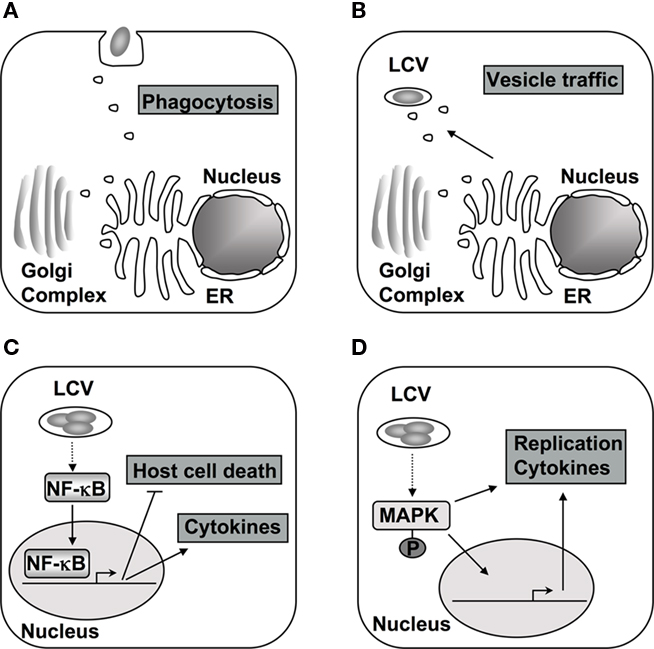
Figure 1. The host phosphorylation system is targeted during different stages of L. pneumophila encounter with host cells. (A) The initial contact and subsequent uptake into the host cell is thought to be dependent on PI3Ks and respective downstream signaling in macrophages (Tachado et al., 2008; Charpentier et al., 2009). (B) Alterations in PI levels at the vacuole contribute to differential protein recruitment and could interfere with endocytic trafficking to vacuole (Weber et al., 2006, 2009b; Ragaz et al., 2008; Brombacher et al., 2009). (C) After contact with cells of higher eukaryotes, the NF-κB pathway is activated, which alters cytokine production, host cell survival, and intracellular replication of L. pneumophila (Losick and Isberg, 2006; Abu-Zant et al., 2007; Shin et al., 2008; Bartfeld et al., 2009; Fontana et al., 2011). (D) The MAPK signaling pathway is also modulated during infection and proper regulation is necessary for L. pneumophila replication in amebae and cytokine production in macrophages (Welsh et al., 2004; Losick and Isberg, 2006; Shin et al., 2008; Li et al., 2009).
Host Factors Involved in Phagocytosis and Establishing a Replication Compartment
Each cycle of infection starts with uptake of L. pneumophila into the host cell by phagocytosis. Uptake into human alveolar macrophages under non-opsonized conditions has been documented to occur by coiling phagocytosis (Horwitz, 1984; Charpentier et al., 2009). The mechanism of phagocytosis of L. pneumophila, however, has been a point of dispute for some time, with the role of PI3Ks being a particular focus of controversy. As mentioned above, pathogens may be taken up in a PI3K-dependent manner (Weber et al., 2009a). Activation of PI3K leads to downstream signaling events that involve synthesis of PI(3,4,5)P3, which is likely to be followed by recruitment of guanidine nucleotide exchange factors that activate Rho family GTPases involved in regulation of actin rearrangements (Lindmo and Stenmark, 2006).
The role of PI3Ks during phagocytosis of L. pneumophila appears to differ between host systems, bacterial strains, and the experimental setup used (see below). Initially, it was postulated that uptake of virulent L. pneumophila JR32 (Philadelphia-1) into U937 human macrophage-like cells does not depend on PI3Ks (Khelef et al., 2001). Phagocytosis of L. pneumophila having an intact Icm/Dot system was not blocked by the PI3K inhibitor wortmannin, and this failure to inhibit uptake was independent of the opsonization conditions used. When actin polymerization was visualized at L. pneumophila entry sites, there appeared to be no affect of inhibitors of PI3K function. However, a mutant having an inactivated Icm/Dot system was taken up in a PI3K-dependent manner indicating that the presence of a functional protein translocation system targets L. pneumophila into a unique uptake pathway (Khelef et al., 2001).
In the ameba Dictyostelium discoideum, inhibition of PI3Ks by wortmannin and LY294002 reduced the uptake of L. pneumophila (Weber et al., 2006; Peracino et al., 2010). In spite of this reduction in uptake, deletion of class I PI3Ks appeared to have a positive influence on intracellular replication (Weber et al., 2006; Peracino et al., 2010). Deletion of genes encoding PI3Ks, as well as chemical inhibition of their activities, changes the morphology of the replication vacuole and it was postulated that this structure might stimulate intracellular replication (Weber et al., 2006). The absence of PI3Ks could result in an altered composition of PIs at the Legionella-containing vacuole (LCV) causing changes in host protein recruitment. Also, improved intracellular replication could be due to enhanced bypass of the endocytic pathway, which requires PI3K activity, and may compete for sequestration of the microorganism into a compartment that is restrictive for intracellular growth (Lindmo and Stenmark, 2006; Weber et al., 2006).
Besides PI3Ks, another enzyme involved in PI metabolism, the inositol polyphosphate-5-phosphatase Dd5p4, influences L. pneumophila infection in D. discoideum. Host cells lacking Dd5p4 showed a defect in uptake, but once internalized, the bacteria showed improved intracellular replication, which is similar to the phenotype observed for loss of PI3K (Weber et al., 2006, 2009b). Dd5p4 is recruited to the LCV in a Icm/Dot-dependent manner and is catalytically active, which putatively leads to conversion from PI(4,5)P2 to PI(4)P (Weber et al., 2009b). PI(4)P is considered a lipid marker for the LCV, that serves as an anchor for Icm/Dot substrates and is detectable at the LCV dependent on the presence of an intact Icm/Dot system (Weber et al., 2006; Ragaz et al., 2008; Brombacher et al., 2009). As in amebae, PI(4)P is also found at the LCV in infected RAW264.7 murine macrophage cells indicating that intracellular replication within ameba and mammalian cells has similar lipid requirements (Weber et al., 2006). Furthermore, the human homolog of Dd5p4, OCRL1, which plays a role in trafficking from endosomes to the trans-Golgi network (Lowe, 2005), could also be detected at the LCV and likely functions similarly to Dd5p4 during intracellular replication (Weber et al., 2009b). Therefore, targeting of PI metabolism seems to occur at different stages of the infectious cycle following uptake and contributes to proper establishment of the LCV (Figure 2).
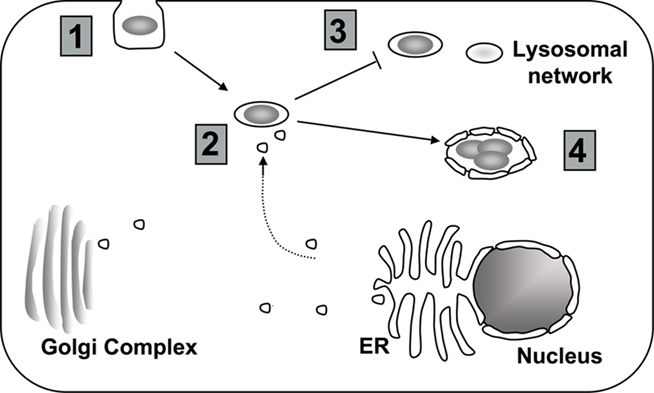
Figure 2. Targeting of host cell PI metabolism by L. pneumophila. (1) Depending on the model system, uptake into the host cell may require PI3K signaling (Khelef et al., 2001; Weber et al., 2006; Tachado et al., 2008; Charpentier et al., 2009; Peracino et al., 2010). (2) To properly establish the LCV, L. pneumophila interferes with host vesicle trafficking. Changes in the composition of PIs at the LCV might contribute to altered trafficking (Weber et al., 2006, 2009b; Isberg et al., 2009). (3) Changes in the PI levels at the vacuole may help L. pneumophila to avoid the endocytic pathway (Lindmo and Stenmark, 2006; Weber et al., 2006, 2009a). (4) Throughout intracellular growth, PIs at the LCV likely provide scaffolding for both L. pneumophila translocated proteins and host proteins (Weber et al., 2006, 2009b; Ragaz et al., 2008; Brombacher et al., 2009).
The notion that PI3Ks do not play a major role in uptake of L. pneumophila was challenged by a study using the J744A.1 murine macrophage cell line (Tachado et al., 2008). In the J744A.1 murine macrophage cell line, phagocytosis of L. pneumophila AA100 (Wadsworth) was reduced more severely by the PI3K inhibitors wortmannin and LY294002 than had been reported with other cell lines (Khelef et al., 2001; Weber et al., 2006; Peracino et al., 2010). In addition, cells expressing a dominant-negative mutant of PI3K were also depressed for uptake of the wild type L. pneumophila strain. Consistent with a role for PI3K during uptake, a downstream signal of PI3K activation, protein kinase B, was activated after challenge with L. pneumophila. Induction of the PI3K pathway could only be observed after contact with L. pneumophila expressing an intact Icm/Dot system (Tachado et al., 2008). One explanation for the conflicting results is that the J744A.1 cell line supports lower levels of L. pneumophila growth than other cell lines. Perhaps in cells in which there is luxurious intracellular growth of the bacterium, uptake is independent of PI3Ks. However, we have observed that wortmannin inhibits uptake of L. pneumophila into permissive mouse macrophages and it has been reported that uptake of L. pneumophila is reduced after treatment of D. discoideum with PI3K inhibitors (Weber et al., 2006; Peracino et al., 2010). There may be subtle differences in cytoskeletal regulatory circuits that determine whether PI3K is involved in uptake in different cell types.
A chemical genetics screen to find host cell functions required for Icm/Dot-dependent substrate translocation also supports the model that PI3Ks are involved in phagocytosis, at least in the J744A.1 cell line (Charpentier et al., 2009). In this study, phagocytosis was found to be a crucial prerequisite for Icm/Dot-promoted protein translocation (Charpentier et al., 2009). Inhibitors of either actin polymerization or PI3K reduced both Icm/Dot-dependent protein translocation and bacterial uptake. Taken together these data indicate that signaling through PI3K plays an important role during phagocytosis in these models.
Other novel targets identified by the chemical genetics screen pointed to the importance of tyrosine phosphatases for phagocytosis of L. pneumophila (Charpentier et al., 2009). The functionally redundant receptor protein tyrosine phosphate phosphatases CD45 and CD148 together with other, as yet unidentified, tyrosine phosphatases were shown to modulate uptake. Uptake of L. pneumophila into bone marrow-derived macrophages isolated from mice lacking CD45 and CD148 was drastically impaired compared to wild type macrophages, without affecting bacterial adhesion to cells. A more severe reduction in uptake relative to the mutant macrophages was observed in the presence of the CD45 inhibitor RWJ-60475, indicating that additional phosphatases may be involved. These results hint at a novel role of tyrosine phosphate phosphatases in phagocytosis of L. pneumophila. This is particularly interesting in light of the fact that there have been few convincing demonstrations that tyrosine phosphatases stimulate phagocytosis, other than the report that the tyrosine phosphatase Shp-1 stimulates Neisseria uptake (Hauck et al., 1999). Generally, tyrosine phosphatase activity has been connected to interference with uptake, as exemplified by the Yersinia YopH tyrosine phosphatase (Adkins et al., 2007).
Activation of the NF-κB Pathway
After phagocytosis, L. pneumophila resides within a membrane-bound compartment in the host cytosol. Consequently, survival of the host cell is necessary for successful replication. One way to prevent cell death involves direct interference of pro-death pathways by Icm/Dot translocated substrates (Laguna et al., 2006; Banga et al., 2007). A second mechanism of preventing host cell death during infection is to exploit proteins that are under the control of the mammalian transcription factor NF-κB, which acts as a positive regulator of genes encoding anti-apoptotic proteins (Karin and Lin, 2002). NF-κB homo- and heterodimers are master regulators of the mammalian innate immune response that control the expression of almost 400 genes (Karin and Lin, 2002; Ahn and Aggarwal, 2005; Hayden and Ghosh, 2008). NF-κB activation can result from sensing of pathogen associated molecular patterns (PAMPs; for example flagellin or peptidoglycan) by pattern recognition receptors (PRRs) that include the membrane-bound toll-like receptors (TLRs) and intracellular nod-like receptors (NLRs; Fritz et al., 2006; Kawai and Akira, 2010). Activation of these receptors triggers a signaling cascade that results in nuclear translocation of NF-κB subunits (Hayden and Ghosh, 2008). Signaling downstream from NLRs involves Rip2 kinase whereas TLR signaling is mediated via the adaptor proteins MyD88 and Trif (Shaw et al., 2008; Kawai and Akira, 2010). Both pathways lead to activation of IκB kinases (IKKs) by phosphorylation. Once activated, IKK phosphorylates IκB family members, resulting in degradation of these inhibitory proteins that are bound to the canonical NF-κB subunits in the cell cytoplasm (Hayden and Ghosh, 2008). Degradation of IκB frees the NF-κB subunits to be translocated into the nucleus (Hayden and Ghosh, 2008). The NF-κB pathway is manipulated by different pathogens such as H. pylori and Rickettsia rickettsii as well as by L. pneumophila (Clifton et al., 1998; Brandt et al., 2005). Challenge of host cells with L. pneumophila results in increased Icm/Dot-dependent transcription of NF-κB subunits as well as NF-κB regulated genes including pro-inflammatory cytokines and antagonists of apoptosis (Losick and Isberg, 2006; Abu-Zant et al., 2007; Shin et al., 2008; Bartfeld et al., 2009; Fontana et al., 2011).
There are probably multiple mechanisms that lead to NF-κB activation during L. pneumophila infection. Besides the engagement of PRRs with PAMPs, direct targeting of the pathway by Icm/Dot translocated substrates, such as LegK1 has been proposed, as pointed out above (Ge et al., 2009). NF-κB activation by L. pneumophila is probably more complex than can be explained by the action of a single effector, and likely occurs via the synergistic interaction of PRR signaling in combination with Icm/Dot-dependent components (Losick and Isberg, 2006; Shin et al., 2008; Bartfeld et al., 2009). At face value, PRR signaling and Icm/Dot activation of NF-κB appear to have different temporal courses of action, with PRR signaling occurring with more rapid kinetics than observed for Icm/Dot activation of the pathway (Bartfeld et al., 2009). It is unclear, however, if Icm/Dot activation of NF-κB ever occurs totally independently of PRR signaling. In macrophages from mice lacking TLR signaling via MyD88 and Nod signaling via Rip2, there appears to be little Icm/Dot-dependent signaling (Shin et al., 2008), although the presence of either PRR pathway is sufficient to support Icm/Dot-dependent NF-κB activation. This argues that although neither the type of bacterial ligand nor its site of encounter within the host cell are important for signaling, there is a requirement for the host cell to sense a PAMP for there to exist a strong Icm/Dot-dependent response.
In the human alveolar epithelial cell line A549, it has been shown that NF-κB activation follows a biphasic pattern. Short-term activation, measured by NF-κB nuclear translocation, depends on TLR5 and MyD88 (Bartfeld et al., 2009). This is followed by a TLR-independent long-term activation for which a functional Icm/Dot system is required. In concert with the data on synergy, these cells still retain Nod signaling, which could facilitate the Icm/Dot-dependent response. During long-term activation, IκB is degraded and anti-apoptotic genes are expressed (Bartfeld et al., 2009). Induction of anti-apoptotic genes appears to be the common theme in different host cell types (Losick and Isberg, 2006; Abu-Zant et al., 2007). The importance of host cell survival to maintain efficient intracellular replication was demonstrated in A/J bone marrow-derived macrophages. Inhibition of NF-κB caused increased host cell death in response to L. pneumophila challenge. However, the presence of NF-κB enhanced cell survival and was necessary for efficient replication (Losick and Isberg, 2006). Furthermore, one of the NF-κB regulated antagonists of apoptosis had a direct positive influence on host cell survival. In bone marrow macrophages derived from mice lacking plasminogen activator inhibitor-2 (PAI-2), there was increased cell death in response to L. pneumophila challenge (Losick and Isberg, 2006).
In bone marrow-derived macrophages from A/J mice, L. pneumophila activates NF-κB via at least two pathways depending on the multiplicity of infection. At low dose infections, there is little nuclear translocation of NF-κB in the absence of Icm/Dot, indicating that PRR recognition is not sufficient to give a robust signal (Losick and Isberg, 2006). For these low dose infections, NF-κB nuclear translocation is still observed in the absence of MyD88 and Trif`, but relies on Icm/Dot (Losick and Isberg, 2006; Losick et al., 2010), and presumably Nod signaling, based on the work from the Roy lab (Shin et al., 2008). At an elevated multiplicity of infection, however, MyD88-dependent NF-κB activation can be observed in the absence of Icm/Dot (Losick and Isberg, 2006).
The data on Nod signaling adds complexity to formulating models on NF-κB signaling, but the basic message is consistent with the idea that as long as at least one PRR signaling pathway is intact, an Icm/Dot-dependent signal can be detected. It is clear that in bone marrow-derived macrophages capable of TLR signaling, Icm/Dot-dependent activation of NF-κB occurs in the absence of Nod1 or Rip2, even under conditions of low multiplicity challenge with L. pneumophila (Losick and Isberg, 2006; Losick et al., 2010). However, in HEK293T cells, which do not express TLRs that efficiently engage L. pneumophila, knockdown of Nod1 reduces NF-κB activation (Losick et al., 2010). These results complement results indicating that Icm/Dot-dependent NF-κB activation in the absence of MyD88 is only seen when Rip2 is present (Shin et al., 2008). Therefore, crosstalk between Icm/Dot translocated substrates and PAMP signaling must exist, but the source of the PAMP or the site in the cell that PAMP signaling is initiated do not appear to be critical (Losick et al., 2010). TLR engagement acts together with Icm/Dot in cells lacking Rip2 signaling, while similarly, Nod signaling collaborates with Icm/Dot in cells that lack the TLR pathway. This indicates that in the case of collaboration with Icm/Dot, the NLR, and TLR pathways could be redundant.
The significant impact of Icm/Dot on the activation of NF-κB led to two investigations to identify translocated substrates that could directly induce activation of this protein. NF-κB can be stimulated as a response to many different cellular insults, with ER stress and disruption of the actin cytoskeleton being two important examples (Nemeth et al., 2004; Ahn and Aggarwal, 2005; Schroder, 2008). Using similar approaches, two laboratories have identified Icm/Dot translocated substrates that are able to induce an NF-κB reporter when ectopically expressed in HEK293T cells. As described above, LegK1 was one of these substrates (Ge et al., 2009). In addition, there were a number of activators that showed modest activation of the reporter (Ge et al., 2009; Losick et al., 2010) as well as another strong inducer, the translocated substrate LnaB (Losick et al., 2010). When bacteria are grown into post-exponential phase, LnaB was shown to be required to fully activate the NF-κB reporter after challenge of HEK293T cells with L. pneumophila (Losick et al., 2010). The C-terminal coiled coil domain of LnaB was required for NF-κB induction. This suggests that this domain of LnaB interacts with a protein in the signaling cascade upstream of NF-κB or it may contribute to a cellular activity that increases NF-κB signaling. However, the role of LnaB in a cellular process leading directly or indirectly to NF-κB activation is not known (Losick et al., 2010). Although there is no evidence that LegK1 induces NF-κB after L. pneumophila challenge (Losick et al., 2010), as described above, in vitro experiments demonstrate a direct interaction with the signaling cascade upstream of NF-κB (Ge et al., 2009), and its pathway of activation could be very different from that observed with LnaB.
Besides LnaB and LegK1, five Icm/Dot translocated inhibitors of host translation exhibited NF-κB inducing activity accompanied by a distinctive transcriptional response including IL23a and Csf2 induction (Fontana et al., 2011). It is possible that many Icm/Dot translocated substrates may lead to activation of the NF-κB pathway via a stress response rather than directly modulating the activity of proteins that regulate the nuclear translocation of this protein. The L. pneumophila proteins Lgt1, Lgt2, Lgt3, SidI, and SidL are known to cause an inhibition of host translation. This interferes with the synthesis of the unstable IκB inhibitory protein, which releases cytoplasmic NF-κB, allowing subsequent translocation into the nucleus (Hayden and Ghosh, 2008; Fontana et al., 2011). This inhibition of host protein synthesis caused by Icm/Dot substrates appears to be a key to causing sustained activation of NF-κB that can synergize with either TLR or Rip2-dependent signaling, as a L. pneumophila mutant lacking several translocated substrates that act as protein synthesis inhibitors is defective for NF-κB activation (Fontana et al., 2011; Figure 3).
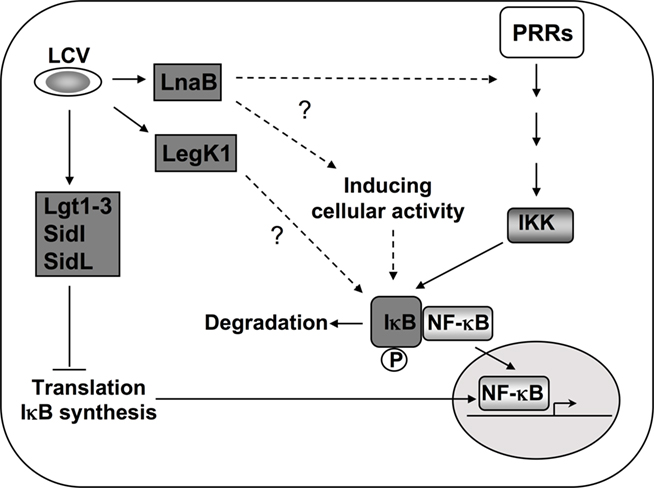
Figure 3. Activation of NF-κB during L. pneumophila infection. In addition to PRR mediated activation, Icm/Dot translocated substrates induce NF-κB by multiple mechanisms. During induction of the NF-κB pathway, the inhibitor IκB is phosphorylated by IKK and degraded, leading to nuclear translocation of the transcription factor (Hayden and Ghosh, 2008). Five Dot/Icm translocated substrates are known to target this pathway by inhibiting host translation (Fontana et al., 2011). This inhibition of translation interferes with the synthesis of the unstable inhibitor IκB and frees NF-κB subunits to translocate into the nucleus (Hayden and Ghosh, 2008; Fontana et al., 2011). The Ser/Thr kinase LegK1 has the ability to act directly on the NF-κB pathway by phosphorylating IκB (Ge et al., 2009), resulting in its degradation. The translocated protein LnaB activates NF-κB by an as yet unknown mechanism (Losick et al., 2010).
Map Kinases and Dual Specificity Phosphatases
In addition to the NF-κB pathway, which is only found in multicellular eukaryotes, the MAP kinase pathway is a second component of the innate immune system that is targeted during L. pneumophila infection (Welsh et al., 2004; Losick and Isberg, 2006; Shin et al., 2008; Li et al., 2009). The natural amebal hosts employ the MAP kinase pathway, so there is good reason to believe that selective pressures for effective interaction with MAP kinases must have taken place to facilitate intracellular replication of the bacterium. MAPKs regulate diverse cellular processes such as gene expression, cytoskeletal integrity, cell death, mitosis, and the induction of inflammatory mediators (Johnson and Lapadat, 2002; Jeffrey et al., 2007; Pullikuth and Catling, 2007; Huang et al., 2009). A cascade of sequentially active kinases, MAPKKKs and MAPKKs, activate MAPKs by threonine and tyrosine phosphorylation (Johnson and Lapadat, 2002; Huang et al., 2009). Activated MAPKs in turn phosphorylate specific substrates such as transcription factors, other kinases, or cytoskeletal proteins (Johnson and Lapadat, 2002). The four best characterized families of MAPKs found in higher eukaryotes, ERK, JNK, p38, and ERK5 respond to various stimuli, and are activated by specific MAPKKKs, resulting in both signal and target-specific responses (Johnson and Lapadat, 2002; Huang et al., 2009). MAPK signaling can be induced by activation of TLRs or NLRs as well as other stress response signals (Johnson and Lapadat, 2002; Huang et al., 2009). Linkage of NLRs to MAPKs occurs via Rip2 and Card9 while TLR signaling to MAPKs is MyD88-dependent (Hsu et al., 2007; Liu et al., 2007; Ting et al., 2010).
In order to ensure a proper balance of activation, MAPKs are regulated on a variety of levels including elaborate feedback loops and spatial separation of signaling (Jeffrey et al., 2007). After activation by the MAP kinase relay, inactivation occurs by dephosphorylation of Thr and Tyr residues of MAPKs by DUSPs (Lang et al., 2006; Jeffrey et al., 2007). DUSP family members are under tight control both at the transcriptional level and by post-translational modifications (Patterson et al., 2009) and in order to ensure specific targeting of MAPKs, they differ in their expression pattern and cellular localization (Lang et al., 2006; Jeffrey et al., 2007). Despite these differences, a common feature of many DUSPs is their transcriptional activation downstream from MAPK signaling, providing important feedback control of MAPK activation. In addition to transcriptional control, DUSP protein stability is influenced by MAPKs (Jeffrey et al., 2007).
Unlike the NF-κB pathway, MAPKs are also found in lower eukaryotes such as yeast and ameba (Molina et al., 2010). D. discoideum contains two enzymes similar to the mammalian ERK family (Gaskins et al., 1994; Segall et al., 1995). In D. discoideum, ERK-1 is phosphorylated shortly after L. pneumophila challenge with either a wild type or a Icm/Dot-deficient mutant (Li et al., 2009). This activation is transient with peak activation 1-h post infection. Inactivation of ERK-1 most likely relies on the tyrosine kinase/dual specificity phosphatase DupA, as there is constitutive activation of the MAPK in strains lacking DupA. The correct temporal regulation of ERK-1 activation has a significant impact on intracellular growth and host gene expression. In a mutant lacking DupA, intracellular replication of L. pneumophila is impaired, and this accompanied by hyperphosphorylation of ERK-1 relative to wild type amebae (Li et al., 2009). The resulting transcriptional response in cells having hyperactivated ERK-1 included over 500 misregulated genes that were also impacted in wild type amebae after challenge with L. pneumophila. Interestingly, these genes include those encoding proteins hypothesized to play a role in the amebal response to pathogens (Li et al., 2009).
As in amebae, MAPK activation was also observed as a response to infection in murine bone marrow-derived macrophages (Shin et al., 2008). Here, ERK is activated independently of Icm/Dot and in the absence of the TLR signaling. Activation of p38 and SAPK/JNK follows a different pattern from mammalian ERK and is composed of an initial MyD88-dependent, Icm/Dot-independent component, as well as a delayed prolonged MyD88-independent component that relies on Icm/Dot. The kinetic data are reminiscent of MAPK activation in amebae, in that p38 and SAPK/JNK activation peaks at 1-h post infection and continues for 4 h. The high and sustained MAPK activity in response to L. pneumophila is necessary for increased cytokine production and requires MyD88-dependent, Icm/Dot-independent and MyD88-independent, Icm/Dot-dependent signaling. As Icm/Dot is required to fully activate MAPK signaling it was proposed that translocated substrates may play a role in this process. This idea was supported by data showing that the pore forming activity of the type IV secretion system alone is not sufficient to activate p38 and SAPK/JNK (Shin et al., 2008). Signaling through p38 and SAPK/JNK may involve Icm/Dot substrates that directly target the MAPK pathway or substrates that exhibit inducing activity by acting on a different host process and thereby indirectly activate MAPKs (Figure 4). However, the most likely Icm/Dot substrates containing Ser/Thr kinase domains, LegK1, Legk2, and LegK3, could be excluded as direct p38 activators (Shin et al., 2008). As described for NF-κB induction (Fontana et al., 2011), Icm/Dot translocated substrates that inhibit host protein synthesis could also explain the Icm/Dot-dependent mechanism of MAPK activation.
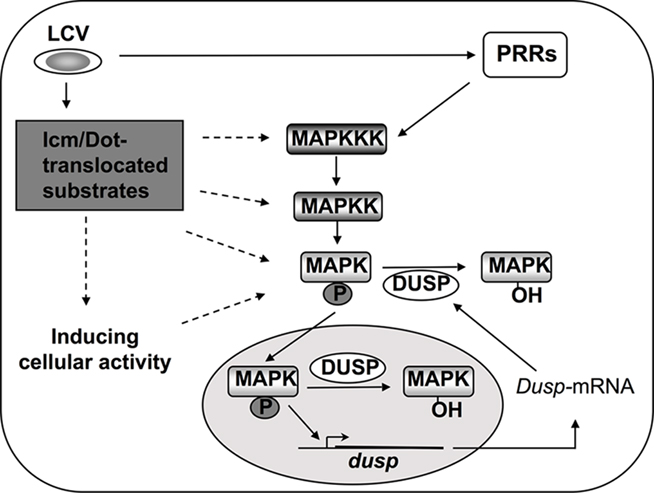
Figure 4. Mechanisms of MAPK activation during L. pneumophila infection. MAPKs are activated by sequentially induced kinases and in turn phosphorylate cellular and nuclear proteins such as transcription factors (Johnson and Lapadat, 2002; Huang et al., 2009). Inactivation of MAPKs results from dephosphorylation by dual specificity phosphatases (DUSPs; Lang et al., 2006; Jeffrey et al., 2007). DUSPs are regulated on many levels to ensure proper signaling through MAPKs, and MAPKs themselves can control DUSPs at the level of transcription and protein stability (Jeffrey et al., 2007). During L. pneumophila challenge of host cells, induction of the MAPK signaling pathway occurs through PRR signaling as well as by Icm/Dot-dependent activity in macrophages, and via unidentified sensors in amebae (Shin et al., 2008; Li et al., 2009). Whether Icm/Dot translocated substrates target kinases upstream of MAPK, or, whether interference with protein synthesis is sufficient to alter MAPK activity, is unknown (Shin et al., 2008).
As observed with DupA in amebae, DUSPs also play a role during infection in mammalian cells. An Icm/Dot-dependent increase in transcription of dusp genes was observed in the human macrophage-like U937 cell line (Losick and Isberg, 2006). In mouse bone marrow-derived macrophages, it was shown that Icm/Dot-dependent induction of dusp1 transcription did not require MyD88 or Rip2 kinase (Shin et al., 2008). Since dusp transcription is upregulated by MAPKs, this increase might be due to enhanced MAPK activity during infection. However, an increase of DUSP protein levels that would be expected to accompany increased transcription of the gene could not be observed, perhaps because of the presence of the L. pneumophila translocated substrates that interfere with protein synthesis in the host cell.
Mitogen-activated protein kinase activation appears to be a common response to L. pneumophila infection in lower and higher eukaryotes. Even though a direct participation of L. pneumophila proteins in MAPK activation seems likely, so far no Icm/Dot substrate was shown to act directly on the MAPK pathway. Concerning the function of DUSPs in regulating MAPKs during infection, the requirement of DUSPs to ensure proper balance of MAPK signaling is obvious from results in D. discoideum, as misregulation of ERK-1 interferes with intracellular growth of the bacterium (Li et al., 2009). The role of DUSPs in mammalian cells is not clear, however, especially since the induction of gene expression does not appear reflected in increased protein levels of DUSPs. In fact, the Icm/Dot-dependent induction of dusp transcription may be the result of MAPK activation caused by interference of host protein synthesis by L. pneumophila, so both transcriptional induction, and the lack of a translational response, are promoted by the same translocated substrates. Dusp expression is no longer elevated as a response to L. pneumophila infection in the absence of the five Icm/Dot translocated inhibitors of host translation, Lgt1, Lgt2, Lgt3, SidI, and SidL.
Concluding Remarks
Legionella pneumophila is known to interfere with many host cell processes such as the ubiquitination machinery (Kubori et al., 2008, 2010; Ivanov and Roy, 2009; Ensminger and Isberg, 2010; Price et al., 2010), host translation (de Felipe et al., 2005; Belyi et al., 2006, 2008), or vesicle trafficking (Murata et al., 2006; Ingmundson et al., 2007; Machner and Isberg, 2007). Here we have presented a selected overview of targeted host cell pathways that mediate signal transduction through changes in the phosphorylation state of proteins and lipids. As phosphorylation and dephosphorylation are among the most common modifications in cell signaling, L. pneumophila exploits host phosphorylation at all stages of infection. Targeting of the host phosphorylation machinery may involve direct modification of host factors by L. pneumophila proteins that act as kinases or phosphatases as well as sensing of cellular processes during infection that indirectly change the phosphorylation state of host proteins. In the case of PI metabolism, the importance of controlling flux and recruitment of this important lipid during intracellular replication in amebae was clearly demonstrated (Weber et al., 2006, 2009b; Ragaz et al., 2008; Brombacher et al., 2009; Peracino et al., 2010), whereas the role of PI3Ks during uptake of L. pneumophila is not yet resolved and differs depending on the model system (Khelef et al., 2001; Weber et al., 2006; Tachado et al., 2008; Charpentier et al., 2009; Peracino et al., 2010). The requirement of Icm/Dot translocation for NF-κB and MAPK activation implies that translocated substrates contribute to these regulatory changes in host cells. Besides the inducing activity of inhibitors of host translation, no other mechanism for this activation has been proposed based on in vivo studies, so in the future it will be very interesting to determine what other factors induce these pathways apart from PRR signaling. With the emerging knowledge on the function of Icm/Dot translocated substrates, details regarding how L. pneumophila manipulates major signaling pathways involving phosphorylation should be forthcoming shortly.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Elizabeth Creasey, Alexander Ensminger, and Sina Mohammadi for their review of the manuscript. Eva Haenssler was supported by the German Academic exchange service (DAAD). Ralph R. Isberg is an investigator at the Howard Hughes Medical Institute.
References
Abu-Zant, A., Jones, S., Asare, R., Suttles, J., Price, C., Graham, J., and Kwaik, Y. A. (2007). Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell. Microbiol. 9, 246–264.
Adkins, I., Koberle, M., Grobner, S., Bohn, E., Autenrieth, I. B., and Borgmann, S. (2007). Yersinia outer proteins E, H, P, and T differentially target the cytoskeleton and inhibit phagocytic capacity of dendritic cells. Int. J. Med. Microbiol. 297, 235–244.
Ahn, K. S., and Aggarwal, B. B. (2005). Transcription factor NF-κB: a sensor for smoke and stress signals. Ann. N. Y. Acad. Sci. 1056, 218–233.
Back, S. H., Scheuner, D., Han, J., Song, B., Ribick, M., Wang, J., Gildersleeve, R. D., Pennathur, S., and Kaufman, R. J. (2009). Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 10, 13–26.
Backert, S., Ziska, E., Brinkmann, V., Zimny-Arndt, U., Fauconnier, A., Jungblut, P. R., Naumann, M., and Meyer, T. F. (2000). Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell. Microbiol. 2, 155–164.
Banga, S., Gao, P., Shen, X., Fiscus, V., Zong, W. X., Chen, L., and Luo, Z. Q. (2007). Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc. Natl. Acad. Sci. U.S.A. 104, 5121–5126.
Bartfeld, S., Engels, C., Bauer, B., Aurass, P., Flieger, A., Bruggemann, H., and Meyer, T. F. (2009). Temporal resolution of two-tracked NF-κB activation by Legionella pneumophila. Cell. Microbiol. 11, 1638–1651.
Belyi, Y., Niggeweg, R., Opitz, B., Vogelsgesang, M., Hippenstiel, S., Wilm, M., and Aktories, K. (2006). Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl. Acad. Sci. U.S.A. 103, 16953–16958.
Belyi, Y., Tabakova, I., Stahl, M., and Aktories, K. (2008). Lgt: a family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J. Bacteriol. 190, 3026–3035.
Beresford, N., Patel, S., Armstrong, J., Szoor, B., Fordham-Skelton, A. P., and Tabernero, L. (2007). MptpB, a virulence factor from Mycobacterium tuberculosis, exhibits triple-specificity phosphatase activity. Biochem. J. 406, 13–18.
Berger, K. H., Merriam, J. J., and Isberg, R. R. (1994). Altered intracellular targeting properties associated with mutations in the Legionella pneumophila dotA gene. Mol. Microbiol. 14, 809–822.
Brandt, S., Kwok, T., Hartig, R., Konig, W., and Backert, S. (2005). NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. U.S.A. 102, 9300–9305.
Brombacher, E., Urwyler, S., Ragaz, C., Weber, S. S., Kami, K., Overduin, M., and Hilbi, H. (2009). Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 284, 4846–4856.
Bruggemann, H., Cazalet, C., and Buchrieser, C. (2006a). Adaptation of Legionella pneumophila to the host environment: role of protein secretion, effectors and eukaryotic-like proteins. Curr. Opin. Microbiol. 9, 86–94.
Bruggemann, H., Hagman, A., Jules, M., Sismeiro, O., Dillies, M. A., Gouyette, C., Kunst, F., Steinert, M., Heuner, K., Coppee, J. Y., and Buchrieser, C. (2006b). Virulence strategies for infecting phagocytes deduced from the in vivo transcriptional program of Legionella pneumophila. Cell. Microbiol. 8, 1228–1240.
Burstein, D., Zusman, T., Degtyar, E., Viner, R., Segal, G., and Pupko, T. (2009). Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 5, e1000508. doi: 10.1371/journal.ppat.1000508
Charpentier, X., Gabay, J. E., Reyes, M., Zhu, J. W., Weiss, A., and Shuman, H. A. (2009). Chemical genetics reveals bacterial and host cell functions critical for type IV effector translocation by Legionella pneumophila. PLoS Pathog. 5, e1000501. doi: 10.1371/journal.ppat.1000501
Clifton, D. R., Goss, R. A., Sahni, S. K., van Antwerp, D., Baggs, R. B., Marder, V. J., Silverman, D. J., and Sporn, L. A. (1998). NF-( B-dependent inhibition of apoptosis is essential for host cellsurvival during Rickettsia rickettsii infection. Proc. Natl. Acad. Sci. U.S.A. 95, 4646–4651.
Cohen, P. (2000). The regulation of protein function by multisite phosphorylation – a 25 year update. Trends Biochem. Sci. 25, 596–601.
Covacci, A., Censini, S., Bugnoli, M., Petracca, R., Burroni, D., Macchia, G., Massone, A., Papini, E., Xiang, Z., Figura, N., and Rappuoli, R. (1993). Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. U.S.A. 90, 5791–5795.
de Felipe, K. S., Glover, R. T., Charpentier, X., Anderson, O. R., Reyes, M., Pericone, C. D., and Shuman, H. A. (2008). Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 4, e1000117. doi: 10.1371/journal.ppat.1000117
de Felipe, K. S., Pampou, S., Jovanovic, O. S., Pericone, C. D., Ye, S. F., Kalachikov, S., and Shuman, H. A. (2005). Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J. Bacteriol. 187, 7716–7726.
Duronio, V. (2008). The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem. J. 415, 333–344.
Ensminger, A. W., and Isberg, R. R. (2010). E3 ubiquitin ligase activity and targeting of BAT3 by multiple Legionella pneumophila translocated substrates. Infect. Immun. 78, 3905–3919.
Farbrother, P., Wagner, C., Na, J., Tunggal, B., Morio, T., Urushihara, H., Tanaka, Y., Schleicher, M., Steinert, M., and Eichinger, L. (2006). Dictyostelium transcriptional host cell response upon infection with Legionella. Cell. Microbiol. 8, 438–456.
Fontana, M. F., Banga, S., Barry, K. C., Shen, X., Tan, Y., Luo, Z. Q., and Vance, R. E. (2011). Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 7, e1001289. doi: 10.1371/journal.ppat.1001289
Fritz, J. H., Ferrero, R. L., Philpott, D. J., and Girardin, S. E. (2006). Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 7, 1250–1257.
Gaskins, C., Maeda, M., and Firtel, R. A. (1994). Identification and functional analysis of a developmentally regulated extracellular signal-regulated kinase gene in Dictyostelium discoideum. Mol. Cell Biol. 14, 6996–7012.
Ge, J., Xu, H., Li, T., Zhou, Y., Zhang, Z., Li, S., Liu, L., and Shao, F. (2009). A Legionella type IV effector activates the NF-κB pathway by phosphorylating the IκB family of inhibitors. Proc. Natl. Acad. Sci. U.S.A. 106, 13725–13730.
Hauck, C. R., Gulbins, E., Lang, F., and Meyer, T. F. (1999). Tyrosine phosphatase SHP-1 is involved in CD66-mediated phagocytosis of Opa52-expressing Neisseria gonorrhoeae. Infect. Immun. 67, 5490–5494.
Hervet, E., Charpentier, X., Vianney, A., Lazzaroni, J. C., Gilbert, C., Atlan, D., and Doublet, P. (2011). The protein kinase LegK2 is a T4SS effector involved in endoplasmic reticulum recruitment and intracellular replication of Legionella pneumophila. Infect. Immun. doi: 10.1128/IAI.00805-10. [Epub ahead of print].
Horwitz, M. A. (1983a). Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158, 1319–1331.
Horwitz, M. A. (1983b). The Legionnaires’ disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J. Exp. Med. 158, 2108–2126.
Horwitz, M. A. (1984). Phagocytosis of the Legionnaires’ disease bacterium (Legionella pneumophila) occurs by a novel mechanism: engulfment within a pseudopod coil. Cell 36, 27–33.
Horwitz, M. A., and Maxfield, F. R. (1984). Legionella pneumophila inhibits acidification of its phagosome in human monocytes. J. Cell Biol. 99, 1936–1943.
Hsu, Y. M., Zhang, Y., You, Y., Wang, D., Li, H., Duramad, O., Qin, X. F., Dong, C., and Lin, X. (2007). The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 8, 198–205.
Huang, G., Shi, L. Z., and Chi, H. (2009). Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine 48, 161–169.
Huang, L., Boyd, D., Amyot, W. M., Hempstead, A. D., Luo, Z. Q., O’Connor, T. J., Chen, C., Machner, M., Montminy, T., and Isberg, R. R. (2010). The E Block motif is associated with Legionella pneumophila translocated substrates. Cell. Microbiol. 13, 227–245.
Ingmundson, A., Delprato, A., Lambright, D. G., and Roy, C. R. (2007). Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450, 365–369.
Ireton, K., Payrastre, B., Chap, H., Ogawa, W., Sakaue, H., Kasuga, M., and Cossart, P. (1996). A role for phosphoinositide 3-kinase in bacterial invasion. Science 274, 780–782.
Isberg, R. R., O’Connor, T. J., and Heidtman, M. (2009). The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24.
Ivanov, S. S., and Roy, C. R. (2009). Modulation of ubiquitin dynamics and suppression of DALIS formation by the Legionella pneumophila Dot/Icm system. Cell. Microbiol. 11, 261–278.
Jeffrey, K. L., Camps, M., Rommel, C., and Mackay, C. R. (2007). Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug Discov. 6, 391–403.
Johnson, G. L., and Lapadat, R. (2002). Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912.
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat. Immunol. 11, 373–384.
Khelef, N., Shuman, H. A., and Maxfield, F. R. (2001). Phagocytosis of wild-type Legionella pneumophila occurs through a wortmannin-insensitive pathway. Infect. Immun. 69, 5157–5161.
Krauss, M., and Haucke, V. (2007). Phosphoinositide-metabolizing enzymes at the interface between membrane traffic and cell signalling. EMBO Rep. 8, 241–246.
Kubori, T., Hyakutake, A., and Nagai, H. (2008). Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol. Microbiol. 67, 1307–1319.
Kubori, T., Shinzawa, N., Kanuka, H., and Nagai, H. (2010). Legionella metaeffector exploits host proteasome to temporally regulate cognate effector. PLoS Pathog. 6, e1001216. doi: 10.1371/journal.ppat.1001216
Kwok, T., Zabler, D., Urman, S., Rohde, M., Hartig, R., Wessler, S., Misselwitz, R., Berger, J., Sewald, N., Konig, W., and Backert, S. (2007). Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449, 862–866.
Laguna, R. K., Creasey, E. A., Li, Z., Valtz, N., and Isberg, R. R. (2006). A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U.S.A. 103, 18745–18750.
Lang, R., Hammer, M., and Mages, J. (2006). DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J. Immunol. 177, 7497–7504.
Li, Z., Dugan, A. S., Bloomfield, G., Skelton, J., Ivens, A., Losick, V., and Isberg, R. R. (2009). The amoebal MAP kinase response to Legionella pneumophila is regulated by DupA. Cell Host Microbe 6, 253–267.
Lindmo, K., and Stenmark, H. (2006). Regulation of membrane traffic by phosphoinositide 3-kinases. J. Cell Sci. 119, 605–614.
Liu, Y., Shepherd, E. G., and Nelin, L. D. (2007). MAPK phosphatases – regulating the immune response. Nat. Rev. Immunol. 7, 202–212.
Losick, V. P., Haenssler, E., Moy, M. Y., and Isberg, R. R. (2010). LnaB: a Legionella pneumophila activator of NF-κB. Cell. Microbiol. 12, 1083–1097.
Losick, V. P., and Isberg, R. R. (2006). NF-κB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203, 2177–2189.
Machner, M. P., and Isberg, R. R. (2007). A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science 318, 974–977.
Manning, G., Plowman, G. D., Hunter, T., and Sudarsanam, S. (2002a). Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 27, 514–520.
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002b). The protein kinase complement of the human genome. Science 298, 1912–1934.
Marra, A., Blander, S. J., Horwitz, M. A., and Shuman, H. A. (1992). Identification of a Legionella pneumophila locus required for intracellular multiplication in human macrophages. Proc. Natl. Acad. Sci. U.S.A. 89, 9607–9611.
Molina, M., Cid, V. J., and Martin, H. (2010). Fine regulation of Saccharomyces cerevisiae MAPK pathways by post-translational modifications. Yeast 27, 503–511.
Moorhead, G. B., De Wever, V., Templeton, G., and Kerk, D. (2009). Evolution of protein phosphatases in plants and animals. Biochem. J. 417, 401–409.
Mostowy, S., and Cossart, P. (2009). Cytoskeleton rearrangements during Listeria infection: clathrin and septins as new players in the game. Cell Motil. Cytoskeleton 66, 816–823.
Murata, T., Delprato, A., Ingmundson, A., Toomre, D. K., Lambright, D. G., and Roy, C. R. (2006). The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat. Cell Biol. 8, 971–977.
Nemeth, Z. H., Deitch, E. A., Davidson, M. T., Szabo, C., Vizi, E. S., and Hasko, G. (2004). Disruption of the actin cytoskeleton results in nuclear factor-κB activation and inflammatory mediator production in cultured human intestinal epithelial cells. J. Cell. Physiol. 200, 71–81.
Nolen, B., Taylor, S., and Ghosh, G. (2004). Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 15, 661–675.
Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., and Mann, M. (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648.
Patterson, K. I., Brummer, T., O’Brien, P. M., and Daly, R. J. (2009). Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem. J. 418, 475–489.
Peracino, B., Balest, A., and Bozzaro, S. (2010). Phosphoinositides differentially regulate bacterial uptake and Nramp1-induced resistance to Legionella infection in Dictyostelium. J. Cell Sci. 123, 4039–4051.
Philips, J. A. (2008). Mycobacterial manipulation of vacuolar sorting. Cell. Microbiol. 10, 2408–2415.
Pidoux, G., and Tasken, K. (2009). Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J. Mol. Endocrinol. 44, 271–284.
Price, C. T., Al-Khodor, S., Al-Quadan, T., and Abu Kwaik, Y. (2010). Indispensable role for the eukaryotic-like ankyrin domains of the ankyrin B effector of Legionella pneumophila within macrophages and amoebae. Infect. Immun. 78, 2079–2088.
Pullikuth, A. K., and Catling, A. D. (2007). Scaffold mediated regulation of MAPK signaling and cytoskeletal dynamics: a perspective. Cell. Signal. 19, 1621–1632.
Ragaz, C., Pietsch, H., Urwyler, S., Tiaden, A., Weber, S. S., and Hilbi, H. (2008). The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell. Microbiol. 10, 2416–2433.
Ribet, D., and Cossart, P. (2010). Post-translational modifications in host cells during bacterial infection. FEBS Lett. 584, 2748–2758.
Ryan, K. R., and Shapiro, L. (2003). Temporal and spatial regulation in prokaryotic cell cycle progression and development. Annu. Rev. Biochem. 72, 367–394.
Scott, J. D., and Pawson, T. (2009). Cell signaling in space and time: where proteins come together and when they’re apart. Science 326, 1220–1224.
Segal, E. D., Cha, J., Lo, J., Falkow, S., and Tompkins, L. S. (1999). Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 96, 14559–14564.
Segal, E. D., Falkow, S., and Tompkins, L. S. (1996). Helicobacter pylori attachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 1259–1264.
Segal, G., Purcell, M., and Shuman, H. A. (1998). Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc. Natl. Acad. Sci. U.S.A. 95, 1669–1674.
Segall, J. E., Kuspa, A., Shaulsky, G., Ecke, M., Maeda, M., Gaskins, C., Firtel, R. A., and Loomis, W. F. (1995). A MAP kinase necessary for receptor-mediated activation of adenylyl cyclase in Dictyostelium. J. Cell Biol. 128, 405–413.
Shaw, M. H., Reimer, T., Kim, Y. G., and Nunez, G. (2008). NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr. Opin. Immunol. 20, 377–382.
Shin, S., Case, C. L., Archer, K. A., Nogueira, C. V., Kobayashi, K. S., Flavell, R. A., Roy, C. R., and Zamboni, D. S. (2008). Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog. 4, e1000220. doi: 10.1371/journal.ppat.1000220
Stein, M., Bagnoli, F., Halenbeck, R., Rappuoli, R., Fantl, W. J., and Covacci, A. (2002). c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol. Microbiol. 43, 971–980.
Tachado, S. D., Samrakandi, M. M., and Cirillo, J. D. (2008). Non-opsonic phagocytosis of Legionella pneumophila by macrophages is mediated by phosphatidylinositol 3-kinase. PLoS ONE 3, e3324. doi: 10.1371/journal.pone.0003324
Ting, J. P., Duncan, J. A., and Lei, Y. (2010). How the noninflammasome NLRs function in the innate immune system. Science 327, 286–290.
Toker, A., and Cantley, L. C. (1997). Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387, 673–676.
Vergne, I., Chua, J., Lee, H. H., Lucas, M., Belisle, J., and Deretic, V. (2005). Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 102, 4033–4038.
Viboud, G. I., and Bliska, J. B. (2005). Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59, 69–89.
Vogel, J. P., Andrews, H. L., Wong, S. K., and Isberg, R. R. (1998). Conjugative transfer by the virulence system of Legionella pneumophila. Science 279, 873–876.
Walburger, A., Koul, A., Ferrari, G., Nguyen, L., Prescianotto-Baschong, C., Huygen, K., Klebl, B., Thompson, C., Bacher, G., and Pieters, J. (2004). Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304, 1800–1804.
Weber, S. S., Ragaz, C., and Hilbi, H. (2009a). Pathogen trafficking pathways and host phosphoinositide metabolism. Mol. Microbiol. 71, 1341–1352.
Weber, S. S., Ragaz, C., and Hilbi, H. (2009b). The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell. Microbiol. 11, 442–460.
Weber, S. S., Ragaz, C., Reus, K., Nyfeler, Y., and Hilbi, H. (2006). Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2, e46. doi: 10.1371/journal.ppat.0020046
Keywords: Legionella, phosphatidylinositol 3-kinase, NF-κB, mitogen-activated protein kinase
Citation: Haenssler E and Isberg RR (2011) Control of host cell phosphorylation by Legionella pneumophila. Front. Microbio. 2:64. doi: 10.3389/fmicb.2011.00064
Received: 24 February 2011;
Paper pending published: 14 March 2011;
Accepted: 24 March 2011;
Published online: 04 April 2011.
Edited by:
Carmen Buchrieser, Pasteur Institute, FranceReviewed by:
Antje Flieger, Robert Koch Institute, GermanyHubert Hilbi, Max von Pettenkofer-Institute, Germany
Copyright: © 2011 Haenssler and Isberg. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Ralph R. Isberg, Department of Molecular Biology and Microbiology, Tufts University School of Medicine, 150 Harrison Avenue, Boston, MA 02111, USA. e-mail: ralph.isberg@tufts.edu